


Abstract
Single-cell whole-genome haplotyping allows simultaneous detection of haplotypes associated with monogenic diseases, chromosome copy-numbering and subsequently, has revealed mosaicism in embryos and embryonic stem cells. Methods, such as karyomapping and haplarithmisis, were deployed as a generic and genome-wide approach for preimplantation genetic testing (PGT) and are replacing traditional PGT methods. While current methods primarily rely on single-nucleotide polymorphism (SNP) array, we envision sequencing-based methods to become more accessible and cost-efficient. Here, we developed a novel sequencing-based methodology to haplotype and copy-number profile single cells. Following DNA amplification, genomic size and complexity is reduced through restriction enzyme digestion and DNA is genotyped through sequencing. This single-cell genotyping-by-sequencing (scGBS) is the input for haplarithmisis, an algorithm we previously developed for SNP array-based single-cell haplotyping. We established technical parameters and developed an analysis pipeline enabling accurate concurrent haplotyping and copy-number profiling of single cells. We demonstrate its value in human blastomere and trophectoderm samples as application for PGT for monogenic disorders. Furthermore, we demonstrate the method to work in other species through analyzing blastomeres of bovine embryos. Our scGBS method opens up the path for single-cell haplotyping of any species with diploid genomes and could make its way into the clinic as a PGT application.
INTRODUCTION
Technologies for single-cell whole-genome analyses allow disclosing inter-cellular genetic heterogeneity, which is fundamentally changing our understanding of DNA mutation in development, ageing and disease (1–3). They also enable novel medical practice, in particular for genetic selection of human preimplantation embryos (4–6). Current methods for single-cell genome analysis require some form of whole genome amplification (WGA) to yield sufficient input material for microarray or next-generation sequencing (NGS) analyses (1,2,7). However, WGA produces artefacts—including locus drop-out (LDO), allelic drop-out (ADO), chimeric DNA molecules, base replication errors and unevenness in amplification—that challenge the detection of genetic variation at the single-cell level (4). An effective way to alleviate WGA artefacts is haplotyping that connects variant alleles present on the same DNA double helix within a single cell. Several family-based and population-based methods for inferring haplotypes of single-nucleotide polymorphisms (SNPs) or SNP B-allele frequency (BAFs) have been developed (5,8–17). However, these methods have a number of shortcomings when it comes to single-cell SNP haplotyping. They go awry on the error-prone single-cell SNP genotypes mainly due to ADO and putative base replication errors, often ignore DNA copy number aberrations and cannot distinguish mitotic from meiotic allelic imbalances (5,8). Additionally, for methods that make use of sequencing data, (ultra-)deep sequencing of genomes is required, which is cost prohibitive and computationally demanding (15).
To overcome those challenges, we developed a SNP array-based single-cell haplotyping workflow termed single-cell haplotyping and imputation of linked disease variants (siCHILD) by combining novel analytical modules such as the core haplarithmisis (6). Haplarithmisis allows for pedigree-based haplotyping through the use of genotypes, B-allele frequency (BAF) and logR values. This methodology has been applied in studies on both human and bovine preimplantation embryos and generated valuable insight into early embryogenesis (18–26). For instance, bovine single-cell haplarithmisis of bovine embryos uncovered segregation of parental genomes into separate lineages, highlighting a possible novel concept for the formation of chimeric and mixoploid embryos (21). Human embryos are now frequently genotyped via SNP arrays and subsequently, haplotyped for the generic and genome-wide identification of disease allele inheritance in the diagnostic setting of preimplantation genetic testing for monogenic disorders (PGT-M) (23,24,26–28). In recent years, multiple approaches for single-cell haplotyping have been developed, yet only a limited number have been clinically implemented (5,6,9,12,28–31). These first approaches are based on the use of SNP arrays to genotype single-cell and bulk DNA. Currently, two main single-cell haplotyping algorithms are available for use with SNP array data: karyomapping and haplarithmisis (6,32). These have been validated and implemented for clinical use as a methodology for PGT-M. Karyomapping utilizes only discrete SNP genotypes, while haplarithmisis implements the use of SNP B-allele frequency (BAF) values. Thereby, karyomapping allows the identification of meiotic trisomies, however on top of that haplarithmisis further identifies and discriminates trisomies of mitotic origin. Concurrent haplotyping and copy-number profiling enables comprehensive PGT effectively combining PGT-M and preimplantation genetic testing for aneuploidy (PGT-A).
With the ever-reducing costs of sequencing, we envision that single-cell SNP arrays may be replaced by sequencing-based approaches. While deep sequencing of amplified single-cell DNA does allow single-cell genotyping, this approach remains prohibitively expensive to type large number of single cells (33,34). Here, we developed a novel cost-efficient single-cell genotyping-by-sequencing (scGBS) approach (Figure 1). The methodology generates reduced genomic representation libraries using a restriction enzyme (RE) that frequently cuts the amplified genome of the cell, followed by size selection and PCR of the shorter fragments, and finally paired-end sequencing (35,36). We converted the SNP-array-based haplarithmisis workflow to allow imputation of sequencing-based data and we established the parameters allowing for equal performance compared with array-based single-cell haplotyping. We demonstrate the use of scGBS and subsequent haplotyping analysis on single-cell whole-genome amplified DNA in human and bovine. Furthermore, we establish quality parameters for PGT-M and embryo-related research.
Figure 1.

scGBS can be performed on DNA extracted from multiple cells, e.g. a cell line, or single cells, e.g. from embryos samples. Parental DNA and DNA from family members, such as grandparents or siblings, with respect to the analysis sample(s), is used for haplotyping. Preparation for scGBS consists of isolation of a single cell or multiple cells followed by a whole genome amplification (WGA) by multiple displacement amplification (MDA). Subsequently, the amplified genomic DNA is digested by a restriction enzyme followed by adapter ligation, size selection and a PCR. Multiplexing of samples is performed after adapter ligation. Processing of scGBS data consists of demultiplexing raw sequencing reads per sample, correction of overlapping reads by FLASH (40) and alignment of the corrected reads to the reference genome with BWA MEM (42). Next, GATK Haplotypecaller is applied to each sample separately and GATK GenotypeGVCFS allows for joint genotype calling per pedigree with samples to analyze combined with parental and phasing reference samples (43). A customized conversion is applied to obtain the desired input for siCHILD analysis such as A/B calling and B-allele frequency (BAF). In parallel, after the alignment step a copy-number profiling is performed per sample with QDNAseq (46). Finally, discrete genotypes, BAF and logR values are combined into one input matrix for siCHILD analysis.
MATERIALS AND METHODS
Cell lines
HapMap Epstein-Barr virus (EBV)-transformed lymphoblastoid cell lines derived from family CEPH/Utah pedigree 1463 were obtained from the NIGMS Human Genetic Cell Repository at the Coriell Institute for Medical Research for individuals GM12877, GM12878, GM12891, GM12892, GM12882 and GM12887. These cell lines were cultured in DMEM/F12 medium (Gibco, Thermo Fisher Scientific Inc., USA) with 10% fetal bovine serum (Thermo Fisher Scientific Inc., USA) at 37°C. Multi-cell samples were collected after culture and DNA was extracted using DNAeasy Blood & Tissue kit (Qiagen, Germany). Single cells of individual GM12882 and GM12887 were isolated by manual pipetting using a STRIPPER pipette with a 75 μm capillary (Origio, CooperSurgical, Inc., USA) and washed three times using polyvinylpyrrolidone in 1× phosphate-buffered saline (1% PVP-PBS). Subsequently, each single cell was transferred into a 0.2 ml PCR tube with 2 μl 1× PBS and stored at −20°C until use. Single cells were whole genome amplified (WGAed) using REPLI-g Single Cell Kit (Qiagen, Germany) according to manufacturer’s protocol with all volumes halved. Multiple displacement amplification (MDA) was performed at 30°C for 2 h followed by 65°C for 10 min for inactivation. DNA concentrations were quantified by Qubit 2.0 Fluorometer (Invitrogen, Thermo Fisher Scientific Inc., USA).
PGT-M samples
The study was approved by the local (S59286, Commissie Medische Ethiek UZ/KU Leuven) and federal ethical committee (ADV_053, Federale Commissie voor medisch en wetenschappelijk onderzoek op embryo’s in vitro). All patients involved in this study signed an informed consent form. Sample collection, SNP array analysis, library preparation and sequencing were conducted at UZ/KU Leuven. For diagnostic PGT-M, a biopsy was performed on human embryos, fertilized via intracytoplasmatic sperm injection (ICSI), with an adequate morphology. Stimulation protocols for hormonal down-regulation and follicular growth were used as described previously (37). In cleavage-stage biopsies, a single blastomere was biopsied from embryos with ≥6 blastomeres on day 3. In day-5/6 trophectoderm biopsies, a small number (five on average) of cells from the trophectoderm were biopsied (38). Embryo biopsy processing was conducted by performing WGA by MDA using the REPLI-g SC kit (Qiagen, Germany) according to the manufacturer’s instructions with full or half reaction volumes and a reduction of the incubation reaction to 2 h and inactivation of the enzyme for 3 min at 65°C (6,23). Familial bulk DNA, i.e. maternal, paternal and grandparents of the prospective embryo or the couple’s offspring, extracted from blood, was also genotyped for subsequent haplotyping analysis. For scGBS-based PGT-M, biopsy material and familial bulk DNA was processed via the protocol for scGBS.
Bovine samples
Single blastomeres of bovine preimplantation embryos were dissociated and whole genome amplified as described previously (21). Briefly, zona pellucida was removed by pronase treatment and single blastomeres were disassociated and picked individually. Single cell processing consisted of performing WGA by MDA using the REPLI-g SC kit (Qiagen, Germany) according to the manufacturer’s instructions with full or half reaction volumes and a 3 h incubation reaction. Familial samples consisted of DNA extracted from the cow’s ovarian tissue, the bull’s sperm (DNeasy Blood and Tissue kit, Qiagen, Germany) and WGA material of a sibling embryo.
SNP array
Genotyping of human cell line and PGT-M samples was carried out using the Infinium HD assay kit with HumanCytoSNP-12v2.1 BeadChips (Illumina Inc., USA) according to manufacturer’s instructions. Bovine samples were genotyped with the BovineHD BeadChips and the Infinium HD assay kit (Illumina Inc., USA) according to the manufacturer’s instructions. As input for the protocol, 600 ng of WGA DNA and 200 ng DNA isolated from a large number of cells or bulk DNA was used. Raw images were obtained by the Illumina iScan system and analyzed by Illumina’s GenomeStudio Genotyping Module with a GenCall threshold of 0.75. The output file containing the information of genotype calls, B-allele frequencies and logR ratios was fed into siCHILD for downstream analysis on both human and bovine samples (6).
siCHILD/haplarithmisis
Concurrent haplotyping and copy-number profiling for SNP array and scGBS was performed with siCHILD/haplarithmisis. Genotype calls, BAF and logR values were fed into the pipeline for both human and bovine samples (6,20,21). In brief, siCHILD relies on pedigree-based haplotyping analysis of genotypes from parents and phasing reference(s) to separate the two haplotypes of the parent(s). Subsequently, BAF values of the sample of interest are plotted across the chromosomes and distinguished across different parental SNP categories (P1/P2 and M1/M2) to evaluate parental haplotype inheritance. Profiles generated from paternal and maternal SNP categories are complementary to each other and the distance between the two paternal/maternal categories is 0.5 in a normal disomic state of the chromosome. Deviations from this distance represent a deviation in the ratio of the haplotypes and thus copy-number or copy-neutral aberrations (6). LogR values are used for copy-number profiling and can thus complement in the distinction of a disomy from a uniparental disomy (Supplementary Figure S1).
Single-cell genotyping-by-sequencing
Restriction enzyme digestion and library preparation was performed as described (36) with the following modifications to the protocol: 500 ng of input DNA was used for both multi-cell DNA and single-cell WGA product, pooling occurred at equal amounts of DNA (100 ng) and an additional size-selection step (140–240 bp) with BluePippin (Sage Science, Inc., USA) was performed prior to a PCR amplification of 8 cycles (Figure 1). Barcodes were generated through the module Barcode Generator of GBSX (39). For in silico restriction enzyme digestion and subsequent determination of the amount of expected target sequence (125 nucleotides surrounding the restriction site), the Restriction Enzyme Predictor tool of GBSX was applied (39). For HapMap samples and three PGT-M families paired-end (2×125 bp) sequencing was performed on a HiSeq2500 system (Illumina Inc., USA) in multiple runs. For the remaining PGT-M samples, paired-end (2×150 bp) sequencing was performed on a NextSeq 550 system (Illumina Inc., USA) with 20 samples on one run in High Output mode. Paired-end sequencing data were demultiplexed using the Demultiplexer module of GBSX. Subsequently, paired-end reads were flashed (i.e. reads of fragments that in size are <2× read length are overlapped and merged) using FLASH (40). Afterwards, both flashed and non-flashed reads could be merged and mapped to the GRCh37 (hg19) and bosTau9 (ARS-UCD1.2, U.S. Department of Agriculture, Agricultural Research Service) reference genomes with BWA MEM (41,42) (Figure 1). We applied GATK Depth of Coverage module for determining the depth across the targeted regions (43). Picard CollectHSMetrics was used to determine the number of reads with a mapping quality > 0 (Supplementary Table S1) (http://broadinstitute.github.io/picard/).
Genotype inference following scGBS
We applied GATK Haplotypecaller (Best Practice) to the BAM files of each sample. A multi-sample genotyping file was created using GATK GVCFs (Figure 1) (43). For performance testing of single-cell WGA material, we compared single-cell with bulk GBS sequences derived from siblings GM12882 (n = 5 single cells) and GM12887 (n = 2 single cells) of the CEPH/Utah 1463 HapMap family for which we also generated parental bulk scGBS data. Comparison analyses were performed with the use of BCFtools, BEDtools and custom scripts (44,45). The following dbSNP information was used: Database of Single Nucleotide Polymorphisms (dbSNP). Bethesda (MD): National Center for Biotechnology Information, National Library of Medicine (dbSNP Build ID: 151) available from: http://www.ncbi.nlm.nih.gov/SNP/. Subsequently, for haplotype reconstruction, we applied only those SNVs having a depth of coverage of ≥7 for multi-cell samples and ≥11 for single-cell samples. The SNV calls were then transformed to bi-allelic calls (i.e. AA, AB and BB) with a custom R script using the following Bioconductor libraries: SNPlocs.Hsapiens.dbSNP144.GRCh37, VariantAnnotation and vcfR. B-allele frequency values were calculated based on allele-specific depth of coverage. Accuracy calculation of haplotype blocks, i.e. haploblocks, was performed between scGBS-derived single-cell and bulk samples, SNP-array-derived single-cell and bulk samples and scGBS-derived versus SNP-array-derived samples, for which either bulk haplotypes or the SNP array-derived haplotypes served as the reference. This accuracy or haplotype concordance was expressed as the percentage of matched haploblock length divided by the total length of haplotypes obtained in the reference and computed by using BEDtools and custom scripts (Supplementary Figure S2) (45).
Copy-number profiling
In addition to haplotype information, copy-number profiling following scGBS was performed. Bioconductor’s package QDNAseq and custom scripts (available via https://github.com/GenomicsCoreLeuven/publications_JV/tree/main/GBS) were used for copy-number profiling with the GRCh37 (hg19) and bosTau9 (ARS-UCD1.2, U.S. Department of Agriculture, Agricultural Research Service) reference genomes, respectively for human and bovine (46). Copy-number profiles were visually compared between scGBS- and SNP-array based data. In order to describe an aneuploidy, the copy-number deviation had to be supported by the corresponding BAF profile and vice versa.
RESULTS
Selection of restriction enzyme
Three different restriction enzymes (REs), ApeKI (G^CWGC), NspI (RCATG^Y) and PstI (CTGCA^G) were assessed for their human genome complexity reduction. An in silico digestion of the GRCh37 reference genome (hg19) was employed to identify the possible target fragments and to determine the distribution of fragment size ranges (Supplementary Figure S3). A total of approximately 4.9, 3.0 and 1.3 million fragments are generated after in silico RE digestion with ApeKI, NspI and PstI, respectively. A further reduction of the genome by size selection (80–200 bp) would yield a theoretical 19.0, 11.1 and 7.1% of the in silico predicted fragments, corresponding to a total number of 935 755, 337 578 and 91 370 fragments to be sequenced for ApeKI, NspI and PstI, respectively. The range for size-selection was guided by a computational process of merging the overlapping paired-end sequencing reads, i.e. fragment sizes shorter than twice the read length, which is called fast length adjustment of short reads or FLASH (40). This step could further minimize the fraction of the genome to be sequenced while in parallel improve base calling accuracy, which is of importance for genotyping. For haplotyping it is crucial that informative SNPs are genotyped, i.e. SNPs that are heterozygous in one parent and homozygous in the other parent (32). A median of 98 980 (± 1784) informative SNPs was identified in bulk DNA samples of both parents from a total of 10 PGT-M families interrogated via the 300 K SNP array platform (in-house data). As a guide in the selection of the RE, an overlap was performed between in silico generated intervals with biallelic SNPs with a minor allele frequency of >0.05 extracted from dbSNP to allow the inclusion of sufficient common variants to be used for downstream haplotyping analysis, which is based on informative SNPs (Supplementary Table S2). This showed in silico that restriction digestion with ApeKI, regardless of size-selection, and with NspI without size-selection would surpass the total number of SNPs covered on the human SNP array (6,23,24,47). Combined with size-selection, the total size of the genome targeted by ApeKI and NspI would result in 125.7 and 47.7 Mb, respectively (Supplementary Table S3).
scGBS on HapMap cell lines
scGBS was first performed on samples of the HapMap cell lines of family CEPH/Utah Pedigree 1463. Joint SNP calling of individuals GM12877 (father) and GM12878 (mother) allowed to identify the number of biallelic SNPs. A comparison was made between size and non-size selection with FLASH correction. Restriction digestion with ApeKI combined with size selection and FLASH had the highest number of informative SNPs (Supplementary Table S4). To allow for pedigree-based haplotyping using bulk as well as single-cell DNA samples, GBS libraries were made from individuals GM12877 (father), GM12878 (mother), GM12882 (sibling 06), GM12887 (sibling 11) with bulk DNA and a total of 7 single cells from GM12882 (n = 5) and GM12887 (n = 2). Each sibling is alternately used as a phasing reference to complete pedigree-based haplotyping. A total of 818.6M raw reads were gathered across all individuals in the pedigree. Following alignment of the GBS data and in silico digestion of the GRCh37 reference genome (hg19), the mean depth and breadth of sequencing coverage for the ApeKI targeted regions in the bulk DNA and single cells were 100.5 X (± 65.02 X SD), 88.65 X (± 5.22 X SD), 25 X (± 10.6 X SD) and 83.69 X (± 5.72 X SD), respectively. Comparison of in silico genomic intervals with the actual sequenced genomic intervals revealed a mean of 56% similarity (48). After joint genotyping, we first compared genotypes from the bulk sequences of individuals GM12877 and GM12878 with the available high-confidence variant calls from the Platinum genomes, which showed that a minimum of 7X depth of coverage produces 98% accurate heterozygous SNV calls (Figure 2A) (49). Subsequently, we determined the minimal depth of coverage for single cells. Comparison of single-cell SNVs with the multi-cell heterozygous SNVs with a coverage of 7X or higher in function of single-cell sequencing coverage found that 11X gives 74.7% (±15.7 SD) accurate heterozygous single-cell SNV calls (Figure 2B and Supplementary Figure S4). These single-cell and bulk SNV genotypes were fed to siCHILD/haplarithmisis, producing single-cell scGBS haplotypes that reached a concordance of ≥99% for most samples with an average of 98.1% when compared to bulk scGBS haplotypes across both individuals. More specifically, 98.6% paternal and 97.7% maternal haplotype concordance on average was reached between scGBS-based haplarithm profiles for single-cell versus bulk haplotypes (Supplementary Table S5). Furthermore, scGBS delivered comparable accuracies to SNP array-derived haplotypes with an average of 98.5% paternal and 97.7% maternal haplotype concordance (Figure 2C and Supplementary Table S6). This is similar to the results of single-cell versus bulk haplotypes after SNP array, which on average resulted in 99.1% and 99.3% maternal and paternal haplotype concordance, respectively (Supplementary Table S7).
Figure 2.

scGBS haplotyping and copy-number profiling on HapMap cell lines. (A) Genotypes from joint genotyping individuals GM12877 (father) and GM12878 (mother) were compared to heterozygote SNV calls from the Platinum Genomes (Illumina Inc., USA) and both accuracy (blue line = mean ± standard deviation) and the total number (red line = mean ± standard deviation) of the genotypes were evaluated against the minimum coverage of the genotype calls in order to select a coverage threshold. The gray line represents the coverage of 7×, which was applied as a threshold to bulk samples in subsequent analyses. (B) Genotypes from single cells of siblings GM12882 and GM12887 were compared to heterozygote SNV calls from its respective bulk sample with a threshold applied of 7× coverage. The accuracy (blue line = mean ± standard deviation) and the total number (red line = mean ± standard deviation) of the calls were evaluated against the minimum coverage of the genotype calls. The gray line represents the coverage of 11×, which was applied as a threshold to single-cell samples in subsequent analyses. (C) For each chromosome an ideogram together with the haplotype blocks after SNP array (top) and scGBS (bottom) is shown. Dark and light blue represent paternal haplotyping, whereas red and light red represent maternal haplotyping. Transition from dark to light color or vice versa represents an homologous recombination site. Genome-wide haplotype block comparison of scGBS- and SNP-array-based haplotyping results for a single blastomere biopsy sample from GM12882 from the HapMap pedigree (GM12882_sc37) are shown. Phasing occurred via sibling GM12887, which results in the availability of both maternal and paternal haplotypes. (D) An example of the genome-wide comparison between scGBS- and SNP-array-based copy-number profiling is shown for two samples from the HapMap pedigree; one bulk (GM12887_MC) and one single-cell (GM12887_sc04). Both genome-wide maternal and paternal haplarithm (Mat-BAF and Pat-BAF tracks) plots together with a copy-number values (logR track) plot for chromosome 1 to X are displayed. Haplarithm plots serve as the visualization of the haplotyping process by haplarithmisis (Supplementary Figure S1). Haplotyping was performed with a sibling (GM12882) and thus, both maternal and paternal haplarithm can be displayed. For the first sample (GM12887_MC), interpretation of the logR profile identified a mosaic paternal trisomy for chromosome X (green box), which was not reported in the karyotype of the cell line. In the single-cell sample, GM12887_sc04, the pure trisomy for chromosome X is present. Copy-number analysis with a combination of haplarithm and logR profiles allows to specify parental origin for the aberrations. In case of disomic chromosomes, red and blue lines for maternal and/or paternal haplarithms are spaced with a distance of 0.5 apart (see legend bottom right). The pure paternal trisomy X in the single cell is indicated indirectly by the increased distance (= 0.67) between the maternal red and blue lines, which represents a lower fraction of maternal compared to paternal chromosomes. Here, the presence of a paternal trisomy for chromosome X in a mosaic state is indicated by a distance of <0.67 between the red and blue lines of the maternal haplarithm. These findings show concordant between scGBS and SNP array.
Copy-number profiling scGBS data
Concurrent haplotyping and copy-number profiling provides a comprehensive view of the chromosomal copy numbers and parental origin. Furthermore, it allows identification of genome-wide ploidy deviations such as some forms of triploidy or genome-wide uniparental disomy. Copy-number profiles generated after scGBS were visually compared with the logR profiles from SNP array. Copy-number aberrations via scGBS were concordant with the combined haplarithm and logR analysis following SNP array for all cells analyzed (Figure 2D and Supplementary Figure S5). The karyotypes from cell lines GM12882 and GM12887 are 46XY and 46XX, respectively. Unexpectedly, the copy number profile of the bulk sample for individual GM12887 revealed the presence of a paternal trisomy X in approximately 25% of the cells. Interestingly, both single cells isolated from the GM12887 cell line carried the paternal trisomy X.
Preimplantation genetic testing of human embryos
To further validate the scGBS methodology for PGT-M, 14 single blastomeres and 3 trophectoderm samples biopsies from human preimplantation embryos were processed. These samples, derived from 6 families with various genetic disorders instigating PGT-M, were previously haplotyped via SNP array. Pedigree-based haplotyping was performed via the family members listed in Supplementary Table S8. Specifically, for scGBS in family 6 phasing was performed with a single grandparent (maternal grandfather) instead of a grandparental duo. Haploblock inheritance was visually assessed at the locus of interest with regard to the modes of inheritance specified per family. Four PGT-M families with a total of 12 embryos underwent investigation for an autosomal dominant mutation. Six embryos did not carry the haplotype linked to the mutation, 5 carried the risk haplotype and for one embryo the result was inconclusive due to the proximity of a recombination site to the border of the inherited haploblock. Analysis for an X-linked recessive mutation in one PGT-M family identified a carrier female and an affected male embryo. Results on three embryos, analyzed for an autosomal recessive disorder in the family, identified two carriers of the mutation and one embryo with an inconclusive result due to the proximity of a recombination site, i.e. <150 SNPs flanking the genetic crossover, based on the threshold applied through the original SNP array analyses as determined by Zamani Esteki et al. (6). All of the scGBS and SNP-array based analysis results concerning the inherited haploblock linked to the mutation for the locus of interest were concordant (Figure 3A–D). Genome-wide haplotype concordance was assessed for all samples either on paternal, maternal or biparental haplotype profiles depending on the analysis following the mode of inheritance (Figure 4A,B). An average of 97.5% paternal and 97.8% maternal haplotype concordance was reached between scGBS- and SNP-array-based haplarithm profiles (Supplementary Table S9). Additionally, copy-number aberrations upon clinical evaluation following SNP array data were compared between scGBS and SNP array. This clinical evaluation marks that a copy-number aberration is only called when it is both supported by a deviation in haplarithm(s) and logR tracks. Small imbalances might be observed on copy-number profiling alone, but are thus not considered. An example of this can be seen for SNP array data in E03 from Family 4 (Supplementary Figure S6) at telomeric regions for some chromosomes and for scGBS data in a centromeric region of chromosome X for E06 from Family 6 (Figure 5). For 17 human preimplantation embryos a total of 11 aneuploidies were identified. In 10 embryos no aneuploidies were detected. Of the remaining embryos, most (n = 4) had one or two aneuploidies, a combination of a single aneuploidy with a segmental aneuploidy (n = 2) or a single segmental aneuploidy (n = 1). Interestingly, two embryos from the same family carried up to two meiotic trisomies per embryo, which haplarithmisis can discriminate from a mitotic error through the use of BAF values. Furthermore, the trisomies were of maternal origin and resulted from a meiosis I error. All findings were concordant through visualization of copy-number profiles following SNP array and scGBS analyses (Figure 5 and Supplementary Figure S6).
Figure 3.

Haplotyping results of the disease loci associated chromosome are shown after scGBS and SNP array processing of the sample. Samples represent a single blastomere biopsied from a day 3 embryo for all except in (D), which represents a trophectoderm biopsy (on average 5 cells from a day-5 blastocyst). Each subfigure consists of a family pedigree with haplotype inheritance information and the haplotyping result (haplotype blocks) of one or two embryos per family. The embryo number is indicated in green or red for a haplotyping analysis result which deemed the embryo eligible or not for transfer, respectively. Haplotype blocks with blue or red coloring are shown for each embryo and methodology (scGBS versus SNP array) and indicate paternal or maternal haplotype inheritance, respectively. Additionally, in (B) a maternal haplarithm, i.e. segmented maternal BAF values track, is shown under the haplotype block. From this haplarithm track, besides the haplotype blocks, also the number of copies per haplotype block can be deduced. In this specific track, the red dotted line shows segmented values of the M1 SNPs and the blue dotted line represents segmented values of M2 SNPs, each a distinct category of maternal SNPs. The distance between the two categories should be 0.5 for a chromosome in a disomic state (1 maternal and 1 paternal) and 0 for the inheritance of only one chromosome (monosomy). The locus of interest is indicated by an orange dashed line corresponding to the position along the chromosome ideogram. (A) Autosomal dominant inheritance linked to the (grand)maternal haplotype. Embryo 1 (E01) inherited the grandpaternal haplotype (red) and is unaffected for the monogenic disorder. Embryo 2 (E02) inherited the grandmaternal haplotype (pink) and is affected for the monogenic disorder. (B) X-linked recessive mutation inheritance linked to the (grand)maternal haplotype of the X-chromosome. Both mother and grandmother of the embryos are a carrier for this mutation. Embryo 1 (E01) inherited the grandmaternal haplotype (pink). Information from the maternal haplarithm shows chrX to be present in a disomic state and hence, an unaffected chrX is inherited from the father of the embryo resulting E01 to be a carrier female. Embryo 2 (E02) inherited the grandmaternal haplotype (pink). The maternal haplarithm profile shows only one chrX to be present and hence, no additional chrX is inherited from the father, but a Y-chromosome instead (not shown). Therefore, E02 results in an affected male. (C) Autosomal recessive inheritance linked to the maternal and paternal haplotypes carried by the affected sibling. Embryo 8 (E08) inherited the same paternal haplotype as the sibling (blue) and consequently the paternal mutation. The maternal haplotype is flanked by a homologous recombination site, which is too close to the locus of interest (orange dashed line) to allow distinguishing inheritance of the same or opposite haplotype compared to the sibling. Hence, haplotyping results for E08 are found inconclusive. Embryo 10 (E10) inherited the same paternal haplotype (blue) as the sibling and the opposite for the maternal haplotype (pink). E10 is thus an unaffected carrier of the paternal mutation. (D) Autosomal dominant inheritance linked to the maternal (grandpaternal) haplotype in red. Embryo 4 (E04) inherited the grandmaternal haplotype (pink) and hence, is unaffected for the monogenic disorder.
Figure 4.
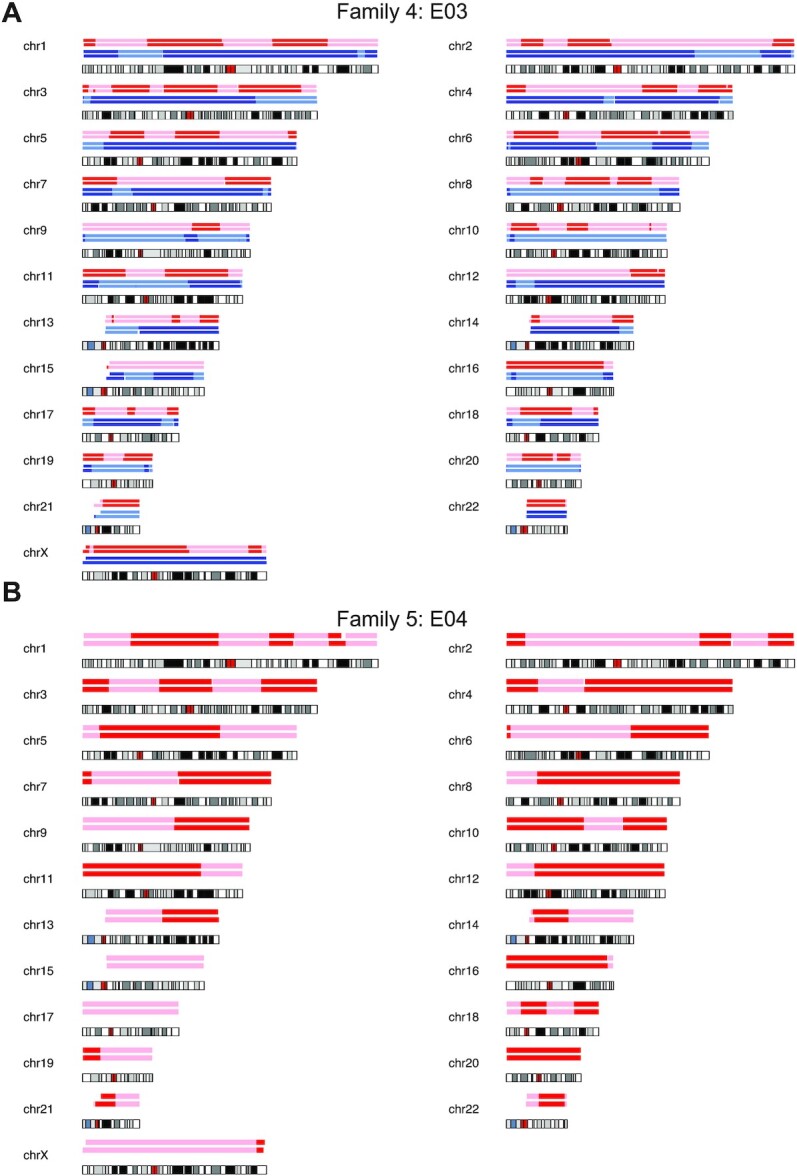
For each chromosome an ideogram together with the haplotype blocks after SNP array (top) and scGBS (bottom) is shown. Dark and light blue represent paternal haplotyping, whereas red and light red represent maternal haplotyping. Transition from dark to light color or vice versa represents an homologous recombination site. (A) Genome-wide haplotype block comparison of scGBS- and SNP array-based haplotyping results for a single blastomere biopsy sample from family 4. Phasing occurred via an affected sibling, which results in the availability of both maternal and paternal haplotypes. (B) Genome-wide haplotype block comparison of scGBS- and SNP array-based haplotyping results for a trophectoderm biopsy sample from family 5. Phasing occurred via maternal grandparents, hence only maternal haplotypes are drawn.
Figure 5.

An example of the genome-wide comparison between scGBS- and SNP-array-based copy-number profiling is shown for two embryo biopsies from two families. Only the genome-wide maternal haplarithm (Mat-BAF) track together with a copy-number values (logR) track are displayed both for SNP array and scGBS data. A legend is provided in the bottom right of the displayed haplarithm profiles in case of disomy and/or deviations from the disomic state from the maternal information according to the principles of haplarithmisis in Figure S1. In both families haplotyping was performed with maternal grandparents and thus, only the maternal haplarithm can be displayed. For the first embryo biopsy (E09 of family 1), interpretation of the logR profile identified a monosomy for chromosome 14 and a segmental trisomy for chromosome 16. In the second embryo biopsy, embryo 8 of family 2, a single trisomy for chromosome 19 is present. Copy-number analysis with a combination of haplarithm and logR profiles allows to specify parental origin for the aberrations. In case of disomic chromosomes, red and blue lines for maternal and/or paternal haplarithms are spaced with a distance of 0.5 apart. Here, in the first embryo, the presence of a maternal monosomy for chromosome 14 (only the maternal copy is remaining) is indicated by a distance of 0 between the red and blue lines of the maternal haplarithm. The segmental trisomy for chromosome 16 is of paternal origin, which is indicated indirectly by the increased distance (= 0.67) between the maternal red and blue lines, which represents a lower fraction of maternal compared to paternal chromosomes. For the second embryo, a meiotic maternal trisomy can be elucidated, since a decreased distance of 0.33 on the maternal haplarithm can be seen. The red and blue lines are centered around 0.5, which indicates the presence of two different haplotypes along the chromosome and hence, corresponds with a meiosis I error. The aberrations were concordant between scGBS and SNP array.
Genetic dissection of bovine IVF embryos
Single-cell haplotyping has been restricted to organisms where SNP arrays are available. To expand the single-cell haplotyping potential, we explored the value of scGBS to study other species. The human scGBS protocol was applied to bovine embryos. Seven single bovine blastomeres from a total of three bovine embryos in three different families were processed by scGBS. Upon comparison of in silico genomic intervals with the sequenced intervals a mean of 61% similarity was obtained. Four of the blastomeres were previously genotyped with bovine SNP array and analyzed with haplarithmisis (20, unpublished results). All 7 blastomeres were evaluated for copy-number changes as well. Upon SNP array analysis, two blastomeres of one day 3 embryo showed a reciprocal chromosome gain and loss for chromosome 6 in blastomere 1 and 2, respectively, and a chromosome loss for chromosome 14 in both (Figure 6A, e.g. E03_Bl001_BRP010). The combined use of haplotyping and copy-number profiling provides strength to detect more complex genomic constitutions, besides single chromosomal aberrations, as well. Following SNP array, blastomere 1 of BAC_E04 revealed a complex genome-wide profile with signs of polyploidy when BAF-profiles were linked with logR-profiles and compared among chromosomes (Supplementary Figure S7). On the other hand, blastomere 3 of BAC_E04 showed an androgenetic profile, i.e. only a paternal genome is present. Thereby, haplarithmisis revealed parental genome segregation profiles across the blastomeres from the two remaining embryos. Comparison of the homologous recombination sites for the paternal haplotypes, i.e. the alternation point of haploblocks, between blastomeres 1 and 3 of BAC_E04 reveals a distinct homologous recombination pattern between the two and hence, shows two paternal genomes are present (Figure 6B). In the third embryo, BAC_E02, three blastomeres were analyzed and revealed the following: one androgenetic and two biparental blastomeres with a reciprocal segmental gain and loss in each (Supplementary Figure S7). Biologically, this reveals one paternal genome is sequestered into a distinct lineage for both embryos BAC_E02 and BAC_E04. This phenomenon was previously discovered in another cohort of bovine embryo samples and termed heterogoneic division (21). All copy-number findings were concordant between SNP array, when available, and scGBS data. For four blastomeres, genome-wide haplotype comparison of scGBS haplotypes with SNP array haplotypes was performed and showed an average of 93.8% paternal and 94.9% maternal haplotype concordance (Supplementary Table S10).
Figure 6.
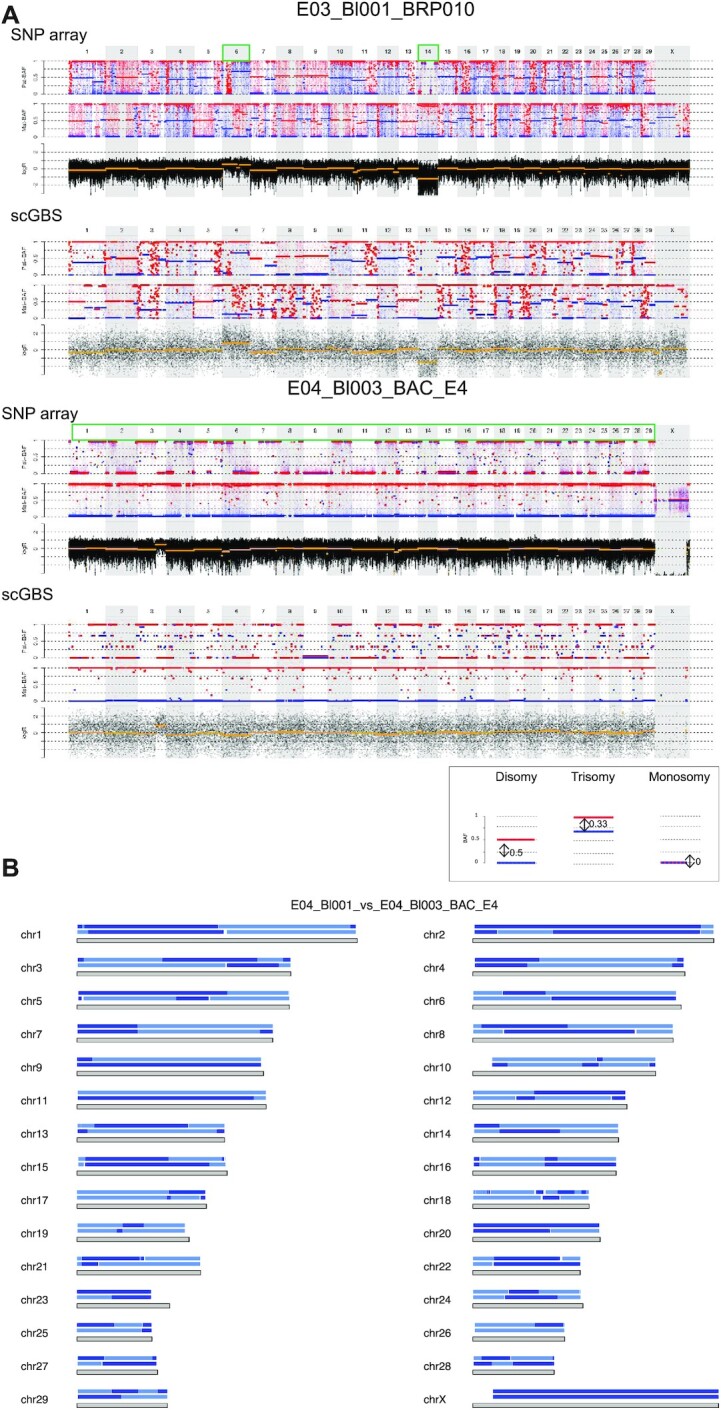
scGBS applied to bovine single blastomeres. Examples of haplotyping and copy-number profiling results from bovine single blastomeres are shown. (A) For each chromosome the length is characterized by a grey block with the haplotype blocks of E04_Bl001 (top) and E04_Bl003 (bottom) from family BAC_E4 is shown. Dark and light blue represent paternal haplotyping. Transition from dark to light color or vice versa represents an homologous recombination site. Genome-wide paternal haplotype block comparison of the two single blastomeres from the same embryo reveals multiple different homologous recombination sites indicating two differential paternal genomes present in the same embryo. (B) Genome-wide comparison between scGBS- and SNP array-based haplotype and copy-number profiling is shown for two bovine embryo biopsies from two families. The genome-wide maternal and paternal haplarithm (Mat-BAF and Pat-BAF, respectively) tracks together with a copy-number values (logR) track are displayed. In both families haplotyping was performed with a sibling embryo, which results in the display of both maternal and paternal haplarithm. A legend is present below the figure to highlight a disomic profile and the specific haplarithm profile for the parent-specific aberration. In case of disomic chromosomes, red and blue lines for maternal and/or paternal haplarithms are spaced with a distance of 0.5 apart. In E03_Bl001_BRP010, comprehensive analysis by using haplotyping and copy-number profiling results in the detection two single aneuploidies, i.e. a paternal trisomy of chromosome 6 (distance Pat-BAF = 0.33 and Mat-BAF = 0.67) and a paternal monosomy for chromosome 14 (distance Pat-BAF = 0 and Mat-BAF = 1). In a second example, a single blastomere from a separate embryo, E04_Bl003_BAC_E4, combined analysis of haplotyping and logR results in the identification of only paternal chromosomes genome-wide, with a segmental gain for chromosome 3. Analysis of logR profile alone would have resulted in the interpretation of a segmental trisomy for chromosome 3. However, the distance of the red and blue lines on the Pat-BAF profile is 0 (Pat-BAF) and 1 for the Mat-BAF profile reveals the more complex genomic constitution of the single blastomere.
DISCUSSION
Here we demonstrate that a reduced representation genome sequencing approach (scGBS), enables haplotype-based profiling and concurrent chromosomal copy-number analysis of single cells without requiring deep whole-genome sequencing. This methodology expands the toolbox of single-cell haplotyping approaches and can be implemented for studying genomic aberrations in single cells, genomic recombination in germ cells, embryos and other tissues. In addition, we demonstrate the potential for clinical applications such as PGT-M and PGT-A.
Conversion of array-based approaches to sequencing will allow for sample multiplexing and hands-on time to be reduced. Additionally, by stepping away from a SNP array-based approach in which a fixed number and set of SNP markers is interrogated in each experiment, a more comprehensive view of the (fractional) genome can be achieved by sequencing. This grants access to the detection of familial-specific markers and/or variations. Finally, for many organisms SNP arrays are not available. The conversion to sequencing-based haplotyping allows single-cell haplotyping in all species. Haploseek combines both sequencing and SNP arrays to profile single cells by utilizing single-cell low-coverage whole-genome sequencing for copy-number profiling and haplotyping, but processes phasing references via SNP arrays to determine maternal and paternal haplotypes (9,12). scHaplotyper demonstrated discrimination and visualization of the inheritance of the haplotypes for PGT-M purposes based on single-cell DNA sequencing by using a Hidden Markov Model on phased SNPs; however, no detailed information was provided on the sequencing process itself (31). Chen et al. utilized whole-genome sequencing to accomplish genome-wide haplotyping for comprehensive PGT in both the presence and absence of a proband for haplotype analysis (50). In the latter case, the embryonic samples itself serve as the phasing reference for haplotyping. An important aspect of parent-only haplotyping is that it can only be applied if direct genotyping of the variant can reliably be performed and multiple, and hence, sufficient embryo samples are available for discriminating the parental disease-causing allele in the offspring. Furthermore, novel sequencing methodologies such as linked-read, long-read and Hi-C sequencing have either been applied or proposed for genome-wide haplotyping (51–54). Nonetheless, these whole-genome sequencing strategies, some more than others, are still prohibitively expensive. Alternatively, genotyping-by-sequencing (GBS) can be used to reduce the genomic size and complexity of the genome as described here and can replace the use of SNP arrays altogether. This principle was previously also developed by Agilent Technologies as OnePGT (29). Both OnePGT and scGBS rely on reduced representation sequencing and are based on the principles of haplarithmisis, which are embedded in the SNP array-based siCHILD. This allows the discrimination of, for example, meiotic from mitotic trisomies and uniparental disomy from monosomy through the interpretation of BAF values, which unite parental information with ploidy assessment, and of logR values, which is also termed comprehensive PGT (6,29). Copy-number profiling alone via QDNAseq after scGBS processing of the samples could offer a valuable alternative to low-coverage WGS for the detection of structural rearrangements. Low-coverage sequencing of GBS libraries was showcased by OnePGT in cases of offspring at risk for unbalanced structural rearrangements from balanced translocation carriers (PGT-SR). Despite a decrease in cost when scGBS libraries are sequenced at a low coverage, the simultaneous detection of haplotypes with their copy-number is dismissed and no distinction between normal embryos or a balanced translocation carrier embryo can be made. Despite their similarities regarding GBS-processing and haplarithmisis-based analysis, OnePGT presents itself as a closed toolbox. Recently, McCoy et al. employed low-coverage sequencing linked to allele frequencies and linkage disequilibrium to build an algorithm for the detection and classification of chromosomal abnormalities informed by haplotypes (55). Like so, the discrimination of meiotic and mitotic aberrations and genome-wide ploidy abnormalities such as haploidy and triploidy is made possible from low-coverage sequencing data. This approach adds an informative layer to standard PGT-A, while remaining cost-effective. However, the lack of use of family members does not allow this workflow to be applied for comprehensive PGT or PGT-M alone. Haplarithmisis is developed to enable correct phasing in sequencing data with high amount of amplification artefacts. Most recently, an improved version of MDA, called Primary Template Directed Amplification or PTA in short, served as a basis for a sequencing-based PGT methodology mainly applied to aneuploidy and direct variant screening, which could eventually enable traditional phasing approaches to be used (56–59).
ApeKI showed versatility for increasing the number of SNPs that could be targeted in the future, through widening the size-selection range or bypassing it completely, if sequencing costs decrease. This offers flexibility/amenability of the assay without the need of using a different RE. Size selection allowed to further reduce the fraction of the genome to be sequenced following the restriction digestion and in silico FLASH correction of the paired-end reads increased SNP calling accuracy moderately. Sonah et al. implemented selective amplification of GBS fragments in their protocol in a similar effort to increase the depth of coverage (60). Such selection offers a possible higher degree of multiplexing without a dramatic loss of SNP calls. However, a trade-off should always be made between the depth of coverage and the amount of SNP calls necessary per application. Hence, scGBS represents a more flexible accessible platform both from the wet lab and bioinformatics aspects. Cost-effectiveness of scGBS versus previously discussed methods can be manifested on different levels. Compared to SNP array, with scGBS hands-on-time is reduced given that SNP array processing generally takes three days. Moreover, as sequencing service facilities emerge, the investment of acquiring SNP array equipment is outweighed by outsourcing samples to these facilities. The use of more high-throughput sequencing platforms, for example, the introduction of the Illumina NovaSeq machines, compared to the use of HiSeq and NextSeq machines here in this study, allows for a higher degree of sample multiplexing and hence, a sequencing cost that is competitive to or even drops below the cost of SNP arrays. Other methodologies depend on the (clinical) application, i.e. PGT-M versus PGT-A, and degree of information, e.g. type of versus parental origin of chromosomal aberrations, that is needed. In a clinical setting, this can vary depending on the counseling and material that is available. Additionally, we showcase a first-time application of concurrent haplotyping and copy-number assessment through reduced representation sequencing on bovine samples without any changes to the wet lab protocol. However, variations exist in the employment of restriction enzymes for identifying SNPs from the sequenced fragments as markers for genotyping. First, there are many different restriction enzymes available, each of them creating a unique digestion pattern of the genome. Moreover, the recognition site where a restriction enzyme will cut can vary in length, symmetry and GC versus AT content. In turn this leads to a scalable reduction of genomic proportion dependent on the choice of restriction enzyme.
Previously, concurrent genome-wide haplotyping and copy-number assessment has allowed to finetune embryo transfer policies regarding embryo prioritization and allows to consider nonpronuclear and monopronuclear (0 and 1PN) embryos for transfer, embryos which are in other circumstances discarded (23,24). The aim of PGT-M is to prevent the transfer of an embryo carrying the risk allele inherited from one or both parents. For this haplotyping is performed to identify to which haplotype the risk allele is linked. OnePGT excels through its automatic delineation of haploblocks and thus, an automatic call for haplotyping at the locus of interest is provided. For all these approaches, genotype information is needed, with respect to the embryo analyzed, from both parents together with a sibling (affected or unaffected for the monogenic disorder) or grandparents from the parental side at risk of transmitting the disease allele. A one-time scGBS process would be performed on samples of the couple and the phasing references prior to the start of the IVF cycle to serve as a diagnostic work-up. This is in concordance with the current practice for both the array-based approaches and the recently developed sequencing-based methodologies. Moreover, OnePGT and siCHILD allow pedigree-based haplotyping with only one grandparental genotype available in case phasing should be performed through the grandparents of the respective embryo. Here, we applied single grandparental haplotyping for one PGT-M family upon scGBS. Recently, an additional feature was incorporated into siCHILD to allow haplotype analysis via parental siblings (47). The features of alternative phasing options can be applied to scGBS data in the future. For bovine samples, pedigree-based haplotyping was achieved by genotyping a sibling embryo to the embryo undergoing analysis. This strategy provides an interesting addition to the scGBS analysis platform by eliminating the need for extensive pedigree knowledge, i.e. grandparental genotype information, in human, cattle or any species for future developments. Recently, advancements for the correction of mutations on the disease-causing allele in human embryos have been made with CRISPR-Cas9 technology (61,62). Application of our scGBS haplotyping-based approach, prior and/or after gene editing, can increase throughput of such analyses and eliminate the need for separate assays to establish mutation detection, copy-number profiling and genotyping, albeit in familial, single blastomere or trophectoderm samples. Furthermore, scGBS could be used in animal breeding programs allowing for haplotyping-based embryo selection.
In conclusion, combination of scGBS with haplarithmisis allows for a sequencing-based concurrent haplotype and copy-number assessment of cell lines or embryo samples in any species with a diploid genome. The application of scGBS will enable single-cell chromosomal instability studies and can potentially be translated to the clinic as a mean for comprehensive PGT for human and bovine preimplantation embryos.
DATA AVAILABILITY
In compliance with the GDPR and the study protocol, the HapMap and PGT-M dataset used in the study is not publicly available. Cell line, embryo, parental and phasing relatives raw genotyping and scGBS data has been deposited at the European Genome-phenome Archive (EGA), which is hosted by the EBI and the CRG, under accession number EGAS00001005401. It is available to academic users upon request to the Data Access Committee (DAC) of KU Leuven via the corresponding author (JRV). The bovine scGBS data for this study have been deposited in the European Nucleotide Archive (ENA) at EMBL-EBI under accession number PRJEB45461 (https://www.ebi.ac.uk/ena/browser/view/PRJEB45461). The bovine SNP array genotyping data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (Edgar et al., 2002) and are accessible through GEO Series accession number GSE178156 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE178156). We have provided the bioinformatical scripts via the following link: https://github.com/GenomicsCoreLeuven/publications_JV/tree/main/GBS
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank the couples who participated in the study.
Contributor Information
Heleen Masset, Laboratory for Cytogenetics and Genome Research, Department of Human Genetics, KU Leuven, Leuven, 3000, Belgium.
Jia Ding, Center of Human Genetics, University Hospitals of Leuven, Leuven, 3000, Belgium.
Eftychia Dimitriadou, Center of Human Genetics, University Hospitals of Leuven, Leuven, 3000, Belgium.
Amin Ardeshirdavani, Department of Electrical Engineering (ESAT), STADIUS Center for Dynamical Systems, Signal Processing and Data Analytics, KU Leuven, Leuven, Belgium.
Sophie Debrock, Leuven University Fertility Center, University Hospitals Leuven, Leuven, 3000, Belgium.
Olga Tšuiko, Laboratory for Cytogenetics and Genome Research, Department of Human Genetics, KU Leuven, Leuven, 3000, Belgium; Center of Human Genetics, University Hospitals of Leuven, Leuven, 3000, Belgium.
Katrien Smits, Department of Internal Medicine, Reproduction and Population Medicine, Ghent University, Merelbeke, 9820, Belgium.
Karen Peeraer, Leuven University Fertility Center, University Hospitals Leuven, Leuven, 3000, Belgium.
Yves Moreau, Department of Electrical Engineering (ESAT), STADIUS Center for Dynamical Systems, Signal Processing and Data Analytics, KU Leuven, Leuven, Belgium.
Thierry Voet, Laboratory of Reproductive Genomics, Department of Human Genetics, KU Leuven, Leuven, 3000, Belgium.
Masoud Zamani Esteki, Department of Clinical Genetics, Maastricht University Medical Center, Maastricht, 6202 AZ, The Netherlands; Department of Genetics and Cell Biology, GROW School for Oncology and Developmental Biology, Maastricht University, Maastricht, 6229 ER, The Netherlands.
Joris R Vermeesch, Laboratory for Cytogenetics and Genome Research, Department of Human Genetics, KU Leuven, Leuven, 3000, Belgium; Center of Human Genetics, University Hospitals of Leuven, Leuven, 3000, Belgium.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Fonds Wetenschappelijk Onderzoek [11A7119N to H.M]. Funding for open access charge: KU Leuven [C14/18/092 to J.R.V., T.V.].
Conflict of interest statement. M.Z.E, J.R.V. and T.V. are co-inventors on patent applications: ZL910050-PCT/EP2011/060211- WO/2011/157846 Methods for haplotyping single cells’ and ZL913096-PCT/EP2014/068315 ‘Haplotyping and copy-number typing using polymorphic variant allelic frequencies’. T.V. and J.R.V. are co-inventors on patent application: ZL912076-PCT/EP2013/070858 ‘High-throughput genotyping by sequencing’. Haplarithmisis (‘Haplotyping and copy-number typing using polymorphic variant allelic frequencies’) has been licensed to Agilent Technologies. The remaining authors declare no conflict of interest.
REFERENCES
- 1. Navin N., Kendall J., Troge J., Andrews P., Rodgers L., McIndoo J., Cook K., Stepansky A., Levy D., Esposito D.et al.. Tumour evolution inferred by single-cell sequencing. Nature. 2011; 472:90–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Voet T., Kumar P., Van Loo P., Cooke S.L., Marshall J., Lin M.-L., Zamani Esteki M., Van der Aa N., Mateiu L., McBride D.J.et al.. Single-cell paired-end genome sequencing reveals structural variation per cell cycle. Nucleic Acids Res. 2013; 41:6119–6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen X., Love J.C., Navin N.E., Pachter L, Stubbington M.J.T., Svensson V., Sweedler J.V., Teichmann S.A.. Single-cell analysis at the threshold. Nat. Biotechnol. 2016; 34:1111–1118. [DOI] [PubMed] [Google Scholar]
- 4. Vermeesch J.R., Voet T., Devriendt K.. Prenatal and pre-implantation genetic diagnosis. Nat. Rev. Genet. 2016; 17:643–656. [DOI] [PubMed] [Google Scholar]
- 5. Natesan S.A., Bladon A.J., Coskun S., Qubbaj W., Prates R., Munne S., Coonen E., Dreesen J.C.F.M., Stevens S.J.C., Paulussen A.D.C.et al.. Genome-wide karyomapping accurately identifies the inheritance of single-gene defects in human preimplantation embryos in vitro. Genet. Med. 2014; 16:838–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zamani Esteki M., Dimitriadou E., Mateiu L., Melotte C., Van der Aa N., Kumar P., Das R., Theunis K., Cheng J., Legius E.et al.. Concurrent whole-genome haplotyping and copy-number profiling of single cells. Am. J. Hum. Genet. 2015; 96:894–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Macaulay I.C., Voet T.. Single cell genomics: advances and future perspectives. PLoS Genet. 2014; 10:e1004126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R.. Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002; 30:97–101. [DOI] [PubMed] [Google Scholar]
- 9. Zeevi D.A., Backenroth D., Hakam-Spector E., Renbaum P., Mann T., Zahdeh F., Segel R., Zeligson S., Eldar-Geva T., Ben-Ami I.et al.. Expanded clinical validation of Haploseek for comprehensive preimplantation genetic testing. Genet. Med. 2021; 23:1334–1340. [DOI] [PubMed] [Google Scholar]
- 10. Kitzman J.O., Mackenzie A.P., Adey A., Hiatt J.B., Patwardhan R.P., Sudmant P.H., Ng S.B., Alkan C., Qiu R., Eichler E.E.et al.. Haplotype-resolved genome sequencing of a Gujarati Indian individual. Nat. Biotechnol. 2011; 29:59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Suk E.-K., McEwen G.K., Duitama J., Nowick K., Schulz S., Palczewski S., Schreiber S., Holloway D.T., McLaughlin S., Peckham H.et al.. A comprehensively molecular haplotype-resolved genome of a European individual. Genome Res. 2011; 21:1672–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Backenroth D., Zahdeh F., Kling Y., Peretz A., Rosen T., Kort D., Zeligson S., Dror T., Kirshberg S., Burak E.et al.. Haploseek: a 24-hour all-in-one method for preimplantation genetic diagnosis (PGD) of monogenic disease and aneuploidy. Genet. Med. 2018; 21:1390–1399. [DOI] [PubMed] [Google Scholar]
- 13. Stephens M., Scheet P.. Accounting for decay of linkage disequilibrium in haplotype inference and missing-data imputation. Am. J. Hum. Genet. 2005; 76:449–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Scheet P., Stephens M.. A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase. Am. J. Hum. Genet. 2006; 78:629–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nik-Zainal S., Van Loo P., Wedge D.C., Alexandrov L.B., Greenman C.D., Lau K.W., Raine K., Jones D., Marshall J., Ramakrishna M.et al.. The life history of 21 breast cancers. Cell. 2012; 149:994–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vattathil S., Scheet P.. Haplotype-based profiling of subtle allelic imbalance with SNP arrays. Genome Res. 2013; 23:152–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Peters B.A., Kermani B.G., Alferov O., Agarwal M.R., McElwain M.A., Gulbahce N., Hayden D.M., Tang Y.T., Zhang R.Y., Tearle R.et al.. Detection and phasing of single base de novo mutations in biopsies from human in vitro fertilized embryos by advanced whole-genome sequencing. Genome Res. 2015; 25:426–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ottolini C.S., Kitchen J., Xanthopoulou L., Gordon T., Summers M.C., Handyside A.H.. Tripolar mitosis and partitioning of the genome arrests human preimplantation development in vitro. Sci. Rep. 2017; 7:9744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ottolini C.S., Rogers S., Sage K., Summers M.C., Capalbo A., Griffin D.K., Sarasa J., Wells D., Handyside A.H.. Karyomapping identifies second polar body DNA persisting to the blastocyst stage: implications for embryo biopsy. Reprod. Biomed. Online. 2015; 31:776–782. [DOI] [PubMed] [Google Scholar]
- 20. Tsuiko O., Catteeuw M., Zamani Esteki M., Destouni A., Bogado Pascottini O., Besenfelder U., Havlicek V., Smits K., Kurg A., Salumets A.et al.. Genome stability of bovine in vivo-conceived cleavage-stage embryos is higher compared to in vitro-produced embryos. Hum. Reprod. 2017; 32:2348–2357. [DOI] [PubMed] [Google Scholar]
- 21. Destouni A., Zamani Esteki M., Catteeuw M., Tsuiko O., Dimitriadou E., Smits K., Kurg A., Salumets A., Van Soom A., Voet T.et al.. Zygotes segregate entire parental genomes in distinct blastomere lineages causing cleavage-stage chimerism and mixoploidy. Genome Res. 2016; 26:567–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zamani Esteki M., Viltrop T., Tšuiko O., Tiirats A., Koel M., Nõukas M., Žilina O., Teearu K., Marjonen H., Kahila H.et al.. In vitro fertilization does not increase the incidence of de novo copy number alterations in fetal and placental lineages. Nat. Med. 2019; 25:1699–1705. [DOI] [PubMed] [Google Scholar]
- 23. Dimitriadou E., Melotte C., Debrock S., Esteki M.Z., Dierickx K., Voet T., Devriendt K., de Ravel T., Legius E., Peeraer K.et al.. Principles guiding embryo selection following genome-wide haplotyping of preimplantation embryos. Hum. Reprod. 2017; 32:687–697. [DOI] [PubMed] [Google Scholar]
- 24. Destouni A., Dimitriadou E., Masset H., Debrock S., Melotte C., Van Den Bogaert K., Zamani Esteki M., Ding J., Voet T., Denayer E.et al.. Genome-wide haplotyping embryos developing from 0PN and 1PN zygotes increases transferrable embryos in PGT-M. Hum. Reprod. 2018; 33:2302–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Turner K.J., Silvestri G., Black D.H., Dobson G., Smith C., Handyside A.H., Sinclair K.D., Griffin D.K.. Karyomapping for simultaneous genomic evaluation and aneuploidy screening of preimplantation bovine embryos: The first live-born calves. Theriogenology. 2019; 125:249–258. [DOI] [PubMed] [Google Scholar]
- 26. Konstantinidis M., Prates R., Goodall N.-N., Fischer J., Tecson V., Lemma T., Chu B., Jordan A., Armenti E., Wells D.et al.. Live births following Karyomapping of human blastocysts: experience from clinical application of the method. Reprod. Biomed. Online. 2015; 31:394–403. [DOI] [PubMed] [Google Scholar]
- 27. Shi D., Xu J., Niu W., Liu Y., Shi H., Yao G., Shi S., Li G., Song W., Jin H.et al.. Live births following preimplantation genetic testing for dynamic mutation diseases by karyomapping: a report of three cases. J. Assist. Reprod. Genet. 2020; 37:539–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Thornhill A.R., Handyside A.H., Ottolini C., Natesan S.A., Taylor J., Sage K., Harton G., Cliffe K., Affara N., Konstantinidis M.et al.. Karyomapping-a comprehensive means of simultaneous monogenic and cytogenetic PGD: comparison with standard approaches in real time for Marfan syndrome. J. Assist. Reprod. Genet. 2015; 32:347–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Masset H., Zamani Esteki M., Dimitriadou E., Dreesen J., Debrock S., Derhaag J., Derks K., Destouni A., Drüsedau M., Meekels J.et al.. Multi-centre evaluation of a comprehensive preimplantation genetic test through haplotyping-by-sequencing. Hum. Reprod. 2019; 34:1608–1619. [DOI] [PubMed] [Google Scholar]
- 30. Handyside A.H., Harton G.L., Mariani B., Thornhill A.R., Affara N., Shaw M.-A., Griffin D.K.. Karyomapping: a universal method for genome wide analysis of genetic disease based on mapping crossovers between parental haplotypes. J. Med. Genet. 2010; 47:651–658. [DOI] [PubMed] [Google Scholar]
- 31. Yan Z., Zhu X., Wang Y., Nie Y., Guan S., Kuo Y., Chang D., Li R., Qiao J., Yan L.. 2020) scHaplotyper: haplotype construction and visualization for genetic diagnosis using single cell DNA sequencing data. BMC Bioinformatics. 21:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Handyside A.H., Harton G.L., Mariani B., Thornhill A.R., Affara N., Shaw M.-A., Griffin D.K.. Karyomapping: a universal method for genome wide analysis of genetic disease based on mapping crossovers between parental haplotypes. J. Med. Genet. 2010; 47:651–658. [DOI] [PubMed] [Google Scholar]
- 33. Lodato M.A., Woodworth M.B., Lee S., Evrony G.D., Mehta B.K., Karger A., Lee S., Chittenden T.W., D’Gama A.M., Cai X.et al.. Somatic mutation in single human neurons tracks developmental and transcriptional history. Science. 2015; 350:94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lodato M.A., Rodin R.E., Bohrson C.L., Coulter M.E., Barton A.R., Kwon M., Sherman M.A., Vitzthum C.M., Luquette L.J., Yandava C.N.et al.. Aging and neurodegeneration are associated with increased mutations in single human neurons. Science. 2018; 359:555–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Davey J.W., Hohenlohe P.A., Etter P.D., Boone J.Q., Catchen J.M., Blaxter M.L.. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 2011; 12:499–510. [DOI] [PubMed] [Google Scholar]
- 36. Elshire R.J., Glaubitz J.C., Sun Q., Poland J.A., Kawamoto K., Buckler E.S., Mitchell S.E.. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One. 2011; 6:e19379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Debrock S., Melotte C., Spiessens C., Peeraer K., Vanneste E., Meeuwis L., Meuleman C., Frijns J.-P., Vermeesch J.R., D’Hooghe T.M.. Preimplantation genetic screening for aneuploidy of embryos after in vitro fertilization in women aged at least 35 years: a prospective randomized trial. Fertil. Steril. 2010; 93:364–373. [DOI] [PubMed] [Google Scholar]
- 38. Capalbo A., Ubaldi F.M., Cimadomo D., Maggiulli R., Patassini C., Dusi L., Sanges F., Buffo L., Venturella R., Rienzi L.. Consistent and reproducible outcomes of blastocyst biopsy and aneuploidy screening across different biopsy practitioners: a multicentre study involving 2586 embryo biopsies. Hum. Reprod. 2016; 31:199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Herten K., Hestand M.S., Vermeesch J.R., Van Houdt J.K.J.. GBSX: a toolkit for experimental design and demultiplexing genotyping by sequencing experiments. BMC Bioinformatics. 2015; 16:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Magoč T., Salzberg S.L.. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011; 27:2957–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Church D.M., Schneider V.A., Graves T., Auger K., Cunningham F., Bouk N., Chen H.-C., Agarwala R., McLaren W.M., Ritchie G.R.S.et al.. Modernizing reference genome assemblies. PLoS Biol. 2011; 9:e1001091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. 2013; arXiv doi:26 May 2013, preprint: not peer reviewedhttps://arxiv.org/abs/1303.3997.
- 43. McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M.et al.. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010; 20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Scheinin I., Sie D., Bengtsson H., van de Wiel M.A., Olshen A.B., van Thuijl H.F., van Essen H.F., Eijk P.P., Rustenburg F., Meijer G.A.et al.. DNA copy number analysis of fresh and formalin-fixed specimens by shallow whole-genome sequencing with identification and exclusion of problematic regions in the genome assembly. Genome Res. 2014; 24:2022–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ding J., Dimitriadou E., Tšuiko O., Destouni A., Melotte C., Van Den Bogaert K., Debrock S., Jatsenko T., Esteki M.Z., Voet T.et al.. Identity-by-state-based haplotyping expands the application of comprehensive preimplantation genetic testing. Hum. Reprod. 2020; 35:718–726. [DOI] [PubMed] [Google Scholar]
- 48. Favorov A., Mularoni L., Cope L.M., Medvedeva Y., Mironov A.A., Makeev V.J., Wheelan S.J.. Exploring massive, genome scale datasets with the GenometriCorr package. PLoS Comput. Biol. 2012; 8:e1002529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Eberle M.A., Fritzilas E., Krusche P., Källberg M., Moore B.L., Bekritsky M.A., Iqbal Z., Chuang H.-Y., Humphray S.J., Halpern A.L.et al.. A reference data set of 5.4 million phased human variants validated by genetic inheritance from sequencing a three-generation 17-member pedigree. Genome Res. 2017; 27:157–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen S., Yin X., Zhang S., Xia J., Liu P., Xie P., Yan H., Liang X., Zhang J., Chen Y.et al.. Comprehensive preimplantation genetic testing by massively parallel sequencing. Hum. Reprod. 2021; 36:236–247. [DOI] [PubMed] [Google Scholar]
- 51. Li Q., Mao Y., Li S., Du H., He W., He J., Kong L., Zhang J., Liang B., Liu J.. Haplotyping by linked-read sequencing (HLRS) of the genetic disease carriers for preimplantation genetic testing without a proband or relatives. BMC Med. Genomics. 2020; 13:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liu S., Wang H., Leigh D., Cram D.S., Wang L., Yao Y.. Third-generation sequencing: any future opportunities for PGT. J. Assist. Reprod. Genet. 2021; 38:357–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Garg S. Computational methods for chromosome-scale haplotype reconstruction. Genome Biol. 2021; 22:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Garg S., Fungtammasan A., Carroll A., Chou M., Schmitt A., Zhou X., Mac S., Peluso P., Hatas E., Ghurye J.et al.. Chromosome-scale, haplotype-resolved assembly of human genomes. Nat. Biotechnol. 2021; 39:309–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ariad D., Yan S.M., Victor A.R., Barnes F.L., Zouves C.G., Viotti M., McCoy R.C.. Haplotype-aware inference of human chromosome abnormalities. Proc. Natl. Acad. Sci. U.S.A. 2021; 118:e2109307118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hinch A.G., Zhang G., Becker P.W., Moralli D., Hinch R., Davies B., Bowden R., Donnelly P.. Factors influencing meiotic recombination revealed by whole-genome sequencing of single sperm. Science. 2019; 363:eaau8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Xia Y., Gonzales-Pena V., Klein D.J., Luquette J.J., Puzon L., Siddiqui N., Reddy V., Park P., Behr B.R., Gawad C.. Genome-wide disease screening in early human embryos with primary template-directed amplification. 2021; bioRxiv doi:31 August 2021, preprint: not peer reviewed 10.1101/2021.07.06.451077. [DOI]
- 58. Gonzalez-Pena V., Natarajan S., Xia Y., Klein D., Carter R., Pang Y., Shaner B., Annu K., Putnam D., Chen W.et al.. Accurate genomic variant detection in single cells with primary template-directed amplification. Proc. Natl. Acad. Sci. 2021; 118:e2024176118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Picher Á.J., Budeus B., Wafzig O., Krüger C., García-Gómez S., Martínez-Jiménez M.I., Díaz-Talavera A., Weber D., Blanco L., Schneider A.. TruePrime is a novel method for whole-genome amplification from single cells based on TthPrimPol. Nat. Commun. 2016; 7:13296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sonah H., Bastien M., Iquira E., Tardivel A., Légaré G., Boyle B., Normandeau É., Laroche J., Larose S., Jean M.et al.. An improved genotyping by sequencing (GBS) approach offering increased versatility and efficiency of SNP discovery and genotyping. PLoS One. 2013; 8:e54603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zuccaro M. V, Xu J., Mitchell C., Marin D., Zimmerman R., Rana B., Weinstein E., King R.T., Palmerola K.L., Smith M.E.et al.. Allele-Specific Chromosome Removal after Cas9 Cleavage in Human Embryos. Cell. 2020; 183:1650–1664. [DOI] [PubMed] [Google Scholar]
- 62. Ma H., Marti-Gutierrez N., Park S.-W., Wu J., Lee Y., Suzuki K., Koski A., Ji D., Hayama T., Ahmed R.et al.. Correction of a pathogenic gene mutation in human embryos. Nature. 2017; 548:413–419. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
In compliance with the GDPR and the study protocol, the HapMap and PGT-M dataset used in the study is not publicly available. Cell line, embryo, parental and phasing relatives raw genotyping and scGBS data has been deposited at the European Genome-phenome Archive (EGA), which is hosted by the EBI and the CRG, under accession number EGAS00001005401. It is available to academic users upon request to the Data Access Committee (DAC) of KU Leuven via the corresponding author (JRV). The bovine scGBS data for this study have been deposited in the European Nucleotide Archive (ENA) at EMBL-EBI under accession number PRJEB45461 (https://www.ebi.ac.uk/ena/browser/view/PRJEB45461). The bovine SNP array genotyping data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (Edgar et al., 2002) and are accessible through GEO Series accession number GSE178156 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE178156). We have provided the bioinformatical scripts via the following link: https://github.com/GenomicsCoreLeuven/publications_JV/tree/main/GBS