

Graphical abstract

Abstract
The discovery of antiviral agents against SARS-CoV-2 is an important step toward ending the COVID-19 pandemic and to tackle future outbreaks. In this context, the main protease (Mpro) represents an ideal target for developing coronavirus antivirals, being conserved among different strains and essential for survival. In this work, using in silico tools, we created and validated a docking protocol able to predict binders to the catalytic site of Mpro. The following structure-based virtual screening of a subset of the ZINC library (over 4.3 million unique structures), led to the identification of a hit compound having a 2-thiobenzimidazole scaffold. The inhibitory activity was confirmed using a FRET-based proteolytic assay against recombinant Mpro. Structure-activity relationships were obtained with the synthesis of a small library of analogs, guided by the analysis of the docking pose. Our efforts led to the identification of a micromolar Mpro inhibitor (IC50 = 14.9 µM) with an original scaffold possessing ideal drug-like properties (predicted using the QikProp function) and representing a promising lead for the development of a novel class of coronavirus antivirals.
The current COVID-19 pandemic has created an unprecedented global health crisis,1 highlighting the need for broad spectrum antivirals to complement the roll out of vaccines. Whereas immunization has been the first-line strategy for the mitigation of the pandemic, vaccines have a few drawbacks, such as loss of efficacy over time and against variants,2, 3 inability to cure active cases; unavailability to immunocompromised subjects; and logistical issues concerning administration and delivery, especially in remote parts of the world. The development of effective small-molecule antiviral therapeutics is thus urgently needed.
SARS-CoV-2 is the etiological agent of COVID-19 and its replicase gene encodes two overlapping polyproteins, pp1a and pp1ab, which mediate all of the functions required for viral replication and transcription.4 Two different cysteine proteases, namely the main or chymotrypsin-like protease (Mpro or 3CLpro) and the papain-like protease (PLpro), are involved in the processing of the replicase gene.5 The Mpro digests the viral polyprotein at more than 11 sites to generate functional proteins, making it an essential enzyme for viral replication and survival.6 Mpro is well-conserved among coronavirus species7, 8 and has a unique substrate preference for glutamine at the P1 position (no human protease are known to have this specificity).9 All these factors make the main protease one of the top targets for the development of broad-spectrum antivirals.8, 10.
Mpro is found in its inactive form in the polyprotein and needs auto-cleavage at both the N- and C-terminal sites for activation.11 The active form is a homodimer made of two 34 kDa protomers, namely A and B, oriented almost at 90° to each other (Fig. 1 A). Only protomer A is catalytically active, whereas the substrate-binding site of B is collapsed, rendering protomer B inactive. The homodimerization, driven by a three-dimensional domain swap at the C-terminal,12 is essential for maintaining the active conformation and has been suggested as a promising alternative target for the identification of inhibitors,13 and recently Günther and co-workers have identified an allosteric inhibitor targeting the dimerization domain.14 Nevertheless, the most advanced compounds in the drug discovery pipeline target the active site and act by either inactivating the enzyme (generally by covalent modification) or by competing with the natural substrate, which are both well-validated mechanism for viral protease inhibition.15 SARS-CoV-2 Mpro catalytic site features a Cys145-His41 dyad,16, 17 instead of the canonical Cys-His-Glu/Asp catalytic triad of enteroviral main proteases. A water molecule is hydrogen-bonded to His41 (Fig. 1A) and can be considered the third component of a catalytic triad.17, 18
Fig. 1.

Structure of Mpro and its inhibitors. (A) X-ray crystal structure of the SARS-CoV-2 Mpro homodimer highlighting the different domains, the active site and the catalytic dyad. (B) Structures of the most advanced inhibitors, covalent and noncovalent, discovered to date.
The Mpro inhibitors so far identified can be divided in two main categories, covalent and noncovalent (Fig. 1B). Covalent inhibitors (comprising the majority of the agents under development) consist of peptidomimetic compounds bearing an electrophilic warhead which binds covalently, reversibly or irreversibly, to Cys145 and inactivates the enzyme. The most advanced compound of this category is nirmaltrevir (PF-07321332, Fig. 1B),19 an orally-active agent developed by Pfizer, which received FDA emergency use authorization (EUA) for the treatment of mild-to-moderate COVID-19. Additional covalent compounds in preclinical stage are lufotrelvir (PF-07304814)20 and GC376,21 although concerns regarding oral bioavailability and short half-life are impairing their progress to the clinic. Other potent peptidomimetic and covalent inhibitors in earlier stages of development are reported in Fig. S1.22– 22, 23, 24, 25, 26, 27, 28 Although the covalent, peptidomimetic strategy has had success in quickly yielding potent drugs to treat SARS-CoV-2, some concerns regarding ADME profiles remain. Specifically, it is well-known that peptide-like structures can suffer from proteolytic degradation and poor bioavailability.29 For example, nirmatrelvir has low bioavailability due to excessive first-pass metabolism30 and, in the clinic, it has to be administered in combination with ritonavir (a potent CYP3A inhibitor) to reach adequate plasma concentration. This can interfere with many commonly prescribed drugs.
To overcome these limitations, researchers in the field have worked to identify noncovalent, more drug-like Mpro inhibitors. The most promising noncovalent inhibitors (Fig. 1B) have been discovered by structure-based design, starting from existing drugs31, 32 or previously-described inhibitors of SARS-CoV-1 main protease.33, 34 Also, very recently, Carlsson and co-workers identified broad spectrum inhibitors following an ultra large virtual screening approach.35 Whereas these compounds have good translation potential to the clinic, it is also essential to identify inhibitors constructed from different scaffolds to add alternatives to our arsenal of therapeutics to fight coronavirus diseases.
We report herein the discovery of a class of SARS-CoV-2 Mpro inhibitors having a 2-thiobenzimidazole scaffold. This novel family of inhibitors was identified by means of an in silico virtual screening protocol supported by a FRET-based biochemical assay against recombinant SARS-CoV-2 Mpro. The following SAR exploration, using a combination of structure-based design and synthetic organic chemistry, yielded a drug-like molecule showing an IC50 in the micromolar range, representing a promising lead compound for further investigation.
The active site of Mpro is comprised of five highly conserved sub-pockets (S1, S1′, S2, S3 and S4), possessing structural and electronic features to specifically recognize the amino acid sequence (P1, P1′, P2, P3 and P4) of the natural substrate. Inhibitors must be designed with molecular features able to strongly interact with the sub-pockets, thus resulting in efficient competition with the substrate. We began our drug discovery campaign by creating a robust molecular docking protocol, aiming to use it to screen commercial databases and identify small-molecule binders. As a starting point, we chose to use a crystal structure of SARS-CoV-2 Mpro (PDB: 6 W63) in complex with the noncovalent, nonpeptidic inhibitor X77. It is well-known that, in comparison to the apo structure, the co-crystallization with a ligand places the residues of the active site in a more appropriate conformation for molecular docking predictions. The pose and interactions of X77 in the active site of Mpro (Fig. 2 ) reveal that the pyridine ring sits in the S1 pocket and its aromatic nitrogen performs an H-bond donation to the protonated Nε of His163, a key interaction also found in the binding mode of the natural substrate through the P1 glutamine. The 4-tert-butyl-benzene fits snugly into the S2 hydrophobic pocket, the cyclohexyl ring performs hydrophobic interaction in the S4 site and one of the amide carbonyls interacts through hydrogen bonding with the backbone of Glu166. Finally, X77 makes two key anchoring H-bond interactions with the backbone of Cys145 and the imidazole ring in the S1′ site performs a π-π stacking with His41 (Fig. 2A).
Fig. 2.

X-ray co-crystal structure of X77 in complex with Mpro (PDB entry 6 W63). (A) Key residues are shown as cyan sticks, H-bond interactions and π-π stackings are depicted as red and green dashes, respectively. (B) Surface rendering noting the binding pockets and the overlapped poses of X77 in the crystal structure (orange sticks) and in the re-docking experiment (purple sticks). The calculated RMSD between the two poses is 0.559.
Our in silico work began with the preparation of the Mpro structure, which was pre-processed and minimized using the Protein Preparation Wizard in the Schrödinger suite (see Experimental Section). The minimized structure was employed to create a docking protocol using the Glide algorithm.36 Before moving forward with the virtual screening, the protocol was validated by redocking experiments and by calculating the enrichment factor using 52 known inhibitors and 100 decoys from the DUD-E protease database.37 As reported by Gavernet and co-workers,38 the lack of adequate validation of docking protocols has been a significant issue in the recently published research on Mpro inhibitors, leading to the disclosure of many false-positive hits. Our docking procedure precisely reproduced the crystal pose of the cognate ligand (RMSD = 0.559, Fig. 2B), and returned an excellent enrichment factor (ROC = 0.90, AUC = 0.92, Fig. S2) when challenged against a database of decoy protease inhibitors, confirming the accuracy of the docking protocol and its ability to predict binders.
A consensus scoring approach was used for the virtual screening of a subset of the ZINC15 database.39 It is well established that consensus docking outperforms single docking for both scoring and pose prediction accuracy.40 The subset of ligands was obtained by filtering the large ZINC15 database using criteria such as molecular weight, LogP, in-stock availability, and absence of reactive groups (Fig. 2). The structures obtained were prepared with the LigPrep function, generating a total of 5.2 million ligands for the virtual screening. Because of its calculation speed, we initially used the HTVS function of Glide to screen the whole dataset and then redocked the top-ranking ligands with the standard precision (SP) setting. Next, docking poses were rescored using the Molecular Mechanics Generalized Born Surface Area (MM-GBSA) function41 to limit the number of false positives. Finally, the top 1,000 ranking hits were screened for PAINs patterns and the poses visually inspected, leading to the selection 15 compounds (Fig. 3 A) showing the highest chemical diversity (Fig. S3), which were purchased for biological evaluation. A table summarizing the calculated docking scores, binding energies, and selected physicochemical properties is given in the Supporting Info (Table S1).
Fig. 3.

(A) Workflow for the virtual screening leading to the identification of 15 virtual hits. The number of structures filtered during each step is given in the left bars. (B) Screening of the virtual hits against SARS-CoV-2 Mpro using the FRET assays. Proteolysis was monitored by cleaved substrate fluorescence following a 15 min incubation of 50 µM compound with 15 nM Mpro. Residual percent enzyme activity was calculated after normalization of the initial velocity to the DMSO negative control. Results are the average standard deviation (error bars) of three repeats. Boceprevir was used as positive control. (C) Concentration-response curves of compound 8. Values were obtained from linear regression analysis of initial velocities and IC50 curves were plotted using the software Prism. The assay was performed also in the presence of 0.02 % of Triton-X (blue values) to rule out aggregation-based inhibitory effects. Values are average of three independent experiments. The structure of 8 is also shown.
Recombinant main protease was expressed in BL21 (D3) E. coli after transfection with a pET-29a(+) vector, containing the gene for SARS-CoV-2 Mpro and harboring a C-terminal His6-tag. The tag was not removed during protein purification having been already shown that native Mpro is as active as C-terminal His6-tag Mpro.21, 42 Protein purification was carried out with HisTrap column (Ni Sepharose) and finally with size-exclusion chromatography to greater than 90 % purity based on Coomassie staining SDS-PAGE analysis (Fig. S4). Native mass spectrometry (Fig. S5) confirmed the molecular weight of His6-tag Mpro without the N-terminal methionine, likely cleaved by E. coli methionine aminopeptidase.
Next, a FRET-based enzymatic assay was established to measure the proteolytic activity of the recombinant enzyme and test the virtual hits. Briefly, kinetic measurements were carried out in reaction buffer (20 mM HEPES, 120 mM NaCl, 0.5 mM TCEP, 0.4 mM EDTA, 20 % glycerol, pH = 7.5) containing Mpro at a final concentration of 15 nM. The inhibitory activity of the purchased compounds was initially investigated at 50 µM and they were pre-incubated with the enzyme for 15 min at 30 °C. The reaction was initiated by adding a dabcyl-edans labeled peptide substrate dissolved in buffer, which generates a fluorescent product after enzymatic cleavage. Fluorescent output was measured at 90 s intervals and the reaction was monitored until completion (around 2.5 h). Baseline fluorescence of the test compounds and substrate were subtracted from the kinetic measurements while DMSO and boceprevir, a known SARS-CoV-2 main protease inhibitor,21, 43 were used as negative and positive controls, respectively. The initial velocity was calculated by linear regression using the data points from the first 30 min of the reaction and normalized to the DMSO negative control, furnishing the residual percentage enzyme activity. All experiments were carried out in triplicate.
Several virtual hits exhibited some level of activity in the FRET assay (Fig. 3B), and above all, two compounds (8 and 10) showed more than 50 % Mpro inhibition at 50 µM. The activity of these two inhibitors was further characterized with concentration–response assays to determine the IC50 values. To rule out non-specific effects due to aggregation-based inhibition, the IC50 experiments were repeated in the presence of the detergent Triton-X, a well-established assay for the detection of promiscuous inhibitors.44 A modest IC50 of 54.0 µM (Fig. S6) was obtained for compound 10, whereas derivative 8 (Fig. 3C) showed an interesting IC50 value of 24.2 µM. For comparison, boceprevir inhibited with an IC50 of 12.1 µM (Fig. S7) in this assay (the reported IC50 in a similar FRET-based assays is 1.5–5.4 µM).,19, 21, 32 Furthermore, the activity of 8 was not attenuated in the presence of Triton-X (Fig. 3C), excluding aggregation effects and corroborating a specific noncovalent interaction with the enzyme.
Compound 8 belongs to the family of 2-thiobenzimidazoles and similar derivatives have been reported as thromboxane receptor antagonists (US Pat. 5,124,336) and, more interestingly, as inhibitor of human chymase (US Pat. 7,268,145), a chymotrypsin-like serine protease found in mast cells.45 Nevertheless, no antiviral activity or inhibition of cysteine proteases has ever been reported for these heterocycles, suggesting that compound 8 is a promising scaffold for the development of a new class of inhibitors of coronavirus main protease.
In the predicted pose (Fig. 4 A-B), the 2-thiobenzimidazole ring of compound 8 sits snugly in the active site of the Mpro and its substituents interact with the different sub-pockets of the enzyme. The calculated docking score is −9.435, the best among the series of virtual hits. The two carbonyl groups perform multiple hydrogen bonding interactions with the residues of the “oxyanion hole” (Gly143, Ser144 and Cys145), a critical substrate recognition region in cysteine proteases. The imidazolidinone ring lies in the S1 site, similarly to the P1 pyrrolidinone found in most of the peptidomimetic inhibitors, although not deeply enough to the perform the canonical H-bonding interactions with Glu166 and Phe140. The N-benzyl substituent occupies the S2 hydrophobic pocket and the C5 chlorine extends in the S4 site. No occupation of the S1′ site is observed in the predicted pose.
Fig. 4.

(A) Docking pose of compound 8 in the active site of SARS-CoV-2 Mpro. Key residues are shown as cyan sticks, H-bond interactions are depicted as red dashes. (B) Surface rendering of the docking pose noting the binding pockets. (C) Overview of the synthetic modification carried out for the hit expansion.
We used these in silico predictions and literature data to direct our synthetic efforts for SAR exploration and hit expansion (Fig. 4C). The first straightforward modification to improve the binding affinity, was the replacement of a hydrogen bond donor group on the imidazolidinone ring with an acceptor to interact with the Nε of His163 in the S1 site, analogously to the contact performed by the pyridine ring in X77 and in other known inhibitors.31, 33 For this reason, we planned to change the imidazolidinone ring to an oxazolidinone group. We also probed the conversion of the C5 chlorine to methyl and tert-butyl, since the S4 pocket is known to be profitably occupied by small lipophilic groups.9 Moreover, as mentioned above, compound 8 does not occupy the S1′ sub-pocket with any of its substituents. Previous structure–activity studies have shown that aromatic rings, in particular phenyl groups and 5-membered heterocycles, fit well in the S1′ site of Mpro increasing the binding affinity of small molecule inhibitors.16, 33 To target this pocket, a benzyl and a 2‑furanyl substituent (with a methylene spacer) were introduced at the α-carbon of the amide group. According to our MM-GBSA calculations, this change leads to a favorable gain in free energy of binding (the average ΔΔGb is −10 kcal/mol) and, in the case of the 2‑furanyl substitution, it adds a π-π stacking interaction with His41 (Fig. S8). This modification generates a chiral center where the R enantiomer has the highest predicted affinity. Finally, following the observation that the distance between the C6 hydrogen and the carbonyl group of Arg188 is 2.7 Å (Fig. 4A), we replaced this hydrogen with chlorine and hydroxy groups expecting to add a halogen and a hydrogen bond, respectively. Being an intermediate for the synthesis of the C6 hydroxy derivative, a methoxy group was also probed in this position. Guided by this rationale, a focused small library of analogues of 8 was designed and synthesized (Table 1 ), comprising 23 derivatives (compounds 16–38) bearing the patterns of substitution described above. Chiral analogues were prepared as racemic mixtures to ease the synthetic work, aiming to address chirality during the hit-to-lead optimization.
Table 1.
Structure and enzymatic activity of the 2-thiobenzimidazole derivatives synthesized.
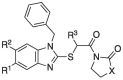 | |||||
|---|---|---|---|---|---|
| Cmpd | X | R1 | R2 | R3 | IC50 (µM) |
| 8 | NH | Cl | H | H | 24.2 |
| 16 | NH | Me | H | H | NAa |
| 17 | NH | t-Bu | H | H | NA |
| 18 | O | Cl | H | H | 62.2 |
| 19 | O | Me | H | H | NA |
| 20 | O | t-Bu | H | H | 35.1 |
| 21 | NH | Cl | H | Bn | 29.9 |
| 22 | NH | Me | H | Bn | 32.4 |
| 23 | O | Cl | H | Bn | 26.7 |
| 24 | O | Me | H | Bn | 36.5 |
| 25 | O | t-Bu | H | Bn | 29.5 |
| 26 | O | Cl | H | CH2-furanyl | 14.9 |
| 27 | O | H | Cl | H | NA |
| 28 | O | H | OMe | H | NA |
| 29 | O | H | OH | H | NA |
| 30 | O | H | Cl | CH2-furanyl | 52.9 |
| 31 | O | H | OMe | CH2-furanyl | 53.9 |
| 32 | O | H | OH | CH2-furanyl | 55.8 |
| 33 | O | Cl | Cl | H | 29.5 |
| 34 | O | Cl | OMe | H | 52.5 |
| 35 | O | Cl | OH | H | 35.8 |
| 36 | O | Cl | Cl | CH2-furanyl | 30.8 |
| 37 | O | Cl | OMe | CH2-furanyl | 27.7 |
| 38 | O | Cl | OH | CH2-furanyl | 46.1 |
Not active (no inhibition observed at 50 µM).
For the synthesis of the hit expansion library (compounds 16–38), we developed a convergent approach that started with the preparation of two separate building blocks: α-substituted chloroacetyl imidazolidinones (40a and 42a) or oxazolidinones (40b, 42b, and 46) (Scheme 1 ) and 2-thiobenzimidazoles 50a-i (Scheme 2 ). These building blocks were coupled in a single final step to generate the target compounds 16–38 (Scheme 2). Three different routes were used to prepare the α-substituted chloroacetyl imidazolidinones, depending on the nature of the substituent on the α-carbon (Scheme 1). The N-chloroacetyl imidazolidinone and oxazolidinones 40a and b were obtained in a single step by reacting chloroacetyl chloride with imidazolidinone 39a or oxazolidinone 39b under basic conditions. On the other side, l-phenylalanine was used as starting material for the synthesis of the α-benzyl analogues. First, a chlorination protocol comprising of a nitrosylation of the primary amine and subsequent chlorination by the acidic media, afforded the α-chloro acid 41. These conditions led to the racemization of the chiral center. Building blocks 42a and b were synthesized by activating the acid in situ with thionyl chloride followed by acylation with 39a or b. The preparation of the α-methylenefuranyl derivatives required a longer synthetic route starting from furfural. In this case only the oxazolidinone compound was prepared because of synthetic accessibility. Knoevenagel condensation with Meldrum’s acid, followed by sodium borohydride reduction, furnished the alkylated β-ketoester 43. Next, hydrolysis and decarboxylation of 43, carried out at high temperature in pyridine/water media, provided 3‑furanyl propionic acid 44. The key oxazolidinone intermediate 45 was obtained by amidation reaction using standard EDC conditions. Lastly, treatment with dibutylboryl triflate generated a stable enolate intermediate which was chlorinated with N-chlorosuccinimide, affording the target building block 46.
Scheme 1.

Synthetic procedures for the synthesis of α-substituted chloroacetyl imidazolidinones and oxazolidinones.
Scheme 2.

Synthetic pathway for the preparation of the 2-thiobenzimidazole building blocks and for the final compounds. Structural data for 16–38 are given in Table 1.
The synthetic pathway for the preparation of the 2-thiobenzimidazoles 50a-i, began with the N-alkylation of the 2-nitroaniline starting materials 47a-e using benzyl bromide in DMF, furnishing N-benzyl anilines 48a-e (Scheme 2). The addition of a strong base (sodium hydride) was necessary due to the poor nucleophilicity of the ortho-nitroaniline group. The 5‑methoxy functionality (compounds 48f-g) was introduced by nucleophilic aromatic substitution of the 5‑chloro derivatives 48d-e with in-situ generated sodium methoxide (Scheme 2). Next, the diamine intermediates 49a-g were obtained after Bechamp reduction of the aromatic nitro group using an iron catalyst and ammonium chloride as a proton source. The following cyclization, promoted by thiocarbonyldiimidazole (TDCI), generated the 2-thiobenzilimidazole core (compounds 50a-g). Phenol derivatives 50 h-i were obtained after deprotection of the corresponding methoxy analogues with HBr at high temperature. A final coupling reaction with one of the appropriate chloroacetyl building blocks afforded the designed targets 16–38.
The small library prepared was tested in the FRET-based enzymatic assay to determine the IC50 values (Table 1) and obtain preliminary structure activity relationships. From this analysis, it would seem that the optimal substituent to target the S4 subpocket is the C5 chloro, since both bigger (tert-butyl) and smaller (methyl) lipophilic groups show decreased potency (e.g., 16, 17, 19 and 20). Also, removal of the C5 substituent resulted in a substantial loss of activity (27–32) and the general impact on potency is Cl > t-Bu > Me ≫ H. The switch from imidazolidinone to oxazolidinone led to divergent results. With a C5 tert-butyl, we observed a positive impact on the inhibitory activity (17 vs 20), whereas in all the other cases no significant difference was noted. Considering also the better synthetic accessibility, we decided to retain the oxazolidinone ring in subsequent derivatizations. As expected, targeting the S1′ subpocket had a beneficial effect on the enzymatic activity of the inhibitors. For example, inactive C5‑methyl derivatives 16 and 19 regained potency with the insertion of the benzyl ring targeting the S1′ (see compounds 22 and 24, showing IC50 values of 32.4 and 36.5 µM, respectively). The best results were obtained with the methyl-2‑furanyl group in this position, and in particular with derivative 26, which has the highest potency in the whole series with an IC50 of 14.9 µM. The predicted docking pose of 26 is shown in Fig. S8, highlighting all the interactions with the active site of Mpro. In particular, the oxazolidinone moiety makes a series of H-bond interactions with the oxyanion hole residues (Gly143, Ser144 and Cy145) and with His163 in the S1 site, while the furanyl ring sits in the S1′ pocket performing a π-π stacking with His41. The last set of derivatives bearing a substituent on the C6 carbon (30–38), did not show an increase in potency, suggesting that they are unable to interact with the Arg188 carbonyl group, contrary to what was initially predicted.
To investigate the drug-likeness of compound 26, we calculated in silico several ADME properties using the software QikProp (Schrödinger Inc.). As a result, all the calculated pharmaceutical properties of 26 fall in the range of 95 % of known drugs (Table S2).
In conclusion, we report a multidisciplinary approach encompassing molecular docking, protein expression, in-vitro assays and synthetic medicinal chemistry leading to the identification of a family of SARS-CoV-2 main protease inhibitors. Our best inhibitor, compound 26, shows a micromolar activity against the recombinant enzyme (IC50 = 14.9 µM) and drug-like properties, suggesting that this novel class of benzimidazoles could be a promising new scaffold for further development.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We would like to express special thanks to Sara A. Thannickal and Kenneth A. Stapleford (NYU Grossman School of Medicine, NY) for helpful discussion in the preparation of this manuscript. This research was supported by New York University Abu Dhabi. Part of this work was carried out using Core Technology Platform resources at New York University Abu Dhabi.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bmcl.2022.128867.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.World Health Organization. WHO Coronavirus (COVID-19) Dashboard. https://covid19.who.int/ (accessed April 26, 2022).
- 2.Dispinseri S., Marzinotto I., Brigatti C., et al. Seasonal betacoronavirus antibodies′ expansion post-BNT161b2 vaccination associates with reduced SARS-CoV-2 VoC neutralization. J Clin Immunol. 2022;42:448–458. doi: 10.1007/s10875-021-01190-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harvey W.T., Carabelli A.M., Jackson B., et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat Rev Microbiol. 2021;19:409–424. doi: 10.1038/s41579-021-00573-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thiel V., Herold J., Schelle B., Siddell S.G. Viral replicase gene products suffice for coronavirus discontinuous transcription. J Virol. 2001;75:6676–6681. doi: 10.1128/jvi.75.14.6676-6681.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Domling A., Gao L. Chemistry and biology of SARS-CoV-2. Chem. 2020;6:1283–1295. doi: 10.1016/j.chempr.2020.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thiel V., Ivanov K.A., Putics A., et al. Mechanisms and enzymes involved in SARS coronavirus genome expression. J Gen Virol. 2003;84:2305–2315. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- 7.Yang H., Xie W., Xue X., et al. Design of wide-spectrum inhibitors targeting Coronavirus main proteases. PLoS Biol. 2005;3:1742–1752. doi: 10.1371/journal.pbio.0030324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ullrich S., Nitsche C. The SARS-CoV-2 main protease as drug target. Bioorg Med Chem Lett. 2020;30 doi: 10.1016/j.bmcl.2020.127377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rut W., Groborz K., Zhang L., et al. SARS-CoV-2 Mpro inhibitors and activity-based probes for patient-sample imaging. Nat Chem Biol. 2021;17:222–228. doi: 10.1038/s41589-020-00689-z. [DOI] [PubMed] [Google Scholar]
- 10.Gil C., Ginex T., Maestro I., et al. COVID-19: drug targets and potential treatments. J Med Chem. 2020;63:12359–12386. doi: 10.1021/acs.jmedchem.0c00606. [DOI] [PubMed] [Google Scholar]
- 11.Muramatsu T., Takemoto C., Kim Y.-T., et al. SARS-CoV 3CL protease cleaves its C-terminal autoprocessing site by novel subsite cooperativity. Proc Natl Acad Sci USA. 2016;113:12997–13002. doi: 10.1073/pnas.1601327113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kang X., Zhong N., Zou P., Zhang S., Jin C., Xia B. Foldon unfolding mediates the interconversion between Mpro-C monomer and 3D domain-swapped dimer. Proc Natl Acad Sci USA. 2012;109:14900–14905. doi: 10.1073/pnas.1205241109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goyal B., Goyal D. Targeting the dimerization of the main protease of coronaviruses: a potential broad-spectrum therapeutic strategy. ACS Comb Sci. 2020;22:297–305. doi: 10.1021/acscombsci.0c00058. [DOI] [PubMed] [Google Scholar]
- 14.Guenther S., Reinke P.Y.A., Fernandez-Garcia Y., et al. X-ray screening identifies active site and allosteric inhibitors of SARS-CoV-2 main protease. Science. 2021;372:642–646. doi: 10.1126/science.abf7945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Patick A.K., Potts K.E. Protease inhibitors as antiviral agents. Clin Microbiol Rev. 1998;11:614–627. doi: 10.1128/cmr.11.4.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang L., Lin D., Sun X., et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved -ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kneller D.W., Phillips G., O'Neill H.M., et al. Structural plasticity of SARS-CoV-2 3CL Mpro active site cavity revealed by room temperature X-ray crystallography. Nat Commun. 2020;11:3202. doi: 10.1038/s41467-020-16954-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang H., He S., Deng W., et al. Comprehensive insights into the catalytic mechanism of middle east respiratory syndrome 3C-Like protease and severe acute respiratory syndrome 3C-like protease. ACS Catal. 2020;10:5871–5890. doi: 10.1021/acscatal.0c00110. [DOI] [PubMed] [Google Scholar]
- 19.Owen D.R., Allerton C.M.N., Anderson A.S., et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science. 2021;374:1586–1593. doi: 10.1126/science.abl4784. [DOI] [PubMed] [Google Scholar]
- 20.Boras B., Jones R.M., Anson B.J., et al. Preclinical characterization of an intravenous coronavirus 3CL protease inhibitor for the potential treatment of COVID19. Nat Commun. 2021;12:6055. doi: 10.1038/s41467-021-26239-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma C., Sacco M.D., Hurst B., et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020;30:678–692. doi: 10.1038/s41422-020-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vandyck K., Abdelnabi R., Gupta K., et al. ALG-097111, a potent and selective SARS-CoV-2 3-chymotrypsin-like cysteine protease inhibitor exhibits in vivo efficacy in a Syrian Hamster model. Biochem Biophys Res Commun. 2021;555:134–139. doi: 10.1016/j.bbrc.2021.03.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qiao J., Li Y.-S., Zeng R., et al. SARS-CoV-2 Mpro inhibitors with antiviral activity in a transgenic mouse model. Science. 2021;371:1374–1378. doi: 10.1126/science.abf1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma C., Xia Z., Sacco M.D., et al. Discovery of Di- and trihaloacetamides as covalent SARS-CoV-2 main protease inhibitors with high target specificity. J Am Chem Soc. 2021;143:20697–20709. doi: 10.1021/jacs.1c08060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dai W., Zhang B., Jiang X.-M., et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020;368:1331–1335. doi: 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hattori S.-I., Higashi-Kuwata N., Hayashi H., et al. A small molecule compound with an indole moiety inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat Commun. 2021;12:668. doi: 10.1038/s41467-021-20900-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chamakuri S., Lu S., Ucisik M.N., et al. DNA-encoded chemistry technology yields expedient access to SARS-CoV-2 Mpro inhibitors. Proc Natl Acad Sci USA. 2021;118 doi: 10.1073/pnas.2111172118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Konno S., Kobayashi K., Senda M., et al. 3CL Protease inhibitors with an electrophilic arylketone moiety as anti-SARS-CoV-2 agents. J Med Chem. 2022;65:2926–2939. doi: 10.1021/acs.jmedchem.1c00665. [DOI] [PubMed] [Google Scholar]
- 29.Vagner J., Qu H., Hruby V.J. Peptidomimetics, a synthetic tool of drug discovery. Curr Opin Chem Biol. 2008;12:292–296. doi: 10.1016/j.cbpa.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eng H., Dantonio A.L., Kadar E.P., Obach R.S., et al. Disposition of PF-07321332 (Nirmatrelvir), an Orally Bioavailable Inhibitor of SARS-CoV-2 3CL Protease, across Animals and Humans. Drug Metab Dispos. 2022;50 doi: 10.1124/dmd.121.000801. Ahead of Print. DOI: 10.1124/dmd.121.000801. [DOI] [PubMed] [Google Scholar]
- 31.Zhang C.-H., Stone E.A., Deshmukh M., et al. Potent noncovalent inhibitors of the main protease of SARS-CoV-2 from molecular sculpting of the drug perampanel guided by free energy perturbation calculations. ACS Cent Sci. 2021;7:467–475. doi: 10.1021/acscentsci.1c00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ghahremanpour M.M., Tirado-Rives J., Deshmukh M., et al. Identification of 14 known drugs as inhibitors of the main protease of SARS-CoV-2. ACS Med Chem Lett. 2020;11:2526–2533. doi: 10.1021/acsmedchemlett.0c00521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kitamura N., Sacco M.D., Ma C., et al. Expedited approach toward the rational design of noncovalent SARS-CoV-2 main protease inhibitors. J Med Chem. 2022;65:2848–2865. doi: 10.1021/acs.jmedchem.1c00509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Han S.H., Goins C.M., Arya T., et al. Structure-based optimization of ML300-derived, noncovalent inhibitors targeting the severe acute respiratory syndrome coronavirus 3CL protease (SARS-CoV-2 3CLpro) J Med Chem. 2022;65:2880–2904. doi: 10.1021/acs.jmedchem.1c00598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luttens A., Gullberg H., Abdurakhmanov E., et al. Ultralarge virtual screening identifies SARS-CoV-2 main protease inhibitors with broad-spectrum activity against coronaviruses. J Am Chem Soc. 2022;144:2905–2920. doi: 10.1021/jacs.1c08402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Friesner R.A., Banks J.L., Murphy R.B., et al. A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 37.Mysinger M.M., Carchia M., Irwin J.J., Shoichet B.K. Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J Med Chem. 2012;55:6582–6594. doi: 10.1021/jm300687e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Llanos M.A., Gantner M.E., Rodriguez S., et al. Strengths and weaknesses of docking simulations in the SARS-CoV-2 Era: the main protease (Mpro) case study. J Chem Inf Model. 2021;61:3758–3770. doi: 10.1021/acs.jcim.1c00404. [DOI] [PubMed] [Google Scholar]
- 39.Sterling T., Irwin J.J. ZINC 15 - ligand discovery for everyone. J Chem Inf Model. 2015;55:2324–2337. doi: 10.1021/acs.jcim.5b00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang R., Wang S. How does consensus scoring work for virtual library screening? An idealized computer experiment. J Chem Inf Comput Sci. 2001;41:1422–1426. doi: 10.1021/ci010025x. [DOI] [PubMed] [Google Scholar]
- 41.Thompson D.C., Humblet C., Joseph-McCarthy D. Investigation of MM-PBSA rescoring of docking poses. J Chem Inf Model. 2008;48:1081–1091. doi: 10.1021/ci700470c. [DOI] [PubMed] [Google Scholar]
- 42.Sacco M.D., Ma C., Lagarias P., et al. Structure and inhibition of the SARS-CoV-2 main protease reveal strategy for developing dual inhibitors against Mpro and cathepsin L. Sci Adv. 2020;6:eabe0751. doi: 10.1126/sciadv.abe0751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fu L., Ye F., Feng Y., et al. Both Boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat Commun. 2020;11:4417. doi: 10.1038/s41467-020-18233-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Feng B.Y., Shoichet B.K. A detergent-based assay for the detection of promiscuous inhibitors. Nat Protoc. 2006;1:550–553. doi: 10.1038/nprot.2006.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Irani A.A., Schechter N.M., Craig S.S., DeBlois G., Schwartz L.B. Two types of human mast cells that have distinct neutral protease compositions. Proc Natl Acad Sci USA. 1986;83:4464–4468. doi: 10.1073/pnas.83.12.4464. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.