

Abstract
New quinazoline derivatives were designed based on the structural modification of the reported inhibitors to enhance their selectivity toward Aurora A. The synthesized compounds were tested over Aurora A, and a cytotoxicity assay was performed over NCI cell lines to select the best candidate for further evaluation. Compound 6e (2-(3-bromophenyl)-8-fluoroquinazoline-4-carboxylic acid) was the most potent compound among the tested derivatives. A Kinase panel assay was conducted for compound 6e over 14 kinases to evaluate its selectivity profile. Further cell cycle and apoptosis analysis were evaluated for compound 6e over the MCF-7 cell line at its IC50 of 168.78 µM. It arrested the cell cycle at the G1 phase and induced apoptosis. Molecular docking was performed to explore the possible binding mode of compound 6e into the active site. It showed significant binding into the main pocket in addition to potential binding interactions with the key amino acid residues. Accordingly, compound 6e can be considered a potential lead for further structural and molecular optimization of the quinazoline-based carboxylic acid scaffold for Aurora A kinase selective inhibition with apoptosis properties.
Keywords: quinazoline, Aurora A, kinases, anticancer, molecular docking, cell cycle, apoptosis
1. Introduction
Cancer is considered the second most common cause of death worldwide and is characterized by uncontrolled cell proliferation [1,2]. Several kinases have been found to have a key role in cell growth and proliferation [3]. Aurora kinases (Aurora A, B and C) play a significant role in cell cycle regulation and mitotic spindle development. Aurora kinase is a class of serine/threonine kinase that regulates cell division. These kinases are regulatory proteins involved in centrosome maturation, chromosome alignment, chromosome segregation and cytokinesis [4]. Aurora kinase dysfunction may lead to critical, unstable chromosome content and may lead to cancer disease [5,6]. Aurora A kinase is a member of the Aurora kinase family that is extensively overexpressed in human malignancies [7]. As a result, Aurora A kinase has been proposed as a promising anticancer target for human cancer treatment. Inhibition of protein kinases via the ATP-binding site (adenosine triphosphate) has emerged as a promising route for treating cancer [1]. Aurora kinases have sparked a lot of interest as anticancer targets throughout recent decades [8,9,10]. The discovery of a large number of small-molecule inhibitors of Aurora kinases, the majority of which inhibit both isoforms A and B, led to the discovery of Aurora kinase modulators as cancer-treating agents. One example of a non-selective Aurora A and B inhibitor is a quinazoline-based derivative (AZD-1152, I) with different binding motifs around the central core [11,12]. However, a recent study found that Aurora B suppression is linked to a number of side effects, including the production of unstable polyploid cells, which could stimulate carcinogenesis [13]. Therefore, the selective inhibition of Aurora A over Aurora B isoform has been considered an attractive anticancer target for the treatment of human cancers [7]. A number of Aurora A kinase inhibitors have been developed (phthalazine-based derivative, II [14] and quinazoline-based derivatives, III [3], which showed a selective profile towards Aurora A over Aurora B isoform (Figure 1).
Figure 1.

Reported Aurora A inhibitors.
It was noticed that the selective inhibitors (II and III) shared a similarly sized fused bicyclic core scaffold with two nitrogen hetero atoms (HBAs, hydrogen bond acceptors) with the non-selective inhibitor (AZD-1152, I). However, the selective agents (II and III) showed structural simplification around the central core compared to non-selective inhibitors (AZD-1152, I). It was suggested that the binding active site of Aurora A kinase has limited-sized access compared to that of Aurora B kinase. As a result, the small-sized derivatives may accommodate the Aurora A active site with appropriate binding forces. Therefore, our research group was interested in investigating the Aurora A inhibitory activity of small-sized substitution-containing quinazoline central scaffolds (5b, 6a–e and 7a,b).
In addition, we designed a series of compounds with differing sized halogen atoms that can afford additional binding(s) to the key amino acids inside the active site of Aurora A kinase through halogen interaction. Furthermore, the terminal free carboxylic group was incorporated in the designed derivatives to be a potential solvent-exposed binding group (6a–e). Additionally, a group of ester-based derivatives (5b and 7a,b) were designed in order to enhance the t-PSA (topological polar surface area) of these derivatives to investigate the effect of enhanced t-PSA on both the inhibitory activity as well as the cellular induction through the lipophilic membrane (Figure 2). In vitro screening (including kinase inhibitory assay, cytotoxic assay, cell cycle arrest analysis and apoptosis assay) and in silico molecular docking screening were conducted on the designed compounds to evaluate the inhibitory activity as well as the selectivity profile.
Figure 2.

Rational design of synthesized compounds. (A) Structural simplification was applied to the quinazoline-based scaffold (I, AZD-1152) to enhance the selectivity profile towards Aurora A over Aurora B kinase enzyme. Both terminal long aliphatic chain and amide spacer were omitted. In addition, the central fused six-membered ring was replaced with its ring isostere (phthalazine ring). Terminal phenyl ring was directly attached to the central scaffold; (B) structural modification was designed to introduce the quinazoline ring as a core central scaffold. An additional halogen group (Cl) was introduced into the quinazoline ring; (C) the current derivatives (5b, 6a–e and 7a,b) were designed to keep the quinazoline ring as the main core scaffold. A small halogen group (F) was introduced to the quinazoline ring to explore any additional binding to the key amino acid residues in the active site of Aurora A kinase without any steric hindrance. Additional groups were introduced into the terminal phenyl ring (Me, Cl and Br). Terminal polar groups were introduced to be a potential binding motif for the solvent-exposure area (free carboxylic acid, 6a–e; and ester-based substitution, 5b and 7a,b).
2. Results and Discussion
2.1. Chemistry
The final target compounds (5b, 6a–e and 7a,b) were synthesized as depicted in Scheme 1 and Scheme 2. Benzanilide derivatives (3a–e) were afforded through the Schotten–Baumann reaction [14]. Where appropriate, strong electrophilic acyl chlorides (2a,b) were coupled with selected primary amines (1a,b) in the presence of triethyl amine as a base. Compounds 3a–e were refluxed with thionyl chloride to afford N-phenylbenzimidoyl chloride derivatives (4a–e). The cyclization of compounds 4a–e was achieved by heating with ethyl cyanoformate in the presence of tin tetrachloride to afford compounds 5a–e. Final target free carboxylic acid derivatives (6a–e) were afforded by basic hydrolysis of corresponding ethyl esters (5a–e). Ester-based derivatives (7a,b) were prepared by Fischer esterification of free carboxylic acid derivative (6b) with appropriate alcohols in the presence of a catalytic amount of sulfuric acid (Table 1).
Scheme 1.

Reagents and conditions. (a) Et3N, DCM, 0 °C, 4 h; (b) SOCl2, reflux, 3 h; (c) EtOOC-CN, SnCl4, o-DCB, 140 °C, 15 min; (d) NaOH, EtOH, reflux, 2 h.
Scheme 2.

Reagents and conditions. (e) MeOH, (7a) i-PrOH, (7b) H2SO4, reflux, 18 h.
Table 1.
Key structure of target compounds.
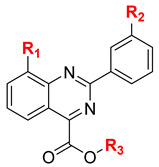
| Compound | R1 | R2 | R3 |
|---|---|---|---|
| 5b | H | Cl | Et |
| 6a | H | H | H |
| 6b | H | Cl | H |
| 6c | F | Me | H |
| 6d | F | H | H |
| 6e | F | Br | H |
| 7a | H | Cl | Me |
| 7b | H | Cl | i-Pr |
2.2. In Vitro Screening
2.2.1. Kinase Inhibitory Assay
An in vitro kinase inhibitory assay was conducted for the free carboxylic acid derivatives (6a–e) at single dose testing (10 µM) against Aurora A kinase enzyme (Table 2). The tested compounds showed different inhibitory activities. The results revealed that the compounds with halogen substitution at the terminal phenyl ring (6b,e) showed higher activities than that of unsubstituted (6a,d) and methyl-substituted derivative (6c). In addition, the fluorine atom in the main quinazoline scaffold possessed an additional inhibitory effect (compounds 6c–e). Compound 6e (bearing F atom in the core quinazoline scaffold and Br group at the terminal phenyl ring) exhibited the most potent inhibitory activity among the tested compounds (6a–e). The results showed that the structural modification that was applied to the reported Aurora A kinase inhibitors (II and II) to afford the current derivatives in this study will retain the inhibitory activity towards Aurora A kinase. The replacement of the terminal pyrazole ring with a free carboxylic group, along with the additional substitution with halogen groups (6e), maintained the ATP competitive binding into the active site of the target kinase (Aurora A). It was suggested that the terminal carboxylic group would provide the same HBD/HBA environment similar to the pyrazole ring, which may result in the same binding interactions with the key amino acid residues in the active site. However, the binding pattern may be altered (which will be discussed in the molecular docking section).
Table 2.
In vitro kinase inhibitory assay (inhibition %) of free carboxylic acid-based derivatives (6a–e) over Aurora A kinase.
| Compound | Aurora A (Inhibition%) |
|---|---|
| 6a | 7.64 ± 1.19 |
| 6b | 28.48 ± 2.40 |
| 6c | 14.83 ± 0.14 |
| 6d | 22.18 ± 1.86 |
| 6e | 51.78 ± 0.38 |
Compound 5b (the ethyl ester precursor of the final free-carboxylic acid derivative 6b) as well as compounds 7a,b were designed, synthesized and tested to investigate the impact of the ester form of the target compounds on the inhibitory activity (Table 3). The results showed that the terminal free carboxylic group is essential for activity as the esterification of this group dropped the activity by five-fold. Compared to the free carboxylic-based derivatives, the masking effect of the free carboxylic group had a deteriorating effect on kinase inhibition (especially compound 7a, which showed negative results). This revealed the crucial role of the free carboxylic group in the ATP competitive binding into the active site. It was suggested that the free carboxylic group share with essential HBD in the interaction with one of the key amino acid residues in the active site. Along with the essential HBD, the additional ethyl (5b), methyl (7a) and isopropyl (7b) groups may contribute to an unfavorable steric effect inside the active site with the core amino acid residue(s).
Table 3.
In vitro kinase inhibitory assay (inhibition%) of ester-based derivatives (5b and 7a,b) over Aurora A kinase.
| Compound | Aurora A (Inhibition%) |
|---|---|
| 5b | 6.53 ± 0.58 |
| 7a | −2.61 ± 0.15 |
| 7b | 3.83 ± 1.38 |
The most active compound (6e) among the tested compounds was selected for further investigation in an in vitro kinase panel assay to explore the selectivity profile of the Aurora A kinase enzyme. Fourteen kinase-related enzymes were selected to conduct the kinase panel assay at single dose testing (10 µM) (Table 4). The results revealed that compound 6e showed poor inhibitory activities against the selected kinases (Activity% > 83%) in contrast to the inhibitory activity against Aurora A kinase enzyme (Activity% = 48.22%).
Table 4.
In vitro kinase inhibitory assay (activity%) of compound 6e over kinase panel.
| Kinase | Compound 6e (Activity%) | Kinase | Compound 6e (Activity%) |
|---|---|---|---|
| ABL-1 | 106.77 | CDK-2/cyclin A | 83.72 |
| AKT-1 | 104.50 | CDK-8/cyclin C | 97.81 |
| ALK | 99.86 | BRAFV599E | 100.92 |
| Aurora A | 48.22 | RAF-1 | 111.64 |
| Aurora B | 92.90 | MAPK APK-2 | 99.55 |
| CAMKK-2 | 83.66 | MEK-1 | 85.42 |
| VRK-2 | 118.72 | mTOR/FRAP-1 | 105.00 |
2.2.2. Cytotoxicity Assay
In vitro cytotoxic screening of target compounds (free carboxylic acid derivatives, 6a–e; and ester-based derivatives, 5b and 7a,b) was performed against NCI (National Cancer Institute) human cancer cell lines using a single dose assay (10 µM) (Table 5 and Tables S1–S8). The results showed that the tested compounds exhibited different cytotoxic activities. Moreover, the human CNS (central nervous system) cancer cell line (SNB-75) was the most sensitive cell line among the tested cell lines. Compound 6e (the most active compound in the in vitro Aurora A kinase inhibitory assay) showed the most potent cytotoxic activities among the tested compounds. In line with their Aurora A kinase inhibitory results, the ester-based compounds (5b and 7a,b) did not show any significant cytotoxic effect in the current study. It was proposed that the high t-PSA of ester-based derivatives failed to improve the cell induction and hence the cytotoxic activity.
Table 5.
In vitro cytotoxicity assay (growth%) of compounds 5b, 6a–e and 7a,b over NCI human CNS cancer cell line (SNB-75).
| Compound | SNB-75 (Growth%) | Compound | SNB-75 (Growth%) |
|---|---|---|---|
| 5b | 75.14 | 6d | 82.18 |
| 6a | 88.17 | 6e | 66.99 |
| 6b | 73.59 | 7a | 80.12 |
| 6c | 79.79 | 7b | 75.14 |
2.2.3. Cell Cycle Assay
In order to explore the effect of compound 6e on the cell cycle, an initial in vitro cytotoxic assay was conducted over two available human cancer cell lines (urinary bladder cancer, T-24; and breast cancer, MCF-7) at five-point concentrations (0.1, 1, 10, 100 and 1000 µM) to calculate the IC50 (half-maximal inhibitory concentration) to be used in the cell cycle assay (Figure 3). Compound 6e showed IC50 values of 257.87 and 168.78 µM over T-24 and MCF-7 cell lines, respectively.
Figure 3.

In vitro cytotoxic assay at 5-point assay of compound 6e over human urinary bladder cancer cell line (T-24, (A)) and human breast cancer cell line (MCF-7, (B)).
The cell cycle cytotoxicity assay was performed by treating the most sensitive cell line (breast cancer cell line, MCF-7) with compound 6e at its IC50 for 24 h using paclitaxel (1 µM) as a positive control (Table 6). Cell cycle distribution was measured using flow cytometric analysis after staining the DNA (deoxyribonucleic acid) content of the treated cells with propidium iodide (Figure 4). Compound 6e induced the increase in the cells of the G1 phase from 51.45% in control cells to 60.68% in the treated cells. Meanwhile, the cells of both S and G2 phases decreased to 17.47 and 18.29%, respectively, compared to that of control cells (22.27 and 21.34%, respectively). The results indicated that the cell cycle of MCF-7 cells was significantly arrested by compound 6e at the DNA replication step rather than mitosis.
Table 6.
In vitro cell cycle assay results of compound 6e in MCF-7 cell line.
| Cell Cycle Phase | Positive Control (%) | Compound 6e (%) |
|---|---|---|
| G1 | 51.45 | 60.68 |
| S | 22.79 | 17.47 |
| G2 | 21.34 | 18.29 |
Figure 4.

In vitro cell cycle cytotoxic assay of compound 6e over human cancer cell line (MCF-7). Compound 6e induced the cell cycle arrest at the G1 phase (B) using paclitaxel as a positive control (A). PE, phycoerythrin.
2.2.4. Apoptosis
The effect of compound 6e on cellular apoptosis in a human breast cancer cell line (MCF-7) was evaluated at its IC50 using doxorubicin as a positive control (10 µM) after 24 h using flow cytometric analysis (Table 7). The results revealed that compound 6e induced better cellular apoptosis (2.16%) compared to that of positive control (1.52%) after 24 h co-culture (Figure 5).
Table 7.
In vitro cellular apoptosis assay of compound 6e in MCF-7 cell line.
| Viability | Positive Control (%) | Compound 6e (%) |
|---|---|---|
| Intact cells | 98.48 | 97.83 |
| Early apoptosis | 0.08 | 0.1 |
| Late apoptosis | 0.68 | 0.81 |
| Necrosis | 0.76 | 1.26 |
| Total death | 1.52 | 2.16 |
Figure 5.

Effect of compound 6e (B) on cellular apoptosis after 24 h co-culture with MCF-7 cell lines using doxorubicin as a positive control (A). FITC, fluorescein isothiocyanate.
2.3. In Silico Screening
A molecular docking experiment was conducted for the tested compounds (5b, 6a–e and 7a,b) into the active site of the kinase domain in Aurora A kinase enzyme in order to explore the possible binding interaction of the newly designed compounds with the key amino acid residues in the ATP binding site (Table 8). An X-ray structure of the active site of Aurora A kinase enzyme with embedded reference ligand (II, phthalazinone derivative) was used in this study (PDB ID: 3P9J) [15]. An initial docking experiment was conducted for the reference ligand (II) in the Aurora A kinase active site to validate the selected molecular docking protocol. The results of the validation step showed that the reference ligand (II) exhibited the same binding mode as reported in the protein data bank [15] with 100% pose occupancy (Figure 6A) with a docking score of −5.34118 kcal/mol. In addition, it revealed that the pyrazole ring occupied the hinge region and interacted with the key amino acid residues at this binding region (Glu 211, Tyr 212 and Ala 213). Moreover, the phthalazinone ring was anchored to the external region near the solvent-exposed area to bind with key aromatic amino acid residue in that region (Leu 139). At the same time, the terminal phenyl ring was pointed towards the solvent-exposed area with no additional visible binding interaction.
Table 8.
In silico molecular docking score (kcal/mol) of the tested compounds (5b, 6a–e and 7a,b) into Aurora A kinase domain (PDB ID: 3P9J).
| Compound | Docking Score (kcal/mol) | Compound | Docking Score (kcal/mol) |
|---|---|---|---|
| 5b | −11.9746 | 6d | −13.4928 |
| 6a | −11.0161 | 6e | −14.0872 |
| 6b | −12.5924 | 7a | −12.6606 |
| 6c | −16.2153 | 7b | −11.3566 |
Figure 6.

Three-dimensional binding mode into Aurora A active site (PDB ID: 3P9J). (A) reference ligand (II); (B) compound 6e.
The tested compounds were then docked into the same prepared binding site to explore the possible virtual binding interactions. The molecular docking results showed that the tested compounds exhibited higher docking scores than the reference ligand. It is proposed that the employment of the quinazoline ring into the active site will enhance binding to the Aurora A kinase active site. However, in the case of reference ligand (II), the quinazoline ring was not fully fitted into the active site and stayed at the gate of the binding site instead. In addition, most of the generated poses of tested compounds shared the same binding pattern. Compound 6e (the most active compound among the tested series in the in vitro kinase assay and cytotoxicity screening) showed an interesting new binding mode (70% pose occupancy) with key amino acid residues in the hinge region of the active site (Val 147, Glu 211, Tyr 212, Ala 213 and Leu 263) with both hydrophobic and halogen interactions (Table 9 and Figure 6B). Moreover, the quinazoline binding motif occupied the hinge region. The Br-substituted terminal phenyl ring engaged the external pocket with additional halogen and hydrophobic interactions. Furthermore, the free carboxylic group was pointed out and waved in the solvent-exposed area. Regarding ester-based derivatives (5b and 7a,b), it was interesting to identify a new binding mode; the compound was flipped in order to anchor the ester group into the binding pocket rather than the solvent area (60% pose occupancy).
Table 9.
Possible binding interactions of compound 6e into Aurora A active site.
| Binding Motif | Amino Acid * | Binding Interaction | Distance (Å) |
|---|---|---|---|
| Central motif (quinazoline) | |||
| Phenyl | Ala 160 Val 147 Leu 263 |
Hydrophobic Hydrophobic Hydrophobic |
- - - |
| Pyrimidine | Leu 139 Val 147 Leu 263 |
Hydrophobic Hydrophobic Hydrophobic |
- - - |
| F | Glu 211 Tyr 212 Ala 213 |
Halogen Halogen Halogen |
3.52 2.61 2.49 |
| Terminal motif | |||
| Phenyl | Leu 139 | Hydrophobic | - |
| Br | Leu 139 Tyr 212 Pro 214 |
Halogen Halogen Halogen |
- - 3.27 |
* Ala, alanine; Val, valine; Leu, leucine; Glu, glutamine; Tyr, tyrosine; Pro, proline.
The docking results revealed that the fluorine group at the quinazoline ring has a significant impact on binding to the hinge region through additional binding along with the small-sized volume of this halogen atom (similar to a hydrogen atom) which prevents any clashes in that crucial region. In addition, the substitution of the terminal phenyl ring with a halogen atom (Br) modulated the orientation of this terminal ring through additional binding to the key amino acid residue in this area (Pro 214).
From the previous results (in vitro assay and in silico screening), the new quinazoline derivatives provide a promising selective inhibition profile toward Aurora A kinase. In addition, it delivers a new challenge to medicinal chemists to improve structural modification in order to enhance the inhibitory effect and maintain the selectivity profile. Moreover, enhancing the cell permeability is considered a key role in the same field to ensure cell induction and inhibition of the intracellular kinase (Aurora A) without any disruption in ATP competitive inhibition effect, as discussed before.
3. Materials and Methods
3.1. Chemistry
3.1.1. General
All solvents and reagents were obtained from commercial suppliers and used without further purification. TLC was performed using glass sheets pre-coated with silica gel 60 F254 purchased by Merk. The NMR spectra were obtained on Bruker Avance 400. Column chromatography was carried out on Merck Silica Gel 60 (230–400 mesh), as previously described [16].
3.1.2. General Procedure for the Synthesis of Derivatives 3a–e
A solution of appropriate acid chloride (2a–d, 1 mmol, 1 Eq) in DCM was added dropwise to a solution of appropriate aromatic amine (1a,b, 1 mmol, 1 Eq) and triethylamine (1 mmol, 1 Eq) in DCM at 0 °C. The reaction mixture was stirred at rt for 4 h or until the TLC showed reaction completion. The organic layer was washed with brine (2 × 5 mL), dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude residue was purified by column chromatography to give the titled product 3a–e [17].
3.1.3. General Procedure for the Synthesis of Derivatives 4a–e
A mixture of compound 3a–e (1 mmol) and thionyl chloride (2 mL) was refluxed for 3 h. The excess thionyl chloride was removed under reduced pressure to obtain the titled product 4a–e to be used in the next step without further purification [18].
3.1.4. General Procedure for the Synthesis of Derivatives 5a–e
Ethyl cyanoformate (1 mmol, 1 Eq) and tin tetrachloride (1 mmol, 1 Eq) were added to a solution of the appropriate benzanilide imidoyl chloride derivative (4a–e, 1 mmol, 1 Eq) in o-dichlorobenzene (1 mL). The reaction mixture was allowed to stir at 140 °C for 15 min. The reaction mixture was cooled to rt and produced alkaline with a cold 0 °C aqueous solution of NaOH (1 M). The mixture was then extracted with DCM, dried over magnesium sulfate and evaporated under reduced pressure. The crude residues were used directly in the next step without further purification except for compound 5b, which was purified using column chromatography (2% EtOAc in hexane) [19].
Ethyl 2-(3-chlorophenyl)quinazoline-4-carboxylate (5b)
Grey solid, yield: 81%, mp: 75.2–76.3 °C, HPLC purity: 25.28 min, 98.39%, 1H NMR (400 MHz, CDCl3): δ 8.65 (s, 1H), 8.55–8.53 (m, 1H), 8.47 (d, J = 8.4 Hz, 1H), 8.16 (d, J = 8.5 Hz, 1H), 7.95 (t, J = 7.9 Hz, 1H), 7.68 (t, J = 7.9 Hz, 1H), 7.48–7.45 (m, 2H), 4.65 (q, J = 7.2 Hz, 2H), 1.54 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 165.10, 158.89, 157.84, 152.31, 139.21, 134.81, 134.61, 130.86, 129.88, 129.27, 128.74, 128.58, 126.83, 125.91, 120.55, 62.69, 14.30. HRMS (ESI) m/z calculated for C17H13ClN2O2 [M + H]+: 313.0744. Found: 313.0732.
3.1.5. General Procedure for the Synthesis of Derivatives 6a–e
A solution of corresponding ethyl esters (5a–e, 1 mmol, 1 Eq) in EtOH was stirred with an aqueous NaOH solution (20%, 5 mL). The reaction mixture was refluxed for 2 h. The reaction mixture was concentrated under reduced pressure, acidified with a citric acid solution (20%, 5 mL) and extracted with EtOAc (10 mL). The organic layer was washed with brine (2 × 5 mL), dried over anhydrous Na2SO4 and concentrated under reduced pressure to give the titled product 6a–e [19].
2-Phenylquinazoline-4-carboxylic acid (6a)
Brown solid, yield: 31%, mp: 146.2–147.2 °C, HPLC purity: 14.20 min, 97.79%, 1H NMR (400 MHz, DMSO-d6): δ 8.59–8.57 (m, 2H), 8.42 (d, J = 8.3 Hz, 1H), 8.18–8.09 (m, 2H), 7.81 (t, J = 7.9 Hz, 1H), 7.61–7.59 (m, 3H).
2-(3-Chlorophenyl)quinazoline-4-carboxylic acid (6b)
Yellow solid, yield: 86%, mp: 175.6–176.6 °C, HPLC purity: 18.41 min, 99.93%, 1H NMR (400 MHz, DMSO-d6): δ 8.55 (s, 1H), 8.52 (d, J = 6.9 Hz, 1H), 8.46 (d, J = 8.3 Hz, 1H), 8.19–8.09 (m, 2H), 7.83 (t, J = 7.6 Hz, 1H), 7.65–7.61 (m, 2H). HRMS (ESI) m/z calculated for C15H9ClN2O2 [M + H]+: 285.0431. Found: 285.0419.
8-Fluoro-2-(m-tolyl)quinazoline-4-carboxylic acid (6c)
White solid, yield: 5%, mp: 349.3–350.3 °C, HPLC purity: 17.40 min, 97.03%, 1H NMR (400 MHz, DMSO-d6): δ 8.38–8.33 (m, 2H), 7.98 (d, J = 8.2 Hz, 1H), 7.79–7.75 (m, 1H), 7.62–7.56 (m, 1H), 7.48–7.44 (m, 1H), 7.39–7.37 (m, 1H), 2.46 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 170.02, 168.67, 160.17, 158.21, 155.67, 141.00, 138.25, 137.97, 131.97, 129.12, 127.00, 125.90, 124.14, 121.02, 118.16, 21.59. HRMS (ESI) m/z calculated for C16H11FN2O2 [M + H]+: 283.0883. Found: 283.0871.
8-Fluoro-2-phenylquinazoline-4-carboxylic acid (6d)
White solid, yield: 7%, mp: 353.3–354.3 °C, HPLC purity: 15.33 min, 99.79%, 1H NMR (400 MHz, DMSO-d6): δ 8.55 (s, 2H), 7.99 (s, 1H), 7.79–7.58 (m, 4H). HRMS (ESI) m/z calculated for C15H9FN2O2 [M + H]+: 269.0726. Found: 269.0714.
2-(3-Bromophenyl)-8-fluoroquinazoline-4-carboxylic acid (6e)
White solid, yield: 4%, mp: 335.4–336.4 °C, HPLC purity: 19.74 min, 99.86%, 1H NMR (400 MHz, DMSO-d6): δ 8.66 (s, 1H), 8.54 (d, J = 7.8 Hz, 1H), 8.01 (d, J = 8.2 Hz, 1H), 7.83–7.76 (m, 2H), 7.66–7.54 (m, 2H).
3.1.6. General Procedure for the Synthesis of Derivatives 7a,b
Sulfuric acid (0.2 mL) was added to a solution of compound 6b (1 mmol) in appropriate alcohol (10 mL). The reaction mixture was refluxed for 18 h. The reaction mixture was cooled, quenched with a saturated solution of NaHCO3 (2 mL) and concentrated under reduced pressure. The crude residue was extracted between EtOAc (10 mL) and water (5 mL). The organic layer was washed with brine (2 × 5 mL), dried over anhydrous Na2SO4 and concentrated under reduced pressure to give the titled product 7a,b.
Methyl 2-(3-chlorophenyl)quinazoline-4-carboxylate (7a)
White solid, yield: 91%, mp: 124.4–125.4 °C, HPLC purity: 23.52 min, 99.96%, 1H NMR (400 MHz, CDCl3): δ 8.64 (s, 1H), 8.54–8.50 (m, 2H), 8.16 (d, J = 8.5 Hz, 1H), 7.98–7.93 (m, 1H), 7.70–7.66 (m, 1H) 7.50–7.44 (m, 2H) 4.15 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 165.46, 158.88, 157.17, 152.41, 139.15, 134.85, 134.67, 130.89, 129.92, 129.28, 128.73, 128.70, 126.80, 125.97, 120.66, 53.32. HRMS (ESI) m/z calculated for C16H11ClN2O2 [M + H]+: 299.0587. Found: 299.0575.
Isopropyl 2-(3-chlorophenyl)quinazoline-4-carboxylate (7b)
Greenish grey solid, yield: 89%, mp: 72.6–73.6 °C, HPLC purity: 26.90 min, 96.79%, 1H NMR (400 MHz, CDCl3): δ 8.65 (s, 1H), 8.59–8.53 (m, 1H), 8.39 (d, J = 8.4 Hz, 1H), 8.14 (d, J = 8.5 Hz, 1H), 7.96–7.92 (m, 1H), 7.68–7.64 (m, 1H), 7.49–7.44 (m, 2H), 5.56–5.46 (m, 1H), 1.54 (d, J = 6.3 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 164.74, 158.89, 158.48, 152.21, 139.24, 134.77, 134.53, 130.83, 129.85, 129.26, 128.74, 128.46, 126.85, 125.79, 120.42, 70.81, 21.92. HRMS (ESI) m/z calculated for C18H15ClN2O2 [M + H]+: 327.0900. Found: 327.0887.
3.2. In Vitro Screening
3.2.1. Kinase Inhibitory Assay
The in vitro kinase inhibitory assay was performed at Reaction Biology Corp (http://www.reactionbiology.com accessed on 15 February 2022) using Kinase HotSpotSM in order to measure the inhibitory activities of tested candidates, as previously performed [20,21]. The experiment contained specific kinase/substrate pairs along with required cofactors. Base reaction buffer: 20 mM Hepes (pH 7.5), 10 mM MgCl2, 1 mM EGTA, 0.02% Brij35, 0.02 mg/mL BSA, 0.1 mM Na3VO4, 2 mM DTT, 1% DMSO. The tested compounds were dissolved in 100% DMSO to a specific concentration. The serial dilution was conducted by Integra Viaflo Assist in DMSO. The reaction mixture containing the examined compound and 33P-ATP was incubated at rt for 2 h, and radioactivity was detected by the filter-binding method. Kinase activity data were expressed as the percentage of remaining kinase activity in test samples compared to vehicle (DMSO) reactions.
3.2.2. Cytotoxicity Assay
The in vitro cytotoxic screening of tested compounds (5b, 6a–e and 7a,b) over nine NCI human cancer cell lines was carried out at the National Cancer Institute (NCI), Bethesda, Maryland, USA (dtp.cancer.gov accessed on 25 February 2022), as previously performed [22]. A 48 h drug exposure protocol was adopted, and a sulforhodamine B (SRB) assay was utilized to assess the cell growth and viability. The human tumor cell lines of the cancer screening panel were grown in RPMI 1640 medium containing 5% fetal bovine serum and 2 mM L-glutamine. For a typical screening experiment, cells were inoculated into 96 well microtiter plates in 100 µL at plating densities ranging from 5000 to 40,000 cells/well, depending on the doubling time of individual cell lines. After cell inoculation, the microtiter plates were incubated at 37 °C, 5% CO2, 95% air and 100% relative humidity for 24 h prior to the addition of experimental drugs.
After 24 h, two plates of each cell line were fixed in situ with TCA to represent a measurement of the cell population for each cell line at the time of drug addition (Tz). Experimental compounds were solubilized in dimethylsulfoxide at 400-fold the desired final maximum test concentration and stored frozen prior to use. At the time of compound addition, an aliquot of frozen concentrate was thawed and diluted to twice the desired final maximum test concentration with a complete medium containing 50 µg/mL gentamicin. Aliquots of 100 µL were added to the appropriate microtiter wells already containing 100 µL of the medium, resulting in the required final compound concentration (10 µM).
Following test compound addition, the plates were incubated for an additional 48 h at 37 °C, 5% CO2, 95% air and 100% relative humidity. For adherent cells, the assay was terminated by the addition of cold TCA. Cells were fixed in situ by the gentle addition of 50 µL of cold 50% (w/v) TCA (final concentration, 10% TCA) and incubated for 60 min at 4 °C. The supernatant was discarded, and the plates were washed five times with tap water and air-dried. Sulforhodamine B (SRB) solution (100 µL) at 0.4% (w/v) in 1% acetic acid was added to each well, and plates were incubated for 10 min at rt. After staining, the unbound dye was removed by washing five times with 1% acetic acid, and the plates were air-dried. The bound stain was subsequently solubilized with 10 mM trizma base, and the absorbance was read on an automated plate reader at a wavelength of 515 nm. For suspension cells, the methodology is the same, except that the assay was terminated by fixing settled cells at the bottom of the wells by gently adding 50 µL of 80% TCA (final concentration, 16% TCA).
The additional in vitro cytotoxicity cell assay for compound 6e was carried out over urinary bladder (T-24) and breast cancer (MCF-7) cell lines to evaluate IC50. Both cell lines were obtained from Nawah Scientific Inc. (Mokatam, Cairo, Egypt). T-24 and MCF-7 cells were maintained in McCoy’s and DMEM, respectively, and supplemented with 100 mg/mL of streptomycin, 100 units/mL of penicillin and 10% of heat-inactivated fetal bovine serum in a humidified, 5% (v/v) CO2 atmosphere at 37 °C. Cell viability was assessed by an SRB assay. Aliquots of 100 μL cell suspension (5 × 103 cells) were placed in 96-well plates and incubated in complete media for 24 h. Cells were treated with another aliquot of 100 μL media containing compound 6e at various concentrations (0.1, 1, 10, 100, 1000 μM as final concentrations). After 72 h of compound 6e exposure, cells were fixed by replacing the media with 150 μL of 10% TCA and incubated at 4 °C for 1 h. The TCA solution was removed, and the cells were washed five times with distilled water. Aliquots of 70 μL SRB solution (0.4% w/v) were added and incubated in a dark place at rt for 10 min. Plates were washed three times with 1% acetic acid and allowed to air-dry overnight. Then, 150 μL of TRIS (10 mM) was added to dissolve the protein-bound SRB stain; the absorbance was measured at 540 nm using a BMG LABTECH®-FLUOstar Omega microplate reader (Ortenberg, Germany) [23,24].
3.2.3. Cell Cycle
Cell cycle distribution was measured after the treatment of MCF-7 cells with compound 6e (at its IC50 value over MCF-7 cell line) and paclitaxel (1 µM) for 24 h as a positive control. Cells (105 cells) were collected by trypsinization and washed twice with ice-cold PBS (pH 7.4). Cells were re-suspended in 2 mL of 60% ice-cold EtOH and incubated at 4 °C for 1 h for fixation. Fixed cells were washed twice again with PBS (pH 7.4) and re-suspended in 1 mL of PBS containing 50 µg/mL RNAase A and 10 µg/mL propidium iodide (PI). After 20 min of incubation in the dark at 37 C, cells were analyzed for DNA contents using flow cytometry analysis using FL2 (λex/em 535/617 nm) signal detector (ACEA Novocyte™ flow cytometer, ACEA Biosciences Inc., San Diego, CA, USA). Twelve thousand events were acquired. Cell cycle distribution was calculated using ACEA NovoExpress™ software (ACEA Biosciences Inc., San Diego, CA, USA) [24,25,26,27,28].
3.2.4. Apoptosis
Apoptosis and necrosis cell populations were determined using the Annexin V-FITC apoptosis detection kit (Abcam Inc., Cambridge Science Park, Cambridge, UK) coupled with 2 fluorescent channels flowcytometry. After treatment with compound 6e (at its IC50 value over MCF-7 cell line) and doxorubicin (10 µM) for 24 h as a positive control, MCF-7 cells (105 cells) were collected by trypsinization and washed twice with ice-cold PBS (pH 7.4). Then, cells were incubated in the dark with 0.5 mL of Annexin V-FITC/PI solution for 30 min at rt according to the manufacturer’s protocol. After staining, cells were injected via an ACEA Novocyte™ flow cytometer (ACEA Biosciences Inc., San Diego, CA, USA) and analyzed for FITC and PI fluorescent signals using FL1 and FL2 signal detectors, respectively (λex/em 488/530 nm for FITC and λex/em 535/617 nm for PI). Twelve thousand events were acquired, and positive FITC and PI cells were quantified by quadrant analysis and calculated using ACEA NovoExpress™ software (ACEA Biosciences Inc., San Diego, CA, USA) [25,27,28].
3.3. In Silico Screening
A molecular docking study was conducted using Discovery Studio Client 21.1.0.20298 (Biovia Corp) (www.3ds.com accessed on 27 February 2022). The X-ray structure of the kinase domain of Aurora A kinase enzyme was downloaded from the protein data bank (www.rcsb.org accessed on 28 February 2022) (PDB ID: 3P9J) [15]. Water molecules were removed, keeping only the kinase domain with its corresponding reference ligand (compound II). The bonds and bond orders of the amino acid chain were checked and corrected. The terminal residues were checked and adjusted.
A CHARMm forcefield was applied to the protein domain. A Momany–Rone forcefield was selected for the partial charge. The binding site was defined using the coordinates of the reference ligand. The CDOCKER algorithm (CHARMm-based molecular dynamic scheme) [29] was used in this study to generate the most stable conformers with the binding sites. Random ligand conformations were generated (10 conformers) from the initial ligand structure through dynamic target temperature (100 K), followed by random rotations. The generated conformers were refined by grid-based (GRID-1) simulated annealing.
The docking protocol was validated by running an initial docking experiment (pre-docking) for the reference ligand (II, PDB ID: P9J). The docking experiment of compound 6e was conducted to generate the 10 most possible conformers with the binding site. The generated conformers were visualized to investigate the binding interaction between the tested compound (6e) and key amino acid residues in the active site of the Aurora A kinase domain.
4. Conclusions
A number of quinazoline-based derivatives were designed and synthesized based on the structural modification of previously reported Aurora A kinase inhibitors (I–III). Free carboxylic acid-bearing derivatives (6a–e) as well as ester-based compounds (5b and 7a,b) were tested for their inhibitory activities over the Aurora A kinase enzyme. Compound 6e exhibited the most potent inhibitory activity among the tested compounds. Furthermore, selectivity profile testing was conducted for compound 6e over 14 kinases (including Aurora B kinase). In addition, an in vitro cytotoxicity assay was conducted against NCI human cancer cell lines. The CNS cancer cell line (SNB-75) was the most sensitive cell line among the tested lines. Both cell cycle and cellular apoptosis analyses were carried out to compound 6e. Compound 6e was able to arrest the cell cycle of the MCF-7 cell line at the G1 phase. In addition, it induced apoptosis compared to the positive control. In silico molecular docking screening was carried out to explore the binding mode of compound 6e into the active site of the Aurora A kinase domain. It exhibited perfect fitting into the active side key regions compared to the reference ligand (compound II). In addition, it showed potential binding interactions with the key amino acid residues in the kinase pocket. The results revealed that both halogens in the central core scaffold and terminal phenyl ring have a significant role in the binding to the active site. Furthermore, the small-sized halogen atom (F) introduced into the central core had the perfect size to avoid any clashes and maintain the perfect fitting into the active site. In conclusion, the new quinazoline derivatives can be considered a potential hit for further structural and molecular optimization in future.
Acknowledgments
The authors are beholden to the support provided by the Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2022R127), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia. M.H.A. thanks Taif University Researchers Project number (TURSP-2020/91), Taif University, Taif, Saudi Arabia. The authors would like to thank the Deanship of scientific research at Umm Al-Qura University for supporting this work by grant code (22UQU4290565DSR27).
Supplementary Materials
The following are available online at https://www.mdpi.com/2075-1729/12/6/876/s1, experimental and spectral analysis of both key intermediates and final target compounds. In vitro cytotoxicity data over NCI human cancer cell lines (Table S1–S8).
Author Contributions
Conceptualization, A.E. and E.J.R.; methodology, A.E. and M.H.E., validation, M.H.A., M.A.S.A. and A.A.; formal analysis, U.M.A., S.F.I., D.A. and A.E.; investigation, M.H.A., M.A.S.A., W.M. and E.J.R.; resources, A.E. and E.J.R.; data curation, M.H.E., S.F.I., D.A. and U.M.A.; writing—original draft preparation, M.H.E. and U.M.A.; writing—review and editing, all authors; supervision, E.J.R.; project administration, A.E.; funding acquisition, E.J.R. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All of the data are contained within the article and the Supplementary Materials.
Conflicts of Interest
The authors declare no conflict of interest.
Funding Statement
This work was supported by the KIST Institutional programs (Grant No. 2E31624) from the Korea Institute of Science and Technology, the Creative Fusion Research Program through the Creative Allied Project funded by the National Research Council of Science & Technology (CAP-12-1-KIST).
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Borisa A.C., Bhatt H.G. A comprehensive review on Aurora kinase: Small molecule inhibitors and clinical trial studies. Eur. J. Med. Chem. 2017;140:1–19. doi: 10.1016/j.ejmech.2017.08.045. [DOI] [PubMed] [Google Scholar]
- 2.Andrews P.D. Aurora kinases: Shining lights on the therapeutic horizon? Oncogene. 2005;24:5005–5015. doi: 10.1038/sj.onc.1208752. [DOI] [PubMed] [Google Scholar]
- 3.Long L., Wang Y.-H., Zhuo J.-X., Tu Z.-C., Wu R., Yan M., Liu Q., Lu G. Structure-based drug design: Synthesis and biological evaluation of quinazolin-4-amine derivatives as selective Aurora A kinase inhibitors. Eur. J. Med. Chem. 2018;157:1361–1375. doi: 10.1016/j.ejmech.2018.08.053. [DOI] [PubMed] [Google Scholar]
- 4.Pradhan T., Gupta O., Singh G., Monga V. Aurora kinase inhibitors as potential anticancer agents: Recent advances. Eur. J. Med. Chem. 2021;221:113495. doi: 10.1016/j.ejmech.2021.113495. [DOI] [PubMed] [Google Scholar]
- 5.Fu S., Li Y., Huang J., Liu T., Hong Z., Chen A., Bast R.C., Kavanagh J.J., Gershenson D.M., Sood A.K. Aurora kinase inhibitor VE 465 synergistically enhances cytotoxicity of carboplatin in ovarian cancer cells through induction of apoptosis and downregulation of histone 3. Cancer Biol. Ther. 2012;13:1034–1041. doi: 10.4161/cbt.21045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lindon C., Grant R., Min M. Ubiquitin-mediated degradation of aurora kinases. Front. Oncol. 2016;5:307. doi: 10.3389/fonc.2015.00307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fan C., Zhong T., Yang H., Yang Y., Wang D., Yang X., Xu Y., Fan Y. Design, synthesis, biological evaluation of 6-(2-amino-1H-benzo [d] imidazole-6-yl) quinazolin-4 (3H)-one derivatives as novel anticancer agents with Aurora kinase inhibition. Eur. J. Med. Chem. 2020;190:112108. doi: 10.1016/j.ejmech.2020.112108. [DOI] [PubMed] [Google Scholar]
- 8.Bavetsias V., Faisal A., Crumpler S., Brown N., Kosmopoulou M., Joshi A., Atrash B., Pérez-Fuertes Y., Schmitt J.A., Boxall K.J. Aurora isoform selectivity: Design and synthesis of imidazo [4,5-b] pyridine derivatives as highly selective inhibitors of Aurora-A kinase in cells. J. Med. Chem. 2013;56:9122–9135. doi: 10.1021/jm401115g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pollard J.R., Mortimore M. Discovery and development of aurora kinase inhibitors as anticancer agents. J. Med. Chem. 2009;52:2629–2651. doi: 10.1021/jm8012129. [DOI] [PubMed] [Google Scholar]
- 10.Green M.R., Woolery J.E., Mahadevan D. Update on aurora kinase targeted therapeutics in oncology. Expert Opin. Drug Dis. 2011;6:291–307. doi: 10.1517/17460441.2011.555395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheung C.H.A., Coumar M.S., Chang J.Y., Hsieh H.P. Aurora kinase inhibitor patents and agents in clinical testing: An update (2009–10) Expert Opin. Ther. Pat. 2011;21:857–884. doi: 10.1517/13543776.2011.574614. [DOI] [PubMed] [Google Scholar]
- 12.Mortlock A.A., Foote K.M., Heron N.M., Jung F.H., Pasquet G., Lohmann J.-J.M., Warin N., Renaud F., De Savi C., Roberts N.J. Discovery, synthesis, and in vivo activity of a new class of pyrazoloquinazolines as selective inhibitors of aurora B kinase. J. Med. Chem. 2007;50:2213–2224. doi: 10.1021/jm061335f. [DOI] [PubMed] [Google Scholar]
- 13.Aliagas-Martin I., Burdick D., Corson L., Dotson J., Drummond J., Fields C., Huang O.W., Hunsaker T., Kleinheinz T., Krueger E., et al. A class of 2,4-bisanilinopyrimidine Aurora A inhibitors with unusually high selectivity against Aurora B. J. Med. Chem. 2009;52:3300–3307. doi: 10.1021/jm9000314. [DOI] [PubMed] [Google Scholar]
- 14.White T.D., Berglund K.D., Groh J.M., Johnson M.D., Miller R.D., Yates M.H. Development of a continuous Schotten–Baumann route to an acyl sulfonamide. Org. Process Res. Dev. 2012;16:939–957. doi: 10.1021/op200344a. [DOI] [Google Scholar]
- 15.Prime M.E., Courtney S.M., Brookfield F.A., Marston R.W., Walker V., Warne J., Boyd A.E., Kairies N.A., von der Saal W., Limberg A., et al. Phthalazinone pyrazoles as potent, selective, and orally bioavailable inhibitors of Aurora-A kinase. J. Med. Chem. 2011;54:312–319. doi: 10.1021/jm101346r. [DOI] [PubMed] [Google Scholar]
- 16.Park J.-E., Elkamhawy A., Hassan A.H., Pae A.N., Lee J., Paik S., Park B.-G., Roh E.J. Synthesis and evaluation of new pyridyl/pyrazinyl thiourea derivatives: Neuroprotection against amyloid-β-induced toxicity. Eur. J. Med. Chem. 2017;141:322–334. doi: 10.1016/j.ejmech.2017.09.043. [DOI] [PubMed] [Google Scholar]
- 17.Miao D., Shi X., He G., Tong Y., Jiang Z., Han S. One-pot synthesis of 2-arylbenzoxazole derivatives via Cu (I) catalyzed C–N/C–O coupling of N-(2-chloro-phenyl)-2-halo-benzamides and primary amines. Tetrahedron. 2015;71:431–435. doi: 10.1016/j.tet.2014.12.008. [DOI] [Google Scholar]
- 18.Zhao H.-B., Hou Z.-W., Liu Z.-J., Zhou Z.-F., Song J., Xu H.-C. Amidinyl radical formation through anodic N−H bond cleavage and its application in aromatic C−H bond functionalization. Angew. Chem. Int. Ed. 2017;56:587–590. doi: 10.1002/anie.201610715. [DOI] [PubMed] [Google Scholar]
- 19.Giardina G.A.M., Sarau H.M., Farina C., Medhurst A.D., Grugni M., Raveglia L.F., Schmidt D.B., Rigolio R., Luttmann M., Vecchietti V., et al. Discovery of a novel class of selective non-peptide antagonists for the human neurokinin-3 receptor. 1. Identification of the 4-quinolinecarboxamide framework. J. Med. Chem. 1997;40:1794–1807. doi: 10.1021/jm960818o. [DOI] [PubMed] [Google Scholar]
- 20.Elkamhawy A., Ammar U.M., Paik S., Abdellattif M.H., Elsherbeny M.H., Lee K., Roh E.J. Scaffold repurposing of in-house small molecule candidates leads to discovery of first-in-class CDK-1/HER-2 dual inhibitors: In vitro and in silico screening. Molecules. 2021;26:5324. doi: 10.3390/molecules26175324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee K., Nada H., Byun H.J., Lee C.H., Elkamhawy A. Hit Identification of a Novel Quinazoline Sulfonamide as a Promising EphB3 Inhibitor: Design, Virtual Combinatorial Library, Synthesis, Biological Evaluation, and Docking Simulation Studies. Pharmaceuticals. 2021;14:1247. doi: 10.3390/ph14121247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Al-Sanea M.M., Elkamhawy A., Zakaria A., Park B.S., Kwon Y., Lee S.H., Lee S.W., Kim I.T. Synthesis and in vitro screening of phenylbipyridinylpyrazole derivatives as potential antiproliferative agents. Molecules. 2015;20:1031–1045. doi: 10.3390/molecules20011031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Skehan P., Storeng R., Scudiero D., Monks A., McMahon J., Vistica D., Warren J.T., Bokesch H., Kenney S., Boyd M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990;82:1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 24.Allam R.M., Al-Abd A.M., Khedr A., Sharaf O.A., Nofal S.M., Khalifa A.E., Mosli H.A., Abdel-Naim A.B. Fingolimod interrupts the cross talk between estrogen metabolism and sphingolipid metabolism within prostate cancer cells. Toxicol. Lett. 2018;291:77–85. doi: 10.1016/j.toxlet.2018.04.008. [DOI] [PubMed] [Google Scholar]
- 25.Fekry M., Ezzat S.M., Salama M.M., AlShehri O.Y., Al-Abd A.M. Bioactive glycoalkaloides isolated from Solanum melongena fruit peels with potential anticancer properties against hepatocellular carcinoma cells. Sci. Rep. 2019;9:1746. doi: 10.1038/s41598-018-36089-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baghdadi M.A., Al-Abbasi F.A., El-Halawany A.M., Aseeri A.H., Al-Abd A.M. Anticancer profiling for coumarins and related O-naphthoquinones from mansonia gagei against solid tumor cells in vitro. Molecules. 2018;23:1020. doi: 10.3390/molecules23051020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alaufi O.M., Noorwali A., Zahran F., Al-Abd A.M., Al-Attas S. Cytotoxicity of thymoquinone alone or in combination with cisplatin (CDDP) against oral squamous cell carcinoma in vitro. Sci. Rep. 2017;7:13131. doi: 10.1038/s41598-017-13357-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mohamed G.A., Al-Abd A.M., El-Halawany A.M., Abdallah H., Ibrahim S.R.M. New xanthones and cytotoxic constituents from Garcinia mangostana fruit hulls against human hepatocellular, breast, and colorectal cancer cell lines. J. Enthnopharmacol. 2017;198:302–312. doi: 10.1016/j.jep.2017.01.030. [DOI] [PubMed] [Google Scholar]
- 29.Wu G., Robertson D.H., Brooks C.L., Vieth M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER—A CHARMm-based MD docking algorithm. J. Comput. Chem. 2003;24:1549–1562. doi: 10.1002/jcc.10306. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All of the data are contained within the article and the Supplementary Materials.