

Abstract
JAK2V617F is the most common mutation in patients with BCR-ABL negative myeloproliferative neoplasms (MPNs). The eradication of JAK2V617F hematopoietic stem cells (HSCs) is critical for achieving molecular remissions and cure. We investigate the distinct effects of two therapies, ruxolitinib (JAK1/2 inhibitor) and interferon-alpha (IFN-α), on the disease-initiating HSC population. Whereas ruxolitinib inhibits Stat5 activation in erythroid progenitor populations, it fails to inhibit this same pathway in HSCs. In contrast, IFN-α has direct effects on HSCs. Furthermore, STAT1 phosphorylation and pathway activation is greater after IFN-α stimulation in Jak2V617F murine HSCs with increased induction of reactive oxygen species, DNA damage and reduction in quiescence after chronic IFN-α treatment. Interestingly, ruxolitinib does not block IFN-α induced reactive oxygen species and DNA damage in Jak2V617F murine HSCs in vivo. This work provides a mechanistic rationale informing how pegylated IFN-α reduces JAK2V617F allelic burden in the clinical setting and may inform future clinical efforts to combine ruxolitinib with pegylated IFN-α in patients with MPN.
Introduction
Myeloproliferative neoplasms (MPNs) are clonal hematological malignancies that arise from mutations in the hematopoietic stem cell (HSC) compartment [1, 2]. JAK2V617F is the most common genetic mutation found in BCR-ABL negative MPN patients identified in ~95% of polycythemia vera (PV) patients, and ~50–60% of patients with essential thrombocythemia and primary myelofibrosis [3–6]. JAK2V617F causes constitutive JAK-STAT signaling, cytokine-independent growth of MPN hematopoietic cells and a bias towards erythroid differentiation [2, 4, 7]. JAK-STAT activation is common to all MPN pathogenesis [8]. MPN is initiated from and maintained by a pool of Jak2V617F mutated long-term HSCs (LT-HSCs) [7, 9, 10]. Eradication of these disease-initiating stem cells is required to achieve long-term disease remission or cure.
Treatment with the JAK1/2 inhibitor, ruxolitinib (Jakavi, Novartis) improves patient disease-related symptoms including relieving constitutional symptoms, decreasing splenomegaly, and reducing blood counts [11, 12]. However, ruxolitinib responses occur in both JAK2V617F mutant and JAK2-WT patients [12, 13]. Furthermore, patients with JAK2V617F show only minor reductions in mutant allelic burden, even after long-term treatment [11, 13–16], suggesting that ruxolitinib is unable to eradicate MPN disease-initiating cells.
Treatment with interferon-alpha (IFN-α) can induce hematological remissions, reduce JAK2V617F allelic burden, and can induce molecular remissions in some patients [17–20], indicating that IFN-α may preferentially deplete the MPN disease-initiating stem cell population. Pegylated versions of IFN-α, peg-IFN-α2a, and peg–IFN-α2b, with longer half-lives have been developed, clinically approved and are frequently better tolerated by patients.
Prior studies have shown that IFN-α has direct effects on HSCs and activates cell cycle in quiescent HSC populations leading to proliferation-associated functional exhaustion in wild-type (WT) mice [21, 22]. IFN-α induced proliferation is mediated through IFNα/β receptor coupled to JAK1 and STAT1-dependent signaling [21, 23]. Sustained stimulation by IFN-α leads to accumulation of DNA damage and reactive oxygen species (ROS) resulting in LT-HSC attrition [24].
We have previously shown that IFN-α preferentially depletes murine Jak2V617F (hereafter Jak2+/VF) LT-HSCs associated with increased exit from quiescence in a Jak2V617F-dependent manner [25]. There is also a growing body of evidence linking JAK2V617F with increased DNA damage, ROS production, replication fork stalling, and impairment in the intra-S checkpoint during cell cycle, suggesting a predisposition for IFN-α induced stem cell exhaustion [26].
The efficacy of ruxolitinib in the amelioration of disease-related symptoms and the preferential selectivity of IFN-α in the depletion of disease-initiating cells presents as a rational combination treatment in Jak2V617F-driven MPN. There are currently two clinical trials assessing the efficacy of this combination [27] (#EudraCT2013-003295-12; NCT02742324). Preliminary results indicate that 63% of PV patients achieved complete hematological response [28]. These promising preliminary clinical responses have generated interest in combining the therapies in the future.
We investigate the distinct effects of ruxolitinib and IFN-α on Jak2+/VF MPN disease-initiating HSCs. We demonstrate that effective targeting of Jak2+/VF LT-HSCs by pegylated IFN-α is linked to preferential activation of JAK1-STAT1 signaling in Jak2+/VF HSC downstream of IFN-α but requires sustained IFN-α pathway activation. Ruxolitinib did not eradicate Jak2+/VF LT-HSCs, nor did it effectively prevent JAK-STAT signaling in these cells. Although ruxolitinib may antagonize IFN-α induced JAK1 signaling, we show that ruxolitinib is not effective at targeting Jak2+/VF HSCs in vivo, and thus these two drugs target different cell populations, thereby permitting combination therapy that retains favorable activity of both treatments. These results deliver important insights into the distinct mechanisms of action of ruxolitinib and IFN-α and their potential to be used in combination for the treatment of MPN patients.
Methods
Mice
Jak2V617F knock-in mice, Jak2V617F/+; E2Acre+ (Jak2 +/VF) [7], and B6.Ifnar1 (hereafter AR1−/−) knockout mice have been described [29]. CD45.1 Ptprca and CD45.2 C57BL/6 J were obtained from Animal Resources Centre, Australia. Mice were maintained in pathogen-free facilities at the QIMR Berghofer Medical Research Institute, ethics protocol A11605M.
Generation of chimeric WT/Jak2+/VF MPN mice
Lethally irradiated male and female recipient mice C57BL/6JxPtprca.F1 (CD45.1/2 double positive) (2 × 5.5 Gy at least 3 h apart, 24 h prior to transplantation), received equal amounts of either Jak2+/VF or Jak2+/VF−;AR1−/− bone marrow (BM) post red cell lysis combined with equivalent amounts of CD45.1 Ptprca helper BM, total 2 × 106 cells per recipient via lateral tail vein. Mice were maintained on antibiotic (Baytril) water for 2 weeks. Hematocrit, blood parameters, and chimerism were measured 4 weeks after transplantation.
In vivo ruxolitinib and IFN-α treatment of mice
WT/Jak2+/VF chimeric mice were treated with ruxolitinib (INCB018424, Chemietek) at 60 mg/kg bis in die (bid) in dH2O [30], 5% N,N-Dimethylacetamide (Sigma) or vehicle by oral gavage for 4 weeks or ten doses as indicated in text.
IFN-α: Chimeric mice were treated with subcutaneous 50,000 IU murine IFN-α (Miltenyi Biotech) (0.1%BSA/PBS) and analyzed 48 h post treatment.
Ropeginterferon-α11 (mP1101, PharmaEssentia): WT/Jak2+/VF chimeric mice were treated with murine ropeginterferon-α (peg-IFN-α; 600 ng in 200 μl 0.1% BSA/PBS, subcutaneous injection), once a week for 4 weeks and analyzed 48 h after the last injection.
Blood count analysis and dissection
Peripheral blood (PB) was analyzed on the Hemavet (Model 950, Drew scientific). BM and spleen were homogenized through a 70 μm filter (Greiner-bio-one, Cat # 542070) and red blood cells lysed (BD Pharmlyse, Cat # 555899).
Flow cytometry analysis
All samples were analyzed by flow cytometry using FACS LSR Fortessa. Postacquisition analyses of data were performed with FlowJo software V10 (Treestar, CA). Percentage chimerism was defined as the proportion of Jak2+/VF cells as a percentage of total donor CD45 expressing cells. Calculation: (%Jak2+/VF)/(%Jak2+/VF + %WT) × 100. Population frequencies were calculated as a percentage of the respective donor CD45 expressing cells (WT-CD45.1; Jak2+/VF-CD45.2).
Details of antibodies and clones included in Supplementary Table 1. For cell cycle, γH2AX, and 2′,7′–dichlorofluorescein diacetate (DCFDA) (carboxy-H2DCFDA, life technologies) analysis BM cells were stained with the lineage cocktail and depleted of lineage positive cells by biotin-binder Dynabeads (Invitrogen)
Cell cycle and γH2AX analysis: lineage-depleted BM cells were stained with CD45.1, Sca1, cKIT, CD48 and CD150, and then fixed and permeabilized (Fix & Perm, GAS-004, Invitrogen). Samples were stained with anti-Ki-67 (B56, BD Bioscience) or γH2AX (S139)(20E3) during the permeabilization stage and then stained with Hoechst 33342 (Invitrogen) at a final concentration 20 μg/ml in 2% FCS/PBS.
DCFDA analysis: lineage-depleted BM cells were incubated with 10 μM DCFDA in 2% FCS/PBS for 45 min at 37°C, then stained with secondary antibodies for 15 min at room temperature, washed and resuspended in 1:5000 sytox blue in 2% FCS/PBS.
pSTAT1 analysis: WT or Jak2+/VF Lin- Sca1 + cKit + (LSKs) were isolated, cultured overnight in RPMI (Gibco; no FCS with mSCF (50 ng/ml), mTPO (10 ng/ml), and mIL-3 (10 ng/ml) (Peprotech), 37 °C, 5% CO2 in a 96 well V bottom plate (Costar) and treated for 30 min with 1000 U/ml IFN-α and/or 300 nM ruxolitinib (dissolved in DMSO; Sigma-Aldrich) for 30 min, stained for cell surface CD48, and intracellular pSTAT1. For in vivo ruxolitinib, LSKs were isolated 1 h after the last dose of ruxolitinib, cultured for 1 h and then stimulated with 1000 U/ml IFN-α for 30 min before staining.
LSKs were lysed and fixed (BD Phosflow Lyse/Fix Buffer, Cat # 558049) as per manufacturer’s instructions, washed (BD Pharmingen Stain Buffer, Cat # 554656), permeabilized with 150 μl of ice cold Perm III (BD Phosflow, Perm III, Cat # 558050) and incubated for 30 min on ice in the dark. Cells were stained in 30 μl staining buffer containing 1:20 dilution of STAT1 antibody for 45 min at room temperature.
pSTAT5 analysis: performed as previously described [31]. Lineage-depleted BM cells were starved for 30 min in 1% bovine serum albumin (BSA)/PBS ex vivo and then stimulated with either murine thrombopoietin (mTPO, PeproTech) at 100 ng/ml or murine interleukin-3 (mIL-3, PeproTech) at 20 ng/ml or vehicle (1% BSA in PBS) for 10 min at 37°C. The cells were then fixed and permeabilized (Fix & Perm, GAS-004, Invitrogen) then stained for intracellular pSTAT5. Median florescence intensity (MFI) fold change was normalized to the average MFI of WT vehicle unstimulated samples.
Immunofluorescence microscopy
WT and Jak2+/VF LT-HSCs (LSK+ CD48− CD150+) and MPPs (LSK+ CD48+) were isolated 48 h post treatment with 50,000 IU IFN-α or vehicle (0.1% BSA/PBS). Cells were resuspended in 30 μl PBS and placed onto polylysine slides (Thermo scientific, Menzel-Glaser Polysine slides). Cells were fixed for 15 min with 4% PFA/PBS; washed 2 × 5 min 4 °C PBS then 2 × 5 min room temperature PBS; permeabilized for 10 min 0.2% Triton/PBS; washed 2 × 5 min PBS; blocked 1 h PBS/1% FCS/0.1% Triton-X-100 at room temperature; stained γH2AX in PBS/1% FCS at 4 °C; washed 2 × 5 min PBS/1% FCS/0.1% Triton-X-100 (Sigma-Aldrich). Cells were then mounted with Prolong Gold antifade reagent with Dapi (Molecular probes Life Technologies). Cells were imaged using the Zeiss 780-NLO Point Scanning Confocal microscope. Foci were enumerated manually and a minimum of 40 cells were counted per sample. Cells with greater than three foci were considered as having greater γH2AX expression compared with baseline.
Gene expression analysis
We analysed previously published microarray data GSE44961 [25] on Jak2VF and WT HSC (CD150+LKS+) from IFNα or vehicle-treated controls (4 weeks of IFNα, n = 4 in each group). In brief, data were analysed in R. Signal intensities were quantile normalized and log2 transformed and were used for heatmaps and GSEA analysis. Differential expression analysis was performed utilizing linear modeling and empirical bayes approach as implemented in the limma R package [32].
Statistical analysis
Statistical analysis was performed with Prism, Student’s t test, Mann–Whitney test for two groups or one-way ANOVA, multiple comparison post-Tukey test for comparison of more than two groups. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001. Statistically significant differences are marked on the graphs to facilitate comparison. Group sizes were calculated to detect 20% difference in median parameters with 80% power, alpha 0.05. Biological replicates were separated by cage number on a nonblinded randomized basis.
Results
Ruxolitinib depletes erythroid precursor cells but does not reduce Jak2V617F allelic burden
In order to characterize the effects of ruxolitinib on Jak2+/VF vs. WT HSC, we generated mixed WT/Jak2+/VF chimeric mice (Supplementary Fig. 1A). These chimeras display an MPN phenotype similar to the disease observed in primary donors with enlarged spleens and elevated hemoglobin. Ruxolitinib treatment (60 mg/kg, bid) [30, 33] for 4 weeks resulted in reduced splenic extramedullary hematopoiesis and BM cellularity (Fig. 1a, Supplementary Fig. 1B). PB analysis revealed fewer monocytes and T-cells but no change in leukocyte count, B-cell numbers, hemoglobin concentration, platelet count or Jak2+/VF allelic burden (Fig. 1b, c, Supplementary Fig. 1C). We reasoned that the prolonged life span of murine erythrocytes (average of 40 days [34]) may mask acute effects of ruxolitinib; therefore, we examined erythroid precursor cells [35]. Ruxolitinib treatment reduced PB red cell precursor reticulocyte frequency (Fig. 1d, Supplementary Fig. 1D), and also reduced erythroid progenitor cells in the spleen and to a lesser extent in the BM (Fig. 1e, Supplementary Fig. 1E–G; RI to RIV using CD71 and Ter119 [35]). The profound effects on splenic erythroid elements suggest that ruxolitinib is effective at targeting Jak2V617F-driven extramedullary erythropoiesis. These findings were present after only 1 week of ruxolitinib (total of ten doses), by which time ruxolitinib has achieved pharmacokinetic steady state [33]. A mild increase in apoptosis was noted in late stage erythroid progenitors from BM and spleen (Fig. 1f, Supplementary Fig. 1H–I).
Fig. 1.
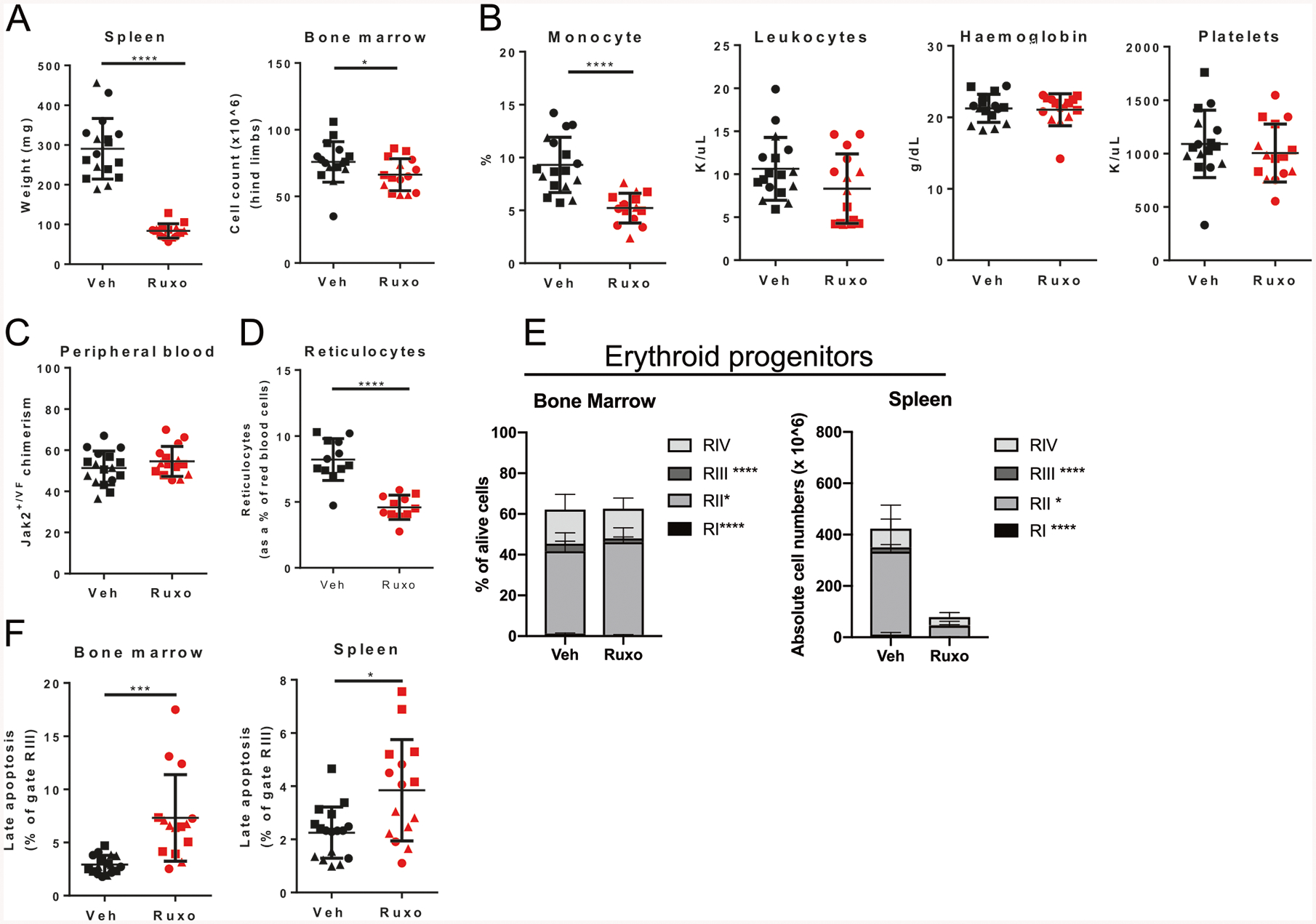
Ruxolitinib depletes erythroid progenitor cells but does not reduce Jak2V617F allelic burden. WT/Jak2+/VF chimeric mice treated for 4 weeks with vehicle (Veh) or ruxolitinib (Ruxo) demonstrating a spleen weight and BM cellularity, b PB leukocyte count, hemoglobin, monocytes, platelets, c Jak2+/VF chimerism and d reticulocytes. WT/Jak2+/VF chimeric mice treated with ten doses of vehicle or ruxolitinib demonstrating frequency of e erythroid progenitors in BM and spleen and f apoptotic RIII erythroid progenitors in the BM and spleen. Each point represents a biological replicate. Statistics are shown for relevant comparisons mentioned in text. Pooled data from three experiments, each experiment identified with a different symbol shape. Student’s t test, *p < 0.05, ***p < 0.001, ****p < 0.0001
Ruxolitinib does not deplete Jak2+/VF MPN disease-initiating LT-HSCs
MPN disease-initiating cells reside exclusively within the immunophenotypically defined LT-HSC population [9] (Supplementary Fig. 2A). Ruxolitinib treatment effected a reduction in WT LSKs and a trend towards reduction in Jak2+/VF LSK cells, in particular WT MPP and ST-HSC populations (Fig. 2a, Supplementary Fig. 2B), but did not alter WT or Jak2+/VF LT-HSC frequencies, nor did it alter committed myeloid progenitor cell populations (Fig. 2a, Supplementary Fig. 2C).
Fig. 2.
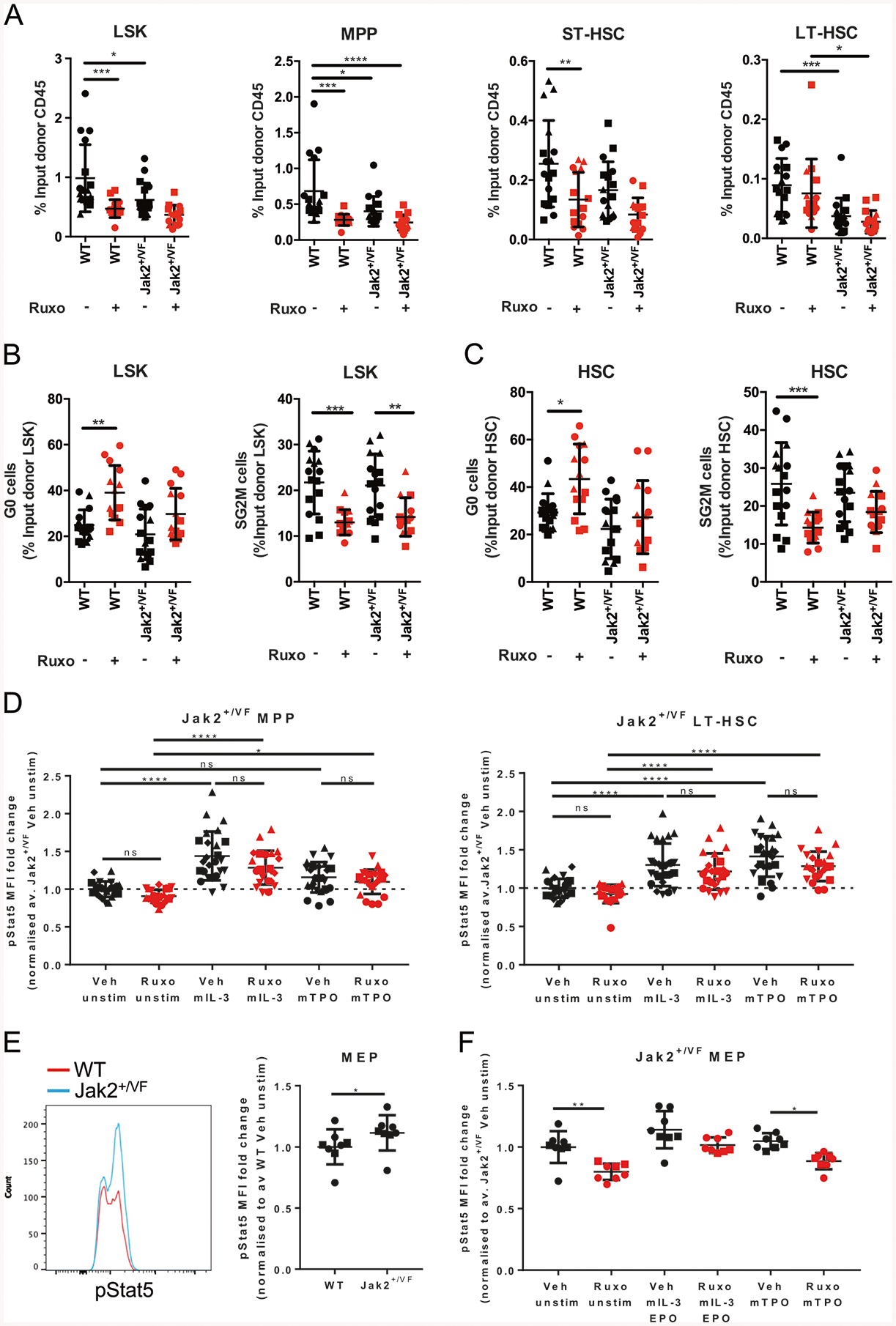
Ruxolitinib does not affect Jak2 MPN disease-initiating LT-HSCs. WT/Jak2+/VF chimeric mice were treated for 4 weeks with vehicle or ruxolitinib. a Frequency of BM WT and Jak2+/VF LSKs, MPPs, ST-HSCs, and LT-HSCs expressed as a percentage of the respective input donor CD45+ population (WT-CD45.1, Jak2+/VF−CD45.2). b Frequency of quiescent (G0) and cycling (SG2M) WT and Jak2+/VF LSKs and c WT and Jak2+/VF HSCs (LSK+ CD150+). d WT/Jak2+/VF chimeric mice were treated with ten doses of vehicle or ruxolitinib (60 mg/kg bid). Mean fluorescence intensity (MFI) of phosphorylated STAT5 (pSTAT5) in Jak2+/VF MPPs and LT-HSCs with or without ex vivo stimulation by murine IL-3 or TPO, expressed as fold change over the vehicle (Veh) unstimulated (unstim) control average (av.). e Representative basal phosphorylated STAT5 histograms of WT and Jak2+/VF bone marrow MEPs and MFI calculated, expressed as fold change over the WT average. f MFI of phosphorylated STAT5 of bone marrow Jak2+/VF MEPs+/− in vivo ruxolitinib and +/− ex vivo stimulation with mIL-3 or mTPO, expressed as fold change over the Veh unstim control average. Each point represents a biological replicate. Pooled data from three experiments for 4 weeks ruxolitinib treatment, and pooled data from five experiments for pSTAT5. Each experiment identified with a different symbol shape. Statistics are shown for relevant comparisons mentioned in text. One-way ANOVA, post-Tukey test, * p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
Both WT and Jak2+/VF LSKs showed reduction in actively cycling cells (cells in S/G2/M) after ruxolitinib treatment when compared with vehicle control (Fig. 2b, Supplementary Fig. 2D). Regarding the HSC compartment, increased quiescence and decreased cycling were only observed in WT HSCs after ruxolitinib when compared with vehicle control, whereas only a trend towards reductions were observed in the Jak2+/VF HSCs treated with ruxolitinib when compared with vehicle (LKS+ CD150+, Fig. 2c, Supplementary Fig. 2D). Together, these results indicate that ruxolitinib has preferential effects on differentiated myeloid cells vs. HSC populations, and limited capacity to target Jak2+/VF HSCs.
Constitutive or hyperactive STAT5 signaling through mutant JAK2V617F signaling is required for the development of JAK2V617F MPN [4, 36–38]. pSTAT5 was induced in Jak2+/VF LT-HSCs and MPPs after stimulation with mTPO and mIL-3 (Fig. 2d), and ruxolitinib was unable to inhibit ex vivo mIL-3 or mTPO-induced pSTAT5 induction in LT-HSC populations (Fig. 2d). We hypothesized that ruxolitinib may be inhibiting STAT5 phosphorylation in differentiated megakaryocyte-erythroid progenitors (MEPs), which are expanded in Jak2+/VF mice [7]. Jak2+/VF MEPs showed mildly increased basal STAT5 phosphorylation (Fig. 2e), and ruxolitinib treatment reduced basal Jak2+/VF MEP STAT5 phosphorylation and also reduced pSTAT5 levels after TPO stimulation (Fig. 2f). These data show that ruxolitinib preferentially targets differentiated progenitor cells, while sparing more primitive LT-HSC and MPPs populations in the BM.
IFN-α induces ROS and DNA damage in Jak2+/VF LT-HSCs
In contrast to ruxolitinib, IFN-α treatment has been shown to reduce JAK2V617F allelic burden in MPN patients [17]. In addition, we have shown that murine Jak2+/VF LT-HSCs are depleted by IFN-α treatment [25]. IFN-α treatment induces WT HSC exhaustion through induction of cell cycle [21, 22] and there are strong links between regulation of ROS and maintenance of quiescence [24, 39]. Interestingly, some models of JAK2V617F-driven MPN have increased DNA damage and ROS but this has not been assessed in our model [26, 40–42]. We therefore sought to determine whether the preferential depletion of Jak2+/VF HSCs by IFN-α may be due to stem cell exhaustion and the accumulation of DNA damage during replicative stress.
We investigated the effects of acute IFN-α, 48 h post administration in WT/Jak2+/VF chimeric mice. Treatment of chimeric mice with subcutaneous 50 K IU murine IFN-α resulted in a dramatic increase in WT and Jak2+/VF MPP frequency but did not alter WT or Jak2+/VF LT-HSC frequency (Fig. 3a, Supplementary Fig. 3A). No effects were observed for Jak2+/VF-AR1−/− cells indicating that the increase in MPP frequency is a cell autonomous response to IFN-α (Fig. 3a). IFN-α treatment reduced MPP and LT-HSC quiescence, and IFN-α treated LT-HSCs had increased accumulation in G1 phase (Fig. 3b, Supplementary Fig. 3B–C). The response to a single dose of IFN-α was similar between WT and Jak2+/VF genotypes (Fig. 3b, Supplementary Fig. 3B–C).
Fig. 3.
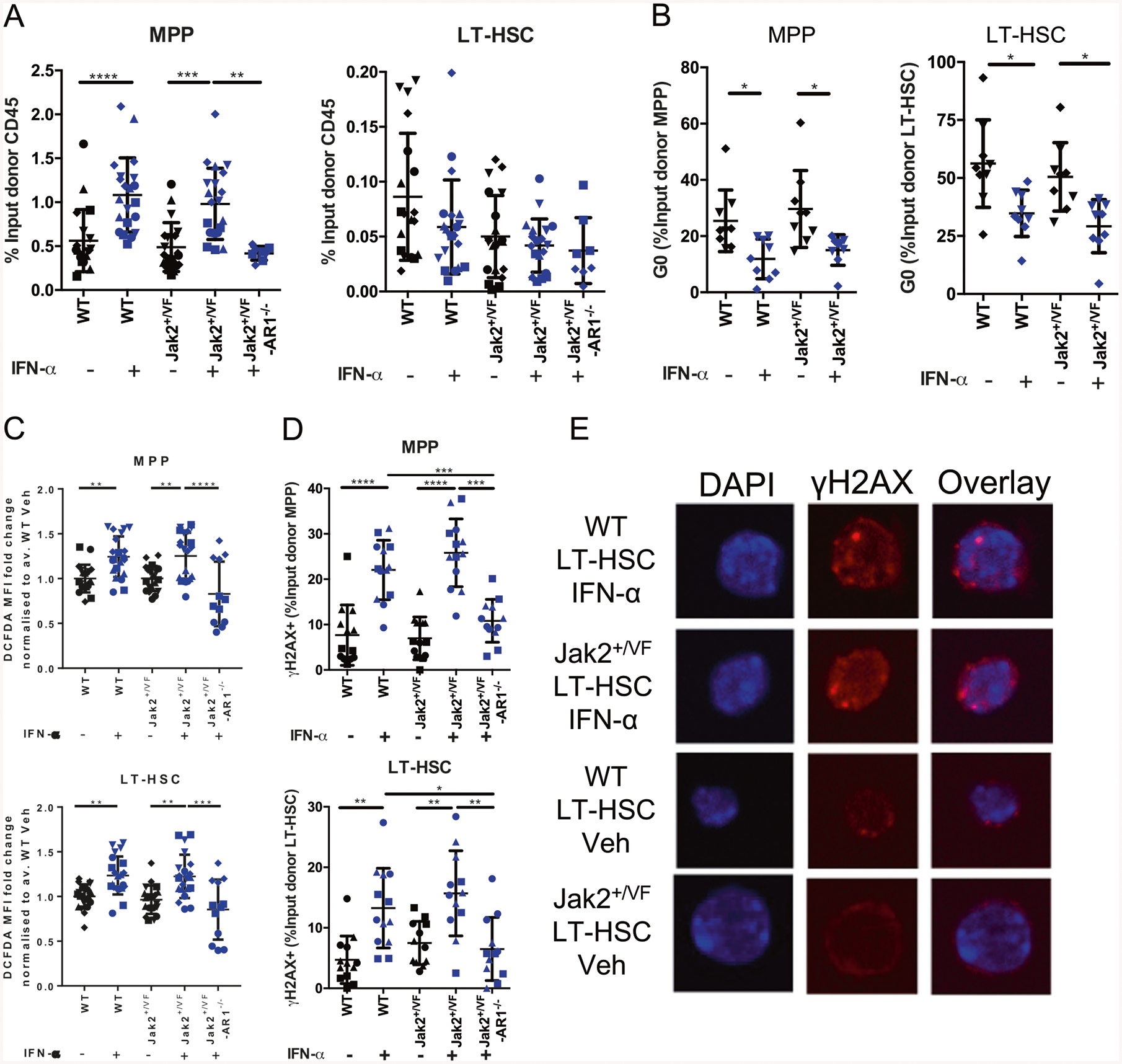
IFN-α induces ROS and DNA damage in Jak2+/VF LT-HSCs. WT/Jak2+/VF chimeric mice treated with 50 K IU murine recombinant IFN-α or vehicle and bone marrow HSPCs analyzed 48 h post injection. a Frequency of BM WT and Jak2+/VF MPPs (LSK+ CD48+) and LT-HSCs (LSK+ CD48−) expressed as a percentage of the respective input donor CD45+ population (WT-CD45.1, Jak2+/VF-CD45.2). b G0, G1, and SG2M cell cycle status of MPPs, and LT-HSCs. c ROS production for MPPs and LT-HSCs shown as DCFDA MFI, expressed as fold change over WT vehicle (Veh) control average. d Frequency of γH2AX+ MPPs and LT-HSCs as determined by flow cytometry. e Immunofluorescence microscopy visualization of γH2AX foci in vehicle and IFN-α treated LT-HSCs. Each point represents a biological replicate. Pooled data from 3 to 5 experiments, each experiment identified with a different symbol shape. Statistics are shown for relevant comparisons mentioned in text. One-way ANOVA, post Tukey test, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
ROS accumulation has been reported after activation of IFN-α signaling in WT HSC populations leading to DNA damage and functional decline of HSCs [24, 43–48]. Consistent with these studies, we observed increases in ROS in IFN-α treated WT and Jak2+/VF MPPs and LT-HSCs compared with vehicle and Jak2+/VF-AR1−/− controls, indicating that ROS accumulation was a cell autonomous response to IFN-α treatment (Fig. 3c, Supplementary Fig. 3D). Again, there were no differences between WT and Jak2+/VF genotypes regarding ROS production in IFN-α or vehicle-treated samples (Fig. 3c, Supplementary Fig. 3D).
IFN-α treated WT and Jak2+/VF MPPs and LT-HSCs showed increased proportion of γH2AX positive cells, indicating the accumulation of DNA damage, compared with vehicle controls and compared with Jak2+/VF-AR1−/− LT-HSCs (Fig. 3d, Supplementary Fig. 3E). In addition, an increase in γH2AX foci was observed in the nuclei of IFN-α treated WT and Jak2+/VF LT-HSCs compared with vehicle (Fig. 3e, Supplementary Fig. 3F), with IFN-α treated WT and Jak2+/VF LT-HSCs having an increased proportion of cells with >3 foci per cell when compared with vehicle controls (Supplementary Fig. 3F). Again, across both assays, no difference in γH2AX accumulation was observed between WT and Jak2+/VF IFN-α treated LT-HSCs.
Altogether, these data show that Jak2+/VF and WT HSPCs exhibit similar kinetics after acute IFN-α treatment, with loss of quiescence, increased ROS production and increased DNA damage.
Jak2+/VF LT-HSCs exhibit STAT1 hypersensitivity after IFN-α treatment
We next sought to determine whether IFN-α signaling pathways are altered between Jak2+/VF and WT HSCs [49]. IFN-α-induced proliferation of HSC and progenitor cells is STAT1 dependent [21], and we have previously observed baseline activation of STAT1 gene expression pathways in Jak2+/VF HSCs (Fig. 4a) [25]. Interestingly, pSTAT1 activation downstream of IFN-α was enhanced in Jak2+/VF LT-HSCs compared with WT LT-HSCs, and this was not observed in MPPs (Fig. 4b, Supplementary Fig. 3G). As we have demonstrated that acute IFN-α has similar effects on WT and Jak2+/VF LT-HSCs, we hypothesized that chronic IFN-α treatment may be required to observe differential responses in WT and Jak2+/VF LT-HSCs. This is consistent with the need for long term IFN-α treatment to achieve molecular responses in patients with JAK2V617F MPN. Unlike other models of JAK2V617F induced MPN [26, 42], we did not observe increased DNA damage at baseline in the Jak2V617F knock-in mouse model (Fig. 3d, e). However, we did observe enrichment of pathways regulating DNA repair and oxidative phosphorylation in chronic IFN-α treated Jak2+/VF compared with IFN-α treated WT HSCs (Fig. 4c). Furthermore, analysis of microarray data revealed that the pathways regulating DNA repair were different between Jak2+/VF and WT, but also highly regulated by IFN-α treatment (including recombinatorial repair, nucleotide excision repair, base excision repair, and nonhomologous end joining) (Fig. 4d, Supplementary Fig. 4–5; Supplementary Table 2). This indicates an increased sensitivity or primed state of Jak2+/VF LT-HSCs to IFN-α induced DNA damage and exhaustion at the transcriptional level. The differential expression analysis comparing the effect of IFN-α within and across Jak2+/VF and WT CD150+ LSK is provided in Supplementary Table 2. Of note, pathways regulating cell adhesion were more highly enriched in WT HSCs post IFN-α treatment (Supplementary Table 3). More specifically Cxcr4 was more highly expressed in WT LT-HSCs (Supplementary Table 2), which has the potential to influence the mobilization of LT-HSCs.
Fig. 4.
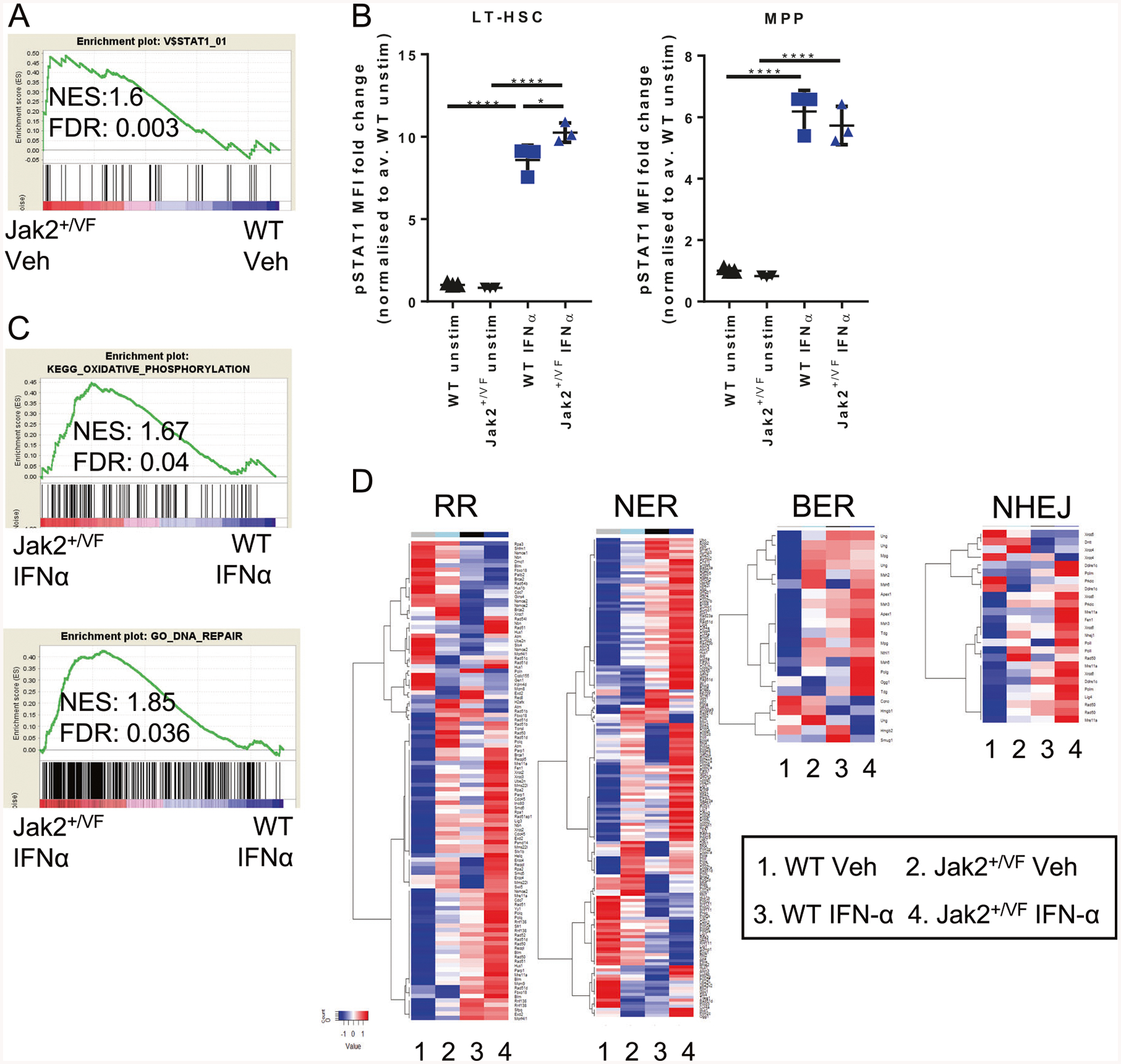
Jak2+/VF LT-HSCs exhibit STAT1 hypersensitivity after IFN-α. a Microarray analysis of WT and Jak2+/VF HSCs (LSK CD150+) shows enrichment of STAT1 signaling in Jak2+/VF HSCs. b Sorted WT and Jak2+/VF LSKs were rested overnight and then treated +/− ex vivo with 1000 U/ml murine recombinant IFN-α and stained for CD48 and intracellular phosphorylated STAT1. MFI of phosphorylated STAT1 in ex vivo treated LT-HSCs and MPPs, expressed as fold change over WT vehicle (Veh) treated unstimulated control average. c Microarray was performed on HSCs (LSK CD150+) isolated from WT/Jak2+/VF chimeric mice treated with 10 K IU murine unmodified IFN-α or vehicle per day for 4 weeks. Jak2+/VF IFN-α treated HSCs are enriched for oxidative phosphorylation and DNA repair gene signatures compared with IFN-α treated WT HSCs. d Heatmaps showing averaged gene expression of genes involved in DNA repair pathways in WT and Jak2+/VF vehicle treated and IFN-α treated HSCs. Each point represents a biological replicate. One-way ANOVA, post-Tukey test, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
Chronic treatment with peg-IFN-α preferentially targets Jak2+/VF LT-HSCs through activation of cell cycle, induction of ROS and the accumulation of DNA damage
We next examined a pegylated form of murine IFN-α (murine ropeginterferon-α, PharmaEssentia (peg-IFN-α)) with prolonged biological half-life. Four weeks of peg-IFN-α treatment resulted in reduced spleen weight, no change in BM cellularity, reduced PB leukocytes, monocytes, and mature T and B-cells; red blood cell count and hematocrit (Fig. 5a, b, Supplementary Fig. 6A–C). Eight weeks of peg-IFN-α treatment resulted in pronounced reduction in hematocrit and white cell count together with reduction in PB Jak2+/VF chimerism, consistent with clinical data suggesting reduction of JAK2+/VF allelic burden requires sustained IFN-α treatment [17] (Fig. 5c, Supplementary Fig. 6D). PB reticulocytes and erythroid progenitor populations in the BM were reduced (Fig. 5d, Supplementary Fig. 6E), and there was reduced extramedullary erythropoiesis (Fig. 5d, Supplementary Fig. 6F). BM LSKs were dramatically expanded in peg-IFN-α treated mice and Jak2+/VF MPPs showed a greater expansion compared with WT MPPs (Fig. 5e, Supplementary Fig. 6G–H) together with a trend to a decrease Jak2+/VF and WT LT-HSC frequencies (Fig. 5e, Supplementary Fig. 6G).
Fig. 5.

Chronic treatment with peg-IFN-α reduces peripheral disease parameters and Jak2V617F allelic burden. WT/Jak2+/VF chimeric mice treated weekly for 4 weeks with peg-IFN-α (600 ng) or vehicle (0.1% BSA/PBS) demonstrating a spleen weights, b PB leukocyte count and hematocrit. WT/Jak2+/VF chimeric mice treated weekly for 8 weeks with peg-IFN-α (600 ng) or vehicle (0.1% BSA/PBS) demonstrating c Jak2+/VF chimerism in the peripheral blood of the vehicle and peg-IFN-α group. WT/Jak2+/VF chimeric mice treated weekly for 4 weeks with peg-IFN-α (600 ng) or vehicle (0.1% BSA/PBS) demonstrating d frequency of erythroid progenitors (RI, RII, RIII, and RIV) in bone marrow and spleen and e frequency of MPPs and LT-HSCs. Each point represents a biological replicate. Pooled data from two experiments, each experiment identified with a different symbol shape. Statistics are shown for relevant comparisons mentioned in text. One-way ANOVA, post-Tukey test, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
Peg-IFN-α treated WT and Jak2+/VF MPPs showed reduced quiescence, increased ROS production and DNA damage compared with vehicle controls (Fig. 6a–c, Supplementary Fig. 7A–C). Peg-IFN-α treated WT and Jak2+/VF MPPs showed similar effects (Fig. 6a–c), however importantly peg-IFN-α treated Jak2+/VF LT-HSCs showed greater reduction in quiescence and greater accumulation in G1 phase, higher ROS production and more DNA damage compared with peg-IFN-α treated WT LT-HSCs (Fig. 6d–f). Peg-IFN-α treated Jak2+/VF MPPs and LT-HSCs demonstrate increased cell cycling (SG2M) compared with vehicle (Fig. 6a, d).
Fig. 6.

Chronic treatment with peg-IFN-α preferentially targets Jak2+/VF LT-HSCs through activation of cell cycle, induction of ROS and the accumulation of DNA damage. WT/Jak2+/VF chimeric mice treated weekly for 4 weeks with peg-IFN-α (600 ng) or vehicle (0.1%BSA/PBS) demonstrating a G0,G1, and SG2M cell cycle status of MPPs, b ROS production for MPPs shown as DCFDA MFI fold change, normalized to average WT vehicle (Veh) treated MFI, c frequency of γH2AX+ MPPs, d G0, G1, and SG2M cell cycle status of LT-HSCs, including representative flow cytometry plots of cell cycle distribution. e ROS production for LT-HSCs shown as DCFDA MFI, expressed as fold change over WT Veh average, including representative DCFDA histograms. f frequency of γH2AX+ LT-HSCs, including representative γH2AX histograms. Each point represents a biological replicate. Pooled data from two experiments, each experiment identified with a different symbol shape. Statistics are shown for relevant comparisons mentioned in text. One-way ANOVA, post-Tukey test, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
Altogether, these data show that Jak2+/VF and WT LT-HSCs have similar kinetics of activation and DNA damage after acute IFN-a treatment; however there is enhanced downstream activation of STAT1 phosphorylation by IFN-α in Jak2+/VF LT-HSCs. Chronic treatment with peg-IFN-α has a greater effect on cell cycle, DNA damage and ROS within Jak2+/VF LT-HSCs compared with WT LT-HSCs. These data provide mechanistic explanation underlying molecular responses in JAK2V617F MPN patients after long-term treatment with peg-IFN-α.
IFN-α and ruxolitinib target different cell populations in vivo
IFN-α and ruxolitinib are both highly effective treatments for MPN, and preliminary results from a clinical trial investigating ruxolitinib and IFN-α treatment combination in PV patients have shown complete hematological remissions in 63% of patients [28]. These data provide a rationale for combining these two drugs in the future, however a potential caveat to this combination arises from IFN-α dependency on activation of JAK1 signaling. Hypothetically, ruxolitinib (a JAK1/2 kinase inhibitor) may antagonize the effects of IFN-α in vivo.
In order to determine the capacity for antagonism of these two compounds, we firstly investigated the effect of ex vivo combination ruxolitinib and IFN-α on STAT1 phosphorylation. Ex vivo stimulation with IFN-α increased pSTAT1 in WT and Jak2+/VF MPPs and LT-HSCs compared with unstimulated controls (Fig. 7a). Again, pSTAT1 levels were higher in Jak2+/VF compared with WT LT-HSCs when stimulated with IFN-α (Fig. 7a). Ex vivo ruxolitinib treatment completely abrogated ex vivo IFN-α induced pSTAT1 upregulation in both MPPs and LT-HSCs (Fig. 7a), consistent with on target inhibition of JAK1 downstream of IFNAR1.
Fig. 7.

IFN-α and ruxolitinib target different cell populations in vivo. a Phosphorylated STAT1 MFI in ex vivo treated MPPs and LT-HSCs, expressed as fold change over WT vehicle (Veh) control average, including representative histograms for LT-HSCs. b Phosphorylated STAT1 MFI in ex vivo treated MPPs and LT-HSCs from WT/Jak2+/VF chimeric mice treated with in vivo ruxolitinib, expressed as fold change over WT Veh unstimulated control average. c Experimental schema for combination Ruxolitinib and IFN-α treatment of WT/Jak2+/VF chimeric mice, demonstrating d frequency of Jak2+/VF LT-HSCs, e G0 and SG2M cell cycle status of Jak2+/VF LT-HSCs, f Jak2+/VF LT-HSC ROS production as measured by DCFDA MFI, expressed as fold change over IFN-α treated average. g Frequency of γH2AX+ Jak2+/VF LT-HSCs. Each point represents a biological replicate. Pooled data from two experiments for pSTAT1 analysis and pooled data from 3 to 4 experiments for combination treatment analysis. Each experiment identified with a different symbol shape. Unequal numbers caused by a mechanical failure of blood analyser during one experiment. Statistics are shown for relevant comparisons mentioned in text. One-way ANOVA, post-Tukey test, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
We next tested whether in vivo ruxolitinib could inhibit IFN-α induced pSTAT1. MPPs and LT-HSCs from vehicle-treated mice that were stimulated with IFN-α showed increased STAT1 phosphorylation compared with unstimulated controls (Fig. 7b). In vivo treatment with ruxolitinib prior to IFN-α stimulation resulted in incomplete inhibition of STAT1 phosphorylation in Jak2+/VF LT-HSCs compared with vehicle treated (Fig. 7b). These results are in line with our previous data demonstrating that ruxolitinib has minimal effects on LT-HSCs (Fig. 2d). The complete inhibition of JAK-STAT signaling after ruxolitinib ex vivo treatment suggests that the inability to inhibit signaling in vivo relates to inadequate intracellular drug concentrations in LT-HSC, rather than cell autonomous resistance or drug efflux.
Finally, we evaluated the effects of ruxolitinib treatment on IFN-α induced HSC proliferation, ROS accumulation and DNA damage. For these experiments, WT/Jak2+/VF chimeric mice were treated with 8–10 doses of vehicle or ruxolitinib (60 mg/kg bid) together with a single dose of IFN-α (50 K IU per mouse; Fig. 7c).
Combined treatment with IFN-α and ruxolitinib in vivo had no effect on Jak2+/VF LT-HSC frequency (Fig. 7d).Ruxolitinib only partially attenuated IFN-α induced loss of quiescence in Jak2+/VF LT-HSCs (Fig. 7e), again demonstrating that LT-HSC pSTAT1 signaling was unable to be completely blocked by ruxolitinib. Furthermore, ruxolitinib did not inhibit IFN-α induced ROS in Jak2+/VF LT-HSCs (Fig. 7f), or IFN-α induced increased γH2AX expression in Jak2+/VF LT-HSCs (Fig. 7g).
Altogether, these results demonstrate that ruxolitinib is a partial inhibitor of IFN-α signaling. However, in vivo treatment with ruxolitinib is unlikely to reach sufficient concentrations in LT-HSCs to inhibit JAK-STAT signaling or be detrimental to downstream consequences of IFN-α induced cell cycle activation and DNA damage induction.
We show no evidence of functional antagonism between ruxolitinib and IFN-α in vivo, a finding that has clinical relevance for the ongoing clinical use of this therapeutic combination.
Discussion
Treatment of MPN patients with either the JAK1/2 inhibitor ruxolitinib or IFN-α can result in hematological remissions; however long-term IFN-α treatment has consistently been shown to reduce in JAK2V617F allelic burden [11, 13, 17, 18, 50]. Our results show that ruxolitinib has maximal effects on peripheral erythroid precursor cells and minimal effects on the Jak2+/VF disease-initiating HSC compartment. In contrast, IFN-α can directly target the MPN disease-initiating population and chronic treatment with murine ropeginterferon-α selectively targets Jak2+/VF HSCs via activation from quiescence, induction of ROS and accumulation of DNA damage.
We demonstrate that 4 weeks treatment with ruxolitinib or IFN-α reduced the release of immature red cells (reticulocytes) into the PB. However the long life span of red cells masks the effects on total red cell mass [34]. Consistent with this assumption, hematological remissions may take upward of 32 weeks of ruxolitinib treatment [14].
Prolonged treatment with long-acting pegylated IFN-α was able to induce hematological remissions and also reduction in Jak2+/VF chimerism, consistent with clinical studies that demonstrate a reduction in JAK2+/VF allelic burden may take many months of continuous treatment.
LT-HSCs are notoriously hard to target with therapeutics because they primarily reside in a quiescent state in order to preserve their self-renewal capacity and to protect themselves from replicative and genotoxic stress [51]. Nevertheless, studies have shown that ~95% of ruxolitinib is absorbed after oral administration and ruxolitinib is not a substrate of MDR p-glycoprotein [52, 53]. The use of ruxolitinib to target the disease-initiating mutation in Jak2VF-driven MPN has parallels with the use of imatinib in the treatment of chronic myeloid leukemia. Complete cytogenetic responses are achieved in the vast majority of CML patients treated with the ABL kinase inhibitor imatinib. However, in most patients a population of BCR-ABL expressing primitive stem cells persists even after chronic therapy. Studies have suggested that CML stem cells do not depend on BCR-ABL kinase activity for survival, only their proliferative advantage [54, 55] and may persist after imatinib treatment. Similarly, it is possible that Jak2+/VF LT-HSCs may not require constitutively active Jak2 activity for their maintained survival.
Long-acting versions of IFN-α can induce molecular remissions in MPN patients [12–14, 17, 18]. We have previously shown that long-term treatment with IFN-α preferentially depletes murine Jak2+/VF LT-HSCs [56], consistent with the trend towards reduction observed in response to chronic peg-IFN-α administration and reduced chimerism in longer-term studies. More recent studies have now linked the acquisition of DNA damage and ROS to functional attrition of stem cells [24]. Our results indicate an increased sensitivity or primed state of Jak2+/VF LT-HSCs to IFN-α induced exhaustion at the transcriptional level with baseline enrichment for STAT1 targets, DNA damage and mitochondrial activation— related gene expression in Jak2+/VF HSCs compared with WT HSCs [40, 41, 57]. Importantly, while acute IFN-α treatment induces cell cycle, DNA damage and ROS production in WT and Jak2+/VF HSCs and progenitors, chronic peg-IFN-α treatment is necessary for preferential reduction in quiescence, increased accumulation in G1 phase, ROS production and DNA damage in Jak2+/VF LT-HSCs compared with WT LT-HSCs, and is consistent with previously published data using unmodified IFN-α [25]. Others have shown that recurrent activation of interferon signaling, induced by poly I:C injection, leads to attenuation of HSC cell cycling response [58]. By using a weekly pegylated IFN-α schedule, this allows HSCs more time to recover between each injection and may help prevent this resistance. As chronic stem cell activation is required, this supports increased replication stress as the mechanism behind Jak2+/VF LT-HSC depletion. These data are in line with previous studies implicating replication stress as a critical factor in the functional decline of stem cells [43] and the observations that JAK2V617F cells have increased replication fork stalling [26].
We also observed sustained production of ROS in LT-HSCs with chronic IFN-α treatment. It would be of interest to examine if ROS scavengers are able to prevent the effects of murine ropeginterferon-α treatment [24]. Of greater clinical interest would be to examine whether activation of ROS pathways could synergize with peg-IFN-α, or whether peg-IFN-α might be combined with drugs that prevent DNA repair, such as the PARP inhibitors [59]. Such an approach has been hypothesized for combination treatment with ruxolitinib and PARP inhibitors [59]; however, we did not observe increased DNA damage in LT-HSCs with ruxolitinib treatment. Current clinical trials are evaluating the effectiveness of the combination of peg-IFN-α with ruxolitinib in MPN patients. Early results indicate that this combination is highly effective in reducing allelic burden (Blood 2018 132: A581), however toxicity has been a limiting factor in published trials [27]. This research will help inform ongoing clinical research that seeks to develop the most effective implementation strategies for these highly effective therapeutic agents, ruxolitinib and IFN-α, in the treatment of MPN patients.
Supplementary Material
Acknowledgements
We are grateful for the assistance of the QIMR Berghofer animal house, flow cytometry facility and business development office. We gratefully acknowledge the support of the MPN Research Foundation, MPN Alliance of Australia, CSL Centenary Fellowship, NHMRC, Gordon and Jessie Gilmour Trust, Cure Cancer Australia Foundation (SWL) and Leukaemia Foundation of Australia (SWL and RA). Murine Ropeginterferon-α (mP1101) was a gift from PharmaEssentia.
Footnotes
Conflict of interest SWL has participated in advisory boards for Novartis. Pegylated murine IFN-α (mP1101) was a gift from Pharmaessentia. FHH has served as a consultant for and has received research funding from Novartis Inc. SJL, NTC, and CWL are employees of PharmaEssentia Co. The other authors declare that they have no conflict of interest.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Ishii T, Bruno E, Hoffman R, Xu M. Involvement of various hematopoietic-cell lineages by the JAK2V617F mutation in polycythemia vera. Blood. 2006;108:3128–34. [DOI] [PubMed] [Google Scholar]
- 2.Jamieson CH, Gotlib J, Durocher JA, Chao MP, Mariappan MR, Lay M, et al. The JAK2 V617F mutation occurs in hematopoietic stem cells in polycythemia vera and predisposes toward erythroid differentiation. Proc Natl Acad Sci USA. 2006;103:6224–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365: 1054–61. [DOI] [PubMed] [Google Scholar]
- 4.James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–8. [DOI] [PubMed] [Google Scholar]
- 5.Kralovics R, Passamonti F, Buser AS, Teo S-S, Tiedt R, Passweg JR, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. New Engl J Med. 2005;352:1779–90. [DOI] [PubMed] [Google Scholar]
- 6.Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387–97. [DOI] [PubMed] [Google Scholar]
- 7.Mullally A, Lane SW, Ball B, Megerdichian C, Okabe R, Al-Shahrour F, et al. Physiological Jak2V617F expression causes a lethal myeloproliferative neoplasm with differential effects on hematopoietic stem and progenitor cells. Cancer Cell. 2010;17:584–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mullally A, Lane SW, Brumme K, Ebert BL. Myeloproliferative neoplasm animal models. hematology/oncology. Clinics. 2012;26:1065–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mullally A, Poveromo L, Schneider RK, Al-Shahrour F, Lane SW, Ebert BL. Distinct roles for long-term hematopoietic stem cells and erythroid precursor cells in a murine model of Jak2V617F-mediated polycythemia vera. Blood. 2012; 120:166–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reinisch A, Thomas D, Corces MR, Zhang X, Gratzinger D, Hong WJ, et al. A humanized bone marrow ossicle xeno transplantation model enables improved engraftment of healthy and leukemic human hematopoietic cells. Nat Med. 2016;22:812–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deininger M, Radich J, Burn TC, Huber R, Paranagama D, Verstovsek S. The effect of long-term ruxolitinib treatment on JAK2p.V617F allele burden in patients with myelofibrosis. Blood. 2015;126:1551–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. New Engl J Med. 2012;366:799–807. 2012/03/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harrison C, Kiladjian J-J, Al-Ali HK, Gisslinger H, Waltzman R, Stalbovskaya V, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. New Engl J Med. 2012;366:787–98. [DOI] [PubMed] [Google Scholar]
- 14.Vannucchi AM, Kiladjian JJ, Griesshammer M, Masszi T, Durrant S, Passamonti F, et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. New Engl J Med. 2015;372:426–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vannucchi AM, Verstovsek S, Guglielmelli P, Griesshammer M, Burn TC, Naim A, et al. Ruxolitinib reduces JAK2 p.V617F allele burden in patients with polycythemia vera enrolled in the RESPONSE study. Ann Hematol. 2017;96:1113–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harrison CN, Vannucchi AM, Kiladjian JJ, Al-Ali HK, Gisslinger H, Knoops L, et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia. 2016;30:1701–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kiladjian JJ, Cassinat B, Chevret S, Turlure P, Cambier N, Roussel M. Pegylated interferon-alfa-2a induces complete hematologic and molecular responses with low toxicity in polycythemia vera. Blood. 2008;112:3065–72. [DOI] [PubMed] [Google Scholar]
- 18.Quintas-Cardama A, Kantarjian H, Manshouri T, Luthra R, Estrov Z, Pierce S. Pegylated interferon alfa-2a yields high rates of hematologic and molecular response in patients with advanced essential thrombocythemia and polycythemia vera. J Clin Oncol. 2009;27:5418–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Them NC, Bagienski K, Berg T, Gisslinger B, Schalling M, Chen D, et al. Molecular responses and chromosomal aberrations in patients with polycythemia vera treated with peg-proline-interferon alpha-2b. Am J Hematol. 2015;90:288–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stauffer Larsen T, Iversen KF, Hansen E, Mathiasen AB, Marcher C, Frederiksen M, et al. Long term molecular responses in a cohort of Danish patients with essential thrombocythemia, polycythemia vera and myelofibrosis treated with recombinant interferon alpha. Leuk Res. 2013;37:1041–5. [DOI] [PubMed] [Google Scholar]
- 21.Essers MA, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, et al. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature. 2009;458:904–8. [DOI] [PubMed] [Google Scholar]
- 22.Sato T, Onai N, Yoshihara H, Arai F, Suda T, Ohteki T. Interferon regulatory factor-2 protects quiescent hematopoietic stem cells from type I interferon-dependent exhaustion. Nat Med. 2009;15:696–700. [DOI] [PubMed] [Google Scholar]
- 23.Kleppe M, Spitzer MH, Li S, Dong L, Papalexi E, Hill C, et al. JAK1 as a convergent regulator of hematopoietic stem cell function and stress hematopoiesis. Blood. 2016;128:722. [Google Scholar]
- 24.Walter D, Lier A, Geiselhart A, Thalheimer FB, Huntscha S, Sobotta MC, et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature. 2015;520:549–52. [DOI] [PubMed] [Google Scholar]
- 25.Mullally A, Bruedigam C, Poveromo L, Heidel FH, Purdon A, Vu T, et al. Depletion of Jak2V617F myeloproliferative neoplasm propagating stem cells by interferon-alpha in a murine model of polycythemia vera. Blood. 2013;121:3692–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen E, Ahn JS, Massie CE, Clynes D, Godfrey AL, Li J, et al. JAK2V617F promotes replication fork stalling with disease-restricted impairment of the intra-S checkpoint response. Proc Natl Acad Sci. 2014;111:15190–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mikkelsen SU, Kjaer L, Bjorn ME, Knudsen TA, Sorensen AL, Andersen CBL, et al. Safety and efficacy of combination therapy of interferon-alpha2 and ruxolitinib in polycythemia vera and myelofibrosis. Cancer Med. 2018;7:3571–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mikkelsen SU, Kjær L, Skov V, Bjørn ME, Andersen CL, Bjerrum OW, et al. Safety and efficacy of combination therapy of interferon-alpha2 + JAK1–2 inhibitor in the philadelphia-negative chronic myeloproliferative neoplasms. Preliminary results from the Danish combi-trial—an open label, single arm, nonrandomized multicenter phase ii study. Blood. 2015;126:824. [Google Scholar]
- 29.Swann JB, Hayakawa Y, Zerafa N, Sheehan KCF, Scott B, Schreiber RD, et al. Type I IFN contributes to NK cell homeostasis, activation, and antitumor function. J Immunol. 2007;178:7540–9. 2007. [DOI] [PubMed] [Google Scholar]
- 30.Bhagwat N, Koppikar P, Keller M, Marubayashi S, Shank K, Rampal R, et al. Improved targeting of JAK2 leads to increased therapeutic efficacy in myeloproliferative neoplasms. Blood. 2014;123:2075–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vu T, Austin R, Paine Kuhn C, Bruedigam C, Song A, Guignes S, et al. Jak2V617F driven myeloproliferative neoplasm occurs independently of interleukin-3 receptor beta common signaling. Haematologica 2016;101:e77–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3. 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 33.Shi JG, Chen X, McGee RF, Landman RR, Emm T, Lo Y, et al. The pharmacokinetics, pharmacodynamics, and safety of orally dosed INCB018424 phosphate in healthy volunteers. J Clin Pharmacol. 2011;51:1644–54. [DOI] [PubMed] [Google Scholar]
- 34.Van Putten LM, Croon F. The life span of red cells in the rat and the mouse as determined by labeling with DFP32 in vivo. Blood. 1958;13:789. [PubMed] [Google Scholar]
- 35.Koulnis M, Pop R, Porpiglia E, Shearstone JR, Hidalgo D, Socolovsky M Identification and analysis of mouse erythroid progenitors using the CD71/TER119 flow-cytometric assay. J Vis Exp. 2017. 10.3791/55305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kato Y, Iwama A, Tadokoro Y, Shimoda K, Minoguchi M, Akira S, et al. Selective activation of STAT5 unveils its role in stem cell self-renewal in normal and leukemic hematopoiesis. J Exp Med. 2005;202:169–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yan D, Hutchison RE, Mohi G. Critical requirement for Stat5 in a mouse model of polycythemia vera. Blood. 2012;119:3539–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rampal R, Al-Shahrour F, Abdel-Wahab O, Patel JP, Brunel JP, Mermel CH, et al. Integrated genomic analysis illustrates the central role of JAK-STAT pathway activation in myeloproliferative neoplasm pathogenesis. Blood. 2014;123:e123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tothova Z, Kollipara R, Huntly BJ, Lee BH, Castrillon DH, Cullen DE, et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128:325–39. [DOI] [PubMed] [Google Scholar]
- 40.Marty C, Lacout C, Droin N, Le Couedic JP, Ribrag V, Solary E, et al. A role for reactive oxygen species in JAK2 V617F myeloproliferative neoplasm progression. Leukemia. 2013;27:2187–95. [DOI] [PubMed] [Google Scholar]
- 41.Plo I, Nakatake M, Malivert L, de Villartay JP, Giraudier S, Villeval JL, et al. JAK2 stimulates homologous recombination and genetic instability: potential implication in the heterogeneity of myeloproliferative disorders. Blood. 2008;112:1402–12. [DOI] [PubMed] [Google Scholar]
- 42.Chen E, Ahn JS, Sykes DB, Breyfogle LJ, Godfrey AL, Nangalia J, et al. RECQL5 suppresses oncogenic JAK2-induced replication stress and genomic instability. Cell Rep. 2015;13:2345–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Flach J, Bakker ST, Mohrin M, Conroy PC, Pietras EM, Reynaud D, et al. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature. 2014;512:198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barzilai A, Yamamoto K. DNA damage responses to oxidative stress. DNA Repair. 2004;3:1109–15. [DOI] [PubMed] [Google Scholar]
- 45.Tasdogan A, Kumar S, Allies G, Bausinger J, Beckel F, Hofemeister H, et al. DNA damage-induced HSPC malfunction depends on ros accumulation downstream of ifn-1 signaling and bid mobilization. Cell Stem Cell. 2017;20:415. [DOI] [PubMed] [Google Scholar]
- 46.Miyamoto K, Araki KY, Naka K, Arai F, Takubo K, Yamazaki S, et al. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell. 2007;1:101–12. [DOI] [PubMed] [Google Scholar]
- 47.Eliasson P, Jönsson J-I. The hematopoietic stem cell niche: Low in oxygen but a nice place to be. J Cell Physiol. 2010; 222:17–22. [DOI] [PubMed] [Google Scholar]
- 48.Ito K, Hirao A, Arai F, Matsuoka S, Takubo K, Hamaguchi I, et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature. 2004;431:997–1002. [DOI] [PubMed] [Google Scholar]
- 49.Czech J, Cordua S, Weinbergerova B, Baumeister J, Crepcia A, Han L, et al. JAK2V617F but not CALR mutations confer increased molecular responses to interferon-α via JAK1/STAT1 activation. Leukemia. 2019;33:995–1010. [DOI] [PubMed] [Google Scholar]
- 50.Ianotto JC, Chauveau A, Boyer-Perrard F, Gyan E, Laribi K, Cony-Makhoul P, et al. Benefits and pitfalls of pegylated interferon-alpha2a therapy in patients with myeloproliferative neoplasm-associated myelofibrosis: a French Intergroup of Myeloproliferative neoplasms (FIM) study. Haematologica. 2018;103:438–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boulais PE, Frenette PS. Making sense of hematopoietic stem cell niches. Blood. 2015;125:2621–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ostojic A, Vrhovac R, Verstovsek S. Ruxolitinib: a new JAK1/2 inhibitor that offers promising options for treatment of myelofibrosis. Future Oncol. 2011;7:1035–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.HIGHLIGHTS OF PRESCRIBING INFORMATION: JAKAFI™ (ruxolitinib) tablets, for oral use. 2011. [cited 2018 10/01/2018]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/202192lbl.pdf.
- 54.Corbin AS, Agarwal A, Loriaux M, Cortes J, Deininger MW, Druker BJ. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J Clin Invest. 2011;121:396–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chu S, McDonald T, Lin A, Chakraborty S, Huang Q, Snyder DS, et al. Persistence of leukemia stem cells in chronic myelogenous leukemia patients in prolonged remission with imatinib treatment. Blood. 2011;118:5565–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mullally A, Bruedigam C, Poveromo L, Heidel FH, Purdon A, Vu T, et al. Depletion of Jak2V617F myeloproliferative neoplasmpropagating stem cells by interferon-α in a murine model of polycythemia vera. Blood. 2013;121:3692–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li J, Spensberger D, Ahn JS, Anand S, Beer PA, Ghevaert C, et al. JAK2 V617F impairs hematopoietic stem cell function in a conditional knock-in mouse model of JAK2 V617F-positive essential thrombocythemia. Blood. 2010;116:1528–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pietras EM, Lakshminarasimhan R, Techner JM, Fong S, Flach J, Binnewies M, et al. Re-entry into quiescence protects hematopoietic stem cells from the killing effect of chronic exposure to type I interferons. J Exp Med. 2014;211:245–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nieborowska-Skorska M, Maifrede S, Dasgupta Y, Sullivan K, Flis S, Le BV, et al. Ruxolitinib-induced defects in DNA repair cause sensitivity to PARP inhibitors in myeloproliferative neoplasms. Blood. 2017;130:2848–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.