

ABSTRACT
Cancer stem cells (CSCs) are the main drivers of disease progression and chemotherapy resistance in breast cancer. Tumor progression and chemoresistance might then be prevented by CSC-targeted therapies. We previously demonstrated that Toll-like Receptor (TLR)2 is overexpressed in CSCs and fuels their self-renewal. Here, we show that high TLR2 expression is linked to poor prognosis in breast cancer patients, therefore representing a candidate target for breast cancer treatment. By using a novel mammary cancer-prone TLR2KO mouse model, we demonstrate that TLR2 is required for CSC pool maintenance and for regulatory T cell induction. Accordingly, cancer-prone TLR2KO mice display delayed tumor onset and increased survival. Transplantation of TLR2WT and TLR2KO cancer cells in either TLR2WT or TLR2KO hosts shows that tumor initiation is mostly sustained by TLR2 expression in cancer cells. TLR2 host deficiency partially impairs cancer cell growth, implying a pro-tumorigenic effect of TLR2 expression in immune cells. Finally, we demonstrate that doxorubicin-induced release of HMGB1 activates TLR2 signaling in cancer cells, leading to a chemotherapy-resistant phenotype. Unprecedented use of TLR2 inhibitors in vivo reduces tumor growth and potentiates doxorubicin efficacy with no negative impact on the host immune system, opening new perspectives for the treatment of breast cancer patients.
KEYWORDS: Breast cancer, Toll-like receptor 2, HER2, HMGB1, chemoresistance
Introduction
Breast cancer (BC) is the most prevalent tumor in women, and its incidence is progressively increasing.1 Remarkable advances in diagnosis and adjuvant and neoadjuvant treatments have improved patient outcomes. However, BC is still the first cause of cancer-associated mortality in women in more than 100 countries.1 More than 30% of patients experience local or distal relapse after surgery, and about 80% of deaths occur in patients whose BC progressed to metastatic disease despite treatment.2
Although endocrine and HER2-targeted therapies are commonly used for estrogen receptor (ER)- and HER2-positive early BCs, respectively, systemic chemotherapy with anthracyclines or cyclophosphamide is the cornerstone of treatment for triple negative (TN) BC, high-risk luminal cancers, and advanced BC. Neoadjuvant chemotherapy using a doxorubicin-based schedule has recently become a common therapeutic option for operable BC.3 However, the emergence of chemoresistance is a common event that often leads to treatment failure and metastasis.4
Resistance to chemotherapy can be attributed to several cancer-cell-intrinsic mechanisms, including altered drug absorption, efflux, or metabolism, the increased activation of pro-survival pathways, enhanced DNA repair, and tumor stemness.4 Furthermore, many chemotherapeutic drugs induce the release of several molecules by dying or injured cancer cells. One class of molecules released or exposed on the cell surface following chemotherapy-induced cell death is represented by damage-associated molecular patterns (DAMPs), which are a wide-ranging group of biomolecules that include (but are not limited to) High Mobility Group Box (HMGB)1, ATP, Heat Shock Protein (HSP)70 and HSP90, Calreticulin, and Annexin A1. Such DAMPs can act as signals by interacting with pattern recognition receptors (PRRs), leading to functional alterations in cells composing the tumor microenvironment (TME).5 Physiologically, PRRs are expressed in immune cells and promote inflammatory responses.6 In this scenario, dying or injured cancer cells release or expose on their surface DAMPs that help recruiting and activating cells of the immune system, including dendritic cells (DCs) that engulf them and initiate an adaptive immune response against the tumor. This type of cell death is called immunogenic cell death (ICD). However, some DAMPs may switch immune cells toward an anti-inflammatory response, depending on the type of cell death that induced their release, with which kinetic (acute vs chronic), and the PRR they engage.7,8 Therefore, surviving tumor cells can exploit TME alterations induced by cancer cell death to stimulate their survival and proliferation, resulting in cancer progression.4,9 In addition, several PRRs have recently been observed in cancer cells as well, where they can activate pro-tumor pathways, and thus act as a double-edge sword in cancer.6,10
We have previously demonstrated that BC cells express Toll-like receptor (TLR)2, a PRR playing a key role in the survival and self-renewal of cancer stem cells (CSCs). CSCs are a small population of cancer cells able to self-renew, differentiate into all the cell lineages that form the tumor bulk and initiate the metastatic cascade.11,12 Breast CSCs are resistant to most current therapies, including chemotherapy, being thus responsible for local or distal relapse after treatment.13–15 Therefore, high levels of CSCs in breast tumors are associated with poor prognosis.16 TLR2 activation in breast CSCs is autocrinally induced by the secretion of the DAMP HMGB1, which leads to the activation of the NF-κB, AKT, and MAPK pathways and the subsequent production of Interleukin (IL)-6, tumor growth factor (TGF)-β, granulocytic-colony stimulating factor (G-CSF), and vascular endothelial growth factor (VEGF).6,13 IL-6 and TGF-β induce STAT3- and Smad3-pathway activation in CSCs, promoting their survival, proliferation, self-renewal, and invasion.13
Besides being actively secreted by CSCs,13 HMGB1 can be released into the TME as a consequence of treatment with chemotherapeutic drugs, including doxorubicin and taxanes, that induce ICD.17 High HMGB1 levels have been associated with clinical progressive disease during neoadjuvant chemotherapy in TNBC patients.18 We therefore hypothesize that ICD-inducing chemotherapy triggers the release of HMGB1 that, engaging TLR2, fosters the appearance of CSC-related features, including chemoresistance. TLR2 inhibition may therefore prevent the tumor-promoting effect of HMGB1, thus preserving the anti-tumor function of ICD-inducing chemotherapy.
Herein, we show that TLR2 promotes BC growth and chemoresistance, mainly via tumor cell-intrinsic mechanisms, and that its inhibition improves the efficacy of doxorubicin in preclinical models of BC. This study therefore identifies TLR2 as a new potential target for combined therapies.
Materials and methods
Cell cultures
MDA-MB-231 and 4T1 cell lines were purchased from American Type Culture Collection (ATCC) in 2018, aliquoted, frozen, and then used within 10 passages after resuscitation in DMEM or RPMI 1640 (ThermoFisher Scientific) 10% FBS (Sigma-Aldrich), respectively. TUBO cell line was derived from a BALB-neuT primary tumor and cultured in DMEM 20% FBS.19 WT-874, WT-879, WT-880, KO-C26, KO-M26, and KO-E26 cell lines were derived, as described in the study of Liu and Chen,20 from mammary tumors isolated from C57Bl6-neuT mice either TLR2 wild-type (neuT-TLR2WT) or TLR2 knock-out (neuT-TLR2KO), respectively, and cultured in DMEM-F12 (ThermoFisher Scientific) with 20% FBS. All cells were negative for mycoplasma.21
Meta-analysis using patient databases
The prognostic value of TLR2 mRNA expression in the 5-y-relapse-free (RFS) and overall survival (OS) of patients with BC, either treated with chemotherapy or not, was analyzed using the 32 breast cancer transcriptomic datasets available through the open access online software Kaplan-Meier Plotter.22 Patients were stratified into two groups according to TLR2 expression using the “auto select best cutoff” function. Removal of redundant samples, exclusion of biased arrays and proportional hazard assumptions check were included in quality control. Hazard ratios and p-values obtained with each dataset, either considering all patients or separating them by molecular subtype, were merged with Fisher’s method23 from the metap package (https://CRAN.R-project.org/package=metap).
The expression levels of TLR2 across BC molecular subtypes were obtained from the transcriptomic datasets collected in the MetaGxBreast R package,24 and PAM50 subtyping was performed through the molecular subtyping function of the genefu R package (“pam50” model).25 Since the log-rank tests were two-sided, one-sided p-values were obtained with the two2one function from the metap package, taking the hazard ratio into account, before applying Fisher’s method. A total of 32 datasets had a probe for TLR2 and allowed the inference of the molecular subtype. TLR2 mRNA expression levels in BC patients that responded to adjuvant chemotherapy and those that did not were analyzed using the datasets and tools available;22,26 patients who relapsed within 5 y of treatment were compared with patients who did not, excluding those censored before the 5 y had passed.
The correlation between TLR2 expression and a CSC signature was computed using the transcriptomic datasets in MetaGxBreast and the cor.test function in R, collapsing the expression of CSC markers16 with single sample GSEA (GSVA R package).27 Correlations’ p-values were merged similarly to log-rank survival tests’ p-values. A total of 33 datasets had probes for TLR2 and for the CSC signature.
Mice
Mice were bred and maintained under saprophytic and pathogen-free conditions at the animal facility of the Molecular Biotechnology Center and treated in accordance with EU and institutional guidelines, with the approval of the Animal Care and Use Committee of the University of Turin and the Italian Ministry of Health (authorizations N° 107/2020-PR and 500/2017-PR).
NeuT-TLR2WT mice were generated by crossing male BALB-neuT19 with C57Bl6 female mice, and then backcrossing neuT male offspring with C57Bl6 mice for 12 generations. NeuT-TLR2WT males were then crossed with B6.129-Tlr2tm1Kir/J female mice (The Jackson Laboratory), and the Tlr2Het offspring intercrossed to obtain neuT-TLR2KO mice. Mice were screened with primers to neuT (Fwd: 5’-GTAACACAGGCAGATGTAGGA-3’; Rev: 5’-ATCGGTGATGTCGGCGATAT-3’), Tlr2 (Fwd: 5’-CTTCCTGAATTTGTCCAGTACA-3’; RevWT: 5’-GGGCCAGCTCATTCCTCCCAC-3’; RevTlr2tm1Kir5’-GAAACGGAATGTTGTGGAGT-3’), and β-casein (Fwd: 5’-GATGTGCTCCAGGCTAAAGTT-3’; Rev: 5’-AGAAACGGAATGTTGTGGAGT-3’), according to Jackson Laboratory website protocols. Tumor growth was monitored twice per week with a caliper (N = 21 neuT-TLR2WT and N = 22 neuT-TLR2KO mice) until all 10 mammary glands displayed a tumor, or a tumor exceeded 10 mm mean diameter.
1 × 106 WT-874, WT-879, WT-880, KO-C26, KO-E26, and KO-M26 cells were injected orthotopically into the fourth mammary gland of 8-week-old syngeneic C57Bl6 mice either TLR2WT or TLR2KO or in NOD/SCID/IL-2 receptor gamma chain deficient (NSG, Charles River Laboratories) female mice (N = 6 per group). Cells were dissociated from 5 to 6 mm mean diameter tumors explanted from neuT-TLR2WT (N = 4) and neuT-TLR2KO (N = 4) mice, using the protocol described in the study of Lanzardo et al.28 Then, 1 × 104 or 1 × 105 cells were injected orthotopically into the fourth mammary gland of 8-week-old C57Bl6 mice (N = 16). Tumor growth was monitored twice per week by caliper. All mice were culled when the WT-874 tumors reached 10 mm in mean diameter.
1 × 104 4T1 cells were transfected with a pool of TLR2-specific or scrambled siRNAs, injected orthotopically, 24 hours later, into 6-week-old female BALB/c mice (Charles River Laboratories), and tumor growth was monitored. In the therapeutic model, 1 × 104 4T1 cells were injected orthotopically into 6-week-old female BALB/c mice (N = 40). When the tumors reached 2 mm mean diameter, mice were blindly randomized into four groups, which received intratumor and intraperitoneal (i.p.) injections of the vehicle (control); 50 µg CU-CPT22 (Tocris Bioscience, Cat#4884) intratumorally and vehicle i.p. (CU-CPT22-treated); 3 mg/Kg doxorubicin (Sigma-Aldrich, Cat#D1515) i.p. and vehicle intratumorally (doxorubicin-treated); or 50 µg CU-CPT22 intratumorally and 3 mg/Kg doxorubicin i.p. (CU-CPT22 + doxorubicin-treated) twice per week. When control tumors reached 10 mm mean diameter, mice were culled, tumors removed and processed for FACS, and lungs fixed in 4% formaldehyde, paraffin embedded, sectioned into four sections spaced by 50 µm and H&E stained. Pictures were acquired using a Nikon SMZ1000 stereomicroscope (Mager Scientific) and metastases counted with ImageJ.28
Ex vivo T regulatory cell (Treg) analysis
Blood of tumor-bearing neuT-TLR2WT mice was collected and processed, and T cells were activated as in the study of Conti et al.,29 and either treated or not with 20 µg/ml peptidoglycan from S. aureus (PGN-SA; InvivoGen, Cat#tlrl-pgns2) or 10 µg/ml HMGB1 (Sigma-Aldrich, Cat#SRP6265). The percentage of Tregs was analyzed 6 d later by FACS.
FACS analysis
Single-cell suspensions were obtained from fresh primary tumors, spleens, and heparinized blood as in the study of Conti et al.30 1 × 106 cells were treated with Fc-receptor blocker (BD Bioscience, Cat#01245B) and stained for 20 min at 4°C with anti-mouse CD45-VioGreen (Cat#130-110-803), CD11b-FITC (Cat#130-110-803), CD3-FITC (Cat#130-119-135), CD4-APC-Vio770 (Cat#130-119-134), CD8-VioBlue (Cat#130-123-865), CD49b-PE (Cat#130-123-702), B220-PE-Vio770 (Cat#130-123-702), γδ TCR-PE/Cy7 (Cat#130-123-290), CD19-PE (Cat#130-123-272), Gr1-VioBlue (Cat#130-102-830), F4/80-PE-Vio770 (Cat#130-118-320) and MHC-II-APC (Cat#130-102-139) from Miltenyi Biotec, and CD44-PE (Cat#103007), CD24-PE/Cy7 (Cat#101822), Sca1-AlexaFluor647 (Cat#122518), CD206-PE (Cat#141706), CD4-PE/Cy7 (Cat#100528), GITR-PE (Cat#126310), TLR2-PE (Cat#148604) and CD25-APC (Cat#101910) from Biolegend, the Aldefluor kit (Stem Cell Technologies, Cat#01700) or the anti-FoxP3-FITC kit (eBioscience, Cat#10191071-5775-40).31 At least 50,000 CD45+ events were acquired per sample on BD-FACSVerse and analyzed with FlowJO10.5.3. Cells were gated according to their physical parameters, and dead cells were excluded following propidium iodide (Sigma-Aldrich, Cat#P4864) or LIVE/DEAD™ Fixable Violet Dead Cell Stain Kit 450 nm (Thermo Fisher Cat#L34963,) staining. The CD45+ leucocytes and CD45− cells were gated, and cell populations analyzed as follows: NK: CD3− CD49b+; CD4+ T cells: CD3+ CD49b− CD4+; CD8+ T cells: CD3+ CD49b− CD8+; γδ TCR+ T cells: CD3+ CD49b− γδ TCR+; Tregs: CD3+ CD4+ CD25high GITR+ Foxp3+; B cells: CD3− B220+; MDSCs: CD11b+ Gr1+; M1 macrophages: CD11b+ F4/80+ MHC-II+ CD206−; M2 macrophages: CD11b+ F4/80+ CD206+;32,33 CSCs: CD45− Sca1+, CD45− CD44+ CD24− and CD45− Aldefluor+.30 Cell lines were characterized by staining with anti-mouse HER2 (Ab4, Cat#OP16, Calbiochem), α smooth muscle actin-FITC (Abcam Cat#ab8211), CD44-PE, CD24-PE/Cy7, Sca1-AlexaFluor647, TLR2-PE (Biolegend, Cat#148604 and Cat#392306), Aldeafluor, Oct-4 (Abcam Cat#ab19857), Musashi-1 (Abcam Cat#ab52865) or cytokeratin (CK)19-FITC (Abcam Cat#ab15463), followed by either rabbit anti-mouse Ig-PE (Cat# R043901), swine anti-rabbit Ig-FITC (Cat#F0205) from Dako or goat anti-rabbit IgG-PE (Thermo Fisher, Cat# P-2771MP). For the staining of intracellular proteins, cells were fixed and permeabilized with the BD Cytofix Cytoperm kit (BDBioscience, Cat# BD 554714). Tumor cells were either treated with 5 or 0.2 µM doxorubicin, 15 µM docetaxel (Hikma) or 12.5 µM cisplatin (Sandoz) and/or with 5 µM CU-CPT22 or 10 µg/ml of the HMGB1 inhibitor BOX-A (HMGBIotech, Cat#HM-014) or 8 µM of the NF-κB inhibitor BAY 11–7082 (Sigma-Aldrich, Cat#B5556) for 48 hours, or left untreated, and then stained with AnnexinV-Apoptosis Kit-APC (eBioscience, Cat#88-8007-72).34 To analyze NF-κB activation, cells were either treated with 5 µM doxorubicin and/or 5 µM CU-CPT22 for 48 hours or starved in serum-free medium for 4 hours and then stimulated for 30 min with or without 10 µg/ml HMGB1. Cells were fixed with 4% formaldehyde and 90% methanol (Sigma-Aldrich), permeabilized and stained for 30 min at 4°C with Alexa Fluor 647-conjugated anti-pNF-κB p65-S536 (Cell Signaling Technology, Cat#4887).13
3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyl Tetrazolium Bromide (MTT) assay
5 × 103 cells were cultured overnight in 96-well plates, and scalar doses of either docetaxel, cisplatin, CU-CPT22 or doxorubicin, alone or with 5 µM CU-CPT22, were then added. Viability was measured 48 hours after by MTT (0.5 mg/ml; Sigma Aldrich, Cat#M2128).30 The combined effect of doxorubicin and CU-CPT22 was determined as in the study of She et al.,35 by Bliss independence analyses, using the equation: C = (A + B) − (A × B), where A and B are the fractional growth inhibitions of doxorubicin and CU-CPT22, respectively, at a given dose. “Delta Bliss” scores (the difference between the observed growth inhibition and the Bliss expectation at the same dose) of the different doses were summed to generate a Bliss score. Bliss score = 0: additive effect, Bliss score > 0: synergy, Bliss score < 0: antagonism.
Tumorsphere-generation assays
Tumorspheres were generated from cell lines or cells dissociated from tumors.13 Dissociated tumorspheres were transfected with a pool of TLR2-specific (MSS216272, MSS216273, MSS280579) or scrambled (Cat#12935110) siRNAs using Lipofectamine 2000 (all from ThermoFisher Scientific). Twenty-four hours later, cells were treated with 20 µg/ml PGN-SA or 10 µg/ml HMGB1 or left untreated. After 4 d, contrast phase images were acquired (10X) and the number of spheres counted (4X) using Zeiss Axio Observer or Leica DMi1 inverted microscope connected to DC120 digital camera.30
ELISA
Supernatants of cells treated with 5 µM or 0.2 µM doxorubicin or left untreated were harvested after 48 hours, and HMGB1, IL-6, TGF-β and VEGF were quantified using ELISA (IBL International GmbH, Cat#ST51011 and R&D Systems, Cat#DY406, DY1679, DY493).
RNA extraction and real-time quantitative PCR (qPCR)
Total RNA was isolated from siRNA-transfected tumorspheres, 48 hours after transfection, using TriZol (ThermoFisher Scientific) according to the manufacturer’s instructions. RNA quality was assessed with the Agilent 2100 Bioanalyzer and quantified with a NanoDrop 2000 Spectrophotometer (ThermoFisher Scientific). Genomic DNA contaminations were removed with a DNA-free kit (Ambion), RNA was retrotranscribed with RETROscript reagents (Ambion), and qPCRs were carried out using gene-specific primers (QuantiTect Primer Assay; Qiagen), SYBR green, and a 7900HT RT-PCR system (Applied Biosystems). Quantitative normalization was performed on the expression of GAPDH. The expression levels in siTLR2-transfected tumorspheres relative to siSCR-transfected tumorspheres were calculated using the comparative ΔCt (threshold cycle number) method and reported as fold changes, as in the study of Conti et al.13
Magnetic resonance imaging
MR images were acquired at 7.1 T on a Bruker Avance300 spectrometer equipped with the Micro2.5 microimaging probe at room temperature. T2W morphological images were acquired using a standard RARE (Rapid Acquisition with Refocused Echoes) sequence with the following parameters: TR = 4000 ms, TE = 40.57 ms, RARE factor = 24, flip angle = 180°, number of averages = 4, FOV = 30 mm x 30 mm, slice thickness 1 mm, matrix size 256 x 256, and spatial resolution = 0.117 × 0.117 mm/pixel. Diffusion-Weighted MR Images (DWI-MRI) were acquired using Spin Echo sequences with TE = 27 ms, TR = 750 ms, number of averages = 1, FOV = 30 x 30 mm, slice thickness = 1 mm, matrix size 128 x 128, spatial resolution = 0.234 × 0.234 mm/pixel. Six different B-values were used (0, 150, 250, 500, 755, and 1000 s/mm2). Apparent diffusion constant (ADC) maps were calculated by fitting DW-MRI signal intensity as a function of B-values using Bruker ParaVision360 software.36 ADC maps were obtained by superimposing the ADC values of the tumor ROI onto T2w MRI (Fiji-Image software).
Statistical analysis
Statistical significance was evaluated using GraphPad8 software. Differences in tumor-free mice and survival were analyzed using Mantel-Cox log-rank test. Differences in sphere formation, FACS and ELISA data, and tumor multiplicity were analyzed using two-tailed unpaired Student’s t test (with Welch’s correction for samples with different variance) and non-parametric Mann–Whitney or Kruskal–Wallis test when the distribution calculated using Shapiro-Wilk or Kolmogorov Smirnov test was not normal. Differences in cell viability and tumor growth were analyzed using two-way ANOVA with Bonferroni posttest. P < 0.05 was considered significant.
Results
TLR2 expression is linked to poor prognosis in BC patients
To investigate whether TLR2 expression correlates with prognosis in BC patients, we performed a meta-analysis on public gene-expression data from human breast tumors.22,26 Patients with high TLR2 expression showed significantly lower RFS (Figure 1a) and a trend in decreased OS (Figure 1b) than those with low TLR2, suggesting that TLR2 may play a role in BC progression, as also confirmed by a meta-analysis in 32 individual datasets (Supplementary Table 1, Fisher’s collapsed p-value = 6.35 x 10−9). We did not find a significant difference in survival analyzing each BC subtype separately, probably due to the lower statistical power in smaller sets of samples. However, using an extensive set of transcriptomic data (33 datasets from the MetaGxBreast database24), we observed that TLR2 displays higher expression in the most aggressive Basal-like subtype with respect to the other subtypes (one-sided Wilcoxon signed-rank test p-value <0.05 in 27/33 datasets; data not shown).
Figure 1.
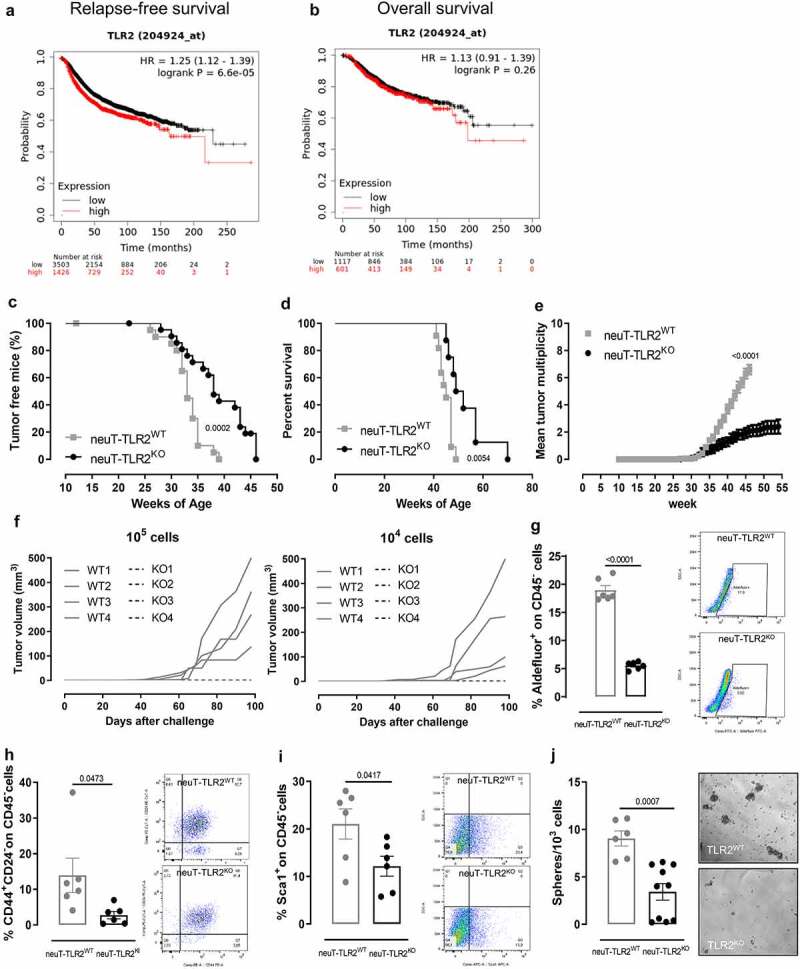
TLR2 correlates with poor prognosis in BC patients, and its deletion hinders HER2-driven mammary carcinogenesis and affects CSCs (a, b) Kaplan–Meier plots displaying (a) RFS and (b) OS in BC patients from all the datasets included in the KMPlotter website, stratified according to TLR2 mRNA expression (N = 4929 and N = 1728 in A and B, respectively). (c) Tumor-free survival, (d) OS, (e) mean tumor multiplicity of neuT-TLR2WT (N = 21) and neuT-TLR2KO (N = 22) mice. (f) Mean tumor volume of C57Bl6 mice injected orthotopically with 1 x 104 or 1 x 105 cells dissociated from tumors explanted from 4 neuT-TLR2WT (solid lines) or 4 neuT-TLR2KO (dashed lines) mice. (g-i) FACS analysis of (g) Aldefluor+ (h) CD44+ CD24− and (i) Sca1+ CSCs in tumors explanted from neuT-TLR2WT or neuT-TLR2KO mice. Representative density plots are shown. Graphs show mean ± SEM of the percentage of CSCs from among CD45− cells (each dot represents a mouse). N = 6 per group. (l) Sphere-generating ability (number of tumorspheres generated every 103 plated cells) of cells dissociated from the tumors of neuT-TLR2WT (N = 6) and neuT-TLR2KO (N = 10) mice. Results come from at least 3 independent experiments. (c-d) Log-rank Mantel-Cox, (e) Mann Whitney, and (g-j) unpaired Student’s t tests with Welch’s correction.
TLR2 deletion hinders HER2-driven mammary carcinogenesis
To study the role of TLR2 in mammary carcinogenesis and progression, we generated neuT-TLR2WT and neuT-TLR2KO mice, which carry the activated rat HER2/neu oncogene under the MMTV promoter and spontaneously develop mammary tumors. Tumor onset was significantly delayed in neuT-TLR2KO mice (Figure 1c), which displayed significantly higher survival (Figure 1d) and a decrease in tumor multiplicity compared to neuT-TLR2WT mice (Figure 1e). This was accompanied by a lower frequency of CSCs functionally defined as tumor-initiating cells, as demonstrated by the inability of cells dissociated from four different neuT-TLR2KO tumors to generate progressively growing masses when injected orthotopically at different amounts (either 1 x 105 or 1 x 104 cells) in syngeneic mice. Cells dissociated from four different neuT-TLR2WT tumors gave rise to tumors with a 100% penetrance at both dilutions (Figure 1f). Moreover, the lower frequency of CSCs in neuT-TLR2KO tumors was confirmed by the lower percentage of cells stained with the Aldefluor reagent, which measures the activity of aldehyde dehydrogenase 1, typically increased in breast CSCs,15 and of cells displaying the prototypical breast CSC phenotype CD44+ CD24−15 or expressing Sca1, a marker of CSCs in the neuT mouse model,37,38 observed in neuT-TLR2KO than in neuT-TLR2WT tumors (Figure 1g-i and Supplementary Fig 1a). Likewise, cells purified from neuT-TLR2KO tumors generated significantly fewer tumorspheres than those from neuT-TLR2WT (Figure 1j), confirming that TLR2 promotes CSC survival and self-renewal, and plays a key role in HER2-driven mammary carcinogenesis.
TLR2 deletion alters Treg frequency without affecting other immune cell populations
Since TLR2 is expressed in many immune cells including neutrophils, myeloid-derived suppressor cells (MDSC), monocytes, DCs, macrophages, NK cells, T (including Treg), and B lymphocytes,6 we assessed the percentage of the main immune cell types in the tumors, blood, and spleens from neuT-TLR2KO and neuT-TLR2WT mice at the experimental endpoint. The FACS gating strategy used is reported in Supplementary Fig 1b. While no significant differences were observed in most of the myeloid and lymphoid populations (Figure 2a-f) in tumors, blood, and spleens, and in intratumor macrophage polarization (Figure 2g), significantly lower Treg frequency was found in the tumors, blood, and spleens from neuT-TLR2KO mice (Figure 2h-j). Since in the neuT mouse model the amount of Treg cells can positively correlate with tumor progression,39 and we have observed an enhanced tumor progression in neuT-TLR2WT mice as compared to neuT-TLR2KO (Figure 1c-e), we analyzed the presence of Tregs in tumor-free TLR2WT and TLR2KO mice, to exclude that the observed difference in this cell population was due to variations in tumor burden and not to the expression of TLR2. Significantly lower levels of circulating Tregs were found in tumor-free TLR2KO than in TLR2WT mice (Figure 2k), suggesting that TLR2 can influence Treg expansion, regardless of the presence of cancer cells. Moreover, in TLR2WT mice, about 50% of Tregs and only 10% of effector T cells were positive for TLR2 staining, with Tregs expressing higher TLR2 levels (measured as MFI) than effector CD4+ T cells (Figure 2l). Interestingly, when circulating T cells from tumor-bearing TLR2WT mice were activated in the presence of TLR2 activators, PGN-SA or HMGB1, the percentage of Tregs significantly increased (Figure 2m), suggesting that TLR2 may directly promote Treg expansion.
Figure 2.
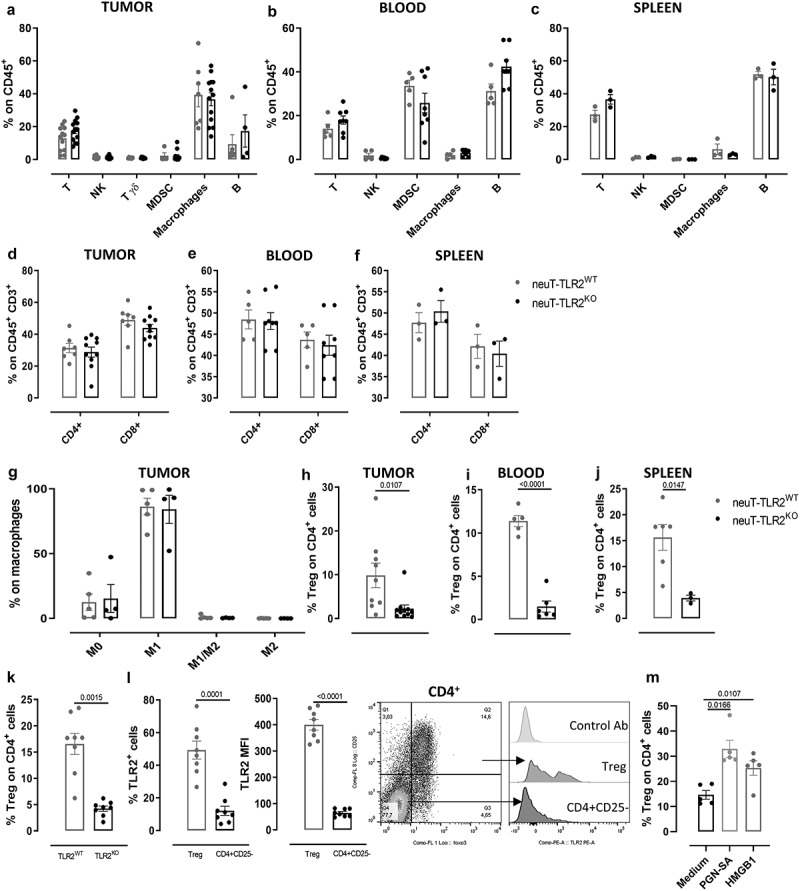
TLR2 deletion alters Treg cell frequency without affecting other immune populations. FACS analysis of immune cells in tumors (N ≥ 6 per group), blood (N = 5 neuT-TLR2KO and 6 neuT-TLR2WT) and spleens (N ≥ 3 per group) from neuT-TLR2KO and neuT-TLR2WT mice. Graphs show mean ± SEM of the percentage of (a-c) lymphocytes and myeloid cells from among CD45+ cells, (d-f) CD4+ and CD8+ from among CD3+ T cells, (g) M0, M1, M1/M2 and M2 from among total tumor macrophages, (h-j) Tregs from among CD4+ T cells. Each dot represents a mouse. (k) FACS analysis of peripheral Tregs in TLR2KO and TLR2WT mice. Graphs show mean ± SEM of the percentage of Tregs from among CD4+ T cells (N = 8 per group). (l) FACS analysis of TLR2 expression on Tregs and CD4+ CD25− cells from TLR2WT mice. Graphs show mean ± SEM of the percentage of TLR2+ cells and of TLR2 MFI (N = 8 per group). A representative density plot of Treg staining on CD4+ T cells and representative histograms of isotype control Ab or TLR2 staining on Treg and CD4+ CD25− effector T cells are shown. (m) FACS analysis of Tregs from TLR2WT mice upon ex vivo stimulation of blood T lymphocytes with TLR2 activating ligands. Graphs show mean ± SEM of the percentage of Tregs from among CD4+ cells. Each dot represents a mouse (N = 5 per group). Results come from at least 3 independent experiments. Unpaired Student’s t test with Welch’s correction.
TLR2KO BC cells display impaired tumorigenic properties
To dissect the contribution of cancer and host cell TLR2 signaling to tumor progression, we generated cell lines from the primary tumors of neuT-TLR2WT and neuT-TLR2KO mice, named WT-874, WT-879, WT-880, and KO-M26, KO-E26, and KO-C26, respectively. WT-874, WT-879, WT-880 express TLR2, which as expected is absent in KO-M26, KO-E26, and KO-C26 cells (Supplementary Fig 2a). All these cells express HER2 and the luminal marker cytokeratin 19 (CK19) but are negative for the basal marker alpha smooth muscle actin (α-SMA) (Supplementary Fig 2a). In vitro, TLR2WT cells displayed a slightly higher proliferation and lower basal apoptosis as compared to TLR2KO cells (Supplementary Fig 2b, c).
We performed orthotopic cross transplantation of WT-874 and KO-M26 cells into TLR2WT and TLR2KO mice. Tumor onset and progression correlated to the presence of TLR2 in cancer cells. Indeed, WT-874 cells generated tumors in 100% of mice, regardless of the recipient mouse genotype, while KO-M26 cells generated tumors in only 20% and 30% of TLR2WT and TLR2KO mice, respectively (Figure 3a). Moreover, the tumors generated by KO-M26 cells remained very small (mean volumes at sacrifice <1.5 mm3), regardless of the genotype of the receiving mice (Figure 3b). The tumors generated by WT-874 cells injected into TLR2KO mice were significantly larger (mean volumes at sacrifice: 127 mm3), and even significantly bigger tumors were generated by the WT-874 cells injected into TLR2WT mice (mean volumes at sacrifice: 500 mm3) (Figure 3b). Similar results were obtained using KO-E26 cells (Supplementary Fig 3).
Figure 3.
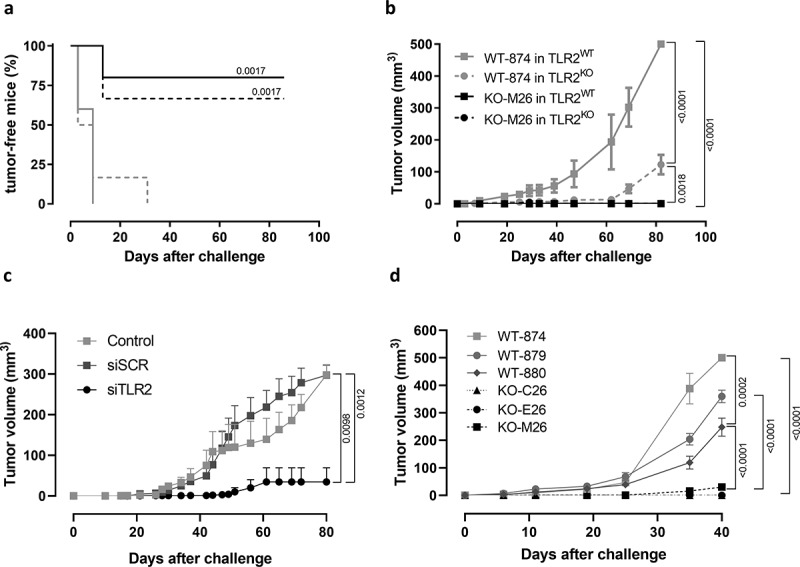
TLR2KO BC cells display impaired tumorigenicity. (a) Tumor-free survival and (b) mean tumor volume of TLR2WT and TLR2KO mice (N = 6 per group, from 2 independent experiments) injected orthotopically with 1 × 106 WT-874 or KO-M26 cells. (c) Mean tumor volume of BALB/c mice orthotopically injected with 1 × 104 4T1 cells left untreated (N = 5), transfected with scrambled siRNAs (N = 7) or siRNAs to TLR2 (N = 7). (d) Mean tumor volume of NSG mice injected orthotopically with 1 × 106 WT-874, WT-879, WT-880, KO-C26, KO-M26 or KO-E26 cells (N = 6 per group from two independent experiments). (a) Log-rank (Mantel-Cox) of KO-M26 cells injected into TLR2WT and TLR2KO mice vs WT-874 cells injected into TLR2WT mice. (b) two-way ANOVA test.
To confirm the TLR2 tumor-cell-intrinsic effects, 4T1 cells were silenced for TLR2 and injected into syngeneic BALB/c mice. TLR2 silencing significantly impaired tumor growth compared to control cells (Figure 3c). Moreover, a transplantation experiment performed in NSG mice indicated that the decreased tumorigenic ability of TLR2KO BC cells is maintained in immunodeficient mice. Indeed, KO-M26, KO-E26, and KO-C26 cells generated smaller tumors than WT-874, WT-879 and WT-880 cells (Figure 3d). These results, and the observations on Tregs, strongly suggest that TLR2 expressed in BC cells promotes BC growth, and that host TLR2-expressing cells provide a partial contribution to tumor progression.
TLR2 promotes CSC self-renewal and its inhibition impairs cell viability
We then exploited TLR2WT and TLR2KO cell lines to characterize the tumor-cell-intrinsic pro-carcinogenic effects of TLR2. When cultured in tumorsphere-forming conditions, WT-874 cells generated significantly more tumorspheres than KO-M26 and KO-E26 (Figure 4a). Moreover, the percentage of Aldefluor+ cells was significantly higher in WT-874 than in KO-M26 and KO-E26 cells, both when cultured in 2D conditions and as CSC-enriched P1 tumorspheres (Figure 4b). Similarly, a higher frequency of cells expressing the CSC markers Musashi-1,40 Oct4,41 Sca1, and CD44+CD24− was measured in WT-874, WT-879 and WT-880 cells than in KO-M26, KO-E26, and KO-C26 cells (Supplementary Fig 2d-g), indicating that TLR2 is involved in CSC self-renewal. Moreover, TLR2 induces the secretion of pro-tumoral cytokines, since, compared to KO-M26 or KO-E26 cells, WT-874 produced significantly higher amounts of TGF-β, IL-6, and VEGF (Figure 4c-e), which are responsible for triggering a pro-tumoral loop that promotes CSC self-renewal, proliferation, and invasion.13
Figure 4.
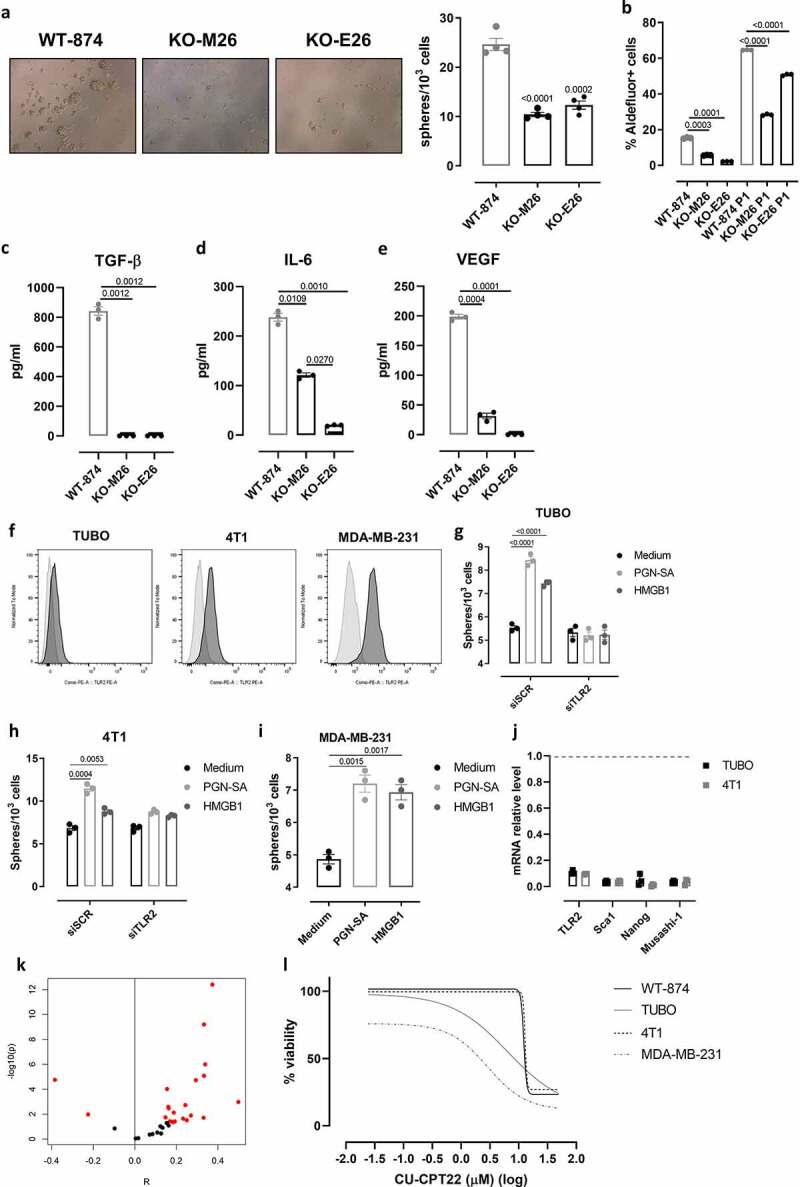
TLR2 promotes breast CSC self-renewal and its inhibition impairs cell viability. (a) Representative images of tumorspheres (Magnification 10X), and graph showing sphere-generating ability (number of tumorspheres generated every 103 plated cells) of WT-874, KO-M26, and KO-E26 cells cultured for 5 d in tumorsphere-forming conditions. (b) Means ± SEM of the percentage of Aldefluor+ cells in WT-874, KO-M26, and KO-E26 cells cultured for 5 d in 2D or tumorsphere-forming conditions. (c-e) ELISA of TGF-β, IL-6 and VEGF released into the supernatant of WT-874, KO-M26, KO-E26 cultured for 48 hours in 2D conditions. (f) Representative FACS histograms of TLR2 (dark gray) and control (light gray) staining on TUBO, 4T1 and MDA-MB-231 cells. (g, h) Tumorsphere-generating ability of TUBO and 4T1 cells transfected with a pool of scrambled siRNAs (siSCR) or siRNAs to TLR2 (siTLR2) treated, 24 hours after transfection, with PGN-SA or HMGB1 for 4 d. (i) Tumorsphere-generating ability of MDA-MB-231 cells treated with PGN-SA or HMGB1 for 5 d. (j) Relative expression by qPCR of TLR2, Sca1, Nanog and Musashi-1 mRNA in TUBO and 4T1 tumorspheres transfected with siTLR2 as compared to those transfected with siSCR (gray dotted line). (k) Plot indicating the correlation between TLR2 expression, and a CSC signature computed with ssGSEA for each of the BC transcriptomic datasets comprised in MetaGxBreast. X axis shows the value of Pearson’s correlation, y axis represents correlation’s significance. Red dots label datasets with a p-value<0.05, showing that significant tests are skewed toward positive values. (l) Concentration-dependent cytotoxicity of CU-CPT22 (50 to 0.02 µM) on WT-874, TUBO, 4T1 and MDA-MB-231 cells, evaluated using MTT after 48 hours of treatment. The graph shows log(inhibitor) vs. response – variable slope (four parameters) non-linear regression of data from three independent experiments, calculated with GrapPad8 software. All graphs report data from 3 independent experiments. Student’s t test.
Several mouse and human BC cell lines, including TUBO, 4T1, and MDA-MB-231, express TLR2 (figure 4f). We therefore assessed the effect of TLR2 activation on their tumorsphere-generation ability. Murine HER2+ TUBO and TNBC 4T1 cells were stimulated with PGN-SA or HMGB1. Under these stimuli, TUBO and 4T1 cells generated significantly more spheres and expressed higher levels of the CSC marker Sca1 than control cells (Figure 4g, h and Supplementary Fig 4). This effect was TLR2-dependent, as was not observed in TLR2-silenced cells (Figure 4g, h). PGN-SA and HMGB1 also increased tumorsphere generation in human TNBC MDA-MB-231 cells (Figure 4i). Moreover, in both TUBO and 4T1 tumorspheres, TLR2 silencing with specific siRNAs decreased the mRNA level of the CSC markers Sca1, Nanog,41 and Musashi-140 as compared to what observed in scrambled siRNA-transfected tumorspheres (Figure 4j), confirming that TLR2 promotes BC stemness, as we previously demonstrated.13 As an independent indication of the relationship between TLR2 and CSC activity, we analyzed 33 BC transcriptomic datasets and reported that TLR2 expression positively correlates with a published breast CSC signature16 in most of them (Figure 4k; Pearson correlation, Fisher’s merged p-value across 33 datasets: 1.4 × 10−38).
Finally, the effect of TLR2 pharmacological inhibition on BC cell viability was assessed. Indeed, the TLR2 inhibitor CU-CPT22 dose-dependently impaired WT-874, TUBO, 4T1, and MDA-MB-231 cell viability (Figure 4l), while it was ineffective on KO-M26 or KO-E26 cells (not shown). Overall, these results demonstrate that TLR2 signaling promotes breast CSC self-renewal and that its inhibition affects BC cell viability.
TLR2 mediates resistance to doxorubicin
High TLR2 expression in BC patients correlates with a significantly impaired response to chemotherapy, as chemotherapy-treated BC patients with TLR2high tumors showed lower RFS than patients with TLR2low tumors (Figure 5a), and TLR2 expression is significantly higher in non-responder than responder patients (Figure 5b). Accordingly, WT-874, WT-879 and WT-880 cells were more resistant to doxorubicin than KO-M26, KO-E26, and KO-C26 cells, which showed lower viability (Figure 5c) and significantly higher apoptosis (Figure 5d) in response to doxorubicin. Similar results in terms of viability and apoptosis were obtained using docetaxel, which – as doxorubicin – can induce ICD42 (Supplementary Fig 5a, c). On the contrary, treatment with cisplatin, which is reported not to induce ICD,42 impaired the viability and induced apoptosis of TLR2WT and TLR2KO cells to a similar extent (Supplementary Fig 5b, c). Interestingly, TLR2 inhibition with CU-CPT22 increased the sensitivity of HER2+ (WT-874 and TUBO) and TN (4T1 and MDA-MB-231) BC cells to doxorubicin (Figure 5e). Significantly, the two drugs in combination exerted synergistic activity, as calculated using the Bliss independence model (figure 5f). To dissect the mechanisms involved in TLR2-dependent resistance to ICD-inducing drugs, we analyzed the supernatants of doxorubicin-treated cells and found that doxorubicin induced the release of HMGB1, which may activate TLR2, in all BC cells tested, with higher amounts released by KO-M26 and KO-E26 cells, which is in line with their higher sensitivity to doxorubicin (Figure 5g). Treatment with HMGB1 induced an increase in the phosphorylated form of the TLR2 downstream signal transducer p65 NF-κB in WT-874 cells but not in KO-M26 and KO-E26 cells (Figure 5h). As expected, doxorubicin increased phospho-NF-κB in WT-874, TUBO, 4T1, and MDA-MB-231 cells, while TLR2 inhibition with CU-CPT22 significantly reduced it. The addition of CU-CPT22 to doxorubicin-treated cells significantly reduced phosphorylated NF-κB in all cell lines (Figure 5i and Supplementary Fig 6a). Moreover, doxorubicin treatment induced the release of the NF-κB-regulated cytokine IL-6, which was abolished by adding CU-CPT22 (Supplementary Fig 7A). Since NF-κB inhibits apoptosis and promotes cell survival,6 treatment with CU-CPT22 slightly induced apoptosis of WT-874, TUBO, 4T1, and MDA-MB-231 cells (Figure 5j and Supplementary Fig 6b). Of note, the level of apoptosis induced by CU-CPT22 was similar to the basal apoptosis observed in TLR2KO cells (Supplementary Fig 2c) and to that induced by siRNA-mediated TLR2 silencing in WT-874 cells (Supplementary Fig 6c). Most importantly, the addition of CU-CPT22 significantly improved doxorubicin apoptotic effect in WT-874, TUBO, 4T1, and MDA-MB-231 cells (Figure 5j and Supplementary Fig 6b). Of note, treatment of MDA-MB-231 cells with doxorubicin, alone or combined with CU-CPT22, increased the release of HMGB1 and other DAMPs that can potentially bind TLR2, such as HSP70, HSP90, Morgana (CHORDC1), HSP60, and Heat shock cognate 71 kDa protein (HSC70)43 (Supplementary Fig 7b), confirming the potential capability of doxorubicin to induce TLR2 activation on cancer cells. The effects of CU-CPT22 were not due to off-target mechanisms, as CU-CPT22 treatment did not exert any activity on KO-M26 or KO-E26 cells (Supplementary Fig 8a, b). Interestingly, CU-CPT22 increased doxorubicin-induced apoptosis to a similar extent as treatment with the NF-κB inhibitor BAY 11–7082 (Supplementary Fig 8c) and HMGB1 antagonist BOX-A (Supplementary Fig 8d).
Figure 5.
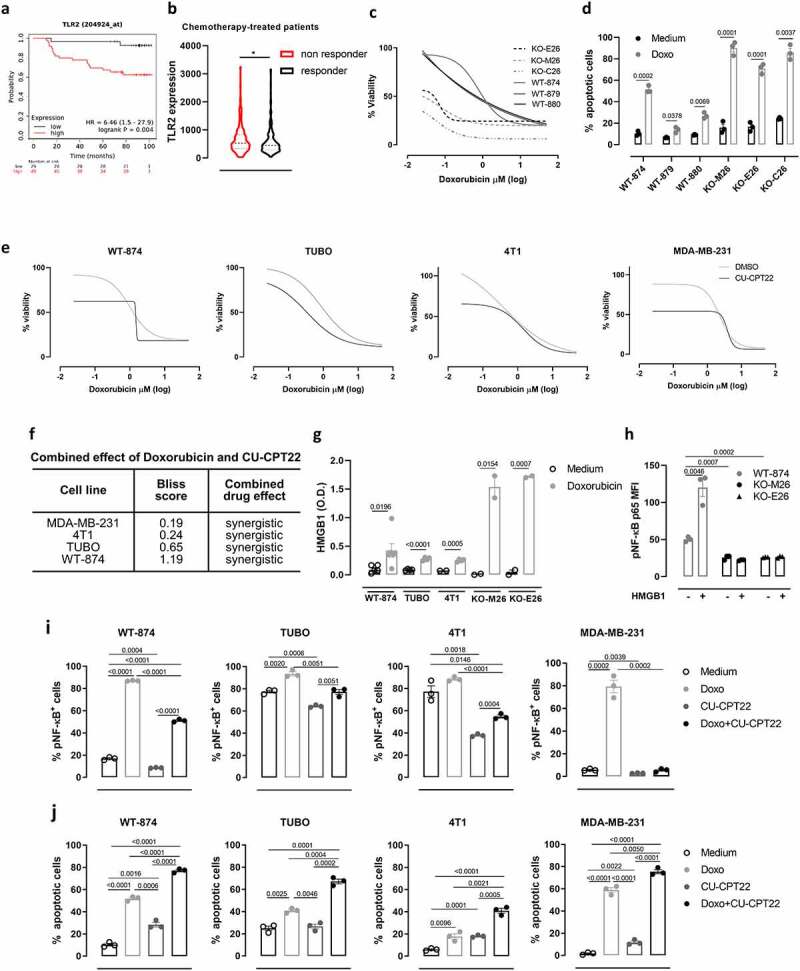
TLR2 promotes BC resistance to doxorubicin. (a) Kaplan–Meier plots displaying 5-y RFS of chemotherapy-treated BC patients stratified according to TLR2 mRNA expression (N = 78). (b) TLR2 mRNA expression levels in the tumors from BC patients that relapsed (non-responders; N = 221) or not (responders; N = 256) within 5 y of adjuvant chemotherapy. (c) Concentration-dependent cytotoxicity of doxorubicin (50 to 0.02 µM) on WT-874, WT-879, WT-880, KO-M26, KO-E26 and KO-C26 cells, evaluated by MTT after 48 hours of treatment. The graph shows log(inhibitor) vs. response – variable slope (four parameters) non-linear regression of data from three independent experiments, calculated using GrapPad8 software. (d) WT-874, WT-879, WT-880, KO-M26, KO-E26 and KO-C26 cells were treated with doxorubicin for 48 hours and analyzed using Annexin-V/propidium iodide FACS staining. The graph shows means ± SEM of the % of Annexin-V+ from three independent experiments. (e) Concentration-dependent toxicity of doxorubicin (50 to 0.02 µM) alone or combined with 5 µM CU-CPT22 on WT-874, TUBO, 4T1 and MDA-MB-231 cells, evaluated via MTT after 48 hours of treatment in three independent experiments. (f) Analysis of the effect of the combination of doxorubicin and CU-CPT22 on BC cell lines. Bliss score > 0: synergistic effect; Bliss score = 0: additive effect; Bliss score < 0: antagonistic effect. (g) ELISA analysis of HMGB1 released into the supernatant by BC cells that were treated with doxorubicin for 48 hours or left untreated. The graph shows means ± SEM of HMGB1 concentration from ≥2 independent experiments. (h) FACS analysis of p65 NF-κB phosphorylation in WT-874, KO-M26 and KO-E26 cells treated with HMGB1 for 30 minutes or left untreated. The means ± SEM of mean fluorescence intensity (MFI) from three independent experiments are reported. (i, j) WT-874, TUBO, 4T1 and MDA-MB-231 cells were treated for 48 hours with or without doxorubicin, CU-CPT22 or a combination of both. (i) Graphs of FACS analysis of p65 NF-κB phosphorylation. (j) Annexin-V/propidium iodide analysis of cell apoptosis. All graphs report the means ± SEM of the percentage of positive cells from three independent experiments. Student’s t tests.
These results demonstrate that TLR2/NF-κB signaling, which is activated by doxorubicin-induced HMGB1 release, is involved in chemoresistance, and that TLR2 inhibition restores BC cell sensitivity to doxorubicin.
Treatment with a TLR2 inhibitor potentiates doxorubicin anti-cancer effects in vivo
To verify whether TLR2 inhibition exerts an antitumor effect in vivo, we treated 4T1 tumor-bearing BALB/c mice with two different doses of CU-CPT22 (10 or 50 µg) intratumorally, twice per week, starting when tumors reached 2 mm in mean diameter. Both CU-CPT22 doses significantly reduced tumor growth, and treatment at 50 µg significantly reduced the number of spontaneous lung metastases (Supplementary Fig 9). We then analyzed whether CU-CPT22 potentiates the activity of doxorubicin. 4T1 tumor-bearing mice were treated with doxorubicin, with and without CU-CPT22 (50 µg), twice per week. Both doxorubicin and CU-CPT22, administered as single treatments, significantly reduced tumor growth compared to control mice (Figure 6a). However, the combination of the two drugs induced a statistically significant reduction in tumor growth compared to the two single treatments (Figure 6a). Moreover, mice treated with CU-CPT22 + doxorubicin showed significantly fewer lung metastases than control or doxorubicin-treated mice (Figure 6b). Mice underwent MRI at the experimental end point to better characterize the effects of the combination treatment. T2w morphological axial and coronal MR images were acquired at B0 = 7 T. As shown in the representative T2w axial images, a significant decrease in tumor volume occurred upon treatment with CU-CPT22 + doxorubicin (Figure 6c, upper). The MRI quantification of tumor volume showed decreases of 38 ± 2%, 47 ± 10%, and 75 ± 16% in mice treated with CU-CPT22, doxorubicin, or both, respectively, compared to control mice (Figure 6d). ADC-MRI maps, which report the degrees of freedom of water, were obtained using DWI-MRI. False color representative axial ADC maps of the tumor region (superimposed onto morphological T2w MR images) are reported in Figure 6c (lower) for control mice and mice treated with doxorubicin and CU-CPT22 simultaneously. A significant higher ADC value, an indication of decreased cellularity, was observed in CU-CPT22 + doxorubicin-treated mice than in control mice, while no significant effect was detected in mice treated with doxorubicin or CU-CPT22 alone (Figure 6e). A cytofluorimetric analysis of tumors showed that the combination of CU-CPT22 and doxorubicin induced a statistically significant decrease in Sca1+ and CD44+ CD24− CSCs compared to tumors from mice treated with doxorubicin alone, demonstrating that CU-CPT22 impairs CSC survival also in vivo (figure 6f, g). Moreover, the combined therapy induced a significant increase in tumor-infiltrating CD8+ T cells and a decrease in Tregs (Figure 6h, i), while no significant differences in CD4+ T, NK cells, and Ly6C+ monocytic MDSCs were observed (Figure 6j-l). Of note, CU-CPT22, alone or combined with doxorubicin, induced a significant reduction in Ly6G+ granulocytic MDSCs (Figure 6m). Despite not inducing variations in the total macrophage population (Figure 6n), the combined treatment also significantly decreased M2 while increasing M1 macrophages (Figure 6o). Overall, these data indicate that treatment with TLR2 inhibitor enhances the efficacy of doxorubicin, reducing breast tumor growth and metastatic dissemination and restoring an immunocompetent TME.
Figure 6.
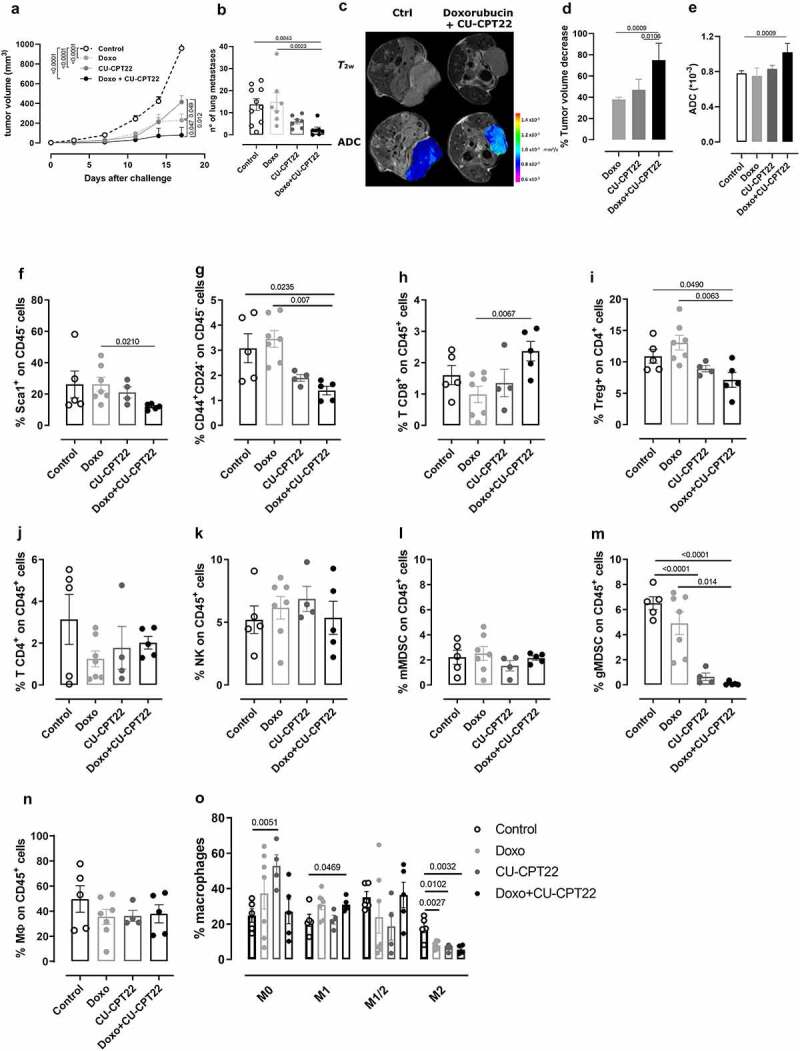
TLR2 inhibition increases doxorubicin anti-cancer activity in vivo. BALB/c mice were orthotopically challenged with 1 × 104 4T1 cells. When the tumors reached 2 mm in diameter, the mice were treated i.p. with 3 mg/kg doxorubicin and intratumorally with 50 µg CU-CPT22, or the vehicle alone as a control (N = 7 per treated groups and N = 10 per control group). The treatment was repeated twice per week. Graphs show (a) mean tumor volume, and (b) the number of spontaneous lung metastases per square mm measured in lungs. (c-e) T2w and DWI axial MR images of mice were acquired at the end point of the treatment. (c) Representative T2w axial images and false color representative axial ADC maps of the tumor region (superimposed on morphological T2w MR images) of control and doxorubicin + CU-CPT22-treated mice. (d) Tumor volume was measured by manually drawing ROIs in the tumor for all axial slices covering the entire tumor (Fiji-ImageJ free software). Decrease of volume in the treated mice was reported as the percentage of the volume of control mice (mean ± SD). (e) Mean ± SD of ADC values from the tumors of treated mice. (f-o) FACS analysis of (f) Sca1+ and (g) CD44+ CD24− cells from among CD45−, (h) CD8+ T cells, (j) CD4 + T cells, (k) NK cells, (l) monocytic MDSCs, (m) granulocytic MDSCs, and (n) macrophages from among CD45+ leucocytes and (i) Tregs from among CD4+ T cells and (o) M1, M2, M1/M2 or M0 polarized macrophages among the macrophage population in the tumors. Graphs show means ± SEM of the percentage of positive cells (N ≥ 4). Each circle represents a mouse. Student’s t test or Mann–Whitney test. In (a), two-way ANOVA analysis.
Discussion
Chemotherapy is still the main option for TNBC and metastatic BC patients, with neoadjuvant and adjuvant chemotherapy becoming mainstays for treating early BC. However, chemoresistance is common in both advanced and early BC, and about 30% of early-stage BC patients relapse despite treatment.3,44 Therefore, the development of combined treatments to counteract chemoresistance and strengthen anti-cancer activity is an urgent medical need.
Since breast CSCs are chemotherapy-resistant and involved in tumor relapse,15 we have previously sought genes that play a key role in breast CSC survival. We identified TLR2 as being overexpressed in breast CSCs and as playing a role in their self-renewal.13 Herein, we have analyzed TLR2ʹs role in mammary carcinogenesis by generating genetically modified mice that, to our knowledge, represent the only model of HER2-driven mammary carcinogenesis on a TLR2KO background. Although the carcinogenic process was not completely abolished, TLR2KO mice displayed delayed tumor onset and decreased tumor multiplicity, suggesting a contribution of TLR2 to BC development. The HER2 transgene acts as a potent tumor driver in neuT mice, not allowing to fully appreciate the contribution of TLR2 in affecting tumor growth. Moreover, we cannot exclude that the constitutive deletion of TLR2 may activate compensatory mechanisms that cannot be observed upon acute inhibition of TLR2. Indeed, while treatment of several BC cell lines with the TLR2 inhibitor CU-CPT22 dose-dependently decreased their viability, the cell lines generated from neuT-TLR2KO tumors displayed only a slight decrease in cell viability as compared to their TLR2WT counterpart. The generation of a conditional, tissue-specific TLR2KO mouse model might contribute to better elucidate the role of TLR2 in tumor progression.
However, data from BC patients, whose tumors express higher TLR2 levels than adjacent normal tissue and in which Wang et al. demonstrated that high tumor TLR2 expression, assessed by qPCR, is associated with poor OS and unresponsiveness to endocrine therapy in the luminal B subtype,45 are in line with what was observed in our preclinical model. Significantly, several genetic alterations that result in increased TLR2-signaling-pathway activation, including mutations that generate constitutively active forms of TLR2 and amplifications in TLR2 downstream effector IRAK1 (observed in 23.8% of patients), have been identified in BC patients.46 In this study, we observed a significant association between high TLR2 expression and poor RFS in BC patients but not with OS. It should be noted that all the microarray datasets we analyzed are from the tumor bulk, where the expression of TLR2 in cancer and immune cells cannot be distinguished. The analysis of single-cell RNA sequencing datasets would allow to better correlate TLR2 expression on cancer cells and patients’ outcome.
The role of TLR2 is, at least partly, mediated by its ability to promote breast-CSC self-renewal, as suggested by the higher frequency of cells that express typical breast CSC markers38 and the enhanced CSC-enriched tumorsphere-generating ability of BC from TLR2-expressing mice compared to their neuT-TLR2KO counterparts. Indeed, we have previously demonstrated that TLR2 stimulation in breast CSCs leads to the activation of the MyD88/NF-κB and AKT pathways, which promote the secretion of several cytokines and growth factors, such as TGF-β and IL-6.13 These cytokines promote CSC survival, self-renewal, and invasion via the autocrine activation of the STAT3 and Smad3 pathways,6,13,47 in accordance with data that show increased TLR2 expression in BC cell lines endowed with metastatic potential.48 However, CSC self-renewal and differentiation are controlled by a complex interplay among different signaling pathways, including those shared with normal stem cells, such as the Notch, Wnt, and Hedgehog pathways.12 Therefore, TLR2 is one of the many players that support CSCs. Future experiments aimed at analyzing the tumorigenic potential of the putative CSC fraction isolated from neuT-TLR2WT or neuT-TLR2KO tumors could further demonstrate the impact of TLR2 deletion on cancer stemness. Nevertheless, our transplantation experiments confirmed that in cancer cells, TLR2 signaling mediates pro-tumorigenic effects as, unlike TLR2WT BC cells, TLR2KO BC cells have a poor ability to generate tumors when injected into both immunocompetent and NSG mice. Moreover, host-cell TLR2 also contributes to tumor progression, as TLR2KO mice represented a more hostile environment than TLR2WT mice for the growth of TLR2+ BC. This suggests that TLR2 expression in immune cells may contribute to their immunosuppressive activity. In fact, several research groups have demonstrated that TLR2 stimulation in MDSCs induces their accumulation in the TME and lymphoid organs49 and the production of IL-6 and IL-10, which polarize M2 macrophages and favor metastatic dissemination.50,51 Moreover, TLR2 has been shown to promote Treg proliferation both directly, through the TLR2-MyD88-NF-κB axis,52,53 and indirectly, via the activation of dysfunctional DCs that produce IL-6 and IL-10.54 Fittingly, we observed a decrease in both tumor-infiltrating and peripheral Tregs in tumor-bearing neuT-TLR2KO, compared to neuT-TLR2WT mice. This decrease was not due to the lower tumor burden characteristic of neuT-TLR2KO mice, since healthy, not neuT, TLR2KO mice showed a lower percentage of circulating Tregs than TLR2WT mice. We confirmed in vitro that TLR2 activation in T cells promotes the expansion of Tregs, which expresses higher TLR2 levels than effector CD4+ T cells, suggesting that TLR2 plays an important role in Treg biology. However, TLR2 deficiency did not affect the percentages of other immune cell populations, whether in the tumor or periphery. Nevertheless, further studies are required to demonstrate their functional integrity.
Besides its role in breast carcinogenesis, TLR2 also plays a role in BC chemoresistance, as we observed that high TLR2 expression correlates with low RFS in chemotherapy-treated BC patients. Furthermore, the deletion or pharmacologic inhibition of TLR2 increases the sensitivity of several BC cell lines to doxorubicin. This mechanism of chemoresistance is mediated by the release of TLR2-activating DAMPs, such as HMGB1, following doxorubicin-induced ICD. Despite our study lacks the analysis of total NF-kB by WB due to technical issues, the increase of phospho-NF-κB in TLR2WT but not in TLR2KO cells treated with HMGB1 for 30 minutes, a time window insufficient to induce de novo NF-κB production, supports the role of NF-κB activation downstream to the HMGB1/TLR2 axis. Although HMGB1 can also bind other receptors such as RAGE and TLR4,6 we did not observe HMGB1-dependent NF-κB phosphorylation in TLR2KO BC cells, and TLR2 inhibition or silencing was sufficient to inhibit HMGB1 activity.13 Thus, TLR2-activation induces the production of IL-6, a well-known promoter of survival, growth, and proliferation in both immunosuppressive and cancer cells,55,56 thus protecting cells from apoptosis. Indeed, high HMGB1 levels correlate with poor response to neoadjuvant chemotherapy in TNBC patients,18 and HMGB1 protects BC cells from chemotherapy and promotes metastasis formation.57 Moreover, other DAMPs that can activate TLR2 may be released into the TME following ICD-inducing chemotherapy,6,7,43 and indeed we observed the release of HSP70, HSP90, HSP60, and HSC70 from doxorubicin-treated MDA-MB-231 cells. Other chaperones, such as Morgana, are known to be constitutively released from MDA-MB-231 cells and to signal through TLR2.43 Interestingly, TLR2 activation may also be induced by bacteria. Specific microbiota have recently been found in mammary glands, with normal and neoplastic samples showing alterations in amount and composition.58 Neoadjuvant chemotherapy changes breast microbiota composition, inducing an increase in Pseudomonas aeruginosa, a potential TLR2 activator that stimulates BC cell proliferation.59 Moreover, many chemotherapy-treated patients suffer from opportunistic infections from Gram-positive bacteria, with the commensal bacteria diffusing to the tumor, which could further activate TLR2.60 Therefore, the administration of TLR2 inhibitors in combination with chemotherapy might not only impair the pro-inflammatory and pro-tumorigenic activity of HMGB1 and other DAMPs but may also counteract some of the detrimental effects of breast dysbiosis. Further experiments are needed to confirm this hypothesis.
Our in vivo therapeutic experiments have demonstrated that treatment with the TLR2 inhibitor CU-CPT22 enhances the capability of doxorubicin to hamper BC progression, potentiating its anti-cancer effects and decreasing metastatic dissemination. The combination of doxorubicin and TLR2 inhibition significantly decreased the frequency of Sca1+ and CD44+ CD24− cells in the tumors. Moreover, MRI analysis identified a significant reduction in tumor volume (about 75%) in mice treated with doxorubicin in combination with CU-CPT22 as compared to control mice. Further insights into tumor features have been gained by using DWI-MRI to obtain ADC maps that report the degree of freedom of water, which is a technique that is extensively applied to assess increases in cellularity during tumor growth.36 Treatment with doxorubicin and CU-CPT22 caused an increase in water freedom, suggesting a lowering of cellularity because of tumor regression.
Significantly, although TLR2 is expressed in immune cells, no detrimental alterations in the immune system were observed in mice treated with CU-CPT22 and in TLR2KO mice, which supports the safety and feasibility of TLR2 targeting. Indeed, innate and acquired immune responses are well preserved in the absence of TLR2,61 and phase I clinical trials with TLR2-targeted therapies showed initial promising results in hematological malignancies.62,63 Importantly, anthracyclines, including doxorubicin, can induce ICD in cancer cells, leading to the promotion of an anti-tumor immune response.64 Besides HMGB1, the main DAMPs that mediate this response are Annexin A1, ATP, and calreticulin, alongside several cytokines released by cancer cells that contribute to immune-stimulation.65 Therefore, it should be emphasized that the determinants of immune stimulation in doxorubicin-induced ICD are diversified, and the inhibition of the HMGB1-TLR2 axis on immune cells could not be sufficient to halt the anti-tumor immune response. Moreover, it has been shown that the expression of TLR4, another HMGB1 receptor, on DCs is required for the induction of a tumor-specific T cell response, while lack of TLR2 did not affect it.66 Conversely, TLR2 activation on MDSCs has been demonstrated to promote pro-tumoral inflammation, while TLR4 was not involved in this mechanism.50 Moreover, activation of TLR2 can lead to the expansion of Tregs, thus suppressing anti-tumor immunity in vivo.52,53 Therefore, TLR2 and TLR4 appear to trigger opposite mechanisms in a cell-type-dependent manner, with TLR2 being dispensable for anti-tumor activity of DCs but required for pro-tumor activity of MDSCs and Tregs, and TLR4 being dispensable for pro-tumor activity of MDSCs but required for antitumor activity of DCs. Nevertheless, the anti-tumor vs pro-tumor effects resulting from TLR2 engagement are a complex matter of debate,67 with TLR2 agonists having been reported as successful adjuvants for anti-cancer immunotherapy,68 and TLR2 inhibitors (monoclonal antibodies or small-molecule inhibitors) successfully used in clinical trials for the treatment of acute myeloid leukemia and myelodysplastic syndrome.62,63 In our 4T1-based mouse model, doxorubicin alone did not induce any significant changes in the proportion of immune cell populations infiltrating the tumor (except for a decrease in M2-polarized macrophages), while CU-CPT22 alone and even more when combined with doxorubicin significantly reduced Tregs, granulocytic MDSCs, and M2 macrophages while increasing CD8+ cytotoxic T cells and M1 macrophages. These results suggest that the pro-tumor function exerted by TLR2 stimulation on cancer cells and immune-suppressive cells may hinder the capability of doxorubicin to generate an anti-tumor immune response. Conversely, the addition of CU-CPT22 unleashes the full immunogenic potential of doxorubicin-induced ICD, leading to a remodeling of the TME. Of note, similar results were observed when blocking the TLR2 ligand HMGB1 in a 4T1 tumor model, which improved the efficacy of immune checkpoint inhibitors.69 The combined treatment with doxorubicin and CU-CPT22 was more effective than doxorubicin monotherapy in hindering 4T1 tumor progression in vivo, but tumor growth was not completely abolished. This could be due to the 4T1 intrinsic features that might lead to an underestimation of the potential effect of the combined protocol proposed. Indeed, the 4T1 model is quite resistant to several combined therapies and immunotherapies, and the combination of cyclophosphamide/doxorubicin with anti-PD-1 or anti-PD-L1 antibodies showed no, or very limited, increase in anti-tumor effect.70,71
The results so far obtained and described in this manuscript demonstrate that TLR2 plays a role in BC chemoresistance (Figure 7), although it cannot be considered a master regulator of BC onset and progression. Its targeting may enhance BC cell vulnerability to standard therapies, similarly to what is observed in BC using immune checkpoint inhibitors.70,71 In conclusion, our data add fresh knowledge on the complex role played by TLR2 in cancer and contribute with a slightly provocative view to the scientific debate on the opportunity of using either TLR2 agonists or inhibitors in cancer therapy.
Figure 7.
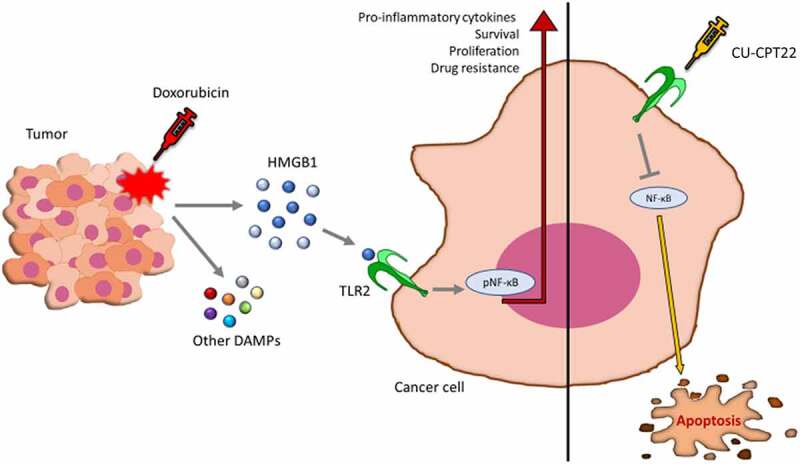
TLR2 inhibition promotes doxorubicin effects in impairing BC growth. Schematic representation of the mechanism of TLR2-dependent chemoresistance and of the effects of CU-CPT22 treatment in combination with doxorubicin.
Supplementary Material
Acknowledgments
We thank Dr. Dale Lawson for his revision and editing of the manuscript.
Funding Statement
The research leading to these results has received funding from AIRC under the IG 2021 – ID. 25766 project – principal investigator L. Conti, IG 2015 – ID. 16724 and IG 2018 – ID. 21468 projects – principal investigator F. Cavallo, IG 2020 – ID. 24930 project – principal investigator M. Brancaccio, from Fondazione Ricerca Molinette Onlus, from the University of Turin (Turin, Italy), and a liberal contribution from Banca d’Italia. The Italian Ministry for University and Research (MUR) is gratefully acknowledged for yearly FOE funding to the Euro-BioImaging Multi-Modal Molecular Imaging Italian Node (MMMI). Roberto Ruiu was supported by a fellowship from Fondazione Umberto Veronesi.
Abbreviations
α-SMA: alpha smooth muscle actin; ADC: Apparent diffusion constant; BC: breast cancer; CK19: cytokeratin 19; CSC: cancer stem cells; DAMP: damage-associated molecular patterns; DC: dendritic cells; DWI-MRI: Diffusion Weighted Magnetic Resonance Imaging; ER: estrogen receptor; G-CSF: granulocyte-colony stimulating factor; HMGB1: high mobility group box 1; HSC70: heat shock cognate 71 kDa protein; HSP: heat shock protein; IL: interleukin; i.p.: intraperitoneal; MDSC: myeloid-derived suppressor cell; MRI: Magnetic Resonance Imaging; NSG: NOD SCID gamma; OS: overall survival; PGN-SA: peptidoglycan from S. aureus; PRR: pattern recognition receptors; RARE: Rapid Acquisition with Refocused Echoes; RFS: relapse-free survival; TGF-β: tumor growth factor; TLR2: Toll-like Receptor 2; TME: Tumor microenvironment; TNBC: triple negative breast cancer; Treg: T regulatory cell; VEGF: vascular endothelial growth factor.
Author’s contributions
L.C. and F.C. designed the project and the experiments. L.C., A.D.L., and E.B. wrote the paper, and F.C. and E.Q. reviewed it. E.B., A.D.L., I.M., F.R., L.A., E.Q., and R.R. performed experiments and analyzed the data. G.F. and E.D.G. performed and analyzed MRI. P.P. performed and M.B. supervised DAMP analysis. A.S. performed bioinformatics analysis. L.C. and F.C. coordinated and directed the study. All authors have read and approved the final version of the manuscript.
Data sharing statement
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Ethics approval and consent to participate
Mice were bred and maintained under saprophytic and pathogen-free conditions at the animal facility of the Molecular Biotechnology Center, and treated in accordance with EU and institutional guidelines, with the approval of the Animal Care and Use Committee of University of Turin and of the Italian Ministry of Health (authorizations N° 107/2020-PR and 500/2017-PR).
Disclosure statement
The authors declare no potential conflicts of interest.
Financial support
The research leading to these results has received funding from AIRC under the IG 2021 – ID. 25766 project – principal investigator L. Conti, IG 2015 – ID. 16724 and IG 2018 – ID. 21468 projects – principal investigator F. Cavallo, IG 2020 – ID. 24930 project – principal investigator M. Brancaccio, from Fondazione Ricerca Molinette Onlus, from the University of Turin (Turin, Italy), and a liberal contribution from Banca d’Italia. The Italian Ministry for University and Research (MUR) is gratefully acknowledged for yearly FOE funding to the Euro-BioImaging Multi-Modal Molecular Imaging Italian Node (MMMI). Roberto Ruiu was supported by a fellowship from Fondazione Umberto Veronesi.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/2162402X.2022.2086752
References
- 1.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F.. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–18. doi: 10.3322/caac.21660 [DOI] [PubMed] [Google Scholar]
- 2.Ayala de la Pena F, Andres R, Garcia-Saenz JA, Manso L, Margeli M, Dalmau E, Pernas S, Prat A, Servitja S, Ciruelos E, et al. SEOM clinical guidelines in early stage breast cancer (2018). Clin Transl Oncol. 2019;21:18–30. doi: 10.1007/s12094-018-1973-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nigdelis MP, Karamouzis MV, Kontos M, Alexandrou A, Goulis DG, Lambrinoudaki I. Updates on the treatment of invasive breast cancer: quo Vadimus? Maturitas. 2021;145:64–72. doi: 10.1016/j.maturitas.2020.11.006 [DOI] [PubMed] [Google Scholar]
- 4.Ji X, Lu Y, Tian H, Meng X, Wei M, Cho WC. Chemoresistance mechanisms of breast cancer and their countermeasures. Biomed Pharmacother. 2019;114:108800. doi: 10.1016/j.biopha.2019.108800 [DOI] [PubMed] [Google Scholar]
- 5.van Schaik TA, Chen KS, Shah K. Therapy-induced tumor cell death: friend or foe of immunotherapy? Front Oncol. 2021;11:678562. doi: 10.3389/fonc.2021.678562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Di Lorenzo A, Bolli E, Tarone L, Cavallo F, Conti L. Toll-like receptor 2 at the crossroad between cancer cells, the immune system, and the microbiota. Int J Mol Sci. 2020;21:9418. doi: 10.3390/ijms21249418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Galluzzi L, Humeau J, Buque A, Zitvogel L, Kroemer G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat Rev Clin Oncol. 2020;17:725–741. doi: 10.1038/s41571-020-0413-z [DOI] [PubMed] [Google Scholar]
- 8.Jinushi M, Yagita H, Yoshiyama H, Tahara H. Putting the brakes on anticancer therapies: suppression of innate immune pathways by tumor-associated myeloid cells. Trends Mol Med. 2013;19:536–545. doi: 10.1016/j.molmed.2013.06.001 [DOI] [PubMed] [Google Scholar]
- 9.Conti L, Ruiu R, Barutello G, Macagno M, Bandini S, Cavallo F, Lanzardo S. Microenvironment, oncoantigens, and antitumor vaccination: lessons learned from BALB-neuT mice. Biomed Res Int. 2014;2014:534969. doi: 10.1155/2014/534969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pandey S, Singh S, Anang V, Bhatt AN, Natarajan K, Dwarakanath BS. Pattern recognition receptors in cancer progression and metastasis. Cancer Growth Metastasis. 2015;8:25–34. doi: 10.4137/CGM.S24314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clarke MF. Clinical and Therapeutic Implications of Cancer Stem Cells. N Engl J Med. 2019;380:2237–2245. doi: 10.1056/NEJMra1804280 [DOI] [PubMed] [Google Scholar]
- 12.Ruiu R, Tarone L, Rolih V, Barutello G, Bolli E, Riccardo F, Cavallo F, Conti L. Cancer stem cell immunology and immunotherapy: harnessing the immune system against cancer’s source. Prog Mol Biol Transl Sci. 2019;164:119–188. doi: 10.1016/bs.pmbts.2019.03.008 [DOI] [PubMed] [Google Scholar]
- 13.Conti L, Lanzardo S, Arigoni M, Antonazzo R, Radaelli E, Cantarella D, Calogero RA, Cavallo F. The noninflammatory role of high mobility group box 1/Toll-like receptor 2 axis in the self-renewal of mammary cancer stem cells. FASEB J. 2013;27:4731–4744. doi: 10.1096/fj.13-230201 [DOI] [PubMed] [Google Scholar]
- 14.Brooks MD, Burness ML, Wicha MS. Therapeutic implications of cellular heterogeneity and plasticity in breast cancer. Cell Stem Cell. 2015;17:260–271. doi: 10.1016/j.stem.2015.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quaglino E, Conti L, Cavallo F. Breast cancer stem cell antigens as targets for immunotherapy. Semin Immunol. 2020;47:101386. doi: 10.1016/j.smim.2020.101386 [DOI] [PubMed] [Google Scholar]
- 16.Pece S, Disalvatore D, Tosoni D, Vecchi M, Confalonieri S, Bertalot G, Viale G, Colleoni M, Veronesi P, Galimberti V, et al. Identification and clinical validation of a multigene assay that interrogates the biology of cancer stem cells and predicts metastasis in breast cancer: a retrospective consecutive study. EBioMedicine. 2019;42:352–362. doi: 10.1016/j.ebiom.2019.02.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bianchi ME, Crippa MP, Manfredi AA, Mezzapelle R, Rovere Querini P, Venereau E. High-mobility group box 1 protein orchestrates responses to tissue damage via inflammation, innate and adaptive immunity, and tissue repair. Immunol Rev. 2017;280:74–82. doi: 10.1111/imr.12601 [DOI] [PubMed] [Google Scholar]
- 18.Tanabe Y, Tsuda H, Yoshida M, Yunokawa M, Yonemori K, Shimizu C, Yamamoto S, Kinoshita T, Fujiwara Y, Tamura K, et al. Pathological features of triple-negative breast cancers that showed progressive disease during neoadjuvant chemotherapy. Cancer Sci. 2017;108:1520–1529. doi: 10.1111/cas.13274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rovero S, Amici A, Di Carlo E, Bei R, Nanni P, Quaglino E, Porcedda P, Boggio K, Smorlesi A, Lollini P-L, et al. DNA vaccination against rat her-2/Neu p185 more effectively inhibits carcinogenesis than transplantable carcinomas in transgenic BALB/c mice. J Immunol. 2000;165:5133–5142. doi: 10.4049/jimmunol.165.9.5133 [DOI] [PubMed] [Google Scholar]
- 20.Liu S, Chen H. Isolation of primary breast cancer cells from HER2 transgenic mice. Bio Protocol. 2016;6:e1956. doi: 10.21769/BioProtoc.1956 [DOI] [Google Scholar]
- 21.Geninatti Crich S, Cadenazzi M, Lanzardo S, Conti L, Ruiu R, Alberti D, Cavallo F, Cutrin JC, Aime S. Targeting ferritin receptors for the selective delivery of imaging and therapeutic agents to breast cancer cells. Nanoscale. 2015;7:6527–6533. doi: 10.1039/C5NR00352K [DOI] [PubMed] [Google Scholar]
- 22.Gyorffy B. Survival analysis across the entire transcriptome identifies biomarkers with the highest prognostic power in breast cancer. Comput Struct Biotechnol J. 2021;19:4101–4109. doi: 10.1016/j.csbj.2021.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Michael D. Metap: meta-analysis of significance values. R package version 1.7., 2021.
- 24.Gendoo DMA, Zon M, Sandhu V, Manem VSK, Ratanasirigulchai N, Chen GM, Waldron L, Haibe-Kains B. MetaGxData: clinically annotated breast, ovarian and pancreatic cancer datasets and their use in generating a multi-cancer gene signature. Sci Rep. 2019;9:8770. doi: 10.1038/s41598-019-45165-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deena MA, Ratanasirigulchai N, Schroeder MS, Pare L, Parker JS, Prat A, Haibe-Kains B. R package version 2.26.0; 2021. http://www.pmgenomics.ca/bhklab/software/genefu Accessed 13 March 2022.
- 26.Fekete JT, Gyorffy B. ROCplot.org: validating predictive biomarkers of chemotherapy/hormonal therapy/anti-HER2 therapy using transcriptomic data of 3,104 breast cancer patients. Int J Cancer. 2019;145:3140–3151. doi: 10.1002/ijc.32369 [DOI] [PubMed] [Google Scholar]
- 27.Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013;14:7. doi: 10.1186/1471-2105-14-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lanzardo S, Conti L, Rooke R, Ruiu R, Accart N, Bolli E, Arigoni M, Macagno M, Barrera G, Pizzimenti S, et al. Immunotargeting of antigen xCT attenuates stem-like cell behavior and metastatic progression in breast cancer. Cancer Res. 2016;76:62–72. doi: 10.1158/0008-5472.CAN-15-1208 [DOI] [PubMed] [Google Scholar]
- 29.Conti L, De Palma R, Rolla S, Boselli D, Rodolico G, Kaur S, Silvennoinen O, Niccolai E, Amedei A, Ivaldi F, et al. Th17 cells in multiple sclerosis express higher levels of JAK2, which increases their surface expression of IFN-gammaR2. J Immunol. 2012;188:1011–1018. doi: 10.4049/jimmunol.1004013 [DOI] [PubMed] [Google Scholar]
- 30.Conti L, Bolli E, Di Lorenzo A, Franceschi V, Macchi F, Riccardo F, Ruiu R, Russo L, Quaglino E, Donofrio G, et al. Immunotargeting of the xCT cystine/glutamate antiporter potentiates the efficacy of Her2-targeted immunotherapies in breast cancer. Cancer Immunol Res. 2020;8:1039–1053. doi: 10.1158/2326-6066.CIR-20-0082 [DOI] [PubMed] [Google Scholar]
- 31.Bandini S, Macagno M, Hysi A, Lanzardo S, Conti L, Bello A, Riccardo F, Ruiu R, Merighi IF, Forni G, et al. The non-inflammatory role of C1q during Her2/neu-driven mammary carcinogenesis. Oncoimmunology. 2016;5:e1253653. doi: 10.1080/2162402X.2016.1253653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Donofrio G, Tebaldi G, Lanzardo S, Ruiu R, Bolli E, Ballatore A, Rolih V, Macchi F, Conti L, Cavallo F, et al. Bovine herpesvirus 4-based vector delivering the full length xCT DNA efficiently protects mice from mammary cancer metastases by targeting cancer stem cells. Oncoimmunology. 2018;7:e1494108. doi: 10.1080/2162402X.2018.1494108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Macagno M, Bandini S, Stramucci L, Quaglino E, Conti L, Balmas E, Smyth MJ, Lollini P-L, Musiani P, Forni G, et al. Multiple roles of perforin in hampering ERBB-2 (Her-2/neu) carcinogenesis in transgenic male mice. J Immunol. 2014;192:5434–5441. doi: 10.4049/jimmunol.1301248 [DOI] [PubMed] [Google Scholar]
- 34.Conti L, Lanzardo S, Ruiu R, Cadenazzi M, Cavallo F, Aime S, Crich SG. L-Ferritin targets breast cancer stem cells and delivers therapeutic and imaging agents. Oncotarget. 2016;7:66713–66727. doi: 10.18632/oncotarget.10920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.She S, Zhao Y, Kang B, Chen C, Chen X, Zhang X, Chen W, Dan S, Wang H, Wang Y-J, et al. Combined inhibition of JAK1/2 and DNMT1 by newly identified small-molecule compounds synergistically suppresses the survival and proliferation of cervical cancer cells. Cell Death Dis. 2020;11:724. doi: 10.1038/s41419-020-02934-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ferrauto G, Di Gregorio E, Lanzardo S, Ciolli L, Iezzi M, Aime S. Generation of multiparametric MRI maps by using Gd-labelled- RBCs reveals phenotypes and stages of murine prostate cancer. Sci Rep. 2018;8:10567. doi: 10.1038/s41598-018-28926-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tallerico R, Conti L, Lanzardo S, Sottile R, Garofalo C, Wagner AK, Johansson MH, Cristiani CM, Kärre K, Carbone E, et al. NK cells control breast cancer and related cancer stem cell hematological spread. Oncoimmunology. 2017;6:e1284718. doi: 10.1080/2162402X.2017.1284718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grange C, Lanzardo S, Cavallo F, Camussi G, Bussolati B. Sca-1 identifies the tumor-initiating cells in mammary tumors of BALB-neuT transgenic mice. Neoplasia. 2008;10:1433–1443. doi: 10.1593/neo.08902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ambrosino E, Spadaro M, Iezzi M, Curcio C, Forni G, Musiani P, Wei W-Z, Cavallo F. Immunosurveillance of Erbb2 carcinogenesis in transgenic mice is concealed by a dominant regulatory T-cell self-tolerance. Cancer Res. 2006;66:7734–7740. doi: 10.1158/0008-5472.CAN-06-1432 [DOI] [PubMed] [Google Scholar]
- 40.Troschel FM, Palenta H, Borrmann K, Heshe K, Hua SH, Yip GW, Kiesel L, Eich HT, Götte M, Greve B, et al. Knockdown of the prognostic cancer stem cell marker Musashi-1 decreases radio-resistance while enhancing apoptosis in hormone receptor-positive breast cancer cells via p21(WAF1/CIP1). J Cancer Res Clin Oncol. 2021;147:3299–3312. doi: 10.1007/s00432-021-03743-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang D, Lu P, Zhang H, Luo M, Zhang X, Wei X, Gao J, Zhao Z, Liu C. Oct-4 and Nanog promote the epithelial-mesenchymal transition of breast cancer stem cells and are associated with poor prognosis in breast cancer patients. Oncotarget. 2014;5:10803–10815. doi: 10.18632/oncotarget.2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Galluzzi L, Vitale I, Warren S, Adjemian S, Agostinis P, Martinez AB, Chan TA, Coukos G, Demaria S, Deutsch E, et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J Immunother Cancer. 2020;8:e000337. doi: 10.1136/jitc-2019-000337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Secli L, Avalle L, Poggio P, Fragale G, Cannata C, Conti L, Iannucci A, Carrà G, Rubinetto C, Miniscalco B, et al. Targeting the extracellular HSP90 co-chaperone morgana inhibits cancer cell migration and promotes anticancer immunity. Cancer Res. 2021;81:4794–4807. doi: 10.1158/0008-5472.CAN-20-3150 [DOI] [PubMed] [Google Scholar]
- 44.Fisusi FA, Akala EO. Drug combinations in breast cancer therapy. Pharm Nanotechnol. 2019;7:3–23. doi: 10.2174/2211738507666190122111224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Y, Liu S, Zhang Y, Yang J. Dysregulation of TLR2 serves as a prognostic biomarker in breast cancer and predicts resistance to endocrine therapy in the luminal B subtype. Front Oncol. 2020;10:547. doi: 10.3389/fonc.2020.00547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scheeren FA, Kuo AH, van Weele LJ, Cai S, Glykofridis I, Sikandar SS, Zabala M, Qian D, Lam JS, Johnston D, et al. A cell-intrinsic role for TLR2-MYD88 in intestinal and breast epithelia and oncogenesis. Nat Cell Biol. 2014;16:1238–1248. doi: 10.1038/ncb3058 [DOI] [PubMed] [Google Scholar]
- 47.Quaglino E, Cavallo F, Conti L. Cancer stem cell antigens as targets for new combined anti-cancer therapies. Int J Biochem Cell Biol. 2020;129:105861. doi: 10.1016/j.biocel.2020.105861 [DOI] [PubMed] [Google Scholar]
- 48.Xie W, Huang Y, Xie W, Guo A, Wu W. Bacteria peptidoglycan promoted breast cancer cell invasiveness and adhesiveness by targeting toll-like receptor 2 in the cancer cells. PLoS One. 2010;5:e10850. doi: 10.1371/journal.pone.0010850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maruyama A, Shime H, Takeda Y, Azuma M, Matsumoto M, Seya T. Pam2 lipopeptides systemically increase myeloid-derived suppressor cells through TLR2 signaling. Biochem Biophys Res Commun. 2015;457:445–450. doi: 10.1016/j.bbrc.2015.01.011 [DOI] [PubMed] [Google Scholar]
- 50.Kim S, Takahashi H, Lin WW, Descargues P, Grivennikov S, Kim Y, Luo J-L, Karin M. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature. 2009;457:102–106. doi: 10.1038/nature07623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol. 2007;179:977–983. doi: 10.4049/jimmunol.179.2.977 [DOI] [PubMed] [Google Scholar]
- 52.Chen YQ, Li PC, Pan N, Gao R, Wen ZF, Zhang TY, Huang F, Wu F-Y, Ou X-L, Zhang J-P, et al. Tumor-released autophagosomes induces CD4(+) T cell-mediated immunosuppression via a TLR2-IL-6 cascade. J Immunother Cancer. 2019;7:178. doi: 10.1186/s40425-019-0646-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yamazaki S, Okada K, Maruyama A, Matsumoto M, Yagita H, Seya T. TLR2-dependent induction of IL-10 and Foxp3+ CD25+ CD4+ regulatory T cells prevents effective anti-tumor immunity induced by Pam2 lipopeptides in vivo. PLoS One. 2011;6:e18833. doi: 10.1371/journal.pone.0018833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tang M, Diao J, Gu H, Khatri I, Zhao J, Cattral MS. Toll-like receptor 2 activation promotes tumor dendritic cell dysfunction by regulating IL-6 and IL-10 receptor signaling. Cell Rep. 2015;13:2851–2864. doi: 10.1016/j.celrep.2015.11.053 [DOI] [PubMed] [Google Scholar]
- 55.Regis G, Icardi L, Conti L, Chiarle R, Piva R, Giovarelli M, Poli V, Novelli F. IL-6, but not IFN-gamma, triggers apoptosis and inhibits in vivo growth of human malignant T cells on STAT3 silencing. Leukemia. 2009;23:2102–2108. doi: 10.1038/leu.2009.139 [DOI] [PubMed] [Google Scholar]
- 56.Masjedi A, Hashemi V, Hojjat-Farsangi M, Ghalamfarsa G, Azizi G, Yousefi M, Jadidi-Niaragh F. The significant role of interleukin-6 and its signaling pathway in the immunopathogenesis and treatment of breast cancer. Biomed Pharmacother. 2018;108:1415–1424. doi: 10.1016/j.biopha.2018.09.177 [DOI] [PubMed] [Google Scholar]
- 57.Sohun M, Shen H. The implication and potential applications of high-mobility group box 1 protein in breast cancer. Ann Transl Med. 2016;4:217. doi: 10.21037/atm.2016.05.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fernandez MF, Reina-Perez I, Astorga JM, Rodriguez-Carrillo A, Plaza-Diaz J, Fontana L. Breast cancer and its relationship with the microbiota. Int J Environ Res Public Health. 2018;15:1747. doi: 10.3390/ijerph15081747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chiba A, Bawaneh A, Velazquez C, Clear KYJ, Wilson AS, Howard-mcnatt M, Levine EA, Levi-Polyachenko N, Yates-Alston SA, Diggle SP, et al. Neoadjuvant chemotherapy shifts breast tumor microbiota populations to regulate drug responsiveness and the development of metastasis. Mol Cancer Res. 2020;18:130–139. doi: 10.1158/1541-7786.MCR-19-0451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Montassier E, Gastinne T, Vangay P, Al-Ghalith GA, Bruley Des Varannes S, Massart S, Moreau P, Potel G, de La Cochetière MF, Batard E, et al. Chemotherapy-driven dysbiosis in the intestinal microbiome. Aliment Pharmacol Ther. 2015;42:515–528. doi: 10.1111/apt.13302 [DOI] [PubMed] [Google Scholar]
- 61.Wooten RM, Ma Y, Yoder RA, Brown JP, Weis JH, Zachary JF, Kirschning CJ, Weis JJ. Toll-like receptor 2 is required for innate, but not acquired, host defense to Borrelia burgdorferi. J Immunol. 2002;168:348–355. doi: 10.4049/jimmunol.168.1.348 [DOI] [PubMed] [Google Scholar]
- 62.Garcia-Manero G, Montalban-Bravo G, Yang H, Wei Y, Alvarado Y, Daver NG, DiNardo CD, et al. A clinical study of OPN-305, a Toll-like Receptor 2 (TLR-2) antibody, in patients with lower risk Myelodysplastic Syndromes (MDS) that have received prior Hypomethylating Agent (HMA). Ther Blood. 2016;128:227. [Google Scholar]
- 63.Kovacsovics TJ, Mims A, Salama ME, Pantin J, Rao N, Kosak KM, Ahorukomeye P, Glenn MJ, Deininger MWN, Boucher KM, et al. Combination of the low anticoagulant heparin CX-01 with chemotherapy for the treatment of acute myeloid leukemia. Blood Adv. 2018;2:381–389. doi: 10.1182/bloodadvances.2017013391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2016;17:97–111. doi: 10.1038/nri.2016.107 [DOI] [PubMed] [Google Scholar]
- 65.Fucikova J, Kepp O, Kasikova L, Petroni G, Yamazaki T, Liu P, Zhao L, Spisek R, Kroemer G, Galluzzi L, et al. Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis. 2020;11:1013. doi: 10.1038/s41419-020-03221-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Apetoh L, Ghiringhelli F, Tesniere A, Criollo A, Ortiz C, Lidereau R, Mariette C, Chaput N, Mira J-P, Delaloge S, et al. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol Rev. 2007;220:47–59. doi: 10.1111/j.1600-065X.2007.00573.x [DOI] [PubMed] [Google Scholar]
- 67.Kaczanowska S, Joseph AM, Davila E. TLR agonists: our best frenemy in cancer immunotherapy. J Leukoc Biol. 2013;93:847–863. doi: 10.1189/jlb.1012501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang Y, Su L, Morin MD, Jones BT, Mifune Y, Shi H, et al. Adjuvant effect of the novel TLR1/TLR2 agonist Diprovocim synergizes with anti-PD-L1 to eliminate melanoma in mice. Proc Natl Acad Sci USA. 2018;115:E8698–E706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hubert P, Roncarati P, Demoulin S, Pilard C, Ancion M, Reynders C, Lerho T, Bruyere D, Lebeau A, Radermecker C, et al. Extracellular HMGB1 blockade inhibits tumor growth through profoundly remodeling immune microenvironment and enhances checkpoint inhibitor-based immunotherapy. J Immunother Cancer. 2021;9:e001966. doi: 10.1136/jitc-2020-001966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Black M, Barsoum IB, Truesdell P, Cotechini T, Macdonald-Goodfellow SK, Petroff M, Siemens DR, Koti M, Craig AWB, Graham CH, et al. Activation of the PD-1/PD-L1 immune checkpoint confers tumor cell chemoresistance associated with increased metastasis. Oncotarget. 2016;7:10557–10567. doi: 10.18632/oncotarget.7235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Grasselly C, Denis M, Bourguignon A, Talhi N, Mathe D, Tourette A, Serre L, Jordheim LP, Matera EL, Dumontet C, et al. The antitumor activity of combinations of cytotoxic chemotherapy and immune checkpoint inhibitors is model-dependent. Front Immunol. 2018;9:2100. doi: 10.3389/fimmu.2018.02100 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.