

ABSTRACT
Antibody-directed nanotherapeutics (ADNs) represent a promising delivery platform for selective delivery of an encapsulated drug payload to the site of disease that improves the therapeutic index. Although both single-chain Fv (scFv) and Fab antibody fragments have been used for targeting, no platform approach applicable to any target has emerged. scFv can suffer from intrinsic instability, and the Fabs are challenging to use due to native disulfide over-reduction and resulting impurities at the end of the conjugation process. This occurs because of the close proximity of the disulfide bond connecting the heavy and light chain to the free cysteine at the C-terminus, which is commonly used as the conjugation site. Here we show that by engineering an alternative heavy chain-light chain disulfide within the Fab, we can maintain efficient conjugation while eliminating the process impurities and retaining stability. We have demonstrated the utility of this technology for efficient ADN delivery and internalization for a series of targets, including EphA2, EGFR, and ErbB2. We expect that this technology will be broadly applicable for targeting of nanoparticle encapsulated payloads, including DNA, mRNA, and small molecules.
KEYWORDS: Antibody fragment, stability, manufacturability, liposome, antibody-drug conjugate, developability, antibody engineering
Introduction
Targeted nanoparticles represent a promising therapeutic strategy for treating receptor-overexpressing cancers, autoimmune disorders, and even in the construction of vaccines. These targeted nanoparticles use antibody fragments rather than full-length IgG molecules in order to minimize Fc-mediated clearance from the circulation.1,2 Although single-chain variable fragments (scFv) have been utilized in multiple immunoliposome constructs,3–7 it is often difficult to identify molecules with the requisite thermal stability (minimum melting temperature 60°C, and preferably >70°C) to allow for their use in a robust manufacturing process. The exclusion of unstable scFv severely limits the number of candidates against any target, including those that may have other desirable characteristics such as high expression levels, species cross-reactivity, and good binding affinity. When compared to scFvs, Fabs are generally more thermally stable, which allows a larger panel of viable antibody choices.8,9 However, the conjugation procedure used for many nanoparticle-based constructs relies on selective reduction of a C-terminal cysteine,6,10–12 and the close proximity of other internal disulfides, specifically the disulfide connecting the heavy and light chain, commonly results in over-reduction of the Fab, resulting in the conjugation of lower molecular weight impurities that are both difficult to characterize and may yield undesirable pharmacologic properties.
Antibodies are adaptable molecules, amenable to a range of modifications to the disulfide bond pairing. Adding disulfide bonds, both between the heavy and light chain of an antibody13,14 or within one of the chains (reviewed by Hagihara and Saereas15) has been shown to improve stability. Single-domain antibodies and antibodies with longer complementarity-determining regions have been found to have additional disulfide bonds, which is thought to have evolved to help stabilize the loop. Alternative disulfide bonds have also been used as a solution to improve light-chain pairing within bispecific antibodies.16,17
Antibodies are also frequently engineered to facilitate the delivery of small molecules (i.e., antibody-drug conjugates (ADCs)) or liposomes. Much work has gone into identifying positions within antibodies to add cysteines for conjugation.18 In conjugating antibody fragments, site-specific conjugations to cysteine are often exploited. Cysteines are used because they are rare in the antibody fragments and are typically remote from the antigen-binding site. Modified antibodies wherein cysteines are engineered at specific locations for conjugation of cytotoxic drugs have been previously described.18–22 Incorporation of unnatural amino acids can be used for unique conjugation handles, but these unnatural antibodies are not generally compatible with the standard manufacturing process.23 In many of these fragments, the native interchain disulfide bond between the heavy and light chain constant regions (CH1 and CL) is absent, either because the interchain cysteines have been used as a site of attachment for cytotoxic drug, or because the interchain cysteines have been replaced by another amino acid to avoid effector molecule attachment to those cysteines.19,24
This report describes an engineering campaign to identify Fabs with alternative disulfide pairing that retained the stability of the parental Fabs. These engineered Fabs have improved conjugations properties, including near-complete elimination of low molecular weight impurities, which are particularly challenging in the manufacturing process. The engineered Fabs have shown excellent stability and excelled in internalizing conjugated liposomes using a variety of cell lines.
Results
Engineering Fabs with alternative disulfide bonds
We first wanted to characterize the role of the disulfide bond between the heavy and light chain on Fab thermal stability, since antibody fragments need to have melting temperatures of at least 70°C to be robustly conjugated to a nanoparticle. An antibody from a human naïve library (with a kappa light chain) was expressed on three different isotypes (IgG1, IgG2, and IgG4) with and without the disulfide bond. Antibodies were also designed with disulfide bonds at alternative positions within the Fab, further away from the C-terminus. The amino acid positions were determined by examining a crystal structure (1HZH) and mutating residues to cysteine when the heavy chain and light chain were within 10 Å of each other (Figure 1). Finally, a well-studied VH-VL (HC: G44C, LC: G100C, Kabat numbering used throughout this report) disulfide pair13 was also included.
Figure 1.
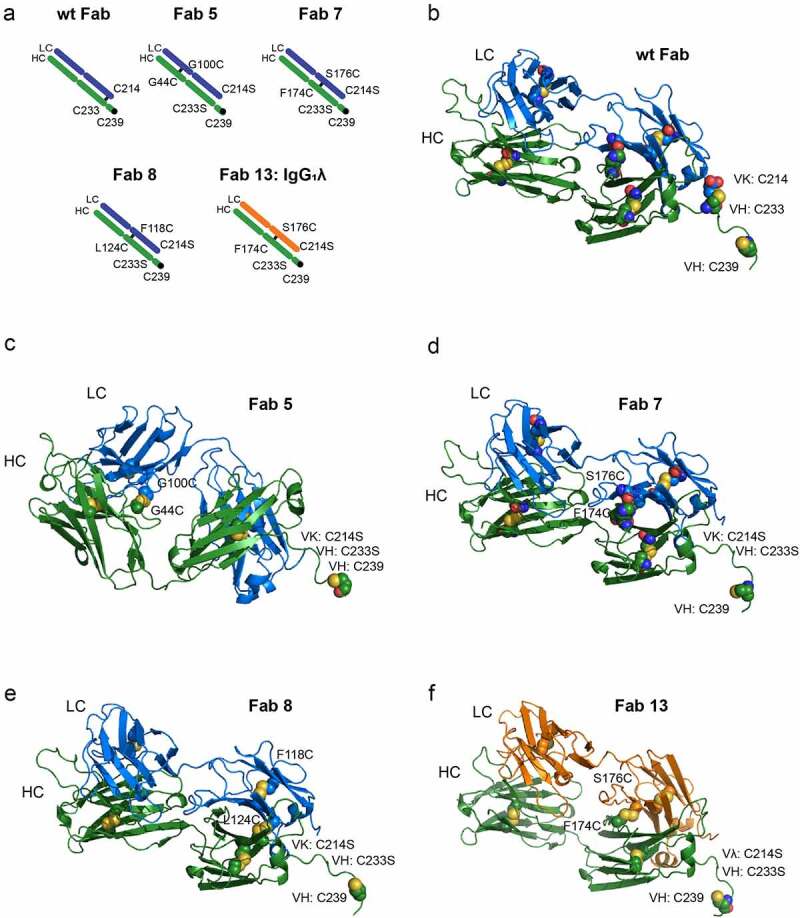
Sites selected for the addition of disulfide bonds (a) shows a cartoon representation of the disulfide bonds on a wild-type Fab or engineered Fabs, including one with a lambda light chain. (b) shows a PyMOL representation of the disulfide bond for a wild-type Fab. The disulfide bonds are in spheres and the free cysteine for conjugation at position 239 is also a sphere. (c-e) show the alternative disulfide pairing for a Fab with a kappa light chain and (f) shows an alternative disulfide pairing with a lambda light chain (Fab 13). The alternative disulfide pair in Fab 7 and Fab 13 (HC: F174C, C233S LC: S176C, C214S) was selected for additional engineering.
These Fabs were transiently expressed in HEK 293 cells and purified using CH1 resin. The melting temperature was then measured using differential scanning fluorimetry (DSF). As observed in Table 1 and Supplemental Figure S1, removing the disulfide bond resulted in a lowering of the melting temperature by approximately 5°C. The two Fabs containing alternative disulfide pairing, Fab 5 (HC: G44C LC: G100C) and Fab 7 (HC: F174C, C233S LC: S176C, C214S) retained melting temperatures above 75°C. In contrast, Fab 8 (HC: L124C, C233S LC: F118C, C214S) had over a 10°C loss in melting temperature. Based on these results, we decided to test the conjugation capability of the Fabs that maintained their thermal stability.
Table 1.
Design of Fabs with melting temperatures
| Construct | Description | Variable domain | Tm (degC) |
|---|---|---|---|
| Fab 1 | Wild-type IgG1 | Naïve kappa | 78.5 |
| Fab 2 | IgG1 without disulfide bonds (HC: C233S LC: C214S) | Naïve kappa | 72.5 |
| Fab 3 | Wild-type IgG2 | Naïve kappa | 75.7 |
| Fab 4 | IgG2 without disulfide bonds (HC: C127S LC: C214S) | Naïve kappa | 70.7 |
| Fab 5 | IgG1 (HC: G44C LC: G100C) | Naïve kappa | 77.7 |
| Fab 7 | IgG1 (HC: F174C, C233S LC: S176C, C214S) | Naïve kappa | 75.7 |
| Fab 8 | IgG1 (HC: L124C, C233S LC: F118C, C214S) | Naïve kappa | 65.7 |
| Fab 9 | Wild-type IgG4 | Naïve kappa | 75.7 |
| Fab 10 | IgG4 without disulfide bonds (HC: C127S LC: C214S) | Naïve kappa | 70.9 |
| Fab 11 | Wild-type IgG1 | MM-310 | 77.3 |
| Fab 12 | IgG1 (HC: G44C LC: G100C) | MM-310 | 76.1 |
| Fab 13 | IgG1 (HC: F174C, C233S LC: S176C, C214S) | MM-310 | 76.1 |
| Fab 14 | IgG1 (HC: G44C, F174C, C233S LC: G100C, S176C, C214S) | MM-310 | 77.5 |
| Fab 15 | IgG1 (HC: H172E, F174C, C233S LC: T162D, S176C, C214S) | MM-310 | 79.6 |
| Fab 16 | IgG1 (HC: H172F, F174C, C233S LC: T162L, S174V, S176C, C214S) | MM-310 | 80.7 |
| Fab 17 | IgG1 (HC: G44L, F174C, C233S LC: S176C, G100L C214S) | MM-310 | 75.1 |
| Fab 18 | IgG1 (HC: G44L, H172E, F174C, C233S LC: G100L, T162D, S176C, C214S) | MM-310 | 73.8 |
| Fab 19 | IgG1 (HC: G44L, H172F, F174C, C233S LC: G100L, T162L, T172V, S176C, C214S) | MM-310 | 74.8 |
Testing of conjugation efficiency
We took two sets of mutations (Fab 5 and Fab 7) and designed Fabs using an anti-EphA2 antibody fragment (MM-310) as the variable domain that had been previously engineered to have high stability.25 This antibody fragment has a lambda light chain, which allowed us to confirm that the alternative disulfide pairing would work for both types of light chains. The melting temperatures of these antibodies (Fab 11 – Fab 14 in Table 1) were similar to the first round of antibody fragments tested. The wild-type Fab (Fab 11) melting temperature was 77.3°C, and all three Fabs with alternative disulfides had melting temperatures that were higher than 75°C.
We then tested these constructs in conjugations assays. Here, various Fab’ dimers were first reduced under mild reducing conditions using cysteine, and subsequently conjugated to a maleimide-terminated PEG2000-distearoylphosphatidylethanolamine lipid anchor (Figure 2), that could subsequently be used in the preparation of immunoliposomes. The results of the SDS-PAGE analysis of Fab 11, Fab 12, Fab 13, and Fab 14 during the conjugation are shown in Figure 3. These gels show the non-reduced and reduced Fabs prior to conjugation, the conjugation mix, the purified conjugation, and the unconjugated fraction. Fab 12 (HC: G44C LC: G100C) produced a high proportion of chain dissociation products, as shown by the lower molecular weight species in lane 9. Fab 13 (HC: F174C, C233S LC: S176C, C214S) shows the best interchain stability (as shown by the intact Fab in the non-reduced sample, lane 12) and purity of the conjugate (lane 15). Fab 14 produced a low proportion of chain dissociation products, but a high proportion of multiple-conjugation products, as evidenced by the presence of the higher molecular weight species in lane 20.
Figure 2.
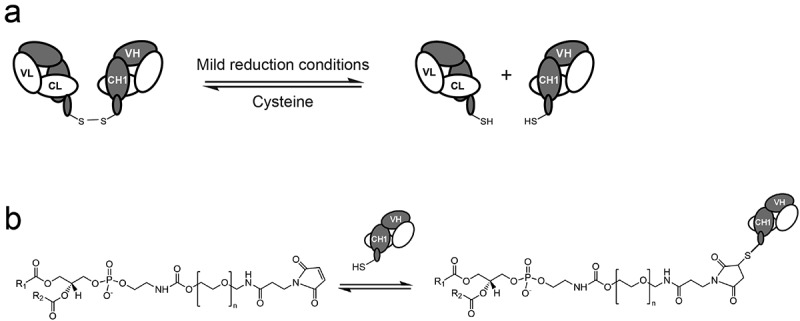
Schematic of the conjugation and subsequent insertion of stabilized Fabs into liposomes or lipid nanoparticles. (a) Fab dimers are initially reduced under mild reducing conditions to generate Fab fragments with a single C-terminal cysteine. (b) These Fabs are subsequently conjugated to maleimde-terminated PEG-distearoylphosphatidylethanolamine (Mal-PEG-DSPE) in the presence of excess PEG-DSPE.
Figure 3.
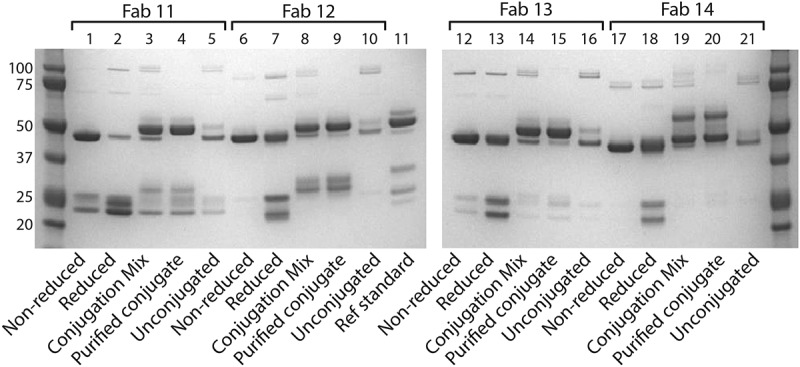
SDS-PAGE analysis of conjugated and unconjugated Fabs Fab 11, Fab 12, Fab 13, and Fab 14 were run on SDS-PAGE to analyze the purity of the Fabs during the conjugation process. Each Fab was run as non-reduced and reduced Fab, and then both the conjugation mixture and purified conjugate were run. Molecular weight markers are included in the first lane and last lanes of the gel. A reference standard is included.
Additional engineering to improve stability
Although Fabs containing the HC: F174C, LC: S176C mutations are improved in the conjugation efficiency to liposomes, they have a slightly lower melting temperature when compared to the wild-type Fab (78.5°C vs 75.7°C for the kappa, 77.3°C vs 76.1°C for the lambda). We wanted to further improve the thermal stability of the Fab by additional engineering. We tested the addition of negative charge (Fab 15), as well as the addition of hydrophobic residues to improve packing (Fab 16, Fab 17). We also combined mutations in various permutations (Fab 17–19). The specific mutations and melting temperatures are shown in Table 1. Adding negative charge and altering packing of the Fab resulted in higher melting temperatures (79.6°C and 80.7°C) when compared to Fab 13 (76.1°C), but interestingly, combining the mutations (Fab 18) did not result in improvements in stability. The analytical SEC profiles of Fab 11 and Fab 16–19 from the initial purifications are shown in Supplemental Figure S2; no improvements in the percentage of monomer were observed.
Figure 4 shows the SDS-PAGE of the additionally engineered Fabs (Fab 15-19) as non-reduced protein, reduced protein, conjugation mixture, and purified conjugates. Among these Fabs, Fab 16 (lane 12) and Fab 19 (lane 24) gave the lowest quantity of dissociated chains; however, all were significantly better than the wild type. This shows that these Fabs have high stability against chain dissociation during conjugation to pegylated lipids.
Figure 4.
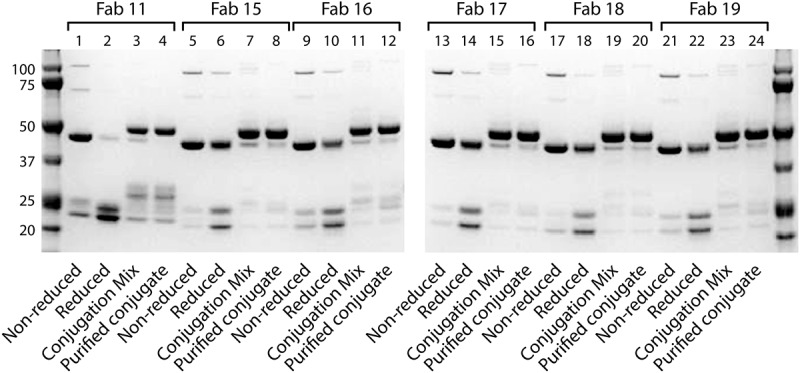
SDS-PAGE analysis of conjugated and unconjugated Fabs Fab 11, Fab 15, Fab 16, Fab 17, Fab 18 and Fab 19 were run on SDS-PAGE to analyze the purity of the Fabs during the conjugation process. Each Fab was run as non-reduced and reduced Fab, and then both the conjugation mixture and purified conjugate were run. Molecular weight markers are included in the first lane and last lanes of the gel. .
Reduction and conjugation of engineered Fabs to Mal-terminated PEG-DSPE
The SH/protein ratios for the reduced Fabs are shown in Table 2. Ideally, the SH/protein ratio would be close to 1. As shown in the table, Fab 11, which contains wild-type disulfide bonds, has an SH/protein ratio of 1.64, suggesting the Fab is being over-reduced. Changing the disulfide pairing resulted in an improved SH/protein ratio for most of the Fabs, indicating the engineered Fabs are not being over-reduced. Interestingly, Fabs with the disulfide bond in the variable domain (Fab 12 and Fab 14) had worse SH/protein ratios, suggesting over-reduction.
Table 2.
Reduction, conjugation yield, and target binding of Fabs
| Fab | Description | SH/protein | Conjugate yield (reduced protein), % |
% Binding to EphA2 (relative to Fab 11) |
|---|---|---|---|---|
| Fab 11 | Wild-type IgG1 | 1.64 | 62.5 | 100 |
| Fab 12 | IgG1 (HC: G44C LC: G100C) | 1.94 | 57.9 | 108.3 |
| Fab 13 | IgG1 (HC: F174C, C233S LC: S176C, C214S) | 1.16 | 66.2 | 96.9 |
| Fab 14 | IgG1 (HC: G44C, F174C, C233S LC: G100C, S176C, C214S) | 1.92 | 48.5 | 105.0 |
| Fab 15 | IgG1 (HC: H172E, F174C, C233S LC: T162D, S176C, C214S) | 1.11 | 69.5 | 86.0 |
| Fab 16 | IgG1 (HC: H172F, F174C, C233S LC: T162L, S174V, S176C, C214S) | 1.10 | 83.1 | 85.0 |
| Fab 17 | IgG1 (HC: G44L, F174C, C233S LC: S176C, G100L C214S) | 1.09 | 79.5 | 89.7 |
| Fab 18 | IgG1 (HC: G44L, H172E, F174C, C233S LC: G100L, T162D, S176C, C214S) | 1.44 | 79.5 | 75.5 |
| Fab 19 | IgG1 (HC: G44L, H172F, F174C, C233S LC: G100L, T162L, T172V, S176C, C214S) | 1.17 | 76.9 | 96.8 |
Additionally, the binding of the anti-EphA2 Fab conjugates to EphA2 was measured relative to Fab 11, which contains wild-type disulfide pairing. One Fab, Fab 18, had poor binding, but the rest had greater than 85% binding to the target, indicating that the engineered Fabs were effectively incorporated into Fab-targeted drug-loaded liposomes. Fab conjugates 16, 17, and 19 showed the best combination of high conjugation efficiency and EphA2 binding of those tested.
Insertion of Fab-PEG-DSPE into liposomes and characterization
As shown in Table 3, the wild-type Fab (Fab 11) had a high percentage of non-product bands: 45.2% for the conjugate and 24% for the conjugate-comprising liposomes. In contrast, the engineered Fabs exhibited a reduction of non-product bands. Using Fab 13, Fab 15, Fab17, Fab 18, Fab 19, Fab 20, Fab 21, or Fab 22 resulted in less than 10% non-product bands. The insertion efficiency was calculated as the percent of protein, per unit of phospholipid that remained associated with the liposomes after purification by size-exclusion chromatography.
Table 3.
Generation of non-product bands and liposome insertion efficiency of Fabs
| |
Non-product bands (%) |
Insertion Efficiency (%) |
||
|---|---|---|---|---|
| Fab | Conjugate | Liposomes | ||
| 11 | 45.2 | 24.0 | 63.4 | |
| 12 | 27.5 | 16.6 | 69.5 | |
| 13 | 6.4 | 2.2 | 84.9 | |
| 14 | 23.4 | 10.1 | 42.4 | |
| 15 | 8.2 | 11.0 | 92.8 | |
| 16 | 15.0 | 14.3 | 93.7 | |
| 17 | 9.4 | 8.8 | 84.2 | |
| 18 | 7.6 | 9.5 | 93.3 | |
| 19 | 8.6 | 8.1 | 87.4 | |
| 20 | 5.3 | 4.5 | 95.5 | |
| 21 | 5.5 | 4.5 | 99.5 | |
| 22 | 5.8 | 3.4 | 90.1 | |
Additional characterization of F174C:S176C mutations
Based on the liposome incorporation results, several engineered Fabs would be appropriate for use as targeted nanoparticle therapies. We thus further characterized the Fabs with the fewest mutations (Fab 7/Fab 13), HC: F174C, C233S LC: S176C, C214S), chosen to minimize potential immunogenicity. In addition to the Fab previously generated using the sequence of the anti-EphA2 scFv 310,25 we also generated Fabs with either wild-type or alternative disulfide pairing with the sequence of P1X, an anti-epidermal growth factor receptor (EGFR) monoclonal antibody and a component of the oligoclonal EGFR inhibitor, MM-151.26,27 We measured the melting temperature and conducted accelerated stability studies (Table 4 and Supplemental Figure S3). Altering the disulfide bond location did not appear to affect the overall stability; all measurements appear similar, except slightly higher aggregation at 60°C for the P1X Fab7.
Table 4.
Stability analysis of Fabs, as measured by % aggregation using SEC
| Tm (°C) | One week 4°C (% change) |
One week 37 °C (% change) | 30 min 60 °C (% change) | |
|---|---|---|---|---|
| Fab 11 | 80.6 | 0.1 | 9.2 | 0.7 |
| Fab 13 | 80.6 | 0.2 | 6.3 | 0.8 |
| P1X_ wild-type DiS | 77.5 | 0.3 | 8.4 | 0.4 |
| P1X (HC: F174C, C233S LC: S176C, C214S) | 76.0 | 0.2 | 8.0 | 2.0 |
Finally, we tested these antibodies in cell-based assays to confirm that the alternative disulfide pair did not affect its ability to induce uptake of the liposomes into cells (Figure 5). MM-310 (EphA2) Fab7, P1X (EGFR) Fab7, and MM-302 (ErbB2) Fab 7 were tested for their ability to induce uptake of corresponding immunoliposomes in three cell lines: A549 (EGFR+, EphA2+, ErbB2-), OVCAR-3 (EGFR+, EphA2+, ErbB2+), and KYSE-410 (EGFR+, EphA2+, ErbB2+). All Fabs could promote significant uptake in multiple cell lines (35-to-481-fold) over nontargeted liposome controls, suggesting that the alternative disulfide pairing is not affecting its ability to actively target immunoliposomes to cancer cell lines overexpressing the receptors.
Figure 5.
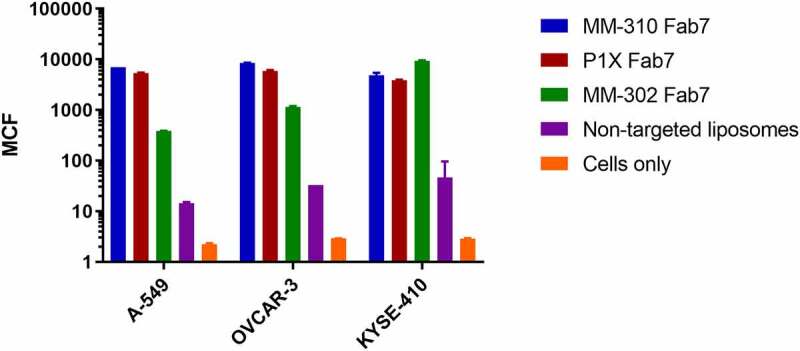
Uptake and internalization of liposomes by Fab variants on various cell lines. Fabs were evaluated for the ability to cause cellular uptake of liposomes in three cell lines using the CLIA high-throughput assay. A549 is Erb2-, EGFR+ and EphA2+, OVCAR-3 is ErbB2+, EGFR+, and EphA2+, and KSYE-410 is also ErbB2+, EGFR+, and EphA2 + .
Discussion
Antibody-directed nanotherapeutics (ADNs), such as immunoliposomes containing DNA, RNA, or small-molecules, provide a unique platform for selectively delivering therapeutic payloads to sites of disease, improving their therapeutic index.2,5,27 They offer a variety of advantages over conventional ADCs, including increased versatility resulting from improved protection of the payload from degradation, to the significantly higher drug loading capacity (10,000 vs 2–8 drugs/mAb) and a second layer of targeting due to the relatively large size of the nanoparticle and its restricted distribution in healthy tissues.28,29
Other differences between the two platforms include the format of the antibody or targeting ligand, and the conjugation strategy. For conventional ADCs, the antibodies typically used are in the full IgG format to take advantage of the FcRn-mediated recycling, and thus longer retention in the circulation, while with ADNs the carefully engineered particle itself provides for the protracted pharmacokinetics, and antibody fragments, such as Fabs or scFv, are routinely used.3,5,6,12 In fact, with ADNs the conjugation of full IgG molecules can result in increased clearance by the liver.1 ADNs hold substantial promise as agents that can improve the delivery and therapeutic index of both small molecule and nucleic acid-based therapeutics.28,30–32 A high Tm, small unilamellar liposome drug carrier platform has been well established for this purpose due to good drug retention properties, low clearance from the circulation and certain selectivity to tumor tissues due to differential vascular permeability and retention (EPR effect).29,31 Liposome nanotherapeutics also afford micellar post-insertion, a versatile and robust “click” method, to be used for attachment of targeting ligands (including proteins) to the particles.7,33 The antibody-targeting ligands are convenient and useful for modulating the internalization and overall microdistribution of the nanoparticle at the site of disease.5,27 Typically, construction of the final ADN on the high-Tm liposome drug carrier platform using membrane post-insertion method requires a high temperature post-insertion or membrane capture step (60–65°C) to incorporate the targeting ligand efficiently,7,12,33 raising the opportunity for denaturation and inactivation of the antibody during the process. Identifying antibody fragments with the requisite stability thus becomes imperative. Maintaining the ability to not only bind to target cells, but also induce internalization in order to enable intracellular processing and drug release, is essential.2,27,34
In addition to immunoliposomes, lipid nanoparticles (LNPs) or polymers delivering nucleic acids have provided for efficient delivery and expression of proteins. Indeed, the current SARS-COV-2 vaccines, mRNA-1273 from Moderna and BNT162b2 from BioNtech, use LNPs to enable highly efficient mRNA vaccines in the treatment of SARS-CoV-2.35–37 Although many of these LNPs are currently nontargeted outside of the tropism provided by the nanoparticle itself, their specificity toward specific cell types can be increased dramatically through a combination of particle surface engineering to reduced nonspecific cell association and the conjugation of targeting ligands, such as antibody fragments. We have previously shown that HER2-targeted LNP could result in a 850-fold increase in gene expression in HER2-overexpressing cancer cells for anionic pegylated LNPs and 167-fold increase for neutral pegylated LNPs.38 More recent studies have conjugated antibody fragments against CD3 or CD5 to polymeric nanoparticles39,40 or LNPs41 in an effort to reprogram immune cells for next-generation CAR-T therapies. The engineering of Fab’ molecules in a format that would allow them to be efficiently conjugated to these various nanoparticles is critical to their successful commercial development.
We have described here the optimization of the Fab format to bury stabilizing disulfides in the interior of the fragment, away from the C-terminal conjugation site introduced for covalent attachment to immunoliposomes through a maleimide-terminated PEG-distearoylphosphatidyl-ethanolamine lipopolymer anchor. Placement of the disulfide bond on the interior of the fragment should also minimize potential immunogenicity. The approach yields a lower level of over-reduction of the Fab during the initial reduction step of the process, and lower molecular weight conjugated species, while maintaining the ability to bind and induce cell uptake of the conjugated immunoliposomes. Next steps for this platform would include testing the alternative disulfide pairing at larger scales for additional CMC assessments, as well as testing on a larger panel of antibody sequences. This significant advancement will broaden the pool of developable antibodies by allowing more routine use of Fabs as targeting ligands to a variety of cellular targets where an antibody is available.
Competing interests
All authors were employees of Merrimack Pharmaceuticals at the time the research was conducted. MR and DCD also own Merrimack stock at the time of publication.
Materials and methods
Design of thermostable Fabs
Constructs were synthesized and subcloned into a pCEP4 mammalian expression vector (Invitrogen). The IgG1 Fab constructs were engineered to include the heavy chain C-terminal sequence DKTHTCAA. The IgG2 Fab constructs (Fab 3 and Fab 4) were engineered to include the heavy chain C-terminal sequence ERKCAA. The IgG4 Fab constructs (Fab 9 and Fab 10) were engineered to have the heavy chain C-terminal sequence ESKYGCAA.
All Fab constructs were transiently expressed using the 293 F system (Invitrogen®). Cells were grown to 600 mL using F17 media supplemented with 4 mM L-glutamine and 0.1% Pluronic® F-68 (BASF®) in 5% CO2 to a density of 1.7 million cells/mL in a 2 L flask, and then transfected with 1 µg of DNA and 2.5 µg high molecular weight polyethyleneimine/mL of cells. After six days, the proteins were harvested by centrifuging the cells at 4000x g and filtered using a 0.22 µm filter.
The filtered supernatant was incubated with CaptureSelect IgG1-CH1 affinity matrix (Life Technologies) for 1 hour at room temperature with agitation. The slurry was filtered, poured into a column, and equilibrated with phosphate-buffered saline (PBS). The bound protein was eluted with 100 mM glycine pH 3.0, neutralized with 1 M Tris to a pH of 5.5, and filtered with a 0.2 μm filter. Purified proteins were then analyzed using SDS-PAGE analysis.
Thermostability analysis using differential scanning fluorescence
Purified Fabs were analyzed to determine their melting temperatures. Melting temperatures were determined by differential scanning fluorescence. For Fabs 1–Fab 14, 10 µM of protein and 1X Sypro Orange (Life Technologies) in 1X PBS was mixed to a final volume of 25 µL and heated from 20°C to 90°C at a rate of 1°C/min using the IQ5 real-time detection system (Bio-Rad). For Fab 15 – Fab 19, 10 µM of protein and 1X of Protein Thermal Shift Buffer and Dye (Life Technologies) was mixed to a final volume of 20 μL and heated from 25°C to 99°C at a rate of 3°C/min using the ViiA7 real-time detection system (Life Technologies). The melting temperature reported is the temperature of the maximum value of the first derivative.
Reduction of Fabs with cysteine
To prepare purified Fabs for conjugation with mal-PEG-DSPE (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethylene glycol)]), Fabs in solution in 0.1 M glycine-HCl or 10 mM citrate, pH adjusted to about 6.0 with Tris-base, were concentrated on a YM-10 diafiltration membrane (Amicon) to about 4–5 mg/mL of the protein. Reduction/activation of the C-terminal cysteine present in the heavy-chain sequences of each Fab was performed by adding ethylenediaminetetraacetic acid (EDTA) to 5 mM and cysteine hydrochloride, pH 5.7 (adjusted with 1 M trisodium citrate) to 15 mM, followed by incubation at 30°C for 1 hour. The solution was passed through a SEPHADEX G-25 (PD-10) column to exchange the protein into conjugation buffer (5 mM citrate, 1 mM EDTA, 140 mM NaCl, pH 6.0). Aliquots of the resulting protein solution were diluted with conjugation buffer, typically 5–10-fold, to a volume of 0.9 mL, mixed with 0.1 mL of 1 M HEPES-Na buffer pH 7.3, and 0.01 mL of 20 mM, 5,5’-dithiobis(2-nitrobenzoic acid) (“Ellman’s reagent”) in dimethyl sulfoxide. Five to 10 minutes after mixing, the absorbance of the solution was measured at 412 nm against a protein-free blank. Concentration of reactive thiol groups was calculated using the molar extinction value of 12,500 L/mol/cm and normalized to the molar concentration of the protein (A280 = 1 molecular weight of kDa) determined by UV spectrophotometry at 280 nm using molar extinction coefficient calculated from the protein’s amino acid sequence (about 1.43) to give SH/protein ratio.
Conjugation of Fabs with mal-PEG-DSPE
Reduced Fabs were conjugated to mal-PEG-DSPE linker in the following way. First, mal-PEG-DSPE (PEG mol. weight 2000, NOF Corp., Japan) and methoxy-PEG-DSPE (PEG mol. weight 2000, Avanti Polar Lipids, USA) were co-dissolved in distilled water, acidified with citric acid to pH 5.7, at a concentration of 10 mg/mL each. The solution was briefly heated to 60°C to effect the formation of mixed micelles containing thiol-reactive and nonreactive PEG-DSPE derivative. Then, the linker solution was added to 1 mL of the reduced protein solution in the conjugation buffer to achieve the mass ratio of the active (mal-PEG-DSPE) linker to the protein of 0.23, and the conjugation mix was stirred at room temperature for about 4 hours. The reaction was stopped by quenching unreacted maleimide groups with 0.5 mM cysteine for 5–10 min, and, after analytical sampling, the mix was applied on a gravity-fed chromatography column with Ultrogel AcA 34 (Sigma Chemical Co, USA), bed volume 17 mL, equilibrated with the conjugate storage buffer (10% w/v sucrose, 10 mM citrate-Na, pH 6.5).
The column was eluted with the same buffer, 0.5-mL fractions were collected, and the protein concentration was determined by spectrophotometry at 280 nm using the same extinction coefficients as for the unconjugated Fabs. Due to micellar character of the Fab-PEG-DSPE conjugate in aqueous solution,7 the conjugate appeared in the fractions near the column void volume (first peak). These fractions were combined and passed through a 0.2-µm polyethersulfone syringe filter to give the purified conjugate. The second (smaller) protein peak, containing unconjugated protein, was detected and sampled for analysis.
Insertion of Fab’ conjugates into liposomes
Liposomes of HSPC-Cholesterol-methoxyPEG(2000)DSPE (3:2:0.3 molar ratio) with an average size of 91 nm (PdI = 0.06) were loaded with doxorubicin hydrochloride at the drug/liposome ratio of 0.13 g/mol phospholipid using ammonium sulfate gradient method (0.25 M ammonium sulfate) essentially as described by Martin.42 The lipids of the liposome were quantified by phosphate assay following acid digestion.43
A solution of Fab-PEG-DSPE [PEG (2000)] conjugate in 10% sucrose-10 mM citrate buffer pH 6.5 was added to a suspension of liposomes in 10% sucrose, 10 mM histidine buffer pH 6.5, along with extra sucrose-citrate buffer to achieve concentrations of 0.16 mg/mL of the Fab and 8 mM of the liposome phospholipid (Fab/liposome ratio of 20 g protein/mol of phospholipid, or about 30 Fab molecules/liposome). The mixture was quickly heated to 60°C and maintained at this temperature for 30 minutes with stirring. Then the mixture was chilled on ice, and the liposomes with membrane-inserted Fab-PEG-DSPE conjugates were separated from the non-inserted conjugate and extra-liposomal drug by size-exclusion chromatography on a Sepharose CL-4B column, eluted with 144 mM NaCl-5 mM HEPES buffer pH 6.5 as described previously.11 The chromatography showed practically no leakage of the drug from the liposomes during the incubation, as judged by the absence of any visually detectable chromatographic band corresponding to free doxorubicin.
Aliquots of the liposomes containing known amounts of phospholipid were solubilized in SDS-PAGE running buffer and separated by SDS-PAGE on the NuPage BT 4–12% gel (Life Technologies). The gels were stained with SimplyBlue Coomassie, and the bands were quantified by densitometry using concurrently run dilutions of bovine serum albumin (Pierce) as standards. Any protein on the gel that was higher or lower molecular weight species than predicted for the conjugate was classified as non-product bands, and the percentage was calculated by comparing it to the density of the correct product band.
Determination of EphA2 binding of Fab-PEG-DSPE conjugates and corresponding immunoliposomes
The Fab-PEG-DSPE conjugates were assayed for EphA2 binding strength using the ForteBIO system to determine whether conjugation or engineering of the Fab affected binding activity. Anti-His5 sensors were first coated with his-tagged recombinant, human EphA at a concentration of 10 µg/mL protein in PBS. The sensors were then incubated in 4 µg/mL of Fab-PEG-DSPE conjugate in PBS. The slope of an association curve between 2 and 10 sec was determined and compared across the variants and to the reference conjugate, Fab 11-PEG-PE, which is the anti-EphA2 antibody with wild-type disulfide pairing.
The purified immunoliposomes were assayed for EphA2 binding strength by ForteBIO Octet Red96 system (Pall) in PBS at 25 µM liposome phospholipid using anti-His5 sensors coated with recombinant human EphA2 with C-terminal hexahistidine. The slope of the association curve from 3 to 20 sec was determined and compared to the slope observed for liposomes with inserted conjugate of the wild-type Fab.
Cell Uptake of Fab-targeted immunoliposomes
Three cell lines (A-549, OVCAR-3, and KYSE-410) plated at a density of 100,000 cells/well overnight and were subsequently incubated with fluorescently labeled (DiIC18(5)-DS) liposomes at a concentration of 25 µM phospholipid at 37°C for 4 hours. The cells were then washed twice with 200 µL of PBS and analyzed using FACS (FL4) to measure the amount of signal associated with the cells. Untreated cells and cells incubated with nontargeted liposomes (i.e., no antibody conjugate included) were included as controls.
Engineered forms of additional antibodies
Fab versions of the anti-EGFR antibody P1X 27 and anti-ErbB2 antibody F512 having the same constant regions as the wild-type Fab or Fab 7 were engineered and expressed essentially as described above for the EphA2 Fab.
Size exclusion chromatography
Sample (50 mg) was injected on a TSKgel SuperSW3000 column using 50 mM sodium phosphate, 400 mM NaClO4, pH 7.0, as running buffer. The run time was 20 min. All measurements were performed on Agilent 1100 HPLC equipped with an auto sampler, binary pump, and diode array detector. Data were analyzed using Chemstation software.
SDS-PAGE
SDS-PAGE was performed using 10 ug of protein on Bio-Rad Mini-PROTEAN TGX gels and Coomassie G-250 staining according to the supplier’s protocol.
Accelerated stability studies
Experiments were conducted with antibodies buffered with 0.1 M citric acid, 50 mM Tris, pH 6.0. Antibodies were incubated at °C or 37°C for 1 week, or for 30 minutes at 60°C. Antibodies were then measured for aggregation using size-exclusion chromatography as described above.
Supplementary Material
Funding Statement
The author(s) reported there is no funding associated with the work featured in this article.
Abbreviations
ADN, antibody-directed nanotherapeutics; CLIA, chelated ligand-induced internalization assay; EphA2, ephrin type-A receptor 2; FACS, fluorescence-activated cell sorting; DSF, differential scanning fluorimetry; HTP, high throughput; scFv, single-chain variable fragment
Disclosure statement
MLG, DBK, NK, TK, BB, MR, DCD, and AAL were all employees of Merrimack Pharmaceuticals at the time the work was completed. MR and DCD currently hold stock in Merrimack Pharmaceuticals.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19420862.2022.2083466
References
- 1.Harding JA, Engbers CM, Newman MS, Goldstein NI, Zalipsky S.. Immunogenicity and pharmacokinetic attributes of poly(ethylene glycol)-grafted immunoliposomes. Biochim Biophys Acta. 1997;1327(2):181–10. doi: 10.1016/S0005-2736(97)00056-4. [DOI] [PubMed] [Google Scholar]
- 2.Noble CO, Kirpotin DB, Hayes ME, Mamot C, Hong K, Park JW, Benz CC, Marks JD, Drummond DC. Development of ligand-targeted liposomes for cancer therapy. Expert Opin Ther Targets. 2004;8(4):335–53. doi: 10.1517/14728222.8.4.335. [DOI] [PubMed] [Google Scholar]
- 3.Cheng WW, Allen TM. The use of single chain Fv as targeting agents for immunoliposomes: an update on immunoliposomal drugs for cancer treatment. Expert Opin Drug Deliv. 2010;7(4):461–78. doi: 10.1517/17425240903579963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheng WW, Das D, Suresh M, Allen TM. Expression and purification of two anti-CD19 single chain Fv fragments for targeting of liposomes to CD19-expressing cells. Biochim Biophys Acta. 2007;1768(1):21–29. doi: 10.1016/j.bbamem.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 5.Kamoun WS, Kirpotin DB, Huang ZR, Tipparaju SK, Noble CO, Hayes ME, Luus L, Koshkaryev A, Kim J, Olivier K, et al. Antitumour activity and tolerability of an EphA2-targeted nanotherapeutic in multiple mouse models. Nat Biomed Eng. 2019;3(4):264–80. doi: 10.1038/s41551-019-0385-4. [DOI] [PubMed] [Google Scholar]
- 6.Mamot C, Drummond DC, Greiser U, Hong K, Kirpotin DB, Marks JD, Park JW. Epidermal growth factor receptor (EGFR)-targeted immunoliposomes mediate specific and efficient drug delivery to EGFR- and EGFRvIII-overexpressing tumor cells. Cancer Res. 2003;63:3154–61. [PubMed] [Google Scholar]
- 7.Nellis DF, Giardina SL, Janini GM, Shenoy SR, Marks JD, Tsai R, Drummond DC, Hong K, Park JW, Ouellette TF, et al. Preclinical manufacture of anti-HER2 liposome-inserting, scFv-PEG-lipid conjugate. 2. Conjugate micelle identity, purity, stability, and potency analysis. Biotechnol Prog. 2005;21(1):221–32. doi: 10.1021/bp049839z. [DOI] [PubMed] [Google Scholar]
- 8.Quintero-Hernandez V, Juarez-Gonzalez VR, Ortiz-Leon M, Sanchez R, Possani LD, Becerril B. The change of the scFv into the Fab format improves the stability and in vivo toxin neutralization capacity of recombinant antibodies. Mol Immunol. 2007;44(6):1307–15. doi: 10.1016/j.molimm.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 9.Rothlisberger D, Honegger A, Pluckthun A. Domain interactions in the Fab fragment: a comparative evaluation of the single-chain Fv and Fab format engineered with variable domains of different stability. J Mol Biol. 2005;347(4):773–89. doi: 10.1016/j.jmb.2005.01.053. [DOI] [PubMed] [Google Scholar]
- 10.Kirpotin D, Park JW, Hong K, Zalipsky S, Li WL, Carter P, Benz CC, Papahadjopoulos D. Sterically Stabilized Anti-HER2 Immunoliposomes: design and Targeting to Human Breast Cancer Cells in Vitro. Biochemistry. 1997;36(1):66–75. doi: 10.1021/bi962148u. [DOI] [PubMed] [Google Scholar]
- 11.Kirpotin DB, Noble CO, Hayes ME, Huang Z, Kornaga T, Zhou Y, et al. Building and characterizing antibody-targeted lipidic nanotherapeutics. Methods Enzymol. 2012;502:139–66. [DOI] [PubMed] [Google Scholar]
- 12.Nellis DF, Ekstrom DL, Kirpotin DB, Zhu J, Andersson R, Broadt TL, Ouellette TF, Perkins SC, Roach JM, Drummond DC, et al. Preclinical manufacture of an anti-HER2 scFv-PEG-DSPE, liposome-inserting conjugate. 1. Gram-scale production and purification. Biotechnol Prog. 2005;21(1):205–20. doi: 10.1021/bp049840y. [DOI] [PubMed] [Google Scholar]
- 13.Brinkmann U, Reiter Y, Jung SH, Lee B, Pastan I. A recombinant immunotoxin containing a disulfide-stabilized Fv fragment. Proc Natl Acad Sci U S A. 1993;90(16):7538–42. doi: 10.1073/pnas.90.16.7538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakamura H, Oda-Ueda N, Ueda T, Ohkuri T. A novel engineered interchain disulfide bond in the constant region enhances the thermostability of Adalimumab Fab. Biochem Biophys Res Commun. 2018;495(1):7–11. doi: 10.1016/j.bbrc.2017.10.140. [DOI] [PubMed] [Google Scholar]
- 15.Hagihara Y, Saerens D. Engineering disulfide bonds within an antibody. Biochim Biophys Acta. 2014;1844(11):2016–23. doi: 10.1016/j.bbapap.2014.07.005. [DOI] [PubMed] [Google Scholar]
- 16.Mazor Y, Oganesyan V, Yang C, Hansen A, Wang J, Liu H, Sachsenmeier K, Carlson M, Gadre DV, Borrok MJ, et al. Improving target cell specificity using a novel monovalent bispecific IgG design. MAbs. 2015;7(2):377–89. doi: 10.1080/19420862.2015.1007816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vaks L, Litvak-Greenfeld D, Dror S, Shefet-Carasso L, Matatov G, Nahary L, Shapira S, Hakim R, Alroy I, Benhar I, et al. Design principles for bispecific IgGs. Opportunities and Pitfalls of Artificial Disulfide Bonds. Antibodies (Basel). 2018;7:27. doi: 10.3390/antib7030027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pillow TH, Tien J, Parsons-Reponte KL, Bhakta S, Li H, Staben LR, Li G, Chuh J, Fourie-O’Donohue A, Darwish M, et al. Site-specific trastuzumab maytansinoid antibody-drug conjugates with improved therapeutic activity through linker and antibody engineering. J Med Chem. 2014;57(19):7890–99. doi: 10.1021/jm500552c. [DOI] [PubMed] [Google Scholar]
- 19.Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov. 2017;16(5):315–37. doi: 10.1038/nrd.2016.268. [DOI] [PubMed] [Google Scholar]
- 20.Jeffrey SC, Burke PJ, Lyon RP, Meyer DW, Sussman D, Anderson M, Hunter JH, Leiske CI, Miyamoto JB, Nicholas ND, et al. A potent anti-CD70 antibody-drug conjugate combining a dimeric pyrrolobenzodiazepine drug with site-specific conjugation technology. Bioconjug Chem. 2013;24(7):1256–63. doi: 10.1021/bc400217g. [DOI] [PubMed] [Google Scholar]
- 21.Junutula JR, Raab H, Clark S, Bhakta S, Leipold DD, Weir S, Chen Y, Simpson M, Tsai SP, Dennis MS, et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol. 2008;26(8):925–32. doi: 10.1038/nbt.1480. [DOI] [PubMed] [Google Scholar]
- 22.Tumey LN, Li F, Rago B, Han X, Loganzo F, Musto S, Graziani EI, Puthenveetil S, Casavant J, Marquette K, et al. Site selection: a case study in the identification of optimal cysteine engineered antibody drug conjugates. Aaps j. 2017;19(4):1123–35. doi: 10.1208/s12248-017-0083-7. [DOI] [PubMed] [Google Scholar]
- 23.Hallam TJ, Wold E, Wahl A, Smider VV. Antibody conjugates with unnatural amino acids. Mol Pharm. 2015;12(6):1848–62. doi: 10.1021/acs.molpharmaceut.5b00082. [DOI] [PubMed] [Google Scholar]
- 24.Jacobsen FW, Stevenson R, Li C, Salimi-Moosavi H, Liu L, Wen J, Luo Q, Daris K, Buck L, Miller S, et al. Engineering an IgG scaffold lacking effector function with optimized developability. J Biol Chem. 2017;292(5):1865–75. doi: 10.1074/jbc.M116.748525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Geddie ML, Kohli N, Kirpotin DB, Razlog M, Jiao Y, Kornaga T, Rennard R, Xu L, Schoerberl B, Marks JD, et al. Improving the developability of an anti-EphA2 single-chain variable fragment for nanoparticle targeting. MAbs. 2017;9(1):58–67. doi: 10.1080/19420862.2016.1259047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kearns JD, Bukhalid R, Sevecka M, Tan G, Gerami-Moayed N, Werner SL, Kohli N, Burenkova O, Sloss CM, King AM, et al. Enhanced targeting of the EGFR network with MM-151, an oligoclonal Anti-EGFR antibody therapeutic. Mol Cancer Ther. 2015;14(7):1625–36. doi: 10.1158/1535-7163.MCT-14-0772. [DOI] [PubMed] [Google Scholar]
- 27.Kirpotin DB, Drummond DC, Shao Y, Shalaby MR, Hong K, Nielsen UB, Marks JD, Benz CC, Park JW. Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models. Cancer Res. 2006;66(13):6732–40. doi: 10.1158/0008-5472.CAN-05-4199. [DOI] [PubMed] [Google Scholar]
- 28.Drummond DC, Noble CO, Hayes ME, Park JW, Kirpotin DB. Pharmacokinetics and in vivo drug release rates in liposomal nanocarrier development. J Pharm Sci. 2008;97(11):4696–740. doi: 10.1002/jps.21358. [DOI] [PubMed] [Google Scholar]
- 29.Maeda H. Toward a full understanding of the EPR effect in primary and metastatic tumors as well as issues related to its heterogeneity. Adv Drug Deliv Rev. 2015;91:3–6. doi: 10.1016/j.addr.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 30.Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Deliv Rev. 2013;65(1):36–48. doi: 10.1016/j.addr.2012.09.037. [DOI] [PubMed] [Google Scholar]
- 31.Drummond DC, Meyer O, Hong K, Kirpotin DB, Papahadjopoulos D. Optimizing liposomes for delivery of chemotherapeutic agents to solid tumors. Pharmacol Rev. 1999;51:691–743. [PubMed] [Google Scholar]
- 32.Setten RL, Rossi JJ, Han SP. The current state and future directions of RNAi-based therapeutics. Nat Rev Drug Discov. 2019;18(6):421–46. doi: 10.1038/s41573-019-0017-4. [DOI] [PubMed] [Google Scholar]
- 33.Iden DL, Allen TM. In vitro and in vivo comparison of immunoliposomes made by conventional coupling techniques with those made by a new post-insertion approach. Biochim Biophys Acta. 2001;1513(2):207–16. doi: 10.1016/S0005-2736(01)00357-1. [DOI] [PubMed] [Google Scholar]
- 34.Sapra P, Allen TM. Internalizing antibodies are necessary for improved therapeutic efficacy of antibody-targeted liposomal drugs. Cancer Res. 2002;62:7190–94. [PubMed] [Google Scholar]
- 35.Baden LR, El Sahly HM, Essink B, Kotloff K, Frey S, Novak R, Diemert D, Spector SA, Rouphael N, Creech CB, et al. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N Engl J Med. 2021;384(5):403–16. doi: 10.1056/NEJMoa2035389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu J, Liu Y, Xia H, Zou J, Weaver SC, Swanson KA, Cai H, Cutler M, Cooper D, Muik A, et al. BNT162b2-elicited neutralization of B.1.617 and other SARS-CoV-2 variants. Nature. 2021;596(7871):273–75. doi: 10.1038/s41586-021-03693-y. [DOI] [PubMed] [Google Scholar]
- 37.Polack FP, Thomas SJ, Kitchin N, Absalon J, Gurtman A, Lockhart S, Perez JL, Pérez Marc G, Moreira ED, Zerbini C, et al. Safety and efficacy of the BNT162b2 mRNA covid-19 vaccine. N Engl J Med. 2020;383(27):2603–15. doi: 10.1056/NEJMoa2034577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hayes ME, Drummond DC, Hong K, Zheng WW, Khorosheva VA, Cohen JA, Park JW, Marks JD, Benz CC, Kirpotin DB, et al. Increased target specificity of anti-HER2 genospheres by modification of surface charge and degree of PEGylation. Mol Pharm. 2006;3(6):726–36. doi: 10.1021/mp060040v. [DOI] [PubMed] [Google Scholar]
- 39.Parayath NN, Stephan SB, Koehne AL, Nelson PS, Stephan MT. In vitro-transcribed antigen receptor mRNA nanocarriers for transient expression in circulating T cells in vivo. Nat Commun. 2020;11(1):6080. doi: 10.1038/s41467-020-19486-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith TT, Stephan SB, Moffett HF, McKnight LE, Ji W, Reiman D, Bonagofski E, Wohlfahrt ME, Pillai SPS, Stephan MT, et al. In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nat Nanotechnol. 2017;12(8):813–20. doi: 10.1038/nnano.2017.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rurik JG, Tombácz I, Yadegari A, Méndez Fernández PO, Shewale SV, Li L, Kimura T, Soliman OY, Papp TE, Tam YK, et al. CAR T cells produced in vivo to treat cardiac injury. Science. 2022;375(6576):91–96. doi: 10.1126/science.abm0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martin F. Case study: DOXIL, the development of pegylated liposomal doxorubicin. In: Burgess D, editor. Injectable dispersed systems: formulation, processing, and performance. New York (NY): Informa Healthcare; 2007. p. 427–80. [Google Scholar]
- 43.Morrison WR. A fast, simple and reliable method for the microdetermination of phosphorus in biological materials. Anal Biochem. 1964;7(2):218–24. doi: 10.1016/0003-2697(64)90231-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.