

ABSTRACT
PARP inhibitors are synthetically lethal with BRCA1/2 mutations, and in this setting, accumulation of DNA damage leads to cell death. Because increased DNA damage and subsequent immune activation can prime an anti-tumor immune response, we studied the impact of olaparib ± immune checkpoint blockade (ICB) on anti-tumor activity and the immune microenvironment. Concurrent combination of olaparib, at clinically relevant exposures, with ICB gave durable and deeper anti-tumor activity in the Brca1m BR5 model vs. monotherapies. Olaparib and combination treatment modulated the immune microenvironment, including increases in CD8+ T cells and NK cells, and upregulation of immune pathways, including type I IFN and STING signaling. Olaparib also induced a dose-dependent upregulation of immune pathways, including JAK/STAT, STING and type I IFN, in the tumor cell compartment of a BRCA1m (HBCx-10) but not a BRCA WT (HBCx-9) breast PDX model. In vitro, olaparib induced BRCAm tumor cell–specific dendritic cell transactivation. Relevance to human disease was assessed using patient samples from the MEDIOLA (NCT02734004) trial, which showed increased type I IFN, STING, and JAK/STAT pathway expression following olaparib treatment, in line with preclinical findings. These data together provide evidence for a mechanism and schedule underpinning potential benefit of ICB combination with olaparib.
KEYWORDS: PARP inhibitor, olaparib, cGAS-STING, immune checkpoint blockade, BRCA
Background
Identification of PD-1/PD-L1 and CTLA-4 as T cell checkpoints, and therefore potential points of intervention to improve anti-tumor immune responses,1,2 has led to the clinical development and regulatory approval of multiple therapeutics targeting PD-1, PD-L1, or CTLA-4. Although these have resulted in clinical responses in multiple indications, including melanoma, non-small cell lung cancer (NSCLC), and mismatch-repair-deficient cancers,3 the majority of patients do not show a clinical response to ICB monotherapy, suggesting potential for additional benefit from combination therapy with tumor-targeted agents (reviewed in Gotwals et al.4).
Advances in the understanding of DNA damage repair pathways have identified synthetic lethality in tumors with BRCA1 or BRCA2 mutations to treatment with PARP inhibitors (PARPi), where accumulation of DNA damage leads ultimately to cell death.5–7 PARPi are now approved clinically for the treatment of multiple cancer indications, including ovarian, breast, prostate, and pancreatic cancers, which are often reported to have low responses to immunotherapy.8,9
DNA damage, which may result in increased levels of cytosolic DNA, can be detected via intracellular anti-viral DNA sensors, including cGAS and IFI16,10 which initiate activation of pro-inflammatory gene expression programs, including type I interferon (IFN) responses.11 Similarly, deficiencies in DNA damage repair pathways, including BRCA1/2 and ATM, are reported to result in increased baseline pro-inflammatory signaling.12,13 Type I IFNs have pleiotropic effects on the immune response, including supporting the maturation of dendritic cells (DCs), which process and present antigens released by dying tumor cells to prime an anti-tumor T cell response.14 Because of the potential for increased DNA damage to stimulate immune priming, there is ongoing clinical investigation of PARPi in combination with ICB, in multiple tumor types.15
To define the activity and elucidate potential mechanisms underpinning combination of the PARPi olaparib with ICB, anti-tumor activity and immune modulation in a Brca1 mutant (Brca1m) syngeneic tumor model were investigated. Additionally, effects on tumor immune infiltrate and development of immune memory were assessed. To confirm activation of tumor-intrinsic pathways by PARPi and impact of BRCA mutation in human tumor cells, effects of olaparib on transcriptional changes in the tumor cell compartment of BRCA WT and mutant patient-derived xenograft (PDX) models were compared. Direct effects of olaparib treatment on human immune cells were evaluated, including in vitro tumor/dendritic cell co-cultures. Finally, consistent with the findings in our preclinical studies, changes in STING pathway expression were observed in clinical pre- and post-olaparib treatment biopsies from the MEDIOLA (NCT02734004) clinical trial.16
Materials and methods
In vivo studies
BR5 cells17 were obtained from Prof. Sandra Orsulic laboratory and grown in DMEM with 10% FBS, under standard conditions. 3 × 106 cells in 50% Matrigel were implanted subcutaneously (SC) into female FVB/N mice (Jackson and Envigo UK). CT26 and MC38 cells were purchased from ATCC and grown in RMPI-1640 and DMEM, respectively, with 10% FCS. 5 × 105 CT26 cells or 1 × 107 MC38 cells were implanted subcutaneously into female Balb/c and C57Bl/6 (Charles River) mice, respectively. HBCx-9 and HBCx-10 PDX studies were performed at Xentech. Tumor fragments were implanted subcutaneously into female athymic Nude-Foxn1nu mice (Envigo).
Animal studies were conducted in accordance with UK Home Office legislation, the Animal Scientific Procedures Act 1986, and the AstraZeneca Global Bioethics policy or Institutional Animal Care and Use Committee guidelines. Experimental work is outlined in project license 70/8894, which has gone through the AstraZeneca Ethical Review Process. Olaparib was formulated in 10% DMSO and 30% Kleptose (Roquette) and administered by oral gavage once daily (QD) at 10 mL/kg final dose volume. Anti-murine PD-L1 and anti-murine CTLA-4 surrogate antibodies for durvalumab and tremelimumab (AstraZeneca) were diluted in saline and administered intraperitoneally twice weekly (BIW) at 5 mL/kg or 10 mL/kg final dose volume. Tumor growth inhibition was analyzed as described before.18,19
Flow cytometry
MC38 and BR5 tumors were prepared and cells stained with antibodies as described.20 Briefly, tumors were harvested and dissociated using a mouse tumor dissociation kit (Miltenyi Biotec). Cells were stained with Live-Dead fixable Zombie UV stain dye (ThermoFisher) and then with a mix of flow cytometry antibodies listed in Supplementary Table 1. Sample data were acquired using a BD LSR Fortessa (BD Bioscience), and data analysis was performed using Flow Jo 10.6 (BD Bioscience). For statistical analysis, the frequency data were processed with a beta regression, treating treatment as a fixed effect. Post hoc testing with no correction for multiple testing was used to compare each treatment group to the reference group. As there are multiple variables for a treatment versus references group, introducing a multiple testing burden, the p values were adjusted using the Benjamini and Hochberg FDR methodology to control the false discovery rate within significant calls to 5% (* p < 0.05, ** p < 0.01, *** p < 0.001, and **** p < 0.0001).
RNA sequencing from human patient samples
All patients provided written informed consent, and ethics committee approvals were as previously described.16 Sequencing was performed as per the manufacturer’s protocol on the Illumina HiSeq platform with 2x150bp-paired end reads using the KAPA whole-transcriptome kit, generating an average of 50 million reads per sample (Illumina).
The RNAseq pipeline implemented in bcbio-nextgen (version 1.1.6) (https://bcbio-nextgen.readthedocs.org/en/latest/) was used for quality control and gene expression quantification. Reads were aligned to the UCSC-build GRCh38 Homo Sapiens genome, augmented with transcript information from Ensembl release 86 using HiSat2.21 Alignments were evaluated for evenness of coverage, rRNA content, genomic context of alignments, and complexity using a combination of FastQC, Qualimap, and custom tools.22
Gene signature analysis of human patient samples
A STING signaling gene signature was developed through the integration of the following gene sets: canonical STING pathway genes (from Dunphy et al.10), STING, and IRF3 downstream genes using an internal causal reasoning database. To reduce the contribution of non-specific STING pathway genes and broad inflammatory genes, NFκB (from internal causal reasoning database) and non-canonical STING pathway genes (from Dunphy et al.10) were removed from the gene signature generation. The STING gene signature was optimized by removing the genes in the signature that did not show positive correlation with the overall gene signature. A further optimization step was used to remove genes that did not show upregulation following treatment with dsDNA and/or downregulation following siSTING in preclinical models (from Abe et al.23). The STING gene signature was then scored using the GSVA method in R.
Results
Combination of olaparib with ICB results in increased in vivo anti-tumor responses and development of immune memory
Combinations of ICB with PARPi are being tested clinically; however, the effects of tumor genetics and contribution of components to response have not been fully explored. Olaparib was first tested in combination with either anti-PD-L1 or anti-CTLA-4 in CT26 and MC38 syngeneic models, which are commonly used for investigation of tumor immune modulation. In these Brca WT models, no olaparib monotherapy anti-tumor activity was observed, nor was olaparib plus anti-PD-L1 combination activity greater than that observed with anti-PD-L1 alone (Fig S1a-b). Similar results were observed with anti-CTLA-4 as the combination partner for olaparib (Fig S1c-d). Olaparib shows robust clinical benefit in patients with germline or somatic mutations in BRCA.24 To assess the activity of olaparib in a disease-relevant context, we used a Brca1 mutated (Brca1m) syngeneic model, BR5. In this model, olaparib dosed concurrently with anti-PD-L1 demonstrated potent and reproducible combination activity (Figure 1a), with increased numbers of complete responders (CRs, defined as tumors ≤14 mm3 at the end of the study) in the combination group (6/9 in a representative study shown) versus either olaparib (1/9 CR) or anti-PD-L1 monotherapy (2/9 CRs). Similar results were observed when olaparib was combined with anti-CTLA-4 in the BR5 model (Fig S2a). Monotherapy olaparib also resulted in significant 58% growth rate inhibition (45% inhibition for anti-PD-L1 alone), while combination treatment resulted in 106% growth rate inhibition (Figure 1b).
Figure 1.

Anti-tumor activity of olaparib alone and in combination with anti-PD-L1 in BR5 Brca1m syngeneic tumor model. (a) Individual animal tumor growth curves or mean tumor volume ± SEM for BR5 model treated with vehicle, 100 mg/kg olaparib daily, 10 mg/kg anti-PD-L1 biweekly, or the combination. Dosing period is indicated by shaded boxes. Data representative of n = 4 independent experiments. (b) Boxplot of the growth rate for BR5 tumors treated as above. Whiskers indicate the minimum and maximum and lines indicate median values. One-way ANOVA was used for statistical analysis (* p < 0.05; **** p < 0.0001). (c) Swimmer lane plots of survival (bars) for mice bearing BR5 tumors following treatment as above. For mice that reached a complete response (CR), times of complete response identification and progression are marked with triangles and circles, respectively. (d) Mice that showed complete regression of tumor following treatment with anti-PD-L1 and olaparib + anti-PD-L1 were rechallenged with BR5 tumor cells in the contralateral flank and monitored for tumor growth.
The responses observed in mice treated with olaparib plus ICB combinations were durable (representative plot for BR5 model shown in Figure 1c). When evaluated across multiple (n = 3) studies, over at least 90 days, olaparib monotherapy treatment resulted in 10/30 CRs, of which 8 recurred prior to the end of the study. Anti-PD-L1 monotherapy resulted in 5/30 CRs, of which 4 were durable. In contrast, olaparib dosed concurrently with anti-PD-L1 resulted in 19/30 CRs, of which only 5 recurred. Time to recurrence off-treatment in the combination treated animals did not differ significantly from olaparib monotherapy-treated mice (range 3–44 days vs. 3–49 days). Thus, the differences between combination and olaparib monotherapy were the frequency of both complete and durable complete responses (Supplementary Table 5). Similar results were observed for olaparib and anti-CTLA-4 combination (Fig S2b).
Olaparib in combination with ICB also resulted in the development of immune memory. Mice that had shown a complete response following either anti-PD-L1 or anti-CTLA-4 alone or in combination with olaparib were rechallenged with implantation of 3 × 106 BR5 cells on the opposite flank. All re-challenged animals showed initial tumor growth, followed by complete and durable tumor rejection, without further treatment (Figure 1d; S2c). Olaparib monotherapy did not show consistently durable complete responses, so these mice were not able to be rechallenged.
Concurrent schedule of olaparib and ICB shows most consistent anti-tumor activity
Most current clinical trials use olaparib and anti-PD-L1 in a concurrent dose scheduling regime (e.g. NCT03334617 and NCT02734004, www.clinicaltrials.gov). However, several trials utilize an anti-PD-L1/chemotherapy lead-in schedule (i.e. NCT03740165 and NCT04191135), and an olaparib lead-in schedule has also been used (NCT02734004). However, there is a lack of preclinical data comparing different dosing sequences. Hence, a comparison of a concurrent schedule of olaparib plus anti-PD-L1 with an olaparib-lead-in or anti-PD-L1 lead-in schedules was tested in the BR5 Brca1m model. When dosing started on day 7 (palpable tumors), the concurrent dosing schedule resulted in a number of durable complete responses. This effect was comparable to the anti-PD-L1 lead-in schedule, where olaparib dosing was delayed for 14 days. These two schedules were superior to the olaparib lead-in (Figure 2a, 2c, S3), where anti-PD-L1 dosing was similarly delayed. Moreover, the concurrent schedule caused 103% growth rate inhibition compared to 81% and 63% for the anti-PD-L1 lead-in and olaparib lead-in schedules, respectively (Figure 2b). These results were consistent even when olaparib dosing was extended from 3 to 5 weeks (Supplementary Table 6). The three dosing schedules were further compared in a study where dosing commenced on day 22. Although no complete responses were reached (Figure 2d), a significant growth rate inhibition was observed in all schedules (Figure 2e), and survival analysis revealed that the concurrent and olaparib lead-in schedules significantly prolonged survival compared to the anti-PD-L1 lead-in schedule (Figure 2f). Taken together, these data suggest that the concurrent schedule of the two agents delivers the best anti-tumor benefit in a Brca1m tumor model.
Figure 2.

Concurrent schedule of olaparib and ICB is the most efficacious in a Brca1m tumor model. (a) Individual animal tumor growth curves for BR5 model treated with vehicle, 100 mg/kg olaparib daily, 10 mg/kg anti-PD-L1 biweekly, or the combination. Dosing period is indicated by shaded boxes and red (olaparib) or blue (anti-PD-L1) lines. Data representative of at least n = 3 independent experiments. (b) Boxplot of the growth rate for BR5 tumors treated as above. Whiskers indicate the minimum and maximum and lines indicate median values. One-way ANOVA was used for statistical analysis (*** p < 0.001; **** p < 0.0001). (c) Swimmer lane plots of survival (bars) for mice bearing BR5 tumors following treatment as above. For mice that reached a complete response, times of complete response identification and progression are marked with triangles and circles, respectively. (d) Individual animal tumor growth curves for BR5 model treated with vehicle, 100 mg/kg olaparib daily, 10 mg/kg anti-PD-L1 biweekly, or the combination. Dosing period is indicated by shaded boxes and red (olaparib) or blue (anti-PD-L1) lines. (e) Boxplot of the growth rate for BR5 tumors treated as in (d). Whiskers indicate the minimum and maximum and lines indicate median values. One-way ANOVA was used for statistical analysis (**** p < 0.0001). (f) Kaplan–Meier survival plot for study in (d). Log rank test was used for statistical analysis (** p < 0.01; **** p < 0.0001).
Olaparib treatment increases CD8+ T cells and NK cells and decreases myeloid cell infiltration
It has previously been reported that inhibition of PARP in BRCAm tumors results in modification of the immune infiltrate;25–28 therefore, we investigated changes in immune cell infiltration in the Brca1m BR5 model (Figure 3a-c). Treatment with olaparib resulted in a significant increase in the frequency of tumor-infiltrating total (CD3+) and CD8+ T cells, which was maintained with anti-PD-L1 combination treatment. Combination treatment also resulted in a significantly increased frequency of granzyme B–positive (GzmB+) CD8+ T cells (1.6-fold increase from control), whereas olaparib or anti-PD-L1 alone did not. Similarly, there was an increase in proliferating Ki67+ CD8+ T cells in the combination group (1.3-fold increase from control, however, not statistically significant). Olaparib treatment alone or in combination with anti-PD-L1 also significantly increased the frequency of tumor NK cells (1.4- and 1.5-fold increase from control, respectively), which was accompanied by an increase in the frequency of GzmB+ NK cells in the combination group (1.4-fold increase). In contrast, neither total T cell frequency nor CD8+ T cell frequency was altered by olaparib monotherapy in the MC38 model (Brca WT), although an increase in CD8+ T cells was observed in the anti-PD-L1 and combination groups (1.7- and 2.1-fold increase, respectively) (Fig S4a-b, S4c: Cd8a, Cd8b1). No significant changes in NK cell frequency were observed with any treatment in the MC38 model (Fig S4a-b, S4c: Ncr1). Finally, decreases in the frequencies of macrophages, CD11b+Ly6C+ cells (M-MDSCs), and CD11b+Ly6G+ MDSCs were observed following olaparib treatment ± anti-PD-L1 in the BR5 model, indicating a potential relative reduction in immunosuppressive myeloid cells (Figure 3c). In contrast, no consistent changes in myeloid cells were seen in the MC38 model with olaparib monotherapy (Fig S4). Thus, increases in immune cells associated with an increased anti-tumor immune response were observed in olaparib-treated Brca1m but not Brca WT tumors, and this effect was further increased in combination with anti-PD-L1.
Figure 3.

Olaparib alone or in combination with anti-PD-L1 alters frequency of tumor infiltrating immune cells towards an immuno-activated phenotype. (a) Schema of flow cytometry experiments. (b) Mean tumor volume ± SEM for representative study. (c) Frequency of CD3+ cells, CD4+ T cells, CD8+ T cells, Ki67+ CD8+ T cells, GzmB+ CD8+ T cells, NK cells, GzmB+ NK cells, macrophages, CD11b+Ly6C+, and CD11b+Ly6G+ MDSCs depicted as percentage of CD45+ cells in BR5 tumors. Representative of n = 2 independent experiments with n = 8 mice/group per experiment. One-way ANOVA was used for statistical analysis (* p < 0.05; ** p < 0.01; **** p < 0.0001).
Immune response pathway expression is increased in vivo in olaparib-treated Brca1m tumors
To identify immune pathways that may be altered in response to olaparib ± checkpoint inhibition and that could potentially mechanistically underpin observed immune changes and anti-tumor activity, we evaluated gene expression in treated BR5 tumors using the Nanostring nCounter pan-cancer immune profiling panel. Mice with established BR5 tumors were treated with olaparib or vehicle plus either anti-PD-L1, anti-CTLA-4, or isotype control antibodies (Figure 3a). Differential gene expression analysis revealed significant upregulation of multiple immune genes following olaparib treatment (Figure 4a), including those associated with T cell infiltration and activation (Cd8a, Zap70, Tbx21, Lck, Hck, Cd4, Fyn, Cd69) and cytotoxic T cell activity (Gzma, Prf1). Genes associated with T cell exhaustion (Ctla4, Lag3, Btla, Tigit, Havcr2 (Tim3)), as well as Pdl1 (Cd274) and Foxp3, were also upregulated, suggesting active suppression of an ongoing immune response, which was also observed in the olaparib + anti-PD-L1-treated tumors (Gene list in Supplementary Table 4). Pathways significantly upregulated in response to olaparib encompassed innate and adaptive immune responses, including T cell signaling, as well as those associated with antigen processing and presentation and IL-12/STAT4 (Figure 4b), suggesting potential for increased dendritic cell activation. In contrast, analysis of gene expression in the Brca WT MC38 tumors indicated no significant transcriptomic changes in the markers of immune cell infiltration, T cell suppression, or Pdl1 with olaparib treatment, although increased expression of some of these genes was seen in the anti-PD-L1-treated tumors (1.8-fold increase for Gzmb (p = 1.92E-05), 1.9-fold increase for Cd8a (p = 0.0151), and 2.1-fold increase for Cd8b1 (p = 0.0146)), consistent with flow cytometry results (Fig S4c and b).
Figure 4.

Transcriptomic changes in the BR5 syngeneic model treated with olaparib ± anti-PDL-1 antibody. (a) Differential expression of genes in BR5 tumors treated as in Figure 2a. Tumor RNA was isolated and analyzed with Nanostring mouse immune 770 CodeSet. Shown are olaparib + isotype control groups vs. isotype control alone (vehicle). (c) Changes in gene expression from (a) for olaparib-treated tumors. Significantly upregulated/downregulated pathways are depicted. (c) Modulation of expression of STING gene signature in tumors treated with olaparib ± anti-PD-L1 / anti-CTLA-4 (vehicle n = 9, olaparib n = 7, anti-PD-L1 n = 9, anti-CTLA-4 n = 8, olaparib + anti-PD-L1 n = 6, olaparib + anti-CTLA-4 n = 7). (d) GSVA score for STING pathway genes in BR5 samples. Two-tailed t-test was used for statistical analysis (* p = 0.0393, ** p = 0.0022).
Increased cellular DNA damage, as a consequence of PARPi treatment of Brca mutant tumors, has been reported to activate the type I IFN pathway via cGAS-STING signaling.25,28–32 To investigate this in the BR5 model, we developed a STING gene signature (see Supplemental Material and Methods) that was able to capture modulation of the cGAS/ STING pathway in preclinical models treated with dsDNA or cGAMP and which was lost upon STING RNA knockdown. In contrast, an IFN-I gene signature showed no modulation in response to dsDNA versus siSTING + dsDNA, highlighting a stark differentiation of this STING signature from an IFN-I signature (Fig S6). A significant upregulation of the STING gene signature was observed in the BR5 model treated with olaparib monotherapy and olaparib plus anti-PD-L1 / anti-CTLA-4 antibodies (t-test p values, 0.002 and 0.03 respectively; Figure 4c left panel). Neither anti-PD-L1 nor anti-CTLA-4 monotherapy showed STING signature upregulation, while olaparib plus anti-PD-L1 showed significant upregulation (Figure 4c). Upregulation of the STING signature by olaparib monotherapy and the combination with anti-PD-L1 was validated in an independent experiment in the BR5 model using gene expression analysis (transcriptomic analysis by qRT-PCR) of the STING pathway (Figure 4d). Consistent with flow cytometry and Nanostring results, expression of a cytotoxic lymphocyte signature 33 was also upregulated in the monotherapy and anti-PD-L1 / anti-CTLA-4 combination groups (Fig S5a). Thus, olaparib treatment of Brca1m but not Brca WT tumors results in upregulation of genes associated with an anti-tumor immune response, and this is further enhanced in combination with ICB.
Olaparib treatment results in increased tumor-cell intrinsic immune activation pathway expression
Activation of type I IFN as a result of DNA damage can occur in tumor cells, or in myeloid cells, as a result of phagocytosis of dead or damaged tumor cells,34–36 via myeloid cell uptake of DNA-containing exosomes37 or secreted cGAMP.38 To evaluate tumor cell-intrinsic transcriptional changes, RNAseq analysis of an olaparib-sensitive BRCA1m triple-negative breast cancer (TNBC) patient-derived xenograft (PDX) (HBCx-10) was compared to a PARPi-insensitive BRCA WT TNBC PDX (HBCx-9) 39 (Figure 5a-b). Differential gene expression analysis of human-specific RNAseq data from olaparib-treated BRCA1m HBCx-10 tumors implanted in immunodeficient mice showed that the dominant upregulated pathways were immune-associated, specifically including those associated with type I IFN response, the STING pathway, as well as JAK/STAT pathway activation (Figure 5a,c, S8a). Analysis of murine genes (stromal compartment) showed less pronounced activation of type I IFN pathway (Fig S7). In contrast, no coordinate upregulation of these immune pathways by olaparib was observed in the BRCA WT HBCx-9 model, although some upregulation of type I IFN was observed at the highest dose of olaparib in HBCx-9 (Figure 5b-c, S8b).
Figure 5.

Immune gene pathway expression in olaparib-treated PDX models and in the TCGA pan-cancer database. (a, b) Heatmaps of gene expression changes in the BRCA1m HBCx-10 (a) or BRCA WT HBCx-9 (b) patient-derived xenograft (PDX) models treated with olaparib. PDX tumors (n = 3–5/group) were treated with 50 or 100 mg/kg of olaparib or with vehicle for 7 days. Tumors were harvested either 6 hours or 24 hours after the last dose of olaparib, and samples were analyzed by RNAseq. (c) GSVA scoring of type I IFN pathway expression in HBCx-9 and HBCx-10 tumors treated with vehicle or the indicated dosage of olaparib (n = 3–5/group). p values refer to aggregated timepoints within each treatment. (d) Causal reasoning analysis of common regulators for gene expression changes in HBCx-10 tumors treated with indicated dosage of olaparib and harvested at the indicated timepoint. Regulators shown are those whose activity was altered across both doses of olaparib and at both timepoints. (e) Changes in expression of activated STING pathway in HBCx-9 and HBCx-10 tumors treated as in a-c. (f) Gene signature scoring (GSVA method) of STING pathway and CD8 T cell signatures across the TCGA pancancer samples database. Tumor type is indicated on the x-axis by the TCGA nomenclature (https://gdc.cancer.gov/resources-tcga-users/tcga-code-tables/tcga-study-abbreviations).
Figure 5.
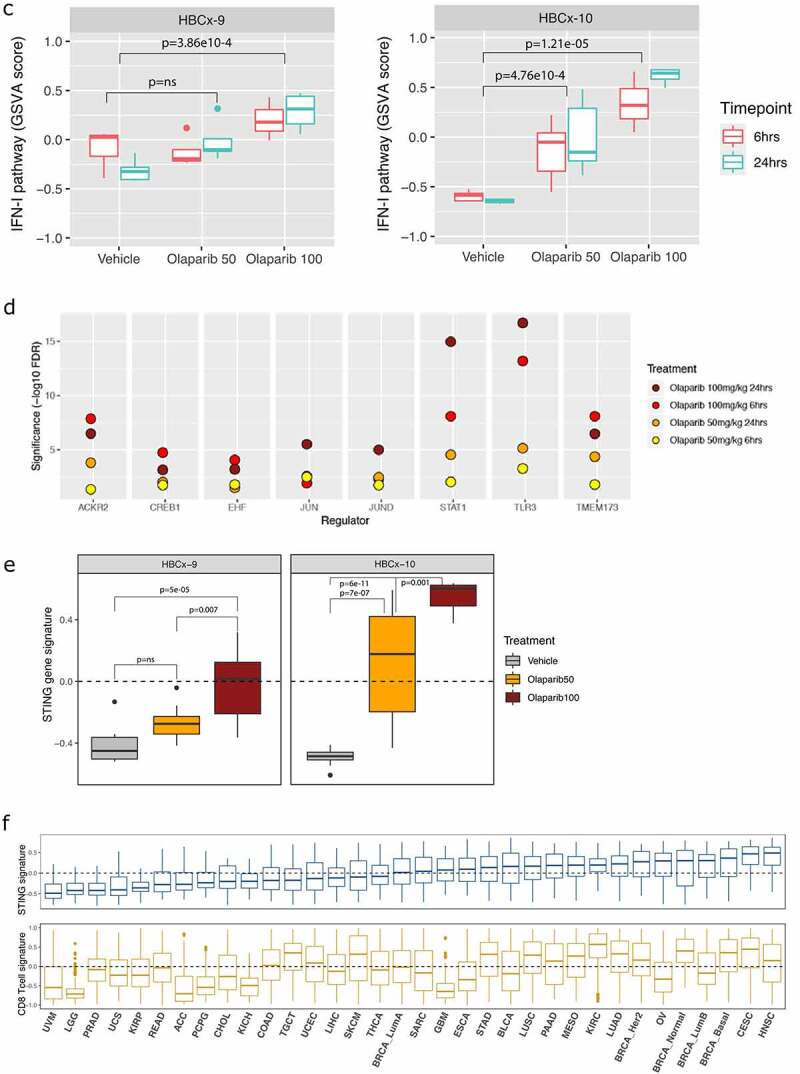
(Continued).
Causal reasoning analysis further identified biological networks linked to immune pathways in the human tumor cell compartment of olaparib-treated HBCx-10 tumors and was compared across two olaparib dose levels (50 mg/kg and 100 mg/kg) and two time points (6 and 24 hours). It revealed significant enrichment in key biological networks associated with immune responses to DNA damage, including TLR3, STING (TMEM173), and STAT1, which is activated in response to type I IFN signaling (Figure 5d), and further support that broad immune signaling is activated by olaparib treatment. STING signature expression was also compared in HBCx-9 and HBCx-10 and showed that both high and low dose of olaparib monotherapy was sufficient to induce activation of STING signaling in a dose-dependent manner in the BRCA1m model (Figure 5e). In contrast, in the BRCA WT HBCx-9 model, 50 mg/kg of olaparib did not cause significant enrichment in biological networks associated with immune pathways, while the higher dose of olaparib modestly upregulated a STING gene signature (Fig S8, S5c). Hence, olaparib treatment induces expression of the immune-associated pathways in BRCAm tumors more potently than in BRCA WT tumors in vivo.
PARP inhibition demonstrates indirect effects on immune cells
Because we had observed increased tumor cell-intrinsic activation of innate immune pathways, we investigated whether olaparib-treated tumor cells could modulate in vitro activation of dendritic cells (DCs). Immature DCs were co-cultured with olaparib-treated or control PARPi-sensitive BRCA1m MDA-MB-436 cells40 or conditioned medium (cMed). Co-culture with olaparib-treated cancer cells or cMed induced CD86 expression on DCs (Figure 6a). Isogenic cell lines (DLD-1 WT and BRCA2−/−) were subsequently used to confirm the specificity of DC transactivation by olaparib for tumor BRCA status. We find that etoposide, olaparib, and talazoparib induce DC transactivation to a similar extent in DLD-1 BRCA2−/− co-cultures. However, in DLD-1 WT co-cultures, both olaparib and talazoparib elicited significantly less DC transactivation, in contrast to etoposide, which showed no specificity for BRCA status (Figure 6c). The modest activity of talazoparib in DLD-1 WT co-cultures may be attributed to its greater PARP trapping activity, which has previously been reported to induce DNA breaks in WT cells, resulting in DC transactivation.31 Importantly, neither olaparib nor talazoparib showed direct activation of DCs (Fig S9a). Cytokines secreted by olaparib-treated cancer cells were evaluated, and surprisingly type I IFN was not detected. However, MDA-MB-436 showed increased secretion of IFN-λ and IL-1β in response to olaparib (Figure 6b), while DLD-1 BRCA2−/− showed increased IL-8 secretion (Fig S9b), suggesting olaparib induced a proinflammatory phenotype. Consistent with in vivo results, both BRCA-deficient cell lines showed activation of STING-TBK1 and NFκB pathways upon PARPi treatment (Figure 6d), as well as activation of a DNA damage repair response, including p-ATM and γH2AX induction and cell death (PARP cleavage; Fig S9c). Interestingly, the PARPi responses may be associated with their PARP-trapping activity, as the weak trapper, veliparib41, shows no significant changes in these pathways. These experiments therefore highlight the selective immune cross-talk elicited by olaparib in BRCA-deficient cells in vitro, as well as distinct patterns of cytokine expression by different tumor cell lines.
Figure 6.

Olaparib induces tumor-immune cross-talk in BRCA-deficient cells. (a) Conditioned medium (cMed) and tumor cells were collected from MDA-MB-436 cultures treated with olaparib (3 µM) or talazoparib (0.1 µM) for 3 days. Immature DCs were incubated with cMed alone or cMed plus tumor cells (co-culture) for 24 hours and quantified for CD86 expression by flow cytometry (n=5 unique DC : MDA-MB-436 combinations from three experiments; mean ± SEM; statistics show > 99.99% confidence interval to controls). (b) Tumor cell cultures were treated for 3days and cMed quantified for the secretion of interferon and pro-inflammatory cytokines. Absolute modulation of IFN-λ and IL-1β from MDA-MB-436 supernatants (mean of four biological replicates ± SEM; data representative of three independent experiments). (c) DLD-1 WT and BRCA2−/− cultures were treated for 3 days with etoposide (10 µM), olaparib (3 µM), or talazoparib (0.1 µM) and then co-cultured with immature DCs (1:1 ratio) for 24 hours. Quantification of CD86 expression is shown for four unique DC : DLD-1 combinations (mean ± SEM; statistics by Student’s t-test; representative n=2). (d) MDA-MB-436 and DLD-1 cultures treated for 3 days and conditioned medium were immunoblotted for the indicated pathway nodes. Densitometry for p-NFκB and p-TBK1 (mean ± SEM; n=3; statistics indicate > 99.9% confidence interval to DMSO). (e) Human CD3+ T cells from 4 blood donors were stimulated with anti-CD3 / CD28 beads and cultured for 4 days in the presence of compounds. Representative Cell Trace Violet traces for CD4+ and CD8+ T cells, proliferation, viability, and IFN-γ secretion (n=4; mean ± SEM; T cell data is representative of three independent experiments using unique blood donors).
Recent reports have suggested that deletion of PARP1/2 in T cells results in impaired T cell activation and reduced anti-tumor immune responses,42 and a role for PARP1 in T cell activation via modulation of NFAT localization or function has also been suggested.43,44 We therefore investigated the tolerance of human T cells to olaparib. Olaparib had no significant impact on proliferation or viability of activated T cells at concentrations up to 10 μM nor on the secretion of IFN-γ (Figure 6e). We also observed no change in the expression of CD25 and CD69 activation markers, although we did quantify a small decrease in the secretion of IL-2 and TNF-α at 3–10 μM (Fig S9d). Finally, we investigated whether olaparib affected the ex vivo differentiation and polarization of human macrophages from peripheral blood monocytes (Fig S9e). Our data show inclusion of olaparib (at 1 and 3 μM) did not modulate the viability or surface expression of CD86, CD206, or PD-L1 on cells cultured under M1 or M2 polarizing conditions, although a slight decrease in CD86 expression was observed when the cells were cultured with M-CSF plus olaparib vs. M-CSF alone. Overall, our data demonstrate that olaparib exhibits a number of favorable characteristics with respect to ICB-combination activity, including tumor cell–mediated innate immune transactivation.
STING signaling is activated in olaparib-treated patient tumors
We then investigated the potential clinical relevance of our STING signaling pathway in human tumor samples by analyzing the STING gene signature expression in PANCANCER TCGA samples. This analysis showed increased gene expression in tumors from head and neck, cervical, breast, ovarian, renal, and lung adenocarcinomas (Figure 5f). Conversely, tumor types such as uveal melanoma, prostate, low-grade glioma, and colorectal showed very low STING signaling pathway activity. This is consistent with previously reported loss of STING in colorectal cancer45 and of prostate cancer as an ‘immune cold’ tumor type.46,47 Interestingly, we observed a broad correlation of the STING gene signature with a CD8 T-cell gene signature (corr = 0.53, pval < 2.2e-16), consistent with its reported role in immune priming.48
Finally, we assessed the ability of olaparib to activate the STING axis in BRCAm tumors from the breast cohort of the MEDIOLA clinical trial (NCT02734004).49 RNA was isolated from paired biopsies from five patients before and after 4-week 300 mg bid olaparib monotherapy treatment (P1 to P5) or one patient before and after 8 weeks of olaparib monotherapy plus 4 weeks of olaparib in combination with durvalumab (P6). GSVA analysis from whole transcriptome RNA-seq revealed that five of the six patients showed an increase of STING and IFN-I pathway activity following olaparib treatment (Figure 7, S10), although this was minimal for one patient who already had high baseline expression. Importantly, P1 who did not show an increase in STING, was a non-responder, and early progressor (defined as patients who demonstrated disease progression in target lesions at or before week 28).49 Furthermore, evaluation of ctDNA collected from this patient at baseline and progression identified a single BRCA1 reversion event of a somatic deletion spanning the original deletion (15bp). The JAK-STAT pathway also showed parallel modulation following olaparib treatment in these patients. Notably, this was consistent across the intrinsic molecular subtypes (TNBC and HR+) of the breast tumors, suggesting a non-subtype-specific program of immune activation within this BRCAm cohort.25
Figure 7.
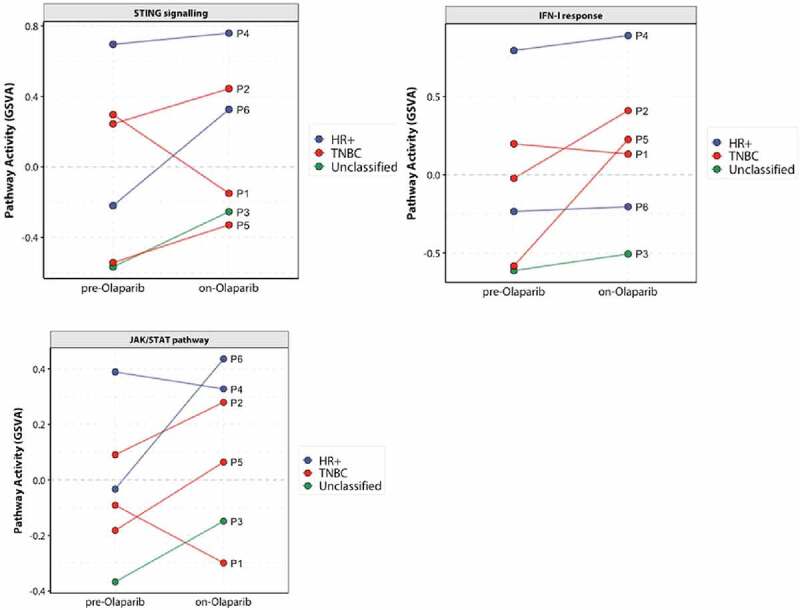
Upregulation in STING, IFN, and JAK/STAT pathway activity after olaparib treatment in BRCA mutant breast cancer patients enrolled in the MEDIOLA clinical trial (N = 6). Whole transcriptome profiling by RNAseq of paired biopsies from each patient before and after at least one cycle of olaparib therapy. Colors indicate the intrinsic molecular subtype of the tumor at baseline.
Discussion/conclusion
We demonstrate that olaparib in combination with ICB results in durable anti-tumor activity and immune memory in a Brca1m model. The efficacy is associated with olaparib-driven changes in immune cell infiltration and activation status, and increased expression of immune pathways, including STING/Type I IFN pathways. Importantly, we demonstrate that these pathways are also modulated in clinical samples from the MEDIOLA (NCT02734004) trial.
Scheduling of combinations of immune checkpoint blockade with small molecule pathway inhibitors, radiation or cytotoxic therapy can have profound effects on anti-tumor activity. Sub-group analysis of a trial combining chemotherapy with the anti-PD-L1 durvalumab (GeparNuevo, NCT02685059) reported improved pCR rate in patients where ICB treatment was started prior to chemotherapy.50 Our data demonstrate that concurrent dosing of olaparib with anti-PD-L1 shows the most consistent effect of several schedules on both tumor growth rate inhibition and depth of response, independent of the day when dosing started.
In the Brca1m BR5 model, olaparib monotherapy treatment resulted in changes in the tumor microenvironment, which were not observed in a Brca WT model. Significant increases in the frequency and activation of tumor infiltrating CD8+ T cells and NK cells, which were further augmented by combination with anti-PD-L1, were observed and were consistent with an increased anti-tumor immune response. It is formally possible that some differences between the Brca1m and Brca WT syngeneic models may have been due to tumor background, as these were not isogenic models. However, it is also important to note the differences in tumor cell line sensitivity to olaparib (BR5 GI50 = 0.23 µM, MC38 GI50 = 10 µM, CT26 GI50 = 10 µM; data not shown), which may result in differences in immune priming, consistent with our observations.
It has been reported that PARP1/2 knockout negatively affects murine T cells;42 however, this is not consistent with our in vivo observations. Furthermore, our in vitro studies show that olaparib does not inhibit human T cell activation or proliferation. Additionally, olaparib treatment does not interfere with generation of immune memory, as rechallenge of mice showing complete response to therapy resulted in complete tumor rejection, following initial tumor growth.
Combination therapy also led to a reduction in potentially suppressive myeloid cell types in the BR5 model. PARP inhibition has previously been reported to cause an increased CD8+ T cell infiltration;25,28 however, changes in the myeloid compartment have varied between reports, and our observation of a reduction in tumor infiltrating macrophages and CD11b+Ly6C+ myeloid cells has not previously been reported. Our result appears to be in contrast to a recent report that PARP1 knockout and sub-clinical low-dose olaparib resulted in increased MDSC infiltration.51 Similarly, treatment with low-dose olaparib for 5 days has been reported to increase tumor infiltration by CD11b+ and F4/80+ myeloid cells and expression of proinflammatory CD80 and CD40 markers in a Brca1m TNBC preclinical model.52 It has also been reported that modest doses of the PARP inhibitor BMN673 (talazoparib) resulted in decreased infiltration of CD11b+Gr1+ MDSC but no changes in F4/80+ macrophages in peritoneal ascites in the BR5 model.53 Differences between these observations may be affected by dose and schedule, as well as activity of the PARP inhibitors; however, effects of the tissue microenvironment studied (e.g. peritoneal wash vs. subcutaneous tumor) and tumor growth kinetics cannot be excluded.20 Because olaparib inhibits the activity of both PARP1 and PARP2,54 it is also formally possible that PARP2 may play a role in remodeling of the myeloid cell infiltrate. Further work is required to understand the potential interplay between PARP1/2 activity, tissue microenvironment, and tumor infiltrate kinetics on myeloid cell recruitment in tumors.
We demonstrate that olaparib alone, and in combination with anti-PD-L1, modulates immune pathway expression and tumor immune infiltration specifically in Brca/BRCAm tumor models and not in the WT models tested. This differs to some previously described effects of PARP inhibition by niraparib and talazoparib.28,31 In these reports, immunomodulatory effects of PARPi ± ICB were observed regardless of BRCA proficiency. In parallel, a greater anti-proliferative effect55 and STING activation56 have been reported for niraparib and talazoparib in proliferating cells, regardless of the BRCA status. Talazoparib and niraparib also have been reported to have higher incidence of toxicity in the clinic (with more dose reductions/discontinuations than olaparib).55 It remains unclear whether the cytotoxicity of these two agents, when combined with IO agents, may represent an additional opportunity or may translate to a higher, more severe incidence of side effects.
Previous studies have reported that PARP inhibition can result in increased DNA damage, micronuclei formation, and upregulation of the cGAS/STING pathway.29 Transcriptomic analyses in both mouse syngeneic tumors and human PDX models demonstrated broad upregulation of multiple immune pathways, in particular STING, IFN-I, and JAK/STAT pathways, upon PARP inhibition. The upregulation of these immune pathways in the HBCx-10 PDX model was particularly striking as this study used immunocompromised mice, and immune pathways were upregulated in the human tumor cell compartment. Thus, these changes were specific to Brca/BRCAm tumor models, in line with the observation of increased T and NK cell infiltration and activation in the BR5 model. In the BRCA WT models, it is possible that a stronger induction of DNA damage, e.g. by combination with a drug blocking an additional DDR pathway, or an independent source of DNA damage, such as radiation or cytotoxic therapy, may trigger a sustained activation of these immune pathways. For example, addition of ceralasertib to olaparib has been reported to increase tumor responses in several preclinical models, including HBCx-9,57–60 and is presently being tested in the clinic in multiple cancer indications (e.g. NCT04065269, NCT03462342, NCT03682289, and NCT03878095).
Our results show combination benefit of olaparib with either anti-PD-L1 or anti-CTLA-4 in the BR5 model. Interestingly, previously published results demonstrated combination activity for another PARPi, veliparib, plus anti-CTLA-4, but none with anti-PD-L1 in the same model.27 This difference can stem from tumor implantation site (subcutaneous vs intraperitoneal) or PARPi used. Veliparib does not significantly increase the DNA damage marker γH2AX in BRCA-deficient cells (Figures 6d, S9C), which may be related to its reported low PARP trapping activity6,41 and so may be mechanistically distinct from olaparib in this model.
Co-culture of human monocyte-derived DCs with olaparib- or talazoparib-treated BRCA-deficient MDA-MB-436 or DLD-1 tumor cells resulted in increased DC activation, whereas etoposide showed no selectivity for tumor genotype. These data therefore highlight the specificity of PARP inhibitors for tumor BRCA mutations in eliciting immune transactivation, as previously demonstrated.26 Conditioned medium from MDA-MB-436 cells treated with either olaparib, talazoparib, or etoposide was also sufficient to activate DCs and contained increased levels of the pro-inflammatory cytokines IFN-λ and IL-1β, suggesting tumor cell activation of cGAS/STING, NFκB, and NLRP3 inflammasome pathways,61,62 which have been reported to play a role in immune priming subsequent to DNA damage.63,64 Increased IL-8 expression was seen upon olaparib treatment in BRCA2−/− DLD-1 cells, suggesting that alternative inflammatory pathways may be activated in tumor cells of different genetic or tissue background. Further work is needed to confirm that increased expression of CD86 in DCs results in increased T cell priming; however, this will require engineering a BRCAm cell line expressing a defined tumor antigen to assay antigen-specific T cell activation.
The STING pathway has been reported to be downregulated in many human cancers;65 however, our analysis of ~10,000 tumors from TCGA shows STING pathway expression heterogeneity across cancer indications. We observed a positive correlation of STING signature and CD8 T cell signatures in head and neck, cervical, basal and HER2+ breast, kidney clear cell, and lung adenocarcinoma tumors. In ovarian, luminal B, and basal breast cancers, there was an enrichment of BRCA1/2 mutations with higher STING signature expression, which is consistent with recent report of higher IFN signature expression in BRCA1m ovarian cancer.66 In contrast, uveal melanoma, prostate cance,r and glioma showed significantly lower levels of both the STING and CD8 T cell signatures. While these results did not show a consistent picture across all cancer types, it suggests that greater baseline STING signaling in some tumor types may associate with potentially increased immune priming and T cell infiltration. Finally, increased STING, type I IFN, and other immune signaling signatures are observed in tumors from 5/6 BRCA mutant patients treated with olaparib (NCT02734004). The patient who did not show this modulation was found to have a BRCA reversion event at baseline and on progression. This may be a consequence of a previous treatment with carboplatin, which has been shown to contribute to BRCA reversion events.67 Future studies could focus on deeper characterization of the immune microenvironment in patients using additional protein or nucleic acid approaches.68
In conclusion, we show that treatment of BRCA-deficient tumors with clinically relevant exposures of olaparib results in anti-tumor efficacy and immune modulation alone and is increased by combination with ICB. Olaparib treatment can drive increased expression of pro-inflammatory gene signatures and modulation of tumor immune infiltrate associated with an anti-tumor immune response. Observation of increased STING/Type I IFN pathway expression in olaparib-treated clinical samples reinforce our preclinical observations of olaparib-mediated immune activation and provide mechanistic support for clinical combination of olaparib with ICB such as anti-PD-L1.69–71
Supplementary Material
Acknowledgments
The authors would like to acknowledge Dr Simon Barry and Dr Alan Lau for critical review of the manuscript; the AZD6738 project team for support of HBCx-10 and HBCx-9 studies; Dr Elizabeth Hardaker and Dr Larissa Carnevalli, LAS, and the In vivo team (TDE Bioscience AstraZeneca UK) for supporting the in vivo studies; Dr Susan Domchek and the patients and principal investigators on the MEDIOLA study, along with the MEDIOLA AstraZeneca clinical team (Dr Laura Opincar, Dr Kassondra Meyer, and Dr Pia Herbolsheimer).
Funding Statement
This study was funded by AstraZeneca.
Disclosure statement
All the authors were employees and shareholders of AstraZeneca at the time of this research.
Data availability statement
PDX RNAseq – Data available from the authors upon reasonable request. MEDIOLA patient RNAseq – Data underlying the findings described in this manuscript cannot be shared due to the content of the Informed Consent forms signed by the patients. Please visit our Disclosure Commitment page for guidance on AstraZeneca Data Sharing Policy: https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/2162402X.2022.2083755
List of abbreviations
| ATM | Ataxia Telangiectasia Mutated |
| BRCAm | BRCA mutant |
| CR | Complete responder |
| CTLA-4 | Cytotoxic T-lymphocyte-associated protein 4 |
| DC | Dendritic cell |
| FBS | Fetal bovine serum |
| FCS | Fetal calf serum |
| GzmB | Granzyme B |
| HR | Homologous recombination |
| ICB | Immune checkpoint blockade |
| IFN | Interferon |
| JAK | Janus kinase |
| MDSC | Myeloid-derived suppressor cells |
| NK | Natural killer |
| NSCLC | Non–small cell lung cancer |
| PARP | Poly (ADP-ribose) polymerase |
| PARPi | PARP inhibitor |
| PD-L1 | Programmed death-ligand 1 |
| PDX | Patient-derived xenograft |
| STAT | Signal transducer and activator of transcription |
| STING | Stimulator of interferon genes |
| TNBC | Triple-negative breast cancer |
| WT | wild-type |
References
- 1.Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative reg ulation of lymphocyte activation. J Exp Med. 2000;192(7):1027–16. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krummel MF, Allison JP.. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995;182(2):459–465. doi: 10.1084/jem.182.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gong J, Chehrazi-Raffle A, Reddi S, Salgia R. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. J Immunother Cancer. 2018;6(1):8. doi: 10.1186/s40425-018-0316-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gotwals P, Cameron S, Cipolletta D, Cremasco V, Crystal A, Hewes B, Mueller B, Quaratino S, Sabatos-Peyton C, Petruzzelli L, et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat Rev Cancer. 2017;17(5):286–301. doi: 10.1038/nrc.2017.17. [DOI] [PubMed] [Google Scholar]
- 5.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 6.Pommier Y, O’Connor MJ, de Bono J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci Transl Med. 2016;8:362ps17. [DOI] [PubMed] [Google Scholar]
- 7.Farmer H, McCabe N, Lord CJ, Tutt ANJ, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 8.Martinez A, Delord J-P, Ayyoub M, Devaud C. Preclinical and Clinical Immunotherapeutic Strategies in Epithelial Ovarian Cancer. Cancers (Basel). 2020;12(7):1761. doi: 10.3390/cancers12071761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim IS, Gao Y, Welte T, Wang H, Liu J, Janghorban M, Sheng K, Niu Y, Goldstein A, Zhao N, et al. Immuno-subtyping of breast cancer reveals distinct myeloid cell profiles and immunotherapy resistance mechanisms. Nat Cell Biol. 2019;21(9):1113–1126. doi: 10.1038/s41556-019-0373-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dunphy G, Flannery SM, Almine JF, Connolly DJ, Paulus C, Jønsson KL, Jakobsen MR, Nevels MM, Bowie AG, Unterholzner L, et al. Non-canonical activation of the DNA sensing adaptor STING by ATM and IFI16 mediates NF-kappaB Signaling after nuclear DNA damage. Mol Cell. 2018;71(5):745–760 e5. doi: 10.1016/j.molcel.2018.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hartlova A, Erttmann SF, Raffi FA, Schmalz AM, Resch U, Anugula S, Lienenklaus S, Nilsson LM, Kröger A, Nilsson JA, et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity. 2015;42(2):332–343. doi: 10.1016/j.immuni.2015.01.012. [DOI] [PubMed] [Google Scholar]
- 12.Heijink AM, Talens F, Jae LT, van Gijn SE, Fehrmann RSN, Brummelkamp TR, van Vugt MATM. BRCA2 deficiency instigates cGAS-mediated inflammatory signaling and confers sensitivity to tumor necrosis factor-alpha-mediated cytotoxicity. Nat Commun. 2019;10(1):100. doi: 10.1038/s41467-018-07927-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reislander T, Lombardi EP, Groelly FJ, Miar A, Porru M, Di Vito S, Wright B, Lockstone H, Biroccio A, Harris A, et al. BRCA2 abrogation triggers innate immune responses potentiated by treatment with PARP inhibitors. Nat Commun. 2019;10(1):3143. doi: 10.1038/s41467-019-11048-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol. 2015;15(7):405–414. doi: 10.1038/nri3845. [DOI] [PubMed] [Google Scholar]
- 15.Brown JS, Sundar R, Lopez J. Combining DNA damaging therapeutics with immunotherapy: more haste, less speed. Br J Cancer. 2018;118(3):312–324. doi: 10.1038/bjc.2017.376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Domchek S, Postel-Vinay S, Im S-A, Park YH, Delord J-P, Italiano A, Alexandre J, You B, Bastian S, Krebs MG, et al. 1191O - Phase II study of olaparib (O) and durvalumab (D) (MEDIOLA): updated results in patients (pts) with germline BRCA-mutated (gBRCAm) metastatic breast cancer (MBC). Annals of Oncology. 2019;30:v477. doi: 10.1093/annonc/mdz253.017. [DOI] [Google Scholar]
- 17.Xing D, Orsulic S. A mouse model for the molecular characterization of brca1-associated ovarian carcinoma. Cancer Res. 2006;66(18):8949–8953. doi: 10.1158/0008-5472.CAN-06-1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karp NA, Wilson Z, Stalker E, Mooney L, Lazic SE, Zhang B, Hardaker E. A multi-batch design to deliver robust estimates of efficacy and reduce animal use - a syngeneic tumour case study. Sci Rep. 2020;10(1):6178. doi: 10.1038/s41598-020-62509-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hather G, Liu R, Bandi S, Mettetal J, Manfredi M, Shyu W-C, Donelan J, Chakravarty A. Growth rate analysis and efficient experimental design for tumor xenograft studies. Cancer Inform. 2014;13(Suppl 4):65–72. doi: 10.4137/CIN.S13974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taylor MA, Hughes AM, Walton J, Coenen-Stass AML, Magiera L, Mooney L, Bell S, Staniszewska AD, Sandin LC, Barry ST, et al. Longitudinal immune characterization of syngeneic tumor models to enable model selection for immune oncology drug discovery. J Immunother Cancer. 2019;7(1):328. doi: 10.1186/s40425-019-0794-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 2019;37(8):907–915. doi: 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garcia-Alcalde F, Okonechnikov K, Carbonell J, Cruz LM, Götz S, Tarazona S, Dopazo J, Meyer TF, Conesa A. Qualimap: evaluating next-generation sequencing alignment data. Bioinformatics. 2012;28(20):2678–2679. doi: 10.1093/bioinformatics/bts503. [DOI] [PubMed] [Google Scholar]
- 23.Abe T, Harashima A, Xia T, Konno H, Konno K, Morales A, Ahn J, Gutman D, Barber G. STING recognition of cytoplasmic DNA instigates cellular defense. Mol Cell. 2013;50(1):5–15. doi: 10.1016/j.molcel.2013.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lord CJ, Ashworth A. PARP inhibitors: synthetic lethality in the clinic. Science. 2017;355(6330):1152–1158. doi: 10.1126/science.aam7344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pantelidou C, Sonzogni O, De Oliveria Taveira M, Mehta AK, Kothari A, Wang D, Visal T, Li MK, Pinto J, Castrillon JA, et al. PARP inhibitor efficacy depends on CD8+T-cell recruitment via intratumoral STING pathway activation in BRCA-deficient models of triple-negative breast cancer. Cancer Discov. 2019;9(6):722–737. doi: 10.1158/2159-8290.CD-18-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ding L, Kim H-J, Wang Q, Kearns M, Jiang T, Ohlson CE, Li BB, Xie S, Liu JF, Stover EH, et al. PARP inhibition elicits STING-dependent antitumor immunity in brca1-deficient ovarian cancer. Cell Rep. 2018;25(11):2972–2980 e5. doi: 10.1016/j.celrep.2018.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Higuchi T, Flies DB, Marjon NA, Mantia-Smaldone G, Ronner L, Gimotty PA, Adams SF. CTLA-4 blockade synergizes therapeutically with PARP inhibition in BRCA1-deficient ovarian cancer. Cancer Immunol Res. 2015;3(11):1257–1268. doi: 10.1158/2326-6066.CIR-15-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Z, Sun K, Xiao Y, Feng B, Mikule K, Ma X, Feng N, Vellano CP, Federico L, Marszalek JR, et al. Niraparib activates interferon signaling and potentiates anti-PD-1 antibody efficacy in tumor models. Sci Rep. 2019;9(1):1853. doi: 10.1038/s41598-019-38534-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chabanon RM, Muirhead G, Krastev DB, Adam J, Morel D, Garrido M, Lamb A, Hénon C, Dorvault N, Rouanne M, et al. PARP inhibition enhances tumor cell-intrinsic immunity in ERCC1-deficient non-small cell lung cancer. J Clin Invest. 2019;129(3):1211–1228. doi: 10.1172/JCI123319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sen T, Rodriguez BL, Chen L, Corte CMD, Morikawa N, Fujimoto J, Cristea S, Nguyen T, Diao L, Li L, et al. Targeting DNA damage response promotes antitumor immunity through STING-mediated T-cell activation in small cell lung cancer. Cancer Discov. 2019;9(5):646–661. doi: 10.1158/2159-8290.CD-18-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen J, Zhao W, Ju Z, Wang L, Peng Y, Labrie M, Yap TA, Mills GB, Peng G. PARPi Triggers the STING-dependent immune response and enhances the therapeutic efficacy of immune checkpoint blockade independent of brcaness. Cancer Res. 2019;79(2):311–319. doi: 10.1158/0008-5472.CAN-18-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim C, Wang XD, Yu Y. PARP1 inhibitors trigger innate immunity via PARP1 trapping-induced DNA damage response. Elife. 2020;9:e60637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tirosh I, Izar B, Prakadan SM, Wadsworth MH, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352(6282):189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu MM, Pu Y, Han D, Shi Y, Cao X, Liang H, Chen X, Li X-D, Deng L, Chen ZJ, et al. Dendritic cells but not macrophages sense tumor mitochondrial DNA for cross-priming through signal regulatory protein alpha signaling. Immunity. 2017;47(2):363–373 e5. doi: 10.1016/j.immuni.2017.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Woo SR, Fuertes M, Corrales L, Spranger S, Furdyna M, Leung MK, Duggan R, Wang Y, Barber G, Fitzgerald K, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41(5):830–842. doi: 10.1016/j.immuni.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klarquist J, Hennies CM, Lehn MA, Reboulet RA, Feau S, Janssen EM. STING-mediated DNA sensing promotes antitumor and autoimmune responses to dying cells. J Immunol. 2014;193(12):6124–6134. doi: 10.4049/jimmunol.1401869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Diamond JM, Vanpouille-Box C, Spada S, Rudqvist N-P, Chapman JR, Ueberheide BM, Pilones KA, Sarfraz Y, Formenti SC, Demaria S, et al. Exosomes shuttle TREX1-sensitive IFN-stimulatory dsDNA from irradiated cancer cells to DCs. Cancer Immunol Res. 2018;6(8):910–920. doi: 10.1158/2326-6066.CIR-17-0581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carozza JA, Böhnert V, Nguyen KC, Skariah G, Shaw KE, Brown JA, Rafat M, von Eyben R, Graves EE, Glenn JS, et al. Extracellular cGAMP is a cancer-cell-produced immunotransmitter involved in radiation-induced anticancer immunity. Nature Cancer. 2020;1(2):184–196. doi: 10.1038/s43018-020-0028-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Riches LC, Trinidad AG, Hughes G, Jones GN, Hughes AM, Thomason AG, Gavine P, Cui A, Ling S, Stott J, et al. Pharmacology of the ATM Inhibitor AZD0156: potentiation of Irradiation and Olaparib Responses Preclinically. Mol Cancer Ther. 2020;19(1):13–25. doi: 10.1158/1535-7163.MCT-18-1394. [DOI] [PubMed] [Google Scholar]
- 40.Jones P, Altamura S, Boueres J, Ferrigno F, Fonsi M, Giomini C, Lamartina S, Monteagudo E, Ontoria JM, Orsale MV, et al. Discovery of 2-{4-[(3S)-Piperidin-3-yl]phenyl}-2H-indazole-7-carboxamide (MK-4827): a novel oral poly(ADP-ribose)polymerase (PARP) inhibitor efficacious in BRCA-1 and −2 Mutant Tumors. J Med Chem. 2009;52(22):7170–7185. doi: 10.1021/jm901188v. [DOI] [PubMed] [Google Scholar]
- 41.Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, Ji J, Takeda S, Pommier Y. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72(21):5588–5599. doi: 10.1158/0008-5472.CAN-12-2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moreno-Lama L, Galindo-Campos MA, Martínez C, Comerma L, Vazquez I, Vernet-Tomas M, Ampurdanés C, Lutfi N, Martin-Caballero J, Dantzer F, et al. Coordinated signals from PARP-1 and PARP-2 are required to establish a proper T cell immune response to breast tumors in mice. Oncogene. 2020;39(13):2835–2843. doi: 10.1038/s41388-020-1175-x. [DOI] [PubMed] [Google Scholar]
- 43.Olabisi OA, Soto-Nieves N, Nieves E, Yang TTC, Yang X, Yu RYL, Suk HY, Macian F, Chow C-W. Regulation of transcription factor NFAT by ADP-ribosylation. Mol Cell Biol. 2008;28(9):2860–2871. doi: 10.1128/MCB.01746-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Valdor R, Schreiber V, Saenz L, Martínez T, Muñoz-Suano A, Dominguez-Villar M, Ramírez P, Parrilla P, Aguado E, García-Cózar F, Yélamos J. Regulation of NFAT by poly(ADP-ribose) polymerase activity in T cells. Mol Immunol. 2008;45(7):1863–1871. doi: 10.1016/j.molimm.2007.10.044. [DOI] [PubMed] [Google Scholar]
- 45.Xia T, Konno H, Ahn J, Barber GN. Deregulation of STING signaling in colorectal carcinoma constrains DNA damage responses and correlates with tumorigenesis. Cell Rep. 2016;14(2):282–297. doi: 10.1016/j.celrep.2015.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bou-Dargham MJ, Sha L, Sang QXA, Zhang J. Immune landscape of human prostate cancer: immune evasion mechanisms and biomarkers for personalized immunotherapy. BMC Cancer. 2020;20(1):572. doi: 10.1186/s12885-020-07058-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vitkin N, Nersesian S, Siemens DR, Koti M. The Tumor Immune Contexture of Prostate Cancer. Front Immunol. 2019;10:603. doi: 10.3389/fimmu.2019.00603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fuertes MB, Woo S-R, Burnett B, Fu Y-X, Gajewski TF. Type I interferon response and innate immune sensing of cancer. Trends Immunol. 2013;34(2):67–73. doi: 10.1016/j.it.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Domchek SM, Postel-Vinay S, Im S-A, Park YH, Delord J-P, Italiano A, Alexandre J, You B, Bastian S, Krebs MG, et al. Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA): an open-label, multicentre, phase 1/2, basket study. Lancet Oncol. 2020;21(9):1155–1164. doi: 10.1016/S1470-2045(20)30324-7. [DOI] [PubMed] [Google Scholar]
- 50.Loibl S, Untch M, Burchardi N, Huober J, Sinn BV, Blohmer J-U, Grischke E-M, Furlanetto J, Tesch H, Hanusch C, et al. A randomised phase II study investigating durvalumab in addition to an anthracycline taxane-based neoadjuvant therapy in early triple-negative breast cancer: clinical results and biomarker analysis of GeparNuevo study. Ann Oncol. 2019;30(8):1279–1288. doi: 10.1093/annonc/mdz158. [DOI] [PubMed] [Google Scholar]
- 51.Ghonim MA, Ibba SV, Tarhuni AF, Errami Y, Luu HH, Dean MJ, El-Bahrawy AH, Wyczechowska D, Benslimane IA, Del Valle L, Al-Khami AA, Ochoa AC, Boulares AH. Targeting PARP-1 with metronomic therapy modulates MDSC suppressive function and enhances anti-PD-1 immunotherapy in colon cancer. J Immunother Cancer. 2021;9(1):e001643. doi: 10.1136/jitc-2020-001643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mehta AK, Cheney EM, Hartl CA, Pantelidou C, Oliwa M, Castrillon JA, Lin JR, Hurst KE, de Oliveira Taveira M, Johnson NT, Oldham WM, Kalocsay M, Berberich MJ, Boswell SA, Kothari A, Johnson S, Dillon DA, Lipschitz M, Rodig S, Santagata S, Garber JE. et al. Targeting immunosuppressive macrophages overcomes PARP inhibitor resistance in BRCA1-associated triple-negative breast cancer. Nat Cancer. 2021;2(1):66–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang J, Wang L, Cong, Z, Amoozgar Z, Kiner E, Xing D, Orsulic S, Matulonis U, Goldberg MS. The PARP1 inhibitor BMN 673 exhibits immunoregulatory effects in a Brca1(-/-) murine model of ovarian cancer. Biochem Biophys Res Commun. 2015;463(4):551–556. doi: 10.1016/j.bbrc.2015.05.083. [DOI] [PubMed] [Google Scholar]
- 54.Menear KA, Adcock C, Boulter R, Cockcroft X, Copsey L, Cranston A, Dillon KJ, Drzewiecki J, Garman S, Gomez S, Javaid H, Kerrigan F, Knights C, Lau A, Loh VM, Matthews ITW, Moore S, O'Connor MJ, Smith GCM, Martin NMB. 4-[3-(4-cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phthalazin- 1-one: a novel bioavailable inhibitor of poly(ADP-ribose) polymerase-1. J Med Chem. 2008;51(20):6581–6591. doi: 10.1021/jm8001263. [DOI] [PubMed] [Google Scholar]
- 55.Pilie PG, Gay CM, Byers LA, O’Connor MJ, Yap TA. PARP Inhibitors: extending Benefit Beyond BRCA-Mutant Cancers. Clin Cancer Res. 2019;25(13):3759–3771. doi: 10.1158/1078-0432.CCR-18-0968. [DOI] [PubMed] [Google Scholar]
- 56.Meng J, Peng J, Feng J, Maurer J, Li X, Li Y, Yao S, Chu R, Pan X, Li J, Zhang T, Liu L, Zhang Q, Yuan Z, Bu H, Song K, Kong B. Niraparib exhibits a synergistic anti-tumor effect with PD-L1 blockade by inducing an immune response in ovarian cancer. J Transl Med. 2021;19(1):415. doi: 10.1186/s12967-021-03073-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim H, George E, Ragland RL, Rafail S, Zhang R, Krepler C, Morgan MA, Herlyn M, Brown EJ, Simpkins F, et al. Targeting the ATR/CHK1 Axis with PARp inhibition results in tumor regression in BRCA-mutant ovarian cancer models. Clin Cancer Res. 2017;23(12):3097–3108. doi: 10.1158/1078-0432.CCR-16-2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim H, Xu H, George E, Hallberg D, Kumar S, Jagannathan V, Medvedev S, Kinose Y, Devins K, Verma P, et al. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat Commun. 2020;11(1):3726. doi: 10.1038/s41467-020-17127-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lau AY, Yates J, Wilson Z, Young LA, Hughes AM, Berges A, Cheung A, Odedra R, Brown E, O'Connor MJ, Hollingsworth S. ATR inhibitor AZD6738 as monotherapy and in combination with olaparib or chemotherapy: defining pre-clinical dose-schedules and efficacy modelling [abstract]. in Proceedings of the American Association for Cancer Research Annual Meeting 2017; 2017 Apr 1-5; Washington, DC. 2017. [Google Scholar]
- 60.Lloyd RL, Wijnhoven PWG, Ramos-Montoya A, Wilson Z, Illuzzi G, Falenta K, Jones GN, James N, Chabbert CD, Stott J, et al. Combined PARP and ATR inhibition potentiates genome instability and cell death in ATM-deficient cancer cells. Oncogene. 2020;39(25):4869–4883. doi: 10.1038/s41388-020-1328-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Osterlund PI, Pietilä TE, Veckman V, Kotenko SV, Julkunen I. IFN regulatory factor family members differentially regulate the expression of type III IFN (IFN-lambda) genes. J Immunol. 2007;179(6):3434–3442. doi: 10.4049/jimmunol.179.6.3434. [DOI] [PubMed] [Google Scholar]
- 62.Gaidt MM, Ebert TS, Chauhan D, Ramshorn K, Pinci F, Zuber S, O’Duill F, Schmid-Burgk JL, Hoss F, Buhmann R, et al. The DNA inflammasome in human myeloid cells is initiated by a STING-cell death program upstream of NLRP3. Cell. 2017;171(5):1110–1124 e18. doi: 10.1016/j.cell.2017.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, Vermaelen K, Panaretakis T, Mignot G, Ullrich E, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. 2009;15(10):1170–1178. doi: 10.1038/nm.2028. [DOI] [PubMed] [Google Scholar]
- 64.Reislander T, Groelly FJ, Tarsounas M. DNA Damage and Cancer Immunotherapy: a STING in the Tale. Mol Cell. 2020;80(1):21–28. doi: 10.1016/j.molcel.2020.07.026. [DOI] [PubMed] [Google Scholar]
- 65.Xia T, Konno H, Barber GN. Recurrent loss of STING signaling in melanoma correlates with susceptibility to viral oncolysis. Cancer Res. 2016;76(22):6747–6759. doi: 10.1158/0008-5472.CAN-16-1404. [DOI] [PubMed] [Google Scholar]
- 66.Bruand M, Barras D, Mina M, Ghisoni E, Morotti M, Lanitis E, Fahr N, Desbuisson M, Grimm A, Zhang H, et al. Cell-autonomous inflammation of BRCA1-deficient ovarian cancers drives both tumor-intrinsic immunoreactivity and immune resistance via STING. Cell Rep. 2021;36(3):109412. doi: 10.1016/j.celrep.2021.109412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lin KK, Harrell MI, Oza AM, Oaknin A, Ray-Coquard I, Tinker AV, Helman E, Radke MR, Say C, Vo L-T, et al. BRCA reversion mutations in circulating tumor DNA predict primary and acquired resistance to the PARP inhibitor rucaparib in high-grade ovarian carcinoma. Cancer Discov. 2019;9(2):210–219. doi: 10.1158/2159-8290.CD-18-0715. [DOI] [PubMed] [Google Scholar]
- 68.Labrie M, Li A, Creason A, Betts C, Keck J, Johnson B, Sivagnanam S, Boniface C, Ma H, Blucher A, et al. Multiomics analysis of serial PARP inhibitor treated metastatic TNBC inform on rational combination therapies. NPJ Precis Oncol. 2021;5(1):92. doi: 10.1038/s41698-021-00232-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chindelevitch L, Ziemek D, Enayetallah A, Randhawa R, Sidders B, Brockel C, Huang ES. Causal reasoning on biological networks: interpreting transcriptional changes. Bioinformatics. 2012;28(8):1114–1121. doi: 10.1093/bioinformatics/bts090. [DOI] [PubMed] [Google Scholar]
- 70.Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013;14(1):7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 2010;8(6):e1000412. doi: 10.1371/journal.pbio.1000412 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
PDX RNAseq – Data available from the authors upon reasonable request. MEDIOLA patient RNAseq – Data underlying the findings described in this manuscript cannot be shared due to the content of the Informed Consent forms signed by the patients. Please visit our Disclosure Commitment page for guidance on AstraZeneca Data Sharing Policy: https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure.