

Abstract
Expression of d-(−)-lactate dehydrogenase (d-LDH) and l-(+)-LDH genes (ldhD and ldhL, respectively) and production of d-(−)- and l-(+)-lactic acid were studied in Lactobacillus helveticus CNRZ32. In order to develop a host for production of pure l-(+)-isomer of lactic acid, two ldhD-negative L. helveticus CNRZ32 strains were constructed using gene replacement. One of the strains was constructed by deleting the promoter region of the ldhD gene, and the other was constructed by replacing the structural gene of ldhD with an additional copy of the structural gene (ldhL) of l-LDH of the same species. The resulting strains were designated GRL86 and GRL89, respectively. In strain GRL89, the second copy of the ldhL structural gene was expressed under the ldhD promoter. The two d-LDH-negative strains produced only l-(+)-lactic acid in an amount equal to the total lactate produced by the wild type. The maximum l-LDH activity was found to be 53 and 93% higher in GRL86 and GRL89, respectively, than in the wild-type strain. Furthermore, process variables for l-(+)-lactic acid production by GRL89 were optimized using statistical experimental design and response surface methodology. The temperature and pH optima were 41°C and pH 5.9. At low pH, when the growth and lactic acid production are uncoupled, strain GRL89 produced approximately 20% more lactic acid than GRL86.
Lactobacillus helveticus is a homofermentative, thermo- and acid-tolerant lactic acid bacterium with the capacity to produce high levels of lactic acid (23). It is widely used in the dairy industry and generally recognized as safe. Further, its behavior in both batch (2, 8) and continuous (1, 19, 21) lactic acid fermentations has been extensively studied. Lactic acid produced by L. helveticus is a racemic mixture of l-(+)- and d-(−)-isomers. l-(+)-Lactic acid is the preferred isomer, since d-(−)-lactic acid is not metabolized in humans, and for many applications, like manufacturing of biodegradable plastics and pharmaceutical products, it is more advantageous than a mixture of both isomers (22).
A few attempts have been made to improve and modify lactic acid production by metabolic engineering in lactobacilli, producing both l-(+)- and d-(−)-lactic acids. In L. helveticus and Lactobacillus plantarum, enhancement of l-(+)-lactic acid production has been tried by the inactivation of ldhD and by increasing the copy number of ldhL, respectively (5, 11). In L. helveticus, inactivation of ldhD led to a twofold increase in the amount of l-(+)-lactic acid, thus restoring the amount of total lactic acid to the level in the wild-type strain. In L. plantarum, overexpression of the ldhL gene increased the amount of l-(+)-lactate dehydrogenase (l-LDH) but had hardly any effect on lactic acid production. Furthermore, in L. plantarum, prevention of d-(−)-LDH (d-LDH) activity by the inactivation of ldhD or l-LDH activity by inactivation of ldhL did not substantially affect the total amount of lactic acid production (11, 12). Only in Lactococcus lactis, in which the ldhL gene is located as part of the las operon, did an increase in the copy number of the whole operon result in a slight increase in lactic acid production (10).
According to Savijoki and Palva (25), the L. helveticus ldhL gene is transcribed as a monocistronic unit, and expression was found to be most abundant in the exponential growth phase. The ldhD gene of the same species is expressed as a monocistronic unit as well (18; T. Rantanen and A. Palva, unpublished data). Production levels of d-(−)- and l-(+)-lactic acid seem to depend on changes in the expression of the ldhD and ldhL genes only to a limited extent. Furthermore, there are no data available revealing whether any coregulation occurs at the transcriptional level between the ldhL and ldhD genes.
In this work our aim was to improve the production of l-(+)-lactic acid by L. helveticus. For this purpose, two stable ldhD-negative L. helveticus derivates were constructed by a gene replacement method, and lactic acid synthesis in these strains was characterized at the enzyme and end product levels. In the first construct, transcription of the ldhD gene was prevented by an internal deletion of the promoter region. For the second construct, the ldhD gene was inactivated by replacing the ldhD structural gene with ldhL, resulting in duplication of the gene dose of ldhL. The goal of this strategy was to enhance and extend the synthesis of l-(+)-lactic acid into the stationary phase with the aid of the ldhD promoter. Finally, the behavior of genetically engineered strains was studied in a bioreactor, and the process conditions for lactic acid production by L. helveticus GRL89 were optimized using experimental design and response surface modeling.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
L. helveticus was routinely cultivated in MRS medium (Difco) at 37 or 42°C without shaking. Erythromycin (4 μg ml−1) was added to the medium when the pSA3 plasmid was used. In bioreactor fermentations of L. helveticus, a whey permeate medium (lactose, 41 g liter−1) supplemented with lactose (40 g liter−1) and yeast extract (20 g liter−1) was used.
Escherichia coli strains TOP10F′ (Invitrogen) DH5α and DH5αF′ (16) were grown in Luria broth. Kanamycin (50 μg ml−1), chloramphenicol (100 μg ml−1), tetracycline (10 μg ml−1), ampicillin (50 μg ml−1), and erythromycin (300 μg ml−1) were added to the growth medium when needed. IPTG (isopropylthiogalactopyranoside) was added to the growth medium to a final concentration of 1 mM when pZeRO-2 vectors with inserts were screened in E. coli. For the pUC19 and pJDC9 vectors in E. coli, IPTG and 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) were at the final concentrations of 0.5 mM and 40 μg ml−1, respectively.
Basic DNA techniques and bacterial transformation methods.
Plasmid DNAs from E. coli clones were isolated by using the Wizard Miniprep (Promega) or FlexiPrep (Pharmacia) kit. Total L. helveticus chromosomal DNA was isolated essentially as described earlier (28) but without guanidine hydrochloride treatment. E. coli and L. helveticus strains were transformed by electroporation using a Bio-Rad Gene Pulser and the methods described by Sambrook et al. (24) and Bhowmik and Steele (4), respectively. All other basic DNA methods were performed according to established procedures (24). The oligonucleotides were synthesized with an Applied Biosystems DNA/RNA synthesizer (model 392) and purified with NAP-10 columns (Pharmacia) or by ethanol precipitation. DNA was amplified by PCR in reaction conditions recommended by the manufacturer of Dynazyme DNA polymerase F-501L (Finnzymes). The labeling of DNA probes was performed with a digoxigenin (DIG)-DNA labeling kit or DIG-High Prime kit (Boehringer Mannheim).
Colony and Southern hybridizations were performed as follows. A positive E. coli clone (ERF573; Table 1) carrying the upstream region of ldhD in pZeRO-2 was screened by colony hybridization (14). Southern hybridizations with chromosomal DNA (2 μg) were performed after gel electrophoresis and transfer to a positively charged nylon membrane (Boehringer Mannheim), followed by hybrid detection with a DIG luminescence detection kit (Boehringer Mannheim). DNA sequencing of the upstream region of the L. helveticus CNRZ32 ldhD gene was done by an A.L.F. DNA sequencer (Pharmacia), and the sequence analysis was done with the PC/GENE software (IntelliGenetics). The BLAST network service was used to search for homologous protein sequences.
TABLE 1.
Plasmids and bacterial strains
| Plasmid or strain | Relevant features | Reference or source |
|---|---|---|
| Plasmids | ||
| pZeRO-2 | Kmr, lacZα-ccdB-based E. coli cloning vector | Invitrogen |
| pSA3 | Emr Tcr Cmr, Ts replicon, Streptococcus-E. coli shuttle vector | 9 |
| pJDC9 | Emr, E. coli cloning vector | 7 |
| pUC19 | Apr, E. coli cloning vector | 30 |
| pKTH2153 | Upstream region of the L. helveticus ldhD gene in pZeRO-2 | This work |
| pKTH2154 | ΔldhD construction in pSA3 | This work |
| pKTH2155 | slpA transcription terminator and downstream region of the ldhD gene in pSA3 | This work |
| pKTH2156 | Promoter region of the ldhD gene and structural gene of ldhL in pJDC9 | This work |
| pKTH2157 | ldhD::ldhL construction in pSA3 | This work |
| E. coli | ||
| Top10F′ | Host strain of pZeRO-2 | Invitrogen |
| DH5α | Transformation host | 16 |
| DH5αF′ | Transformation host | 16 |
| ERF573 | E. coli Top10F′ with pKTH2153 | This work |
| ERF574 | E. coli DH5αF′ with pKTH2154 | This work |
| ERF575 | E. coli DH5αF with pKTH2155 | This work |
| ERF576 | E. coli DH5αF′ with pKTH2156 | This work |
| ERF577 | E. coli DH5αF′ with pKTH2157 | This work |
| L. helveticus | ||
| GRL32 | Wild-type strain, l-LDH+d-LDH+ | CNRZ collectiona |
| GRL86 | ΔldhD mutant of CNRZ32 | This work |
| GRL89 | ldhD::ldhL mutant of CNRZ32 | This work |
CNRZ, Centre National de Recherches Zootechniques, Jouy-en-Josas, France.
For RNA isolations and primer extension work, total RNA was isolated from L. helveticus cells essentially as described by Vesanto et al. (26) or by using the RNeasy Mini and Midi kits (Qiagen). The primer extension analysis with a 5′-end-labeled fluorescein oligonucleotide (O1; Table 2) was performed with the A.L.F. DNA sequencer (Pharmacia) according to Vesanto et al. (27).
TABLE 2.
Oligonucleotides used
| Oligonucleotide | Nucleotide sequence (5′→3′) | Usea |
|---|---|---|
| O1 | CTTGAAGAGTATCTGCAGTGTAGTC | ldhD mRNA 5′-end mapping |
| O2 | ATTACGAATTCCAGTTAATGCCGCATTC | PCR |
| O3 | CTTAGGATCCGCTGGAATGATTATCGTG | PCR |
| O4 | GATAGGATCCTTACGCTATTCGAAAAGACG | PCR |
| O5 | TTGTCTGGGAATTCTTTACCTTC | PCR |
| O6 | CCCGCGGATCCTAATAATTATTATTTAGGTGA | PCR |
| O7 | GCATATCGATGTTTTTCCTAACAAAGGC | PCR |
| O8 | CGTATCTAGAATCTGGCTTTTCGCCG | PCR |
| O9 | TGCACGCAACTTAGTCTCTTG | PCR |
| O10 | GATAATTTTAACCATTCCTCATTATACGCTTC | R-PCR for ldhD-ldhL mRNA joint |
| O11 | TGAGGAATGGTTAAAATTATCACTAATAAAAAG | R-PCR for ldhD-ldhL mRNA joint |
| O12 | GAACGGATCCTTATTGACGAACCTTAACGC | PCR |
R-PCR, recombinant PCR.
Construction of an integration vector for L. helveticus ΔldhD strain.
An ldhD promoter deletion strain of L. helveticus was constructed using a gene replacement method (3). The strategy was to construct an integration vector containing the ldhD gene and its upstream region with a 0.6-kb deletion covering the ldhD promoter region. Briefly, a 0.9-kb fragment from the ldhD upstream region was amplified using primers O2 and O3 (Table 2), and a 0.8-kb downstream fragment from the ldhD structural gene was amplified with primer pair O4 and O5 (Table 2). The 0.9- and 0.8-kb fragments were ligated at their BamHI sites and amplified by PCR, followed by EcoRI digestion and ligation with pSA3. The resulting integration vector was designated pKTH2154 after cloning into E. coli.
Construction of an integration vector for L. helveticus ldhD::ldhL strain.
For the expression of ldhL under the ldhD promoter, the SphI-XbaI insert of the integration vector was formed from four separate DNA fragments (Fig. 1), which were the two flanking regions of ldhD needed for homologous recombination, the L. helveticus CNRZ32 ldhL fragment, and a transcription terminator of the Lactobacillus brevis slpA gene (28). The joint between the ldhD promoter and ldhL fragment was constructed at the transcription start sites of these two genes. In detail, the steps in constructing the ldhD::ldhL strain were as follows. The transcription termination region of slpA (0.1 kb) was amplified by PCR using primers O6 and O7 (Table 2). The amplified terminator fragment was ligated with a PCR fragment (0.9 kb) generated with primers O4 and O8 (Table 2) from the ldhD structural gene lacking the first 79 nucleotides of the structural gene after ClaI (authentic site) digestion. The resulting fragment was cloned into pSA3 as a BamHI-XbaI fragment, resulting in plasmid pKTH2155.
FIG. 1.

Replacement of the ldhD structural gene with ldhL. The overlapping oligonucleotides used in constructing the mRNA joint between the ldhD promoter region and the ldhL structural gene are shown. PldhD and tslpA refer to the ldhD promoter region and slpA transcription terminator, respectively.
To create an exact joint at the mRNA start sites of the ldhD and ldhL fragments, the recombinant PCR technique (17) was applied. The data derived from the primer extension of ldhD mRNA and previously published L. helveticus ldhL (25) were used to construct primers for the joint region. The DNA fragments to be annealed were synthesized by PCR with a Perfect Match PCR-Enhancer (Stratagene) and subsequently gel purified. The ldhD fragment (1.5 kb) was synthesized using primer pair O9 and O10 (Table 2) and the ldhL fragment (1.0 kb) was synthesized using oligonucleotides O11 and O12 (Table 2). The complementary bases between the two oligonucleotides used for the construction of the exact mRNA joint are presented in Fig. 1. In the second step of the recombinant PCR, the two DNA fragments were mixed in an equimolar ratio, denatured at 95°C for 30 s, and allowed to anneal for 1 min (first three cycles at 54°C and the rest at 65°C) followed by filling in of the 3′ ends by DNA polymerase (72°C for 3 min). This second step was repeated nine more times. PCR amplification of the double-stranded recombinant molecules was done immediately after the second step using primer pair O2 and O12 (Table 2) with 35 cycles. The amplified PCR fragment was then digested with SphI (authentic site) and BamHI enzymes. The resulting 1.9-kb insert was ligated with SphI- and BamHI-treated pKTH2155, and the integration vector formed was designated pKTH2157. The mRNA joint region was DNA sequenced in order to confirm its correctness.
Bioreactor cultivations.
Stock cultures of bacteria were stored in milk-MRS medium with 10% glycerol at −80°C. Precultures for bioreactor cultivations were grown in 10% skim milk medium at 42°C for 24 h. Four preinoculation steps were required before bioreactor cultivations. Bioreactor cultivations were performed in a 2-liter bioreactor (Biostat B2; Braun) using 1.5 liters of modified whey medium and 3% skim milk inoculation at 42°C. During the course of pH-controlled batch fermentations, the temperature was kept at 42°C and an agitation speed of 200 rpm was maintained. The pH was kept at 5.9 with automatic addition of 7 M NH4OH. Separating the supernatant from cells by centrifugation and storing them at −70°C prepared samples for lactic acid assays. The cell pellet was washed once with water, frozen in liquid nitrogen, and stored at −70°C for later use for enzymatic and protein assays. The cells for RNA isolations were harvested separately, frozen in liquid nitrogen, and stored at −70°C.
Optimization of pH and temperature was carried out in Biostat Q (B. Braun, Melsungen, Germany) multiple fermentor unit bioreactors with a 600-ml working volume. Agitation speed was 900 rpm. By using automatic addition of 6 M NaOH, the pH was maintained at desired values. Cultures were not aerated.
Statistical experimental design.
A central composite circumscribed 22 experimental design with two variables, temperature and pH, four star points, and four replicates at the center point, resulting in a total of 12 experiments, was used. The real and coded values of variables are shown in Table 3. Modde for Windows version 4.0 (Umetri AB, Stockholm, Sweden) software was used for statistical experimental design, analysis of the results, and drawing of the contour plots. Lactic acid (Y1) and biomass (Y2) production could be expressed as a polynomial model of the form:
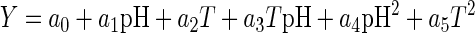 |
1 |
where dependent variable Y is the lactic acid concentration or biomass of L. helveticus GRL89, a0 to a5 are regression coefficients that are to be calculated with linear regression, and pH and T (temperature) are independent variables. The model was fitted using multiple linear regression. The calculated model was validated by MODDE using so-called R2 and Q2 values. The coefficient of determination, R2, describes the goodness of the fit of the model. Goodness of prediction, Q2, is a result of cross-validation, where part of the data is left out of the model and then predicted by the model.
TABLE 3.
Real and coded values of independent variables in optimization of pH and temperature for lactic acid production by L. helveticus GRL89
| Variable | −α | −1 | 0 | +1 | +α |
|---|---|---|---|---|---|
| Temp (°C) | 32.9 | 35 | 40 | 45 | 47.1 |
| pH | 4.85 | 5.1 | 5.7 | 6.3 | 6.55 |
Enzyme, protein, and lactic acid assays.
L. helveticus cell-free extracts from fermentations were prepared by sonicating samples in potassium phosphate buffer (10 mM, pH 6.5) followed by removal of cell debris by centrifugation and use of the supernatants in LDH assays. The LDH enzyme assays were performed spectrophotometrically (340 nm) at 30°C using lactate as the substrate. The l-LDH assays were performed in HEPES buffer (50 mM, pH 8.0) containing 10 mM NAD+ and 150 mM l-(+)-lactic acid, and the d-LDH assays were performed in Tris-HCl buffer (50 mM, pH 9.0) containing 10 mM NAD+ and 100 mM d-(−)-lactic acid. Protein content of the same cell-free samples was determined by the method of Bradford (6) with bovine serum albumin as the standard. For screening of d-(−)-lactic acid-negative phenotype, the assay of von Krusch and Lompe (29) was used. All results are based on average values measured from two parallel samples.
Total lactic acid and lactose were determined by high-performance liquid chromatography (HPLC) using a 35-cm HPX-87H+ cation-exchange column (Bio-Rad Laboratories) at a column temperature of 40°C with UV and RI detectors. The eluent used was 0.5 mM H2SO4 at a flow rate of 0.6 ml min−1. The injection volume of the sample was 20 μl. Furthermore, l-(+)- and d-(−)-lactic acid concentrations of samples were analyzed enzymatically using Boehringer Mannheim's kit (catalog no. 1 112 821). Biomass was determined as dry weight after centrifuging 10 ml of fermentation medium, washing once with distilled water, and drying at 115°C overnight.
Analysis of the level of ldhD and ldhL transcripts.
Isolation of total RNA was performed as described above from the samples withdrawn from bioreactor cultivation of L. helveticus wild-type strains. The isolated RNA was DNase treated before dot blotting. A constant amount of total RNA from different growth phases was used. The DIG-labeled probes chosen were targeted to hybridize with either the ldhL or ldhD structural gene of L. helveticus. Hybridizations were done as described by Sambrook et al. (24) and Hames and Higgins (15). After DIG detections, intensities of the positive hybridization signals were quantified from scanned films with Fluor-S equipment and Multianalyst software (Bio-Rad). The ldh mRNA dot blot analyses were performed in triplicate.
RESULTS AND DISCUSSION
Cloning of the upstream region of L. helveticus CNRZ32 ldhD.
For construction of a promoter deletion vector for ldhD, its upstream region was cloned. The ldhD upstream region was localized in a HindIII fragment of L. helveticus CNRZ32 chromosomal DNA by Southern analysis. The 0.3-kb probe used in the Southern hybridization was designed according to the L. helveticus CNRZ32 ldhD nucleotide sequence published previously (GenBank accession number UO7604). A HindIII- digested DNA fragment pool with a positive hybridization signal (2.6 kb) was ligated with the vector and used to transform E. coli cells, followed by screening of the positive clones by colony hybridization. One positive E. coli clone was chosen for further examination by DNA sequencing. The sequence analysis confirmed that the plasmid contained the upstream region of the L. helveticus CNRZ32 ldhD. The E. coli clone and the positive plasmid were designated ERF573 and pKTH2153, respectively.
Sequence analysis of the 2.6-kb insert in pKTH2153 revealed two open reading frames (ORF1 and ORF2) in the orientation opposite that of the ldhD gene (data not shown). The putative amino acid sequence of ORF1 had almost 70% identity with the Streptococcus pneumoniae exodeoxyribonuclease (P21998), whereas ORF2, located immediately upstream of the ldhD promoter region, had approximately 30% identity with a hypothetical Synechocystis sp. protein (BAA18702).
Construction of L. helveticus CNRZ32 ΔldhD strain.
An ldhD promoter deletion strain of L. helveticus was constructed using a gene replacement method (3). After the integration vector pKTH2154 was constructed (see Materials and Methods) in E. coli, it was transferred to L. helveticus, and integration by the first homologous recombination step was obtained by a temperature shift from 37 to 45°C under erythromycin selection. The second homologous recombination was achieved by growing the cells for 100 generations at 37°C without selection, resulting in 5% erythromycin-sensitive clones. One clone, designated GRL86, of 15 erythromycin-sensitive clones tested showed a d-(−)-lactic acid-negative phenotype.
Determination of the transcription start site of L. helveticus CNRZ32 ldhD.
In order to express the ldhL structural gene under the ldhD promoter, applying the exact joint of the ldhL and ldhD transcription start sites, the transcription initiation site of the ldhD gene was determined by primer extension. Transcription of ldhD was found to start from base −34 (a G) upstream of the first nucleotide of the start codon in the CNRZ32 ldhD sequence (data not shown).
Construction of L. helveticus CNRZ32 ldhD::ldhL strain.
Construction of an L. helveticus CNRZ32 ldhD::ldhL strain was achieved using the gene replacement strategy described above. The integration vector, designated pKTH2157, was constructed from four separate fragments (Fig. 1).
The integration of pKTH2157 in the L. helveticus CNRZ32 chromosome and gene replacement events were achieved as described above after confirmation that the first homologous recombination had taken place only in the region of the ldhD gene. After growing cells for approximately 100 generations at 37°C without selection, about 5% of clones were erythromycin sensitive. One clone (designated GRL89) of 25 erythromycin-sensitive clones tested was unable to produce d-(−)-lactic acid.
Southern hybridization.
To confirm that the desired gene replacements had taken place, Southern hybridizations were performed with chromosomal DNAs isolated from L. helveticus strains CNRZ32, GRL86, and GRL89 using both ldhD and ldhL probes. The same 0.8- and 1.0-kb fragments used for construction of the GRL86 and GRL89 strains, respectively, were utilized. The Southern blot data shown in Fig. 2 confirmed that the desired modifications had taken place. In GRL86 there is a 0.6-kb deletion of the ldhD gene (Fig. 2, lanes 1 and 2), and in GRL89 the 1-kb insertion can be demonstrated (Fig. 2, lanes 3 and 4). Furthermore, in GRL89 (Fig. 2, lanes 5 and 6), the extra copy of the ldhL gene is present in the chromosome, and no changes had occurred in the authentic ldhL locus.
FIG. 2.

Analysis of gene replacements in the ldhD locus of L. helveticus GRL86 and GRL89 chromosomal DNAs by Southern hybridization. Chromosomal DNAs were from the wild-type L. helveticus CNRZ32 (lane 1) and mutant GRL86 (lane 2) digested with SphI and XbaI and from CNRZ32 (lane 3) and GRL89 (lane 4) digested with DraI. DNAs in lanes 1 to 4 were hybridized with an ldhD gene probe. In lanes 5 and 6, the ldhL copies in L. helveticus CNRZ32 and GRL89, respectively, are shown after digestion of the DNAs with DraI and XbaI and hybridization in the presence of an ldhL gene probe.
Lactic acid production.
To assay the expression kinetics of the ldhL and ldhD genes and production of l-(+)- and d-(−)-lactic acid as a function of growth, mRNA analysis, LDH assays, and lactic acid assays were conducted with samples withdrawn from L. helveticus fermentation. The ratio of l-(+)- and d-(−)-lactic acids produced by the wild-type strain was 4:3 at the end of growth (Fig. 3). Production of l-(+)-lactic acid was mainly at the exponential phase of growth, whereas that of d-(−)-lactic acid started later and reached its maximum at the stationary phase (Fig. 3). This is in accordance with earlier observations of homofermentative lactobacilli, where d-(−)-lactic acid production was associated with the stationary phase and low pH of the medium and l-(+)-lactic acid production was associated with earlier growth phases (13). Since the enzyme assays for l-LDH and d-LDH were performed under different conditions, the specific activities obtained could not be quantitatively compared. However, the profiles of l-LDH and d-LDH activities are in agreement with those of lactic acid production. There seemed to be, however, a difference in the stability of d-LDH and l-LDH activities, with a sharp decrease in the d-LDH activity at the stationary phase (Fig. 3). The relative amounts of ldhL mRNA decreased prior to ldhD transcripts. The decreases in the relative amounts of both mRNAs preceded the rises in amounts of their respective acids (Fig. 3). The abrupt cessation of l-(+)-lactic acid production when about 85% of the maximum level of lactic acid production is reached cannot be explained by the lack of l-LDH at that time point. This would rather suggest that the intracellular conditions have changed in such a way that either the affinity of l-LDH for pyruvate is diminished or the catalytic activity of l-LDH is inhibited, resulting in a flow of pyruvate through d-LDH.
FIG. 3.

Expression of the ldh genes and production of lactic acid in L. helveticus CNRZ32 as a function of growth. Symbols: ●, cell density; ▵, l-LDH activity; ◊, d-LDH activity; ■, l-(+)-lactic acid; □, d-(−)-lactic acid; ▴, relative ldhL mRNA intensities with an ldhL gene probe; ⧫, relative ldhD mRNA intensities with an ldhD gene probe.
To study the effects of the gene replacements on growth and lactic acid production in the recombinant L. helveticus strains GRL86 and GRL89, they were cultivated in a pH-controlled bioreactor similarly to the wild-type strain. Minor, insignificant differences between the growth profiles of the recombinant strains and that of the wild type were found (Fig. 4A). Also, the total amount of lactic acid produced, as assayed by HPLC, was very similar in all cases (Fig. 4A). Furthermore, the enzymatic lactic acid assay confirmed that no d-(−)- lactic acid was produced by either of the mutant strains. This in agreement with earlier results obtained with the ldhD insertion inactivation strain by Bhowmik and Steele (5) and further confirms the lack of a lactic acid isomerase in L. helveticus. Similar results have been obtained by inactivation of either the ldhL or ldhD gene in L. plantarum (11, 12).
FIG. 4.

Growth, l-LDH activity, and lactic acid production in L. helveticus strains. Circles, CNRZ32; triangles, GRL86; squares, GRL89. (A) Open symbols refer to growth, and solid symbols refer to total lactic acid. (B) l-LDH activity.
The promoter deletion strain GRL86 grew consistently slightly faster than the wild-type strain, with an accompanying increase in the production rate of l-(+)-lactic acid. This may be due to a lower energy burden of the cell as the result of the lack of ldhD transcription and/or translation. Alternatively, this faster growth may be due to inactivation of the unknown orf2 gene. The production kinetics of l-(+)-lactic acid in mutant strains GRL86 and GRL89 did not differ significantly from each other or from that of total lactic acid in the wild-type CNRZ32 in these cultivation conditions. However, the production phase of l-(+)-lactic acid in the mutant strains was prolonged compared to l-(+)-lactic acid production by the wild type (Fig. 3 and 4). Thus, there was no decrease in the rate of l-(+)-lactate synthesis at a lactic acid concentration at which l-lactate excretion ceased completely in the wild-type strain. This suggests that the rate of l-LDH catalysis is not likely to depend on the external lactic acid concentration. Instead, the change of flow from pyruvate to d-lactate may be due to changes in substrate binding of these two enzymes.
Assays of LDH activity showed that the maximum levels of l-LDH activity in GRL86 and GRL89 were 53 and 93%, respectively, higher than that in CNRZ32 (Fig. 4B). This is in contrast to what was observed earlier with l-LDHs from L. plantarum. In this host, inactivation of neither the ldhL nor the ldhD gene seemed to affect the remaining LDH activity markedly (11, 12). In all three strains, maximum l-LDH activity was reached at the end of the exponential growth phase. Furthermore, as expected, with mutant strains GRL86 and GRL89 no d-LDH activity could be shown.
Optimization by response surface methodology.
In order to determine the effect of pH and temperature on lactic acid production, a response surface methodology was applied for the GRL89 strain. The results of the experimental design are summarized in Table 4. These results were fitted to equation 1 using multilinear regression. Resulting polynomes are plotted as the response surface in Fig. 5.
TABLE 4.
Lactic acid production by L. helveticus GRL89 at 25 h of fermentation using pH and temperature as independent variables
| Real values
|
Coded values
|
Production (g/liter)
|
|||
|---|---|---|---|---|---|
| pH | Temp (°C) | pH | Temp (°C) | Lactic acid | Biomass |
| 5.1 | 35 | −1 | −1 | 4.41 | 1.17 |
| 6.3 | 35 | +1 | −1 | 3.7 | 1.41 |
| 5.1 | 45 | −1 | +1 | 2.6 | 1.34 |
| 6.3 | 45 | +1 | +1 | 42.6 | 2.62 |
| 4.9 | 40 | −α | 0 | 11.85 | 1.69 |
| 6.6 | 40 | +α | 0 | 1.84 | 1.84 |
| 5.7 | 32.9 | 0 | −α | 2.72 | 0.58 |
| 5.7 | 47.1 | 0 | +α | 3.14 | 1.73 |
| 5.7 | 40 | 0 | 0 | 50.08 | 5.28 |
| 5.7 | 40 | 0 | 0 | 50.64 | 4.82 |
| 5.7 | 40 | 0 | 0 | 50.16 | 4.81 |
| 5.7 | 40 | 0 | 0 | 45.08 | 5.28 |
FIG. 5.

Effect of pH and temperature on lactic acid production (A) and biomass formation (B) in L. helveticus GRL89, shown as response surfaces.
A response surface for lactic acid production as a function of pH and temperature by L. helveticus GRL89 is presented in Fig. 5A. As shown, the optimal conditions for lactic acid production are pH 5.6 to 6.0, and the optimal temperature is 40 to 42°C. The statistical values for the model were R2 = 0.875 and Q2 = 0.504. Significant terms in the model are the second-order term for pH, with a probability (P) value of 0.005, and the second-order term for temperature (P = 0.0024).
Figure 5B shows the response surface for the model predicting L. helveticus GRL89 biomass formation as function of temperature and pH. In this model also, both temperature and pH were very significant (P = 0.00035 and 0.00018, respectively). The optimum conditions for biomass were at the center of the experimental domain, 41°C and pH 5.8. These values are very close to the conditions used in the primary batch fermentations and are also in good agreement with earlier studies (20) where unmodified L. helveticus strains have been used. The steep curvature of the response surface is due to high values of the coefficients of second-order terms. Consequently, the optimum area for the growth of L. helveticus GRL89 is quite small. The statistical values for this model were R2 = 0.9435 and Q2 = 0.6197.
Lactic acid production at low pH.
Coupling of growth and acid production in L. helveticus has been shown to be controlled mainly by cultivation pH (2). To test whether there is any difference in the behavior of the ldhD and ldhL promoters under such conditions, strains GRL86 and GRL89 were cultivated at 44°C and pH 5.4. Our preliminary experiments indicated (results not shown) that in these conditions, biomass growth and lactic acid production are uncoupled.
Lactic acid production started in strain GRL86 slightly earlier than in strain GRL89 (Fig. 6A). This was due to faster growth of the deletion strain, as shown by the biomass data from these fermentations (Fig. 6B). The maximal volumetric productivities of the strains were 3.34 and 3.21 g liter−1 h−1, respectively. However, a distinctive increase in lactic acid production with GRL89 was obtained at the late stationary phase. The final lactic acid yield with strain GRL89 was 91.6%, whereas that of strain GRL86 was only 76.2% (Fig. 6A).
FIG. 6.

Batch fermentations of genetically engineered L. helveticus strains GRL86 (open symbols) and GRL89 (solid symbols): concentrations of lactose (circles) and lactic acid (squares) (A) and change in dry weight (B) as a function of time in two parallel cultivations at pH 5.4 and 44°C.
ACKNOWLEDGMENTS
This work was supported by the National Technology Agency, Finland.
We are grateful to Ilkka Palva for valuable discussion and critical reading of the manuscript. We also thank Anneli Virta for the sequencing work and Jaana Jalava for technical assistance.
REFERENCES
- 1.Amrane A, Prigent Y. A novel concept of bioreactor: specialized function two-stage continuous reactor, and its application to lactose conversion into lactic acid. J Biotechnol. 1996;45:195–203. [Google Scholar]
- 2.Amrane A, Prigent Y. Differentiation of pH and free lactic acid effects on the various growth and production phases on Lactobacillus helveticus. J Chem Technol Biotechnol. 1999;74:33–40. [Google Scholar]
- 3.Bhowmik T, Fernández L, Steele J L. Gene replacement in Lactobacillus helveticus. J Bacteriol. 1993;175:6341–6344. doi: 10.1128/jb.175.19.6341-6344.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhowmik T, Steele J L. Development of an electroporation procedure for gene disruption in Lactobacillus helveticus CNRZ 32. J Gen Microbiol. 1993;139:1433–1439. [Google Scholar]
- 5.Bhowmik T, Steele J L. Cloning, characterization and insertional inactivation of the Lactobacillus helveticusd-(−)-lactate dehydrogenase. Appl Microbiol Biotechnol. 1994;41:432–439. doi: 10.1007/BF00939032. [DOI] [PubMed] [Google Scholar]
- 6.Bradford M M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 7.Chen J D, Morrison D A. Construction and properties of a new insertion vector, pJDC9, that is protected by transcriptional terminators and useful for cloning of DNA from Streptococcus pneumoniae. Gene. 1988;64:155–164. doi: 10.1016/0378-1119(88)90489-1. [DOI] [PubMed] [Google Scholar]
- 8.Chiarini L, Mara L, Tabacchioni S. Influence of growth supplements on lactic acid production in whey ultrafiltrate by Lactobacillus helveticus. Appl Microbiol Biotechnol. 1992;36:461–464. [Google Scholar]
- 9.Dao M L, Ferretti J J. Streptococcus-Escherichia coli shuttle vector pSA3 and its use in the cloning of streptococcal genes. Appl Environ Microbiol. 1985;49:115–119. doi: 10.1128/aem.49.1.115-119.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davidson B E, Llanos R M, Cancilla M R, Redman N C, Hillier A J. Current research on the genetics of lactic acid production in lactic acid bacteria. Int Dairy J. 1995;5:763–784. [Google Scholar]
- 11.Ferain T, Garmyn D, Bernard N, Hols P, Delcour J. Lactobacillus plantarum ldhL gene: overexpression and deletion. J Bacteriol. 1994;176:596–601. doi: 10.1128/jb.176.3.596-601.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferain T, Hobbs J N, Jr, Richardson J, Bernard N, Garmyn D, Hols P, Allen N E, Delcour J. Knockout of the two ldh genes has a major impact on peptidoglycan precursor synthesis in Lactobacillus plantarum. J Bacteriol. 1996;178:5431–5437. doi: 10.1128/jb.178.18.5431-5437.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garvie E I. Bacterial lactate dehydrogenases. Microbiol Rev. 1980;44:106–139. doi: 10.1128/mr.44.1.106-139.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grünstein M, Hogness D S. Colony hybridization: a method for the isolation of cloned DNAs that contain a specific gene. Proc Natl Acad Sci USA. 1975;72:3961–3965. doi: 10.1073/pnas.72.10.3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hames B, Higgins S. Nucleic acid hybridization: a practical approach. Oxford, England: IRL Press; 1985. [Google Scholar]
- 16.Hanahan D. Studies on transformation of Escherichia coli with plasmids. J Mol Biol. 1983;166:557–580. doi: 10.1016/s0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
- 17.Higuchi R. Recombinant PCR. In: Innis M A, Gelfand D H, Sninsky J J, White T J, editors. PCR protocols. San Diego, Calif: Academic Press; 1990. pp. 177–183. [Google Scholar]
- 18.Kochhar S, Hottinger H, Chuard N, Taylor P G, Atkinson T, Scawen M D, Nicholls D J. Cloning and overexpression of Lactobacillus helveticusd-lactate dehydrogenase gene in Escherichia coli. Eur J Biochem. 1992;208:799–805. doi: 10.1111/j.1432-1033.1992.tb17250.x. [DOI] [PubMed] [Google Scholar]
- 19.Kulozik U. Physiological aspects of continuous lactic acid fermentations at high dilution rates. Appl Microbiol Biotechnol. 1998;49:506–510. [Google Scholar]
- 20.Kulozik U, Wilde J. Rapid lactic acid production at high cell concentrations in whey ultrafiltrate by Lactobacillus helveticus. Enzyme Microbiol Technol. 1999;24:297–302. [Google Scholar]
- 21.Øyaas J, Storrø I, Levin D W. Uptake of lactose and continuous lactic acid fermentation by entrapped non-growing Lactobacillus helveticus in whey permeate. Appl Microbiol Biotechnol. 1996;46:240–249. [Google Scholar]
- 22.Potera C. Natural lactic acid produced from whey by Ecochem. Genet Eng News. 1992;12:1–22. [Google Scholar]
- 23.Roy D, Goulet J, Le Duy A. Batch fermentation of whey ultrafiltrate by Lactobacillus helveticus for lactic acid production. Appl Microbiol Biotechnol. 1986;24:206–213. [Google Scholar]
- 24.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 25.Savijoki K, Palva A. Molecular genetic characterization of the l-lactate dehydrogenase gene (ldhL) of Lactobacillus helveticus and biochemical characterization of the enzyme. Appl Environ Microbiol. 1997;63:2850–2856. doi: 10.1128/aem.63.7.2850-2856.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vesanto E, Varmanen P, Steele J L, Palva A. Characterization and expression of the Lactobacillus helveticus pepC gene encoding a general aminopeptidase. Eur J Biochem. 1994;24:991–997. doi: 10.1111/j.1432-1033.1994.00991.x. [DOI] [PubMed] [Google Scholar]
- 27.Vesanto E, Savijoki S, Rantanen T, Steele J L, Palva A. An X-prolyl dipeptidyl aminopeptidase (pepX) gene from Lactobacillus helveticus. Microbiology. 1995;141:3067–3075. doi: 10.1099/13500872-141-12-3067. [DOI] [PubMed] [Google Scholar]
- 28.Vidgren G, Palva I, Pakkanen R, Lounatmaa K, Palva A. S-layer protein gene of Lactobacillus brevis: cloning by polymerase chain reaction and determination of the nucleotide sequence. J Bacteriol. 1992;174:7419–7427. doi: 10.1128/jb.174.22.7419-7427.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.von Krusch U, Lompe A. Schnelltest zum qualitativen Nachweis von l-(+)- und d-(−)-Milchsäure für die Bestimmung von Milchsäurebakterien. Milchwissenschaft. 1982;37:65–68. [Google Scholar]
- 30.Yanisch-Perron C, Vieira J, Messing J. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene. 1985;33:103–119. doi: 10.1016/0378-1119(85)90120-9. [DOI] [PubMed] [Google Scholar]