

Abstract
Astrocytes are critical regulators of the immune/inflammatory response in several human central nervous system (CNS) diseases. Emerging evidence suggests that dysfunctional astrocytes are crucial players in seizures. The objective of this study was to investigate the role of transient receptor potential vanilloid 4 (TRPV4) in 4-aminopyridine (4-AP)-induced seizures and the underlying mechanism. We also provide evidence for the role of Yes-associated protein (YAP) in seizures. 4-AP was administered to mice or primary cultured astrocytes. YAP-specific small interfering RNA (siRNA) was administered to primary cultured astrocytes. Mouse brain tissue and surgical specimens from epileptic patient brains were examined, and the results showed that TRPV4 was upregulated, while astrocytes were activated and polarized to the A1 phenotype. The levels of glial fibrillary acidic protein (GFAP), cytokine production, YAP, signal transducer activator of transcription 3 (STAT3), intracellular Ca2+([Ca2+]i) and the third component of complement (C3) were increased in 4-AP-induced mice and astrocytes. Perturbations in the immune microenvironment in the brain were balanced by TRPV4 inhibition or the manipulation of [Ca2+]i in astrocytes. Knocking down YAP with siRNA significantly inhibited 4-AP-induced pathological changes in astrocytes. Our study demonstrated that astrocytic TRPV4 activation promoted neuroinflammation through the TRPV4/Ca2+/YAP/STAT3 signaling pathway in mice with seizures. Astrocyte TRPV4 inhibition attenuated neuroinflammation, reduced neuronal injury, and improved neurobehavioral function. Targeting astrocytic TRPV4 activation may provide a promising therapeutic approach for managing epilepsy.
Supplementary Information
The online version contains supplementary material available at 10.1007/s13311-022-01198-8.
Keywords: TRPV4, Seizures, Astrocyte, Inflammation, 4-Aminopyridine (4-AP)
Introduction
Approximately 60% of epileptic disorders have unknown etiology [1]. Neuroinflammation promotes the occurrence of seizures and neuronal damage after seizures [2], which is accompanied by the release of cytokines, including interleukin-1β (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor-α (TNF-α) [3]. Proinflammatory cytokines are primarily released by abnormal glia [4]. Thus, inhibiting neuroinflammation is thought to be of great clinical value in alleviating neuropathological changes that follow seizures, such as neuronal damage.
Transient receptor potential (TRP) channels are a group of nonselective cationic ion channels. These channels are expressed in a wide range of systems in humans. As a member of the TRP family, TRPV4 is activated by a wide range of stimuli, including physical factors (cell swelling [5–7], heat [7], and mechanical stimuli [8]), endogenous and exogenous chemical factors (endocannabinoids [9], arachidonic acid and its metabolites [9], 4α-phorbol esters and its derivatives [10]), GSK1016790A [11], low pH (pH < 6) [12], and natural compounds isolated from Andrographis paniculata [13]. TRPV4 is extensively expressed in the CNS, including hippocampal neurons [14], astrocytes [15], microglia [16], and oligodendrocytes [17], but it is mainly enriched in astrocytes [15]. Recent studies have shown that TRPV4 activation enhances inflammatory responses and cell damage. A reduction in tissue osmolarity activates TRPV4 and subsequently enhances the production of the proinflammatory cytokines IL-6 and IL-1β [18]. In fetal epithelial cells, the TRPV4 agonist GSK1016790A increases the release of IL-6 via the p38 and ERK pathways [19]. TRPV4 activation stimulates neuropeptide secretion from afferent nerves and induces neurogenic inflammation [20]. During cerebral ischemia, TRPV4 activation induces neuronal apoptosis in the hippocampus by downregulating the expression of phosphatidylinositol 3 kinase (PI3K)/protein kinase B (Akt) and NR2B-N-methyl-d-aspartate glutamate (NMDA) receptors and upregulating p38 mitogen-activated protein kinase (MAPK) signaling pathways [21, 22]. Pharmacological inhibition of TRPV4 reduces lipopolysaccharide (LPS)-induced proinflammatory cytokine production [23]. TRPV4 inhibition decreases infrasound‑induced IL-1β and TNF-α release in cultured glial cells, attenuating neuronal apoptosis [24].
Recent studies have shown that TRPV4 is closely related to seizures [25, 26]. In larval zebrafish, the TRPV4 antagonist RN-1734 almost completely inhibited hyperthermia-induced seizures [27]. Intracerebroventricular (Icv.) injection of the TRPV4 agonist GSK1016790A increased the activation of microglia and astrocytes, neuronal death, and the release of proinflammatory cytokines. TRPV4 antagonists markedly attenuate neuronal death after status epilepticus (SE) [28]. Activation of TRPV4 decreased Kv1.2 and Kv2.1 expression and delayed rectifier potassium current (IK), which are involved in the pathological changes after pilocarpine-induced SE (PISE) [29]. Compared with wild-type mice, TRPV4-KO mice had significantly lower frequencies of epileptic electroencephalographs (EEGs) [25]. These results indicate that TRPV4 is involved in seizure pathogenesis, but the mechanisms underlying seizures are complex and unclear. More studies are needed to explore the mechanism of TRPV4 in seizures.
The Hippo pathway was first discovered in Drosophila [30] and is highly conserved from Drosophila to mammals [31]. This pathway is a master regulator of tissue homeostasis, organ development, and tumor progression [31, 32]. YAP is the crucial transcriptional coactivator of the Hippo signaling pathway. Activation of this pathway enhances the phosphorylation of YAP and its subsequent proteasomal degradation or cytoplasmic retention, while inhibiting this pathway increases the dephosphorylation of YAP and its nuclear translocation, where YAP interacts with TEAD family proteins to initiate gene expression, inhibiting proliferation and inducing apoptotic effects under different pathophysiological conditions [33, 34]. YAP controls organ size in mammals by regulating cell differentiation, proliferation, and apoptosis [35–37]. Previous studies on YAP have mainly focused on its role in the immune response and tumorigenesis [38–40]. However, the function of YAP in the CNS is still poorly understood. In the CNS, YAP is selectively expressed by astrocytes and neural stem cells [41]. A previous study showed that YAP upregulation and activation promote the formation of glial scars after spinal cord injury (SCI) [42]. TRPV4 knockdown increased YAP nuclear translocation via the rho kinase pathway. YAP nuclear translocation regulates astroglial-mesenchymal transition induced by hemoglobin and increases astrocyte activation [43]. Another study showed that TRPV4 promoted matrix stiffness and TGFβ1-induced YAP expression and nuclear translocation [44]. These findings strongly suggest the presence of a signaling pathway involving TRPV4 and YAP.
In the present study, we investigated the role of TRPV4 in 4-AP-induced mice and primary cultured astrocytes and examined the upregulation of TRPV4 expression in epileptic patient brain slices. The results showed that trpv4 was a seizure-associated gene that was upregulated in 4-AP-induced mice. However, whether TRPV4 is involved in inflammatory responses and neuronal damage in 4-AP-induced mice is still unclear. This study focused on astrocytic TRPV4 to investigate the mechanism, including the signaling pathway involved in the TRPV4-induced inflammatory response and neuronal damage, in 4-AP-induced mice.
Materials and Methods
Bioinformatics Analysis
Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov) datasets were used to examine specifically dysregulated genes in seizures. Differentially expressed genes (DEGs) in the epilepsy and control groups were screened with the conditions P < 0.05 and |log2 FC|≥ 1.5. Gene Ontology (GO) enrichment analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis were performed based on the target genes using the Database for Annotation, Visualization and Integration Discovery (DAVID, https://david.ncifcrf.gov).
Animals
Eight-week-old male C57BL/6 wild-type mice (weighing 20 ± 2 g) were approved by the Hubei Province Center for Animal Experiments for use in this study. All animal care and experiments were performed according to the Institutional Animal Care and Use Committee of Wuhan University Medical School and the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 8023, revised 1978). Mice were grouped randomly and maintained with a 12-h light–dark cycle at 25 ± 1 °C with a relative humidity of 60∼80%; food and water were available ad libitum in the animal biosafety level III laboratory (ABSL-III) of Wuhan University. The exact number and groups of mice used in each experiment are described in the relevant results sections.
Human Tissue Specimens and Blood Samples
All procedures involving humans were performed according to the Helsinki Declaration of 1975, as revised in 2000 (World Medical Association Declaration of Helsinki 2000), and were approved by the Medical Ethics Committee of Zhongnan Hospital of Wuhan University. After informed consent was obtained, human brain specimens were collected from patients with epilepsy (n = 7) (1 female, 6 male) (25–58 years) who had undergone surgery at the Department of Neurosurgery of Zhongnan Hospital (Wuhan University, Wuhan, China). After identifying the epileptogenic zone, all patients underwent surgery according to electroclinical data and MRI evaluations. Zhongnan Hospital provided us with control samples (n = 9) (2 female, 7 male) (26–57 years) obtained from autopsies performed within 24 h of death on individuals who died of diverse causes but had no history of seizures or neurological diseases. Serum samples from epilepsy patients (n = 10, including the 7 epilepsy patients mentioned above) (3 female, 7 male) (25–66 years) were provided by the Department of Neurosurgery of Zhongnan Hospital. Ten patients were focal drug-resistant epilepsy patients. The control group consisted of healthy volunteers (n = 5) (5 male) (27–59 years) who did not have any neurological deficits or CNS disease antecedents and were enrolled at Zhongnan Hospital (Wuhan University, Wuhan, China).
4-AP-Induced Seizures in Mice and EEG Recordings
The 4-AP model was established as described previously [45]. Freely moving mice were anesthetized by isoflurane inhalation (RWD, Shenzhen, Guangdong, China). Then, the mice were placed in a stereotaxic instrument (RWD, Shenzhen, Guangdong, China), and two twisted silver steel electrodes were implanted into the bilateral hippocampal CA3 region (2.3 mm posterior to bregma, 2.1 mm lateral lobe, 2.0 mm deep from the pial surface). One week later, the TRPV4 antagonist HC-067047 (10 μM/mouse) or agonist GSK1016790A (1 μM/mouse) was injected into the lateral cerebral ventricle (0.3 mm posterior to bregma, 1.0 mm lateral lobe, and 2.5 mm deep from the pial surface). Twenty-four hours later, the seizure model was induced by intraperitoneal (i.p.) injection of 4-AP (5.6 mg/kg), and typical brain discharge was recorded by EEG for 2 h after 4-AP administration. Ten mice were examined in each group. The EEG signals were digitized with Lab Chart software (AD Instruments, Bella Vista, New South Wales, Australia). Seizure severity was evaluated according to the Racine scale and was classified into five levels: I, facial twitch; II, head nodding; III, unilateral forelimb clonus; IV, bilateral forelimb clonus; and V, tonic–clonic seizure, rearing, and failing. The brains were collected from the mice 24 h after 4-AP injection.
Primary Mouse Cortical Astrocyte Culture
Primary mouse cortical astrocytes were prepared as previously reported [46]. Briefly, trypsin enzymatic digestion and trituration were performed, and cortical cells were resuspended in complete medium (1 × Dulbecco’s modified Eagle’s medium (DMEM)/F12 (HyClone, Logan, Utah, USA), 10% fetal bovine serum (FBS) (GIBCO, Carlsbad, CA, USA), 1% l-glutamine (Biosharp, Hefei, Anhui, China), and 1% penicillin/streptomycin (Beyotime, Shanghai, China)) and seeded on poly-d-lysine-coated Petri dishes. Then, the cells were cultured in an incubator (37 °C, 5% CO2), and the medium was changed every 3 days to ensure adequate nutrition.
Western Blot Analysis
Proteins were extracted by homogenization in RIPA lysis buffer (Beyotime, Shanghai, China) with phenylmethanesulfonyl fluoride (PMSF; Biosharp, China) and phosphatase inhibitors (Roche, Switzerland). A BCA protein assay (BCA, Beyotime, Shanghai, China) was used to quantify the protein concentrations of the supernatants. After that, equal amounts of protein samples were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and transferred to a polyvinylidene fluoride membrane (PVDF; Millipore, UK). The PVDF membranes were then blocked with 5% BSA and incubated with primary antibodies overnight at 4 °C. Detailed information of the primary and secondary antibodies is listed in Table 1. After being incubated, the membranes were washed with Tris-buffered saline containing 0.2% Tween-20 (TBST) and subsequently incubated with a corresponding HRP-conjugated secondary antibody. Then, the membranes were washed with TBST. Finally, the immunoreactive bands were detected by an enhanced chemiluminescence (ECL, Santa Cruz, Dallas, Texas, USA) detection reagent. Band intensity was quantified by spot densitometric analysis using ImageJ software (version 1.41), and the results were normalized to β-actin levels and are reported as the relative intensity of the control.
Table 1.
Antibodies for Western blot and Immunofluorescence
| Antibody | Host | Company | Cat. No | Dilution | Duration |
|---|---|---|---|---|---|
| TRPV4 | Rabbit | Abcam | ab39260 | 1:1000 (1:200) | Overnight 4 °C |
| GFAP | Mouse | Cell signaling | #3670 | 1:1000 (1:100) | Overnight 4 °C |
| YAP | Mouse | Protein tech | 66,900–1-Ig | 1:1000 (1:400) | Overnight 4 °C |
| p-YAP(Ser127) | Rabbit | Cell signaling | #13,008 | 1:1000 | Overnight 4 °C |
| STAT3 | Rabbit | Proteintech | 10,253–2-AP | 1:1000 (1:200) | Overnight 4 °C |
| p-STAT3(Tyr705) | Rabbit | Cell signaling | #9145 | 1:2000 | Overnight 4 °C |
| SOCS3 | Rabbit | Proteintech | 14,025–1-AP | 1:1000 (1:200) | Overnight 4 °C |
| IL-6 | Rabbit | Bioss | bs-0782R | 1:1000 | Overnight 4 °C |
| TNF-α | Rabbit | Abclonal | A11534 | 1:1000 | Overnight 4 °C |
| IL-1β | Rabbit | Abcam | ab9722 | 1:1000 | Overnight 4 °C |
| β-Actin | Mouse | Proteintech | HRP-60008 | 1:10,000 | Overnight 4 °C |
| Anti-mouse IgG-HRP | Goat | PMK Biotech | PMK-014-091 M | 1:20,000 | 2 h RT |
| Anti-rabbit IgG-HRP | Goat | PMK Biotech | PMK-014–090 | 1:20,000 | 2 h RT |
|
594 goat anti-mouse IgG 594 goat anti-rabbit IgG 488 goat anti-mouse IgG 488 goat anti-rabbit IgG |
Goat Goat Goat Goat |
Abbkine Abbkine Abbkine Abbkine |
A23410-1 A23420 A23210 A23220 |
1:500 1:500 1:500 1:500 |
2 h RT 2 h RT 2 h RT 2 h RT |
The antibody dilution used for immunofluorescence is enclosed in brackets
Quantitative Real-Time PCR
Total RNA was extracted from isolated mouse brain tissue or primary cultured astrocytes using TRIzol reagent (Invitrogen Life Technologies Corporation, USA). RNA concentration and quality were quantified by a spectrophotometer (ND-100; Thermo Fisher Scientific, Waltham, MA, USA). First-strand complementary DNA (cDNA) was synthesized by reverse RNA transcription using a Revert Aid First Strand cDNA Synthesis Kit (Thermo Scientific, Rockford, IL, USA). Quantitative real-time PCR (qRT–PCR) was performed with a SYBR Green Real-Time PCR (RT–PCR) master mix kit (Vazyme, Nanjing, Jiangsu, China) according to the manufacturer’s instructions. The primers used are listed in Table 2. The expression of target genes is presented as the fold change normalized to the mRNA level of the internal control β-actin. The 2−ΔΔCt relative quantification method was used to analyze the expression of target genes.
Table 2.
q-PCR primer sequences applied
| Primer | Forward primer 5–3′ | Reverse primer 5–3 |
|---|---|---|
| Trpv4 | TCAACTCGCCCTTCAGAGACA | AAGTAGCCTCCCTCATCCTTGG |
| Gfap | TAAAGACTGTGGAGATGCGGG | CAGAAGGAAGGGAAGTGCTGG |
| Socs3 | CAAGGCCGGAGATTTCGCTTC | TCACGGCGCTCCAGTAGAATC |
| Yap | AGACAACAACATGGCAGGACC | GGATGTGGTCTTGTTCTTATGGCT |
| Il-6 | TAGTCCTTCCTACCCCAATTTCC | TTGGTCCTTAGCCACTACTTC |
| Tnf | GAGGCACTCCCCCAAAAGAT | GGCCATTTGGGAACTTCTCATC |
| Il-1β | TGCCACCTTTTGACAGTGATG | ATGTGCTGCTGCGAGATTTG |
| inos | GGGTCACAACTTTACAGGGAGT | CTCCGTTCTCTTGCAGTTGAC |
| β-actin | CACGATGGAGGGGCCGGACTCATC | TAAAGACCTCTATGCCAACACAGT |
| Cd109 | GACCCGGAGCAAATGTGACTA | AGGTAGTGCCGGAAGAACGAG |
| S100a10 | CATGATGCTTACGTTTCACAGGTT | TGGTCCAGGTCCTTCATTATTTTG |
| Emp1 | GTGCTACTGGCTGGTCTCTTT | ACCACAAGGGAGATGATGGAG |
| Amigo2 | ATCCCGGTGTCGTTTGTCAAG | AGCTCTTGGAATGTGGCACTC |
| C3 | GCAGAGTTTGAGGTGAAGGAA | GTAATAAAATGTCTCTGTGGGCTC |
| Gbp2 | CTGACCCTGAAAAAAGGAGCTGA | CGTGGCCCATTGACTATGATGC |
Immunofluorescence Analysis
Cellular immunofluorescence was used to illustrate the experimental procedure. Primary cultured astrocytes were washed and fixed with 4% paraformaldehyde (PFA, Biosharp) at room temperature for 30 min. After being washed, the cells were permeabilized with filtered PBS containing 0.05% Triton-X 100 (Sigma-Aldrich) for 20 min. The primary cultured astrocytes were incubated with a blocking solution (5% FBS) and then incubated overnight at 4 °C in 3% BSA (Sigma-Aldrich, St. Louis, MO, USA) containing primary antibodies. On the second day, the cells were washed and incubated with a corresponding secondary antibody for 2 h at 37 °C. Detailed information on the primary and secondary antibodies is listed in Table 1. Then, DAPI (300 nM, Sigma-Aldrich) (3 min at 37 °C in the dark) was used to label the nuclei. Finally, immunofluorescence images were obtained using a confocal laser scanning microscope (Leica-LCS-SP8-STED, Leica, Wetzlar, Hesse, Germany).
Nissl Staining
Nissl staining was used to assess neuronal damage and loss. After fixation, paraffin embedding, and sectioning (4 μm), the brain tissue was deparaffinized, hydrated, and stained with toluidine blue solution (Boster Biotech, China).
Immunohistochemistry
Fixed mouse brains were paraffin-embedded and sliced into 4-μm sections. After antigen retrieval was performed, the sections were blocked with 10% normal goat serum and then stained overnight with GFAP (1:100, Cell Signaling Technology, #3670) or YAP (1:100, Protein tech, 66,900–1-Ig) antibodies. On the second day, the sections were incubated with a horseradish peroxidase (HRP)–conjugated anti-mouse antibody (1:500, Abbkine, China) and developed using DAB peroxidase substrate (Beyotime Biotechnology, China). Finally, an Olympus AH-2 light microscope (× 200; Olympus) was used to photograph the sections, and ImageJ software was used to count the number of visualized cells.
Transfection
YAP-siRNA was designed and synthesized by GenePharma Company (Shanghai, China). The siRNA was transfected according to the protocol provided by the manufacturer (Polyplus, Strasbourg, France). Briefly, primary astrocytes were cultured, the transfection mixture was prepared, and the cells were transfected. Primary astrocytes were seeded on a 6-well plate in DMEM/F12 containing 10% FBS and incubated in 95% O2 and 5% CO2 at 37 °C. Astrocytes were transfected when they reached 60–80% confluence. The siRNA was diluted in jetPRIME buffer, vortexed for 10 s, and centrifuged. Next, jetPRIME reagent was added, and the mixture was vortexed for 10 s, centrifuged, and incubated for 10 min at room temperature. The siRNA transfection mixture was then added to the plate, and astrocytes were harvested 24 h after transfection.
Fluorescent Calcium Imaging
[Ca2+]i were measured by Fluo-4 AM (Beyotime, Shanghai, China). Primary cultured astrocytes were seeded in confocal dishes, stimulated with 4-AP (5 mM) for 24 h, and then incubated with Fluo-4 AM at 37 °C for 30 min in the dark. Then, the astrocytes were washed with HBSS (HyClone, Logan, Utah, USA) 3 times and incubated for 40 min in HBSS at 37 °C. Images were obtained by fluorescence microscopy (Leica). The ΔF/F0 ratio was calculated as (F–F0)/F0. F0 is the mean value of F before GSK1016790A (200 nM) treatment (baseline), and ΔF was the difference between the F value at each time point of the experiment and the corresponding F0.
Enzyme-Linked Immunosorbent Assay
The protein concentrations of GFAP and C3 in the serum of normal and epileptic patients were quantified using ELISA (Newqidi Biological Technology, Wuhan, China) kits according to the manufacturer’s instructions.
Statistical Analysis
All data are presented as the mean ± standard error of the mean (SEM). GraphPad Prism v7.00 (GraphPad, La Jolla, CA, USA) was used to analyze the data and construct the graphs. Student’s t test and one-way analysis of variance (ANOVA) were performed to determine the significant differences between groups. For multiple groups, one-way ANOVA was used for statistical analysis. Fisher’s exact test was performed to compare the mortality of mice in the different groups. For in vivo experiments, the n values refer to the number of individual animals in each group. For in vitro studies, the n values indicate the number of times the experiment was independently replicated in cultures derived from different mice, as indicated in the figure legends. P < 0.05 indicated that the difference was statistically significant.
Results
Trpv4 Is a Seizure-Associated Gene
The gene chips GSE88992, GSE40490, and GSE44903 were selected from GEO datasets, and DEGs in the epileptic and control groups were screened by GEO tool online analysis with the screening criteria P < 0.05 and |log2 FC|≥ 1.5. Figure 1a–c shows the volcano plots of the DEGs in epileptic patients and healthy individuals in these 3 gene chips. Compared with those in the control group, 6895, 3778, and 962 DEGs were screened from the GSE88992, GSE40490, and GSE44903 datasets, respectively (Fig. 1d). A total of 31 DEGs were screened from the 3 gene chips (Fig. 1e). KEGG pathway analysis (Fig. 1f) and GO enrichment analysis (Fig. 1g) were performed on these 31 genes using DAVID. KEGG pathway analysis showed enrichment in 18 signaling pathways, including inflammatory mediator regulation of TRP channels (Fig. 1f). GO analysis showed significant enrichment in 11 processes, including the regulation of calcium ion import, which had the most significant difference (Fig. 1g), indicating that Ca2+ may play a vital role in seizures. We investigated intracellular and extracellular Ca2+ in subsequent experiments.
Fig. 1.

TRPV4 is a seizure-associated gene. (a–c) Volcano plots of the 3 gene chips: GSE88992, GSE40490, and GSE44903. (d) Upregulated and downregulated genes in the 3 gene chips. (e) Venn diagrams of the overlapping DEGs. (f–g) Images of KEGG pathway enrichment analysis (f) and biological processes determined by GO analysis (g) in the overlapping DEGs. (h–i) Members of the TRP family (h) and the Venn diagram showing the TRP family and 31 DEGs (i). (j) The protein–protein interaction network (PPI network) of 31 common DEGs. (k) Expression of Trpv4 in GSE88992, GSE40490, and GSE44903
Since most TRP channels are involved in [Ca2+]i regulation, we examined whether TRP family members were differentially expressed in animals with seizures. There are 28 members in the TRP family in mammals (Fig. 1h). Venn diagram analysis of the TRP family members and the 31 DEGs showed that only Trpv4 was common to the two groups (Fig. 1i). To explore the underlying relationships between DEGs, the Cytoscape, DAVID, and STRING platforms were used to analyze the protein–protein interaction network (PPI network) of the 31 DEGs. Furthermore, we found that 18 seizure susceptibility genes including Trpv4 constituted the PPI network (Fig. 1j). Therefore, we selected Trpv4 as the target gene and analyzed its expression in the GSE88992, GSE40490, and GSE44903 gene chips (Fig. 1k). The data showed that Trpv4 was a seizure-associated gene, and its expression was modified in seizures.
TRPV4 Inhibition Reduces 4-AP-Induced Seizure Susceptibility in Mice
We then verified the role of TRPV4 in the 4-AP seizure model and the underlying mechanisms. A TRPV4 agonist (GSK1016790A, 1 μM) and antagonist (HC-067047, 10 μM) were injected into the lateral cerebral ventricle (from bregma: −0.3 mm posterior, + 1.0 mm lateral, −2.5 mm ventral) in mice using a stereotaxic instrument. A mouse seizure model was established by intraperitoneal injection of 4-AP (5.6 mg/kg).
Spontaneous EEG recordings and neurobehavioral observations were performed to evaluate the model’s success and assess seizure severity (Fig. 2a–e, n = 10). The behavior of the mice was observed and recorded for 2 h after 4-AP injection. 4-AP-induced mice showed intermittent but not continuous seizures within 120 min, and the seizure grade of the mice was restored to level 2 before the cessation of the last seizure. During the seizures, the mice gradually recovered from grades 4–5 to grades 2–3 and eventually returned to baseline levels. The EEG recordings showed that compared with control mice, mice in the 4-AP and 4-AP + GSK1016790A groups had higher seizure severity (Fig. 2b), shorter seizure latency (Fig. 2c), and longer seizure duration (Fig. 2d). Mortality in the 4-AP + GSK1016790A group was higher than that in the 4-AP and 4-AP + HC-067047 groups (Fig. 2e), but the difference was not significant. These results suggested that TRPV4 inhibition decreased 4-AP-induced seizure susceptibility.
Fig. 2.

TRPV4 inhibition decreased 4-AP-induced spontaneous EEG seizures and neuronal damage in mice. a The behavior of mice was recorded for 2 h after 4-AP injection. Representative traces of EEG recordings and the corresponding enlarged part in control, 4-AP, 4-AP + HC-067047, and 4-AP + GSK1016790A mice at the middle of the 2 h window (n = 10). b–e Seizure severity (b), seizure latency (c), seizure duration (d), and mortality (e) of mice after 4-AP treatment in the presence or absence of a TRPV4 agonist and antagonist (n = 10). f HC-067047 markedly attenuated 4-AP-induced hippocampal neuronal damage (n = 6). g–i Nissl staining in the hippocampal DG, hilar, and CA3 regions in the 6 groups of mice. The brains were collected 24 h after 4-AP injection. The data are presented as the means ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001, Tukey’s test after one-way analysis of variance (ANOVA)
Then, we performed Nissl staining to investigate the effect of TRPV4 on neuropathic changes after seizures (Fig. 2f–i, n = 6). Compared with those in the sham group, many neurons in the DG, hilar, and CA3 regions of the hippocampus exhibited darker Nissl staining, cytoplasmic shrinkage, and triangulated pyknotic nuclei in the 4-AP, GSK1016790A, and 4-AP + GSK1016790A groups. In contrast, most of the neurons in the 4-AP + HC-067047 group were evenly stained light blue and had regularly shaped cell bodies. This result indicated that the TRPV4 antagonist HC-067047 dramatically attenuated 4-AP-induced hippocampal neuronal damage.
Overall, TRPV4 inhibition reduced seizure susceptibility and neuronal damage in 4-AP-induced mice. Next, we investigated the underlying molecular mechanism.
Astrocyte Activation and Proinflammatory Cytokine Production in 4-AP-Induced Mice Is TRPV4-Dependent
Our bioinformatics results suggested that inflammatory mediator regulation by TRP channels was involved in seizures (Fig. 1f), and TRPV4 was upregulated in the three gene chips (Fig. 1k). We next examined the expression of TRPV4 in 4-AP-induced mice and explored inflammatory changes in the brain microenvironment and the subcellular localization of TRPV4. To verify the role of TRPV4 in the occurrence and development of seizures and whether astrocytes were altered in 4-AP-induced mice, we first examined astrocyte activation and TRPV4 expression in the hippocampus of 4-AP-induced mice (in vivo). Then, we examined TRPV4 expression and astrocyte activation in 4-AP (5 mM)-induced primary cultured astrocytes (in vitro).
The expression of TRPV4 was upregulated at 6, 12, and 24 h after 4-AP injection, as demonstrated by immunofluorescence analysis (Figs. 3a and 4a, n = 3), Western blotting (Figs. 3b, d and 4b, d, n = 3), and qPCR (Figs. 3c and 4c, n = 3) in vivo and in vitro. However, there was no difference in TRPV4 expression in 4-AP-induced primary cultured hippocampal neurons and BV2 microglia (Additional File 1). The expression of GFAP was gradually increased at 12 and 24 h, as demonstrated by immunofluorescence analysis (Figs. 3a and 4a, n = 3), q-PCR (Figs. 3e and 4e, n = 3), and Western blotting (Figs. 3b, f and 4b, f, n = 3) in vivo and in vitro. The inflammatory status of 4-AP-induced mice worsened at 12 and 24 h because the mRNA levels of the proinflammatory factors IL-6, TNF, IL-1β, and iNOS (Fig. 3g–j) were upregulated at these time points (n = 3). In addition, 4-AP significantly increased IL-6, TNF, IL-1β, and iNOS mRNA levels (Fig. 4g–j) at 6, 12, and 24 h in primary cultured astrocytes (n = 3).
Fig. 3.

TRPV4 and GFAP expression levels and proinflammatory cytokine production were upregulated. a Confocal images demonstrate TRPV4 and GFAP expression in the hippocampus (n = 3). b–d The protein (b, d) and mRNA levels (c) of TRPV4 at 3, 6, 12, and 24 h after intraperitoneal injection of 4-AP (5.6 mg/kg) in mice (n = 3). b, e, f The protein (b, f) and mRNA levels (e) of GFAP at 3, 6, 12 h, and 24 h in 4-AP-induced mice (n = 3). g–j The mRNA expression levels of IL-6 (g), TNF-α (h), IL-1β (i), and iNOS (j) at 3, 6, 12, and 24 h in 4-AP-induced mice (n = 3). The data are presented as the means ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001, Tukey’s test after ANOVA
Fig. 4.

The expression of TRPV4, GFAP, and proinflammatory cytokines in 4-AP-induced primary cultured astrocytes. a Representative confocal imaging showing TRPV4 and GFAP expression at 3, 6, 12, and 24 h after 4-AP (5 mM) administration in primary cultured astrocytes (n = 3). b–d The protein (b, d) and mRNA levels (c) of TRPV4 at 3, 6, 12, and 24 h after 4-AP administration in primary cultured astrocytes (n = 3). b, e, f The protein (b, f) and mRNA levels (e) of GFAP at 3, 6, 12, and 24 h after 4-AP administration in primary cultured astrocytes (n = 3). g–j The mRNA levels of IL-6 (g), TNF-α (h), IL-1β (i), and iNOS (j) at 3, 6, 12, and 24 h after 4-AP administration in primary cultured astrocytes (n = 3). The data are presented as the means ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001, Tukey’s test after ANOVA
Taken together, these results suggested that 4-AP significantly increased TRPV4 expression, astrocyte activation, and proinflammatory cytokine production in vivo and in vitro. Since the expression levels of GFAP and TRPV4 gradually increased during the experimental period (3, 6, 12, and 24 h) after 4-AP treatment, we chose 24 h for subsequent experiments.
Signaling Pathway Involved in 4-AP-Induced Astrocyte Activation and Proinflammatory Cytokine Release
The abundant expression of TRPV4 in 4-AP-induced astrocytes and the brain tissue of 4-AP-induced mice suggested that TRPV4 may play a significant role in astrocyte activation. To verify whether cerebral cortical astrocytes were activated, mouse cerebral cortex astrocytes were isolated and cultured. Primary cultured astrocytes were exposed to 4-AP for 24 h. The TRPV4 agonist GSK1016790A (200 nM) and TRPV4 antagonist HC-067047 (10 μM) were used.
In astrocytes treated with 4-AP and TRPV4 agonist GSK1016790A, individually or in combination, the expression of the astrocyte activation marker GFAP was upregulated. The expression of SOCS3, a negative regulator of the STAT3 inflammatory pathway, was downregulated. Moreover, the TRPV4 antagonist HC-067047 suppressed 4-AP- and GSK1016790A-induced astrocyte activation and SOCS3 downregulation (Fig. 5a–b, e–f, n = 3). Additionally, the mRNA and protein expression levels of the proinflammatory cytokines IL-6 (Fig. 5b, g–h), TNF-α (Fig. 5i), IL-1β (Fig. 5j), and iNOS (Fig. 5k) were increased (n = 3). Furthermore, the STAT3 signaling pathway, which is a classic inflammatory pathway associated with astrocyte activation, was elevated in 4-AP- and GSK1016790A-treated astrocytes (Fig. 6a, e) (n = 3). However, inhibiting TRPV4 expression reversed the 4-AP- and GSK1016790A-induced phosphorylation of STAT3 (p-STAT3), astrocyte activation, and proinflammatory cytokine production.
Fig. 5.

TRPV4 promoted astrocyte activation, the production, and the release of proinflammatory cytokines in 4-AP-induced astrocytes. a Representative confocal imaging showing GFAP (green) and SOCS3 (red) expression in primary cultured astrocytes after treatment for 24 h with 4-AP, the TRPV4 antagonist HC-067047 (10 μM), or the TRPV4 agonist GSK1016790A (200 nM) (n = 3). b Representative immunoblots showing the protein expression of GFAP, SOCS3, and IL-6 in primary cultured astrocytes after the different treatments (n = 3). c–d Quantitative analysis of the mRNA (c) and protein (d) levels of GFAP (n = 3). e–f Quantitative analysis of the mRNA (e) and protein (f) levels of SOCS3 (n = 3). g, h Quantitative analysis of the mRNA (g) and protein (h) levels of IL-6 (n = 3). i–k The mRNA levels of TNF-α (i), IL-1β (j), and iNOS (k) (n = 3). The data are presented as means ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001, Tukey’s test after ANOVA
Fig. 6.

TRPV4 promoted astrocyte activation and proinflammatory cytokine production via YAP/STAT3 signaling pathway activation. a Representative immunoblots showing the inflammatory signaling pathways in primary cultured astrocytes after the different treatments (n = 3). b, c Quantitative analysis of the protein (b) and mRNA (c) levels of YAP (n = 3). d, e Quantitative analysis of the absolute p-YAP level (normalized to total YAP) (d) and p-STAT3 level (normalized to total STAT3) (e) (n = 3). f Representative confocal images showing that 4-AP or the TRPV4 agonist GSK1016790A increased YAP nuclear localization. The TRPV4 antagonist reversed this effect (n = 3). The data are presented as means ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001, Tukey’s test after ANOVA
To understand the role of YAP in 4-AP-induced astrocytes, we further examined its expression and phosphorylation. YAP expression was significantly increased (Fig. 6a–c), whereas the phosphorylation of YAP (p-YAP) (normalized to total YAP) was reduced (Fig. 6d) after 4-AP administration (n = 3). We then examined whether TRPV4 promoted the activation of YAP by enhancing YAP nuclear localization. Immunofluorescence staining demonstrated that 4-AP and GSK1016790A mediated the translocation of YAP from the cytoplasm to the nucleus, and this effect was reversed by HC-067047 (Fig. 6f, n = 3).
To further explore the role of YAP, YAP-specific siRNA was administered to astrocytes. YAP-specific siRNA effectively reduced the expression of YAP (Fig. 7a–b) in astrocytes (n = 3). YAP-specific siRNA abolished 4-AP-induced GFAP upregulation (Fig. 7c, d, f, and g, n = 3), suggesting that YAP played a vital role in 4-AP-induced astrocyte activation. In addition, 4-AP downregulated the protein and mRNA levels of SOCS3, and this effect could be reversed by YAP siRNA (Fig. 7d, h–i, n = 3). Moreover, YAP siRNA inhibited the 4-AP-induced upregulation of p-STAT3 protein expression (Fig. 7d, j, n = 3). Furthermore, the upregulated mRNA and protein levels of the proinflammatory cytokines IL-6 (Fig. 7e, k–l), TNF-α (Fig. 7e, m–n), IL-1β (Fig. 7e, o–p), and iNOS (Fig. 7q) were attenuated by YAP siRNA (n = 3), indicating that knocking down YAP could reduce 4-AP-induced activation of inflammation-related signaling pathways and proinflammatory cytokine production in astrocytes and subsequently improve the inflammatory state of astrocytes. These results demonstrate that YAP is essential for 4-AP-induced astrocyte activation and proinflammatory cytokine production, while YAP knockdown inhibits this process.
Fig. 7.

Activation of YAP signaling promoted 4-AP-induced astrocyte activation and proinflammatory cytokine production. a, b YAP protein expression was examined in the NC, si-YAP, and MOCK groups. (NC: negative control, transfected with scrambled siRNA; MOCK: mock transfection). YAP expression was significantly knocked down by the specific siRNA compared with that in the NC and MOCK groups (n = 3). c Double immunostaining of SOCS3 (red) and GFAP (green) in astrocytes after transfection with YAP-specific siRNA (n = 3). d Western blot showing that knocking down YAP reversed the upregulation of GFAP and p-STAT3 and the downregulation of SOCS3 induced by 4-AP in astrocytes (n = 3). e Western blot showing that knocking down YAP reduced the release of proinflammatory cytokines (IL-6, TNF-α, and IL-1β) from astrocytes after 4-AP administration (n = 3). f–j Quantitative analysis of the protein and mRNA levels of GFAP (f, g), SOCS3 (h, i), and p-STAT3 (normalized to total STAT3) (j) (n = 3). k, l Quantitative analysis of the mRNA and protein levels of IL-6 (k, l), TNF-α (m, n), and IL-1β (o, p) (n = 3). q Quantitative analysis of the mRNA level of iNOS (n = 3). The data are presented as means ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001, Tukey’s test after ANOVA
Calcium Manipulation Modifies the YAP/STAT3 Inflammatory Signaling Pathway and Affects Astrocyte Activation and Proinflammatory Cytokine Production
Since TRPV4 is a nonselective cation channel that is mainly permeable to Ca2+, our bioinformatics results suggested that Ca2+ may play a vital role in seizures (Fig. 1g). We hypothesized that Ca2+ acted downstream of TRPV4 activation. We next studied whether the TRPV4 channel was functional in astrocytes. As shown in Fig. 8a–c, GSK1016790A (200 nM), a selective agonist of TRPV4, increased the [Ca2+]i in astrocytes (n = 10). Compared with that in the control group, the increase in [Ca2+]i caused by GSK1016790A in the 4-AP group was significantly enhanced. These results indicated that the TRPV4 channel was functional and that its activation could increase [Ca2+]i in astrocytes.
Fig. 8.

TRPV4 is a functional calcium channel in astrocytes, and chelating [Ca2+]i inhibited 4-AP-induced pathological changes. a Representative images of astrocytes loaded with Fluo-4 AM before (left) and after (right) acute application of GSK1016790A (200 nM) (n = 10). b Traces of astrocytes responding to GSK1016790A (n = 10). The ΔF/F0 was calculated as (F–F0)/F0. F0 was the mean value of F during pretreatment. c The amplitude of Ca2+ influx in astrocytes in the control (black) and 4-AP (red) groups (n = 10). The bar chart corresponds to b. d Astrocytes were pretreated with BAPTA-AM (10 μM) for 30 min before 4-AP or HC-067047 treatment. Representative immunoblots showing the expression levels of inflammation-related signaling factors in primary cultured astrocytes (n = 3). e Representative immunoblots showing the expression levels of proinflammatory cytokines in primary cultured astrocytes (n = 3). f, g Quantitative analysis of the mRNA (f) and protein (g) levels of GFAP. h, i Quantitative analysis of the mRNA (h) and protein (i) levels of SOCS3 (n = 3). j–l Quantitative analysis of the protein level of YAP (j), the absolute p-YAP level (normalized to total YAP) (k), and the p-STAT3 level (normalized to total STAT3) (l) (n = 3). m, n Quantitative analysis of the mRNA (m) and protein (n) levels of IL-6 (n = 3). o, p Quantitative analysis of the mRNA (o) and protein (p) levels of TNF-α (n = 3). q, r Quantitative analysis of the mRNA levels of IL-1β (q) and iNOS (r) (n = 3). The data are presented as means ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001, Tukey’s test after ANOVA
Then, we incubated primary cultured astrocytes with BAPTA-AM (10 μM) or Ca2+-free medium for 30 min before administering 4-AP or the TRPV4 antagonist HC-067047. Ca2+ chelation significantly inhibited 4-AP-induced GFAP upregulation (Figs. 8d, f–g and 9a, c–d), increased p-STAT3 (Figs. 8d, l and 9a, i), and SOCS3 downregulation (Figs. 8d, h–i and 9a, e–f), while the TRPV4 antagonist HC-067047 did not further reverse this process in the presence of BAPTA-AM (10 μM) or Ca2+-free medium (n = 3). The 4-AP-induced upregulation of YAP protein expression (Figs. 8d, j and 9a, g) and downregulation of p-YAP (Figs. 8k and 9h) were reversed by BAPTA-AM (10 μM) or Ca2+-free medium (n = 3). In addition, Ca2+ chelation could also inhibit 4-AP-induced proinflammatory cytokine release (Figs. 8e, m–r and 9b, j–n), while the TRPV4 antagonist HC-067047 did not further reverse this process in the presence of BAPTA-AM (10 μM) or Ca2+-free medium (n = 3).
Fig. 9.

Ca2+-free medium inhibited 4-AP-induced astrocyte activation and proinflammatory cytokine production. a Astrocytes were pretreated with Ca2+-free medium for 30 min before the administration 4-AP or the TRPV4 antagonist HC-067047. Representative immunoblots showing the expression levels of inflammation-related signaling factors in primary cultured astrocytes (n = 3). b Immunoblots showing the expression levels of IL-6 in primary cultured astrocytes (n = 3). c, d Quantitative analysis of the mRNA (c) and protein (d) levels of GFAP (n = 3). e, f Quantitative analysis of the mRNA (e) and protein (f) levels of SOCS3 (n = 3). g–i Quantitative analysis of the protein level of YAP (g), the absolute p-YAP level (normalized to total YAP) (h), and the p-STAT3 level (normalized to total STAT3) (i) (n = 3). j, k Quantitative analysis of the mRNA (j) and protein (k) levels of IL-6 (n = 3). l–n Quantitative analysis of the mRNA levels of TNF-α (l), IL-1β (m), and iNOS (n) (n = 3). The data are presented as means ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001, Tukey’s test after ANOVA
These results indicated that in 4-AP-induced astrocytes, TRPV4 activation could lead to astrocyte activation, upregulate the YAP/STAT3 inflammatory signaling pathway, and increase proinflammatory cytokine production through Ca2+.
Next, the brain tissue of 4-AP-treated mice was examined to confirm the presence of astrocyte activation (Fig. 10a–e); the expression of C3 (the A1-type astrocyte marker) (Fig. 10c, f) and proinflammatory cytokines (IL-6, TNF-α, IL-1β, and iNOS) (Fig. 10h–j) were evaluated, and the A2-type astrocyte marker S100A10 (Fig. 10c, g) was downregulated in 4-AP-, GSK1016790A-, and 4-AP + GSK1016790A-treated mice (n = 3). 4-AP and GSK1016790A promoted astrocyte conversion to the A1 phenotype (Fig. 10f) (neurotoxic) but inhibited the A2 (Fig. 10g) (neuroprotective) phenotype. However, HC-067047 significantly ameliorated these neuropathic changes (n = 3). In addition, the YAP/STAT3 pathway was involved in this process (Additional File 2), which was consistent with the previous in vitro results (Fig. 6). In 4-AP-induecd mice, YAP expression was significantly upregulated (Additional File 3a). In addition, the colocalization of YAP with astrocytes was also increased (Additional File 3b), while YAP colocalization with microglia or neurons was not observed, which has been previously verified by others [41, 42, 47].
Fig. 10.

TRPV4 inhibition improved the inflammatory microenvironment of the brain 24 h after 4-AP injection. (a, b) Immunohistochemical analysis of GFAP in the hippocampal CA3 region of each group (n = 4). (c) Western blot analysis of GFAP, S100A10, and C3 expression in the hippocampus of control and 4-AP-, HC-067047-, 4-AP + HC-067047-, GSK1016790A-, and 4-AP + GSK1016790A-treated mice (n = 3). (d, e) Quantitative analysis of GFAP protein (d) and mRNA (e) levels in the hippocampus (n = 3). (f, g) qPCR analysis of the mRNA levels of A1 (f) and A2 (g) genes in the hippocampus of 4-AP-induced mice (n = 3). (h, i) Western blot and quantification showing the protein expression of IL-6 (n = 3). (j) Relative mRNA levels of IL-6, TNF-α, IL-1β, and iNOS (n = 3). The data are presented as means ± SEM, *P < 0.05, **P < 0.01, *P < 0.001, Tukey’s test after ANOVA
Enhanced Coexpression of TRPV4 and GFAP in the Brain Tissues of Epileptic Patients
Finally, we examined the expression of TRPV4 and GFAP in normal and surgical specimens from epileptic patients. Compared with that in the normal group (n = 9), TRPV4 expression was significantly upregulated in astrocytes in surgical specimens from epileptic patients (n = 7) (Fig. 11a, b), which was consistent with our results in mice. The expression and colocalization of GFAP and C3 were also increased in surgical specimens from epileptic patients (Fig. 11c, d) (n = 5), indicating the presence of A1 astrocytes in surgical specimens from epileptic patients. We measured GFAP and C3 levels by ELISA and found that GFAP was upregulated (Fig. 11e), but C3 was not altered (Fig. 11f) in the serum of epileptic patients (n = 5).
Fig. 11.
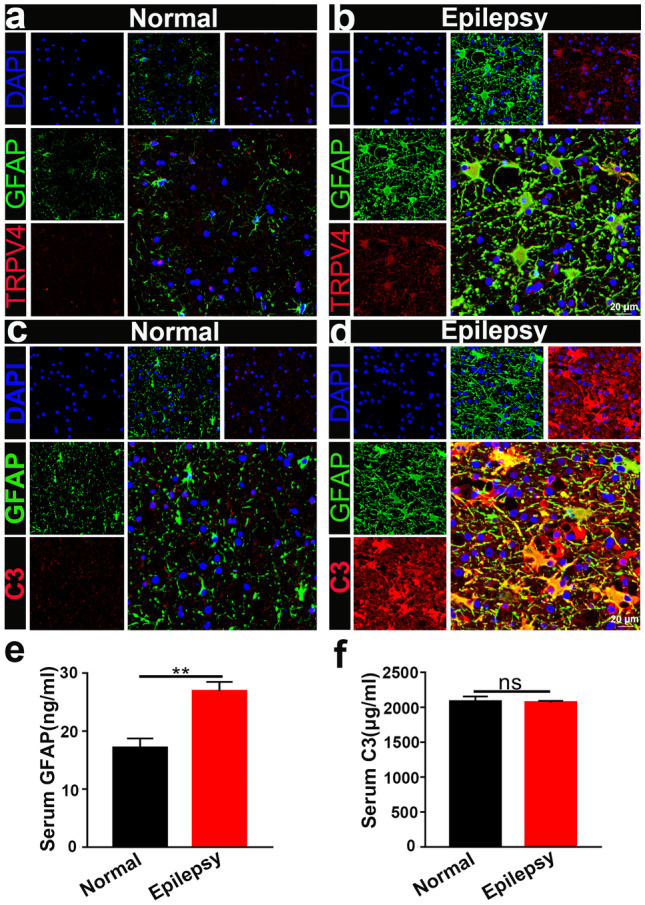
TRPV4 and GFAP expression in the brain tissues of normal and epileptic patients. a, b Compared with that in the normal group (n = 9), TRPV4 was significantly upregulated in astrocytes in the surgical brain specimens of epileptic patients (n = 7). c, d Compared with those in the normal group, GFAP and C3 were increased in the brains of epileptic patients (n = 5). e, f GFAP and C3 in the serum of normal (n = 5) and epileptic patients (n = 10) was measured by ELISA. The data are presented as means ± SEM, **P < 0.01. Unpaired Student’s t test
In conclusion, we found that TRPV4 was upregulated in epileptic patients and played an important role in 4-AP-induced seizures.
Discussion
In this study, we used the 4-AP-induced seizure model to explore the role of TRPV4 in seizures. 4-AP is a K+ channel blocker that can induce high-frequency seizure discharges in animal models and brain slices by increasing the excitability of neurons [48] and the release of neurotransmitters [49–52], which makes this agent suitable for investigating seizures [53]. Moreover, recent studies have shown that 4-AP can directly induce astrocyte activation. Grimaldi et al. [54] showed that 4-AP could increase [Ca2+]i in astrocytes in a concentration-dependent manner. This effect was not caused by the blockade of voltage-gated K+ channels. In the absence of neurons, 4-AP potently induces Ca2+ signaling in cultured astrocytes [54, 55]. However, tetrodotoxin (TTX) did not reduce the frequency or amplitude of 4-AP-induced Ca2+ oscillations in astrocytes [55]. Based on our previous studies using different epilepsy animal models [56–58], we chose 4-AP because Shao et al. [45] showed that 4-AP could increase astrocyte activation in a concentration-dependent manner. Therefore, the direct effect of 4-AP on astrocytes in vivo could be simulated by stimulating primary astrocytes with 4-AP (in vitro).
Moreover, increase in astrocyte [Ca2+]i induces glutamate release from astrocytes that leads to NMDA receptor–mediated increase in neuronal excitation [59]. Paroxysmal depolarization shifts (PDSs) are characterized by interictal discharges in the EEG and represent the cellular correlates of epileptic discharges [60]. Tian et al. [55] showed that 4-AP-induced PDSs were largely TTX-insensitive. The researchers also showed that 4-AP increased sequestered Ca2+ in astrocytes and subsequently induced glutamate release. Prolonged episodes of neuronal depolarization stimulated by astrocytic glutamate contribute to epileptiform discharges, indicating that astrocyte activation contributes to seizure activity. Based on these findings, we chose the 4-AP animal model.
In this work, we found that trpv4 was a seizure-associated gene through bioinformatics screening (Fig. 1) and then validated the upregulation of TRPV4 in 4-AP-induced mice (Fig. 3) and epileptic patients (Fig. 11). In the brain, TRPV4 is most abundantly expressed in astrocytes, and the activation of astrocytic TRPV4 leads to an increase in [Ca2+]i [15, 61]. Our results showed that TRPV4 was expressed mainly in astrocytes and was significantly increased by 4-AP. Moreover, astrocyte activation (Figs. 3, 4, and 11) and proinflammatory cytokine release were significantly increased (Figs. 3 and 4).
Numerous studies have shown that TRPV4 is closely related to inflammation and seizures. TRPV4 activation increased epileptic discharge by enhancing neuronal excitability [25], glutamatergic transmission [62], postsynaptic NMDA receptor [63], and α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptor activity [62] and inhibiting γ-aminobutyric acid (GABA)–activated current (IGABA) [64] and IK [29]. TRPV4 activation and upregulation are associated with neuronal damage after infrasound exposure [24], cerebral ischemia [22], TBI [65], and amyloid β40 treatment [66] and enhanced production of proinflammatory cytokines [18, 19], while TRPV4 inhibition reduces LPS and infrasound‑induced proinflammatory cytokine production [23, 24]. Recently, the relationship between neuroinflammation and seizures has received increasing attention. Icv. injection of the TRPV4 agonist GSK1016790A increased neuronal damage and neuroinflammation caused by microglial cell and astrocyte activation, which is involved in neuronal damage in PISE mice [28]. Consistently, in our study, icv. injection of a TRPV4 agonist increased 4-AP-induced seizure susceptibility, while TRPV4 inhibition significantly decreased seizure susceptibility (Fig. 2). Sustained or prolonged seizures lead to significant neuronal damage and cell death [67, 68]. In our 4-AP-induced mice, hippocampal neuronal damage mainly manifested in the DG and hilar region of the hippocampus. Moreover, 4-AP- and GSK1016790A-induced neuronal damage, astrocyte activation, A1 phenotypic polarization, and proinflammatory cytokine production were markedly blocked by the TRPV4 inhibitor HC-067047 (Figs. 2 and 10). These results suggested that TRPV4 activation-induced neuroinflammation resulted in 4-AP-induced seizures and neuronal damage after seizures.
Reducing Ca2+ influx in astrocytes during brain demyelination significantly attenuated brain inflammation and astrocyte reactivity [69]. In the CNS, the activation of astrocytic TRPV4 leads to an increase in [Ca2+]i through the influx of extracellular Ca2+ [61]. We hypothesize that TRPV4 activation in astrocytes leads to astrocyte activation and neuroinflammation by increasing [Ca2+]i. Therefore, we modified intracellular and extracellular Ca2+ levels with BAPTA-AM and Ca2+-free medium, respectively, and found that Ca2+ chelation inhibited 4-AP-induced astrocyte activation, STAT3 inflammatory signaling pathway activation, and proinflammatory cytokine production (Figs. 8 and 9). Thus, Ca2+ is vital in TRPV4-induced astrocyte activation, STAT3 inflammatory signaling pathway activation, and the production of proinflammatory cytokines.
YAP is selectively expressed by astrocytes and neural stem cells [41]. A previous report showed that YAP regulates the differentiation and proliferation of astrocytes [41]. YAP upregulation and activation promote glial scar formation by increasing astrocytic proliferation after SCI [42]. YAP nuclear translocation regulates astroglial-mesenchymal transition induced by hemoglobin and increases astrocyte activation [43]. In contrast, during ischemia/stroke, YAP nuclear localization in astrocytes reduced reactive astrogliosis, neuronal death,and proinflammatory cytokine release (IL-1β, IL-6, and TNF-α) by inhibiting STAT3 signaling [70]. YAP deletion contributes to hyperactivation of the JAK/STAT inflammatory pathway and reactive astrogliosis [71]. These studies showed that YAP regulates the proliferation and activation of astrocytes but plays different roles in different models of CNS diseases.
Therefore, we examined whether YAP was involved in the regulation of astrocyte activation during seizures. Our results showed that YAP was upregulated and activated by 4-AP or GSK1016790A in vivo and in vitro (Fig. 6 and Additional File 2). The nuclear translocation of YAP was robustly increased in 4-AP- and GSK1016790A-treated astrocytes (Fig. 6). Knocking down YAP with a specific siRNA reduced 4-AP-induced astrocyte activation, STAT3 signaling, and proinflammatory cytokine production (Fig. 7). These results indicated that in 4-AP-induced mice, YAP promoted astrocyte activation and proinflammatory cytokine release. However, Huang et al. [43] reported that YAP-deficient astrocytes showed reactive astrogliosis and induced neuroinflammation. These results differ from ours, probably due to the different models of CNS diseases compared with genetic ablation. Thus, in addition to the classic nuclear factor kappa B (NF-κB) and nucleotide-binding oligomerization domain–like receptor pyrin domain containing 3 (NLRP3)/IL-1β proinflammatory signaling pathways [28, 72], we revealed a new signaling pathway (TRPV4/Ca2+/YAP/STAT3) that also plays a vital role in seizures.
Finally, we obtained similar results in surgical specimens from epileptic patient brain tissue and mouse brain tissue. We found that compared with that in healthy controls, TRPV4 expression was significantly upregulated, and astrocytes were activated and polarized to the A1 phenotype in surgical specimens from patients with epilepsy (Fig. 11), which was consistent with our results in 4-AP-induced mice and cultured astrocytes. A previous study showed that A1 reactive astrocytes could impair hippocampal neurons by secreting complement C3 [73]. Thus, we hypothesized that A1 astrocytes might be related to neuronal damage in the context of 4-AP-induced seizures. However, this hypothesis needs to be confirmed by additional experiments. Although the 4-AP mouse model results were similar to those of the clinical samples, some shortcomings still exist. For example, the sample size was insufficient to draw an absolute conclusion. No seizure types in animal models can fully mimic the pathophysiological changes in epileptic patients. Sometimes animal models tend to have acute seizures, while epileptic patients usually have a chronic pathological change with acute seizures. Therefore, more animal experiments and clinical studies are needed to guide clinical practice.
In summary, we investigated the brains of 4-AP-induced mice and found that TRPV4 was activated and upregulated in astrocytes, which resulted in increased [Ca2+]i in astrocytes, increased nuclear localization of YAP, and increased p-STAT3 and led to the upregulation of proinflammatory cytokines and the markers of A1 astrocytes and the downregulation of A2 astrocyte markers, indicating A1 polarization and activation in astrocytes. A1 astrocytes can release more proinflammatory cytokines than A2 astrocytes and subsequently increase neuroinflammation, leading to perturbations in the microenvironment of the mouse brain, which increases seizure severity and neuronal damage after seizures. Chemical inhibition of TRPV4 balanced the intracerebral immune microenvironment of mice, protecting neurons from massive damage and reducing seizure severity (Fig. 12).
Fig. 12.
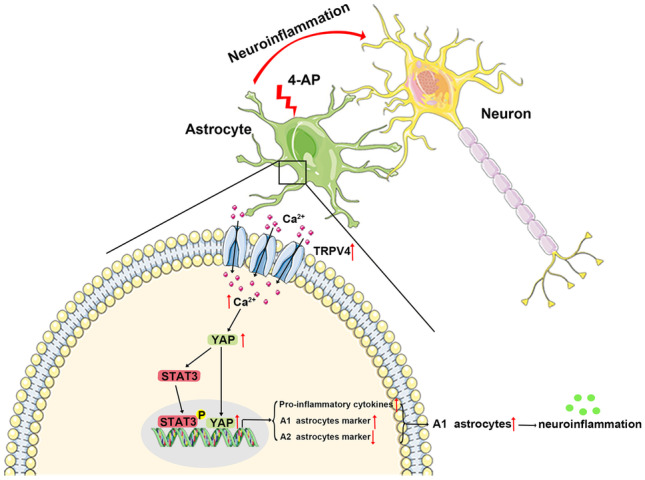
A working model illustrating the role of astrocytic TRPV4 in 4-AP-induced seizures. In this model, TRPV4 is upregulated and activated in astrocytes. Activated TRPV4 promotes the upregulation and activation of YAP by increasing [Ca2+]i, which contributes to the phosphorylation of STAT3 and further leads to the activation and A1 polarization of astrocytes. A1 astrocytes release many proinflammatory cytokines, leading to neuroinflammation and disruption of the immune microenvironment in the brain, thereby increasing seizure susceptibility
In summary, our study showed that YAP promoted astrocyte activation and implicated the STAT3 pathway in promoting proinflammatory cytokine release in 4-AP-induced mice. To our knowledge, this is the first study to report the role of YAP in seizures. In addition, we provide evidence for the role of TRPV4 in promoting astrocyte activation and polarization, the release of proinflammatory cytokines, and neuronal damage in the context of 4-AP-induced seizures. TRPV4 may be an important therapeutic target for seizure treatment.
Conclusion
This study examined mouse brain tissue and surgery specimens from epileptic patient brains and obtained similar results showing that TRPV4 was upregulated and astrocytes were activated and polarized to the A1 phenotype. TRPV4 promoted astrocyte activation and A1 polarization and induced astrocyte-derived IL-6, TNF-α, IL-1β, and iNOS production via Ca2+/YAP/STAT3 signaling. We provide evidence that TRPV4 increases seizure susceptibility in 4-AP-induced mice. We propose that astrocytes undergo activation and A1 phenotype polarization after 4-AP-induced seizures, and TRPV4 activation facilitates YAP nuclear translocation, which is a novel mechanism in this process. Furthermore, we reveal a novel signal transduction pathway (TRPV4/Ca2+/YAP/STAT3) in seizures. Thus, astrocytic TRPV4 may be an important therapeutic target for seizures.
Supplementary Information
Below is the link to the electronic supplementary material.
Additional File 1 The expression level of TRPV4 in primary cultured neurons and BV2 microglia (a) The immunoblot shows the protein expression level of TRPV4 protein in primary cultured neurons before and after 4-AP administration (5 mM, 24 h). (b) The immunoblot shows the TRPV4 protein expression level in BV2 microglia before and after 4-AP administration (5 mM, 24 h). (TIF 3406 kb)
Additional File 2 YAP/STAT3 signaling was activated in 4-AP-induced mice (a) Representative immunoblot showing the expression levels of inflammation-related signaling pathways in the hippocampus (n=3). (b-c) Quantitative analysis of the mRNA (b) and protein (c) levels of SOCS3 (n=3). (d-e) Quantitative analysis of the mRNA (d) and protein (e) levels of YAP (n=3). (f-g) Quantitative analysis of the protein levels of p-YAP (normalized to total YAP) (f) and p-STAT3 (normalized to total STAT3) (g) (n=3). The data are as means±SEM, *P < 0.05, **P < 0.01, ***P < 0.001, Tukey’s test after ANOVA. (TIF 19818 kb)
Additional File 3 Immunohistochemistry and immunofluorescence analysis of YAP in the brain tissues of sham and 4-AP-induced mice (a) Compared with that in the sham group, YAP expression was significantly upregulated in the hippocampus of 4-AP-induced mice (n =3). Red arrows indicate the nuclear localization of YAP. (b) Compared with those in the sham group, the expression levels of GFAP and YAP were increased in the hippocampus of 4-AP-induced mice (n =3). The coexpression of YAP and GFAP (indicated by white arrows) in astrocytes was significantly increased in 4-AP-induced mice. (TIF 9003 kb)
Acknowledgements
We thank Lin Shao, Guang-tong Jiang, Xin Wang, Ying-cheng Zheng, Jun Yan, Ying-lian Tang, Bao-hua Hou, Ya-hui Zhang, Pei-yu Liang, Xiao-min Yu, Xin Lu, Zhi-cheng Zheng, Qian-wen Ma, and Yi-fan Wu for the technical assistance during the experiments. We appreciate Guang-tong Jiang, Lin Shao, and Wei Xu for their valuable comments. We are also grateful to Ying Zhou from the University of Wuhan for sharing her expertise in the confocal operation.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Abbreviations
- CNS
Central nervous system
- TRPV4
Transient receptor potential vanilloid 4
- 4-AP
4-Aminopyridine
- SE
Status epilepticus
- GFAP
Glial fibrillary acidic protein
- SOCS3
Suppressor of cytokine signaling 3
- YAP
Yes-associated protein
- STAT3
Signal transducer activator of transcription 3
- EEG
Electroencephalogram
- [Ca2+]i
Intracellular Ca2+ concentration
- C3
The third component of complement
- ELISA
Enzyme-linked immunosorbent assay
- ip.
Intraperitoneal
- icv.
Intracerebroventricular
- IL-1β
Interleukin-1β
- IL-6
Interleukin-6
- TNF-α
Tumor necrosis factor-α
- iNOS
Inducible nitric oxide synthase
- TRP
Transient receptor potential
- PI3K
Phosphatidyl inositol 3 kinase
- Akt
Protein kinase B
- NMDA
NR2B-N-methyl-d-aspartate glutamate
- MAPK
Mitogen-activated protein kinase
- LPS
Lipopolysaccharide
- IK
Delayed rectifier potassium current
- PISE
Pilocarpine-induced status epilepticus
- WT
Wild type
- HBSS
Hank’s balanced salt solution
- DMEM
Dulbecco’s modified Eagle’s medium
- FBS
Fetal bovine serum
- PMSF
Phenylmethanesulfonyl fluoride
- WB
Western blot
- SDS-PAGE
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- PVDF
Polyvinylidene fluoride membrane
- PFA
Paraformaldehyde
- IF
Immunofluorescence
- siRNA
Small interfering RNA
- GEO
Gene Expression Omnibus
- DEGs
Differentially expressed genes
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- GO
Gene Ontology
- DAVID
Database for Annotation, Visualization and Integration Discovery
- PPI network
Protein-protein interaction network
- JAK2/STAT3
Janus kinases/signal transducer of transcription factor-3
- TBI
Traumatic brain injury
- AMPA
α-Amino-3-hydroxy-5-methyl-4-isoxazole propionic acid
- GABA
γ-Aminobutyric acid
- IGABA
γ-Aminobutyric acid-activated current
- SCI
Spinal cord injury
- HI
Hypoxia-ischemia
- TTX
Tetrodotoxin
- NF-κB
Nuclear factor kappa B
- NLRP3
Nucleotide-binding oligomerization domain-like receptor pyrin domain containing 3 (NLRP3)
Author Contribution
Meng-liu Zeng and Bi-wen Peng conceived and designed the experiments. Meng-liu Zeng, Jing-jing Cheng, and Shuo Kong performed the experiments. Meng-liu Zeng, Xing-ling Yang, Ling Chen, Xiang-lei Jia, and Xue-lei Cheng analyzed the data. Fang-gang He, Yu-min Liu, Lanzi Gongga, Tao-xiang Chen, Wan-hong Liu, and Xiao-hua He contributed to the reagents, materials, and analysis tools. Meng-liu Zeng and Bi-wen Peng wrote the manuscript. All authors reviewed and approved the final manuscript.
Funding
This research is supported by the Natural Science Foundation of China (Grant No. 82171452, No. 82060588), the Medical Science Advancement Program of Wuhan University (No. TFJC2018001, TFLC2018001), and the Translational Medicine and Interdisciplinary Research Joint Fund of Zhongnan Hospital of Wuhan University (Grant NO. ZNLH201909).
Availability of Data and Materials
All data used in this manuscript are available from the corresponding author on reasonable request.
Declarations
Ethics Approval and Consent to Participate
The animal study was reviewed and approved by the Care and Use Committee of Wuhan University Medical School and the National Institutes of Health guide for the care and use of laboratory animals. After giving informed consent, human brain specimens were collected at the Department of Neurosurgery of Zhongnan Hospital (Wuhan University, Wuhan, China). All experiments involving humans were performed following the Declaration of the Helsinki Declaration of 1975, as revised in 2000 (World Medical Association Declaration of Helsinki 2000), and have been approved by the Medical Ethics Committee, Zhongnan Hospital of Wuhan University (Scientific Ethical Approval [2019059]).
Conflict of Interest
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Meng-liu Zeng, Email: 2018103010005@whu.edu.cn.
Jing-jing Cheng, Email: 2018203010001@whu.edu.cn.
Shuo Kong, Email: 2019203010002@whu.edu.cn.
Xing-liang Yang, Email: dracy@whu.edu.cn.
Xiang-lei Jia, Email: 2021103010001@whu.edu.cn.
Xue-lei Cheng, Email: ChengXLwhu@163.com.
Ling Chen, Email: 976818572@qq.com.
Fang-gang He, Email: hefg007@whu.edu.cn.
Yu-min Liu, Email: wb001792@whu.edu.cn.
Yuan-teng Fan, Email: fanyuanteng@163.com.
Lanzi Gongga, Email: qeblanzi@hotmail.com.
Tao-xiang Chen, Email: chentaoxiang@whu.edu.cn.
Wan-hong Liu, Email: liuwanhong@whu.edu.cn.
Xiao-hua He, Email: hexiaohua@whu.edu.cn.
Bi-wen Peng, Email: pengbiwen@whu.edu.cn.
References
- 1.Hauser RM, Henshall DC, Lubin FD. The epigenetics of epilepsy and its progression. The Neuroscientist : a review journal bringing neurobiology, neurology and psychiatry. 2018;24(2):186–200. doi: 10.1177/1073858417705840. [DOI] [PubMed] [Google Scholar]
- 2.Vezzani A, French J, Bartfai T, Baram TZ. The role of inflammation in epilepsy. Nat Rev Neurol. 2011;7(1):31–40. doi: 10.1038/nrneurol.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ravizza T, Rizzi M, Perego C, et al. Inflammatory response and glia activation in developing rat hippocampus after status epilepticus. Epilepsia. 2005;46(Suppl 5):113–117. doi: 10.1111/j.1528-1167.2005.01006.x. [DOI] [PubMed] [Google Scholar]
- 4.Wetherington J, Serrano G, Dingledine R. Astrocytes in the epileptic brain. Neuron. 2008;58(2):168–178. doi: 10.1016/j.neuron.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Strotmann R, Harteneck C, Nunnenmacher K, Schultz G, Plant TD. OTRPC4, a nonselective cation channel that confers sensitivity to extracellular osmolarity. Nat Cell Biol. 2000;2(10):695–702. doi: 10.1038/35036318. [DOI] [PubMed] [Google Scholar]
- 6.Liedtke W, Choe Y, Marti-Renom MA, et al. Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell. 2000;103(3):525–535. doi: 10.1016/S0092-8674(00)00143-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vriens J, Watanabe H, Janssens A, Droogmans G, Voets T, Nilius B. Cell swelling, heat, and chemical agonists use distinct pathways for the activation of the cation channel TRPV4. Proc Natl Acad Sci USA. 2004;101(1):396–401. doi: 10.1073/pnas.0303329101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liedtke W, Tobin DM, Bargmann CI, Friedman JM. Mammalian TRPV4 (VR-OAC) directs behavioral responses to osmotic and mechanical stimuli in Caenorhabditis elegans. Proc Natl Acad Sci USA. 2003;100(Suppl 2):14531–14536. doi: 10.1073/pnas.2235619100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Watanabe H, Vriens J, Prenen J, Droogmans G, Voets T, Nilius B. Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature. 2003;424(6947):434–438. doi: 10.1038/nature01807. [DOI] [PubMed] [Google Scholar]
- 10.Watanabe H, Davis JB, Smart D, et al. Activation of TRPV4 channels (hVRL-2/mTRP12) by phorbol derivatives. J Biol Chem. 2002;277(16):13569–13577. doi: 10.1074/jbc.M200062200. [DOI] [PubMed] [Google Scholar]
- 11.Pankey EA, Zsombok A, Lasker GF, Kadowitz PJ. Analysis of responses to the TRPV4 agonist GSK1016790A in the pulmonary vascular bed of the intact-chest rat. Am J Physiol Heart Circ Physiol. 2014;306(1):H33–40. doi: 10.1152/ajpheart.00303.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.White JP, Cibelli M, Urban L, Nilius B, McGeown JG, Nagy I. TRPV4: molecular conductor of a diverse orchestra. Physiol Rev. 2016;96(3):911–973. doi: 10.1152/physrev.00016.2015. [DOI] [PubMed] [Google Scholar]
- 13.Vriens J, Owsianik G, Janssens A, Voets T, Nilius B. Determinants of 4 alpha-phorbol sensitivity in transmembrane domains 3 and 4 of the cation channel TRPV4. J Biol Chem. 2007;282(17):12796–12803. doi: 10.1074/jbc.M610485200. [DOI] [PubMed] [Google Scholar]
- 14.Shibasaki K, Tominaga M, Ishizaki Y. Hippocampal neuronal maturation triggers post-synaptic clustering of brain temperature-sensor TRPV4. Biochem Biophys Res Commun. 2015;458(1):168–173. doi: 10.1016/j.bbrc.2015.01.087. [DOI] [PubMed] [Google Scholar]
- 15.Dunn KM, Hill-Eubanks DC, Liedtke WB, Nelson MT. TRPV4 channels stimulate Ca2+-induced Ca2+ release in astrocytic endfeet and amplify neurovascular coupling responses. Proc Natl Acad Sci USA. 2013;110(15):6157–6162. doi: 10.1073/pnas.1216514110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Konno M, Shirakawa H, Iida S, et al. Stimulation of transient receptor potential vanilloid 4 channel suppresses abnormal activation of microglia induced by lipopolysaccharide. Glia. 2012;60(5):761–770. doi: 10.1002/glia.22306. [DOI] [PubMed] [Google Scholar]
- 17.Ohashi K, Deyashiki A, Miyake T, et al. TRPV4 is functionally expressed in oligodendrocyte precursor cells and increases their proliferation. Pflugers Arch. 2018;470(5):705–716. doi: 10.1007/s00424-018-2130-3. [DOI] [PubMed] [Google Scholar]
- 18.Walter BA, Purmessur D, Moon A, et al. Reduced tissue osmolarity increases TRPV4 expression and pro-inflammatory cytokines in intervertebral disc cells. Eur Cell Mater. 2016;32:123–136. doi: 10.22203/eCM.v032a08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nayak PS, Wang Y, Najrana T, et al. Mechanotransduction via TRPV4 regulates inflammation and differentiation in fetal mouse distal lung epithelial cells. Respir Res. 2015;16:60. doi: 10.1186/s12931-015-0224-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vergnolle N, Cenac N, Altier C, et al. A role for transient receptor potential vanilloid 4 in tonicity-induced neurogenic inflammation. Br J Pharmacol. 2010;159(5):1161–1173. doi: 10.1111/j.1476-5381.2009.00590.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jie P, Hong Z, Tian Y, et al. Activation of transient receptor potential vanilloid 4 induces apoptosis in hippocampus through downregulating PI3K/Akt and upregulating p38 MAPK signaling pathways. Cell Death Dis. 2015;6:e1775. [DOI] [PMC free article] [PubMed]
- 22.Jie P, Lu Z, Hong Z, et al. Activation of transient receptor potential vanilloid 4 is involved in neuronal injury in middle cerebral artery occlusion in mice. Mol Neurobiol. 2016;53(1):8–17. doi: 10.1007/s12035-014-8992-2. [DOI] [PubMed] [Google Scholar]
- 23.Dalsgaard T, Sonkusare SK, Teuscher C, Poynter ME, Nelson MT. Pharmacological inhibitors of TRPV4 channels reduce cytokine production, restore endothelial function and increase survival in septic mice. Sci Rep. 2016;6:33841. doi: 10.1038/srep33841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi M, Du F, Liu Y, et al. Glial cell-expressed mechanosensitive channel TRPV4 mediates infrasound-induced neuronal impairment. Acta Neuropathol. 2013;126(5):725–739. doi: 10.1007/s00401-013-1166-x. [DOI] [PubMed] [Google Scholar]
- 25.Shibasaki K, Yamada K, Miwa H, et al. Temperature elevation in epileptogenic foci exacerbates epileptic discharge through TRPV4 activation. Lab Invest. 2020;100(2):274–284. doi: 10.1038/s41374-019-0335-5. [DOI] [PubMed] [Google Scholar]
- 26.Chen X, Yang M, Sun F, et al. Expression and cellular distribution of transient receptor potential vanilloid 4 in cortical tubers of the tuberous sclerosis complex. Brain Res. 2016;1636:183–192. doi: 10.1016/j.brainres.2016.02.012. [DOI] [PubMed] [Google Scholar]
- 27.Hunt RF, Hortopan GA, Gillespie A, Baraban SC. A novel zebrafish model of hyperthermia-induced seizures reveals a role for TRPV4 channels and NMDA-type glutamate receptors. Exp Neurol. 2012;237(1):199–206. doi: 10.1016/j.expneurol.2012.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Z, Zhou L, An D, et al. TRPV4-induced inflammatory response is involved in neuronal death in pilocarpine model of temporal lobe epilepsy in mice. Cell Death Dis. 2019;10(6):386. doi: 10.1038/s41419-019-1612-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou L, Xu W, An D, et al. Transient receptor potential vanilloid 4 activation inhibits the delayed rectifier potassium channels in hippocampal pyramidal neurons: an implication in pathological changes following pilocarpine-induced status epilepticus. J Neurosci Res. 2021;99(3):914–926. doi: 10.1002/jnr.24749. [DOI] [PubMed] [Google Scholar]
- 30.Justice RW, Zilian O, Woods DF, Noll M, Bryant PJ. The Drosophila tumor suppressor gene warts encodes a homolog of human myotonic dystrophy kinase and is required for the control of cell shape and proliferation. Genes Dev. 1995;9(5):534–546. doi: 10.1101/gad.9.5.534. [DOI] [PubMed] [Google Scholar]
- 31.Zhao B, Li L, Lei Q, Guan KL. The Hippo-YAP pathway in organ size control and tumorigenesis: an updated version. Genes Dev. 2010;24(9):862–874. doi: 10.1101/gad.1909210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qiao Y, Li T, Zheng S, Wang H. The Hippo pathway as a drug target in gastric cancer. Cancer Lett. 2018;420:14–25. doi: 10.1016/j.canlet.2018.01.062. [DOI] [PubMed] [Google Scholar]
- 33.Maugeri-Sacca M, De Maria R. The Hippo pathway in normal development and cancer. Pharmacol Ther. 2018;186:60–72. doi: 10.1016/j.pharmthera.2017.12.011. [DOI] [PubMed] [Google Scholar]
- 34.Deng Q, Jiang G, Wu Y, et al. GPER/Hippo-YAP signal is involved in Bisphenol S induced migration of triple negative breast cancer (TNBC) cells. J Hazard Mater. 2018;355:1–9. doi: 10.1016/j.jhazmat.2018.05.013. [DOI] [PubMed] [Google Scholar]
- 35.Cai J, Zhang N, Zheng Y, de Wilde RF, Maitra A, Pan D. The Hippo signaling pathway restricts the oncogenic potential of an intestinal regeneration program. Genes Dev. 2010;24(21):2383–2388. doi: 10.1101/gad.1978810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pan D. The hippo signaling pathway in development and cancer. Dev Cell. 2010;19(4):491–505. doi: 10.1016/j.devcel.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnson R, Halder G. The two faces of Hippo: targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat Rev Drug Discovery. 2014;13(1):63–79. doi: 10.1038/nrd4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lau AN, Curtis SJ, Fillmore CM, et al. Tumor-propagating cells and Yap/Taz activity contribute to lung tumor progression and metastasis. EMBO J. 2014;33(5):468–481. doi: 10.1002/embj.201386082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the roots of cancer. Cancer Cell. 2016;29(6):783–803. doi: 10.1016/j.ccell.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Taha Z, Janse van Rensburg HJ, Yang X. The Hippo Pathway: immunity and cancer. Cancers. 2018;10(4). [DOI] [PMC free article] [PubMed]
- 41.Huang Z, Hu J, Pan J, et al. YAP stabilizes SMAD1 and promotes BMP2-induced neocortical astrocytic differentiation. Development. 2016;143(13):2398–2409. doi: 10.1242/dev.130658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xie C, Shen X, Xu X, et al. Astrocytic YAP promotes the formation of glia scars and neural regeneration after spinal cord injury. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2020;40(13):2644–2662. doi: 10.1523/JNEUROSCI.2229-19.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang Y, Ren J, Sun Y, et al. A connexin43/YAP axis regulates astroglial-mesenchymal transition in hemoglobin induced astrocyte activation. Cell Death Differ. 2018;25(10):1870–1884. doi: 10.1038/s41418-018-0137-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sharma S, Goswami R, Zhang DX, Rahaman SO. TRPV4 regulates matrix stiffness and TGFbeta1-induced epithelial-mesenchymal transition. J Cell Mol Med. 2019;23(2):761–774. doi: 10.1111/jcmm.13972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shao L, Jiang GT, Yang XL, et al. Silencing of circIgf1r plays a protective role in neuronal injury via regulating astrocyte polarization during epilepsy. FASEB J: official publication of the Federation of American Societies for Experimental Biology. 2021;35(2):e21330. [DOI] [PubMed]
- 46.Kong W, Wang X, Yang X, et al. Activation of TRPV1 contributes to recurrent febrile seizures via inhibiting the microglial M2 phenotype in the immature brain. Front Cell Neurosci. 2019;13:442. doi: 10.3389/fncel.2019.00442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu H, Cao X, Li W, et al. Targeting connexin 43 provides anti-inflammatory effects after intracerebral hemorrhage injury by regulating YAP signaling. J Neuroinflammation. 2020;17(1):322. doi: 10.1186/s12974-020-01978-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Storm JF. Action potential repolarization and a fast after-hyperpolarization in rat hippocampal pyramidal cells. J Physiol. 1987;385:733–759. doi: 10.1113/jphysiol.1987.sp016517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kovacs A, Mihaly A, Komaromi A, et al. Seizure, neurotransmitter release, and gene expression are closely related in the striatum of 4-aminopyridine-treated rats. Epilepsy Res. 2003;55(1–2):117–129. doi: 10.1016/S0920-1211(03)00113-X. [DOI] [PubMed] [Google Scholar]
- 50.Salazar P, Tapia R. Allopregnanolone potentiates the glutamate-mediated seizures induced by 4-aminopyridine in rat hippocampus in vivo. Neurochem Res. 2012;37(3):596–603. doi: 10.1007/s11064-011-0649-1. [DOI] [PubMed] [Google Scholar]
- 51.Segovia G, Porras A, Mora F. Effects of 4-aminopyridine on extracellular concentrations of glutamate in striatum of the freely moving rat. Neurochem Res. 1997;22(12):1491–1497. doi: 10.1023/A:1021958613125. [DOI] [PubMed] [Google Scholar]
- 52.Tapia R, Sitges M. Effect of 4-aminopyridine on transmitter release in synaptosomes. Brain Res. 1982;250(2):291–299. doi: 10.1016/0006-8993(82)90423-1. [DOI] [PubMed] [Google Scholar]
- 53.Fragoso-Veloz J, Massieu L, Alvarado R, Tapia R. Seizures and wet-dog shakes induced by 4-aminopyridine, and their potentiation by nifedipine. Eur J Pharmacol. 1990;178(3):275–284. doi: 10.1016/0014-2999(90)90106-G. [DOI] [PubMed] [Google Scholar]
- 54.Grimaldi M, Atzori M, Ray P, Alkon DL. Mobilization of calcium from intracellular stores, potentiation of neurotransmitter-induced calcium transients, and capacitative calcium entry by 4-aminopyridine. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2001;21(9):3135–3143. doi: 10.1523/JNEUROSCI.21-09-03135.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tian GF, Azmi H, Takano T, et al. An astrocytic basis of epilepsy. Nat Med. 2005;11(9):973–981. doi: 10.1038/nm1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zheng Z, Liang P, Hou B, et al. The effect of dipeptidyl peptidase IV on disease-associated microglia phenotypic transformation in epilepsy. J Neuroinflammation. 2021;18(1):112. doi: 10.1186/s12974-021-02133-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kong WL, Min JW, Liu YL, Li JX, He XH, Peng BW. Role of TRPV1 in susceptibility to PTZ-induced seizure following repeated hyperthermia challenges in neonatal mice. Epilepsy & behavior : E&B. 2014;31:276–280. doi: 10.1016/j.yebeh.2013.10.022. [DOI] [PubMed] [Google Scholar]
- 58.Hu JJ, Yang XL, Luo WD, et al. Bumetanide reduce the seizure susceptibility induced by pentylenetetrazol via inhibition of aberrant hippocampal neurogenesis in neonatal rats after hypoxia-ischemia. Brain Res Bull. 2017;130:188–199. doi: 10.1016/j.brainresbull.2017.01.022. [DOI] [PubMed] [Google Scholar]
- 59.Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, Haydon PG. Glutamate-mediated astrocyte-neuron signalling. Nature. 1994;369(6483):744–747. doi: 10.1038/369744a0. [DOI] [PubMed] [Google Scholar]
- 60.Hotka M, Kubista H. The paroxysmal depolarization shift in epilepsy research. Int J Biochem Cell Biol. 2019;107:77–81. doi: 10.1016/j.biocel.2018.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Benfenati V, Amiry-Moghaddam M, Caprini M, et al. Expression and functional characterization of transient receptor potential vanilloid-related channel 4 (TRPV4) in rat cortical astrocytes. Neuroscience. 2007;148(4):876–892. doi: 10.1016/j.neuroscience.2007.06.039. [DOI] [PubMed] [Google Scholar]
- 62.Li L, Yin J, Jie PH, et al. Transient receptor potential vanilloid 4 mediates hypotonicity-induced enhancement of synaptic transmission in hippocampal slices. CNS Neurosci Ther. 2013;19(11):854–862. doi: 10.1111/cns.12143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li L, Qu W, Zhou L, et al. Activation of transient receptor potential vanilloid 4 increases NMDA-activated current in hippocampal pyramidal neurons. Front Cell Neurosci. 2013;7:17. doi: 10.3389/fncel.2013.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hong Z, Tian Y, Qi M, et al. Transient receptor potential vanilloid 4 inhibits gamma-aminobutyric acid-activated current in hippocampal pyramidal neurons. Front Mol Neurosci. 2016;9:77. doi: 10.3389/fnmol.2016.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lu KT, Huang TC, Tsai YH, Yang YL. Transient receptor potential vanilloid type 4 channels mediate Na-K-Cl-co-transporter-induced brain edema after traumatic brain injury. J Neurochem. 2017;140(5):718–727. doi: 10.1111/jnc.13920. [DOI] [PubMed] [Google Scholar]
- 66.Bai JZ, Lipski J. Involvement of TRPV4 channels in Abeta(40)-induced hippocampal cell death and astrocytic Ca(2+) signalling. Neurotoxicology. 2014;41:64–72. doi: 10.1016/j.neuro.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 67.Trinka E, Brigo F, Shorvon S. Recent advances in status epilepticus. Curr Opin Neurol. 2016;29(2):189–198. doi: 10.1097/WCO.0000000000000307. [DOI] [PubMed] [Google Scholar]
- 68.Chen SD, Chang AY, Chuang YC. The potential role of mitochondrial dysfunction in seizure-associated cell death in the hippocampus and epileptogenesis. J Bioenerg Biomembr. 2010;42(6):461–465. doi: 10.1007/s10863-010-9321-8. [DOI] [PubMed] [Google Scholar]
- 69.Zamora NN, Cheli VT, Santiago Gonzalez DA, Wan R, Paez PM. Deletion of voltage-gated calcium channels in astrocytes during demyelination reduces brain inflammation and promotes myelin regeneration in mice. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2020;40(17):3332–3347. doi: 10.1523/JNEUROSCI.1644-19.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang L, Li S, Dai Q, et al. Astrocytic Yes-associated protein attenuates cerebral ischemia-induced brain injury by regulating signal transducer and activator of transcription 3 signaling. Exp Neurol. 2020;333:113431. [DOI] [PubMed]
- 71.Huang ZH, Wang Y, Hu GQ, Zhou JL, Mei L, Xiong WC. YAP Is a critical inducer of SOCS3, preventing reactive astrogliosis. Cereb Cortex. 2016;26(5):2299–2310. doi: 10.1093/cercor/bhv292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rosciszewski G, Cadena V, Auzmendi J, et al. Detrimental effects of HMGB-1 require microglial-astroglial interaction: implications for the status epilepticus -induced neuroinflammation. Front Cell Neurosci. 2019;13:380. doi: 10.3389/fncel.2019.00380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hou B, Zhang Y, Liang P, et al. Inhibition of the NLRP3-inflammasome prevents cognitive deficits in experimental autoimmune encephalomyelitis mice via the alteration of astrocyte phenotype. Cell Death Dis. 2020;11(5):377. doi: 10.1038/s41419-020-2565-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional File 1 The expression level of TRPV4 in primary cultured neurons and BV2 microglia (a) The immunoblot shows the protein expression level of TRPV4 protein in primary cultured neurons before and after 4-AP administration (5 mM, 24 h). (b) The immunoblot shows the TRPV4 protein expression level in BV2 microglia before and after 4-AP administration (5 mM, 24 h). (TIF 3406 kb)
Additional File 2 YAP/STAT3 signaling was activated in 4-AP-induced mice (a) Representative immunoblot showing the expression levels of inflammation-related signaling pathways in the hippocampus (n=3). (b-c) Quantitative analysis of the mRNA (b) and protein (c) levels of SOCS3 (n=3). (d-e) Quantitative analysis of the mRNA (d) and protein (e) levels of YAP (n=3). (f-g) Quantitative analysis of the protein levels of p-YAP (normalized to total YAP) (f) and p-STAT3 (normalized to total STAT3) (g) (n=3). The data are as means±SEM, *P < 0.05, **P < 0.01, ***P < 0.001, Tukey’s test after ANOVA. (TIF 19818 kb)
Additional File 3 Immunohistochemistry and immunofluorescence analysis of YAP in the brain tissues of sham and 4-AP-induced mice (a) Compared with that in the sham group, YAP expression was significantly upregulated in the hippocampus of 4-AP-induced mice (n =3). Red arrows indicate the nuclear localization of YAP. (b) Compared with those in the sham group, the expression levels of GFAP and YAP were increased in the hippocampus of 4-AP-induced mice (n =3). The coexpression of YAP and GFAP (indicated by white arrows) in astrocytes was significantly increased in 4-AP-induced mice. (TIF 9003 kb)
Data Availability Statement
All data used in this manuscript are available from the corresponding author on reasonable request.