

ABSTRACT
Macrophages rapidly undergo glycolytic reprogramming in response to macroautophagy/autophagy, inflammasome activation and pyroptosis for the clearance of bacteria. Identification the key molecules involved in these three events will provide critical potential therapeutic applications. Upon S. typhimurium infection, FLT4/VEGFR3 and its ligand VEGFC were inducibly expressed in macrophages, and FLT4 signaling inhibited CASP1 (caspase 1)-dependent inflammasome activation and pyroptosis but enhanced MAP1LC3/LC3 activation for elimination of the bacteria. Consistently, FLT4 mutants lacking the extracellular ligand-binding domain increased production of the proinflammatory metabolites such as succinate and lactate, and reduced antimicrobial metabolites including citrate and NAD(P)H in macrophages and liver upon infection. Mechanistically, FLT4 recruited AMP-activated protein kinase (AMPK) and phosphorylated Y247 and Y441/442 in the PRKAA/alpha subunit for AMPK activation. The AMPK agonist AICAR could rescue glycolytic reprogramming and inflammasome activation in macrophages expressing the mutant FLT4, which has potential translational application in patients carrying Flt4 mutations to prevent recurrent infections. Collectively, we have elucidated that the FLT4-AMPK module in macrophages coordinates glycolytic reprogramming, autophagy, inflammasome activation and pyroptosis to eliminate invading bacteria.
Abbreviations: 3-MA: 3-methyladenine; AICAR: 5-aminoimidazole-4-carboxamide1-β-D-ribofuranoside; AMP: adenosine monophosphate; AMPK: AMP-activated protein kinase; ATP: adenosine triphosphate; BMDM: bone marrow-derived macrophage; CASP1: caspase 1; CFUs: colony-forming units; FLT4/VEGFR3: FMS-like tyrosine kinase 4; GFP: green fluorescent protein; LDH: lactate dehydrogenase; LPS: lipopolysaccharide; MAP1LC3/LC3: microtubule-associated protein 1 light chain 3; PEM: peritoneal exudate macrophage; PRKAA1/AMPKα1: protein kinase, AMP-activated, alpha 1 catalytic subunit; PYCARD/ASC: PYD and CARD domain containing; ROS: reactive oxygen species; SQSTM1/p62: sequestosome 1; TLR4: toll-like receptor 4; ULK1: unc-51 like autophagy activating kinase 1; VEGFC: vascular endothelial growth factor C; WT: wild type
KEYWORDS: AMPK, FLT4/VEGFR3, glycolytic reprogramming, inflammasome, pyroptosis
Introduction
As the first line of host defense against invading pathogens, macrophages recognize pathogen-associated molecular patterns through pattern-recognition receptors to rapidly and efficiently remove pathogens [1]. Various processes are involved during clearance of the intracellular bacteria, including phagocytosis and autophagy, production of pro-inflammatory cytokines or chemokines, as well as macrophage pyroptosis, which might be coordinated with changes of cell metabolism [2]. Upon infection with Gram-negative bacteria, macrophage activation by lipopolysaccharide (LPS) is accompanied by marked metabolic changes including upregulation of aerobic glycolysis and the pentose phosphate pathway, and disruption of the tricarboxylic acid cycle [3]. These metabolic pathways not only provide energy, but also support macrophage function. LPS-activated inflammatory macrophages (M1) utilize Warburg metabolism for rapid adenosine triphosphate (ATP) production to fuel production of cytokines and anti-bacterial defense. The pentose phosphate pathway and citrate metabolism pathways generate NAD(P)H for nitric oxide and reactive oxygen species (ROS) production, which play crucial roles in killing phagocytosed bacteria [4,5]. Moreover, disruption of the tricarboxylic acid cycle leads to increased accumulation of succinate, resulting in the stabilization of HIF1A (hypoxia inducible factor 1, alpha subunit) and the induction of IL1B/IL1β [6]. IL1B maturation is associated with inflammasome activation and is characterized by IL1B cleavage by CASP1, leading to pyroptosis. Balance of these events results in different fates of the host cells during infection [7,8], and therefore it is fundamental to understand the key molecules that coordinate glycolytic reprogramming, inflammasome activation, and macrophage pyroptosis during anti-bacterial defense.
Due to competition from invading microorganisms, deficiency of intracellular nutrients is thought to be a danger signal that leads to activation of autophagy. Autophagy maintains cellular metabolic homeostasis by enabling the removal of defective organelles, protein aggregates, or intracellular microorganisms [9,10]. When autophagy targets non-self origin such as microbe, it is also referred to as xenophagy. We use the term “autophagy” that contains “xenophagy” throughout this manuscript. The autophagic process is regulated by a number of critical gene products identified by previous studies, including MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3) [11,12]. MAP1LC3 plays crucial roles in the formation of globular autophagosomes which would transport bacteria to lysosome for degradation. Interestingly, glucose metabolism is intimately related to cellular autophagy and glucose deprivation results in increased autophagy. This is brought about by the decreased cellular ATP levels, leading to the increased adenosine monophosphate (AMP):ATP ratio that results in activation of the energy sensor AMPK [13]. AMPK promotes autophagy by activation of ULK1 (unc-51 like autophagy activating kinase 1) [14,15]. It is particularly intriguing that AMPK can be activated not only by AMP:ATP ratio, but also by cell surface receptors, hinting those extracellular signals might regulate both autophagy and metabolism [16–18]. We therefore asked which cell surface receptors can sense extracellular signals to activate AMPK and coordinate cellular metabolism with autophagy, inflammasome activation, pyroptosis during bacterial infection.
Our previous finding has demonstrated that the tyrosine kinase receptor FLT4/VEGFR3 (FMS-like tyrosine kinase 4) and the ligand VEGFC (vascular endothelial growth factor C) increase their expression in macrophages upon bacterial infection [19]. In addition, other studies have demonstrated the critical roles of the FLT4-mediated pathway in the regulation of lymphatic and blood vascular development [20]. Of particular interest, FLT4 mutations in patients not only show defective lymphedema [21], but also cause frequently recurrent infections [22] and the underline mechanism has not been fully understood. In this study, we investigated how FLT4 and TLR4 (toll-like receptor 4) sensed their extracellular ligands to link glycolytic metabolism, autophagy, inflammasome activation as well as macrophage pyroptosis together for bacterial elimination.
Results
Glycolytic reprogramming and bacteria clearance are controlled by macrophage surface receptor FLT4
To understand how glycolytic reprogramming changes are linked with bacteria clearance, we examined the glucometabolic profile and the amount of S. typhimurium in the target organ liver, after Flt4WT/WT (wild type) mice were infected with the intracellular bacteria S. typhimurium strain SL1344. The products Citrate and NAD(P)H, which were previously reported to have direct antimicrobial activity [5], were abundantly increased in livers upon infection. In contrast, we noticed that concentrations of succinate and lactate, which could promote proinflammatory macrophage function, were decreased in livers of SL1344-infected Flt4WT/WT mice compared to the PBS-treated Flt4WT/WT mice (Figure 1A). Previous studies suggest that although increased aerobic glycolytic activity can increase lactate production [23], and high levels of succinate and lactate might also be associated with uncontrolled inflammation that would contribute to the induction of sepsis. Since we previously demonstrated that the Flt4 mutant mice lacking the extracellular ligand-binding domain (LBD: lack of immunoglobulin-like domains 2 and 3, named flt4∆LBD/ΔLBD) were much more susceptible to infection [19], we therefore compared glycolytic reprogramming changes between Flt4WT/WT mice and flt4ΔLBD/ΔLBD mice. flt4ΔLBD/ΔLBD mice specifically expressing the mutant flt4ΔLBD/ΔLBD in myeloid cells including macrophages, were generated that showed higher mortality in response to S. typhimurium infection [19]. Unlike Flt4WT/WT mice, flt4ΔLBD/ΔLBD mice failed to increase the production of Citrate and NAD(P)H upon infection. Also, Flt4WT/WT mice decreased the production of succinate and lactate, while the mutant mice showed an increased production of these metabolites (Figure 1B-F). In addition, flt4ΔLBD/ΔLBD mice displayed a substantially increased amount of S. typhimurium bacteria in livers and spleens (Figure 1G). FLT4 is a typical tyrosine kinase receptor, and mice expressing the tyrosine kinase inactive mutant Flt4WT/TKmut (also named Flt4Chy) were generated as previously described [19,20,24]. Similarly, Flt4WT/TKmut mice failed to effectively clear S. typhimurium bacteria in liver and spleen compared to the infected Flt4WT/WT mice (Fig. S1A). We wondered whether the defective antimicrobial defense phenotype in Flt4-deficient mice was caused by the difference in glucose metabolism as evident by higher levels of succinate and lactate, but lower levels of citrate and NAD(P)H compared to infected Flt4WT/WT mice. This might help us to find new strategies to treat the recurrent infections in patients carrying the FLT4 mutations [25].
Figure 1.
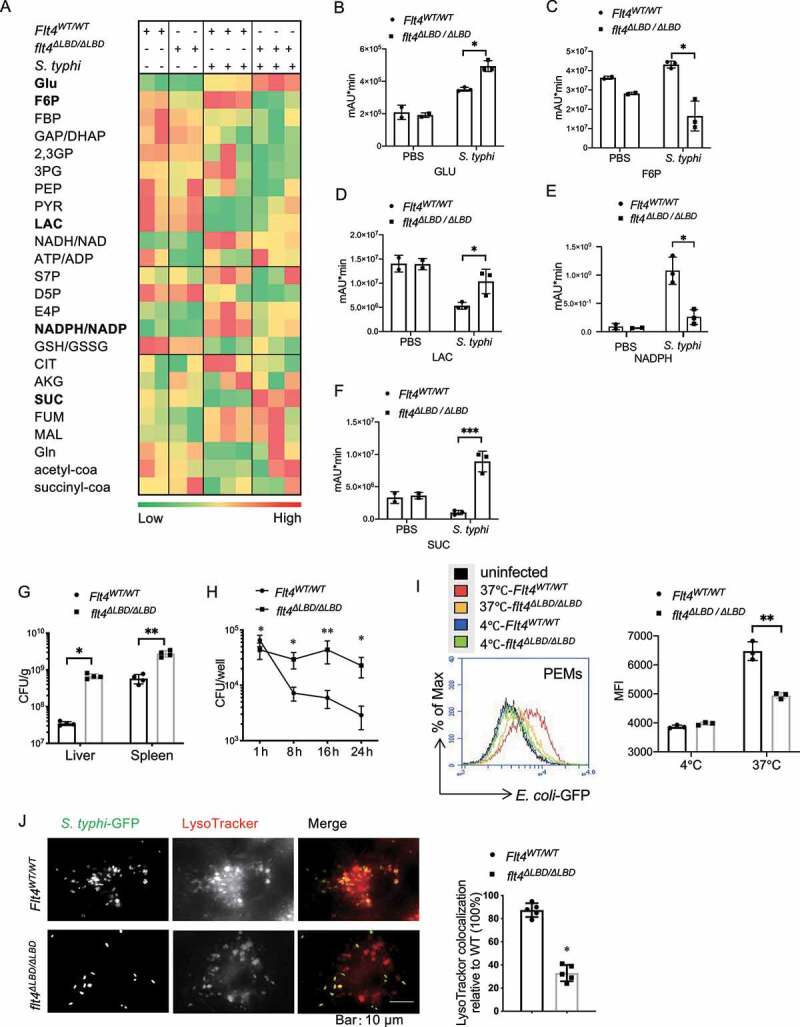
Glycolytic reprogramming and bacteria clearance are controlled by macrophage surface receptor FLT4. (A) S. typhimurium was injected into the tail veins of Flt4WT/WT and flt4∆LBD/∆LBD mice. The glucometabolites profile in livers by LC-MS metabolomics assay. (B-F) Statistical analyses of specific metabolite across treatment groups. *p < 0.05, **p < 0.01 (G) Flt4WT/WT and flt4∆LBD/∆LBD mice were injected in the tail vein with S. typhimurium. bacterial colony-forming units (CFU) in livers and spleens on day 4 were assessed. Data represents 4 mice per group. *p < 0.05, **p < 0.01. (H) Flt4WT/WT and flt4∆LBD/∆LBD PEMs were infected with S. typhimurium for 24 h to determine bacterial CFU. Data are presented as the mean ± SEM of triplicate experiments. *p < 0.05, **p < 0.01. (I) Flt4WT/WT and flt4∆LBD/∆LBD PEMs were infected for 30 min with GFP-E. coli to measure phagocytosis by FACS. Histogram plots of one representative experiment. Quantification of phagocytosis was done by comparing the geometric mean fluorescence intensity (MFI). Data are presented as the mean ± SEM of triplicate experiments. *p < 0.05, **p < 0.01 (J) Flt4WT/WT and flt4∆LBD/∆LBD BMDMs loaded with LysoTracker Red were infected with S. typhimurium-GFP for 1 h, and imaged by confocal microscopy, the percentages of LysoTracker-positive phagosomes were quantified at least 200 cells, Data are presented as the mean ± SEM *p < 0.05, **p < 0.01. 2,3 GP, 2,3-bisphosphoglycerate; 3PG, 3-phosphoglycerate; acetyl-coa, acetyl coenzyme A; AKG, a-ketoglutarate; ATP/ADP, adenosine triphosphate/adenosine diphosphate; CIT, citrate; D5P, D-ribose 5-phosphate; E4P, erythrose 4-phosphate; F6P, fructose-6-phosphate; FBP, fructose 1,6-bisphosphatase; FUM, fumarate; GAP/DHAP, glyceraldehyde-3-phosphate-dihydroxyacetone phosphate; Gln, glutamine; Glu, glucose; GSH:GSSG, The reduced glutathione:oxidized glutathione ratio; LAC, lactate; MAL, malate; NAD, nicotinamide adenine dinucleotide; NADP, nicotinamide adenine dinucleotide phosphate; PEP, phosphoenolpyruvate; PYR, pyruvate; S7P, 7-phosphate sedoheptulose; SUC, succinate; succinyl-coa, succinyl coenzyme A.
To answer this, we investigated how FLT4 affected bacteria clearance by assessment of the colony-forming units (CFUs) of intracellular S. typhimurium in macrophages. Similar to the defective in vivo anti-bacteria response, flt4ΔLBD/∆LBD macrophages harbored much higher amounts of S. typhimurium compared to the Flt4WT/WT macrophages (Figure 1H). Since phagocytes including macrophages could engulf solid particles, a critical process to kill bacteria [26,27], we first checked whether FLT4 signaling affected engulfing ability. GFP (green fluorescent protein)-Escherichia coli (E. coli-GFP) was incubated with PEM (peritoneal exudate macrophage) from flt4∆LBD/∆LBD mice or the Flt4WT/WT littermates, and flow cytometry revealed that flt4∆LBD/∆LBD PEMs phagocytosed less amount of E. coli-GFP than Flt4WT/WT PEMs (Figure 1I). To specifically measure the engulfing ability and rule out potential effect due to lysis of bacteria, we incubated macrophages with fluorescence-labeled latex beads. Bone marrow-derived macrophages (BMDMs) isolated from flt4∆LBD/∆LBD mice showed reduced engulfing ability compared to Flt4WT/WT cells (Fig. S1B). Similar results were obtained using macrophages expressing the mutant Flt4WT/TKmut, which phagocytized much lesser amounts of fluorescence-labeled latex beads when compared to that of the Flt4WT/WT macrophages (Fig. S1C). In addition, because we observed altered NADPH/NADP levels in the infected flt4∆LBD/∆LBD mice, we therefore asked whether FLT4 also affected ROS production that contributed to bacteria clearance. The production of ROS was enhanced in macrophages upon S. typhimurium infection, and flt4∆LBD/∆LBD macrophages displays slightly reduced production of ROS compared to that of the Flt4WT/WT macrophages (Fig. S1D). These results indicate that FLT4 promotes engulfing ability and ROS production to clear bacteria. However, considering the substantial changes of the bacteria CFUs, other mechanism might be also involved in bacterial clearance.
Lysosomal enzymes can contribute to the elimination of bacteria by macrophages. We next assessed whether phago-lysosomal maturation was regulated by FLT4. The internalized S. typhimurium-GFP colocalized with LysoTracker Red in wild-type BMDMs, while this colocalization was decreased in flt4∆LBD/∆LBD BMDMs (Figure 1J). Also, PEMs expressing flt4∆LBD/∆LBD reduced the colocalization of S. typhimurium-GFP and the lysosomes (Fig. S1E). To visualize the whole process of how PEMs clear S. typhimurium-GFP, we used live-cell spinning disk confocal microscopy. In Flt4WT/WT macrophages, S. typhimurium was located within phagolysosomes. In contrast, flt4∆LBD/∆LBD macrophages failed to recruit lysosome (LysoTracker Green) to the S. typhimurium-RFP containing phagosome (Fig. S1F, Video S1-S2), which is critical for subsequent acidification. Taken together, our data have demonstrated that FLT4 could promote bacteria clearance via increasing engulfment, ROS production and the maturation of autophagosome, which was accompanied with the glucometabolic changes.
FLT4 protects macrophages from pyroptosis during clearance of S. typhimurium.
S. typhimurium infection could result in macrophage pyroptosis, characterized by cell swelling with membranous balloons rapidly expanding, rupturing and nuclear condensation. In the movies (Video S1-S2) and still images (Figure 2A), a significantly higher number of flt4∆LBD/∆LBD PEMs display increased percentages of cell death with swelling shapes during S. typhimurium infection. Further analysis using transmission electron microscopy revealed that the infected flt4∆LBD/∆LBD PEMs displayed numerous vacant vacuoles with condensed nuclei and more intracellular intact bacteria (Figure 2B, S2A). In Flt4WT/WT macrophages, there were numerous phago-lysosomal vacuoles containing bacteria remnants. The cell death rate of the infected flt4∆LBD/∆LBD macrophages was higher than that of Flt4WT/WT cells (Figure 2C, D) or flt4WT/∆LBD macrophages, which was measured by PI staining and analyzed by FACS (Fig. S2B) or by fluorescent microscopy (Fig. S2C). As previously reported, glycine treatment can reduce the released levels of cytosolic LDH (lactate dehydrogenase) and protect cells from pyroptosis, and we used this assay to identify pyroptosis [28,29]. We found that glycine treatment could protect flt4∆LBD/∆LBD or Flt4WT/TKmut macrophages from pyroptosis at a comparable level to WT cells following S. typhimurium infection (Figure 2E, S2D). In addition, glycine treatment reduced LDH release to similar levels in WT PEMs and FLT4-silenced PEMs (Fig. S2E). Together, these data indicate that FLT4 protects macrophages from SL1344-infection induced pyroptosis.
Figure 2.
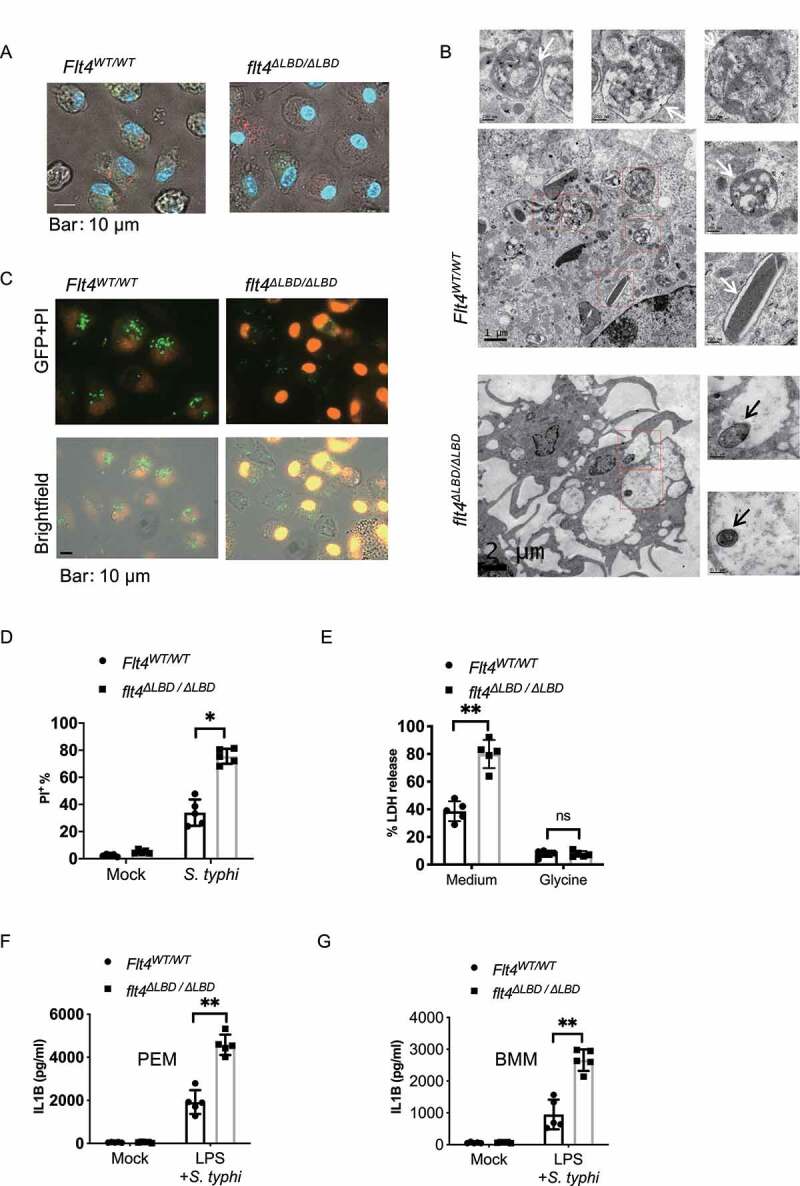
FLT4 protects macrophages from pyroptosis during clearance of S. typhimurium. (A and B) Representative images of S. typhimurium-RFP infected Flt4WT/WT and flt4∆LBD/∆LBD PEMs for 2 h by live-cell spinning disk confocal laser microscopy (A) or by electron microscopy analysis (B), numerous phago-lysosomal vacuoles with membrane containing bacteria remnants were highlighted by white arrows. The intact bacteria in flt4∆LBD/∆LBD PEMs were highlighted by black arrows. Representative images from three independent experiments. Scale bar: 500 nm. (C and D) Flt4WT/WT and flt4∆LBD/∆LBD PEMs were infected with S. typhimurium-GFP for 1 h, followed by staining with PI. The percentages of PI+ cell was analyzed from at least 200 cells. Data are the mean ± SEM Representative images were shown. *p < 0.05, **p < 0.01. (E) Flt4WT/WT and flt4∆LBD/∆LBD PEMs were infected with S. typhimurium in the presence of the medium control or glycine that nonspecifically inhibits ion fluxes. LDH released by dying cells was quantified at least 200 cells. Data are presented as the mean ± SEM *p < 0.05, **p < 0.01 (F and G) Flt4WT/WT and flt4∆LBD/∆LBD PEMs (F) or BMDM (G) were treated with LPS (1 μg/ml, 4 h) followed by S. typhimurium infection for 30 min. IL1B concentrations in supernatants were examined by ELISA. Data are presented as the mean ± SEM *p < 0.05, **p < 0.01.
Inflammasome activation and subsequent pyroptosis are key defenses against bacterial infections. We therefore investigated inflammasome activation by analyzing mature IL1B concentrations in cell supernatants by ELISA. Indeed, flt4∆LBD/∆LBD PEMs or BMDMs secreted IL1B at much higher levels compared to their control cells (Figure 2F, G). Moreover, the enhanced IL1B production in flt4∆LBD/∆LBD macrophages was consistent with the increase in glucometabolic products including succinate and lactate from the infected flt4∆LBD/∆LBD mice (Figure 1A) which have both been suggested to promote inflammation. Previous studies have reported that CASP1 and GSDMD (gasdermin D)-induced pyroptotic cell death [30,31], which might have a protective role against many bacterial species [32]. However, excessive activation of inflammasome signaling and pyroptosis in vivo may be deleterious to the host [7,33]. In our study, we have observed higher concentrations of IL1B and the increased pyroptosis vs. more bacteria counts in SL1344-infected flt4∆LBD/∆LBD macrophages. We next examined how flt4∆LBD/∆LBD macrophages could result in excessive inflammasome activation in S. typhimurium infection.
FLT4 inhibits inflammasome and CASP1 activation in S. typhimurium infected macrophages
The release of mature IL1B during inflammasome assembly requires CASP1 activation to cleave the precursor pro-IL1B. Another readout for inflammasome activation is the formation of PYCARD/ASC (PYD and CARD domain containing /apoptosis-associated speck-like protein containing a CARD) speck which serves as a platform for CASP1 activation. We found that higher percentages of PYCARD specks were formed in flt4∆LBD/∆LBD macrophages (Figure 3A), Flt4WT/TKmut macrophages (Fig. S3A), and FLT4 siRNA knockdown (KD) macrophages (Fig. S3B) upon S. typhimurium infection, compared to their respective controls. In addition, flt4∆LBD/∆LBD macrophages enhanced levels of PYCARD oligomers when compared to that in S. typhimurium infected Flt4WT/WT control cells as measured by the oligomerization state of proteins (Figure 3B). These data indicate that FLT4 signaling inhibits PYCARD speck formation in macrophages upon bacterial infection.
Figure 3.
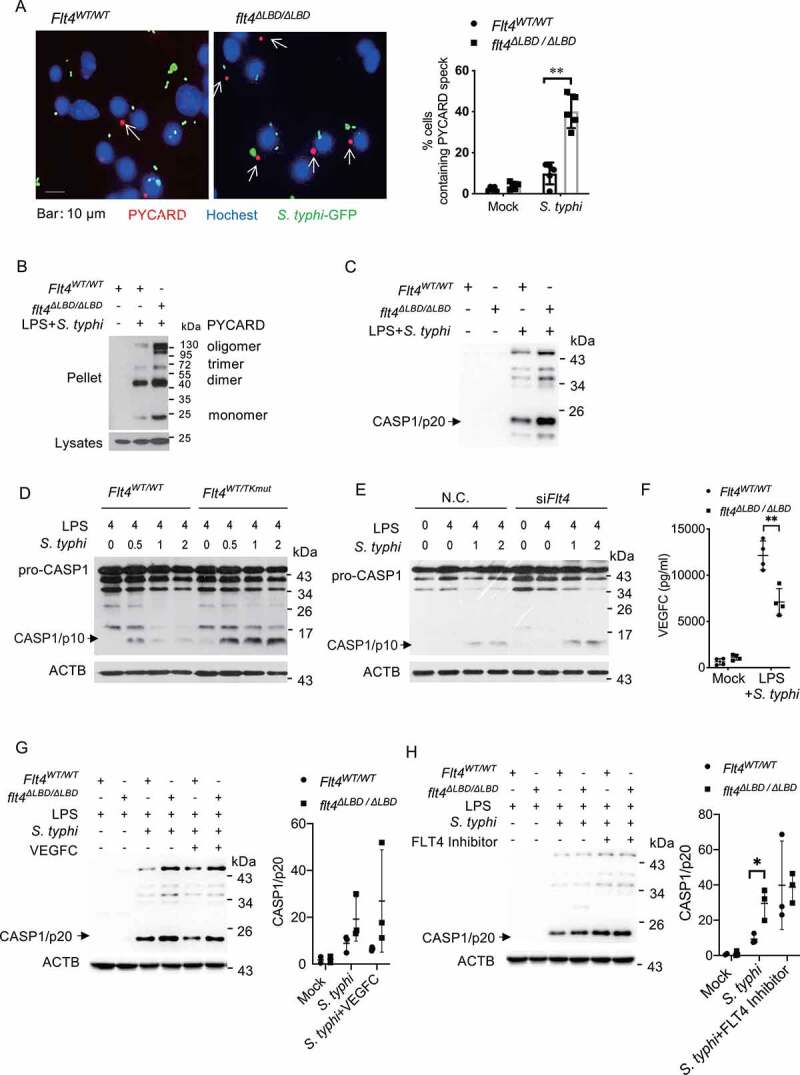
FLT4 inhibits inflammasome and CASP1 activation in S. typhimurium infected macrophages. (A) Flt4WT/WT and flt4∆LBD/∆LBD PEMs were pretreated with LPS followed by S. typhimurium infection, and immunostained for PYCARD (red) and Hoechst (blue). Images are representative of three independent experiments (scale bar: 10 μm). The percentages of macrophages containing PYCARD foci were analyzed from at least 200 cells in each experiment and data are presented as the mean ± SEM **p < 0.01(B) Flt4WT/WT and flt4∆LBD/∆LBD PEMs were pretreated with LPS followed by S. typhimurium infection to assess PYCARD oligomerization by immunoblotting. Flt4WT/WT and flt4∆LBD/∆LBD PEMs (C), in the presence or absence of exogenous VEGFC (G) or the FLT4 inhibitor MAZ51 (10 μM) (H), or Flt4WT/WTand Flt4WT/TKmut BMDMs (D), or the control and Flt4 siRNA transfected PEMs (E) were pretreated with LPS followed by S. typhimurium infection for the indicated times. The levels of pro-CASP1 (inactive CASP1), CASP1/p10 (active CASP1) in cell lysates (D, E) or CASP1/p20 in supernatants (C, G, H) were examined by immunoblotting. The immunoblots were statistically quantified by densitometry. Representative data from three independent experiments were shown. (F) Flt4WT/WT and flt4∆LBD/∆LBD PEMs were primed with 1 μg/ml LPS followed by S. typhimurium infection. VEGFC concentrations in supernatants were examined by ELISA. Data are presented as the mean ± SEM *p < 0.05, **p < 0.01.
We next measured how FLT4 affected CASP1 activation. CASP1 is a critical enzyme which cleaves the pro-IL1B/interleukin-1β protein into the active IL1B. Association of pro-CASP1 with inflammasome induces its autocatalytic activity and self-cleavage that generates the catalytical active subunits CASP1/p10 and CASP1/p20. Compared to the control cells, flt4∆LBD/∆LBD macrophages showed increased levels of the cleaved CASP1/p20 form of CASP1 in response to S. typhimurium infection (Figure 3C). Similarly, the kinase dead mutant Flt4WT/TKmut or knock-down of FLT4 significantly enhanced the cleaved CASP1/p10 form of CASP1 in S. typhimurium-infected PEMs (Figure 3D, E). These data indicate that FLT4 signaling inhibits inflammasome activation.
LPS priming and S. typhimurium infection rapidly induced VEGFC protein expression (Figure 3F). Importantly, treatment with exogenous VEGFC could decrease CASP1 activation in Flt4WT/WT macrophages (Figure 3G, S3C), while treatment with the FLT4 inhibitor enhanced CASP1 activation in Flt4WT/WT macrophages (Figure 3H, Fig. S3D). Critically, treatment using both exogenous VEGFC and the FLT4 inhibitor did not further affect CASP1 activation in flt4∆LBD/∆LBD macrophages (Figure 3G, H). The immortalized bone-marrow-derived macrophage (iBMDM) were transduced to stably overexpressing FLT4 (i.e., iBMDM-FLT4), which decreased CASP1 activation after S. typhimurium infection (Fig. S3E). These data together provide evidence that in S. typhimurium-infected macrophages, activation of FLT4 signaling suppresses inflammasome-triggered PYCARD specks formation and CASP1 activation.
FLT4 enhances bacteria elimination and prevents inflammasome activation in an autophagy-dependent manner
During macrophage recognition of pathogens or engulfment of cytoplasmic bacteria, the cytosolic form of MAP1LC3 is conjugated to phosphatidylethanolamine to form an MAP1LC3–phosphatidylethanolamine conjugate (MAP1LC3-II), which is recruited to phagophore membranes. Upon maturation, autophagosomes fuse with lysosomes to degrade and clear invading pathogens, which is also termed an autolysosome including some types of xenophagy [34]. We detected bacteria degradation inside these macrophages by electron microscopy, showing the irregular edges within wild-type macrophages (Figure 4A, right panel) in contrast to flt4∆LBD/∆LBD macrophages. The process of autolysosome formation involves a phagosome and components of the autophagic machinery. We therefore measured MAP1LC3-I and MAP1LC3-II levels in S. typhimurium infected wild-type or flt4∆LBD/∆LBD macrophages by immunoblotting. S. typhimurium infection enhanced the ratio of MAP1LC3-II: MAP1LC3-I in wild-type BMDMs, while flt4∆LBD/∆LBD BMDMs shoed substantially reduced MAP1LC3-II levels (Figure 4B). Both overexpressing FLT4 in iBMDM cells and exogenous VEGFC treatment enhanced MAP1LC3-II levels (Figure 4C, D). In agreement with this, treatment with the FLT4 inhibitor or the pharmacological autophagy inhibitor 3-methyladenine (3-Ma) decreased MAP1LC3-II levels (Figure 4E, F). These findings suggest that FLT4 signaling enhances autolysosome formation in a MAP1LC3-dependent manner. Compared to Flt4WT/WT BMDMs, the amount of MAP1LC3 puncta was decreased in flt4∆LBD/∆LBD BMDMs, which was consistent with the immunoblotting results. In addition, flt4∆LBD/∆LBD BMDMs showed decreased percentages of colocalization of LAMP1 with MAP1LC3. These data indicate that FLT4 could promote phago-lysosomal maturation (Figure 4G). To further determine whether the FLT4 signaling enhances autophagolysosomal formation, Flt4WT/WT and flt4∆LBD/∆LBD macrophages were pre-treated with or without bafilomycin A1 to block autophagolysosomal fusion, followed by infection. We noticed that flt4∆LBD/∆LBD macrophages displayed increased SQSTM1/p62 (sequestosome 1) protein levels upon infection compared to Flt4WT/WT macrophages. This suggests that FLT4 is involved in the regulation of autophagolysosomal formation (Figure 4H). LGALS3/galectin 3 has been reported to function during lysosomal damage-involved autophagy. We therefore analyzed LGALS3 puncta formation in Flt4WT/WT and flt4∆LBD/∆LBD PEMs. Immunofluorescence data showed an increase in LGALS3 puncta formation in Flt4WT/WT macrophages upon infection not seen in flt4∆LBD/∆LBD macrophages (Fig. S4A).
Figure 4.
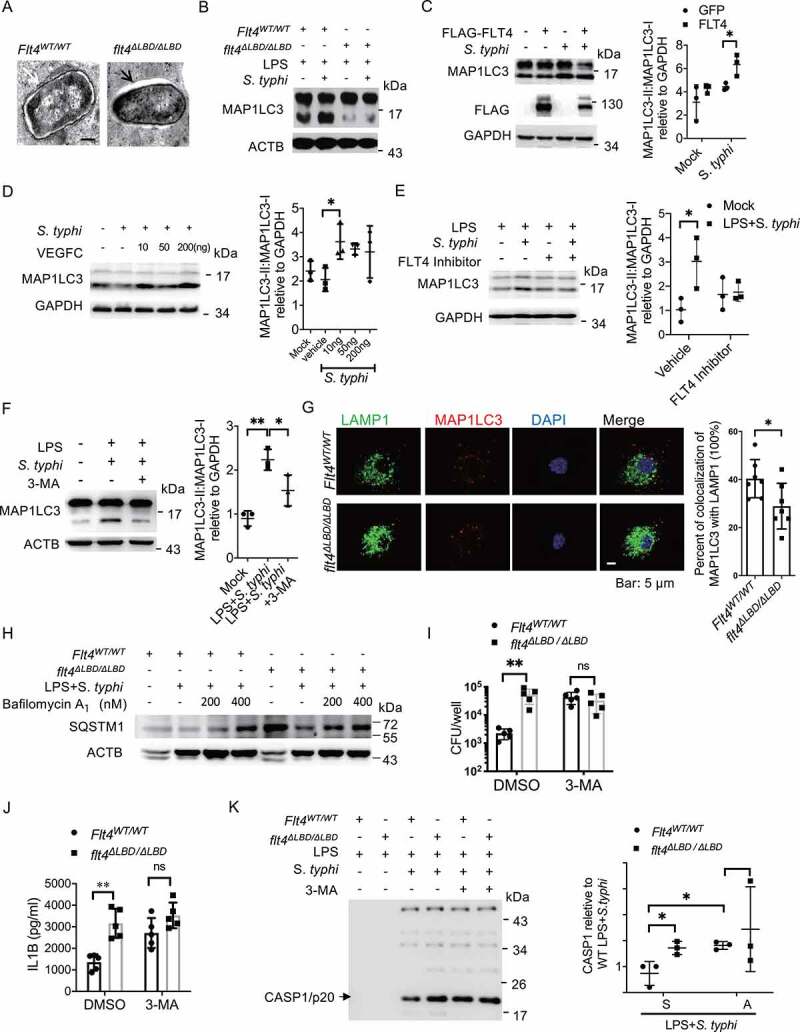
FLT4 enhances bacteria elimination and prevents inflammasome activation in an autophagy-dependent manner. (A) S. typhimurium infected Flt4WT/WT and flt4∆LBD/∆LBD BMDMs were prepared for electron microscopy analysis. Representative images are from three independent experiments (Scale bar 500 nm). (B-F) Flt4WT/WT and flt4∆LBD/∆LBD PEMs (B), or iBMDM cells overexpressing GFP and FLT4 (C), or exogenous VEGFC-treated PEMs (D), or the FLT4 inhibitor MAZ51 (10 μM)-treated PEMs (E), or 3-MA (5 mM)-treated PEMs (F) were primed with LPS (1 μg/ml) followed by S. typhimurium infection. MAP1LC3-I and MAP1LC3-II levels were examined by immunoblotting. The immunoblots were statistically quantified by densitometry. (G) Flt4WT/WT and flt4∆LBD/∆LBD PEMs were pretreated with LPS followed by S. typhimurium infection, and immunostained for LAMP1 (green), MAP1LC3 (red) and DAPI (blue). The percentages of MAP1LC3 and LAMP1 colocalization were analyzed from at least 200 cells. Data were the mean ± SEM Representative images were shown. *p < 0.05, **p < 0.01.(H) Flt4WT/WT and flt4∆LBD/∆LBD PEMs were pretreated with LPS followed by S. typhimurium infection in the absence or presence of bafilomycin. The levels of SQSTM1 were examined by immunoblotting. (I) Flt4WT/WT and flt4∆LBD/∆LBD PEMs were infected with S. typhimurium in the absence or presence of 3-MA (5 mM), CFUs were enumerated. Data are presented as the mean ± SEM *p < 0.05, **p < 0.01(J-K) Flt4WT/WT and flt4∆LBD/∆LBD PEMs were primed with LPS (1 μg/ml) followed by S. typhimurium infection in the absence or presence of 3-MA (5 mM), IL1B concentrations in supernatants were examined by ELISA, Data are presented as the mean ± SEM *p < 0.05, **p < 0.01(J), and the levels of CASP1/p20 in supernatants (K) were examined by immunoblotting. The immunoblots were statistically quantified by densitometry.
Since we observed that FLT4 affected both autophagy and inflammasome activation in S. typhimurium-infected macrophages, we asked whether autophagy could regulate inflammasome activation. We used the autophagy inhibitor 3-MA to treat S. typhimurium-infected macrophages, which decreased S. typhimurium clearance in Flt4WT/WT macrophages to a similar level as that in flt4∆LBD/∆LBD macrophages (Figure 4I). Interestingly, 3-MA treatment significantly increased mature IL1B secretion (Figure 4J) as well as enhanced the levels of PYCARD oligomers (Fig. S4B) and CASP1 cleavage (Figure 4K). Furthermore, 3-MA treatment resulted in comparable amounts of intracellular S. typhimurium (Figure 4I) in wild-type and flt4∆LBD/∆LBD infected macrophages, while also eliminating the differences in mature IL1B secretion, PYCARD oligomerization and CASP1 activation (Figure 4J-K, S4B). Collectively, we have elucidated that FLT4 enhances bacteria elimination and attenuates excessive inflammasome activation in an autophagic machinery-dependent manner.
FLT4 interacts with AMPK to coordinate glycometabolic reprogramming, autophagy and inflammasome activation
FLT4 is a tyrosine kinase receptor on the cell surface, and we investigated how its tyrosine kinase activity is crucial to its ability to target downstream effector(s) to regulate autophagy and inflammasome activation. To identify novel binding partners, we overexpressed Flag-tagged FLT4 in the RAW264.7 cell line and employed mass spectrometry. We identified 11 potential target proteins, depending on their potential tyrosine modification sites and functional relevance to autophagy (Fig. S5A). Immunoprecipitation assays were performed to ascertain the interaction between FLT4 and these candidates (Fig. S5B, S5C), and PRKAA1/AMPKα1 (protein kinase, AMP-activated, alpha 1 catalytic subunit) was reciprocally immunoprecipitated with FLT4 in HEK293 cells (Figure 5A). Moreover, the interaction between FLT4 and PRKAA1 was enhanced in response to S. typhimurium infection in RAW264.7 cells (Figure 5B).
Figure 5.
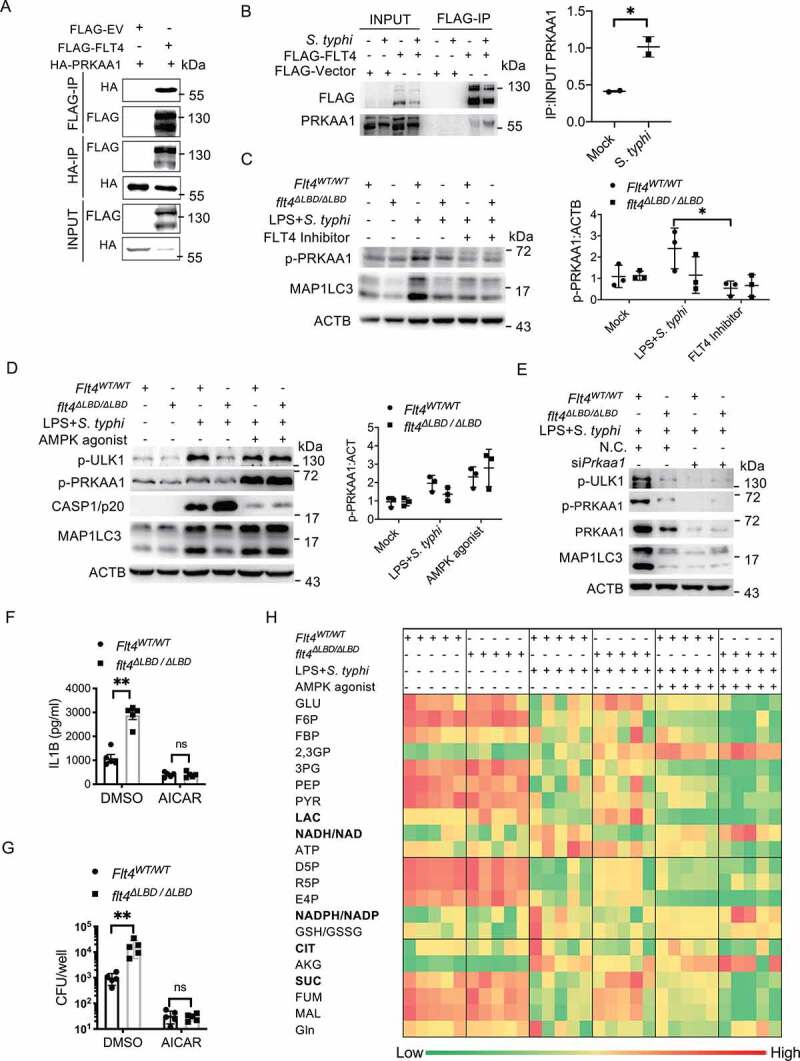
FLT4 interacts with AMPK to coordinate glycometabolic reprogramming, autophagy and inflammasome activation. (A) HEK293T cells were transfected with plasmids encoding FLAG-IRF3 and HA-PRKAA1 and prepared for immunoprecipitation and immunoblotting assays with the indicated antibodies. (B) RAW264.7 cells overexpressing FLAG-FLT4 or the FLAG control were infected with S. typhimurium to assess the endogenous binding of FLT4 to PRKAA1. The immunoblots were statistically quantified by densitometry. (C-E) The FLT4 inhibitor MAZ51 (10 μM)-treated (C), the AMPK agonist AICAR (100 µM)-treated (D), or Prkaa siRNA-treated (E) Flt4WT/WT and flt4∆LBD/∆LBD PEMs were primed with LPS (1 μg/ml) followed by S. typhimurium infection. The levels of PRKAA1 phosphorylation, ULK1 phosphorylation, CASP1/p20 in supernatants and MAP1LC3 lipidation were examined by immunoblotting. The immunoblots were statistically quantified by densitometry. (F-H) Flt4WT/WT and flt4∆LBD/∆LBD PEMs were primed with LPS (1 μg/ml) followed by S. typhimurium infection in the absence or presence of the AMPK agonist AICAR (100 µM). IL1B concentrations in supernatants by ELISA (F), bacterial CFUs (G), and the glucometabolites profile (H) by LC-MS were checked. Data are presented as the mean ± SEM *p < 0.05, **p < 0.01.
Given that AMPK is the main sensor and regulator of cell metabolism [13], we were thus interested in the FLT4-AMPK module. Previous studies have determined that phosphorylation at Thr172 on the PRKAA1 leads to AMPK activation, which directly phosphorylates and activates ULK1, the mammalian homolog of yeast Atg1, for autophagosome formation [14,15]. We therefore examined whether FLT4 signaling could regulate the kinase activity of AMPK for autophagy. Salmonella-infection enhanced phosphorylated PRKAA1 (p-PRKAA1) levels and MAP1LC3 lipidation (i.e. MAP1LC3-II levels) in Flt4WT/WT macrophages, which was substantially reduced by the FLT4 inhibitor treatment (Figure 5C). By contrast, the FLT4 inhibitor failed to further attenuate these effects in flt4∆LBD/∆LBD macrophages (Figure 5C). In agreement with this, exogenous VEGFC treatment slightly enhanced p-PRKAA1 levels and MAP1LC3 lipidation in salmonella-infected Flt4WT/WT macrophages, but not in flt4∆LBD/∆LBD macrophages (Fig. S5D).
We next assessed whether FLT4 induced autophagy via AMPK. Upon salmonella infection, flt4∆LBD/∆LBD macrophages showed decreased amount of p-PRKAA1, p-ULK1(Ser555) and MAP1LC3 lipidation (Figure 5D). Interestingly, treatment with the pharmacological AMPK activator AICAR (5-aminoimidazole-4-carboxamide ribonucleoside) significantly enhanced the levels of p-ULK1, p-PRKAA1 and MAP1LC3 lipidation in flt4∆LBD/∆LBD macrophages, which reached to a similar degree as that in Flt4WT/WT macrophages (Figure 5D). This indicates that the activated AMPK is the downstream effector of FLT4 signaling to regulate autophagy. Moreover, knock-down of PRKAA1 decreased phosphorylation levels of ULK1 and MAP1LC3 lipidation to a similar degree between Flt4WT/WT and flt4∆LBD/∆LBD macrophages upon infection (Figure 5E). We also generated iBMDM cells stably overexpressing FLT4, which were pre-treated with or without the AMPK inhibitor followed by LPS treatment and SL1344 infection. FLT4 overexpression inhibited CASP1 activation but enhanced MAP1LC3 lipidation. Upon treated with the AMPK inhibitor, the changes of MAP1LC3 lipidation were no longer significant and the differences of CASP1 activation became smaller between WT and FLT4 overexpressing cells (Fig. S5E-F). Critically, we observed that treatment with the AMPK agonist AICAR substantially suppressed the production of mature IL1B, suggesting that AMPK inhibits inflammasome activation (Figure 5F). And also, in response to AICAR treatment, Flt4WT/WT and flt4∆LBD/∆LBD macrophages displayed similar amount of mature IL1B (Figure 5F) and intracellular S. typhimurium (Figure 5G). These data suggest that AMPK is downstream target of FLT4 in inflammasome activation and autophagic elimination of intracellular bacteria.
Since we observed that the AMPK agonist could reverse flt4∆LBD/∆LBD macrophages’ phenotype in the regulation of autophagy and inflammasome activation, we asked whether FLT4 regulates glucose metabolism via downstream AMPK. To answer this, we activated AMPK in Flt4WT/WT and flt4∆LBD/∆LBD macrophages upon infection (Figure 5H) using agonist AICAR. In resting state, Flt4WT/WT and flt4∆LBD/∆LBD macrophages showed similar products of glucose metabolism (left two columns). Upon bacterial infection, compared to that in Flt4WT/WT macrophages, flt4∆LBD/∆LBD macrophages display significantly reduced NADH/NAD and NADPH/NADP levels, but enhanced lactate and succinate levels (middle two columns) in agreement with earlier findings from the infected livers of Flt4WT/WT vs. flt4∆LBD/∆LBD mice (Figure 1A). Notably, treatment with AICAR profoundly enhanced NADH/NAD and NADPH/NADP levels, and inhibited lactate levels in both Flt4WT/WT and flt4∆LBD/∆LBD macrophages (right two columns). These data collectively suggest that FLT4 and AMPK function together in macrophages to reprogram glycolytic metabolism, which is linked to inflammasome activation and autophagy.
FLT4-induced PRKAA1 tyrosine phosphorylation is indispensable for autophagy and inflammasome activation
Next, we investigated which tyrosine(s) in PRKAA1 could be phosphorylated by the tyrosine kinase receptor FLT4. FLT4 and PRKAA1 were co-expressed in HEK293 cells, and PRKAA1 was enriched by immunoprecipitation followed by immunoblotting with the 4 G10 antibody recognizing phosphorylated tyrosine sites. Indeed, overexpression of FLT4 promoted PRKAA1 tyrosine phosphorylation (Figure 6A). Based on the mass spectrometry results from the UniPort database, we identified five possible phosphorylated tyrosine sites including Y247 (A1), Y294 (A2), Y441/442 (A3), Y463 (A4), and Y500 (A5) in PRKAA1 (Figure 6B). We generated point mutations at each of these sites to phenylalanine (Phe, F) (named A15, Figure 6B), and transfected each of these into HeLa cells. Compared to the wild-type control (A0), A1 and A3 mutations could significantly reduce MAP1LC3 lipidation (Figure 6C, Figure 6D left panel). We further overexpressed FLT4 with these PRKAA1 mutations, and the A1 and A3 mutations also significantly blocked FLT4-induced MAP1LC3 lipidation (Figure 6D, right panel). These data indicate that phosphorylation of Y247 and Y441/442 in PRKAA1 plays a critical role for autophagy.
Figure 6.
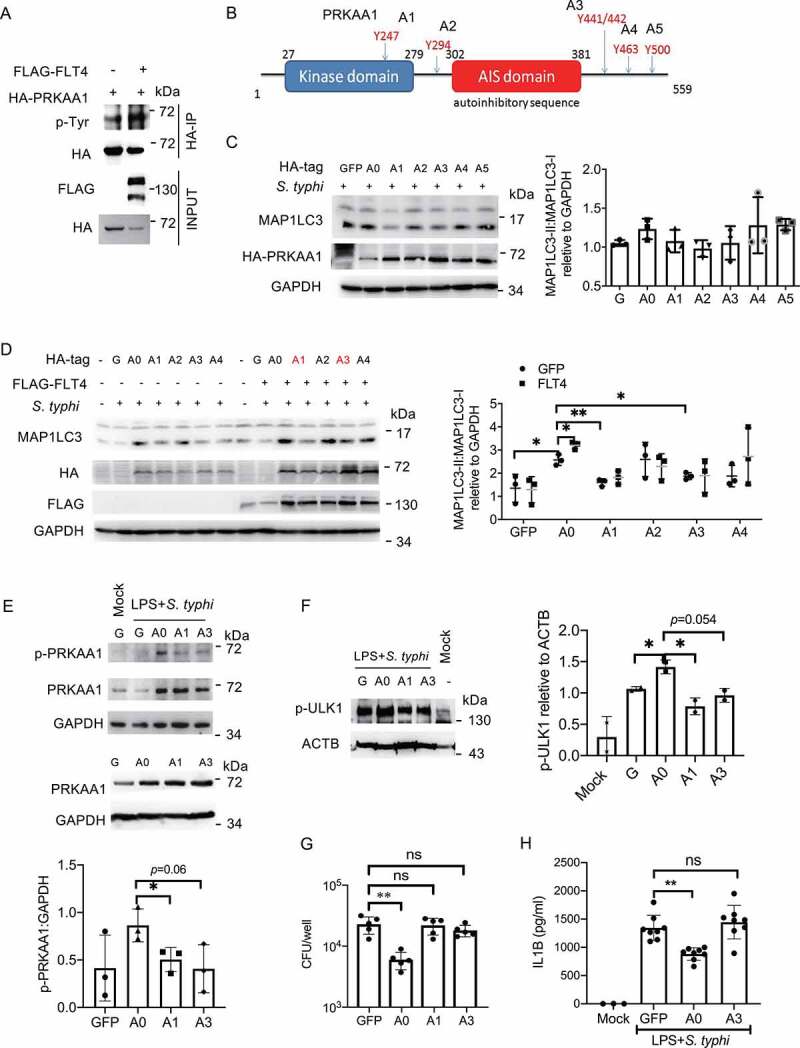
FLT4-induced PRKAA1 tyrosine phosphorylation is indispensable for autophagy and inflammasome activation. (A) HEK293T cells were transfected with FLAG-IRF3 and HA-PRKAA1 and prepared for immunoprecipitation and immunoblotting. (B) The diagram of PRKAA1 domains and tyrosine sites. (C and D) HeLa cells overexpressing wild-type PRKAA1 (A0) or the PRKAA1 mutants (A1, A2, A3, A4, A5) (C), or together with FLAG-FLT4 (D) were infected with S. typhimurium to check the levels of MAP1LC3-I and MAP1LC3-II. The immunoblots were statistically quantified by densitometry. (E-H) iBMDMs stably overexpressing the GFP control, wild-type PRKAA1 (A0) or the mutants (A1 or A3) were primed with LPS (1 μg/ml) followed by S. typhimurium infection. The PRKAA1-ULK1 phosphorylation levels by immunoblotting (E and F), bacterial CFUs (G) or IL1B concentrations in supernatants by ELISA (H), were examined. The immunoblots were statistically quantified by densitometry. Data are presented as the mean ± SEM *p < 0.05, **p < 0.01.
To further identify other previously unknown functions of PRKAA1 tyrosine phosphorylation at Y247 and Y441/442, we stably expressed the wild-type PRKAA1 and the A1 and A3 mutations into iBMDM (Figure 6E lower panel). Upon S. typhimurium infection, the phosphorylation of PRKAA1 and ULK1 was increased in A0 iBMDMs, while the A1 or A3 mutants display notably decreased levels of p-PRKAA1 and p-ULK1 (Figure 6E upper panel, Figure 6F). In agreement with this, compared to the GFP control, PRKAA1 overexpression enhanced bacterial clearance (Figure 6G), but not the A1 and A3 mutants. To determine whether PRKAA1 also regulated inflammasome activation, iBMDM expressing PRKAA1 or the A1 or A3 mutants were infected with S. typhimurium. We noticed that PRKAA1 overexpression reduced mature IL1B production in macrophages, but the A3 mutant (Y441/442 F) failed to display the inhibitory effect (Figure 6H). Taken together, our findings have provided the first evidence that phosphorylation of Y247 and Y441/442 in PRKAA1 by FLT4 is critical for glycolytic reprogramming, bacterial clearance and inflammasome activation.
Discussion
Macrophages express various surface and intracellular receptors to initiate phagocytosis, autophagy, inflammatory responses and cell pyroptosis, which are essential for the host to eliminate bacteria. Among these innate immune responses, phagocytosis and autophagy are essential for macrophages to defend against microbes. TLR4 is a classical receptor that can control both phagocytosis and autophagy. Our previous findings have discovered that TLR4 signaling increases expression levels of the tyrosine kinase receptor FLT4 and its ligand VEGFC in bacterial-infected macrophages [19]. This study further indicates that FLT4 enhances phagocytosis and participates in autophagosome maturation. Furthermore, VEGFC treatment indeed enhances LC3-associated autophagy, suggesting that the enhanced VEGFC concentrations in the site of infection might boost the host to defend against pathogen infections. In contrast, the FLT4 mutants (lacking ligand binding domain, or lacking tyrosine kinase activity) show defective phagocytosis and MAP1LC3 lipidation, which fail to eliminate bacteria. Therefore, this study suggests that in addition to the previously identified function in vascular development, FLT4 also functions critically as an infection-inducible surface receptor to facilitate phagocytosis and autophagy for clearance of bacteria.
Previous studies suggest that autophagy inhibits inflammasome activation [35,36], but the underlying mechanism is not fully understood. In this study, we found that inhibition of autophagy indeed increased inflammasome activation. Moreover, the surface receptor FLT4 is able to link these two functions, i.e. increasing autophagy vs. downregulating inflammasome activation and cell pyroptosis. The accumulating evidence also suggests the inverse relationship between autophagy vs. inflammasome activation and pyroptosis [37]. In this study, we have provided evidence that FLT4 protects macrophages against pyroptosis and inhibits inflammasome activation. These events, combined with the enhanced phagocytosis and autophagy induced by FLT4, enable the host cells to better control infections (Figure 7). In agreement with this model, human patients carrying the inherited FLT4 missense mutations (R1044P, L1044P or P1114L) not only show lymphedema [21], but also display re-occurring infections [38]. The Flt4WT/TKmut mouse model similarly displays lymphedema phenotype comparable to human patients [21,39], and here we provide evidence that Flt4WT/TKmut mice also failed to clear bacteria in vivo.
Figure 7.
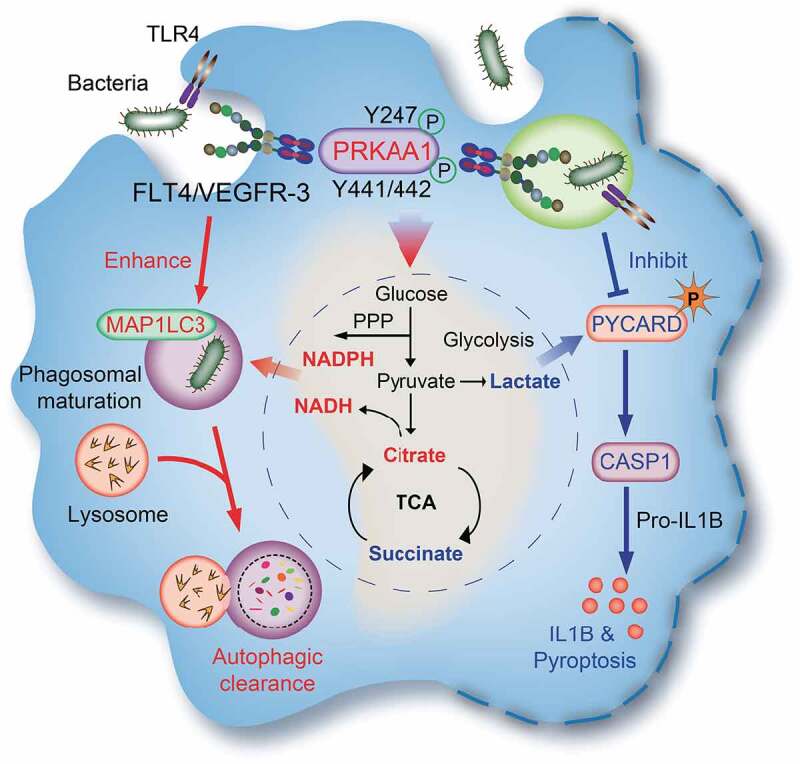
The model: The FLT4-AMPK module regulates macrophage glycolytic metabolism to coordinate autophagy, pyroptosis, and inflammasome activation during bacterial infection. Upon infection, TLR4 recognizes bacterial LPS and enhance the expression levels of FLT4 and the ligand VEGFC. Activated FLT4 via binding to VEGFC could recruit PRKAA1 and phosphorylates its Y247 and Y441/442 to activate AMPK. This signaling pathway regulates macrophage glycolytic metabolism (center), which coordinates autophagy (left) as well as inflammasome activation and pyroptosis (right), resulting in better protection against bacterial infection.
Mechanistically, we provided the first evidence that FLT4 binds and activates the master regulator AMPK, which might explain how FLT4 can coordinate glucose metabolism with inflammasome activation and bacterial elimination. In this scenario, TLR4 is regarded as one example that plays critical role to link metabolic reprogramming with autophagy, inflammasome activation in macrophages. In this study, we have uncovered that the tyrosine kinase receptor FLT4 could bind and activate PRKAA1, which promotes pathogen clearance by increasing autophagy and producing the metabolic intermediates with direct antimicrobial activity. Moreover, the FLT4-AMPK module reduces the pro-inflammatory metabolic intermediates and decreases CASP1-mediated IL1B maturation as well as inflammasome-associated pyroptosis.
AMPK is a pivotal sensor for monitoring cellular energy state and nutrient supply, and its kinase activity is mainly regulated by different phosphorylation events. The upstream kinases like CAMKK2 (calcium/calmodulin-dependent protein kinase kinase 2, beta) and STK11/LKB1 (serine/threonine kinase 11) could increase the phosphorylation of Thr172, which results in AMPK activation [40,41]. Conversely, phosphorylation of Thr479 [42] and Ser485 [43] in the PRKAA1 subunit through AKT- and GSK3B-associated pathways was reported to trigger Thr172 dephosphorylation to inactivate AMPK. A recent report suggests that FYN can phosphorylate the PRKAA1 subunit at Y436 and inhibits its activity [44]. Here, our study has uncovered that FLT4 can phosphorylate Y247 and Y441/442 in PRKAA1, which are crucial for AMPK activation. Mutations in Y247F and Y441/442 F decrease AMPK activation, i.e. reducing the phosphorylation levels of PRKAA1 Thr172 and its downstream target ULK1 (Figure 6E and 6F). Furthermore, activation of AMPK using AICAR could reverse the effects in macrophages expressing the mutant FLT4 (Figure 5F-H), indicating that AMPK is the downstream effector of FLT4 in glycolytic reprogramming for CASP1 mediated inflammasome activation and autophagic elimination of intracellular bacteria. Caspases are related aspartic-serine proteases that regulate inflammation and cell death. A recent report [45] has demonstrated that steatosis-induced decline in AMPK-catalyzed phosphorylation permits CASP6 (caspase 6) activation, leading to hepatocyte death. Interestingly, we have observed that macrophages expressing the mutant FLT4 are more susceptible to CASP1-induced inflammasome activation and pyroptosis (Figures 2, 3). These findings together indicate that AMPK might be critical in linking caspase activation with cell death in various types of cells. In response to infection, macrophages TLR4 recognizes LPS to induce FLT4 expression, which can sense the enhanced concentrations of VEGFC in the microenvironment, resulting in AMPK activation to prevent CASP1-induced pyroptosis and excessive inflammation, as well as to enhance phagocytosis and autophagy to clear bacteria.
Previous studies highlight that targeting VEGFC and FLT4 is crucial to inhibiting lymphangiogenesis and blocking lymphatic tumor metastasis [46–48]. Autophagy is an intracellular recycling process that maintains basal levels of metabolites and biosynthetic intermediates under starvation or other forms of stress and is frequently exploited by cancer cells as an important mechanism for metabolic adaptation. Furthermore, the Warburg effect of aerobic glycolysis is a key metabolic hallmark of cancer. Moving forward, it is important to further investigate whether targeting FLT4 signaling could also reduce autophagy and alter glucose metabolism in tumor cells, which will help us to understand better whether FLT4 is a possible drug target for anti-tumor therapies.
Materials and methods
Animal experiments
Mice were kept and bred under specific pathogen-free conditions at Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences (SIBCB, CAS). The Flt4 mutant mice were generated as previously described [20]. Flt4flox/flox mice were generated with the two loxP sites flanked at exon 4–6 of Flt4. Flt4flox/flox mice were cross-bred with flt4WT/ΔLBD; Lyz2/LysM-Cre+/+ mice to obtain flt4WT/ΔLBD or flt4∆LBD/∆LBD mice on C57BL/6 background. Genotypes were verified by PCR and sequencing. All mice were used around 6–8 weeks old, and the same sex, age was used to avoid bias. The sample size was determined based on individual experiments and is indicated in each experiment. All animal experiments were performed in compliance with National Institutes of Health Guidelines and were approved by the Institutional Animal Care and Use Committee of SIBCB.
Macrophage isolation
Peritoneal elucidated macrophages (PEMs) from mice on the C3H or C57BL/6 background were harvested 4 days after intraperitoneal inoculation of 2.5 ml 3% Brewer thioglycollate medium (Sigma, B2551). To generate bone marrow-derived macrophages (BMDMs), bone marrow cells were collected and cultured in complete DMEM medium supplemented with L929-conditioned medium for a week.
In vivo and in vitro S. typhimurium infection model
The S. typhimurium strain SL1344 (a kind gift from Dr. WH Fang, Zhejiang University, China) was grown in Luria-Bertani (LB) broth (ThermoFisher, 12,780,052) at 37°C overnight and diluted in PBS (ThermoFisher,10,010,049) to infect Flt4WT/WTand Flt4WT/TKmut mice (5 x 104 CFU/ml), or to infect flt4∆LBD/∆LBD and flt4∆LBD/∆LBD mice (5 x 103 CFU/ml). Aliquots (200 μl) were injected into the tail veins of these adult mice. Infected mice were monitored for survival at the indicated time points or killed by cervical dislocation. Spleens and livers were removed, homogenized and placed into 10 ml sterile distilled water to determine viable bacterial counts (series dilutions of the homogenates and then plating on LB agar plates).
Late-log S. typhimurium cultures were prepared using a method optimized for bacterial invasion or infection of cells. Briefly, S. typhimurium were grown for 16 h at 37°C with shaking and then subcultured (1:33) in LB without antibiotics for 3 h. Bacteria were harvested by centrifugation at 1000 x g for 5 min, washed with serum-free cell culture medium twice and resuspended in 1 ml medium. Cells were infected with S. typhimurium at a multiplicity of infection of 10 and infections were synchronized by centrifugation at 100 g for 10 min. Infected cells were washed 30 min post-infection three times with warm PBS to remove extracellular bacteria and fresh medium containing 100 μg/ml gentamicin (Sangon Biotech, A506614) was added for 1 h. Medium was then replaced with medium containing 10 μg/ml gentamicin and the infection continued for indicated times. Where indicated, the chemical inhibitors or agonists were added together with LPS (Sigma, L-2880) treatment for 4 h.
Cell culture and reagents
HEK293T (SCSP-502) were obtained from the Cell Bank of SIBCB, CAS. The murine macrophage cell line RAW264.7 was a gift from Dr. B.X. Ge (Tongji University, Shanghai, China). These cell lines were tested and determined to be mycoplasma free. Primary macrophages and RAW264.7 cells were maintained in a humidified 37°C, 5% CO2 incubator with the complete Dulbecco’s modified Eagle’s medium (ThermoFisher, 11,960,069), which was supplemented with 10% (vol:vol) heat-inactivated fetal bovine serum (FBS; ThermoFisher, A3160901), penicillin and streptomycin (100 U/ml each; ThermoFisher, 15,070,063). Antibodies include anti-Flag (Sigma, A8592), anti-FLT4 (eBioscience,14–5988), anti-MAP1LC3/LC3 (Sigma, L7543), anti-phospho-PRKAA1/AMPKα (Thr172; Cell Signaling Technology, 2531), anti-PRKAA1/AMPKα (Cell Signaling Technology, 5831) anti-CASP1/p10 (Santa Cruz Biotechnology, sc-514), anti-CASP1/p20 (AdipoGen, AG-20B-0042), anti-NLRP3 (Santa Cruz Biotechnology, sc-34,410), anti-PYCARD/ASC (Santa Cruz Biotechnology, sc-22,514-R), anti-ULK1 (Cell Signaling Technology, 5869S), anti-ACTB/β-Actin (Abcam, ab49900), anti-TLR4 (BD Pharmingen, 558,293), anti-TUBA1B (Proteintech, 11,2241-AP).
Plasmid transfection and RNA interference
The full-length murine FLT4 (WT, TKmut and ΔLBD) contains a Flag-tag sequence at the 3ʹ tail and were subcloned into the pMX-IRES-GFP vector (youbio, VT1547). Also, the pShuttle-IRES-hr GFP1 (Stratagene, 240,081) vector carrying each form of FLT4 was used. For retrovirus particle packaging, pCL-10A (Novus Biologicals, NBP229,542) was co-transfected with the pMX-IRES-GFP plasmids. To generate stably RAW264.7 cells overexpressing WT FLT4 or FLT4TKmut, GFP-positive cells were sorted by flow cytometry (AriaII, BD). For knocking down FLT4 or PRKAA1 expression, RAW264.7 macrophage or primary peritoneal macrophage cells were transfected with siRNAs (GenePharma) together with Lipofectamine 2000 (ThermoFisher, 11,668,019), and RNAi efficiency was determined by quantitative RT-PCR (DBI Bioscience), according to manufacturer’s instructions.
Metabolomics analysis
The intracellular levels of metabolites were measured with previously described methods [49]. Briefly, approximately 1 × 107 cells were treated with cold aqueous methanol solution (80%, v:v) to stop cell metabolism quickly. Samples were centrifuged at 12,000 g for 15 min at 4°C and the supernatants were collected to lyophilize and then reconstituted in 500 μl of methanol/water (10:90, v:v). The metabolite extracts were resuspended in 15 mM CH3COONH4 and analyzed using LC-MS/MS. The LC-MS/MS was operated in multiple reaction monitoring (MRM) mode with AB SCIEX 4000QTRAP (AB SCIEX, Canada).
Flow cytometry and immunoblot analysis
Cell staining was analyzed with a FACS Calibur four-color cytometer and cell sorting was performed with a FACS Aria Cell-Sorter. Data were analyzed with FlowJo software (Tree Star, Inc.). After washing with ice-cold PBS, cells were lysed with NP-40 Lysis Buffer (Beyotime Biotechnology, P0013F) supplemented with protease inhibitor cocktail (Roche, 4,693,116,001), 1 mM PMSF (ThermoFisher, 36,978), 1 mM Na3VO4, 50 mM NaF and 5 mM EDTA. Equal amounts of cell lysates were subjected for immunoblotting analysis.
Cell staining and confocal microscopy
Cells were fixed with 4% PFA in PBS (pH 7.4) and permealized with 0.1% Triton X-100 (Beyotime Biotechnology, P0096). After staining with the primary antibodies and fluorescent-labeled secondary antibodies, samples were mounted on slides using fluorescence mounting medium (DAKO, S3023). Olympus BX-81 microscope and Leica TCS MP5 confocal microscope were used. To record the video of cell pyroptosis, the indicated cells were seeded in glass-bottom culture dishes (cellvis, D35 C420-1-N) at about 30–40% confluence. Videos were taken on the ZEISS Cell Observer spinning disk confocal microscope. All image data shown are representative of at least three randomly selected fields.
Phagocytosis of macrophages
Fluorescent yellow-green-labeled latex beads (Sigma, L1030) were opsonized with IgG-containing FBS for 60 min at 37°C. Isolated peritoneal macrophages were further purified by adherence on a 12-well plate. Adherent macrophages were exposed to the opsonized-latex beads for 15–45 min. After washing, macrophages were detached by scraping with a cell scraper (ThermoFisher, 179,693). The percentage of macrophages internalizing beads were detected by flow cytometry.
Electron microscopy
Macrophages grown on coverslips were fixed in 2.5% glutaraldehyde (dissolved in 0.1 M Tousimis phosphate sodium buffer, pH 7.35 (Tousimis, 1051) for 1 h at room temperature. The coverslips were washed twice with 0.1 M sodium cacodylate buffer, treated with 1% tannic acid in 0.1 M sodium cacodylate buffer for 60 min, and then submerged in 1% osmium tetroxide in 0.1 M sodium cacodylate buffer for 60 min. After dehydration in a graded ethanol series, the cells were cleared with propylene oxide for 10 min and dried overnight. The next day the samples were embedded, and pictures were taken with a FEI Tecnai G2 Spirit transmission electron microscope.
Macrophage apoptosis and LDH release assay
Cell death was assessed using ANXA5 (annexin A5) and PI (propidium iodide) staining (BD Biosciences, 556,463). LDH release was assayed using the Cytotoxicity Detection Kit as described by the manufacturer’s protocol (Roche, 11,644,793,001). 10% Triton-X-treated cells were served as 100% for LDH release.
Immunoprecipitation analysis
HEK293T cells were transfected with the Flag-tagged FLT4 together with empty plasmid or the appropriate expression plasmids. At 36 h after transfection, cells were lysed in 1% NP-40 lysis buffer (Beyotime Biotechnology, P0013 F) supplemented with protease inhibitor cocktail (Roche, 4,693,116,001), 1 mM PMSF (ThermoFisher, 36,978), 1 mM Na3VO4, 50 mM NaF and 5 mM EDTA. The cell lysates were then centrifuged for 15 min at 12,000 g and 4°C. Following the incubation of cell lysates with Protein A/G PLUS-Agarose (Santa Cruz, sc-2003) coated with the indicated antibodies and rotation at 4°C for 2 h, the beads were washed five times with lysis buffer and resuspended in SDS–PAGE loading buffer for western blot analysis.
Statistical analysis
Data are represented as the mean ± SEM of at least three experiments. Statistical analyses were performed using the GraphPad Prism8 software, version 8. Statistical significance was calculated using a Student’s two-tailed unpaired t-test for comparisons between groups with equal variances. Comparisons among three or more groups were performed with two-way ANOVA, followed by multiple comparison. ns, not significant (p > 0.05); *p < 0.05, **p < 0.01, ***p < 0.01.
Supplementary Material
Acknowledgments
We are grateful to Dr. Feng Shao (NIBS, Beijing) for his helpful discussion and suggestion. We thank Dr. Baoxue Ge (Tongji University, Shanghai) for providing RAW264.7 cells. We thank Dr. Zhihuan Chew’s help to correct the language. We would like to thank the Core Facility of Chemical Biology, Core Facility of Molecular Biology, Core Facility of Cell Biology, and Animal Core Facility in SIBCB for technique help. We thank National Facility for Protein Science in Shanghai, Zhangjiang Lab, China for MS data collection and analysis.
Funding Statement
This work was supported by grants from the Ministry of Science and Technology of the People’s Republic of China (2016YFD0500407, 2016YFC0905902, 2018YFA0800702, 2016YFD0500207, 2016YFC0905900), National Natural Science Foundation of China (81930038, 81825011, 81630043, 81671572, 81571552, 81701569, 31700781, 81801575, 30671038, 81961160738), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB19000000), and the Priority Academic Program Development of Jiangsu Higher Education Institutions. Dr. Hongyan Wang is supported by the Hundred Talents Program of the Chinese Academy of Sciences.
Disclosure statement
The authors declare no competing financial interests.
Supplementary material
Supplemental data for this article can be accessed here
References
- [1].Kawai T, Akira S.. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010. May;11(5):373–384. [DOI] [PubMed] [Google Scholar]
- [2].Kono H, Rock KL.. How dying cells alert the immune system to danger. Nat Rev Immunol. 2008. Apr;8(4):279–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Van den Bossche J, O’Neill LA, Menon D. Macrophage immunometabolism: where are we (going)? Trends Immunol. 2017. June;38(6):395–406. [DOI] [PubMed] [Google Scholar]
- [4].Nagy C, Haschemi A. Time and demand are two critical dimensions of immunometabolism: the process of macrophage activation and the pentose phosphate pathway. Front Immunol. 2015;6:164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].O’Neill LA. A critical role for citrate metabolism in LPS signalling. Biochem J. 2011. Sep;438(3):e5–6. [DOI] [PubMed] [Google Scholar]
- [6].Tannahill GM, Curtis AM, Adamik J, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 2013. Apr;496(7444):238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].von Moltke J, Trinidad NJ, Moayeri M, et al. Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature. 2012. Oct;490(7418):107–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cohen TS, Prince AS. Activation of inflammasome signaling mediates pathology of acute P. aeruginosa pneumonia. J Clin Invest. 2013. Apr;123(4):1630–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Baxt LA, Garza-Mayers AC, Goldberg MB. Bacterial subversion of host innate immune pathways. Science. 2013. May;340(6133):697–701. [DOI] [PubMed] [Google Scholar]
- [10].Gomes LC, Dikic I. Autophagy in antimicrobial immunity. Mol Cell. 2014. Apr;54(2):224–233. [DOI] [PubMed] [Google Scholar]
- [11].Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000. Dec 1;290(5497):1717–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011. Jan 20;469(7330):323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012. Mar;13(4):251–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kim J, Kundu M, Viollet B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011. Feb;13(2):132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Egan DF, Shackelford DB, Mihaylova MM, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011. Jan;331(6016):456–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Liu Y, Nguyen PT, Wang X, et al. TLR9 and beclin 1 crosstalk regulates muscle AMPK activation in exercise. Nature. 2020. February;578(7796):605–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Starling S. Role for toll-like receptor 9 in muscle AMPK activation. Nat Rev Endocrinol. 2020. April;16(4):197. [DOI] [PubMed] [Google Scholar]
- [18].Peng Z, Luo R, Xie T, et al. Erythrocyte adenosine A2B receptor-mediated AMPK activation: a missing component counteracting CKD by promoting oxygen delivery. J Am Soc Nephrol. 2019. August;30(8):1413–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhang Y, Lu Y, Ma L, et al. Activation of vascular endothelial growth factor receptor-3 in macrophages restrains TLR4-NF-κB signaling and protects against endotoxin shock. Immunity. 2014. Apr 17;40(4):501–514. [DOI] [PubMed] [Google Scholar]
- [20].Zhang L, Zhou F, Han W, et al. VEGFR-3 ligand-binding and kinase activity are required for lymphangiogenesis but not for angiogenesis. Cell Res. 2010. Dec;20(12):1319–1331. [DOI] [PubMed] [Google Scholar]
- [21].Karkkainen MJ, Ferrell RE, Lawrence EC, et al. Missense mutations interfere with VEGFR-3 signalling in primary lymphoedema. Nat Genet. 2000. Jun;25(2):153–159. [DOI] [PubMed] [Google Scholar]
- [22].Almasy E, Szederjesi J, Grigorescu BL, et al. The diagnostic and prognostic role of vascular endothelial growth factor C in sepsis and septic shock. J Crit Care Med (Targu Mures). 2020. Jul;6(3):152–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Van Wyngene L, Vandewalle J, Libert C. Reprogramming of basic metabolic pathways in microbial sepsis: therapeutic targets at last? EMBO Mol Med. 2018. Aug;10(8). DOI: 10.15252/emmm.201708712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Karkkainen MJ, Saaristo A, Jussila L, et al. A model for gene therapy of human hereditary lymphedema. Proc Natl Acad Sci U S A. 2001. Oct;98(22):12677–12682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bar-Or D, Carrick M, Tanner A 2nd, et al. Overcoming the warburg effect: is it the key to survival in sepsis? J Crit Care. 2018. Feb;43:197–201. [DOI] [PubMed] [Google Scholar]
- [26].Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity. 2014. Jul;41(1):21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wong CO, Gregory S, Hu H, et al. Lysosomal degradation is required for sustained phagocytosis of bacteria by macrophages. Cell Host Microbe. 2017. Jun;21(6):719–730.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Fink SL, Bergsbaken T, Cookson BT. Anthrax lethal toxin and Salmonella elicit the common cell death pathway of caspase-1-dependent pyroptosis via distinct mechanisms. Proc Natl Acad Sci U S A. 2008. Mar 18;105(11):4312–4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol. 2006. Nov;8(11):1812–1825. [DOI] [PubMed] [Google Scholar]
- [30].Liu X, Zhang Z, Ruan J, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016. Jul 7;535(7610):153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ding J, Wang K, Liu W, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016. Jun 8;535(7610):111–116. [DOI] [PubMed] [Google Scholar]
- [32].Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009. Feb;7(2):99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Jorgensen I, Miao EA. Pyroptotic cell death defends against intracellular pathogens. Immunol Rev. 2015. May;265(1):130–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Tanida I, Ueno T, Kominami E. LC3 and autophagy. Methods Mol Biol. 2008;445:77–88. [DOI] [PubMed] [Google Scholar]
- [35].Saitoh T, Akira S. Regulation of innate immune responses by autophagy-related proteins. J Cell Biol. 2010. Jun 14;189(6):925–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Nakahira K, Haspel JA, Rathinam VAK, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011. Mar;12(3):222–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Abdelaziz DHA, Khalil H, Cormet-Boyaka E, et al. The cooperation between the autophagy machinery and the inflammasome to implement an appropriate innate immune response: do they regulate each other? Immunol Rev. 2015. May;265(1):194–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Karkkainen MJ, Petrova TV. Vascular endothelial growth factor receptors in the regulation of angiogenesis and lymphangiogenesis. Oncogene. 2000. Nov 20;19(49):5598–5605. [DOI] [PubMed] [Google Scholar]
- [39].Alitalo K, Tammela T, Petrova TV. Lymphangiogenesis in development and human disease. Nature. 2005. Dec 15;438(7070):946–953. [DOI] [PubMed] [Google Scholar]
- [40].Zhang YL, Guo H, Zhang CS, et al. AMP as a low-energy charge signal autonomously initiates assembly of AXIN-AMPK-LKB1 complex for AMPK activation. Cell Metab. 2013. Oct;18(4):546–555. [DOI] [PubMed] [Google Scholar]
- [41].Woods A, Dickerson K, Heath R, et al. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005. Jul;2(1):21–33. [DOI] [PubMed] [Google Scholar]
- [42].Suzuki T, Bridges D, Nakada D, et al. Inhibition of AMPK catabolic action by GSK3. Mol Cell. 2013. May;50(3):407–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Horman S, Vertommen D, Heath R, et al. Insulin antagonizes ischemia-induced Thr172 phosphorylation of AMP-activated protein kinase α-subunits in heart via hierarchical phosphorylation of Ser485/491. J Biol Chem. 2006. Mar;281(9):5335–5340. [DOI] [PubMed] [Google Scholar]
- [44].Yamada E, Okada S, Bastie CC, et al. Fyn phosphorylates AMPK to inhibit AMPK activity and AMP-dependent activation of autophagy. Oncotarget. 2016. Nov;7(46):74612–74629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Zhao P, Sun X, Chaggan C, et al. An AMPK–caspase-6 axis controls liver damage in nonalcoholic steatohepatitis. Science. 2020. Feb 7;367(6478):652–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Secker GA, Harvey NL. VEGFR signaling during lymphatic vascular development: from progenitor cells to functional vessels. Dev Dyn. 2015. Mar;244(3):323–331. [DOI] [PubMed] [Google Scholar]
- [47].Makinen T, Jussila L, Veikkola T, et al. Inhibition of lymphangiogenesis with resulting lymphedema in transgenic mice expressing soluble VEGF receptor-3. Nat Med. 2001. Feb;7(2):199–205. [DOI] [PubMed] [Google Scholar]
- [48].He Y, Kozaki K, Karpanen T, et al. Suppression of tumor lymphangiogenesis and lymph node metastasis by blocking vascular endothelial growth factor receptor 3 signaling. J Natl Cancer Inst. 2002. Jun 5;94(11):819–825. [DOI] [PubMed] [Google Scholar]
- [49].Sun W, Liu Y, Glazer CA, et al. TKTL1 is activated by promoter hypomethylation and contributes to head and neck squamous cell carcinoma carcinogenesis through increased aerobic glycolysis and HIF1alpha stabilization. Clin Cancer Res. 2010. Feb 1;16(3):857–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.