

Abstract
In this study, FDA-approved HCV antiviral drugs and their structural analogues—several of them in clinical trials—were tested for their inhibitory properties toward the SARS-CoV-2 spike protein bound to angiotensin-converting enzyme 2 (6M0J) using a virtual screening approach and computational chemistry methods. The most stable structures and the corresponding binding affinities of thirteen such antiviral compounds were obtained. Frontier molecular orbital theory, global reactivity descriptors, molecular docking calculations and electrostatic potential analysis were used to hypothesize the bioactivity of these drugs against 6M0J. It is found that an increased affinity for the protein is shown by inhibitors with large compound volume, relatively higher electrophilicity index, aromatic rings and heteroatoms that participate in hydrogen bonding. Among the tested drugs, four compounds 10–13 showed excellent results—binding affinities − 11.2 to − 11.5 kcal mol−1. These four top scoring compounds may act as lead compounds for further experimental validation, clinical trials and even for the development of more potent antiviral agents against the SARS-CoV-2.
Graphical abstract
Approved HCV drugs and analogues were tested for their bioactivity towards the SARS-CoV-2 (6M0J) using virtual screening, ESP and MD analysis.

Supplementary Information
The online version contains supplementary material available at 10.1007/s11030-022-10469-7.
Keywords: COVID-19, HCV drugs, SARS-CoV-2, 6M0J, Molecular docking, ESP analysis, MD analysis, Drug repurposing, In silico screening
Introduction
In December 2019, the first cases of infection from a novel coronavirus were reported. Later, the causal organism was reported as a newly mutated strain of SARS coronavirus and termed SARS-CoV-2. This new coronavirus was implicated in an outbreak of a severe pneumonia like illness COVID-19 [1, 2] and has led to a worldwide pandemic with more than 372,000,000 cases and 5,600,000 deaths reported so far [3–8].
In the past eight months, several pharmaceutical companies have announced hugely successful trials of their COVID-19 vaccines [9]. Even though these vaccines are promising, there is no guarantee that they will cure all those vaccinated, and as a consequence there is need for other treatments. Therefore, the development of antiviral agents is an urgent priority even though it usually takes many years for new drugs to be discovered, clinically tested and approved. A good strategy would be trying to find already approved drugs that have some efficacy against similar type of viruses [10] and then test the efficacy of these drugs against SARS-CoV-2 using computational chemistry methods and molecular docking [11–23]. The most effective of these drugs can then be clinically tested and approved.
Researchers are attempting to repurpose a wide variety of existing medications for COVID-19, including HCV, HIV and influenza drugs. Although not closely related, hepatitis C virus (HCV) and the new coronavirus SARS-CoV-2 are both single-stranded RNA viruses. This has led some scientists to suggest that the same antiviral drugs might work against both. Recently, sofosbuvir and daclatasvir, two antiviral drugs used to treat hepatitis C, were associated with faster recovery, shorter hospitalization and improved survival among people with moderate or severe COVID-19 [24]. If larger studies confirm these findings, generic versions of sofosbuvir and daclatasvir could potentially be an affordable and widely accessible treatment for the new coronavirus.
In a previous study, FDA-approved antiviral drugs and lopinavir analogues in clinical trials were tested for their inhibitory properties toward the SARS-CoV-2 spike protein bound to angiotensin-converting enzyme 2 (ACE2) (6M0J) using a virtual screening approach and computational chemistry methods [25]. Among them, four compounds showed excellent results—binding interactions − 9.0 to − 9.3 kcal mol−1—for use against the newly emerged strain of coronavirus.
In the present work, approved HCV drugs and their structural analogues in clinical trials are tested for their inhibition toward the COVID-19 protein (6M0J) using in silico methods. The present work has the following objectives: (1) to obtain the ground-state optimized structures of selected HCV drugs and their analogues (Tables 1 and 2) at a semiempirical level (PM3) [26–29] and subsequently calculate global reactivity descriptors—chemical potential (μ), electrophilicity index (ω)—to identify differences in reactivity. (2) To calculate the energy gap between the highest occupied and the lowest unoccupied molecular orbital (HOMO–LUMO energy gap) of these drugs at the ground state since small energy gaps are associated with higher chemical reactivity and low kinetic stability [30–33]. (3) To investigate the interaction of the ground-state optimized structure of the above-approved drugs and their analogues (Table 2) with the SARS-CoV-2 spike protein (6M0J) [34] using computational chemistry methods and molecular docking. (4) To determine the binding affinities of these drugs (ligands) with the SARS-CoV-2 spike protein (6M0J).
Table 1.
List of HCV drugs and analogues docked against the SARS-CoV-2 spike protein 6M0J


Table 2.
Binding affinity data of inhibitors 1–13 against 6M0J, global reactivity descriptors, Pkd (kd dissociation constants) and binding free energies (ΔGbind)
| # | Compounda | HOMO—LUMO Energy Gap (a.u) | Hardness (n) | Softness (s) | Chemical potential (μ) | Electrophilicity index (ω) | Pkd | Binding affinity (ADFR) (kcal mol−1) | Binding affinity (Vina) (kcal mol−1) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | CID 132,111,205 | 0.2776 | 0.1388 | 7.2037 | − 0.1754 | 0.1109 | 7.34 | − 7.9 | − 9.5 |
| 2 | Daclatasvir | 0.2779 | 0.1389 | 7.1972 | − 0.1708 | 0.1050 | 6.53 | − 9.0 | − 9.7 |
| 3 | CID 135,195,163 | 0.2805 | 0.1403 | 7.1293 | − 0.1697 | 0.1026 | 8.30 | − 9.0 | − 10.2 |
| 4 | Setrobuvir | 0.2986 | 0.1493 | 6.6989 | − 0.1770 | 0.1049 | 6.39 | − 9.6 | − 10.2 |
| 5 | Paritaprevir | 0.2999 | 0.1499 | 6.6692 | − 0.1824 | 0.1109 | 6.77 | − 10.2 | − 10.2 |
| 6 | CID 117,896,834 | 0.2980 | 0.1490 | 6.7108 | − 0.1962 | 0.1292 | 5.98 | − 9.0 | − 10.6 |
| 7 | CID 135,976,538 | 0.2932 | 0.1466 | 6.8211 | − 0.1901 | 0.1232 | 6.53 | − 9.4 | − 10.7 |
| 8 | Elbasvir | 0.2766 | 0.1383 | 7.2297 | − 0.1711 | 0.1059 | 7.55 | − 10.3 | − 10.8 |
| 9 | CID 122,523,225 | 0.2745 | 0.1373 | 7.2850 | − 0.1748 | 0.1113 | 6.04 | − 11.6 | − 10.8 |
| 10 | CID 132,182,097 | 0.2769 | 0.1384 | 7.2235 | − 0.1667 | 0.1004 | 8.50 | − 9.1 | − 11.2 |
| 11 | CID 131,982,844 | 0.2938 | 0.1469 | 6.8077 | − 0.1833 | 0.1144 | 6.86 | − 10.4 | − 11.3 |
| 12 | CID 117,860,584 | 0.3011 | 0.1506 | 6.6413 | − 0.1893 | 0.1190 | 7.22 | − 10.0 | − 11.5 |
| 13 | CID 132,247,343 | 0.2969 | 0.1484 | 6.7374 | − 0.1844 | 0.1145 | 6.13 | − 9.5 | − 11.5 |
| Acetaminophenb | − 5.4 | − 6.6 | |||||||
| Darunavirc | − 8.2 | − 8.5 |
aCiD’s are compound identifier numbers in PubChem
bNegative control
cPositive control
It is known that the virus enters the host cell by binding of the viral spike glycoprotein to the host receptor, angiotensin-converting enzyme 2 (ACE2) [35] therefore (6M0J) seems to be a biologically meaningful receptor.
Computational methods
The semiempirical calculations were carried out using the ArgusLab software [36]. Ab initio molecular orbital calculations were carried out using the ORCA 4.1 quantum chemistry program package [37]. The most stable optimized geometries and frequency calculations of the studied compounds were obtained from the PM3 method at the semiempirical level and from B97-3c/def2-mTZVP [38] methods and basis set at the ab initio level.
The recent resolved three-dimensional crystal structure of SARS-CoV-2 spike protein bound to angiotensin-converting enzyme 2 (ACE2) (PDB ID: 6M0J) [34] was retrieved from the Protein Data Bank with a resolution of 2.45 Å. Approved drugs and their analogues were downloaded from PubChem. The most stable optimized geometries were obtained as described above and were subjected to molecular docking simulation against the SARS-CoV-2 spike protein (PDB ID: 6M0J) using the AutoDock Vina [39] and AutoDock ADFR software. The binding dissociation constant kd between protein and ligand was calculated using KDEEP [40]. In molecular docking simulations [41, 42], flexible–ligand/rigid–receptor docking was performed and accurate docking conditions were selected. All hetero atoms and water molecules were eliminated before docking. The grid box mapping parameters for AutoDock Vina were chosen as follows: Box dimension (Å) x = 64 y = 66 z = 72 and Center (Å) x =− 23.088 y = 18.676 z = − 27.106 along x, y and z directions, respectively. Electrostatic potentials on molecular/vdW surfaces were computed using the Multifunctional Wavefunction Analyzer Multiwfn [43]. Discovery Studio Visualizer was used to depict protein–ligand interactions [44].
Results and discussion
The HCV-studied drugs in this work are listed in Table 1. Computed binding affinities of these compounds using the procedures described above are collected in Table 2. The binding affinity values (kcal mol−1) computed by AutoDock Vina are averages of ten independent trials. The AutoDock ADFR values (kcal mol−1) are averages of the best binding affinity values of each ligand with the protein in the five highest scoring pockets. A plot of the Vina and ADFR binding values is shown in Graph A1 (Online Appendix A). The former values were used throughout this study. The binding dissociation constants kd were singly determined. Global reactivity descriptors of the tabulated compounds—hardness (n), softness (s), chemical potential (μ)—were calculated using the PM3 version of SCF MO and the ArgusLab software [36]. It may be pointed out that in SAR studies the semiempirical SCF methods are more reliable than ab initio methods [45]. The HOMO and LUMO energy values were obtained for all molecules, and then, the global reactivity descriptors were calculated from these values considering Koopmans’ theorem according to the following equations [46, 47]:
| 1 |
| 2 |
| 3 |
| 4 |
The obtained results show that best binding energies (< − 10.6 kcal/mol) are observed in most cases for drugs that exhibit both relatively higher electrophilicity indices ω (electrophilicity index ω > 0.11) and chemical potential μ (μ > 7.6) (Table 2). The electrophilicity index ω encompasses the tendency of an electrophile to acquire an extra amount of electron density. The chemical potential μ is associated with the feasibility of a system to exchange electron density with the environment. Assuming that the HCV drugs of Table 2 can be divided into two groups with respect to binding affinities (strong inhibitors ≤ − 10.6 kcal mol−1, weak inhibitors > − 10.6 kcal mol−1), a logistic regression test was performed with covariates ω (electrophilicity index) and μ (chemical potential) using SPSS [48]. The likelihood ratio tests (Fig. 1) check the contribution of each variable and reveal that ω is significant (sig. < 0.05). Higher ω values characterize the strong inhibitor group (binding affinities.
Fig. 1.
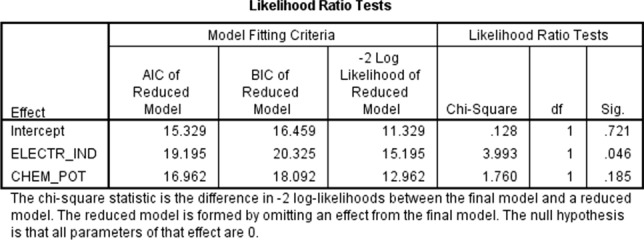
Logistic regression Likelihood Ratio Tests to check the contribution of the covariates electrophilicity index ω and chemical potential μ to the observed binding affinity of the molecules in Table 2. The electrophilicity index ω variable (ELECTR_IND) is significant (sig. < 0.05)
< − 10.6 kcal mol−1). Elbasvir and paritaprevir analogues 10, 11–13 (Tables 1 and 2) fall at the high end of the strong inhibitor category. Elbasvir 8 and its analogue 9, paritaprevir analogue 6 and setrobuvir analogue 7 fall at the low end of this category. Compounds 1–5 belong to the weak inhibitor group. The higher binding affinity observed for 6–13 can be attributed mainly to noncovalent interactions. The formed ligand (drug in Table 2)–receptor (6M0J) complexes reveal that Pi-alkyl, Pi–Pi stacking, conventional hydrogen bonding and halogen bonds are able to increase the binding affinity and explain the differences in binding energies (Table A1, Online Appendix A). It has been reported that particularly hydrogen bonds < 2.3 Å are able to increase the binding affinity considerably and that halogen bonds have almost similar importance as hydrogen bonds in chemical and biological systems [49, 50]. Among all sort of interactions such as CH/O, CH/N, OH/π and NH/π, the CH/π is the most prominent interaction found between drugs-proteins. Docking interactions of paritaprevir analogues 12 and 13 (Tables 1, 2 and A1) with 6M0J are shown in Figs. 2 and 3, respectively. Several Pi–Pi stacked, Pi-alkyl interactions and at least one hydrogen bond < 2.3 Å are observed. Three Pi–Pi stacked interactions are observed between the fluorine-substituted phenanthridine ring of 12 and PHE390 and four Pi-alkyl interactions with ARG393, PHE40, TRP349 and HIS378. Four hydrogen bonds are formed with the protein. The hydrogen bond with ARG393 is at a distance of 2.25 Å. The phenanthridine and pyrazine rings of 13 interact by forming a total of four Pi–Pi stacked interactions with PHE390 and TRP349, respectively. Hydrogen bonds are observed between the oxygen atoms attached to the sulfur atom and ARG514 and the pyrazine nitrogens and SER47. The hydrogen bond with ARG514 is at a distance of 2.02 Å providing increased binding affinity. A Pi-Alkyl interaction is observed between the phenanthridine pi-orbitals and ARG393 (Fig. 3, Table A1). Compounds at the low end of the strong inhibitor category show hydrogen bonding—including hydrogen bonds < 2.3 Å—alkyl and Pi alkyl bonding but with the exception of 7 and elbasvir 8 lack Pi-Pi stacked interactions (Table A1). Elbasvir shows seven conventional hydrogen bond interactions with 6M0J, while three of them with ARG273, HIS345 and HIS505 are < 2.3 Å. A total of six carbon–hydrogen bond interactions are observed with ALA348, PRO346, GLU375, ASP382, TYR385, PHE390. A Pi–Pi stacked bonding interaction is formed between the p orbitals of the imidazole ring of 8 and the p orbitals of HIS345. Pi-Pi–T-shaped interactions are observed between the fused aromatic and 2-pyrroline rings of 8 and HIS378, TRP349 and HIS401 of the protein. Pi-alkyl interactions are formed between the 2-pyrroline ring of 8 and PHE390 and PHE40 and an alkyl bonding interaction with ARG393 (Fig. 4 and Table A1). Compounds in the low inhibitor category show less prominent interactions with the protein comparing with those in the other category. Hydrogen bonding—including hydrogen bonds < 2.3 Å—alkyl and Pi alkyl bonding is also observed but with the exception of 3 and 4 lack Pi–Pi stacked interactions (Table A1). Daclatasvir 2 is a typical example of the compounds in this category (Fig. 5). Several hydrogen bonds are observed with the one between the N–H hydrogen of the acetamido group and GLU208 at a distance of 2.0 Å. A Pi–alkyl interaction is formed between the pyrolidine ring of the ligand and HIS195 of the protein.
Fig. 2.

Docking interactions of compound 12 (Tables 1 and 2) with 6M0J (expanded version in Online Appendix A)
Fig. 3.

Docking interactions of compound 13 (Tables 1 and 2) with 6M0J (expanded version in Online Appendix A)
Fig. 4.

Docking interactions of elbasvir compound 8 (Tables 1 and 2) with 6M0J (expanded version in Online Appendix A)
Fig. 5.

Docking interactions of daclatasvir 2 (Tables 1 and 2) with 6M0J
Drug (ligand) interactions with 6M0J (protein) were further studied by electrostatic potential analysis on the drugs molecular surface. The value of electrostatic calculations for understanding and predicting molecular properties has been recognized for decades. It is well known that molecular electrostatics can be predictive of a molecule’s chemical reactivity and its ability to form certain types of interactions. Electrostatic potential surfaces (ESPs) are used to visualize the electrostatic nature of molecules [51, 52]. Drugs from the strong (compound 12) and weak inhibitor categories (daclatasvir 2) with respect to their binding affinity toward 6M0J are selected from Table 2 and their wavefunctions produced at the B97-3c/def2-mTZVP [38] level. The electrostatic potential of the above compounds on their vdW surfaces was computed using the above wavefunctions. As the final part of ESP analysis, the molecular surface area in each ESP range was calculated in order to quantitatively determine ESP distribution on the whole molecular surface [43]. The obtained results of surface areas (Å2) and corresponding electrostatic potentials ESP (kcal mol−1) were used to plot histogram graphs of 2 and 12 (Figs. 6 and 7, respectively).
Fig. 6.

Surface area (Å2) in each electrostatic potential (kcal mol−1) range on the vdW surface of daclatasvir 2
Fig. 7.

Surface area (Å2) in each electrostatic potential (kcal mol−1) range on the vdW surface of compound 12
From the graphs in Figs. 6 and 7, it can be seen that there is a large portion of molecular surface having low ESP values, namely from − 20 to 20 kcal mol−1. There are also small areas having remarkable positive and negative ESP values, corresponding to the regions closed to the global ESP minimum and maximum, respectively. These global surface maxima and minima were found to be 0.06617912 a.u. (41.528059 kcal mol−1) and − 0.07777287 a.u. (− 48.803254 kcal mol−1) for compound 12, and 0.05207833 a.u. (32.679670 kcal mol−1) and − 0.05769414 a.u. (− 36.203650 kcal mol−1) for compound 2, and their observed differences suggested that the ESP distribution on the vdW surface fluctuates more remarkably in the former. Compound 12 shows higher positive charge density than daclatasvir 2 and a higher overall average charge equal to 0.00300125 a.u. (1.88332 kcal mol−1) compared to an overall average charge equal to 0.00061890 a.u. (0.38837 kcal mol−1) of 2 (Table 3). This higher observed overall average charge—consistent with the higher electrophilicity shown (Table 2)—coupled with other factors may explain the higher attraction shown by 12 toward or inside the negatively charged protein binding pocket (Fig. 8). The ESP plots of compounds 2 and 12 mapped onto the electron density surface for the ground state are shown in Figs. 9 and 10, respectively. Electron-rich (negative ESP) and electron-deficient areas (positive ESP) are indicated with red and white colors, respectively. Electron-rich areas are over oxygen, sulfur, nitrogen and fluorine atoms of 12 (Fig. 10), and these atoms participate in the docking interactions of this drug with 6M0J (Fig. 2). Positive potential appears over hydrogen atoms and the alkyl group, and this is also consistent with the reactivity shown and the hydrogen bonds formed (Fig. 2). Similarly, the ESP plot of compound 2 is very effective to predict the reactive sites of the molecule with the target protein 6M0J [52] (Figs. 5 and 9). Electron-rich areas appear over oxygen and nitrogen, and these atoms participate in hydrogen bonds. Electron-poor areas are observed over hydrogen atoms and alkyl groups. These results demonstrate that weak interactions between molecules, including H bonds and halogen bonds, can be predicted and explained by analyzing the magnitude and positions of the minima and maxima in an electrostatic potential (ESP) on the molecular vdW surface [53].
Table 3.
Surface analysis of electrostatic potential values (ESP) (units a.u.) computed for compounds 2 and 12 (Table 2) using the Multiwfn analyzer [43]
| ESP values (a.u.) | Compound 2 | Compound 12 |
|---|---|---|
| Maximum value | 0.052078 | 0.066179 |
| Minimum value | − 0.057694 | − 0.077772 |
| Overall average value | 0.00061890 | 0.00300125 |
| Positive average value | 0.01292542 | 0.02232525 |
| Negative average value | − 0.01804650 | − 0.02198169 |
| Internal charge separation | 0.01484047 | 0.02193806 |
Fig. 8.

Compound 12 inside the negatively charged 6M0J binding pocket
Fig. 9.

Electrostatic potential (ESP) mapped electron density surface of compound 2 at the PM3 level
Fig. 10.

Electrostatic potential (ESP) mapped electron density surface of compound 12 at the PM3 level
The formed ligand (drug in Table 2)–receptor (6M0J) complexes reveal that pi–pi stacking, pi–alkyl and halogen bonds are able to increase the binding affinity and explain the differences in binding energies. Furthermore, to evaluate the stability of the formed protein–ligand complexes, these structures were subjected to fully solvated atomistic molecular dynamics (MD) simulations using VMD [54] and NAMD [55]. MD simulations were conducted at 310 K for 30 ns. Structural fluctuations of protein and the ligands are indicated by variation in the root-mean-square variation (RMSD). The RMSD of the free protein remained stable between 8 and 22 ns at 2 Å and then slightly increased and fluctuated around 2.1 Å from 22 to 30 ns (Fig. 11). The RMSD of the 6M0J and 13 complex was balanced after 24 ns and slightly fluctuated between 2.2 and 2.3 Å, while the RMSD plot of the 6M0J and 12 initially increased for about 10 ns and then slightly fluctuated around 2 Å. The RMSD of 6M0J and 12 showed that this complex had the greatest effect on protein’s stability (Fig. 11). The RMSD of 6M0J and 11 increased for about 25 ns and then fluctuated between 2.2 and 2.4 Å. As depicted in Fig. 11, the RMSD of the 6M0J and 10 complex was balanced after 20 ns and then slightly fluctuated around 2.10 Å. The study of the RMSF curves of the free 6M0J and its complexes showed fluctuations between 0.5 and 2.5 Å, which indicate that the studied compounds kept close contact with their binding pockets during the simulations (Fig. 12A). However, the 6M0J and 13 complex showed a higher fluctuation for residue 321 (3.4 Å), and this observation is consistent with the RMSD profile of the complex. The radius of the gyration graph, which measures protein compactness, was also examined, and the 6M0J and 10 complex was found to be the most stable, followed by the 11, 13 and 12 (Fig. 12B). The MD analysis revealed that the studied complexes have shown structural stability during the runs.
Fig. 11.
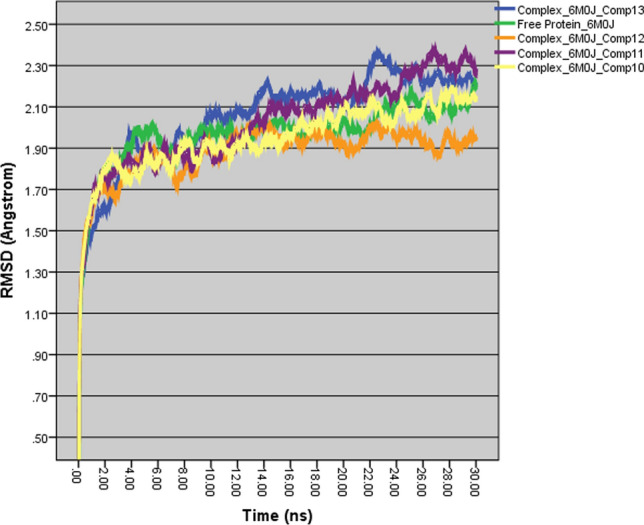
MD simulation RMSD plots (30 ns, 310 K) of free 6M0J and of complexes of 6M0J with compounds 10–13
Fig. 12.
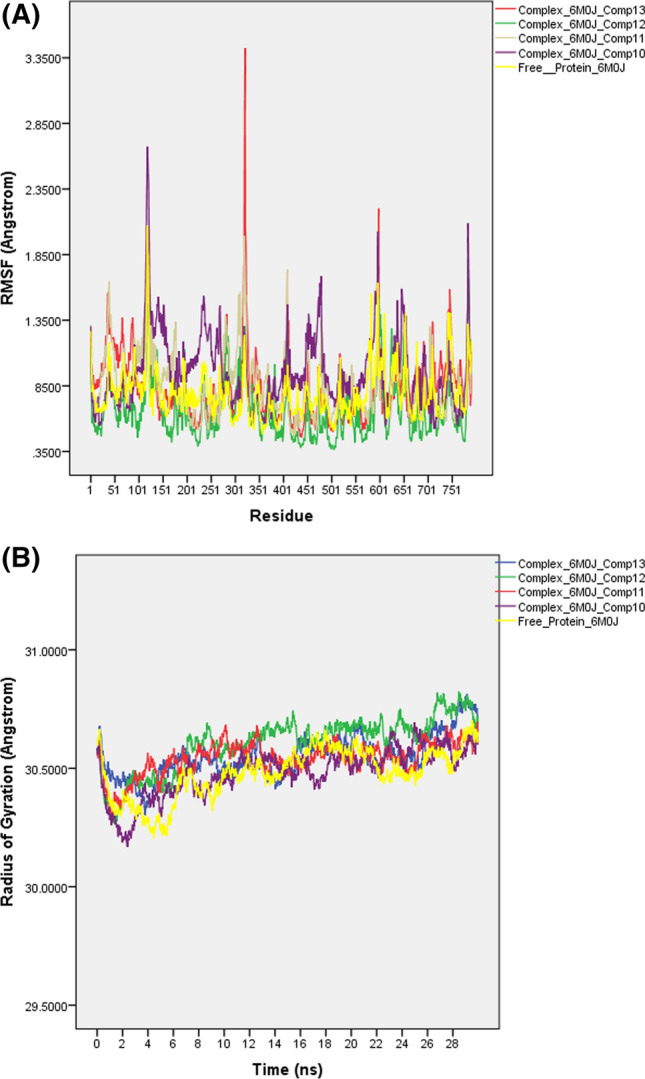
MD simulation: A RMSF and B Radius of Gyration plots (30 ns, 310 K) of free 6M0J and of complexes of 6M0J with compounds 10–13
The ADMET profile of the hit compounds 10–13 was computed in silico using the web-based tool admetSAR [56]. These compounds were found to be within the reference range considering the rule of five, their water solubility (log S), human colon adenocarcinoma (Caco-2) permeability, blood-to-brain barrier (BBB) and human intestinal absorption (HIA) (Table A2, Online Appendix A).
In summary, among the drugs of Table 2 increased affinity for 6M0J exhibit all those with large compound volume, relatively higher electrophilicity index, higher overall average charge, with aromatic rings and heteroatoms that participate in hydrogen bonding. Based on the binding energy, the best compounds were discovered to be the elbavir analogue 10 (− 11.2 kcal mol−1) and the patitaprevir analogues 11, 12 and 13 (− 11.3, − 11.5, − 11.5 kcal mol−1, respectively). Further optimization of these compounds can result in a more effective drug able to stop this newly emerged infection.
Conclusions
The infectious respiratory disease COVID-19 is rapidly expanding throughout the world and has become a serious threat to global health. Considering the time required to develop a new approved drug, drug repurposing seems the most appealing, safe and straightforward approach. In this study, FDA-approved HCV antiviral drugs and their structural analogues in clinical trials were tested for their inhibitory properties toward the COVID-19 protein (6M0J) using a virtual screening approach and computational chemistry methods. The most stable structures and the corresponding binding affinities of thirteen such antiviral compounds were obtained. Molecular docking calculations, ESP analysis, frontier molecular orbital theory and global reactivity descriptors were used to hypothesize the bioactivity of these drugs against the COVID-19 protein (6M0J). Compounds 6–9—among them elbasvir—showed remarkable binding affinities (− 10.6 to − 10.8 kcal mol−1) with 6M0J. Moreover, four compounds 10–13 (Table 2)—elbasvir and paritaprevir analogues—showed excellent binding interactions − 11.2 to − 11.5 kcal mol−1—for use against the newly emerged strain of coronavirus. Furthermore, trajectories analysis revealed that the studied complexes have shown structural stability during the MD runs. Therefore, it is concluded that these four top scoring compounds may act as lead compounds for further experimental validation, for clinical trials and for the development of more potent antiviral agents against the SARS-CoV-2.
Supplementary Information
Below is the link to the electronic supplementary material.
Declarations
Conflict of interest
No potential conflict of interest is reported by the author.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Gorbalenya AE, Baker SC, Baric RS, et al. The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol. 2020;5:536–544. doi: 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kupferschmidt K, Cohen J. Will novel virus go pandemic or be contained? Science. 2020;367:610–611. doi: 10.1126/science.367.6478.610. [DOI] [PubMed] [Google Scholar]
- 3.Coronavirus Disease (COVID-2019) Situation reports 1–45; World Health Organization, (2020)
- 4.Anthony SJ, Johnson CK, Greig DJ, Kramer S, Che X, Wells H, Hicks AL, Joly DO, Wolfe ND, Daszak P, Karesh W, Lipkin WI, Morse SS, Mazet JK, Goldstein T. Global patterns in coronavirus diversity. Virus Evol. 2017;3:1–15. doi: 10.1093/ve/vex012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Su S, Wong G, Shi W, Liu J, Lai AK, Zhou J, Liu W, Bi Y, Gao GF. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. 2016;24:490–502. doi: 10.1016/j.tim.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, Zhao X, Huang B, Shi W, Lu R, Niu P, Zhan F, Ma X, Wang D, Xu W, Wu G, Gao GF, Tan W. A novel coronavirus from patients with pneumonia in China. N Engl J Med. 2019;382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tang B, Bragazzi NL, Li Q, Tang S, Xiao Y, Wu J. An updated estimation of the risk of transmission of the novel coronavirus (2019-nCov) Infect Dis Model. 2020;5:248–255. doi: 10.1016/j.idm.2020.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.BBC News Covid: will there be more than one coronavirus vaccine? https://www.bbc.com/news/health-51665497
- 10.Ghosh AK, Brindisi M, Shahabi D, Chapman ME, Mesecar AD. Drug development and medicinal chemistry efforts toward SARS-coronavirus and covid-19 therapeutics. ChemMedChem. 2020;15:1–27. doi: 10.1002/cmdc.202000223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elfiky AA. Ribavirin, remdesivir, sofosbuvir, galidesivir, and tenofovir against SARS-CoV-2 RNA dependent RNA polymerase (RdRp): a molecular docking study. Life Sci. 2020;253:117592–117598. doi: 10.1016/j.lfs.2020.117592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shah B, Modi P, Sagar SR. In silico studies on therapeutic agents for COVID-19: drug repurposing approach. Life Sci. 2020;252:117652–117664. doi: 10.1016/j.lfs.2020.117652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elfiky AA. Anti-HCV, nucleotide inhibitors, repurposing against COVID-19. Life Sci. 2020;248:117477–117483. doi: 10.1016/j.lfs.2020.117477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumar S, Sharma PP, Shankar U, Kumar D, Joshi SK, Pena L, Durvasula R, Kumar A, Kempaiah P, Poonam RB. Discovery of new hydroxyethylamine analogs against 3CLpro protein target of SARS-CoV-2: molecular docking, molecular dynamics simulation, and structure-activity relationship studies. J Chem Inf Model. 2020;60:5754–5770. doi: 10.1021/acs.jcim.0c00326. [DOI] [PubMed] [Google Scholar]
- 15.Elmezayen AD, Al-Obaidi A, Şahin AT, Yelekçi K. Drug repurposing for coronavirus (COVID-19): in silico screening of known drugs against coronavirus 3CL hydrolase and protease enzymes. J Biomol Struct Dyn. 2020;382:727–733. doi: 10.1080/07391102.2020.1758791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Celik I, Erol M, Duzgun Z. In silico evaluation of potential inhibitory activity of remdesivir, favipiravir, ribavirin and galidesivir active forms on SARS-CoV-2 RNA polymerase. Mol Divers. 2021 doi: 10.1007/s11030-021-10215-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kanhed AM, Patel DV, Teli DM, et al. Identification of potential Mpro inhibitors for the treatment of COVID-19 by using systematic virtual screening approach. Mol Divers. 2021;25:383–401. doi: 10.1007/s11030-020-10130-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang J. Fast Identification of possible drug treatment of coronavirus disease-19 (COVID-19) through computational drug repurposing study. J Chem Inf Model. 2020;60:3277–3286. doi: 10.1021/acs.jcim.0c00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pant S, Singh M, Ravichandiran V, Murty USN, Srivastava HK. Peptide-like and small-molecule inhibitors against Covid-19. J Biomol Struct Dyn. 2020;39:2904–2913. doi: 10.1080/07391102.2020.1757510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Joshi S, Joshi M, Degani MS. Tackling SARS-CoV-2: proposed targets and repurposed drugs. Future Med Chem. 2020;12:1579–1601. doi: 10.4155/fmc-2020-0147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adebambo K. Computational investigation of the interaction of anti-influenza drugs with CoVID-19 protein. Comput Mol Biosci. 2020;10:45–60. doi: 10.4236/cmb.2020.102003. [DOI] [Google Scholar]
- 22.Sharp K, Dange DS (2020) In-silico FDA-approved drug repurposing to find the possible treatment of coronavirus disease-19 (COVID-19). ChemRxiv Preprint 10.26434/chemrxiv.12340718.v1
- 23.Rajbhar P, Singh D, Yadav R (2020) Repurposing of sars inhibitors against COVID19. ChemRxiv Preprint. 10.26434/chemrxiv.12155361.v1
- 24.Sadeghi A, Asgari A, Norouzi A, Kheiri Z, Anushirvani A, Montazeri M, Hosamirudsai H, Afhami S, Akbarpour E, Aliannejad R, Radmard AR, Davarpanah AH, Levi J, Wentzel H, Qavi A, Garratt A, Simmons B, Hill A, Merat S. Sofosbuvir and daclatasvir compared with standard of care in the treatment of patients admitted to hospital with moderate or severe coronavirus infection (COVID-19): a randomized controlled trial. J Antimicrob Chemother. 2020;75:3379–3385. doi: 10.1093/jac/dkaa334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kalamatianos K (2020) Drug repurposing for (COVID-19): in silico screening of known drugs against the SARS-CoV-2 spike protein bound to angiotensin converting enzyme 2 (6M0J). ChemRxiv Preprint, 10.26434/chemrxiv.12857678.v1 [DOI] [PMC free article] [PubMed]
- 26.Stewart JJP. Optimization of parameters for semiempirical methods I. Method. J Comput Chem. 1989;10:209–220. doi: 10.1002/jcc.540100208. [DOI] [Google Scholar]
- 27.Stewart JJP. Optimization of parameters for semiempirical methods II. Appl J Comput Chem. 1989;10:221–264. doi: 10.1002/jcc.540100209. [DOI] [Google Scholar]
- 28.Stewart JJP. Optimization of parameters for semiempirical methods. III Extension of PM3 to Be, Mg, Zn, Ga, Ge, As, Se, Cd, In, Sn, Sb, Te, Hg, Tl, Pb, and Bi. J Comput Chem. 1991;12:320–341. doi: 10.1002/jcc.540120306. [DOI] [Google Scholar]
- 29.Stewart JJP. Optimization of parameters for semiempirical methods IV: extension of MNDO, AM1, and PM3 to more main group elements. J Mol Model. 2004;10:155–164. doi: 10.1007/s00894-004-0183-z. [DOI] [PubMed] [Google Scholar]
- 30.Sinha L, Prasad O, Narayan V, Shukla SR. Raman, FT-IR spectroscopic analysis and first-order hyperpolarisability of 3-benzoyl5-chlorouracil by first principles. J Mol Simul. 2011;37:153–163. doi: 10.1080/08927022.2010.533273. [DOI] [Google Scholar]
- 31.Lewis DFV, Loannides C, Parke DV. Interaction of a series of nitriles with the alcohol-inducible isoform of P450: computer analysis of structure-activity relationships. Xenobiotica. 1994;24:401–408. doi: 10.3109/00498259409043243. [DOI] [PubMed] [Google Scholar]
- 32.Kosar B, Albayrak C. Spectroscopic investigations and quantum chemical computational study of (E)-4-methoxy-2-[(ptolylimino)methyl]phenol. Spectrochim Acta. 2011;78:160–167. doi: 10.1016/j.saa.2010.09.01. [DOI] [PubMed] [Google Scholar]
- 33.Fukui K. Role of frontier orbitals in chemical reactions. Science. 1982;218:747–754. doi: 10.1126/science.218.4574.747. [DOI] [PubMed] [Google Scholar]
- 34.Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, Zhang Q, Shi X, Wang Q, Zhang L, Wang X. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581:215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- 35.Du L, He Y, Zhou Y, Liu S, Zheng B, Jiang S. The spike protein of SARS-CoV – A target for vaccine and therapeutic development. Nat Rev Microbiol. 2009;7:226–236. doi: 10.1038/nrmicro2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thompson MA (2004) Molecular docking using ArgusLab, an efficient shape-based search algorithm and AScore scoring function, In: Proceedings of the ACS meeting, Philadelphia, Pa, USA
- 37.Neese F. The Orca program system. Comput Mol Sci. 2012;2:73–78. doi: 10.1002/wcms.81. [DOI] [Google Scholar]
- 38.Brandenburg JG, Bannwarth C, Hansen A, Grimme S. B97–3c: a revised low cost variant of the B97-D density functional method. J Chem Phys. 2018;148:064104. doi: 10.1063/1.5012601. [DOI] [PubMed] [Google Scholar]
- 39.Trott O, Olson AJ. Software news and update AutoDockVina: Improving the speed and accuracy of docking with a new scor-ing function, efficient optimization, and multithreading. J Comput Chem. 2009;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jiménez J, Škalič M, Martínez-Rosell G, De Fabritiis G. KDEEP: protein-ligand absolute binding affinity prediction via 3D-convolutional neural networks. J Chem Inf Model. 2018;58:287–296. doi: 10.1021/acs.jcim.7b00650. [DOI] [PubMed] [Google Scholar]
- 41.Seeliger D, De Groot BL. Conformational transitions upon ligand binding: holo-structure prediction from apo conformations. PLoS Comput Biol. 2010;6:e1000634–e1000643. doi: 10.1371/journal.pcbi.1000634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morris GM, Lim-Wilby M. Molecular docking. In: Kukol A, editor. Molecular modeling of proteins. Totowa: Humana Press; 2008. pp. 365–382. [Google Scholar]
- 43.Tian L, Feiwu C. Multiwfn: a multifunctional wavefunction analyzer. J Comput Chem. 2012;33:580–592. doi: 10.1002/jcc.22885. [DOI] [PubMed] [Google Scholar]
- 44.Discovery Studio Visualizer (2019) Version 20.1, Dassault systems
- 45.Karelson M (2000) Molecular descriptors in QSAR/QSPR. Wiley-Inter-Science
- 46.Scrocco E, Tomasi J (1973) The electrostatic molecular potential as a tool for the interpretation of molecular properties. In new concepts II, Springer, pp 95–170. 10.1007/3-540-06399-4_6
- 47.Petrolongo C. Quantum chemical study of isolated and interacting molecules with biological activity. Gazz Chim Ital. 1978;108:445–478. [Google Scholar]
- 48.IBMCorp. Released (2011) IBMSPSSStatistics for windows, Version 20.0. Armonk, NY: IBMCorp
- 49.Wade RC, Goodford PJ. The role of hydrogen-bonds in drug binding. Prog Clin Biol Res. 1989;289:433–444. [PubMed] [Google Scholar]
- 50.Sarwar MG, Ajami D, Theodorakopoulos G, Petsalakis ID, Rebek J. Amplified halogen bonding in a small space. JACS. 2013;135:13672–13675. doi: 10.1021/ja407815t. [DOI] [PubMed] [Google Scholar]
- 51.Weiner PK, Langridge R, Blaney JM, Schaefer R, Kollman PA. Electrostatic potential molecular surfaces. Proc Natl Acad Sci. 1982;79:3754–3758. doi: 10.1073/pnas.79.12.3754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Okulik N, Jubert AH. Theoretical study on the structure and reactive sites of non-steroidal anti-inflammatory drugs. J Mol Struct. 2004;682:55–59. doi: 10.1016/j.theochem.2004.04.069. [DOI] [Google Scholar]
- 53.Tian X, Liu Y, Zhu J, Yu Z, Han J, Wang Y. Probing inhibition mechanisms of adenosine deaminase by using molecular dynamics simulations. PLoS One. 2018;13:e0207234. doi: 10.1371/journal.pone.0207234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 55.Phillips JC, Hardy DJ, Maia JDC, Stone JE, Ribeiro JV, Bernardi RC, Buch R, Fiorin G, Hénin J, Jiang W, McGreevy R, Melo MCR, Radak BK, Skeel RD, Singharoy A, Wang Y, Roux B, Aksimentiev A, Luthey-Schulten Z, Kalé LV, Schulten K, Chipot C, Tajkhorshid E. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J Chem Phys. 2020;153:44130–44138. doi: 10.1063/5.0014475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang H, Lou C, Sun L, Li JCY, Wang Z, Li W, Liu G, Tang Y. AdmetSAR 2.0: Web-service for prediction and optimization of chemical ADMET properties. Bioinformatics. 2019;35:1067–1069. doi: 10.1093/bioinformatics/bty707. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.