

Abstract
Introduction: Biomarkers that reflect pathologic processes affecting neuronal function during preclinical and early stages of Alzheimer's disease (AD) are needed to aid drug development.
Methods: A targeted, stable isotope, quantitative mass spectrometry‐based investigation of longitudinal changes in concentrations of previously identified candidate biomarkers was performed in cerebrospinal fluid (CSF) of Alzheimer's Disease Neuroimaging Initiative participants who were classified as cognitively normal (CN; n = 76) or with mild cognitive impairment (MCI; n = 111) at baseline.
Results: Of the candidate biomarkers, the CSF concentration of neuronal pentraxin 2 (NPTX2), a protein involved in synaptic function, exhibited rates of change that were significantly different between three comparison groups (i.e., CN vs. MCI participants; AD pathology positive vs. negative defined by phosphorylated tau181/amyloid beta1‐42 ratio; and clinical progressors vs. non‐progressors). The rate of change of NPTX2 also significantly correlated with declining cognition.
Discussion: CSF NPTX2 concentration is a strong prognostic biomarker candidate of accelerated cognitive decline with potential use as a therapeutic target.
Keywords: Alzheimer's disease, dementia, longitudinal cerebrospinal fluid, mild cognitive impairment, neuropathology, prognostic biomarker, stable isotope‐based quantitative mass spectrometry
1. BACKGROUND
Alzheimer's disease (AD) is a neurodegenerative disorder characterized by the development of a gradually increasing burden of amyloid plaque and tau tangle pathology resulting in the loss of synapses and degeneration of neurons. The disease is characterized by extended pre‐clinical and prodromal (mild cognitive impairment [MCI]) stages prior to dementia—the terminal phase of the disease . 1 , 2 , 3 Despite its outsized societal burden, 4 therapeutics that slow down or reverse the progression of the disease have been elusive. 5
One of the obstacles to drug discovery efforts is the lack of early prognostic biomarkers for AD. Phosphorylated tau (p‐tau) and structural and metabolic brain imaging provide a reliable set of diagnostic biomarkers that strongly correlate with hallmark pathologic changes in the brain and can therefore be used to follow brain pathology only at more advanced stages of AD. 6 While amyloid plaque burden is recognized as an early abnormality in the overall trajectory of development of AD, 7 it is insufficient to accurately predict the time course for disease progression. 8 Also, ≈40% of Alzheimer's Disease Neuroimaging Initiative (ADNI) individuals with MCI did not conform to the National Institute on Aging/Alzheimer's Association (NIA‐AA) framework amyloid/tau/neurodegeneration (AT[N]) classification scheme for defining AD biologically, 9 and their cognitive status could therefore be attributed to other factors. 10 Additional measures that track early pathophysiologic processes involved in the progression of patients classified as MCI are needed to help determine whether AD pathology is truly present, and to estimate with greater accuracy the time of dementia onset. 11 Specifically, early detection of neuro‐dysfunction, such as synapse damage or loss, could provide an improved ability to predict future decline caused by multiple ongoing pathophysiologic processes occurring in the AD brain. 12
Most studies of candidate biomarkers that used cerebrospinal fluid (CSF) have examined samples collected at a single point in time, failing to capture the potentially important feature of within‐individual changes in relation to disease progression. Because longitudinal studies could improve our ability to detect early changes in disease pathology, we evaluated the time course of five candidate biomarkers in cognitively normal (CN) patients at baseline and patients with a baseline diagnosis of MCI. The five biomarkers—(1) chromogranin A (CMGA), (2) fatty acid binding protein (FABPH), (3) neuronal pentraxin 2 (NPTX2), (4) secretogranin (SCG2), and (5) neurosecretory protein VGF (VGF)—were selected based on evidence gathered from prior research including a cross‐sectional mass spectrometry (MS)‐based proteomic study of CSF samples from the ADNI cohort using a multiplexed MRM panel, developed with Caprion, and published previously by our team. 13 Given the goal of ruling in or out significant rates of change in this group of analytes in MCI subjects with a quantitative MS assay, we did not include the many MCI/AD relevant biomarkers already identified with immunoassays, mainly in cross‐sectional studies, such as neurogranin, Vilip‐1, neurofilament light, and YKL‐40. To the extent that these have been evaluated in longitudinal studies, none have shown marked rates of change that consistently relate to the trajectory of pathology and/or symptoms. 11 , 14 , 15
1.1. CMGA
Two longitudinal CSF studies using MS reported 7% to 15% annualized decreases in relatively small groups of participants (30 and 45) with AD. 16 , 17 In the Wildsmith et al. study using MS, CSF CMGA was robustly correlated with CSF tau (r = 0.69) 17 raising the question of how much additional information it would contribute as a predictor of disease trajectory.
1.2. FABPH
Several studies have reported fairly large group differences for CSF FABPH using radioimmunologic assays, 18 , 19 , 20 and this finding was replicated in our earlier study 13 using the Caprion multiplex MRM panel. FABPH may play a role in hippocampal loss in AD. 17
1.3. NPTX2
NPTX2 is a member of the family of neuronal pentraxins that include NPTX1 and NPTXR, proteins secreted from pyramidal neurons that can oligomerize to form mixed NPTX complexes that bind to and modulate post‐synaptic AMPA type glutamate receptors on GABAergic parvalbumin interneurons. 21 , 22 Notably, NPTX2 was reported by both Wildsmith et al. 17 and Hendrickson et al. 16 as showing annualized decreases of 7% or more in AD participants. It also emerged as a very strong predictor of “progression” from MCI to AD in the cross‐sectional ADNI samples analyzed by the MRM assay. 13
HIGHLIGHTS
A quantitative mass spectrometry assay was developed to measure five candidate biomarkers in cerebrospinal fluid.
Longitudinal (within‐subject) changes were necessary to inform on the utility and function of these new candidate biomarkers (e.g., neuronal pentraxin 2 [NPTX2]).
NPTX2 showed robust longitudinal decreases in mild cognitive impairment participants, phosphorylated tau181 /amyloid beta1‐42 ratio positive participants, and participants who progressed.
Decreases of NPTX2 correlated with rates of cognitive decline even in the absence of brain pathology.
NPTX2 is an additional promising marker with potential applications for Alzheimer's disease prognosis and treatment outside of the current biomarker paradigm.
RESEARCH IN CONTEXT
Systematic review: The authors reviewed longitudinal within‐subject studies of cerebrospinal fluid (CSF) analytes in control and mild cognitive impairment (MCI) participants using traditional sources. Many promising biomarkers have been assessed cross‐sectionally, but most do not have robust longitudinal studies looking at change over time and relevant, clinically meaningful outcomes. Research to date on neuronal pentraxin 2 (NPTX2) has supported its potential use; none has been successful in reliably measuring the analyte in CSF in a well‐characterized data set.
Interpretation: Our findings support the current evidence that NPTX2 is a robust prognostic biomarker of cognitive decline and a promising new therapeutic target.
Future directions: Further work should confirm our suggestions of a relationship between NPTX2 changes and cognitive decline that is independent of pathology. Based on our finding, NPTX2 could be explored as a therapeutic target in a subset of MCI subjects to be more precisely defined with future research. Additional assay development is needed to identify a highly sensitive and specific immunoassay for CSF, and possibly blood, samples.
1.4. SCG2 and VGF
These members of the dense core protein families are typically transported in synaptic vesicles and may also serve as markers of synaptic loss and neuronal injury/degeneration. 23 Both emerged as strong predictors from the Spellman et al. 13 cross‐sectional study of participants with MCI who progressed to dementia. In the one longitudinal study in AD from Hendrickson et al. 16 that reported VGF values, annualized decreases of 15% to 20% per year were reported in patients with moderate to severe AD. Additionally, VGF has been identified as the single most common “candidate target” in the Accelerating Medicines Partnership‐Alzheimer's Disease consortium effort to date (personal communication; unpublished results).
The availability of a detailed clinical characterization of participants and longitudinally collected CSF samples provided by the ADNI resource in conjunction with using a rigorous method of absolute quantification of analyte concentration using targeted MS, allowed us to estimate the shape of the mean within‐subject trajectory of the five analytes’ concentration in participants with various clinical features and biomarker profiles. Correlating these trajectories with the evolution of the participants’ clinical characteristics, we were able to assess the suitability of each analyte as an early prognostic biomarker in patients meeting clinical criteria for MCI.
2. METHODS
2.1. Participant characteristics
The study subjects consisted of 187 participants from the ADNI I, II, and GO cohorts that participated in a minimum of three longitudinal CSF sample collections and clinical assessments over a minimum of 3 years. The requirement of at least three longitudinal samples and assessments rather than two was intended to provide a more reliable estimate of the within‐subject rate of change in analyte concentration as well as clinical and physiological parameters. ADNI is a public–private partnership aimed at evaluating known biomarkers and facilitating the discovery of novel biomarkers in early stages of AD to support the development of drug treatments. 24 Procedures for participant recruitment and sample processing followed standardized operating procedures that can be reviewed at www.adni‐info.org. The participants ranged across the disease spectrum from those diagnosed at baseline as CN (n = 76) to those diagnosed as MCI (n = 111) as defined in ADNI. Thirteen percent of participants who were initially diagnosed as CN were later diagnosed as MCI and 26% initially diagnosed as MCI were later diagnosed with dementia (n = 42). Participants’ data were downloaded from the ADNI database (adni.loni.usc.edu) on June 6, 2019. Key clinical and demographic characteristics of the study subjects are summarized in Table 1. Data for approximately 80% of these participants (n = 144) included fluorodeoxyglucose‐positron emission tomography (FDG‐PET) measures obtained within 6 months of their respective baseline visit, and these participants were also categorized within the AT(N) framework (Table 1).
TABLE 1.
Demographic, cognitive, and biomarker characteristics of the study participants
| Cognitively normal (n = 76) | MCI (n = 111) | Total (n = 187) | |
|---|---|---|---|
| Age (y; mean ± SD) | 75.5 ± 5.5 | 71 ± 7.3 | 73 ± 6.9 |
| Sex (% female) | 49% | 40% | 43% |
| Education (y; mean ± SD) | 16.1 ± 2.9 | 16.3 ± 2.7 | 16.2 ± 2.8 |
| % APOE ε4 carriers | 22% | 50% | 39% |
| Number of visits (median (range)) | 3 (3–7) | 3 (3–8) | 3 (3–8) |
| Length of follow‐up (y; mean ± SD; median (range)) | 5.1 ± 2.0; 4.1 (3–10.2) | 4.5 ± 1.4; 4 (2.8–10.1) | 4.7 ± 1.7; 4 (2.8–10.2) |
| Progressors (%) | 13% | 26% | 21% |
| p‐tau181/Aβ1‐42 ratio at baseline (mean ± sd) | 0.026 ± 0.021 | 0.042 ± 0.039 | 0.036 ± 0.034 |
| p‐tau181/Aβ1‐42 ratio status at baseline (% positive) | 32% | 55% | 46% |
| MMSE (mean ± SD) | 29.3 ± 1.1 | 27.7 ± 1.8 | 28.4 ± 1.7 |
| ADAS‐cog (mean ± SD) | 8.7 ± 4.5 | 15.4 ± 6.2 (N = 110) | 12.6 ± 6.5 (N = 186) |
| Cognitively normal (N = 53) | MCI (N = 91) | Total (N = 144) | |
|---|---|---|---|
| A–T–N– | 32% | 26% | 28% |
| A+T–N– | 15% | 15% | 15% |
| A+T+N– | 11% | 21% | 17% |
| A+T+N+ | 4% | 22% | 15% |
| A+T–N+ | 2% | 4% | 3% |
| A‐T+N– | 23% | 7% | 13% |
| A–T–N+ | 9% | 3% | 6% |
| A–T+N+ | 4% | 1% | 2% |
Abbreviations: Aβ, amyloid beta; ADAS‐Cog: Alzheimer's Disease Assessment Scale–Cognitive Subscale; APOE, apolipoprotein E gene; CN, cognitively normal; FDG‐PET, fluorodeoxyglucose positron emission tomography; MCI, mild cognitive impairment; MMSE: Mini‐Mental State Examination; p‐tau, phosphorylated tau; SD, standard deviation; SUVR, standardized uptake value ratio.
Notes: Progressors: participants who were initially diagnosed as CN (resp. MCI) and who were diagnosed within 4 years and 1 month after the initial diagnosis with MCI or dementia (resp. dementia).
p‐tau181/Aβ1‐42 ratio status: participants with p‐tau181/Aβ1‐42 ≥ 0.025 were classified as “ratio positive,” all others were “ratio negative.” 25
A+: Aβ1‐42 ≤ 980 pg/mL; A–: Aβ1‐42 > 980 pg/mL. 25
T+: p‐tau181 ≥ 24 pg/mL; T–: p‐tau181 < 24 pg/mL. 25
N+: FDG‐PET SUVR value < 1.21; N–: FDG‐PET SUVR value > 1.21. 6
2.2. CSF samples
A total of seven hundred and fifty (750) unique CSF samples were included in the study. Seven hundred and thirty (730) longitudinal samples were from the ADNI‐1, ADNI‐2, and ADNI‐GO studies representing 198 participants (187 participants used in the analyses provided three or more samples). Additionally, 20 blinded replicate aliquots, one for each of 20 participants, were distributed throughout the analysis runs and used to assess assay reproducibility. Sample aliquots were stored at –80˚C until use. Due to the presence of endogenous levels of the five absolute quantitation target proteins in CSF, standard curve samples were prepared using recombinant proteins: CMGA (AbCam, #AB85486), FABPH (Sigma Aldrich, SRP4503), NPTX2 (R&D Systems, 7816‐NP‐050), SCG2 (LSBio, #LS‐G25659), and VGF (Origene, TP309477) in 0.2 mg/mL bovine serum albumin in water. Quality control (QC) samples were prepared using the recombinant proteins diluted in a CSF pool from 300 individual donors, representative of the study samples and supplied by ADNI. Aliquots of the standard and QC samples were prepared and frozen at –80°C until use. The multiplexed MRM panel was composed of peptides representing five proteins: CMGA (EDSLEAGLPLQVR), FABPH (SLGVGFATR), NPTX2 (TNYLYGK), SCG2 (THLGEALAPLSK), and VGF (VLEYLNQEK) for absolute quantitation. Purified synthetic stable isotope (15N and 13C) labeled (SIL) peptides were from CPC Scientific (EDSLEAGLPLQVR [CMGA]; SLGVGFATR [FABPH]; VLEYLNQEK [SCG2]; and THLGEALAPLSK [VGF]); and JPT Peptide Technologies (TNYLYGK [NPTX2]).
2.3. CSF sample processing
The 750 CSF samples were processed in four batches, keeping longitudinal samples from each individual participant in the same batch. Fifty (50) μL of CSF were denatured with trifluoroethanol (Sigma) followed by proteolytic digestion with trypsin (Promega) at an approximate 1:25 protease to protein ratio overnight at 37°C. Digestion was stopped by acidification with trifluoroacetic acid. SIL peptides (300 fmol) were spiked into the peptide samples. Peptides were subsequently desalted using Oasis MCX desalting plates (Waters), aliquoted into two replicate mass spectrometry plates, dried by vacuum evaporation, and stored at –20°C prior to MS analysis. A flowchart summarizing the sample processing steps is presented in Figure S1 in supporting information.
2.4. LC‐MRM/MS analysis of CSF samples
Samples were injected by processing batch. The processed samples were re‐solubilized with 10 μL of 97/3 (v/v) water/acetonitrile, containing five internal standard peptides (ISPs): FSDISAAK (ISP‐1), ASSILAT (ISP‐2), NVDQSLLELHK (ISP‐3), QNNGAFDETLFR (ISP‐4), and ELWFSDDPDVTK (ISP‐5), each at 100 ng/mL. The ISP peptides elute at different retention times during the chromatographic gradient and were used to monitor instrument performance during sample analysis. Seven (7) μL of material was injected, per sample, onto a NanoAcquity UPLC (Waters) coupled to a 6500 QTRAP mass spectrometer (SCIEX). Peptide separation was achieved using a 500 μm x 10 mm, 2.7 μm particle size Halo Peptides ES C18 column (Canada Life Science Inc.) at a flow rate of 18 μL/min (Table S4 in supporting information). Two 6500 QTRAP (SCIEX) mass spectrometers were used in the analysis of the samples. Two runs were analyzed on one instrument and the other two runs were analyzed on a separate instrument. Prior to analysis, the two mass spectrometers were cross validated by testing the backup plates from three precision and accuracy runs. Further details are described in the Methods section in supporting information.
2.5. Absolute quantitation
Absolute quantitation was performed using the surrogate peptide approach. A single peptide/transition was used for quantitation and was selected based on the following criteria: sensitivity, chromatographic performance, lack of interference, linearity, precision, and accuracy (Table S3 in supporting information). Peak integration was performed using MultiQuant software (version 2.1, SCIEX). The peak area ratio (endogenous peptide signal/SIL peptide ratio) was used to back‐calculate the concentration of the respective target protein from standard curves created using recombinant versions of the target proteins.
2.6. Data QC and normalization
Each sample batch consisted of three digestion/MS plates. The 20 replicated samples spanned 11 of the 12 analysis plates providing a means of interrogating replication between both plate level processing and run order effects. Block and batch effects due to sample processing or injection run order are common with liquid chromatography (LC)‐MS based assays. The standards and calibration curves used in this study to generate absolute concentrations for the targeted analytes, however, were designed to minimize or eliminate these biases. Simca‐P 15 (Umetrics Inc.) with seven‐fold cross‐validation was used to generate multivariate quantitative partial‐least‐squares (PLS) and class discriminant models (PLS‐DA) to test for residual block effects in the final quantitative data. The data from the five targeted analytes were used as the independent block to predict the noted outcomes. All data processing was performed on Z‐normed log‐2 scaled data.
2.7. Primary statistical analysis
Our primary goal was to investigate longitudinal changes in the concentrations of five candidate analytes (CMGA, FABPH, NPTX2, SCG2, and VGF) in the CSF of individuals from the ADNI cohorts. Specifically, we estimated and compared the mean rates of change in analyte concentrations between study participants categorized at baseline as CN and MCI. In addition to estimating the rates of change in analyte concentration, we tested the hypothesis that the rate of change in each subgroup was different from zero, and that the rates of change differed between the subgroups. We also compared the rates of change between participants with baseline CSF p‐tau181/amyloid beta (Aβ)1‐42 ratio below 0.025 (ratio negative), and participants with baseline p‐tau181/Aβ1‐42 ratio at or above 0.025 (ratio positive). 25 Last, we compared the rates of change between participants whose diagnosis changed from CN to MCI or from MCI to dementia within 4 years and 1 month (progressors) and participants who maintained their diagnosis of CN or MCI for longer than 4 years and 1 month (non‐progressors). The cutoff of 4 years and 1 month was established based on examining the data; approximately 20% of the participants’ diagnoses changed within 4 years, with a handful of participants progressing just a few days after this cutoff. The proportion of participants who progressed after 4 years and 1 month declined sharply (68 study participants [36%] did not progress from their baseline diagnosis during the follow‐up period and 11 of those 68 reverted to their earlier diagnosis). The statistical methodology used in the data analysis had been defined in a formal statistical analysis plan before the final data became available.
The endpoint was defined as the change from baseline in the estimated concentration of each of the five candidate proteins. To estimate the mean rates of change and test the hypotheses, we used linear mixed effects (LME) modelling with participant and recruitment site as random factors. Three separate models were fitted to compare CN and MCI participants, p‐tau181/Aβ1‐42 ratio positive and negative participants, as well as clinical progressors and non‐progressors:
The first LME model involved time (continuous), baseline diagnosis (CN or MCI), and their interaction as predictors; baseline concentration of the analyte; sex; apolipoprotein E (APOE) ε4 carrier status; education level; age at study entry; sample position on plate; sample injection order; and sample storage time as covariates.
The second model featured time (continuous), p‐tau181/Aβ1‐42 ratio status (positive or negative) and their interaction as predictors, and all the covariates listed in Model 1 with baseline diagnosis as an additional covariate.
The final model included time (continuous), progression status (“progressor” or “non‐progressor”) and their interaction as predictors, and all the covariates listed in Model 1 with baseline p‐tau181/Aβ1‐42 ratio and baseline diagnosis as additional covariates.
Post hoc analyses examining the relationship between the rates of change in NPTX2 concentration and the rates of change in known biomarkers of neurodegeneration (p‐tau181, FDG‐PET, hippocampal volume) and measures of cognitive ability (Mini‐Mental State Examination [MMSE], Alzheimer's Disease Assessment Scale–Cognitive 13‐Item Subscale [ADAS‐Cog13]) involved assessing the correlation between pairs of mean yearly slopes obtained from the same participant using Pearson's product moment correlation coefficient, and testing the null hypothesis of the correlation coefficient being equal to zero. All reported P‐values are unadjusted for multiplicity.
2.8. Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its supporting information. Per the data‐sharing ADNI requirement, all data associated with this study has been uploaded to the central ADNI data site (LONI).
3. RESULTS
3.1. Primary results models of longitudinal NPTX2 changes across clinical subsets
The goal of the present study was to examine the rate of change in the concentration of five proteins—CMGA, FABPH, NPTX2, SCG2, and VGF—in the CSF, and to compare these rates between participants stratified on the basis of clinical diagnosis, ratio positivity and clinical progression. Specifically, we fitted three longitudinal linear models to statistically test whether (1) the repeated within‐subject measurements of analyte concentration exhibited robust changes in time across all participants in our analysis set or their pre‐defined subsets; and (2) whether the rate of change in the analyte concentrations was associated with differences in clinical characteristics such as diagnosis, p‐tau181/Aβ1‐42 ratio, and clinical progression.
The results of the longitudinal modeling effort, which allowed us to adjust for important covariates such as each participant's age, gender, APOE ε4 carrier status, and education level, suggested that the concentration of CMGA, NPTX2, SCG2, and VGF proteins in the participants’ CSF tended to decrease over time. These declines were especially evident among participants who were p‐tau181/Aβ1‐42 ratio positive at baseline (Figure 1 and Figure 2B for NPTX2, Table 2, Figures S4‐S7 in supporting information). However, the largest declines, which were also supported by the strongest statistical evidence, were observed for NPTX2. The concentration of NPTX2 among participants with a baseline clinical diagnosis of MCI declined by 0.08 ng/mL per year on average (P < .0001), and by 0.09 ng/mL (P < .0001) among participants with the biomarker profile indicative of AD (baseline p‐tau181/Aβ1‐42 ratio ≥ 0.025; i.e., ratio positive). The declines in NPTX2 concentration were statistically significantly larger among participants classified as MCI compared to CN participants (P = .008; Figure 2A); as well as between p‐tau181/Aβ1‐42 ratio positive participants at baseline compared to ratio negative participants (P = .001; Figure 2B; see Table 2 for details); that is, among groups of participants that are further along the disease continuum.
FIGURE 1.
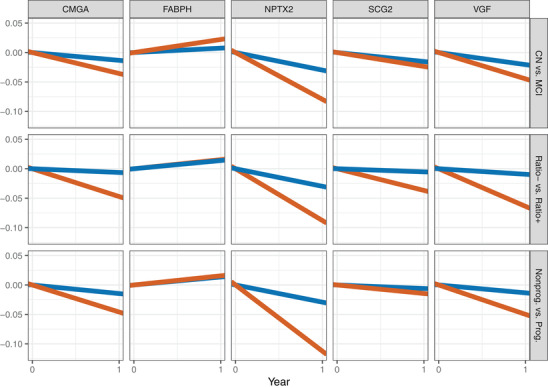
Mean yearly rates of change in the cerebrospinal fluid concentration of each of the five studied analytes (columns) in subgroups of study participants defined by baseline diagnosis (Model 1; first row), baseline phosphorylated tau (p‐tau)181/amyloid beta(Aβ)1‐42 ratio positivity (Model 2; second row) and progression (Model 3; third row). CN: cognitively normal (blue); MCI: mild cognitive impairment (red); Ratio+: participants with p‐tau181/Aβ1‐42 ≥ 0.025 (red); Ratio–: participants with p‐tau181/Aβ1‐42 < 0.025 (blue); Prog.: progressors (red), that is, participants who were initially diagnosed as CN (resp. MCI) and who were diagnosed within 4 years and 1 month after the initial diagnosis with MCI or dementia (resp. dementia). Nonprog.: non‐progressors (blue). The x‐axis corresponds to a period of 1 year.
FIGURE 2.
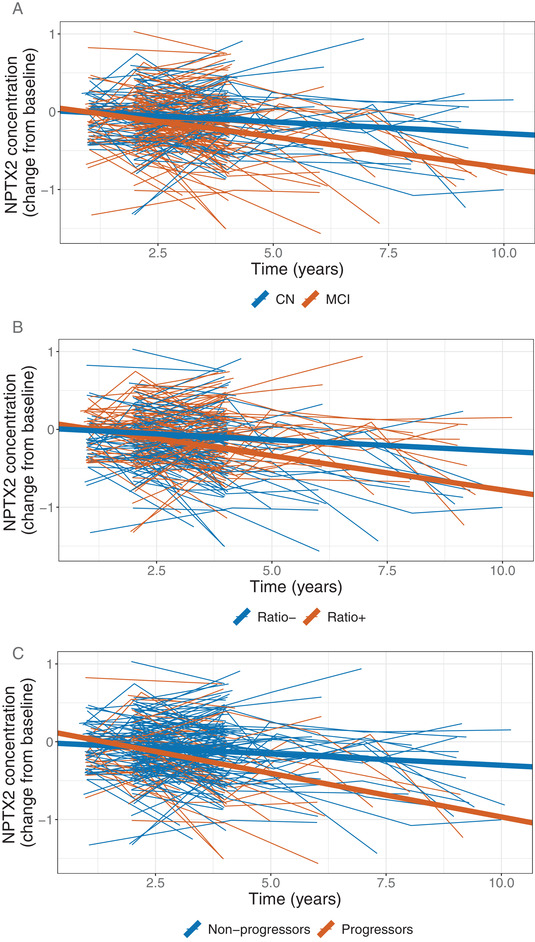
Longitudinal changes from baseline in cerebrospinal fluid concentration of neuronal pentraxin 2 (NPTX2) (raw data from individual participants: thin lines; mean changes resulting from the linear mixed effects (LME) model: thick lines, also shown in Figure 1 under “NPTX2”). A, Participants categorized as cognitively normal (CN) at baseline (blue); participants categorized as mild cognitive impairment (MCI) at baseline (red). B, Participants categorized as phosphorylated tau (p‐tau)181/amyloid beta (Aβ)1‐42 ratio positive at baseline (red); participants categorized as p‐tau181/Aβ1‐42 ratio negative at baseline (blue). C, Participants categorized as progressors (red); participants categorized as non‐progressors (blue)
TABLE 2.
Mean yearly rates of change in the CSF concentration of the five analytes under study resulting from the mixed‐effects modeling and their differences among various subsets of study participants
| CMGA | FABPH | NPTX2 | SCG2 | VGF | ||
|---|---|---|---|---|---|---|
| Model 1 | CN at baseline vs. zero | –0.01 ± 0.01 | 0.01 ± 0.01 | –0.03 ± 0.02 | –0.02 ± 0.01 | –0.02 ± 0.02 |
| MCI at baseline vs. zero | –0.04 ± 0.01* | 0.02 ± 0.01* | –0.08 ± 0.02‡ | –0.02 ± 0.01 | –0.04 ± 0.02* | |
| CN vs. MCI at baseline | 0.02 ± 0.01 | 0.01 ± 0.01 | 0.05 ± 0.02* | 0.01 ± 0.01 | 0.02 ± 0.02 | |
| Model 2 | Ratio– at baseline vs. zero | –0.01 ± 0.01 | 0.02 ± 0.01 | –0.03 ± 0.02 | 0 ± 0.01 | –0.01 ± 0.02 |
| Ratio+ baseline vs. zero | –0.05 ± 0.01† | 0.02 ± 0.01 | –0.09 ± 0.02‡ | –0.04 ± 0.01* | –0.06 ± 0.02‡ | |
| Ratio+ vs. Ratio– at baseline | 0.04 ± 0.01† | 0 ± 0.01 | 0.06 ± 0.02† | 0.03 ± 0.01* | 0.05 ± 0.02‡ | |
| Model 3 | Non‐progressors vs. zero | –0.02 ± 0.01 | 0.01 ± 0.01 | –0.03 ± 0.02 | –0.02 ± 0.01 | –0.02 ± 0.02 |
| Progressors vs. zero | –0.05 ± 0.02* | 0.01 ± 0.02 | –0.11 ± 0.02‡ | –0.03 ± 0.02 | –0.06 ± 0.02* | |
| Progressors vs. Non‐progressors | 0.03 ± 0.02 | 0 ± 0.01 | 0.08 ± 0.02‡ | 0.01 ± 0.02 | 0.04 ± 0.02 |
Notes: P‐values (unadjusted for multiplicity) are associated with the tests of the hypothesis that mean rate of change is equal to zero, or the hypothesis that the two rates of change estimated for two participant groups are the same.
Ratio+: participants with p‐tau181/Aβ1‐42 ≥ 0.025. 25
Ratio–: participants with p‐tau181/Aβ1‐42 < 0.025. 25
Progressors: participants who were initially diagnosed as CN (resp. MCI) and who were diagnosed within 4 years and 1 month after the initial diagnosis with MCI or dementia (resp. dementia).
* < 0.05; † < 0.005; ‡ < 0.0005.
Abbreviations: Aβ, amyloid beta; CMGA, chromogranin A; CN, cognitively normal; CSF, cerebrospinal fluid; FABPH, x fatty acid binding protein; FDG‐PET, fluorodeoxyglucose positron emission tomography; MCI, mild cognitive impairment; NPTX2, neuronal pentraxin 2; p‐tau, phosphorylated tau; SCG2, secretogranin; SUVR, standardized uptake value ratio; VGF, neurosecretory protein VGF.
The magnitude of the declines in NPTX2 concentrations also varied between progressors and non‐progressors (–0.11 vs. –0.03 ng/mL; P = .0004, Figure 2C and Table 2), further validating the association of NPTX2 concentration and clinical prognosis. To put these differences in context, consider that the average baseline concentration of NPTX2 in the study participants was 4.7 ± 0.6 ng/mL (mean ± standard deviation). Thus, over a 5‐year period, a decline of 0.1 ng/mL per year amounts to a cumulative decrease of around 10% or one standard deviation (Figure S8 in supporting information). To validate these findings, we analyzed a restricted dataset involving 110 (59%) participants with at least three measures of NPTX2 concentration in the first 4 years since their first visit to rule out potential undue influence of a small number of measurements from a minority of participants with longer than usual follow‐up. This analysis yielded very similar results to those presented here (data not shown).
3.2. Correlations of the rate of change in NPTX2 concentration with cognitive measures and AD biomarkers
To elucidate the potential role of NPTX2 in the pathophysiologic processes involved in the progression of AD, we examined the relationship between the rates of change in NPTX2 concentration and the rates of change in known biomarkers of neurodegeneration (p‐tau181, FDG‐PET, hippocampal volume) and measures of cognitive ability (Table S7 in supporting information). These were exploratory post hoc analyses. Among the observed relationships, there was a modest positive relationship between the rates of change in NPTX2 and p‐tau181 concentrations in CSF, such that individuals with high rates of decline in NPTX2 had either no change in p‐tau181 or slight decreases over the same period (Figure S12 in supporting information). This relationship was especially evident among participants with baseline p‐tau181/Aβ1‐42 ≥ 0.025 (i.e., ratio positive).
Declines in NPTX2 concentration were also correlated with cognitive declines primarily among participants classified as MCI or p‐tau181/Aβ1‐42 ratio positive (Figure 3). These correlations appear fairly robust; even after excluding potentially influential observations (i.e., mean yearly changes in MMSE < —4 and ADAS‐Cog13 > 10) the correlations remained statistically significant (MMSE: P = 0.016, ADAS‐Cog13: P = .012), albeit moderate in strength (MMSE: r = 0.26, ADAS‐Cog13: r = –0.28). The statistically significant correlation observed between NPTX2 concentration and ADAS‐Cog13 cognitive measure and hippocampal volume among CN participants (Table S7), may point to NPTX2 as a potential prognostic marker of impending brain degeneration and cognitive decline.
FIGURE 3.
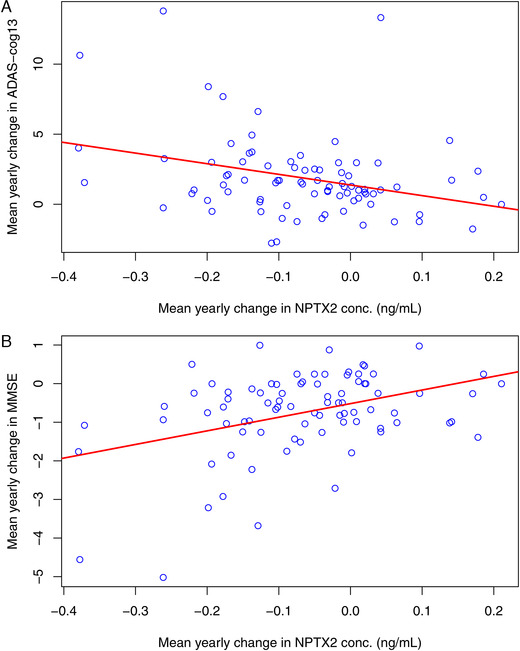
Correlations between neuronal pentraxin 2 (NPTX2) slopes and Mini‐Mental State Examination (MMSE), Alzheimer's Disease Assessment Scale–Cognitive 13‐item Subscale (ADAS‐Cog13) cognitive measures
4. DISCUSSION
Out of the five studied candidate biomarkers, NPTX2 showed the most significant within‐subject changes over the course of 3 or more years. While within‐subject trajectories of CSF concentrations of the five proteins of interest in ADNI CN and MCI participants were highly variable over a period of 3 or more years, group‐level trajectories based on baseline diagnosis, baseline p‐tau181/Aβ1‐42 ratios in CSF, and clinical progression showed differential patterns of change, with those in NPTX2 standing out. CMGA, NPTX2, SCG2, and VGF all showed steeper declines in ratio‐positive participants, classified as exhibiting p‐tau181/Aβ1‐42 baseline ratio above 0.025. 25 In contrast, a hypothesized decline in FABPH across the subgroups based on earlier cross‐sectional studies comparing control, MCI, and AD participants 13 was not observed. This finding highlights the importance of within‐subject longitudinal studies for validating or refuting inferences from cross‐sectional comparisons between diagnostic groups. This study specifically focused on assessing within‐MCI participant changes over time in selected analytes that had not previously been followed longitudinally. Baseline values in MCI participants of neurogranin, VILIP‐1, and others are predictive of subsequent clinical worsening. However, given their very modest (< 5%) annualized rates of change at the group level and small number of participants with large changes, their longitudinal trajectories have not emerged as likely to provide much additional biologically relevant information. 14 , 26 , 27 , 28 , 29 , 30 , 31
From the perspective of identifying a novel biomarker that might apply to drug development, we required a sufficiently accurate and precise assay to interpret individual‐level changes as reflecting biologic processes. Therefore, we developed a quantitative MS assay with labeled analyte internal standards, building on our earlier semi‐quantitative one. 13 Moreover, a minimum of three CSF samples at roughly annual or longer intervals allowed for more reliable within‐subject calculations of rates of change. Using this refined methodology, we identified NPTX2 as the analyte that, at a group level, exhibited statistically significant rates of decline in its CSF concentration far greater than any of the other four analytes among MCI participants versus CN; among progressors versus non‐progressors; as well as in participants with positive baseline CSF values of p‐tau181/Aβ1‐42 versus ratio‐negative participants. Second, we observed that NPTX2 declined in 77% (30 out of 39) of progressing and 72% (61 out of 85) of ratio‐positive participants, a substantial proportion (see Figures S10 and S11 in supporting information). These results extend those from cross‐sectional studies, which supported that “baseline” values of NPTX2 concentration in the CSF of MCI participants when added to Aβ42 provided for improved early prognosis compared to Aβ42 alone, and that when added to Aβ42 and p‐tau181, 22 this information provided for a more accurate prediction of disease progression using the NIA/AA AT(N) classification scheme. 9 The important extension that our longitudinal study provides is the presence of a substantially accelerated clinical progression as indexed by meeting AD diagnostic criteria or by measures of cognitive decline in those individuals in whom NPTX2 decreases regardless of its baseline value.
From the perspective of multiple processes contributing to clinical progression beyond the degree of AD pathologic changes, our findings provide a rationale for exploring modulation of NPTX2 as a therapeutic target for slowing clinical progression in at least a subset of MCI patients who are biologically positive for AD according to the AT(N) classification.
The biological role of NPTX2 has been of wide interest and, at least in animals, one relevant to neuronal function related to cognition based on its involvement in activity‐dependent plasticity. 32 NPTX2 is a member of a family of “long” neuronal pentraxins that traffic to the extracellular surface at excitatory synapses. 32 They have been shown to bind AMPA‐type glutamate receptors and contribute to both developmental and adult synaptic plasticity. 33 , 34 , 35 , 36 , 37 , 38 , 39 , 40 Xiao et al. 21 found that NPTX2 was downregulated in the post mortem brain of human participants with AD and reduced in the CSF of AD patients, which correlated with diminished cognitive function. Xiao et al. 21 interpreted their findings as supporting the hypothesis “that NPTX2 downregulation … represents a previously unrecognized mechanism important for human cognitive dysfunction and progression in Alzheimer's disease.” Furthermore, they noted that because NPTX2 is not downregulated in a widely used model of mouse amyloidosis, 41 and is distinct from other CSF markers attributed to neurodegeneration including tau and p‐tau, NPTX2 is an indicator of specific rather than general decrease in excitatory synapse function. This stands in contrast to the more general synaptic marker neurogranin, increased concentrations of which are highly correlated with tau and overall pathology. 11 , 14 , 26 , 27 A subsequent study in a University of California San Diego cohort showed that the CSF concentration of NPTX2 correlated with cognitive function in MCI participants and was predictive of progression to dementia using the MS data generated by our team and available online. 22 An independent follow‐up analysis of the same cross‐sectional dataset further demonstrated the strong predictive power of decreased NPTX2 concentration in memory decline and medial temporal lobe atrophy. 42 Overall, these studies and analyses show that lower values of NPTX2 in the CSF are associated with poor cognitive function consistent with the possibility that NPTX2 concentrations reflect the function of a discrete population of excitatory synapses in the brain. 21 Recently, NPTX2 decline has also been reported to be associated with the clinical progression of frontotemporal dementia, 43 which suggests that it taps into a process that may be relevant to interactions with other primary pathophysiologic processes.
Our findings on the relationship of longitudinal changes in NPTX2 to the degree of AD pathology as indexed by the p‐tau181/Aβ1‐42 ratio; stable or progressing MCI; and cognitive function address a related question: To what extent do within‐subject decreases of CSF NPTX2 in MCI participants independent of baseline values predict or correlate with various clinical trajectories? Because within‐subject declines of NPTX2 in the present study correlate with declines in cognitive function of patients beyond what could be explained by Aβ1‐42, tau, and structural measures, we explored whether these changes could be clearly related to other processes that were documented over time in the same ADNI participants. If the loss of NPTX2 was simply a reflection of the general loss of gray matter (an aspect of AD), then it would not be as useful as a biomarker with direct relevance.
The patterns of change (Figure 2 and Figures S9‐S11 in supporting information) argue strongly against an overall loss of tissue or extent of brain pathology as an explanation for the decline in NPTX2. For instance, a substantial proportion of individuals who progress or have high p‐tau181/Aβ1‐42 ratios did not show a decline in NPTX2 concentration. Furthermore, no correlations were observed between declines in NPTX2 and hippocampal volume among the studied participants.
Unexpectedly, individuals who showed the greatest rates of decline in NPTX2 were those in whom p‐tau181 did not change or slightly decreased. This is in the opposite direction of what would be expected if worsening pathology, as indexed by increased p‐tau181, were the main determinant of decline in NPTX2. This raises the possibility that in some participants who are positive for AD pathology, a process underlying the declines in NPTX2, accelerates a cognitive decline that is not simply a cascading downstream consequence of AD pathology. Put another way, the present findings indicate that no matter what an MCI participant's starting NPTX2 concentration is in the CSF, if it decreases over a period of several subsequent years, cognitive function will decrease more rapidly, supporting our findings that the presence of AD pathology as indicated by p‐tau181/Aβ1‐42 ratios is not a necessary or sufficient determinant of decreases in NPTX2. This further strengthens the speculation, raised by Xiao et al., 21 that NPTX2 could be a therapeutic target for intervention with the added perspective that this might be the case in only a subset of MCI patients.
CONFLICTS OF INTEREST
Susan A. Baker, Angus C. Nairn, and Daniel S. Spellman have nothing to declare. Leslie M. Shaw receives research support from NIH/NIA U19 AG024904, ADNI3 grant; NIH/NIA P30 AG010124, UPENN ADCC grant; and the Michael J. Fox Foundation for Parkinson's Research. He is a consultant for Biogen and Roche Diagnostics and is on the speaker's bureaus for Biogen and Fujirebio. Angus C. Nairn receives research support from NIH/NIA P30 AG066508. Clifford R. Jack Jr. serves on an independent data monitoring board for Roche, has consulted for and served as a speaker for Eisai, and consulted for Biogen, but he does not receive personal compensation from any commercial entities. Additionally, Clifford R. Jack Jr. receives research support from the NIH and the Alexander Family Alzheimer's Disease Research Professorship of the Mayo Clinic. Ondrej Libiger receives salary and company stock as compensation for his employment with Janssen Research & Development. William Z. Potter was previously employed by the National Institute of Mental Health, and he is a stockholder in Merck & Co., Inc.. He is a Co‐Chair Emeritus for the FNIH Biomarkers Consortium Neuroscience Steering Committee. Currently residing in Philadelphia, PA, he serves on DSMBs for AgeneBio and Regenacy and as a consultant for Karuna, Otsuka, Neurocrine, Eliem, and Emerald Lake Safety. Additionally, he receives grant support from the NIA. Nandini Raghavan is employed by Janssen Research & Development and owns stock in Johnson & Johnson. Kelly L. Umaña, Michael C. Biarnes, and Rosa M. Canet‐Avilés were previously employed by the FNIH. Daniel Chelsky and Yannick‐André Breton were previously employed by Caprion Biosciences. Laetitia Cortes and Mark Watson are employed by Caprion Biosciences. The FNIH provided financial support to Caprion Biosciences to perform the submitted work.
COLLABORATORS
Data used in preparation of this article were obtained from the ADNI database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp‐content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf
Supporting information
Supplementary information
ACKNOWLEDGMENTS
The results of the study represent the work of the Foundation for the National Institutes of Health (FNIH) Biomarkers Consortium “Longitudinal Proteomic Changes in Cerebrospinal Fluid from Alzheimer's Disease Neuroimaging Initiative (ADNI): Towards Better Defining the Trajectory of Prodromal and Early Alzheimer's Disease” project. The study was made possible through the scientific and financial support of government, industry, and academia partners. We are grateful for the contributions of the following Biomarkers Consortium CSF Proteomics Project team members: Justyna Maria Bahl, Ph.D. (Lundbeck); Susan Baker, Ph.D. (Janssen); Michael C. Biarnes, M.S. (FNIH); Rosa M. Canet‐Avilés, Ph.D. (FNIH); Daniel Chelsky, Ph.D. (Caprion Biosciences); Pascal Croteau, M.S. (Caprion Biosciences); Mikkel Nors Harndahl, Ph.D. (Lundbeck); Lee Honigberg, Ph.D. (Genentech); Katherine Horn, Ph.D. (Caprion Biosciences); Wesley Horton, M.S. (FNIH); John Hsiao, M.D. (NIA); Hartmuth Kolb, Ph.D. (Janssen); Ondrej Libiger, Ph.D. (Janssen); Yuqun Luo, Ph.D. (FDA); Angus Nairn, Ph.D. (Yale School of Medicine); Jan Torleif Pedersen, Ph.D. (Lundbeck); Niels Plath, Ph.D. (Lundbeck); William Potter, M.D., Ph.D.; Nandini Raghavan, Ph.D. (Janssen); Laura Rosen, M.D., Ph.D. (Takeda); Erin Rosenbaugh, Ph.D. (FNIH); Leslie Shaw, Ph.D. (University of Pennsylvania); Helle Sickmann, Ph.D. (Lundbeck); Daniel Spellman, Ph.D. (Merck & Co., Inc.); Kelly L. Umaña (FNIH); Mark Watson, Ph.D. (Caprion Biosciences); Kristin Wildsmith, Ph.D. (Genentech); Stephen Zicha, Ph.D. (Takeda). Private funding partners of the project include Genentech, a member of the Roche Group; H. Lundbeck A/S; Janssen Research & Development, LLC; Merck & Co., Inc.; and Takeda Pharmaceutical Company Limited. Private‐sector funding for the study was managed by the Foundation for the National Institutes of Health.
We would additionally like to acknowledge ADNI for the cerebrospinal fluid samples and data analyzed in this study. Data used in the preparation of this article were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). The ADNI was launched in 2003 as a public–private partnership, led by Principal Investigator Michael W. Weiner, M.D. The primary goal of the ADNI has been to test whether serial magnetic resonance imaging (MRI), positron emission tomography (PET), other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of MCI and early Alzheimer's disease. For up‐to‐date information, see www.adni‐info.org. Data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U19 AG024904) and DOD ADNI (Department of Defense award number W81XWH‐12‐2‐0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie; Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol‐Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann‐La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC; Johnson & Johnson Pharmaceutical Research & Development LLC; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company Limited; and Transition Therapeutics. The Canadian Institutes of Health Research provides funds to support ADNI clinical sites in Canada. Private‐sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for NeuroImaging (LONI) at the University of Southern California.
Libiger O, Shaw LM, Watson MH, et al., Alzheimer's Disease Neuroimaging Initiative (ADNI), Foundation for the National Institutes of Health (FNIH) Biomarkers Consortium CSF Longitudinal Proteomics Project Team . Longitudinal CSF proteomics identifies NPTX2 as a prognostic biomarker of Alzheimer's disease. Alzheimer's Dement. 2021;17:1976‐1987. 10.1002/alz.12353
Funding information This study, through multi‐institutional collaboration as a project of the Foundation for the National Institute of Health (FNIH) Biomarkers Consortium, Longitudinal Proteomic Changes in Cerebrospinal Fluid from Alzheimer’s Disease Neuroimaging Initiative (ADNI): Towards Better Defining the Trajectory of Prodromal and Early Alzheimer’s Disease, received funding support provided to the FNIH by Genentech, a member of the Roche Group; H. Lundbeck A/S; Janssen Research & Development, LLC; Merck & Co., Inc.; and Takeda Pharmaceutical Company Limited.
[The copyright line for this article was changed on May 26th, 2021 after original online publication.]
REFERENCES
- 1. Goedert M, Spillantini MG. A century of Alzheimer's disease. Science. 2006;314:777‐781. [DOI] [PubMed] [Google Scholar]
- 2. Beeri MS, Haroutunian V, Schmeidler J, et al. Synaptic protein deficits are associated with dementia irrespective of extreme old age. Neurobiol Aging. 2012;33:1125.e1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Robinson JL, Molina‐Porcel L, Corrada MM, et al. Perforant path synaptic loss correlates with cognitive impairment and Alzheimer's disease in the oldest‐old. Brain J Neurol. 2014;137:2578‐2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shah H, Albanese E, Duggan C, et al. Research priorities to reduce the global burden of dementia by 2025. Lancet Neurol. 2016;15:1285‐1294. [DOI] [PubMed] [Google Scholar]
- 5. Cummings J, Lee G, Mortsdorf T, Ritter A, Zhong K. Alzheimer's disease drug development pipeline: 2017. Alzheimers Dement Transl Res Clin Interv. 2017;3:367‐384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Veitch DP, Weiner MW, Aisen PS, et al. Understanding disease progression and improving Alzheimer's disease clinical trials: recent highlights from the Alzheimer's Disease Neuroimaging Initiative. Alzheimers Dement J Alzheimers Assoc. 2019;15:106‐152. [DOI] [PubMed] [Google Scholar]
- 7. Jack CR, Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 2010;9:119‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Buchhave P, Minthon L, Zetterberg H, Wallin AK, Blennow K, Hansson O. Cerebrospinal fluid levels of β‐amyloid 1‐42, but not of tau, are fully changed already 5 to 10 years before the onset of Alzheimer dementia. Arch Gen Psychiatry. 2012;69:98‐106. [DOI] [PubMed] [Google Scholar]
- 9. Jack CR, Bennett DA, Blennow K, et al. NIA‐AA Research Framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement J Alzheimers Assoc. 2018;14:535‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shaw LM, Figurski M, Landau S, Jagust W, Jack CR, Aisen PS. Predictive performance of CSF and imaging biomarkers in ADNI1/GO/2 MCI participants using the NIA‐AA Research Framework. Barcelona, Spain. 2018. [Google Scholar]
- 11. Molinuevo JL, Ayton S, Batrla R, et al. Current state of Alzheimer's fluid biomarkers. Acta Neuropathol (Berl). 2018;136:821‐853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jack CR, Wiste HJ, Weigand SD, et al. Amyloid‐first and neurodegeneration‐first profiles characterize incident amyloid PET positivity. Neurology. 2013;81:1732‐1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Spellman DS, Wildsmith KR, Honigberg LA, et al. Development and evaluation of a multiplexed mass spectrometry based assay for measuring candidate peptide biomarkers in Alzheimer's Disease Neuroimaging Initiative (ADNI) CSF. Proteomics Clin Appl. 2015;9:715‐731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tarawneh R, D'Angelo G, Crimmins D, et al. Diagnostic and prognostic utility of the synaptic marker neurogranin in Alzheimer disease. JAMA Neurol. 2016;73:561‐571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Olsson B, Lautner R, Andreasson U, et al. CSF and blood biomarkers for the diagnosis of Alzheimer's disease: a systematic review and meta‐analysis. Lancet Neurol. 2016;15:673‐684. [DOI] [PubMed] [Google Scholar]
- 16. Hendrickson RC, Lee AYH, Song Q, et al. High resolution discovery proteomics reveals candidate disease progression markers of Alzheimer's disease in human cerebrospinal fluid. PloS One. 2015;10:e0135365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wildsmith KR, Schauer SP, Smith AM, et al. Identification of longitudinally dynamic biomarkers in Alzheimer's disease cerebrospinal fluid by targeted proteomics. Mol Neurodegener. 2014;9:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Craig‐Schapiro R, Kuhn M, Xiong C, et al. Multiplexed immunoassay panel identifies novel CSF biomarkers for Alzheimer's disease diagnosis and prognosis. PloS One. 2011;6:e18850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Desikan RS, Thompson WK, Holland D, et al. Heart fatty acid binding protein and Aβ‐associated Alzheimer's neurodegeneration. Mol Neurodegener. 2013;8:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chiasserini D, Biscetti L, Eusebi P, et al. Differential role of CSF fatty acid binding protein 3, α‐synuclein, and Alzheimer's disease core biomarkers in Lewy body disorders and Alzheimer's dementia. Alzheimers Res Ther. 2017;9:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xiao M‐F, Xu D, Craig MT, et al. NPTX2 and cognitive dysfunction in Alzheimer's disease. ELife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Galasko D, Xiao M, Xu D, et al. Synaptic biomarkers in CSF aid in diagnosis, correlate with cognition and predict progression in MCI and Alzheimer's disease. Alzheimers Dement N Y N. 2019;5:871‐882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Soares HD, Potter WZ, Pickering E, et al. Plasma biomarkers associated with the apolipoprotein E genotype and Alzheimer disease. Arch Neurol. 2012;69:1310‐1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Weiner MW, Veitch DP, Aisen PS, et al. The Alzheimer's Disease Neuroimaging Initiative: a review of papers published since its inception. Alzheimers Dement J Alzheimers Assoc. 2013;9:e111‐194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Blennow K, Shaw LM, Stomrud E, et al. Predicting clinical decline and conversion to Alzheimer's disease or dementia using novel Elecsys Aβ(1‐42), pTau and tTau CSF immunoassays. Sci Rep. 2019;9:19024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Portelius E, Zetterberg H, Skillbäck T, et al. Cerebrospinal fluid neurogranin: relation to cognition and neurodegeneration in Alzheimer's disease. Brain J Neurol. 2015;138:3373‐3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kester MI, Teunissen CE, Sutphen C, et al. Cerebrospinal fluid VILIP‐1 and YKL‐40, candidate biomarkers to diagnose, predict and monitor Alzheimer's disease in a memory clinic cohort. Alzheimers Res Ther. 2015;7:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kvartsberg H, Portelius E, Andreasson U, et al. Characterization of the postsynaptic protein neurogranin in paired cerebrospinal fluid and plasma samples from Alzheimer's disease patients and healthy controls. Alzheimers Res Ther. 2015;7:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Headley A, De Leon‐Benedetti A, Dong C, et al. Neurogranin as a predictor of memory and executive function decline in MCI patients. Neurology. 2018;90:e887‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Toledo JB, Xie SX, Trojanowski JQ, Shaw LM. Longitudinal change in CSF Tau and Aβ biomarkers for up to 48 months in ADNI. Acta Neuropathol (Berl). 2013;126:659‐670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sutphen CL, McCue L, Herries EM, et al. Longitudinal decreases in multiple cerebrospinal fluid biomarkers of neuronal injury in symptomatic late onset Alzheimer's disease. Alzheimers Dement J Alzheimers Assoc. 2018;14:869‐879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xu D, Hopf C, Reddy R, et al. Narp and NP1 form heterocomplexes that function in developmental and activity‐dependent synaptic plasticity. Neuron. 2003;39:513‐528. [DOI] [PubMed] [Google Scholar]
- 33. Chang MC, Park JM, Pelkey KA, et al. Narp regulates homeostatic scaling of excitatory synapses on parvalbumin‐expressing interneurons. Nat Neurosci. 2010;13:1090‐1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cho RW, Park JM, Wolff SBE, et al. mGluR1/5‐dependent long‐term depression requires the regulated ectodomain cleavage of neuronal pentraxin NPR by TACE. Neuron. 2008;57:858‐871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gu Y, Huang S, Chang MC, Worley P, Kirkwood A, Quinlan EM. Obligatory role for the immediate early gene NARP in critical period plasticity. Neuron. 2013;79:335‐346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee S‐J, Wei M, Zhang C, et al. Presynaptic neuronal pentraxin receptor organizes excitatory and inhibitory synapses. J Neurosci Off J Soc Neurosci. 2017;37:1062‐1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. O'Brien RJ, Xu D, Petralia RS, Steward O, Huganir RL, Worley P. Synaptic clustering of AMPA receptors by the extracellular immediate‐early gene product Narp. Neuron. 1999;23:309‐323. [DOI] [PubMed] [Google Scholar]
- 38. O'Brien R, Xu D, Mi R, Tang X, Hopf C, Worley P. Synaptically targeted narp plays an essential role in the aggregation of AMPA receptors at excitatory synapses in cultured spinal neurons. J Neurosci Off J Soc Neurosci. 2002;22:4487‐4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pelkey KA, Barksdale E, Craig MT, et al. Pentraxins coordinate excitatory synapse maturation and circuit integration of parvalbumin interneurons. Neuron. 2015;85:1257‐1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pelkey KA, Barksdale E, Craig MT, et al. Pentraxins coordinate excitatory synapse maturation and circuit integration of parvalbumin interneurons. Neuron. 2016;90:661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Borchelt DR, Ratovitski T, van Lare J, et al. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19:939‐945. [DOI] [PubMed] [Google Scholar]
- 42. Swanson A, Willette AA. Alzheimer's Disease Neuroimaging Initiative. Neuronal Pentraxin 2 predicts medial temporal atrophy and memory decline across the Alzheimer's disease spectrum. Brain Behav Immun. 2016;58:201‐208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. van der Ende EL, Xiao M, Xu D, et al. Neuronal pentraxin 2: a synapse‐derived CSF biomarker in genetic frontotemporal dementia. J Neurol Neurosurg Psychiatry. 2020;91:612‐621. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its supporting information. Per the data‐sharing ADNI requirement, all data associated with this study has been uploaded to the central ADNI data site (LONI).