


Abstract
Poly(ADP-ribose) polymerase-1 (PARP-1) is a DNA damage sensor and contributes to both DNA repair and cell death processes. However, how PARP-1 signaling is regulated to switch its function from DNA repair to cell death remains largely unknown. Here, we found that PARP-1 plays a central role in alkylating agent-induced PARthanatic cancer cell death. Lysine demethylase 6B (KDM6B) was identified as a key regulator of PARthanatos. Loss of KDM6B protein or its demethylase activity conferred cancer cell resistance to PARthanatic cell death in response to alkylating agents. Mechanistically, KDM6B knockout suppressed methylation at the promoter of O6-methylguanine-DNA methyltransferase (MGMT) to enhance MGMT expression and its direct DNA repair function, thereby inhibiting DNA damage-evoked PARP-1 hyperactivation and subsequent cell death. Moreover, KDM6B knockout triggered sustained Chk1 phosphorylation and activated a second XRCC1-dependent repair machinery to fix DNA damage evading from MGMT repair. Inhibition of MGMT or checkpoint response re-sensitized KDM6B deficient cells to PARthanatos induced by alkylating agents. These findings provide new molecular insights into epigenetic regulation of PARP-1 signaling mediating DNA repair or cell death and identify KDM6B as a biomarker for prediction of cancer cell vulnerability to alkylating agent treatment.
INTRODUCTION
Poly(ADP-ribose) polymerase-1 (PARP-1) is a ubiquitously expressed nuclear enzyme and plays a critical role in DNA damage response in human cancers (1–3). Upon DNA damage, PARP-1 senses DNA single-strand breaks and utilizes NAD+ as the substrate to catalyze the addition of poly(ADP-ribose) (PAR) toward different acceptor proteins, including PARP-1 itself (1–3). As a result, PARP-1 activation leads to the recruitment of DNA repair proteins and nucleases to sites of DNA damage, thereby facilitating DNA damage repair (4). Inhibition of PARP-1 impairs DNA single-strand break repair and eventually causes DNA double-strand breaks, which are primarily repaired through homologous recombination and/or non-homologous end joining mechanisms (5). Blockade of DNA double-strand break repair in BRCA-deficient cancer cells can synergize with PARP inhibitor to induce cancer cell death (6). Accumulating studies reveal a dual function of PARP-1 in cell death and survival. PARP-1 hyperactivation promotes cell death, also known as PARP-1-dependent cell death (PARthanatos), in neurological diseases (7). After PARP-1 is activated by oxidative stress under neuronal injury, PAR functions as a death signal triggering apoptosis-inducing factor (AIF) translocation from mitochondria to the nucleus, where it interacts with the nuclease macrophage migration inhibitory factor (MIF) leading to large DNA fragmentation and neuronal cell death (8, 9). However, whether PARthanatos also occurs in cancer cells and how PARP-1 signaling is regulated and switched between DNA repair and cell death remain poorly understood.
Alkylating agents are the most common and oldest chemotherapy drugs currently used in the clinic to kill cancer (10–12). Alkylating agents are divided into five categories: nitrogen mustard, nitrosoureas, alkyl sulfonates, triazines, and ethylenimines. N-Methyl-N′-nitro-N-nitrosoguanidine (MNNG), belonging to the nitrosourea category, was first discovered in 1950s as an anti-cancer drug through a national cooperative cancer drug screen (12). MNNG contains the most basic structure similar to several commonly used alkylating chemotherapy drugs Temozolomide (TMZ), Streptozotocin, Lomustine and Carmustine (10). It acts by producing an intermediate product methyldiazonium ion and adds alkyl groups specifically to the O6 of guanine and O4 of thymine, leading to DNA damage and cell death (12). Previous studies identified that mitochondrial dysfunction and NAD+ depletion contribute to alkylating agent-induced cell death (13,14). Human AlkB homolog 7 (ALKBH7) promotes loss of mitochondrial function and energy depletion in response to alkylating agents, leading to cell death (14). DNA damage repair proteins including O6-methylguanine-DNA methyltransferase (MGMT), exonuclease 1, and DNA mismatch repair (MMR) protein MutS Homolog 6 (MSH6) also play an important role in alkylating agent-induced cell death (15). Alkylating agents induce PARP-1 activation, but an early report suggests that PARP-1 activation is not sufficient to mediate cell death (13). Interestingly, alkyladenine DNA glycosylase (MPG) is involved in alkylating agent-induced tissue damage in vivo, which is prevented by PARP-1 loss (16). Together, the precise role of PARP-1 in alkylating agent-induced cancer cell death and its signaling regulation are far not clear.
In the present study, we showed that PARP-1 is required for alkylating agents-induced PARthanatic cancer cell death and identified lysine demethylase 6B (KDM6B) as a key regulator of PARthanatos. Upon alkylating agent treatment, KDM6B increased DNA damage by repressing both MGMT-mediated direct DNA repair and checkpoint response associated DNA repair, leading to PARP-1 hyperactivation and subsequent PARthanatic cell death. Inhibition of MGMT or checkpoint response sensitized cells to PARthanatos induced by alkylating agents. These findings unravel an epigenetic mechanism that controls PARP-1-dependent DNA repair vs. cell death pathways and predicts cancer cell vulnerability to alkylating agents.
MATERIALS AND METHODS
Plasmid constructs
Full-length and C-terminal truncated KDM6B cDNA was amplified by PCR from KDM6B plasmids (Addgene, #21212 and #21214) and subcloned into pLVX-Ubc-FLAG vector. Full-length MGMT cDNA was amplified by PCR and cloned into cFUGW-3xFlag-N vector. Catalytically inactive MGMT mutant was generated by site-directed mutagenesis PCR. Full-length human X-Ray Repair Cross Complementing 1 (XRCC1) cDNA was amplified by PCR and cloned into pLVX-Ubc-FLAG vector. DNA oligonucleotides of the single guide RNA (sgRNA) targeting human KDM6B, PARP-1, MGMT or XRCC1 (Supplementary Table S1) were annealed and ligated into BsmBI-linearized lentiCRISPRv2 vector (Addgene, #52961). Other plasmids have been described previously (8,17). All recombinant plasmids were verified by Sanger sequencing.
Cell culture
HeLa (ATCC, CCL-2), MCF-7 (ATCC, HTB-22), MDA-MB-231 (gift from Rolf Brekken, UT Southwestern, Dallas, TX, USA), HEK293T and SUM159 cells (gifts from Gregg L. Semenza, Johns Hopkins University, Baltimore, Maryland, USA) were cultured in DMEM or DMEM/Ham's F-12 supplemented with 10% heat-inactivated fetal bovine serum at 37°C in a 5% CO2/95% air incubator. All KO cell lines were generated using the CRISPR/Cas9 technique and genotyped as described (18). All cell lines were annually tested to be mycoplasma-free and authenticated by STR DNA profiling analysis during 2016–2017.
Clonogenic assay
300 cells/well were seeded on a 24-well plate and treated with indicated drugs for 10 days. Colonies were washed with phosphate-buffered saline (PBS), fixed with 100% methanol, and stained with 0.01% crystal violet. Colony numbers were counted for quantification.
Cell death assay
2 × 105 cells/well were seeded on a 12-well plate and pretreated with vehicle, cell death inhibitors, PARP inhibitors, or Chk1 inhibitor GDC0575 (50 nM) for 30 min, or MGMT inhibitor O6-benzylguanine (BG, 200 μM) for 24 h, followed by the treatment of MNNG (50 μM, 15 min) or methyl methanesulfonate (MMS, 2 mM, 1 h) in the presence or absence of indicated inhibitors. After the treatment with alkylating agents, cells were changed to fresh medium with or without indicated inhibitors for continuous incubation for 24–96 h. Cells were stained with 2 μM propidium iodide (PI) and 7 μM Hoechst 33342 (ThermoFisher) for 5–10 min and imaged under a Zeiss Observer Z1 microscope. The numbers of total and PI-positive cells were quantified as described (8,9).
CRISPR screening and bioinformatics analysis
HeLa cells were transduced overnight at a MOI of 0.4 with a pooled genome-wide CRISPR KO (GeCKO v2) library A or B (Addgene) containing a total of 122 411 sgRNAs (6 sgRNAs per gene, 4 sgRNAs per miRNA and 1000 control sgRNAs). After puromycin (1 μg/ml) selection, cells were treated with MNNG (75 μM) for 15 min, followed by continuous incubation with fresh medium for 3 days. The majority of cells died 24 h after treatment. The survived cells were pooled and subjected to genomic DNA extraction. The sgRNA library was amplified by two steps of PCR as described previously (19). Samples were sequenced on Illumina NextSeq 500 with read configuration as 150 bp, single end. The fastq files were subjected to quality check using fastqc (version 0.11.2, http://www.bioinformatics.babraham.ac.uk/projects/fastqc) and fastq_screen (version 0.4.4, http://www.bioinformatics.babraham.ac.uk/projects/fastq_screen), and adapters trimmed using an in-house script. The reference sgRNA sequences for human GeCKO v2.0 (A and B) were downloaded from Addgene (https://www.addgene.org/pooled-library/). The trimmed fastq files were mapped to reference sgRNA library using MAGeCK (20). Further, read counts for each sgRNA were generated and median normalization was performed to adjust for library sizes. Positively and negatively selected sgRNA and genes were identified using the default parameters of MAGeCK.
RNA-seq
RNA-seq was performed as described previously (17). Total RNA was isolated from parental, KDM6B KO2, and KDM6B rescue HeLa cells using the RNeasy mini kit (Qiagen) and treated with DNase (Qiagen). The quality of total RNA was confirmed with a RNA integrity number score 8.5 or higher by the Agilent Tapestation 4200. mRNAs were used for library preparation with a Kapa mRNA HyperPrep kit (Roche) and sequenced on the Illumina NextSeq 500 with the read configuration as 76 bp, single end. Reads were mapped to the hg19 (UCSC version from igenomes) using Tophat and annotated using a custom R script using the UCSC known Genes table as a reference. Read counts were generated using feature Counts and the differential expression analysis was performed using edgeR (false discovery rate (FDR) < 0.05 and mRNA fold change > 2 as cutoffs), as described previously (17).
Immunoblot assay
Cells were lyzed in modified lysis buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 1 mM β-mercaptoethanol, 1% Igepal and protease inhibitor cocktail) on ice for 30 min. The equal amount of proteins were separated on SDS-PAGE gel and transferred to nitrocellulose membrane. The membrane was blocked with 5% milk and incubated overnight with primary antibodies: anti-Flag antibody (Sigma, F3165), anti-MGMT antibody (Proteintech, 17195-1-AP), anti-PAR antibody (Trevigen, 4336-BPC-100), anti-PARP-1 antibody (Proteintech, 22999-1-AP), anti-γH2AX antibody (Millipore, 05-636), anti-H2AX antibody (Proteintech, 10856-1-AP), anti-phospho-Chk1 (Ser345) antibody (Cell Signaling Technology, 2348), anti-phospho-RPA32 (Ser33) (Fisher, 50-155-7198), anti-RPA70 (Santa Cruz, SC166023), anti-phospho-ATR (Thr1989) antibody (Cell Signaling Technology, 30632S), anti-ATR antibody (Cell Signaling Technology, 13934S), anti-phospho-ATM (Ser1981) (D6H9) (Cell Signaling Technology, 5883S), anti-ATM (Cell Signaling Technology, 92356S), anti-phospho-Chk1 (Ser296) antibody (Cell Signaling Technology, 2349S), anti-Chk1 antibody (Santa Cruz, sc-8408), anti-phospho-Chk2 (Thr68) (C13C1) antibody (Cell Signaling Technology, 2197S), anti-phospho-Chk2 (D9C6) antibody (Cell Signaling Technology, 6334S), anti-PP2A antibody (Proteintech, 10321-1-AP), anti-H3K27me3 (Cell Signaling Technology, 9733S), anti-XRCC1 antibody (Proteintech, 21468-1-AP), anti-MSH2 antibody (25D12) (Santa Cruz, sc-56163), anti-MSH6 antibody (F-1) (Santa Cruz, sc-271979), or anti-actin antibody (Proteintech, 66009-1-AP) at 4°C, followed by donkey anti-mouse or goat anti-rabbit IgG conjugated to HRP for 1 h at room temperature. After washing, the immune complexes were detected by the SuperSignal West Pico Chemiluminescent Substrate (Fisher, P134578) and imaged by ChemiDoc system (Bio-Rad).
Immunostaining assay
Cells were fixed with 4% paraformaldehyde, permeabilized with 0.05% Triton X-100, and blocked with 3% BSA in PBS. For O6 methylguanine (O6 MeG) staining, cells were fixed with methanol for 15 min at room temperature, rehydrated in saline sodium citrate buffer (300 mM NaCl, 30 mM sodium citrate), and treated with RNase A (200 μg/ml) plus RNase T (50 units/ml) for 1 h at 37°C. Cells were then washed with NaCl (140 mM) and denatured by alkali treatment (70 mM NaOH, 140 mM NaCl in 40% ethanol) for 5 min at 0°C. After washing with PBS, cells were blocked with 20% BSA in PBS for 20 min. Next, cells were incubated overnight with anti-Flag antibody (Sigma, F3165,1:2000), anti-MGMT antibody (Proteintech, 67476-1-lg, 1:500), anti-apoptosis-inducing factor (AIF) E-1 (Santa Cruz, sc-13116) or anti-O6-Methyl-2-deoxyguanosine (EM2-3) (AxxoRA, SQX-SQM003.1) at 4°C, washed with PBS with 0.1% Tween-20 for 3 times, and incubated with Alexa488 donkey anti-mouse IgG (1:1000) or Cy3 donkey anti-rabbit IgG (1:1000) for 90–120 min in dark. After washing for three times, cells were incubated with DAPI (1:1000) for 10 min. Cells were washed again and mounted with anti-fade mounting medium. Immunofluorescent analysis was carried out with a Zeiss Observer Z1 fluorescence microscope.
MGMT promoter methylation assay
MGMT promoter methylation was determined as described previously (21). Briefly, genomic DNA was isolated from parental and KDM6B KO2 HeLa cells and subjected to bisulfite modification by EpiMark® Bisulfite Conversion Kit according to the manufacturer's instructions (New England Biolabs, E3318S). The converted DNA was eluted in DNase/RNase-free water and examined by PCR using SYBR Green PCR Master Mix (Bio-Rad) and primers listed in Supplementary Table S2. Genomic DNA treated with a CpG Methyltransferase M.SssI (New England Biolabs) served as positive control. Genomic DNA without bisulfite conversion was used as a negative control. PCR products were separated on nondenaturated 6% polyacrylamide gels and imaged by ChemiDoc system (Bio-Rad).
In vitro MGMT methyltransferase activity assay
MGMT methyltransferase activity was determined by MGMT assay kit (MD0100, Sigma-Aldrich). Briefly, scrambled control (SC) and KDM6B KO2 HeLa cells were pretreated with vehicle or BG (200 μM) for 30 min and lysed in the modified lysis buffer (50 mM Tris, pH 7.5, 1mM EDTA, 1 mM DTT, 5% Glycerol, 50 mM NaCl). The resulting lysates were incubated with a 23-bp customized biotin-labeled DNA substrate containing an O6 MeG next to the PstI cleavage site (Sigma) for 2 h at 37°C. Then the reaction was stopped at 65°C for 5 min. The resulting DNA purified via phenol/chloroform/isoamyl alcohol (25:24:1) with 10 μg yeast tRNA serving as a carrier was incubated with PstI at 37°C for 1 h (10 μl reaction system). The reaction was stopped by 5 μl FBXE buffer (90% Formamide, 0.1% (w/v) Bromophenol blue, 0.1% xylene cyanole, 20 mM EDTA), heated at 95°C for 5 min and quickly chilled on ice, followed by separation on 20% 7 M urea SDS-PAGE gel and immunoblot assay with anti-Biotin antibody (Thermo Fisher Scientific, 692033).
Cell cycle analysis
HeLa cells (1.5 × 105/well) were seeded overnight onto a 12-well plate. Next day, cells were treated with 50 μM MNNG for 15 min at 37°C, washed, and incubated with fresh complete media. Then cells were trypsinized and fixed at 2, 4, 6, 8, 15 and 22 h after MNNG treatment. All cells were labeled with 10 μM EdU at 37°C for 2 h right before the fixation and further conjugated to Alexa Fluor 488 azide by CuSO4-mediated click chemistry reaction for 30 min at room temperature in dark using the Click-iT Plus EdU Alexa Fluor 488 Flow Cytometry Assay Kit in a 500 μl reaction volume per sample (Thermo Fisher Scientific, C10632). After washing, cells were resuspended in 500 μl buffer containing 50 μg/ml RNase (Sigma, 70856) and 2 μg/ml PI (Sigma, P4864), and incubated for 30 min at room temperature in dark. Cells were analyzed on a CytoFlex Flow Cytometer (Beckman Coulter), and the acquired cell cycle data were analyzed using FlowJo version 10.
Quantitative reverse transcription-polymerase chain reaction (qRT-PCR) assay
Total RNA was isolated using TRIzol reagent (ThermoFisher) and cleaned with RNase-free DNase I (ThermoFisher). cDNA was synthesized with M-MuLV Reverse Transcriptase (200 U/μl, New England Biolabs). Real-time PCR was performed by a CFX-96 real-time system (Bio-Rad) using iTaq universal SYBR green supermix (Bio-Rad) with primers listed in Supplementary Table S2. mRNA expression was quantified as described previously (17).
Statistical analysis
Statistical evaluation was performed by unpaired two-tailed Student's t test between two groups and by one- or two-way ANOVA with Tukey's multiple comparisons, Dunnett's multiple comparisons or Sidak's multiple comparisons within multiple groups as indicated using GraphPad Prism 8.0 software. Data are shown as mean ± SEM. P < 0.05 is considered significant.
RESULTS
PARP-1 is required for alkylating agent-induced cell death in cancer cells
Alkylating agents including SN1-type alkylating agent MNNG and SN2-type alkylating agent MMS induced DNA damage and cell death in cancer cells (Supplementary Figure S1A–D). To determine which cell death pathway is responsible for alkylating agent-induced cancer cell death, we screened cell death inhibitors in human cervical cancer HeLa cells treated with DMSO or MNNG (50 μM). Inhibitors for apoptosis (z-VAD, 10 μM), necroptosis (necrostatin-1, 10 μM), and autophagy (3-methyladenine [3-MA], 10 mM) all failed to inhibit MNNG-induced cell death (Figure 1A). In contrast, MNNG-induced cell death was fully prevented by PARP inhibitor DPQ (30 μM) or Olaparib (10 μM) (Figure 1A). PARP-1 senses DNA damage for its activation. PAR formation determined by anti-PAR antibody showed that MNNG treatment for 15 min robustly induced PARP-1 activation in HeLa cells, which was completely blocked by DPQ (30 μM) (Figure 1B). To further confirm the role of PARP-1 in alkylating agent-induced cell death, we generated and validated PARP-1 protein knockdown (KD) in HeLa cells using its small interfering RNAs (siRNAs) (Figure 1C). Similar to pharmacological interventions, PARP-1 KD also fully blocked MNNG-induced HeLa cell death (Figure 1D and E).
Figure 1.

PARP-1 is required for alkylating agent-induced cancer cell death. (A) Representative cell death images in HeLa cells treated with DMSO or MNNG (50 μM, 15 min) for 24 h in the presence or absence of cell death inhibitors DPQ (30 μM), Olaparib (10 μM), z-VAD (10 μM), necrostatin-1 (10 μM), 3-methyladenine (3-MA, 10 mM). Scale bar, 20 μm. (B) Immunoblot analysis of PARP-1 activation in HeLa cells treated with vehicle (−) or MNNG (50 μM) for 15 min in the presence or absence of DPQ (30 μM). (C) Immunoblot analysis of PARP-1 knockdown (KD) in HeLa cells (top). PARP-1 protein levels are quantified (bottom, mean ± SEM, n = 3). ****P < 0.0001 versus control by one-way ANOVA Dunnett's multiple comparisons test. CTL, control. NT, non-target. (D and E) Representative cell death images in control, NT, and PARP-1 KD HeLa cells treated with DMSO or MNNG for 24 h (D). Cell death is quantified in E (mean ± SEM, n = 3). Scale bar, 20 μm. ****P < 0.0001 vs. control by two-way ANOVA Sidak's multiple comparisons test. (F) Representative cell death images in MDA-MB-231 cells treated with DMSO or MNNG (75 μM, 15 min) for 24 h in the presence or absence of DPQ. Scale bar, 20 μm. PI/H, propidium iodide/Hoechst staining. TL, transmission light. (G) Immunoblot analysis of PARP-1 activation in MDA-MB-231 cells treated with vehicle (−) or MNNG for 15 min in the presence or absence of DPQ. (H) Representative cell death images in wildtype (WT) and PARP-1 KO MDA-MB-231 cells treated with MNNG (75 μM, 15 min) for 24 h. Scale bar, 20 μm. (I) Immunoblot analysis of PARP-1 activation in WT and PARP-1 KO MDA-MB-231 cells treated with MNNG for 15 min.
We next studied whether this PARP-1-dependent cell death is also applied to other human cancer cells. Human breast cancer MDA-MB-231 cells were treated with MNNG (100 μM) for 15 min in the presence or absence of PARP inhibitor DPQ (30 μM) and cell death was assessed by PI/Hoechst staining 24 h after treatment. In line with results in HeLa cells, MNNG treatment killed MDA-MB-231 cells, which was blocked by DPQ (Figure 1F). Similar effects were also observed in MCF-7 and SUM159 cells (Supplementary Figure S1C). DPQ treatment also abolished MNNG-induced PARP-1 activation in MDA-MB-231 cells (Figure 1G). Again, genetic knockout (KO) of PARP-1 prevented MNNG-induced PAR formation and cell death in MDA-MB-231 cells (Figure 1H and I). Finally, we studied the effect of PARP-1 on other alkylating agents-induced cell death. To this end, we treated MDA-MB-231 cells with MMS (2 mM) or vehicle. Treatment of MMS rapidly increased PAR formation in a time-dependent manner with peak observed at 40 min and caused DNA damage as γH2AX was remarkable increased 6 h after treatment (Supplementary Figure S1B and E). Pretreatment of DPQ (30 μM) blocked MMS-induced PARP-1 activation and cell death in MDA-MB-231 cells (Supplementary Figure S1D and F). Collectively, these findings indicate that alkylating agents activates PARP-1 to induce cancer cell death (PARthanatos).
Identification of KDM6B as a key cell death regulator of PARthanatos
To unbiasedly identify key mediators controlling PARthanatos in response to alkylating agents, we performed a genome-wide CRISPR/Cas9 screening in HeLa cells (Figure 2A). HeLa cells were transduced with pooled genome-wide CRISPR KO (GeCKO v2) libraries A and B containing 122 411 sgRNAs. After puromycin selection, cells were then treated with MNNG (75 μM) for 15 min to trigger PARthanatos. The survived cells resistant to PARthanatos were harvested for genomic DNA isolation and deep sequencing, which identified 18 enriched sgRNA hits (Figure 2B). Given that two highly enriched KDM6B sgRNAs were identified from the screening, KDM6B, which is a histone H3K27 demethylase, was selected as a top candidate for further studies. To validate the screening results, we generated two independent KDM6B KO HeLa cell lines by the CRISPR/Cas9 technique and these KO cells were confirmed by Sanger sequencing (Figure 2C; Supplementary Figure S2A). KDM6B KO1 or KO2 significantly inhibited MNNG-induced PARthanatos in HeLa cells (Supplementary Figure S2B and C).
Figure 2.

Identification of KDM6B as a key regulator of PARP-1-dependent cell death. (A) The scheme of the CRISPR screening. (B) Top 18 hits including KDM6B were identified from the screening. Red, >105 000 reads. Pink, > 200 reads. Purple, >10 reads. Gray,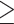 1 read. (C) Genotyping of KDM6B KO2 HeLa cells. (D) Immunoblot analysis of scrambled control (SC), KDM6B KO, and C-terminal WT-KDM6B (WT-C) as well as its H1390A mutant (mut-C) rescue HeLa cells. Numbers indicate the signal intensity. (E and F) Representative cell death images in SC, KDM6B KO2, and rescued HeLa cells 24 h after the treatment with DMSO or MNNG (50 μM, 15 min) (E). PI-positive cells are quantified in (F) (mean ± SEM, n = 4–7). Scale bar, 20 μm. PI/H, propidium iodide/Hoechst staining. TL, transmission light. ****P < 0.0001 by two-way ANOVA Sidak's multiple comparisons test. (G and H) Representative colony survival in SC, KDM6B KO2, and rescued HeLa cells treated with vehicle, MNNG (2 μM), TMZ (250 μM), Cyclophosphamide (CP, 500 μM), and Carmustine (25 μM) for 10 days (G). Colony numbers are quantified in H (mean ± SEM, n = 2–12). *P < 0.05; ***P < 0.001; ****P < 0.0001 versus DMSO by two-way ANOVA Tukey's multiple comparisons test.
1 read. (C) Genotyping of KDM6B KO2 HeLa cells. (D) Immunoblot analysis of scrambled control (SC), KDM6B KO, and C-terminal WT-KDM6B (WT-C) as well as its H1390A mutant (mut-C) rescue HeLa cells. Numbers indicate the signal intensity. (E and F) Representative cell death images in SC, KDM6B KO2, and rescued HeLa cells 24 h after the treatment with DMSO or MNNG (50 μM, 15 min) (E). PI-positive cells are quantified in (F) (mean ± SEM, n = 4–7). Scale bar, 20 μm. PI/H, propidium iodide/Hoechst staining. TL, transmission light. ****P < 0.0001 by two-way ANOVA Sidak's multiple comparisons test. (G and H) Representative colony survival in SC, KDM6B KO2, and rescued HeLa cells treated with vehicle, MNNG (2 μM), TMZ (250 μM), Cyclophosphamide (CP, 500 μM), and Carmustine (25 μM) for 10 days (G). Colony numbers are quantified in H (mean ± SEM, n = 2–12). *P < 0.05; ***P < 0.001; ****P < 0.0001 versus DMSO by two-way ANOVA Tukey's multiple comparisons test.
To determine whether the H3K27 demethylase activity of KDM6B is required for alkylating agent-induced PARthanatos, we next generated KDM6B rescue cells expressing C-terminal wildtype (WT-C) or catalytically inactive (H1390A) KDM6B truncate (mut-C) (Figure 2D, Supplementary Figure S2D) (22). KDM6B KO2 increased H3K27me3 in HeLa cells (Supplementary Figure S2E). In line with previous studies (23), WT-C but not H1390A KDM6B mut-C showed the H3K27 demethylase activity because it eliminated H3K27me3 in HeLa cells (Figure 2D), indicating the specificity of these KO and rescued cell lines. As expected, MNNG treatment robustly induced PARthanatos in HeLa cells, which was blocked by KDM6B KO2 (Figure 2E and F). Ectopic expression of KDM6B WT-C sensitized KO2 cells to MNNG treatment, whereas catalytically inactive KDM6B-C mutant failed to do so (Figure 2E and F).
Clonogenic assay showed that KDM6B KO preserved HeLa cell survival against MNNG treatment, which was reversed by KDM6B WT-C but not H1390A mut-C (Figure 2G and H). Similarly, alkylating agents, which are currently used in the clinic, including TMZ (250 μM), Cyclophosphamide (500 μM), and Carmustine (25 μM), significantly reduced colony numbers. KDM6B KO conferred HeLa cells resistance to the treatment of these alkylating agents (Figure 2G and H). Overexpression of WT, but not catalytically inactive KDM6B-C H1390A mutant, re-sensitized KDM6B KO cells to these alkylating agents (Figure 2G and H). In parallel, we also generated HeLa cells expressing empty vector (EV) or full-length WT-KDM6B or catalytically inactive KDM6B H1390A (Supplementary Figure S2D and F). Consistent with H1390A mut-C, full-length KDM6B H1390A mutant lost its H3K27 demethylase activity as indicated by high H3K27me3 staining in cells and blocked MNNG-induced PARthanatos in HeLa cells (Supplementary Figure S2G and H). Taken together, these findings identified KDM6B as a key regulator of PARthanatos, which requires its H3K27 demethylase activity.
KDM6B suppresses MGMT expression via its H3K27 demethylase activity and regulates alkylating agent-induced PARP-1 activation and DNA damage
To dissect the mechanism by which KDM6B facilitates PARthanatos in response to alkylating agents, we analyzed KDM6B transcriptome in parental, KDM6B KO2, and rescued WT or mutant KDM6B HeLa cells by RNA-seq. KDM6B upregulated 811 genes but downregulated 690 genes (FDR < 0.05 and fold change > 2 as cutoffs, Figure 3A). 290 out of 811 upregulated genes and 250 out of 690 downregulated genes were rescued by WT KDM6B (fold change > 1.5 as cutoff, Figure 3B). Importantly, the majority of rescued genes were regulated by H3K27 demethylase activity (Figure 3C). In parallel with RNA-sequencing study, we found that KDM6B regulated PARP-1 activation following MNNG treatment in a time-dependent manner in HeLa SC cells (Figure 3D). KDM6B KO significantly reduced PARP-1 activation and PAR formation (Figure 3D). In line with that, DNA damage marker γH2AX was also elevated in a time-dependent manner after MNNG treatment, which was greatly inhibited in KDM6B KO2 cells (Figure 3D). Thus, MGMT was identified as a top gene candidate that was repressed by KDM6B through the H3K27 demethylase activity (Figure 3B). MGMT is a key de-alkylating enzyme that transfers a methyl group from the O6 MeG to its own active residue cysteine 145 leading to a direct repair of guanine, and subsequently methylated MGMT protein is degraded at the proteasome (24). We speculated that MGMT impairs the effect of alkylating agents on DNA damage response, PARP-1 activation, and PARthanatos in KDM6B KO cells.
Figure 3.

KDM6B regulates PARP-1 activation and MGMT expression. (A and B) Volcano plots of KDM6B target genes in HeLa cells. Log2FC represents the fold change of mRNA expression in SC/KDM6B KO. n = 2. (C) Venn diagram of KDM6B rescued target genes in HeLa cells. (D) Immunoblot analysis of PAR and DNA damage-related proteins in SC and KDM6B KO2 HeLa cells treated with vehicle (−) or MNNG for indicated time. (E) RT-qPCR analysis of indicated genes in SC and KDM6B KO2 HeLa cells (mean ± SEM, n = 3–6). **P < 0.01 by unpaired two-tailed Student's t test. (F) RNA-seq analysis of MGMT expression in SC, KDM6B KO2, and rescued HeLa cells (mean ± SEM, n = 4). ****P < 0.0001 by one-way ANOVA Sidak's multiple comparisons test. (G) Immunoblot analysis of MGMT in SC, KDM6B KO2, and rescued HeLa cells. (H) RT-qPCR analysis of MGMT expression in HeLa cells expressing EV, WT KDM6B, or catalytically mutant (mut) KDM6B (mean ± SEM, n = 6–12). *P < 0.05; ****P < 0.0001 vs. EV by one-way ANOVA Dunnett's multiple comparisons test. (I) Immunoblot analysis of indicated proteins in HeLa cells expressing full-length WT or H1390A KDM6B. (J) Representative KDM6B and MGMT immunostaining images in HeLa cells expressing WT or catalytically mutant KDM6B. Scale bar, 20 μm. (K) Immunoblot analysis of MGMT in HeLa cells treated with or without GSK-J4 for 72 h. (L and M) Representative DNA PAGE gels of methylated and unmethylated MGMT promoters in WT and KDM6B KO2 HeLa cells (L). DNA intensity is quantified in M (mean ± SEM, n = 4). ****P < 0.0001 by two-way ANOVA Sidak's multiple comparisons test. (N) In vitro MGMT activity assay. The assay strategy is shown on the top. Immunoblot analysis of biotin is shown at the bottom. Numbers indicate the signal intensity.
To validate RNA-seq results, we performed qRT-PCR assay in SC and KDM6B KO2 HeLa cells and found KDM6B KO2 remarkably increased MGMT mRNA expression, which was not altered upon MNNG treatment (Figure 3E; Supplementary Figure S3A). MGMT protein levels were also upregulated in KDM6B KO2 HeLa cells but declined 4 h after MNNG treatment (Figure 3D), which is consistent with the previous finding that MGMT is degraded at the proteasome after repairing damaged DNA (24). In contrast, other nucleic acid demethylases including ALKBH1-9, DNA glycosylase MPG, and endonuclease APE1, which are also known to be involved in alkylating agent-induced DNA damage (16), were not regulated by KDM6B in HeLa cells (Figure 3E). Further, our rescue study showed that expression of WT KDM6B-C but not H1390A mutant reversed induction of MGMT mRNA and protein conferred by KDM6B KO2 in HeLa cells (Figure 3F and G). Similarly, ectopic expression of full-length KDM6B but not H1390A mutant inhibited MGMT mRNA and protein expression (Figure 3H and I). Immunostaining assay further confirmed inhibition of MGMT expression at the nucleus by KDM6B through its H3K27 demethylase activity (Figure 3J). We next employed KDM6B inhibitor GSK-J4 and found that treatment of GSK-J4 (1–10 μM) increased MGMT protein levels in both HeLa and A549 cells (Figure 3K; Supplementary Figure S3B). In line with these in vitro results, KDM6B expression was negatively correlated with MGMT expression in 1210 human pan tumors (r = −0.20) and 160 glioblastoma multiforme tumors (r = −0.32) from the TCGA cohort as determined by Pearson correlation analysis (P < 0.0001) (Supplementary Figure S3C and D). These findings support that KDM6B suppresses MGMT expression via its H3K27 demethylase activity.
KDM6B regulates DNA methylation at the MGMT promoter and inhibits MGMT expression and methyltransferase activity
To understand the mechanism underlying KDM6B-repressed MGMT transcription, we assessed whether KDM6B controls DNA methylation at the MGMT promoter. Strong DNA methylation was detected at the MGMT promoter in WT HeLa cells, but not KDM6B KO2 cells (Figure 3L and M). Consistently, the PCR product of the unmethylated MGMT promoter was significantly increased in KDM6B KO2 cells (Figure 3L and M). KDM6B KO2 had no effect on promoter methylation of the ACTB gene (encoding β-actin), indicating the specific role of KDM6B in MGMT methylation (Figure 3L). Together, these findings indicate that KDM6B increases methylation of the MGMT promoter to repress MGMT expression.
We next studied whether MGMT repression by KDM6B leads to a reduction of MGMT methyltransferase activity. Whole cell lysates from KDM6B KO2 and SC HeLa cells were incubated with a biotin-labeled DNA substrate with a methylated guanine at its O6-position. Once the methylated DNA substrate is converted by MGMT into un-methylated form, it can be cut by the restriction enzyme PstI, leading to reduced biotin signal. We found that biotin-labeled DNA levels were decreased by about 40% when incubated with KDM6B KO2 cell lysates as compared with SC cell lysates, which was completely reversed by a specific MGMT inhibitor BG, a pseudo substrate for MGMT (Figure 3N). These results indicate that KDM6B loss leads to the increased expression and methyltransferase activity of MGMT.
MGMT inhibition re-sensitizes KDM6B-deficient cells to alkylating agent-induced PARthanatos
To determine the role of MGMT in KDM6B KO-induced resistance to PARthanatos in response to alkylating agents, we treated SC and KDM6B KO2 HeLa cells with MGMT inhibitor BG (200 μM) with or without MNNG. As expected, pre-treatment of BG for 24 h specifically depleted MGMT protein in HeLa cells, as MNNG treatment did (Figure 4A). MNNG effectively caused death of SC but not KDM6B KO2 HeLa cells 24 h after treatment (Figure 4B and C). MGMT inhibition by BG treatment robustly killed MNNG-resistant KDM6B KO2 HeLa cells at 72 h, but not 24 h, after MNNG treatment (Figure 4B and C). Treatment of BG alone for up to 96 h had the marginal effect on death of both SC and KDM6B KO2 cells (Figure 4B and C). In line with pharmacological inhibition, genetic deletion of MGMT also significantly reversed HeLa cell survival conferred by KDM6B loss under MNNG treatment conditions (Figure 4D–F). In contrast, MNNG-induced PARthanatos was significantly reduced by overexpression of WT MGMT but not DNA methyltransferase inactive MGMT C145A mutant (Supplementary Figure S4A and B). In parallel, we found that BG treatment reduced endogenous MGMT protein levels and also re-sensitized KDM6B KO2 cells to MMS (Supplementary Figure S4C and D). Likewise, genetic deletion of MGMT also significantly re-sensitized KDM6B KO cells to MMS treatment (Supplementary Figure S4E and F). However, MMS treatment alone had little if any on MGMT or MPG protein levels (Supplementary Figure S4G). Together, these findings indicate that MGMT contributes to KDM6B KO cell resistance to alkylating agents.
Figure 4.

Pharmacological inhibition or deletion of MGMT sensitizes KDM6BKO cells to alkylating agents. (A) Immunoblot analysis of MGMT in SC and KDM6B KO2 HeLa cells 6 h after the treatment of MNNG (50 μM, 15 min) and/or BG (200 μM). (B and C) Representative cell death images in SC and KDM6B KO2 HeLa cells 72 h after treatment with MNNG and/or BG (B). PI-positive cells are quantified in C (mean ± SEM, n = 3). ****P < 0.0001 by two-way ANOVA Tukey's multiple comparisons test. ns, not significant. (D) Immunoblot analysis of MGMT in SC, KDM6B KO2, MGMT KO, and KDM6B/MGMT DKO HeLa cells 4 h after MNNG treatment. Numbers indicate the signal intensity. (E and F) Representative cell images in SC, KDM6B KO2, MGMT KO, KDM6B/MGMT DKO HeLa cells 24 h after MNNG treatment (E). Cell death is quantified in F (mean ± SEM, n = 3). ****P < 0.0001 by two-way ANOVA Sidak's multiple comparisons test. (G, H) Representative images (G) and quantification (H) of AIF nuclear translocation in SC and KDM6B KO cells in the presence or absence of BG (200 μM, pretreatment 24 h) and PARP inhibitor Olaparib (10 μM, 30 min pretreatment) at 4 h post MNNG (50 μM, 15 min) treatment. Red, AIF staining; Blue, DAPI staining; Purple, overlay of AIF and DAPI in the nuclei (mean ± SEM, n = 5). ****P < 0.0001 by two-way ANOVA Tukey's multiple comparisons test. Scale bar, 10 μm.
AIF translocation from mitochondria to nucleus following PARP-1 hyperactivation is a hallmark of PARthanatos (7,8,25). To further study whether KDM6B represses MGMT expression to promote alkylating agent-induced PARthanatos, AIF nuclear translocation was studied in KDM6B KO cells in the presence or absence of MGMT inhibitor BG following MNNG treatment. As expected, MNNG treatment (50 μM, 15 min) caused AIF nuclear translocation 4 h post treatment as indicated by the purple overlay of AIF (red) and a nuclear marker DAPI (blue) (Figure 4G and H), which is highly consistent with our previous reports (8,26). KDM6B KO significantly reduced AIF nuclear translocation, which was robustly reversed by the treatment of MGMT inhibitor BG (200 μM, pretreatment for 24 h) (Figure 4G and H). The treatment of PARP inhibitor Olaparib (10 μM) inhibited MNNG-induced AIF nuclear translocation in both SC and KDM6B KO cells with BG treatment (Figure 4G and H). These data support that KDM6B regulates PARthanatos via repressing MGMT expression.
KDM6B regulates O6 MeG-triggered PARP-1 activation and PARthanatos via controlling MGMT expression
We next studied the effect of low and high doses of alkylating agents including MNNG, MMS and TMZ on O6 MeG levels (Supplementary Figure S5A and B). We found that MNNG (25 μM, 15 min) efficiently increased O6 MeG levels in all HeLa cells, whereas MMS (2 mM) increased O6 MeG to a much lesser degree (<10% positive cells, Supplementary Figure S5A and B). The low dose of MNNG (2 μM, 15 min) had a comparable capacity with TMZ at the concentration of 250 μM, but not 2 μM, which moderately increased O6 MeG levels 2 h post treatment (Supplementary Figure S5A and B). Further, we studied the timeline of O6 MeG generation at 0 min, 20 min, 45 min, 2 h and 6 h post 25 μM MNNG treatment in SC cells (Figure 5A and B, Supplementary Figure S5A and B). We found that O6 MeG levels were immediately increased at 0 min post MNNG treatment and cannot be completely cleared for at least 6 h in SC cells (Figure 5A and B, Supplementary Figure S5A and B). At 20 min post 25 μM MNNG treatment, KDM6B KO, which enhanced MGMT expression and methyltransferase activity, significantly reduced O6 MeG levels and further reduced at 2 and 6 h post MNNG treatment (Figure 5A and B). Inhibition of MGMT by BG (200 μM, 24 h pre-treatment) or genetic deletion completely prevented O6 MeG removal in KDM6B KO HeLa cells during 0–6 h post treatment (Figure 5A and B). In contrast, PARP inhibitor Olaparib had no obvious effect on MNNG-induced O6 MeG levels (Figure 5A and B). These data indicate that KDM6B suppresses O6 MeG removal after alkylating agent exposure via repressing MGMT expression.
Figure 5.

MGMT regulates O6 MeG levels and O6 MeG-triggered PARP-1 activation. (A and B) Representative images of O6 MeG staining (A) and quantification (B) in SC, KDM6B-KO2, MGMT (MT) KO, KDM6B/MGMT DKO cells with or without MGMT inhibitor BG (200 μM) or PARP inhibitor Olaparib (10 μM) at 0 min, 2 h and 6 h after MNNG treatment (25 μM, 15 min). *P < 0.05, ***P < 0.001, ****P < 0.0001 by one-way ANOVA Sidak's multiple comparisons test. Scale bar, 20 μm. (C) Immunoblot analysis of PARP-1 activation and γH2AX in SC and MGMT KO HeLa cells treated with vehicle (−) or MNNG (25 μM, 15 min) for indicated time. (D) Immunoblot analysis of PARP-1 activation and γH2AX in SC, KDM6B KO2, MGMT KO, KDM6B/MGMT DKO HeLa cells treated with vehicle (−) or MNNG (50 μM, 15 min) for indicated time. Numbers on the blot indicate the relative signal intensity.
Next, we studied whether MGMT blocks PARP-1 activation by directly eliminating alkylating agent-induced O6 MeG in KDM6B KO cells. To this end, PARP-1 activation was determined in MGMT-deficient and MGMT-upregulated (KDM6B KO) cells as well as KDM6B/MGMT double KO (DKO) cells at different time points post MNNG treatment. PAR formation was increased immediately (0 min) after the treatment of MNNG (25 μM, 15 min) and then quickly decreased after 20 min in HeLa SC cells (Figure 5C). PAR levels were much higher in MGMT KO cells at 20 min (>2-fold increase) and 40 min (1.4-fold increase) post MNNG treatment, whereas PAR levels at 0 min induced by MNNG treatment in MGMT KO cells were saturated with only moderate increase than that of SC cells (Figure 5C), indicating that inhibition of O6 MeG removal by MGMT KO prolongs PARP-1 activation and PAR clearance. We then employed a high dose of MNNG (50 μM) that was used for cell death assay. MNNG-induced PAR formation was robustly decreased in KDM6B KO2 HeLa cells that mimics MGMT overexpression, which was reversed by MGMT KO 30 min after MNNG treatment (Figure 5D). A weak PAR signal was even detected in KDM6B/MGMT DKO cells 4 h after MNNG treatment (Figure 5D). Likewise, BG treatment moderately increased PAR levels in KDM6B KO2 cells immediately after MNNG treatment (Supplementary Figure S5C), although BG or MGMT KO alone marginally increased PAR formation (Figure 5D; Supplementary Figure S5C). In line with PARP-1 activation, MNNG treatment increased DNA damage in SC cells as indicated by higher γH2AX levels 2–4 h after treatment, which was enhanced by MGMT KO but attenuated by KDM6B KO (Figure 5C and D). Importantly, MGMT KO or BG treatment restored MNNG-induced γH2AX levels in KDM6B KO2 cells (Figure 5D; Supplementary Figure S5D). Lastly, we found that treatment of PARP-1 inhibitor DPQ dramatically inhibited MNNG-induced PAR formation in SC and KDM6B KO2 HeLa cells even in the presence of BG and prevented cell death not only from MNNG alone but also from MNNG plus BG in HeLa cells (Supplementary Figure S5C and E), indicating that PARP-1 activation is a downstream event of O6 MeG formation. Collectively, these data indicate that alkylating agent-induced O6 MeG triggers PARP-1 activation and subsequent PARthanatos, which is augmented by KDM6B through repressing MGMT expression and O6 MeG removal.
KDM6B interferes with alkylating agent-altered cell cycle progression
Our results above showed that death of KDM6B KO cells upon BG treatment was significantly delayed as compared with that of SC cells and KDM6B KO cells were also resistant to MMS treatment that modestly increased O6 MeG levels. These findings suggest that KDM6B KO might trigger a second mechanism independent of MGMT conferring cell resistance to alkylating agents-induced PARthanatos, especially when DNA alkylation evades from MGMT-mediated direct DNA repair. Therefore, we first studied whether KDM6B KO alters cell cycle progression post MNNG treatment. Both EdU incorporation (2 h incubation before the test) and DNA content were measured to determine the cell cycle status in SC and KDM6B KO HeLa cells at 2–22 h post MNNG treatment (50 μM, 15 min) (Supplementary Figure S6). We found that MNNG treatment quickly reduced both S and G1 phases and caused G2 arrest during 2–22 h post the treatment in SC cells (Figure 6A and B). Interestingly, some of SC cells continued to enter the second S phase that was increased at 15 and 22 h post-MNNG treatment (Figure 6A and B). In contrast, KDM6B KO significantly increased S phase especially during 6–22 h post MNNG treatment. In parallel, G1 phase was robustly reduced in KDM6B KO cells at 2–22 h post MNNG treatment (Figure 6A and B). Although cells in G2 phase were also increased in KDM6B KO cells after MNNG treatment, it was significantly less than that in SC (Figure 6A and B). These data indicate that MNNG treatment at the high dose quickly causes G2 arrest in SC cells but KDM6B KO alters cell cycle by increasing S phase.
Figure 6.

Cell cycle distribution in SC and KDM6B KO cells following alkylating agent MNNG treatment. Cells were treated with 50 μM MNNG for 15 min, stained with EdU/PI, and analyzed by flow cytometry at 2, 4, 6, 8, 15 and 22 h post the treatment. (A) Representative flow cytometry images. (B) Cell cycle distribution was quantified (mean ± SEM, n = 3). ****P < 0.0001 vs. identical cell cycle in DMSO group, #P < 0.0001 versus S phase in KDM6B KO cells at the identical timepoint, by two-way ANOVA Tukey's multiple comparisons test.
KDM6B suppresses alkylating agent-induced prolonged activation of Chk1 kinase and its associated DNA repair
To further determine whether DNA damage checkpoint response is involved in resistance to alkylating agents in KDM6B KO cells, we next studied if KDM6B regulates phosphorylation of Chk1 as well as its well-defined upstream factors ataxia telangiectasia mutated protein (ATM), ataxia telangiectasia and Rad3-related protein (ATR), and p32 subunit of replication protein A (RPA32), which all regulate DNA damage response and cell cycle checkpoint response (27), after exposure to MNNG. We found that Chk1 was phosphorylated at both Ser345 and Ser296 upon MNNG treatment, which was first detected 30 min post MNNG treatment and remained for 2 h after treatment in SC cells (Figure 7A). In contrast, constitutive phosphorylation of Chk1 up to at least 6–24 h was observed in KDM6B KO2 HeLa cells after MNNG treatment (Figure 7A; Supplementary Figure S7A). Similarly, ATM was phosphorylated starting at 30 min post MNNG treatment and remaining for at least 2 h in SC and KDM6B KO cells (Figure 7A). Interestingly, both RPA32 and ATR were phosphorylated starting at 2 h post MNNG treatment behind ATM phosphorylation. Moreover, the activation of RPA32 and ATR in KDM6B KO cells was much stronger than that in SC cells (Figure 7A). Chk1 activation has been known to phosphorylate its downstream factor Cdc25A, a step required for Cdc25A ubiquitination and degradation, thereby leading to cell cycle arrest (28). In line with acute Chk1 activation in SC cells, Cdc25A protein was quickly degraded at 4 h post MNNG treatment while DNA damage was quickly increased as indicated by γH2AX in SC cells from 1 to 6 h after MNNG treatment, indicating cell cycle arrest and deficiency of DNA repair (Figure 7A). In contrast, levels of Cdc25A remained relatively stable in KDM6B KO2 HeLa cells at the late period (2–24 h after MNNG treatment) and DNA damage was not further accumulated at least from 1 to 6 h after MNNG treatment, indicating that prolonged Chk1 activation enhanced checkpoint associated repair without causing G2 phase arrest (Figures 6 and 7A). Distinct to phospho-Chk1, Chk2 phosphorylation showed a similar pattern in both SC and KDM6B KO2 HeLa cells (Figure 7A). The expression of protein phosphatase 2A (PP2A), which is involved in Cdc25A dephosphorylation, was not obviously altered in KDM6B KO2 HeLa cells after MNNG treatment (Figure 7A), excluding a possibility of increased dephosphorylation of Cdc25A at the late period in KDM6B KO cells. These data indicate that KDM6B KO causes prolonged phosphorylation of Chk1 and enhances DNA repair.
Figure 7.

KDM6B KO promotes sustained Chk1 activation for DNA repair following alkylating agent treatment. (A) Immunoblot analysis of checkpoint response and DNA damage in SC and KDM6B KO2 HeLa cells 0–6 h after MNNG treatment. (B and C) Effects of MGMT and MGMT C145A mutant overexpression (B) and MGMT KO (C) on checkpoint response in SC and KDM6B KO HeLa cells 1 h and 4 h after MNNG treatment. Asterisk indicates endogenous MGMT and the top band in the MGMT blot designates ectopic MGMT (B). (D) Effects of checkpoint inhibition by GDC0575 (50 nM) on checkpoint response and DNA damage in SC, KDM6B KO2, and KDM6B/MGMT DKO HeLa cells 1 h and 6 h after MNNG treatment. (E and F) Effects of checkpoint inhibition by GDC0575 (50 nM) on MNNG-induced cell death in SC, KDM6B KO2, and KDM6B/MGMT DKO HeLa cells 24 h after MNNG treatment. Representative cell death images are in SC, KDM6B KO2, KDM6B/MGMT DKO HeLa cells 24 h after MNNG treatment (E). Cell death is quantified in F (mean ± SEM, n = 3). ****P < 0.0001 by two-way ANOVA Tukey's multiple comparisons test. (G) Immunoblot analysis of checkpoint response in SC and KDM6B KO2 HeLa cells 0–6 h after hydroxyurea (HU, 2 mM) treatment.
We next assessed whether MGMT alters MNNG-induced Chk1 phosphorylation by overexpression of WT-MGMT or methyltransferase inactive C145A mutant (29). Overexpression of WT-MGMT mimicked the effects of KDM6B KO and was sufficient to cause prolonged activation of Chk1, whereas C145A mutant MGMT failed to do so (Figure 7B). In line with prolonged Chk1 activation in WT-MGMT expressing cells, the level of Cdc25A at 4 h after MNNG treatment was similar to that at 2 h after MNNG treatment. However, Cdc25A levels were reduced in SC and MGMT C145A mutant cells at 4 h after MNNG treatment (Figure 7B). To further study whether MGMT is necessary for the prolonged activation of Chk1, we knocked out MGMT in KDM6B KO cells and found that MGMT KO failed to reverse KDM6B KO-induced prolonged activation of Chk1 (Figure 7C), indicating that other factors other than MGMT might contribute to DNA repair and prevent cell death in response to alkylating agents.
To study the role of checkpoint associated DNA repair besides MGMT-mediated direct DNA repair in KDM6B KO-induced resistance to alkylating agents, DNA damage was determined in KDM6B KO and KDM6B/MGMT DKO cells in the presence or absence of the specific Chk1 inhibitor GDC0575 (30) that blocks Chk1 activation thereby bypassing the checkpoint response and its associated DNA repair. As expected, KDM6B KO cells showed reduced DNA damage and cell death induced by MNNG treatment (Figure 7D–F; Supplementary Figure S7B-D). MGMT KO clearly increased γH2Ax levels 1–6 h after MNNG treatment in KDM6B KO cells and elevated subsequent cell death (Figure 7D–F; Supplementary Figure S7D), indicating that MGMT indeed plays a role in the resistance of KDM6B KO to alkylating agents. GDC0575 treatment inhibited Chk1 activation via reducing the phosphorylation of Chk1 at Ser296 position in SC, KDM6B KO and KDM6B/MGMT DKO cells at both 1 and 6 h after MNNG treatment, although GDC0575 treatment did not obviously reduced the phosphorylation of Chk1 at Ser345 position, which is usually phosphorylated by its upstream factor ATR (Figure 7D; Supplementary Figure S7B and C). The treatment of GDC0575 clearly increased DNA damage in KDM6B KO cells (Figure 7D; Supplementary Figure S7D). In line with this, inhibition of Chk1 activation by GDC0575 also significantly increased the sensitivity of KDM6B KO cells to MNNG-induced cell death (Figure 7E and F), indicating that the sustained Chk1 kinase activity-associated DNA repair also mediates KDM6B KO cell resistance to alkylating agents. Interestingly, the effects of KDM6B were specific to alkylating agents as both KDM6B WT and KO cells showed no difference of Chk1 phosphorylation in response to hydroxyurea (HU) treatment (Figure 7G).
To study a possible DNA repair pathway that is associated with sustained Chk1 activation, we quantified mRNA expression of MSH2, MSH6, MutL homolog 1 (MLH1) and PMS1 homolog 2 (PMS2), which are the key components in the MMR pathway, in SC and KDM6B KO HeLa cells. No obvious difference of these mRNAs was detected in SC and KDM6B KO cells (Supplementary Figure S8A). The protein expression of MSH2 and MSH6 was also not altered by KDM6B KO or MGMT KO (Supplementary Figure S8B). In contrast, the expression of XRCC1 protein, which is a key partner of PARP-1 involved in base-excision repair (BER), was increased by KDM6B KO, but not affected by MGMT KO (Supplementary Figure S8C). We next studied whether XRCC1 controls DNA repair and cell survival in KDM6B KO cells after treatment of alkylating agent. Besides XRCC1 overexpression cell line, two mixed XRCC1 KO subclones and two mixed KDM6B/XRCC1 DKO subclones were established by CRISPR/Cas9-based sgRNAs targeting two different regions on XRCC1 (Figure 8A–C; Supplementary Table 1). Deletion of XRCC1 restored MNNG-induced γH2AX levels in KDM6B KO cells, although overexpression or KO of XRCC1 alone did not obviously affect MNNG (50 μM)-induced γH2AX levels in SC cells (Figure 8A–C). Overexpression of XRCC1 partially prevented cell death at 24 h after MNNG treatment, whereas XRCC1 KO partially reversed KDM6B KO cell resistance to MNNG at 72 h post MNNG treatment (Figure 8D and E). These data indicate that XRCC1-dependent repair pathway but not the MMR pathway confers KDM6B KO cell resistance to PARthanatos.
Figure 8.
XRCC1 regulates MNNG-induced DNA damage and cell death. (A) Immunoblot analysis of XRCC1 overexpression (OE) and DNA damage in HeLa cells 4 h after MNNG treatment (50 μM, 15 min). SC, scrambled control. (B) Immunoblot analysis of XRCC1 KO (mixed clones) and DNA damage in HeLa cells 4 h after MNNG treatment. (C) Immunoblot analysis of XRCC1 expression and DNA damage in SC, KDM6B KO and KDM6B/XRCC1 (6B/XR) DKO (mixed clones) HeLa cells 4 h after MNNG treatment. (D and E) Effects of XRCC1 on MNNG-induced cell death in SC, XRCC1 OE, XRCC1 KO, KDM6B KO, and KDM6B/XRCC1 (6B/XR) DKO HeLa cells 24 or 72 h after MNNG treatment. Representative cell death images are shown in (D). Cell death is quantified in (E) (mean ± SEM, n = 3). ****P < 0.0001 by two-way ANOVA Sidak's multiple comparisons test.
Taken together, KDM6B specifically suppresses alkylating agents-induced prolonged Chk1 activation and its associated DNA repair thereby promoting PARthanatic cell death in response to alkylating agents.
DISCUSSION
In the present study, we dissected the PARthanatos pathway in response to alkylating agents and identified the epigenetic regulator KDM6B as a key regulator of alkylating agent-induced PARthanatos in cancer cells. PARthanatos is one type of programmed necrotic cell death distinct from apoptosis, necrosis, necroptosis and autophagy. PARP-1 is activated when it detects DNA damage and plays a central role in PARthanatic cell death pathway. Genetic deletion or pharmacological inhibition of PARP-1 blocks alkylating agent-induced PARthanatos, which cannot be prevented by inhibitors for apoptosis, necrosis, necroptosis and autophagy.
KDM6B is a well-known histone demethylase that demethylates the gene repressive marker H3K27me3 (31). We found here that KDM6B functions as an upstream regulator of PARP-1 and controls PARP-1 activation in response to alkylating agents via repressing MGMT expression. KDM6B KO increases MGMT expression and activity, which is responsible for the direct removal of DNA adduct upon alkylating agent treatment, leading to cell resistance to DNA damage and PARthanatos. Importantly, we showed that KDM6B increases MGMT promoter methylation and reduces MGMT transcription through its H3K27 demethylase activity. Consistently, overexpression of a demethylase-dead KDM6B mutant increases MGMT expression, indicating that inactive KDM6B mutant might function as a dominant-negative regulator and phenocopy KDM6B KO effects. Our results suggest that KDM6B may increase the chromatin accessibility at the MGMT promoter to facilitate the binding of DNA methyltransferase and subsequent methylation of the MGMT promoter in cancer cells. In line with our findings, a recent report showed lack of MGMT promoter methylation in H3.3K27M mutant glioma, in which H3K27me3 enrichment is increased locally at hundreds of genes (32). Alternatively, KDM6B may demethylate and increase the activity of DNA methyltransferases, which methylates and suppresses MGMT expression. Nevertheless, our rescue as well as inactive demethylase studies showed that KDM6B H3K27 demethylase activity is critical for MGMT repression, which has been further supported by a negative correlation of their expression in human tumors.
MGMT controls the first step in the DNA repair pathway by direct removal of O6 MeG DNA adducts added by alkylating agents. Our study provided direct evidence that alkylating agent rapidly increases O6 MeG levels and activates PARP-1 within minutes, leading to PARthanatos. These events are compromised by increased expression of MGMT in KDM6B KO cells, which can be further reversed by MGMT KO in KDM6B KO cells. O6 MeG generation was observed immediately after MNNG treatment in our study, which is highly consistent with the timeline of PAR formation at 0 min post the treatment. PARP-1 is a DNA damage sensor and detects damaged DNA within seconds to minutes to produce PAR (3,33). Moreover, our findings reveal that MGMT regulates PARP-1 activation and PAR formation in HeLa cells during 0–40 min after MNNG treatment, which is also consistent with the timeline of O6 MeG partial removal by MGMT, although the speed and capacity of PAR clearance by poly(ADP-Ribose) glycohydrolase (PARG) following PARP-1 hyperactivation is much faster and stronger than that of O6 MeG removal and MGMT degradation. The level of MGMT was not obviously changed at 30 min after MNNG treatment but it was gradually reduced at 4 h after treatment in SC cells. Given that MGMT inhibition increases PAR formation whereas PARP-1 inhibition cannot interfere with O6 MeG levels, O6 MeG is an upstream factor triggering PARP-1 activation and the PARP-1 activation process is highly dependent on the amount of O6 MeG produced (doses of alkylating agents used), the speed of O6 MeG removal (the expression level of MGMT), and PAR clearance by PARG. These findings support the importance of MGMT in regulation of PARP-1 activity and KDM6B KO cell resistance to alkylating agents.
Interestingly, KDM6B KO cells with BG treatment is significantly delayed as compared with that of SC cells, suggesting that, besides MGMT, a second independent DNA repair mechanism might also contribute to KDM6B KO cell resistance to alkylating agents, when DNA alkylation evades from MGMT-mediated direct DNA repair. Indeed, the treatment of alkylating agents in KDM6B KO cells causes a prolonged S phase and induces sustained Chk1 activation and its associated DNA repair at least for 24 h, thereby preventing DNA damage accumulation and subsequent cell death. These processes are reversed by checkpoint inhibitor treatment. In contrast, the treatment of alkylating agents in SC cells causes a robust increase of G2 phase and reduction of S phase under conditions we used. Accordingly, transient and acute activation of ATM and Chk1 is induced from 30 min to 2 h post MNNG treatment in SC cells, which subsequently leads to DNA damage increase and cell cycle arrest as indicated by levels of Cdc25A and γH2Ax. Chk1 can be activated during both S and G2 phases (34). Our data implicate that G2 arrest likely contributes to the acute and transient ATM-Chk1 activation immediately post the treatment, whereas prolonged S phase mainly contributes to RPA32-ATR-Chk1 activation, which occurs only in KDM6B KO cells at 2–6 h post MNNG treatment slightly behind G2 arrest-associated Chk1 activation. Checkpoint response in S phase is likely associated with DNA repair as indicated by reduced DNA damage in KDM6B KO cells. Thus, upon alkylating agent treatment, HeLa cells mainly have G2 arrest-associated Chk1 activation lacking DNA repair, whereas KDM6B KO cells have S phase associated prolonged Chk1 activation besides G2 associated acute Chk1 activation, which allows DNA repair during S phase. Future studies are needed to understand the functional difference of checkpoint response in G2 and S phases. Our data support that enhanced prolonged checkpoint response-associated DNA repair is a second step in DNA repair to confer KDM6B KO cell resistance to alkylating agents.
It is important to know how MGMT affects the second step checkpoint response. Our findings revealed that overexpression of MGMT is sufficient but not necessary to promote the prolonged Chk1 activation contributing to HeLa cell resistance to alkylating agents. In line with that, it is not a surprise that inhibition or deletion of MGMT re-sensitizes KDM6B KO cells to MMS treatment, although MMS produces very modest levels of O6 MeG.
MMR has been implicated to function as a DNA repair mechanism associated with cell cycle progression when DNA damage evades from the direct repair (35,36). Our study showed that the expression of base excision repair protein XRCC1 but not MMR proteins is increased in KDM6B KO cells and contributes to KDM6B KO cell resistance to alkylating agents, which is highly consistent with a previous report showing that XRCC1 KO sensitizes CHO cells to MNNG-, TMZ- and MMS-induced DNA damage and cell death (37). Given the tight functional link of XRCC1 with checkpoint response (38), our findings suggest that XRCC1-mediated base excision repair may contribute to checkpoint associated DNA repair in KDM6B KO cells following MNNG treatment. Future studies are required to understand if additional checkpoint associated repair is involved in KDM6B KO cell resistance to alkylating agents.
Accumulating evidence supports that PARP-1 facilitates DNA repair in response to mild DNA damage. Blockage of PARP-1 activity sensitizes cancer cells to death. This concept has been well supported by PARP inhibitors used in the clinic for cancer therapy (39). In contrast, in response to severe DNA damage, excessive activation of PARP-1 causes large DNA fragments and caspase-independent cell death designated PARthanatos, which occurs in many organ systems and is widely involved in different neurologic and non-neurologic diseases, including ischemia-reperfusion injury after stroke and myocardial infarction, glutamate excitotoxicity, neurodegenerative diseases, inflammatory injury, reactive oxygen species–induced injury (7,25). This type of cell death is profoundly prevented by pharmacological inhibition or genetic deletion of PARP-1. The importance of PARP-1 in neuronal cell death and cancer cell death has also been strongly supported by our previous as well as current studies (7–9,25). Our current study identified the epigenetic factor KDM6B as a key cell death regulator to switch PARP-1 function and signaling from DNA repair/cell survival to DNA damage/cell death addressing a huge knowledge gap of PARP-1 functions under different contexts.
Alkylating agents are the most common chemotherapy drugs currently used in the clinic to kill cancer (10–12). However, cancer cell resistance to alkylating agents remains to be one of the biggest challenges in the clinic. We showed that KDM6B deficiency enhances MGMT-mediated DNA repair and checkpoint associated DNA repair (Figures 3–7), thereby blocking DNA damage and PARP-1 hyperactivation, which confers cancer cell resistance to alkylating agents. Our study provides evidence of KDM6B functions in regulation of DNA repair, PARP-1 activation and cell death sensitivity to alkylating agents.
Collectively, our study elucidates an epigenetic mechanism underlying PARP-1 activation and cell death in response to alkylating agents (Figure 9). KDM6B regulates PARthanatos via controlling two steps of sequential DNA repair including MGMT-mediated direct DNA repair and checkpoint associated DNA repair (Figure 9). Low levels of KDM6B first increase direct DNA repair via elevating MGMT expression and further promote sustained checkpoint activation and its associated DNA repair, both of which confer cell resistance to alkylating agents. In contrast, high levels of KDM6B suppress MGMT-mediated DNA repair and checkpoint associated DNA repair, thereby causing PARP-1 hyperactivation and cell death. KDM6B may serve as a biomarker to predict the sensitivity of cancer cells to alkylating agents. Targeting both MGMT and checkpoint associated DNA repair might be a potential therapeutic strategy to re-sensitize cancer cells to alkylating agent treatment.
Figure 9.
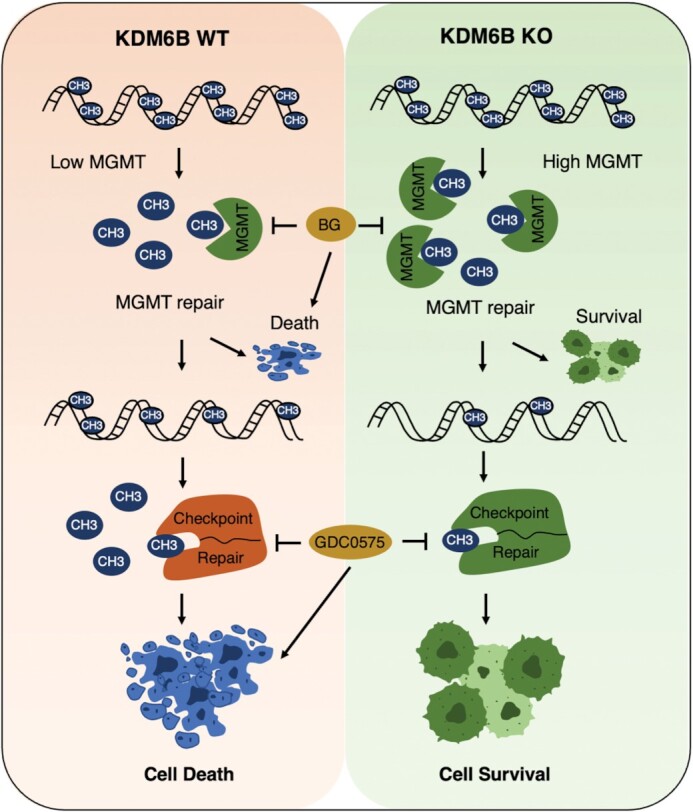
KDM6B is a key cell death regulator of PARthanatos induced by alkylating agents. KDM6B regulates two sequential DNA repair processes including MGMT direct DNA repair and checkpoint associated repair. Loss of KDM6B on one hand enhances MGMT direct DNA repair to promote cell survival, on the other hand triggers sustained Chk1 activation and increases checkpoint associated DNA repair to fix DNA evading from the first step MGMT repair process leading to cell survival. High levels of KDM6B suppress both DNA repair processes leading to cell death. Blocking MGMT and checkpoint associated repair re-sensitizes cancer cells to alkylating agents.
DATA AVAILABILITY
All datasets used/analyzed during the current study have been included in this article and its supplementary materials. The RNA-seq data were deposited at the GEO database with accession number GSE191196 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE191196).
Supplementary Material
ACKNOWLEDGEMENTS
Authors’ contributions: Y.W. and W.L. conceived the idea, designed research, analyzed the data and wrote the paper; M.Y. and C.W. performed most experiments. M.Z. performed RNA-seq and AIF nuclear translocation study. L.B. performed initial cell death assays and CRISPR screening validation. Y.N.W. constructed KDM6B overexpression vectors. A.K. and X.C. performed CRISPR screening and RNA-seq bioinformatics analysis. All authors read and approved the final manuscript.
Contributor Information
Mingming Yang, Department of Pathology, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA.
Chenliang Wang, Department of Pathology, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA.
Mi Zhou, Department of Pathology, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA.
Lei Bao, Department of Pathology, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA.
Yanan Wang, Department of Pathology, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA.
Ashwani Kumar, Eugene McDermott Center for Human Growth and Development, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA.
Chao Xing, Eugene McDermott Center for Human Growth and Development, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA; Department of Bioinformatics, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA; Department of Population and Data Sciences, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA.
Weibo Luo, Department of Pathology, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA; Department of Pharmacology, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA.
Yingfei Wang, Department of Pathology, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA; Department of Neurology, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA; Peter O’Donnell Brain Institute, UT Southwestern Medical Center, Dallas, TX 75390, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health (NIH) [R35GM124693, R00NS078049, R01AG066166]; Welch foundation [I-1939]; University of Texas (UT) Southwestern Medical Center Startup funds; UT Rising Stars (to Y.W.); NIH [R01CA222393]; CRPIT [RP190358]; Welch Foundation [I-1903 to W.L.]. Funding for open access charge: NIH [R35GM124693].
Conflict of interest statement. None declared.
REFERENCES
- 1. Ray Chaudhuri A., Nussenzweig A.. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017; 18:610–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Luo X., Kraus W.L.. On PAR with PARP: cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev. 2012; 26:417–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bai P. Biology of poly(adp-ribose) polymerases: the factotums of cell maintenance. Mol. Cell. 2015; 58:947–958. [DOI] [PubMed] [Google Scholar]
- 4. Beck C., Robert I., Reina-San-Martin B., Schreiber V., Dantzer F.. Poly(ADP-ribose) polymerases in double-strand break repair: focus on PARP1, PARP2 and PARP3. Exp. Cell Res. 2014; 329:18–25. [DOI] [PubMed] [Google Scholar]
- 5. Wang Y., Luo W., Wang Y.. PARP-1 and its associated nucleases in DNA damage response. DNA Repair (Amst). 2019; 81:102651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bryant H.E., Schultz N., Thomas H.D., Parker K.M., Flower D., Lopez E., Kyle S., Meuth M., Curtin N.J., Helleday T.. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005; 434:913–917. [DOI] [PubMed] [Google Scholar]
- 7. Liu S., Luo W., Wang Y.. Emerging role of PARP-1 and PARthanatos in ischemic stroke. J. Neurochem. 2021; 160:74–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang Y., An R., Umanah G.K., Park H., Nambiar K., Eacker S.M., Kim B., Bao L., Harraz M.M., Chang C.et al.. A nuclease that mediates cell death induced by DNA damage and poly(ADP-ribose) polymerase-1. Science. 2016; 354:aad6872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang Y., Kim N.S., Haince J.F., Kang H.C., David K.K., Andrabi S.A., Poirier G.G., Dawson V.L., Dawson T.M.. Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos). Sci. Signal. 2011; 4:ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fu D., Calvo J.A., Samson L.D.. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat. Rev. Cancer. 2012; 12:104–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maanen M.J., Smeets C.J., Beijnen J.H.. Chemistry, pharmacology and pharmacokinetics of N,N',N" -triethylenethiophosphoramide (ThioTEPA). Cancer Treatment Reviews. 2000; 26:257–268. [DOI] [PubMed] [Google Scholar]
- 12. Lawley P.D., Thatcher C.J.. Methylation of deoxyribonucleic acid in cultured mammalian cells by N-methyl-N'-nitro-N-nitrosoguanidine. The influence of cellular thiol concentrations on the extent of methylation and the 6-oxygen atom of guanine as a site of methylation. Biochem. J. 1970; 116:693–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zong W.X., Ditsworth D., Bauer D.E., Wang Z.Q., Thompson C.B.. Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes Dev. 2004; 18:1272–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fu D., Jordan J.J., Samson L.D.. Human ALKBH7 is required for alkylation and oxidation-induced programmed necrosis. Genes Dev. 2013; 27:1089–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Klapacz J., Meira L.B., Luchetti D.G., Calvo J.A., Bronson R.T., Edelmann W., Samson L.D.. O6-methylguanine-induced cell death involves exonuclease 1 as well as DNA mismatch recognition in vivo. Proc. Natl Acad. Sci. U.S.A. 2009; 106:576–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Calvo J.A., Moroski-Erkul C.A., Lake A., Eichinger L.W., Shah D., Jhun I., Limsirichai P., Bronson R.T., Christiani D.C., Meira L.B.et al.. Aag DNA glycosylase promotes alkylation-induced tissue damage mediated by parp1. PLoS Genet. 2013; 9:e1003413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen Y., Zhang B., Bao L., Jin L., Yang M., Peng Y., Kumar A., Wang J.E., Wang C., Zou X.et al.. ZMYND8 acetylation mediates HIF-dependent breast cancer progression and metastasis. J. Clin. Invest. 2018; 128:1937–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang B., Chen Y., Shi X., Zhou M., Bao L., Hatanpaa K.J., Patel T., DeBerardinis R.J., Wang Y., Luo W.. Regulation of branched-chain amino acid metabolism by hypoxia-inducible factor in glioblastoma. Cell Mol. Life Sci. 2021; 78:195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shalem O., Sanjana N.E., Hartenian E., Shi X., Scott D.A., Mikkelson T., Heckl D., Ebert B.L., Root D.E., Doench J.G.et al.. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014; 343:84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li W., Xu H., Xiao T., Cong L., Love M.I., Zhang F., Irizarry R.A., Liu J.S., Brown M., Liu X.S.. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 2014; 15:554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hattermann K., Mehdorn H.M., Mentlein R., Schultka S., Held-Feindt J.. A methylation-specific and SYBR-green-based quantitative polymerase chain reaction technique for O6-methylguanine DNA methyltransferase promoter methylation analysis. Anal. Biochem. 2008; 377:62–71. [DOI] [PubMed] [Google Scholar]
- 22. De Santa F., Totaro M.G., Prosperini E., Notarbartolo S., Testa G., Natoli G.. The histone H3 lysine-27 demethylase jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell. 2007; 130:1083–1094. [DOI] [PubMed] [Google Scholar]
- 23. Yang L., Zha Y., Ding J., Ye B., Liu M., Yan C., Dong Z., Cui H., Ding H.F.. Histone demethylase KDM6B has an anti-tumorigenic function in neuroblastoma by promoting differentiation. Oncogenesis. 2019; 8:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cabrini G., Fabbri E., Lo Nigro C., Dechecchi M.C., Gambari R.. Regulation of expression of O6-methylguanine-DNA methyltransferase and the treatment of glioblastoma (Review). Int. J. Oncol. 2015; 47:417–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang Y., Dawson V.L., Dawson T.M.. Poly(ADP-ribose) signals to mitochondrial AIF: a key event in parthanatos. Exp. Neurol. 2009; 218:193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang Y., Kim N.S., Li X., Greer P.A., Koehler R.C., Dawson V.L., Dawson T.M.. Calpain activation is not required for AIF translocation in PARP-1-dependent cell death (parthanatos). J. Neurochem. 2009; 110:687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Moiseeva T.N., Yin Y., Calderon M.J., Qian C., Schamus-Haynes S., Sugitani N., Osmanbeyoglu H.U., Rothenberg E., Watkins S.C., Bakkenist C.J.. An ATR and CHK1 kinase signaling mechanism that limits origin firing during unperturbed DNA replication. Proc. Natl Acad. Sci. U.S.A. 2019; 116:13374–13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jin J., Shirogane T., Xu L., Nalepa G., Qin J., Elledge S.J., Harper J.W.. SCFbeta-TRCP links chk1 signaling to degradation of the Cdc25A protein phosphatase. Genes Dev. 2003; 17:3062–3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hazra T.K., Roy R., Biswas T., Grabowski D.T., Pegg A.E., Mitra S.. Specific recognition of O6-methylguanine in DNA by active site mutants of human O6-methylguanine-DNA methyltransferase. Biochemistry. 1997; 36:5769–5776. [DOI] [PubMed] [Google Scholar]
- 30. Li M., Huang T., Li X., Shi Z., Sheng Y., Hu M., Song K.. GDC-0575, a CHK1 inhibitor, impairs the development of colitis and colitis-associated cancer by inhibiting CCR2(+) macrophage infiltration in mice. Onco Targets Ther. 2021; 14:2661–2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chung N., Bogliotti Y.S., Ding W., Vilarino M., Takahashi K., Chitwood J.L., Schultz R.M., Ross P.J.. Active H3K27me3 demethylation by KDM6B is required for normal development of bovine preimplantation embryos. Epigenetics. 2017; 12:1048–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chan K.M., Fang D., Gan H., Hashizume R., Yu C., Schroeder M., Gupta N., Mueller S., James C.D., Jenkins R.et al.. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 2013; 27:985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Caron M.C., Sharma A.K., O'Sullivan J., Myler L.R., Ferreira M.T., Rodrigue A., Coulombe Y., Ethier C., Gagne J.P., Langelier M.F.et al.. Poly(ADP-ribose) polymerase-1 antagonizes DNA resection at double-strand breaks. Nat. Commun. 2019; 10:2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gorecki L., Andrs M., Korabecny J.. Clinical candidates targeting the ATR-CHK1-WEE1 axis in cancer. Cancers (Basel). 2021; 13:795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kondo N., Takahashi A., Ono K., Ohnishi T.. DNA damage induced by alkylating agents and repair pathways. J. Nucleic Acids. 2010; 2010:543531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schroering A.G., Williams K.J.. Rapid induction of chromatin-associated DNA mismatch repair proteins after MNNG treatment. DNA Repair (Amst). 2008; 7:951–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Horton J.K., Watson M., Stefanick D.F., Shaughnessy D.T., Taylor J.A., Wilson S.H.. XRCC1 and DNA polymerase beta in cellular protection against cytotoxic DNA single-strand breaks. Cell Res. 2008; 18:48–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chou W.C., Wang H.C., Wong F.H., Ding S.L., Wu P.E., Shieh S.Y., Shen C.Y.. Chk2-dependent phosphorylation of XRCC1 in the DNA damage response promotes base excision repair. EMBO J. 2008; 27:3140–3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rose M., Burgess J.T., O’Byrne K., Richard D.J., Bolderson E.. PARP inhibitors: clinical relevance, mechanisms of action and tumor resistance. Front. Cell Dev. Biol. 2020; 8:564601. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All datasets used/analyzed during the current study have been included in this article and its supplementary materials. The RNA-seq data were deposited at the GEO database with accession number GSE191196 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE191196).