

Abstract
Reverse transcriptases (RTs) are enzymes capable of synthesizing DNA using RNA as a template. Within the last few years, a burst of research has led to the discovery of novel prokaryotic RTs with diverse antiviral properties, such as DRTs (Defense-associated RTs), which belong to the so-called group of unknown RTs (UG) and are closely related to the Abortive Infection system (Abi) RTs. In this work, we performed a systematic analysis of UG and Abi RTs, increasing the number of UG/Abi members up to 42 highly diverse groups, most of which are predicted to be functionally associated with other gene(s) or domain(s). Based on this information, we classified these systems into three major classes. In addition, we reveal that most of these groups are associated with defense functions and/or mobile genetic elements, and demonstrate the antiphage role of four novel groups. Besides, we highlight the presence of one of these systems in novel families of human gut viruses infecting members of the Bacteroidetes and Firmicutes phyla. This work lays the foundation for a comprehensive and unified understanding of these highly diverse RTs with enormous biotechnological potential.
INTRODUCTION
Reverse transcriptases (RTs, also known as RNA-directed DNA Polymerases) are enzymes present in all three domains of life whose main function is to polymerize DNA strands using RNA as a template. Although they were first discovered by Temin & Baltimore in 1970 (1,2), prokaryotic RTs were not observed until 1989 when they were found to be the main component of retrons (3). Later research revealed that most reverse transcriptases (80%) can be phylogenetically clustered into three major lineages: group II introns, diversity-generating retroelements (DGRs), and retrons, which are the best known due to their ecological implications and biotechnological applications. Other minor clades of RTs include abortive infection (Abi) RTs, CRISPR-Cas-associated RTs, Group II-like (G2L), the unknown groups (UG) and rvt elements (4–6).
Comprehensive and systematic analysis of prokaryotic RTs (7), identified the association of RTs with CRISPR-Cas systems and 5 novel gene families (D, E, F1, F2, and G, now known as UG9, UG6, UG1, UG5 and UG3 + UG8, respectively). Further research revealed the existence of other uncharacterized RTs from distinct clades (UG1-UG16 and Group II-like, including those described by Kojima & Kanehisa) (6,8,9), and a more recent work disclosed 11 additional UG RT groups (UG17-UG28), pointing out that UG and Abi RTs may form a novel major lineage branching off from a common node (5). Although it was initially thought that UG and Abi RTs were not very common, it is known that they represent at least 11% of all prokaryotic RTs, showing an enormous diversity and holding great promise for the development of new biotechnological tools (4).
Abi systems can function as prokaryotic defense mechanisms against certain phages (10,11). They are generally constituted by a sensing module that recognizes a phage-specific signal and an effector module that generates a response, either by blocking the viral infection cycle, halting host metabolism, or causing cell death (12). Although there are >20 different Abi systems, only a few have been well characterized. Among them, AbiA, AbiK and AbiP2 share an N-terminal RT domain (13–17), with AbiA harboring an additional C-terminal HEPN domain (higher eukaryotes and prokaryotes nucleotide-binding domain) with predicted RNAse activity (18). Both AbiA and AbiK are commonly found in plasmids and have been shown to protect Lactococcus spp. from diverse phage infections (13), whereas AbiP2 is commonly found in a hypervariable region of P2 prophages in E. coli and confers resistance against T5 phage (17). It has been hypothesized that AbiA and AbiK could have a similar mechanism of action, as both of them confer protection against the same phages either by blocking DNA replication or targeting functionally-related proteins (11). Furthermore, phages escaping AbiK and AbiA interference have been shown to harbor point mutations in single-strand annealing proteins (SSAPs) involved in DNA replication (19). AbiK is the best characterized, and it has been hypothesized to have protein-primed untemplated DNA-polymerase activity (14). The residues responsible for this activity are thought to be located at the C-terminal region (14) that along with the RT domain, is essential for its biological role (13,20).
Although Abi and UG RTs are phylogenetically related, they were thought to be functionally unrelated as they bear distantly related RT domains. Also, previous analyses pointed out the high divergence at the sequence level between AbiK, AbiA and AbiP2 (6). That notwithstanding, recent research that employed a systematic methodology to search for novel antiphage systems (21) highlighted that some members of UG RTs (UG1, UG2, UG3-UG8, UG15, UG16, named DRTs type 1–5 respectively for Defense-associated RTs) act as defense mechanisms against bacterial viruses. This suggests a functional link between UG and Abi RTs and supports the idea that different families of RTs may be implicated in immunity against bacteriophages (4).
Even though some UG and Abi RTs members have been reported to have antiphage functions and associated domain(s) required for this function, some others remain poorly characterized due to insufficient information on their genomic context, associated genes, or biological roles. Considering the great diversity of these RTs, their possible common origin, and the recently disclosed role of retrons and DRTs (21–24), we hypothesize that the lineage composed of UG and Abi RTs (UG/Abi) may constitute a novel family of defense-related RTs with high divergence and a plethora of associated genes. In this work, we performed a systematic analysis of UG/Abi RTs and their neighborhood in search of associated genes and defense hallmarks. As a result, we expanded the number and diversity of UG/Abi RTs with novel groups, of which most are associated with other protein domain(s) and located within defense islands/hotspots. Based on this information, the UG/Abi RTs could be classified into three major classes, a first class of RTs fused to HEAT-like repeats, a second class of highly diverse RTs not fused to any known domain, and a third class commonly associated with C-N hydrolase (carbon-nitrogen hydrolase, also known as nitrilase) or phosphohydrolase domains. Besides, we demonstrate the antiphage activity of three Class 1 members and an additional Class 2 member. Moreover, we reveal that UG27, a Class 2 UG/Abi RT, is commonly encoded in several groups of predicted human gut viruses infecting members of the Bacteroidetes and Firmicutes phyla, which encode a putative non-coding RNA (ncRNA) with a common secondary structure. Finally, different UG groups (UG2, UG3 + UG8 and UG28) that have been described to possess antiphage properties (DRT type 2, DRT type 3 and DRT type 9 respectively) (21) also encode ncRNAs with conserved secondary structures, thought to be essential for the functioning of the systems. Altogether, these findings reveal that the UG/Abi RTs family is a highly diverse and widespread lineage of prokaryotic reverse transcriptases associated with defense functions that would play a very important role in virus-host conflicts.
MATERIALS AND METHODS
Construction of a comprehensive dataset of representative UG/Abi RTs dataset
To increase the number of UG/Abi RT sequences, the most up-to-date phylogenetic tree of prokaryotic RTs based on an alignment of the RT domain of 9141 RTs (5) was used as a reference. Custom HMM profiles for every phylogenetic group (group II introns, retrons, DGRs, CRISPR-associated RTs, G2L and Abi/UG RTs) were built using hmmbuild from the suite HMMER 3.3 (25). Then, the NR database (ftp://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/) was searched (February 2021) with all profiles using hmmsearch (E-value < 1e–05) (25). Only sequences in which the E-value using the UG/Abi profile was at least one order of magnitude lower than the E-value using other profiles were kept. After this, sequences present in Eukaryotic genomes or with unassigned taxonomy were removed using the NCBI API (26), which resulted in a dataset containing 12209 potential UG RT sequences present in Bacteria, Archaea, and their viruses. To remove sequence redundancy, the 12209 sequences were clustered using CD-HIT (27) with default parameters and the option -c 0.85 (85% AAI; average amino acid identity), which resulted in 5727 clusters that were further filtered based on quality and contig completeness criteria to perform a neighborhood analysis. Sequences with no associated NCBI nucleotide accession were discarded from the analysis. Further, the NCBI Identical Protein Groups (IPG) database (26) was used to retrieve information about the completeness of the assemblies in which each of the sequences was found. For each entry, the best IPG candidate was selected based on reference assignments and/or completeness (reference genomes > complete genomes > scaffolds/contigs). If not found in any complete genome, sequences in contigs with the greater neighborhood information (i.e. large contigs where the RT is not located close to their ends) were prioritized by choosing those with the highest W value, a parameter that illustrates the length of the contig weighted by how centered the ORF is in the contig; described below.
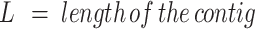 |
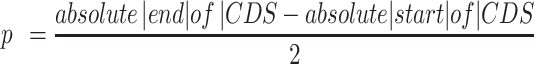 |
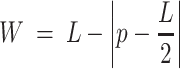 |
Phylogenetic and network analysis of UG/Abi RTs
The cd-hit-2d tool (27) was used to label sequences highly similar (>95% AAI) to the reference entries. After this, a multiple sequence alignment (MSA) of RT1-7 motifs was performed using the MAFFT software (28) with default parameters, and a phylogenetic tree was built using FastTree (29) with the WAG evolutionary model, and discrete gamma model with 20 rate categories. A phylogenetic tree was also constructed with IQ-TREE v1.6.12, with 1000 ultra-fast bootstraps (UFBoot) and SH-like approximate likelihood ratio test (SH-aLRT) with 1000 replicates (-bb 1000 -alrt 1000 options) (30), using the LG + R10 model identified as the best model by Modelfinder (31). To compare full-length sequences, a sequence-similarity network (SSN) of these sequences was also built using the EFI-EST resource (32) and visualized using Cytoscape (33) with the force-directed layout using the BLAST score as a weight.
Retrieval and clustering of neighbor proteins
Coding sequences (CDS) located within ±10 kb of the start and the end of our query proteins were retrieved using the feature table resource from the NCBI Entrez API (26). Due to the frequent misannotation of ORFs, nucleotide sequences of the intergenic regions were also retrieved, and ORFs were predicted using the Prodigal tool with -c -m -n -p meta parameters (34). Neighbor CDS and ORFs predicted in the intergenic sequences were joined and clustered using MMseqs2 (35) with a profile-based deep clustering method previously described (36) using 10 iterations, which rendered 3240 neighbor clusters.
Prediction and annotation of functionally associated genes
Genes functionally associated with UG/Abi RTs were predicted using a methodology previously described (36). Briefly, a presence/absence matrix of neighbor clusters surrounding UG/Abi RT entries was analyzed in search of non-random patterns of association. Based on the distribution of clusters across the tree and the average amino acid identity (AAI) of the co-located RTs, 193 out of 3240 protein clusters with more than 5 members were selected as potentially linked. After this, MSAs and HMM profiles were built using MAFFT (28) with default parameters and hmmbuild (25), respectively. Domain annotation of protein clusters was done using HHsearch against the hh-formatted PFAM (37), CDD (38), COG (39), ECOD (40) and pdb30 databases jointly distributed with HH-suite (41) (Supplementary Table S2). In addition, we also performed comparisons against profiles built using the eggNOG (Bacteria, Archaea and Viruses) (42), pVOG (43) and mMGE (44) databases.
Group adscription and refinement of UG/Abi RTs
Group adscription of sequences was manually assigned based on the phylogeny, the sequence-similarity network, the presence of labeled reference sequences, and the presence/absence matrix. Individual sequences that were difficult to classify, with low support, or with little information about the neighborhood were manually removed. After performing this task iteratively, and rebuilding MSAs, phylogenetic trees, and SSNs as described above, 5022 UG/Abi RT bona fide representative sequences were retained.
Sequence and structure-based annotation of UG/Abi RTs domains
For every UG/Abi RT group, MSAs were built using MAFFT-einsi (28) with default parameters. Groups with bimodal length distribution were subdivided into small and large variants, and MSAs were built independently. Annotation of UG/Abi RTs was done using hhsearch and PFAM, COG, CDD and ECOD databases. We then performed structural predictions employing trRosetta (45) using previously built MSAs as input for modeling. After this, predicted models were compared against the PDB database using the DALI webserver (46). For αRep domains found in Class 1 UG/Abi RTs, motif boundaries were obtained from trRosetta using contact maps and predicted structure models as a reference. Then, trimmed MSAs were used as a query to perform further trRosetta structure predictions. A multiple protein structure alignment was built for every Class1 predicted repeated structure model, by using the mTM-align web server (47) and PyMOL Molecular Graphics System (Schrödinger, LLC) Cealign command with UG8 αRep domain as an anchor.
Taxonomy assignment
To determine the taxonomic distribution of every UG/Abi RT group, every representative sequence was queried against the NCBI taxonomy database (26), and information about the domain, phylum, class, order, family, and genus was retrieved for every associated genome (Supplementary Table S1). Then, relative abundances of phyla across the different UG/Abi RT groups were calculated and plotted using the ggplot2 R package (48). For every group, those phyla with < 1% of relative abundance were removed to improve the visualization.
Prediction of RT defense association
Defense genes were predicted as previously described (21). Briefly, a total of 174 080 bacterial and archaeal genomes were downloaded from GenBank in 2018, and highly similar proteins (at least 98% sequence identity and coverage) were discarded using the linclust option in MMseqs2 with parameters –min-seq-id 0.98 -c 0.98. To identify homologs of each of the 42 groups of RTs within this data set, representatives from each RT group (5022 total sequences) were used as search seeds for MMseqs2. To be labeled as an RT homolog, proteins were required to have a minimum of 80% sequence identity and 80% coverage (–min-seq-id 0.8 -c 0.8) to at least one RT search seed. Following homolog identification, the defense association frequency (defined as the proportion of homologs within 5 kb or 5 ORFs from the nearest annotated known defense system) for each RT group was calculated as previously described (21) (Supplementary Table S3). Groups with defense association frequencies or number homologs below threshold were manually examined for signatures of strong defense association. In particular, groups with one or more homologs that are operonized with known defense genes, or flanked by known defense genes on both 5′ and 3′ ends, were predicted to have a defense function (Supplementary Figure S2).
Cloning
Genes were chemically synthesized as gene fragments (GENEWIZ) or amplified with Q5 (New England Biolabs) or Phusion Flash (Thermo Scientific) polymerase. The native promoter and ORF sequences were retained in all cases except for UG15, which was recoded (21). Inserts were cloned into a low copy pACYC184-derived empty vector (Addgene # 157879) between the HindIII and EcoRI restriction sites using NEBuilder HiFi DNA Assembly mix (New England Biolabs). The full sequences of all plasmids were verified as previously described (21,49). Briefly, ∼25 ng of each plasmid was incubated with purified Tn5 transposome (pre-loaded with Illumina adapters) at 55°C for 10 min in the presence of 5 mM MgCl2 and 10 mM TAPS, resulting in an average fragment size of ∼400 bp. Reactions were treated with 0.5 volume of 0.1% sodium dodecyl sulfate for 5 min at room temperature and amplified with KAPA HiFi HotStart polymerase using primers containing 8 nt i7 and i5 index barcodes. Barcoded amplicons were purified with SPRIselect beads (New England Biolabs) and sequenced with a MiSeq kit (Illumina).
Competent cell production
Escherichia coli K-12 (ATCC 25404) was obtained from the American Type Culture Collection. Cells were cultured in ZymoBroth and made competent using a Mix & Go buffer kit (Zymo) according to the manufacturer's recommended protocol.
Phage plaque assays
Escherichia coli harboring a candidate defense system or a pACYC184 empty vector were cultured in terrific broth at 37°C in the presence of 25 μg/ml chloramphenicol. To 10 ml top agar (10 g/l tryptone, 5 g/l yeast extract, 10 g/L NaCl, 7 g/l agar) was added chloramphenicol (final concentration 25 μg/ml) and 0.5 mL E. coli culture, and the mixture was poured on 10 cm LB-agar plates containing 25 μg/ml chloramphenicol. Ten-fold dilutions of phages T2 (ATCC11303-B2), T5 (ATCC11303-B5) and ZL-19 in phosphate buffered saline were spotted onto the plates at 3 μl per spot. After overnight incubation at 37°C, plates were photographed with a white backlight.
Presence of UG/Abi RTs in mobile genetic elements
Previously built HMM profiles for every UG/Abi group were used to retrieve sequences encoded in the PLSDB plasmid database (50). Firstly, ORFs were predicted for every nucleotide entry in the PLSDB database using Prodigal with default parameters and the -p meta option. After this, hmmsearch (E-value < 1e–20) was used with every profile against the predicted ORFs. For every ORF, group assignment was done based on the top-scoring profile with at least > 80% of sequence coverage. Sequences were aligned against the representative ones and manually inspected to remove false positives. The same procedure was employed against the mMGE and GPD databases (44,51). Further, phage prediction using the genomic neighborhood information was performed using the upstream and downstream nucleotide sequences (±10 kb) of every representative sequence as a query for the metasoftware WhatThePhage (52). Only sequences predicted to be encoded in phages/prophages by at least three different tools included in WhatThePhage were labeled as so.
Analysis of viral genomes harboring UG27: annotation, taxonomy assignment and host prediction
To search for UG27 systems in viral genomes, three different viral databases, IMG/VR (53), GPD (51), and mMGE (44) were used. First, searches with HMM profiles corresponding to UG27 RT, cluster 337, and cluster 346 were done using an E-value cut-off lower than 1e–20 against the viral proteins. Only genomes harboring RT, cluster 337, and cluster 346 were further considered, which rendered 3861 predicted viral genomes. Genomes were dereplicated at 95% sequence identity using dRep with default parameters as previously described (54), resulting in 1447 distinct genomes. In addition, dereplicated viral genomes were searched for Large Terminases (TerL) as a viral marker using the PFAM profiles PF03354, PF04466, PF03237 and PF05876 and a profile built from an alignment of TerL derived from a recent phylogenetic analysis (54). To identify these putative viral sequences and assign taxonomy, whole-genome comparisons and gene-content networks were made using novel phage families (54–56) as references. For whole-genome comparisons, fastANI (57) with parameter –fragLen 500 and default parameters were used. For gene-content networks, genes were called using a modified version of Prodigal (34) to allow for amber (TAG) or opal (TGA) stop codon reassignment (56) in those genomes with the corresponding suppressor tRNAs searched using tRNA-scan-SE (58), and/or an increase in the reassigned-coding density above 10%. After this, vContact2 (59) was used with the –db ‘None’ option and default parameter. To increase the resolution at the gene sharing level, an aggregate protein similarity tree was built as described earlier (55). To obtain family-level viral clades, the tree was rooted and cut at the levels reproducing the six proposed Crassvirales families (56). Finally, viral genomes harboring UG27 systems were annotated using Prokka (60) and the Phrog database (61) of viral orthologous proteins. GFF files produced by Prokka were then imported to R and depicted using gggenes (62) colored by Phrog functional categories (61). Host taxonomy prediction of UG27 viral genomes was done by performing CRISPR spacers comparisons against a reference database using CrisprOpenDB (63). As most of the taxonomy predictions pointed out towards bacteria present in the gut microbiome, viral genomes were compared against recent human gut metagenomic spacers database (64) and a custom collection of spacers found in CRISPR arrays from the human gut microbiome; compiled by running CRISPRCasFinder (65) on all metagenomic contigs from the HMP2-IBD database (66). For the comparison, BLASTn (67) with -task ‘blastn-short’ option was used, and only hits with ≤2 mismatches and >95% sequence identity were kept.
UG28 system RNAseq
E. coli K-12 (ATCC25404) containing a plasmid encoding RT (UG28) was grown to saturation at 37°C in terrific broth in the presence of 25 μg/ml chloramphenicol. RNA was extracted using TRIzol (Thermo Fisher Scientific) and purified with a Direct-zol RNA MiniPrep Plus kit (Zymo). The purified RNA was then treated with 20 units of T4 polynucleotide kinase (NEB) for 3 h at 37°C. Following column purification, the sample was treated with 20 units of 5′ RNA polyphosphatase (Lucigen) for 30 min at 37°C. After an additional round of column purification, the RNA sample was used as input for an NEBNext Small RNA Library Prep for Illumina kit (NEB). Barcoded amplicons were sequenced on a MiSeq (Illumina) with 200 cycles for the forward read. Adaptors were trimmed using CutAdapt (68) with parameters –trim-n -q 20 -m 20 -a AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC, and trimmed reads were aligned to the reference plasmid sequence using Geneious.
Structured ncRNAs prediction
Paired RNAseq reads from SRX814863 were downloaded from the European Nucleotide Archive (www.ebi.ac.uk) and mapped to the RT (UG2) locus from Klebsiella pneumoniae ST23 (nucleotide accession number CP037742.1, corresponding to 100% nucleotide identity relative to the RNAseq reads) (Supplementary Figure S5) (69). Conserved structured ncRNA prediction was done on UG2 (DRT type 2) and UG3 + UG8 (DRT type 3) UG/Abi groups. Due to the existence of a constant intergenic region upstream of the RT, and in line with the RNA-seq data, for UG2 members, nucleotide sequences 300 bp upstream of the RT were retrieved. However, for UG3 + UG8, nucleotide sequences 300 bp downstream of UG8 RTs were retrieved, as this region was previously described (21) to encode a ncRNA. Using the information derived from the clustering and phylogenetic and taxonomic criteria, we divided UG2 and UG8 putative ncRNA sequences into subgroups. These subgroups were aligned using MAFFT-qinsi (28) and further examined in search of structure conservation in the neighborhood using CMfinder04 (70) and R-scape (71), as described previously (36). In groups of UG8 belonging to Terrabacteria phylum, no downstream structure or sequence conservation was observed, and predictions were made retrieving sequences 300 bp upstream instead, which showed broader structure and sequence conservation. Sequence logos for UG8 motifs were created using WebLogo 3 (72). For the prediction of structured RNAs in UG27 systems, the intergenic sequences located between UG27 RTs and cluster 336 were retrieved in representative sequences. Conserved RNAs structures were predicted on these sequences using CMfinder04 (70). Then, covariance models were built using cmbuild from the Infernal suite (73) and were compared against UG27 viral genomes described in the previous section using cmsearch from the same suite. The result of this search was then evaluated for statistically significant co-varying base pairs using R-scape (71).
RESULTS AND DISCUSSION
Expansion and classification of UG/Abi RTs
Recent phylogenetic analysis of prokaryotic reverse transcriptases revealed that UG/Abi RTs constituted a significant proportion (11%) of the whole RT landscape with 991 representative sequences (clustered at 85% of sequence identity) (5). By searching the NCBI public databases on complete or partial genomes and performing phylogenetic analyses of the most informative sequences (Methods), we could expand the number of UG/Abi RTs from 991 up to 5022 representative members, which represents a ∼5-fold increase compared to previously described UG/Abi RTs. Of these 5022 representative sequences, 325 belong to previously undescribed groups (Figure 1 and Supplementary Table S1). It is worth mentioning that by following this methodology, we did not obtain any representative sequence of the UG11 group, consistent with the fact that recent analyses (5,74) highlighted that UG11 does not belong to the UG/Abi lineage, but to the retron lineage. To investigate the relationships between these novel sequences, we performed a phylogenetic reconstruction of the RT domain present in all UG/Abi RT representative sequences, increasing from 28 to 39 the number of well-supported UG RT groups and revealing the presence of three main clades (Figure 1).
Figure 1.

Phylogenetic tree of 5022 representative UG/Abi RT sequences. Alignment of the RT domains 1–7 was used as an input for FastTree (Methods). For visualization, midpoint rooting was applied using FigTree, and clades were collapsed to the UG/Abi group level according to the presence of reference sequences showing a FastTree support > 0.70 (Supplementary File S1) with UFBoot and SH-aLRT support values in the IQ-TREE (not shown) >90%. Due to the presence of reference sequence in two groups, UG13 UG/Abi group was subdivided (UG13a and UG13b) according to phylogenetic criteria and protein length distribution. Numbers in brackets and segments sizes represent the absolute number of representative sequences in each group. Branches corresponding o UG/Abi Classes 1, 2 and 3 are colored in brick red, turquoise, and olive, respectively. The FastTree phylogenetic tree and Multiple Sequence Alignment of the RT domain can be found in NEXUS format as Supplementary File S1.
Of these groups, UG29, UG32, UG35, UG37 and UG38 are entirely new, as they do not contain close homologs present in a previous large RT dataset (5). Other novel groups such as UG30, UG31, UG34 and UG36 encompass sequences that remained unclassified in the referred dataset. Finally, some previously described groups have experienced a great expansion in the number of available sequences and have been redefined. This is the example of UG23 (some sequences present in this group have clustered within the UG33 group), UG13 (subdivided in UG13a and UG13b) or UG39 (now including 15 sequences previously clustered within UG28). Some of these changes were already anticipated by Sharifi and Ye (74), who also pointed out that the 15 sequences within UG28 constituted a new clade that was named UG28b. However, evolutionary analyses indicate that this group is phylogenetically independent and is specifically associated with the protein cluster 64 (VirE + PriCT-2 domains). Given the two lines of evidence, we decided to name this group UG39.
We then performed an exhaustive domain annotation of every UG/Abi group based on profile-profile searches and structural predictions, and a systematic analysis of the neighborhood of UG/Abi RTs in search of putative neighbor associated genes that were grouped into clusters (Figure 2, Supplementary Table S2 and Supplementary File S4). This combined approach revealed that most of the UG/Abi RT groups (40 out of 42, including AbiA, AbiK and AbiP2) are fused or functionally related to other genes or domains, thus disclosing a wide variety of genetic systems that were previously unnoticed. Besides, the annotation of domains, RT phylogeny, and prediction of associated genes made it possible to group UG/Abi RTs into three main classes described in the following sections (see Figures 1 and 2).
Figure 2.

Graphical summary and classification of the various UG/Abi RT systems. Genes are represented as arrows of size proportional to their length. For clarity, the UG3 + UG8 system is included within Class 1, but notice that the UG3 RT is phylogenetically placed within Class 2. The identified domains are represented in different colors. The NCBI Protein and Nucleotide accession IDs of chosen representative sequences are shown to the left. Numbers inside genes indicate the cluster to which they belong, and the names above indicate domain annotation. Genes with dotted lines represent genes that are occasionally present. Vertical dashed lines are represented to be 600 nucleotides (200 aminoacids) apart. Shadows between genes of different variants indicate similar domains. Icons on the left of each group's name represent those that have been experimentally validated as defense systems and those that are considered to be novel. Predicted ncRNAs are indicated by dots with differentiated colors according to the group.
The representative dataset reveals that UG/Abi RTs predominate in Bacteria with only a few examples found in Archaea where they may have arrived by horizontal gene transfer. In Bacteria, these RTs are well distributed in Proteobacteria, Bacteroidetes, Actinobacteria, and Firmicutes as the main phyla with some groups mostly restricted to a particular phylum, (Supplementary Table S1 and Supplementary Figure S1) suggesting host-associated functional dependence.
Class 1 UG/Abi RTs are fused to repeat-containing domains
Profile-profile searches of UG/Abi RTs against the COG database revealed that C-terminal regions of UG20, UG21, and UG23 members share remote homology with proteins containing HEAT-like repeats (Supplementary Table S2). To further investigate this feature, we performed structure predictions using trRosetta (45) and multiple sequence alignments (MSAs) of every UG/Abi RT group (see Methods). Interestingly, we detected alpha-helix repeats at the C-terminal regions of sequences belonging to a large clade comprising up to 24 UG/Abi RT groups, proposed to be denoted as Class 1 UG/Abi RTs. Despite the lack of overall detectable sequence similarity, structural alignments of those models highlighted a common structural motif consisting of a variable number of alpha-helix HEAT-like repeats (Figure 3) that will be named hereafter as αRep domain.
Figure 3.

(A) Multiple structural alignment of predicted protein structures of Class 1 UG/Abi RTs C-terminal regions visualized using using cealign feature from Pymol and visualized using PyMol2Rainbow rainbow spectrum. (B) Heatmap of the pairwise TM-scores obtained using the mTM-align algorithm. The file containing the multiple structural alignment can be found as Supplementary File S3.
Further analyses revealed that other members of this Class are also fused to putative methylase (UG25, UG35), protease (UG9), primase (UG10), HTH (UG32), HEPN (AbiA), or unknown domains (UG10, UG19) (Supplementary Table S2), which points out a huge diversity in these systems, both at the mechanistic and biological level. Those fused domains can be sometimes encoded in a separate open reading frame (ORF) or absent, which allows a further division of some groups (UG10, UG19, UG9 and AbiA) into subgroups consisting of large (fused) or small variants (unfused) (Figure 2 and Materials and Methods). This phenomenon has been previously described for other RT-containing systems such as retrons, in which effector domains can be found both fused or adjacent to the RT (36). Genes in a separated ORF were also recovered by the gene-neighborhood systematic analysis (Methods), indicating that Class 1 UG/Abi RT groups constitute multi-domain systems, either encoded in a single ORF, or associated with other co-located genes.
The only characterized multi-gene system involving a Class 1 UG/Abi RTs is constituted by the association of UG8 with UG3, a Class 2 UG/Abi RT (see below, and Figures 1 and 2). These two genes constitute DRT type 3, which has been described to provide immunity against certain phages (21). Other uncharacterized multi-gene systems include genes with HEPN (UG22), NUDIX (UG23), SLATT (UG23, AbiA), UvrD helicase (UG35) domains, which are commonly found as part of prokaryotic defense systems such as CRISPR-Cas, retrons, RADAR (restriction by an adenosine deaminase acting on RNA), CBASS (Cyclic-oligonucleotide-based anti-phage signaling systems), Gabija, Abi or toxin-antitoxin pairs (18,21,22,36,75,76). On the other hand, as previously described, UG9 members are associated with a Family A DNA-polymerase (7), whereas a previously undescribed UG9 variant is associated with an MBL-fold metallohydrolase and a trypsin-like serine protease domain. Finally, the rest of the UG/Abi groups are also associated with unknown genes (UG10, UG25) or function as multi-domain stand-alone proteins, some of which (UG15, UG16, AbiK, AbiP2) have been described to provide immune functions (15,21).
Class 1 UG/Abi multi-gene systems including hydrolases
Some Class 1 UG/Abi groups are predicted to be associated or fused to enzymes that belong to the class of hydrolases (EC 3), such as nucleases, proteases, or helicases.
UG22 is predicted to be associated with neighbor protein cluster 79, which shares remote homology with HEPN domains (Figure 2 and Supplementary Table S2), a family of RNA-binding proteins that commonly function as endoribonucleases (77). This domain is also found at the C-terminal region of AbiA-large, which is thought to mediate immunity through phage RNA degradation or cell-dormancy induction. HEPN domains are commonly enriched in prokaryotic defense islands (78) and have been described as essential components of other defense systems/genes such as CRISPR-Cas, different Abi, and toxin-antitoxin systems, and ApeA (18,21). They also constitute type IX retrons together with a retron RT, a non-coding RNA, and a winged-helix domain-containing protein (36), which points out that RT and HEPN domains may have been co-opted several times across evolution to perform immune functions. Likewise, UG19 is associated with an unknown domain, either fused at the C-terminal region (UG19-large) or encoded in a co-located gene belonging to cluster 1465 (UG19-small) that shares very distant homology with HEPN domains (Supplementary Table 2). Other UG/Abi systems associated with nucleases include UG7 and UG34 (Figure 2). UG7 can be divided into two phylogeny-congruent variants. The first variant (150 out of 385 members) is found to be associated with cluster 17, which shares remote homology with PD-(D/E)XK nucleases, a highly ubiquitous superfamily of nucleases related to Holliday junction resolvases (79). However, the second variant (165 out of 358) does not appear associated with any gene, suggesting a likely accessory function of these nuclease-like proteins or the need for other nucleases that may act in trans. On the other hand, some (7 out of 17) UG34 members are co-located with cluster 485, which contains a C-terminal helicase domain and an N-terminal nuclease-related (NERD) domain. In addition to their widespread distribution, both PD-(D/E)XK and NERD domains are enriched in prokaryotic defense islands (78). More specifically, PD-(D/E)XK-like domains are found in CRISPR-Cas and R-M systems nucleases, whereas it has been recently described that NERD nucleases can be found associated with prokaryotic Viperins (pVips) (80).
Other Class 1 UG/Abi groups are associated with hydrolases that are present in previously described defense systems. UG23 members are predicted to be associated with cluster 350, which encodes a SMODS (Second Messenger Oligonucleotide or Dinucleotide Synthetase)-associated protein containing two transmembrane helices at the N-terminal region and a C-terminal NUDIX (Nucleoside diphosphate linked moiety X) hydrolase domain (Figure 2), which is thought to cleave nucleoside diphosphate molecules (81). This protein is known for being an effector protein of CBASS systems (76,81). Similar to Abi systems, CBASS possesses a sensor module that detects phage presence. In this case, it produces a cyclic-oligonucleotide signal and an effector module that subsequently recognizes this signal and triggers programmed cell death. Interestingly, in the case of AbiA and UG23, when the effector domain is absent (HEPN at the C-terminal region of AbiA-large or NUDIX hydrolase co-located with UG23) they are replaced by a SLATT protein, which is also sometimes co-located with the aforementioned UG3 + UG8 system. The SLATT proteins contain 2–3 transmembrane helices and are predicted to function as pore-forming effector proteins associated with defense and conflict systems such as CBASS (76,81) and have been recently described to be part of novel defense systems such as RADAR, in which their accessory role contributes to increasing the range of defense against different phages (21). These findings highlight the modularity of the different domains within these systems, being the sensor or effector domains easily replaceable by others with similar functions. Although associated with a wide variety of domains, the factors limiting the capacity of Class 1 UG/Abi RT and αRep domains to perform specific biological functions are yet to be explored, and it is possible that, although they are often associated with other genes or domains, these may have an accessory role, as in the case of the SLATT proteins in RADAR or DRT type 3 systems.
Class 1 UG/Abi systems associated with transferases
Whereas some Class 1 UG/Abi multi-gene systems include hydrolases as putative effector proteins or domains, some other UG/Abi RTs are associated with enzymatic activities belonging to the class of transferases (EC 2), such as nucleotidyltransferases (DNA polymerases and DNA primases) or methyltransferases (also known as methylases).
UG9 RTs function together with a Family-A DNA polymerase (PolA, cluster 58) (7) and can be divided into two different variants. The first, less frequent variant (19 out of 79 members) is associated with hydrolases, with the RT being fused to an N-terminal putative trypsin-like serine protease and an MBL-fold metallohydrolase found upstream (Figure 2). This architecture is reminiscent of Avs1 defense systems, in which the protease is fused at the N-terminal region of a protein containing a STAND ATPase domain and the MBL gene is also located upstream (21). The second UG9 variant (60 out of 79) is not associated with an MBL-fold metallohydrolase and the RT does not harbor an N-terminal protease domain, thus pointing out that both the metallohydrolase and protease domains would operate together. In this way, the MBL/protease pair may function as an accessory or effector module of different defense systems, thus being frequently exchanged among different antiphage systems in a similar way to NUDIX hydrolases or SLATT proteins.
UG10 RTs are very large proteins (>800aa) that also associate with replication-related domains, as they harbor an N-terminal PriS domain (COG4951) that belongs to the AEP (archaeo-eukaryotic primase) superfamily (82,83). The fusion of AEP primases to RTs has been previously described as part of defense systems such as CRISPR-Cas (5) or Class 3 UG/Abi member DRT type 1 (21) and suggests that similar to HEPN domains, both the RT and primase domains may have been co-opted together several times across evolution to perform immune functions. In addition to the N-terminal PriS domain, UG10 members are associated with an unknown domain with ∼800aa that can be either fused to the C-terminal region (UG10-large) or encoded in a separate CDS that belongs to cluster 1038 (UG10-small).
Finally, both UG35 and UG25 RTs harbor an N-terminal methyltransferase domain. To our knowledge, this is the first reported case of a predicted methylase-RT fusion in prokaryotes, although the association of methylase and other bacterial RTs has been previously described (88). Apart from being fused to the methylase domain, UG35 RTs are associated with cluster 22, which is identified as a putative UvrD-like helicase. However, UG25 members are not associated with a helicase but with cluster 579, a cluster containing a DUF1848 domain of unknown function.
Class 2 UG/Abi RTs do not harbor any additional known domain
While Class 1 UG/Abi RTs are fused to αRep and other known domains, Class 2 UG/Abi RTs do not bear any additional known domain. Instead, the 9 different groups belonging to this class harbor the RT domain and are sometimes associated with other gene(s) with DNA-binding or DNA-modifying activities that seem to play an accessory role. Although their C-terminal domains do not appear to have a conserved sequence or predicted structure, further genetics, and structural investigations can make it possible to reveal their possible shared characteristics.
UG2 is the largest group and it can be classified into three different variants (Figure 2). The first variant is a single-gene UG2 RT, whereas the second and third variants are co-located with genes (clusters 315 and 103, respectively, see Supplementary Table S2) that share an N-terminal YodC (COG5475) domain. Besides YodC, cluster 103 presents a WYL-like domain at the C-terminus. Although very little is known about YodC domains, the WYL domain is known to be a group of diverse transcription factors that can regulate a response when binding to RNA molecules (84). This domain is enriched in defense islands (21) and it has been proved to regulate some CRISPR/Cas systems and other defense systems such as the abortive infection AbiG system (84,85). Some of the second variant members have another associated cluster in the neighborhood (cluster 433 in Supplementary Table S2), which contains an N-terminal TM (transmembrane) domain followed by a COG1322 domain (RmuC DNA anti-recombination protein) with a restriction endonuclease-like fold and coiled-coil regions (79) (Supplementary Table S2). For UG2, it has been demonstrated that the sole presence of the RT domain (along with an ncRNA) is sufficient to confer anti-phage functions, thus suggesting that in this case, the associated clusters may have an accessory role.
Other UG/Abi systems possibly associated with accessory proteins include UG28 and UG26. UG28 is sometimes (7 out of 124 members) co-located with cluster 163 which is identified as a SLATT protein, whereas UG26 can be co-located (9 out of 35 members) with cluster 1531, which is a COG2852 domain-containing protein annotated as YcjD, a Very-short-patch-repair endonuclease (Figure 2 and Supplementary Table S2) commonly associated with DNA repair mechanisms and linked to PD-(D/E)XK nucleases (19). RTs belonging to the UG39 group are predicted to be associated with the protein cluster 64 (Figure 2) which contains an N-terminal VirE domain and a C-terminal PriCT-2 domain (Supplementary Table S2). Both domains are commonly found in PrimPol proteins (primase-polymerases) from the AEP superfamily (82,83).
UG4 RTs were previously described to be fused to fimbrial domains (6). In this case, we found a subgroup of proteins from the UG4 group widely associated with cluster 90, cluster 107, cluster 114, cluster 121, cluster 231, and cluster 573 which are predicted to be fimbrial proteins (Figure 2). However, we were unable to find fimbrial domains fused to any of the representative RTs classified as UG4 in our dataset, suggesting that previous identification of the fimbrial domain in UG4 RTs may be due to an inaccurate prediction of the ORF boundaries (6).
RTs from the UG27 group are putatively associated with clusters 346 and 337. Profile-profile searches revealed that none of these clusters showed homology to known domains. This suggests that they may represent novel gene families with unknown functions. The same happens with UG18, which is sometimes co-located (6 out of 10 members) with cluster 2007 that does not harbor any identifiable domain. Nevertheless, the validity of UG18 RTs, similar to UG38, cannot be fully tested, since both of them are small and infrequent groups present in partial genomes from metagenomic sources. Whereas UG18 is harbored by Gammaproteobacteria species (Supplementary Table S1) UG38 members are present in Parcubacteria, Nealsonbacteriaand Atribacteriaspecies,which belong to the Candidate Phyla Radiation (CPR), a recently described group of mostly unculturable nanobacteria that constitute a novel lineage with extremely reduced genomic repertoires due to their predicted symbiotic and/or parasitic lifestyles (86). Because of the nature of their genomic sources, both UG18 and UG38 UG/Abi groups could be significantly expanded by performing exhaustive metagenomic searches.
Finally, reverse transcriptases belonging to UG17 are strongly associated with clusters 7, 163 and 158 which are identified as SLATT (SMODS and SLOG-associating 2TM) proteins. Experimental approaches (87) have described that, under certain conditions, UG17 system (H120-RT + SLATT) influence SbcB (exodeoxyribonuclease I) essentiality in some E. coli strains, as an insertion within the RT abrogates SbcB essentiality. This implies that the presence of UG17 makes SbcB essential, thus denoting that UG17 systems may have a toxic effect possibly mediated by the SLATT gene. Similar to Class 1 UG/Abi RTs, different Class 2 UG/Abi RT groups are associated with SLATT proteins. As described above, these proteins are present in different scenarios, having an effector role (UG17 and CBASS), an accessory role (UG3 + UG8, UG28, RADAR), or an effector-replacement role (AbiA, UG23). In a context of high evolutionary pressure and gene turnover, the gap left behind by outgoing effector modules may be filled with the recruitment of highly mobile and adaptable SLATT proteins that can eventually lead to a long-lasting stable association such as UG17.
Class 3 UG/Abi RTs are associated with (phospho)hydrolase domains
Finally, Class 3 UG/Abi RTs are large proteins (generally larger than 1000 aa) with an RT domain associated or fused to C-N hydrolase (also known as nitrilase) or phosphohydrolase domains. UG1, UG5 and UG29 RTs share a C-terminal C-N hydrolase domain. In the case of UG1 (1200–1300 aa), the characteristic C-N hydrolase domain is thought to be essential for their function, as mutations in this domain abrogate immunity against phages provided by DRT type 1 in Klebsiella pneumoniae (21). In addition, UG1 can be frequently (33 out of 175 members) found to be associated with cluster 85, which is identified as a transmembrane (TM) protein, and UG5 can be found in two different variants, the first of which (UG5-large) shares a similar domain architecture with UG1 with a fused C-terminal C-N hydrolase domain and a TM protein (cluster 85 and 1858, Supplementary Table S2) sometimes encoded downstream (31 out of 299 members). However, the second variant (UG5-small) presents the C–N hydrolase domain encoded in a separate ORF (cluster 249) and it is not co-located with a TM protein.
UG29 RTs are by far the largest proteins in our dataset, with almost 2500 amino acids. These proteins display a characteristic DnaG-type primase at the N-terminus, followed by an RT domain, and a C-terminal hydrolase domain. Although encoded in different CDS, UG6 presents the same hydrolase and DnaG-type primase domains in associated clusters 285 and 192 (respectively), thus indicating a possible common mechanism of action. However, in this case, UG6 also displays an unknown C-terminal domain that shares remote homology (Supplementary Table S2) with kinase domains but lacks the characteristic catalytic amino acids. DnaG-type primases, which contains a CHC2-type zinc-finger followed by a TOPRIM (Topoisomerase-primase) domain, were traditionally thought to be only involved in bacterial chromosome replication whereas AEP primases such as that found in Class 1 UG10 were thought to mediate the same mechanism in archaeal and eukaryotic species (83). Although functionally related, both the AEP and DnaG-type primases are evolutionarily and structurally distinct, revealing that the association of UG10 RTs to AEP primase is an event independent from the acquisition of DnaG-type primases by Class 3 UG/Abi RTs. This similarity of domains explains why members of UG29 have been previously mislabeled as UG10 (21).
On the other hand, UG24 members encode a predicted C-terminal phosphohydrolase domain (COG3294) instead of the above-mentioned hydrolase domain, thus pointing out that the enzymatic reaction that gives rise to the immunity could be carried out indistinctly by C–N hydrolase or phosphohydrolase domains. Furthermore, depending on the genes to which it is associated, UG24 can be divided into two variants, one of them associated with cluster 192 (the same DnaG-type primase found to be associated with UG6) and one of them associated with two genes of unknown function (clusters 1808, 1299).
Although the C-N hydrolase family generally performs cellular functions related to nucleotide metabolism and small molecule biosynthesis, the hydrolases found in Class 3 appear to be more closely related to each other and form a clade distinct from the other nitrilases, leaving open the possibility that their biological function is completely different and may be more related to immune functions (21). In addition to the hydrolase, phosphohydrolase, and primase domains, all these proteins share an extension of amino acids generally located between the RT domain and the respective C-terminal domains with unknown function. This domain could provide these large RTs with sufficient molecular flexibility to orchestrate the different enzymatic reactions catalyzed by their different domains and the associated genes.
Unclassified UG/Abi RTs
Other RTs belonging to the UG/Abi RT family, namely UG36 and UG37, remain unclassified due to the absence of characteristic domains or their phylogenetic positioning, although future expansions in number and diversity may clarify their classification. In the case of UG36, we found that it is associated with genes that present a methylase domain grouped indistinctly in clusters 169 or 2186 (Supplementary Table S2). The domain architecture of this system is reminiscent of UG25 and UG35 RTs, in which the methylase domain is fused to the N-terminal end of the RT-containing protein. Thus, UG36 and UG25/UG35 systems may share a common ancestor. However, there is no evidence that they present a C-terminal αRep or similar domain. Similar to the associations of HEPN or primase with RTs, the methylase domain has been associated with an RT at different points in evolution. A less tight link between RTs and methylases appears to occur in Lactococcus lactis, in which rRNA methylation acts as a mechanism to prevent retrotransposition of endogenous group II introns (88).
On the other side, UG37 corresponds to rvt elements. The rvt are single-copy genes of unknown function that, in addition to the RT domain, contain an N-terminal coiled-coil domain that is responsible for its multimerization in vitro (89). They can be found in all eukaryotic kingdoms and a few bacterial genomes and, although they show a patchy distribution, they are not components of retrotransposons or viruses (90). In vitro studies suggest that rvt are the first chromosomal RTs described to have a protein-priming DNA synthesis initiation mechanism, which is thought to be dependent on the unknown C-terminal domain and the RT catalytic domain itself (89). Protein-priming has been described also for Class 1 AbiK, where the C-terminal domain has been also hypothesized to play this role and it is essential for abortive phage infection (14).
High occurrence of UG/Abi RTs in defense Islands
As we highlighted in the above sections, it has been previously shown that different RT families are involved in defense against phages (4). Recently, some UG/Abi RT groups have been demonstrated to function as defense systems named as DRTs and are located within defense islands (21,78). Besides AbiK, AbiA, AbiP2, and DRTs, it was previously shown that UG1, UG5-large, UG29 (named as UG10 in (21)), UG7 and UG9 are also enriched in defense islands but their immune function has not been experimentally validated (21). Interestingly, a recent work (91) has revealed that P2 bacteriophages and P4-like satellites possess variable genomic regions encoding previously known anti-phage systems and 14 novel characterized defense systems, including a UG5-large system (RT-nitrilase + TM) found in E. coli E101. Within these variable genomic regions, we also found, in addition to UG5-large, type II-A1 and II-A2 retrons (36) and RTs from UG/Abi groups such as UG7, UG3 + UG8 (DRT type 3), UG15 (DRT type 4) and UG17 (Supplementary Table S4), thus suggesting that UG7 and UG17 may also provide immune functions.
In the case of UG5-large, the associated cluster 1858 (TM protein) is necessary for the system to perform immune functions (91). However, previous attempts to describe these immune functions did not take into account this protein and the same may occur in other groups in which associated genes were overlooked. Particularly, this seems to occur also in the case of UG7, in which the tested sequence falls into the variant that is associated with a PD-(D/E)XK nuclease (cluster 17).
Given that many of the UG/Abi RT groups have members that have been validated as defense systems or have been described to be enriched in defense islands, we were interested to know if all UG/Abi RTs are predicted defense genes. To investigate their defense association, we searched for the presence of previously reported defense genes in the vicinity of homologs from each RT group (Materials and Methods). We observed that the majority of UG/Abi RT groups are indeed enriched in known or predicted defense islands and are likely to perform immune functions (Supplementary Table 3). Of the 42 UG/Abi RT groups, members of at least ten groups (AbiA, AbiK, AbiP2, UG1, UG2, UG3, UG5, UG8, UG15 and UG16) have been experimentally shown to confer anti-phage defense activity (13–17,21,91), consistent with all of them having high defense association frequencies. Seven other groups (UG7, UG9, UG12, UG14, UG17, UG28 and UG29) also had strong defense enrichment, with 0.15 association frequency or greater across a minimum of 50 homologs; therefore, they are predicted as novel defense genes (Supplementary Table 3). The remaining groups had either somewhat lower defense association frequencies or fewer than 50 homologs. For these groups, individual genomic loci were examined for instances with strong defense association signatures (Methods and Supplementary Figure S2). Based on this evidence, 7 additional groups (UG10, UG13b, UG19, UG24, UG30, UG31 and UG36) were predicted to have a defense function.
No defense enrichment was detected for UG4 and UG27, consistent with their predicted non-defense function (see below section). On the other hand, defense predictions were inconclusive for the remaining 16 groups (UG6, UG13a, UG18, UG20, UG21, UG22, UG23, UG25, UG26, UG32, UG33, UG34, UG35, UG37, UG38 and UG39), thus requiring further investigation (e.g. search for more homologs or experimental observation).
To confirm that RTs with a high defense score were involved in defense, we heterologously reconstituted some candidate systems (UG12, UG10, UG7 and UG28, plus UG2 and UG15 as a positive control) in E. coli K-12 and challenged them with T2, T5 and ZL-19 coliphages following a similar procedure to that previously described (21). Phage sensitivity of the RT-containing bacteria was compared to that of an empty vector control vector by performing phage plaque assays (Methods). We observed anti-phage activity for UG12, UG10, UG7 and UG28 (Figure 4A). In particular, UG10 was found to be quite active against T2 and ZL-19 phages, whereas UG28 was quite active against T2 and T5 phages. On the other hand, UG7 offered robust protection against T5 phage, whilst UG12 showed a moderate immune effect against T5 and ZL-19 viruses. Following the previous nomenclature for UG/Abi RTs involved in defense (21), we propose to name UG12, UG10, UG7 and UG28 as DRT type 6, 7, 8 and 9 respectively (Figures 2 and 4A).
Figure 4.

(A) Phage plaque assays showing resistance of E. coli harboring plasmids with different UG/Abi RTs against coliphages T2, T5 and ZL-19. Variants with mutations in the catalytic domains of RT and/or effector proteins, as well as deletions of the C-terminal αRep in UG15 are also included. RT, reverse transcriptase; PrimS, primase. (B) Predicted consensus structure of ncRNAs found in UG28 systems.
In addition, we also proved the essentiality of certain components of these systems in the performance of the immune function. For UG2 and UG15 (DRT types 2 and 4), it was previously demonstrated that the mutation of the RT catalytic domain abrogates immunity (21). Here we demonstrate that the same occurs with UG12, UG10, UG7 and UG28 (DRTs type 6–9) (Figure 4A). Besides, the absence of the associated ncRNA in UG2 has the same effect, thus indicating that both the enzymatic action of RT and ncRNA are necessary for the immune function (Figure 4A). The same applies to UG28 (DRT type 9), in which both the ncRNA and the RT domain are needed for the defense phenotype (Figure 4A). Moreover, we also demonstrate that the Class 1 UG/Abi C-terminal αRep domain is required for immunity, as deletion of a small portion (38aa) from the C-terminus of UG15 abolishes immunity (Figure 4A). Other domains such as the PrimS found at the C-terminal region of UG10 RT (DRT type 7) or the PD-(D/E)XK domain found in the effector protein of UG7 (DRT type 8) are also required for the defense against phages (Figure 4A), as mutations in their active sites result in the loss of their immune capacity.
These results reveal that the UG/Abi RTs lineage constitutes a family of RTs largely involved in defense against viruses, as previously highlighted for other prokaryotic RT families such as retrons (21–24). In addition, the enormous diversity of domain architecture and associated genes suggests that the RT domain activity might require other enzymatic activities and/or domains to play an immune role, as we have demonstrated for αRep, PrimS and PD-(D/E)XK domains.
Presence of UG/Abi RTs in mobile genetic elements and viral genomes
It has been previously described that some UG/Abi RTs can be present in MGEs, including plasmids (AbiK and AbiA), prophages (AbiP2 and other groups in P2-like prophages), among others (17,91,92). In addition, UG15 members have been also found within the Helicobacter pylori accessory genome (HPSJM_07740 gene of H. pylori SJM180) (93). To find which UG/Abi RTs were encoded in MGEs, we analyzed the genomic context of every representative sequence in search of phage signatures using the metasoftware WhatThePhage (52) and searched MGE databases (50) with all prokaryotic RTs HMM profiles (Methods).
As a result, we obtained that some UG/Abi groups tend to localize in putative MGEs. Particularly notable is the case of UG27, most of whose members (15 out of 29) are classified as viral by WhatThePhage and whose presence in the GPD and mMGE databases is remarkable (more than 500 instances in each database) (44,51) (Supplementary Table S4). Other groups such as AbiP2, UG2, UG3 + UG8, UG5, UG7, UG12, UG15 and UG17 had several (from 4 to 15) representative sequences predicted to have viral origin according to WhatThePhage. Of them, AbiP2 and UG2 were found to be frequently encoded in plasmids too (48 and 16 instances in PLSDB, respectively), whereas AbiK and UG4 were mainly present only in plasmids (116 and 22 instances in PLSDB, respectively). This suggests that AbiP2 and UG2 are highly mobile across different MGEs, and points out that the lack of detection of UG4 and UG27 in defense islands may be due to the fact that they are located in plasmids and viruses, respectively. Both may have been recruited by MGEs due to their shared evolutionary dynamics (94) and may perform a distinct biological role, such as acting as an addiction module or as an anti-defense system.
To verify the presence of UG27 in viral genomes, we expanded the number of UG27 systems by searching on IMG/VR, GPD, and mMGE databases (44,51,53) obtaining 1447 dereplicated genomes harboring the complete system (781 of which also encode a TerL homolog, Supplementary Figure S3). In an attempt to assign taxonomy to these viral sequences or test whether they belong to a known family, we compared them against reference databases and a compendium of new phage families (54–56) using ANI (average nucleotide identity) and gene-content network information (Methods). At the ANI level, some UG27-containing viral genomes share similarities with recently proposed orders of bacterial viruses, including Crassviralesand Friedlandervirales(55) and viruses from the proposed Quimbyviridaefamily (54) (Supplementary Table S5). Most of the UG27-containing genomes however showed very distant to zero similarity to any currently classified bacteriophage family even at the protein level, so we constructed an aggregate protein similarity (APS) tree (55) to determine whether the phages belonged to known or novel viral families. After cutting the APS tree at the viral family level, by using the six proposed Crassvirales families (56) as references, some of the UG27 containing phages co-clustered with Quimbyviridae (54) and various families within the proposed Crassvirales, Twortvirales and Freidlandervirales viral orders (55) (Supplementary Table S6). Four main clusters (205, 206, 255 and 265) accounted for 1350 out of 1447 UG27-containing genomes. Of these, family cluster 255 was co-clustered with genomes belonging to diverse families within the Friedlandervirales order, whereas family cluster 265 was co-clustered with members of the Sylversterivirdae family belonging to the Crassvirales order. On the other hand, half of the UG27-containing phage genomes were found within three separate family-level clusters (205, 206, and 291) that likely represent novel viral families (Figure 5). The largest of the clusters, with 444 members was more expansive than any other known dsDNA viral family data, whereas the second largest, at 252 members was around the same size as the largest crass-like family δ-crassviridae. The gene content of those clusters (Supplementary Figures S4 and S5) was characteristic of Caudoviricetes class head-tail bacteriophages. Despite the predominance of these two family-level clades in gut metagenomic data worldwide, the families appear to have gone unnoticed so far, and we propose naming them Astarteviridae and Habisviridae, respectively. (Families 206 and 205 in Figure 5, Supplementary Table S6 and Supplementary Figures S4 and S5). Given the low number of members of viral cluster 291 (14 sequences), we did not include this group as a new family, although a future increase in the number of viral genomes from metagenomes could help to resolve this issue.
Figure 5.

Gene-sharing network of UG27-containing viral genomes dereplicated at 95% and similar viruses found in reference databases (Materials and Methods). Nodes represent viral genomes and edges connect genomes with similar gene content. Edge opacity is proportional to vContact2 weights which represent the significance of the relationships. Node colors indicate the family to which every genome belongs based on the APS-tree, and circle color shades around nodes represent the four major families found. Nodes with no connections to UG27-containing nodes were removed to improve the visualization.
According to spacer-based host taxonomy assignment and viral genomes metadata (Materials and Methods), we obtained that most of the predicted hosts of these viral genomes belong to the Bacteroidetes and Firmicutes phyla (Supplementary Table S7), within species highly prevalent in human gut microbiomes, which could be influenced by the origin of the data. Next, we tried to infer whether these putative phages were predicted to be active or not by comparing them with databases of CRISPR spacers derived from human gut metagenomes. To this end, we used a publicly available collection of spacers extracted from 11 817 human gut metagenome datasets (64) and an in-house spacer database that was built by running CRISPRCasFinder (65) on the Integrative Human Microbiome Project – Inflammatory Bowel Disease metagenomic dataset (66) (Methods). By doing this, we obtained that 1346 out of 1447 dereplicated viral genomes harboring UG27 (93,02%) were targeted by at least 1 spacer, with 1197 (82%) being targeted by five or more spacers, suggesting a recent active role of these viral genomes in their natural environment. We also noticed that, in some cases, spacers targeted the intergenic sequence located between UG27 RT and the associated cluster 336, highlighting a possible functional role of this non-coding region (Supplementary Figure S6).
Group-specific structure of ncRNAs in DRTs and UG27
The ncRNA present at the 3′ end of the UG3 + UG8 system has been described to be essential for immune functions (21). Since most UG2 and UG28 homologs are associated with a sizable predicted non-coding sequence in the genomic DNA, located at the 5′ end of the RT ORF, we hypothesized that these sequences also encode for an ncRNA. To investigate the presence of UG2 and UG28-associated ncRNAs, we paired RNAseq reads to UG2 and UG28 (Materials and Methods) systems. This data revealed highly-expressed ncRNA at the 5′ end of both locus with read coverage >1000-fold greater than that of their respective RT mRNA (Supplementary Figure S7)
After this, we predicted conserved secondary structures in the vicinity of UG2, UG8 and UG28 using a methodology similar to the one previously used to uncover some ncRNA present in retrons (36). Due to low sequence and structure conservation, UG2 and UG8 upstream/downstream sequences were grouped into 13 and 20 groups, respectively, according to the RT phylogeny and taxonomic criteria (Supplementary Figures S8 and S9). For UG28, however, the high degree of conservation allowed all sequences to be grouped into a single cluster. Then, consensus RNA structure predictions were made on every group.
Overall, some UG2 upstream sequence groups showed evident secondary structure conservation, whereas we were unable to detect consensus structures in some others, possibly due to a great divergence both at the sequence and the structure level and the low number of homologs (Supplementary Figures S8 and S10). Generally, the predicted secondary structures in predicted UG2 ncRNA greatly vary among the different groups, and there are no clear signs of inter-group sequence conservation, thus suggesting that the interaction between the UG2 RTs and the predicted ncRNAs may be highly specific.
On the other hand, the RNA structure conservation is clearer in UG8, yet maintains a high divergence at the sequence level. Furthermore, there is a clear distinction into two groups within UG8; those with the canonical UG3-UG8-ncRNA architecture in Proteobacteria and Firmicutes, and those present in Terrabacteria, which have a different architecture (UG3 and UG8 frequently encoded in opposite strands and the ncRNA upstream of UG8) (Supplementary Figure S9). In the first case (groups 1–17), the existence of a ‘motif’ that could be related to the function of the RNA is evident. This ‘CACACA’-like motif is globally conserved among these groups and it seems to be exposed in the RNA structure predictions, but the distance to the end of the RT varies greatly (Supplementary Figures S11 and S12). We speculate that this motif could be a recognition/attachment motif or a binding site that promotes the association of the UG8 RT. Although all of them share this motif, there are no signs of inter-group sequence similarity in other regions, highlighting that the importance of this possible ncRNA may be more dependent on its structure than in its sequence.
In contrast to the ncRNAs in UG2 and UG8, conservation at both the structural and sequence level in the predicted ncRNAs for UG28 was surprisingly high (Figure 4B), suggesting that the response mechanism of this particular system might be much more conserved and nonspecific.
Furthermore, we also analyzed the intergenic region located between UG27 RTs and cluster 336 in search of structurally-conserved ncRNAs revealing the presence of a putative conserved ncRNA (Supplementary Figure S6) with three long stem-loops that may be relevant for the functioning of UG27 systems within viral genomes. Due to the nature of the data used for the prediction (mostly coming from metagenomic sources and viral genomes predictions), the existence of this ncRNA should be validated experimentally, although it highlights its importance and the possible requirement for such a system to be functional.
The presence of ncRNAs in Class 1 (UG3 + UG8) and Class 2 (UG2 and UG27) UG/Abi systems highlights the possible existence of non-coding RNAs in other groups of UG/Abi groups. In the case of UG3 + UG8 (DRT type 3), ncRNA plays an indispensable role in achieving immunity against viruses, suggesting that their specific interaction with the RT may be tightly regulated and is responsible for triggering a signal or generating modifications such that the host can cope with a bacteriophage invasion by a yet unknown biological mechanism.
CONCLUSION
Through computational methodologies, in this work we have proposed the existence of a new family of RTs, hereafter proposed to be named UG/Abi RTs. By expanding the number and diversity of these RTs, we attempted to clarify their relationships and confirm their evolutionary and biological similarities. In addition, we built a phylogeny-congruent categorization of UG/Abi RTs in three major Classes, and we have experimentally demonstrated the defensive function of four new groups grouped into Class 1 and Class 2 UG/Abi RTs.
In the context of UG/Abi RTs, the phylogeny of the RTs correlates well with the presence/absence of fused or associated modules, which suggest some degree of co-evolution and a functional limitation to operate with non-specific modules. That notwithstanding, in the highly dynamic evolutionary context of defense systems, these barriers are easily overcome and exchanges of modules/genes between different systems can be observed. The promiscuity of UG/Abi RTs in associating with different effector modules reveal their vast diversity as well as the plasticity of these systems, which likely expands their target molecules and anti-phage actions, possibly compatible with other defense systems, acting along and/or in coordination with some of them, as previously reported for some retrons that serve as a ‘second line of defense’ (22). However, even though different RT families (retrons, CRISPR-associated RTs, UG/Abi RTs) associate with the same type of proteins (HEPN, SLATT, Primases), the way in which they operate and their evolutionary origins can be completely different. This suggests that the role of UG/Abi RTs may lie not only in the domains they contain or the genes to which they are associated but in the specific cooperation of these components in their given biological context and against specific signals that are yet to be determined.
Although there are still many other biological and mechanistic enigmas to be solved, the different UG/Abi RTs described in this work disclosed an enormous diversity of associated genes and domains. That along with the possibility of modifying non-coding RNAs and their involvement in defense functions could make the UG/Abi family RTs a prominent element in the phage-host arms race and a highly valuable source for the development of promising biotechnological and gene-editing tools.
Supplementary Material
ACKNOWLEDGEMENTS
We are specially thankful to Rafael Pinilla-Redondo for his careful review of the manuscript and his valuable contributions and discussions.
Contributor Information
Mario Rodríguez Mestre, Departamento de Bioquímica, Universidad Autónoma de Madrid (UAM) and Instituto de Investigaciones Biomédicas Alberto Sols (CSIC-UAM), Madrid, Spain.
Linyi Alex Gao, Howard Hughes Medical Institute, Cambridge, MA, USA; Broad Institute of MIT and Harvard, Massachusetts Institute of Technology, Cambridge, MA, USA; McGovern Institute for Brain Research, Massachusetts Institute of Technology, Cambridge, MA, USA; Society of Fellows, Harvard University, Cambridge, MA 02138, USA.
Shiraz A Shah, Copenhagen Prospective Studies on Asthma in Childhood, Copenhagen University Hospital, Herlev-Gentofte, Ledreborg Allé 34, DK-2820 Gentofte, Denmark.
Adrián López-Beltrán, Centro de Biotecnología y Genómica de Plantas, Universidad Politécnica de Madrid (UPM) – Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria (INIA), Madrid, Spain.
Alejandro González-Delgado, Department of Soil Microbiology and Symbiotic Systems, Estación Experimental del Zaidín, Consejo Superior de Investigaciones Científicas, Structure, Dynamics and Function of Rhizobacterial Genomes, Grupo de Ecología Genética de la Rizosfera, Spain.
Francisco Martínez-Abarca, Department of Soil Microbiology and Symbiotic Systems, Estación Experimental del Zaidín, Consejo Superior de Investigaciones Científicas, Structure, Dynamics and Function of Rhizobacterial Genomes, Grupo de Ecología Genética de la Rizosfera, Spain.
Jaime Iranzo, Centro de Biotecnología y Genómica de Plantas, Universidad Politécnica de Madrid (UPM) – Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria (INIA), Madrid, Spain; Institute for Biocomputation and Physics of Complex Systems (BIFI), University of Zaragoza, Zaragoza, Spain.
Modesto Redrejo-Rodríguez, Departamento de Bioquímica, Universidad Autónoma de Madrid (UAM) and Instituto de Investigaciones Biomédicas Alberto Sols (CSIC-UAM), Madrid, Spain.
Feng Zhang, Howard Hughes Medical Institute, Cambridge, MA, USA; Broad Institute of MIT and Harvard, Massachusetts Institute of Technology, Cambridge, MA, USA; McGovern Institute for Brain Research, Massachusetts Institute of Technology, Cambridge, MA, USA; Department of Brain and Cognitive Sciences, Massachusetts Institute of Technology, Cambridge, MA, USA; Department of Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA, USA.
Nicolás Toro, Department of Soil Microbiology and Symbiotic Systems, Estación Experimental del Zaidín, Consejo Superior de Investigaciones Científicas, Structure, Dynamics and Function of Rhizobacterial Genomes, Grupo de Ecología Genética de la Rizosfera, Spain.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
MCIN/AEI/10.13039/501100011033/FEDER ‘Una manera de hacer Europa’ [BIO2017-82244-P to N.T]; MCIN/AEI /10.13039/501100011033 [PID2020-113207GB-I00 to N.T and F.M.A.]; M.R.M. is supported by the FPI-UAM program from Universidad Autónoma de Madrid; L.G. is supported by the Society of Fellows of Harvard University; S.A.S. is a recipient of a Novo Nordisk Foundation project grant in basic bioscience [NNF18OC0052965]; A.L.B. is a recipient of FPI fellowship [PRE2020-092935] from Spanish Ministry of Science and Innovation; J.I. is supported by the ‘Ramón y Cajal’ program from the Spanish Ministry of Science [RYC-2017-22524]; Agencia Estatal de Investigación of Spain [PID2019-106618GA-I00]; ‘Severo Ochoa Program for Centres of Excellence in R&D’ from the Agencia Estatal de Investigación of Spain [SEV-2016–0672 (2017–2021) to the C.B.G.P.]; M.R.R. is funded by grants from the Spanish Ministry of Science, Innovation and Universities [PGC2018-093723-A-100, AEI/FEDER]; Fundación Ramón Areces (VirHostOmics); F.Z. is supported by NIH [1DP1-HL141201 and 2R01HG009761-05]; Howard Hughes Medical Institute; Open Philanthropy, the Edward Mallinckrodt, Jr. Foundation; Poitras Center for Psychiatric Disorders Research at MIT; Hock E. Tan and K. Lisa Yang Center for Autism Research at MIT; Yang-Tan Molecular Therapeutics Center at McGovern; Phillips family and J. and P. Poitras. Funding for open access charge: [PID2020-113207GB-I00] from the MCIN/AEI/10.13039/501100011033.
Conflict of interest statement. None declared.
REFERENCES
- 1. Temin HM., Mizutani S.. RNA-dependent DNA polymerase in virions of rous sarcoma virus. Nature. 1970; 226:1211–1213. [DOI] [PubMed] [Google Scholar]
- 2. Baltimore D. RNA-dependent DNA polymerase in virions of RNA tumour viruses. Nature. 1970; 226:1209–1211. [DOI] [PubMed] [Google Scholar]
- 3. Inouye M. The first demonstration of the existence of reverse transcriptases in bacteria. Gene. 2017; 597:76–77. [DOI] [PubMed] [Google Scholar]
- 4. González-Delgado A., Mestre MR., Martínez-Abarca F., Toro N.. Prokaryotic reverse transcriptases: from retroelements to specialized defense systems. FEMS Microbiol. Rev. 2021; 45:fuab025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Toro N., Martínez-Abarca F., Mestre MR., González-Delgado A.. Multiple origins of reverse transcriptases linked to CRISPR-Cas systems. RNA Biol. 2019; 16:1486–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zimmerly S., Wu L.. An unexplored diversity of reverse transcriptases in bacteria. Microbiol Spectr. 2015; 3:MDNA3–A0058–2014. [DOI] [PubMed] [Google Scholar]
- 7. Kojima K.K., Kanehisa M.. Systematic survey for novel types of prokaryotic retroelements based on gene neighborhood and protein architecture. Mol. Biol. Evol. 2008; 25:1395–1404. [DOI] [PubMed] [Google Scholar]
- 8. Simon DM., Zimmerly S.. A diversity of uncharacterized reverse transcriptases in bacteria. Nucleic Acids Res. 2008; 36:7219–7229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Toro N., Nisa-Martínez R.. Comprehensive phylogenetic analysis of bacterial reverse transcriptases. PLoS One. 2014; 9:e114083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lopatina A., Tal N., Sorek R.. Abortive infection: bacterial suicide as an antiviral immune strategy. Annu Rev Virol. 2020; 7:371–384. [DOI] [PubMed] [Google Scholar]
- 11. Chopin MC., Chopin A., Bidnenko E.. Phage abortive infection in lactococci: variations on a theme. Curr. Opin. Microbiol. 2005; 8:473–479. [DOI] [PubMed] [Google Scholar]
- 12. Isaev AB., Musharova OS., Severinov KV.. Microbial arsenal of antiviral defenses. Part iI. Biochemistry (Mosc). 2021; 86:449–470. [DOI] [PubMed] [Google Scholar]
- 13. Fortier LC., Bouchard JD., Moineau S.. Expression and site-directed mutagenesis of the lactococcal abortive phage infection protein abiK. J. Bacteriol. 2005; 187:3721–3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang C., Villion M., Semper C., Coros C., Moineau S., Zimmerly S.. A reverse transcriptase-related protein mediates phage resistance and polymerizes untemplated DNA in vitro. Nucleic Acids. Res. 2011; 39:7620–7629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tangney M., Fitzgerald G.F.. Effectiveness of the lactococcal abortive infection systems AbiA, AbiE, AbiF and AbiG against P335 type phages. FEMS Microbiol. Lett. 2002; 210:67–72. [DOI] [PubMed] [Google Scholar]
- 16. Dinsmore PK., Klaenhammer TR.. Molecular characterization of a genomic region in a lactococcus bacteriophage that is involved in its sensitivity to the phage defense mechanism abiA. J. Bacteriol. 1997; 179:2949–2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Odegrip R., Nilsson AS., Haggård-Ljungquist E.. Identification of a gene encoding a functional reverse transcriptase within a highly variable locus in the P2-like coliphages. J. Bacteriol. 2006; 188:1643–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Anantharaman V., Makarova KS., Burroughs AM., Koonin EV., Aravind L.. Comprehensive analysis of the HEPN superfamily: identification of novel roles in intra-genomic conflicts, defense, pathogenesis and RNA processing. Biol. Direct. 2013; 8:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Steczkiewicz K., Prestel E., Bidnenko E., Szczepankowska AK.. Expanding diversity of firmicutes single-strand annealing proteins: a putative role of bacteriophage-host arms race. Front. Microbiol. 2021; 12:644622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Emond E., Holler BJ., Boucher I., Vandenbergh PA., Vedamuthu ER., Kondo JK., Moineau S.. Phenotypic and genetic characterization of the bacteriophage abortive infection mechanism AbiK from lactococcus lactis. Appl. Environ. Microbiol. 1997; 63:1274–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gao L., Altae-Tran H., Böhning F., Makarova KS., Segel M., Schmid-Burgk JL., Koob J., Wolf YI., Koonin EV., Zhang F.. Diverse enzymatic activities mediate antiviral immunity in prokaryotes. Science. 2020; 369:1077–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Millman A., Bernheim A., Stokar-Avihail A., Fedorenko T., Voichek M., Leavitt A., Oppenheimer-Shaanan Y., Sorek R.. Bacterial retrons function in anti-phage defense. Cell. 2020; 183:1551–1561. [DOI] [PubMed] [Google Scholar]
- 23. Bobonis J., Mateus A., Pfalz B., Garcia-Santamarina S., Galardini M., Kobayashi C., Stein F., Savitski MM., Elfenbein JR., Andrews-Polymenis H.et al.. Bacterial retrons encode tripartite toxin/antitoxin systems. 2020; bioRxiv doi:22 June 2020, preprint: not peer reviewed 10.1101/2020.06.22.160168. [DOI]
- 24. Bobonis J., Mitosch K., Mateus A., Kritikos G., Elfenbein JR., Savitski MM., Andrews-Polymenis H., Typas A.. Phage proteins block and trigger retron toxin/antitoxin systems. 2020; bioRxiv doi:22 June 2020, preprint: not peer reviewed 10.1101/2020.06.22.160242. [DOI]
- 25. Eddy S.R. Accelerated profile HMM searches. PLoS Comput. Biol. 2011; 7:e1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sayers EW., Beck J., Brister JR., Bolton EE., Canese K., Comeau DC., Funk K., Ketter A., Kim S., Kimchi A.et al.. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2020; 48:D9–D16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fu L., Niu B., Zhu Z., Wu S., Li W.. CD-HIT: accelerated for clustering the next-Generation sequencing data. Bioinformatics. 2012; 28:3150–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Katoh K., Standley DM.. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013; 30:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Price MN., Dehal PS., Arkin AP.. FastTree 2–Approximately maximum-likelihood trees for large alignments. PLoS One. 2010; 5:e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nguyen LT., Schmidt HA., von Haeseler A., Minh BQ.. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015; 32:268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kalyaanamoorthy S., Minh BQ., Wong TKF., von Haeseler A., Jermiin LS.. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods. 2017; 14:587–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zallot R., Oberg N., Gerlt JA.. The EFI web resource for genomic enzymology tools: leveraging protein, genome, and metagenome databases to discover novel enzymes and metabolic pathways. Biochemistry. 2019; 58:4169–4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shannon P., Markiel A., Ozier O., Baliga NS., Wang JT., Ramage D., Amin N., Schwikowski B., Ideker T.. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003; 13:2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hyatt D., Chen GL., Locascio PF., Land ML., Larimer FW., Hauser LJ.. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010; 11:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mirdita M., Steinegger M., Söding J.. MMseqs2 desktop and local web server app for fast, interactive sequence searches. Bioinformatics. 2019; 35:2856–2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mestre MR., González-Delgado A., Gutiérrez-Rus LI., Martínez-Abarca F., Toro N.. Systematic prediction of genes functionally associated with bacterial retrons and classification of the encoded tripartite systems. Nucleic Acids Res. 2020; 48:12632–12647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mistry J., Chuguransky S., Williams L., Qureshi M., Salazar GA., Sonnhammer ELL., Tosatto S.C.E., Paladin L., Raj S., Richardson LJ.et al.. Pfam: the protein families database in 2021. Nucleic Acids Res. 2021; 49:D412–D419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang M., Derbyshire MK., Yamashita RA., Marchler-Bauer A.. NCBI’s conserved domain database and tools for protein domain analysis. Curr Protoc Bioinformatics. 2020; 69:e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Galperin MY., Wolf YI., Makarova KS., Vera A.R., Landsman D., Koonin EV.. COG database update: focus on microbial diversity, model organisms, and widespread pathogens. Nucleic Acids Res. 2021; 49:D274–D281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schaeffer RD., Liao Y., Cheng H., Grishin NV.. ECOD: new developments in the evolutionary classification of domains. Nucleic Acids Res. 2017; 45:D296–D302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Steinegger M., Meier M., Mirdita M., Vöhringer H., Haunsberger SJ., Söding J.. HH-suite3 for fast remote homology detection and deep protein annotation. BMC Bioinformatics. 2019; 20:473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Huerta-Cepas J., Szklarczyk D., Heller D., Hernández-Plaza A., Forslund SK., Cook H., Mende DR., Letunic I., Rattei T., Jensen LJ.et al.. EggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019; 47:D309–D314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Grazziotin AL., Koonin EV., Kristensen D.M.. Prokaryotic virus orthologous groups (PVOGs): a resource for comparative genomics and protein family annotation. Nucleic Acids Res. 2017; 45:D491–D498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lai S., Jia L., Subramanian B., Pan S., Zhang J., Dong Y., Chen WH., Zhao XM.. mMGE: a database for human metagenomic extrachromosomal mobile genetic elements. Nucleic Acids Res. 2021; 49:D783–D791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yang J., Anishchenko I., Park H., Peng Z., Ovchinnikov S., Baker D.. Improved protein structure prediction using predicted interresidue orientations. Proc. Natl. Acad. Sci. U.S.A. 2020; 117:1496–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Holm L. DALI and the persistence of protein shape. Protein Sci. 2020; 29:128–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dong R., Pan S., Peng Z., Zhang Y., Yang J.. MTM-Align: a server for fast protein structure database search and multiple protein structure alignment. Nucleic Acids Res. 2018; 46:W380–W386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wickham H. ggplot2: Elegant Graphics for Data Analysis. 2021; NY: Springer-Verlag. [Google Scholar]
- 49. Picelli S., Björklund ÅK., Reinius B., Sagasser S., Winberg G., Sandberg R.. Tn5 transposase and tagmentation procedures for massively scaled sequencing projects. Genome Research. 2014; 24:2033–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Galata V., Fehlmann T., Backes C., Keller A.. PLSDB: a resource of complete bacterial plasmids. Nucleic Acids Res. 2019; 47:D195–D202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Camarillo-Guerrero LF., Almeida A., Rangel-Pineros G., Finn RD., Lawley TD.. Massive expansion of human gut bacteriophage diversity. Cell. 2021; 184:1098–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Marquet M., Hölzer M., Pletz MW., Viehweger A., Makarewicz O., Ehricht R., Brandt C.. What the phage: a scalable workflow for the identification and analysis of phage sequences. 2020; bioRxiv doi:25 July 2020, preprint: not peer reviewed 10.1101/2020.07.24.219899. [DOI] [PMC free article] [PubMed]
- 53. Roux S., Páez-Espino D., Chen IA., Palaniappan K., Ratner A., Chu K., Reddy TBK., Nayfach S., Schulz F., Call L.et al.. IMG/VR v3: an integrated ecological and evolutionary framework for interrogating genomes of uncultivated viruses. Nucleic Acids Res. 2021; 49:D764–D775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Benler S., Yutin N., Antipov D., Rayko M., Shmakov S., Gussow AB., Pevzner P., Koonin EV.. Thousands of previously unknown phages discovered in whole-community human gut metagenomes. Microbiome. 2021; 9:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shah SA., Deng L., Thorsenm J., Pedersen AG., Dion MB., Castro-Mejía JL., Silins R., Romme FO., Sausset R., Ndela EO.et al.. Hundreds of viral families in the healthy infant gut. 2021; bioRxiv doi:24 July 2021, preprint: not peer reviewed 10.1101/2021.07.02.450849. [DOI] [PMC free article] [PubMed]
- 56. Yutin N., Benler S., Shmakov SA., Wolf YI., Tolstoy I., Rayko M., Antipov D., Pevzner PA., Koonin E.V.. Analysis of metagenome-assembled viral genomes from the human gut reveals diverse putative crass-like phages with unique genomic features. Nat. Commun. 2021; 12:1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jain C., Rodriguez-R LM., Phillippy AM., Konstantinidis KT., Aluru S.. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018; 9:5114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chan PP., Lin BY., Mak AJ., Lowe TM.. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021; 49:9077–9096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bin JH., Bolduc B., Zablocki O., Kuhn JH., Roux S., Adriaenssens EM., Brister JR., Kropinski AM., Krupovic M., Lavigne R.et al.. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol. 2019; 37:632–639. [DOI] [PubMed] [Google Scholar]
- 60. Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014; 30:2068–2069. [DOI] [PubMed] [Google Scholar]
- 61. Terzian P., Olo NE., Galiez C., Lossouarn J., Pérez Bucio RE., Mom R., Toussain A., Petit MA., Enault F.. PHROG: families of prokaryotic virus proteins clustered using remote homology. NAR Genomics Bioinformatics. 2021; 3:lqab067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wilkins D., Kurtz Z.. gggenes: Draw gene arrow maps in ggplot2. 2019; [Google Scholar]
- 63. Dion MB., Plante PL., Zufferey E., Shah SA., Corbeil J., Moineau S.. Streamlining CRISPR spacer-based bacterial host predictions to decipher the viral dark matter. Nucleic Acids Res. 2021; 49:3127–3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sugimoto R., Nishimura L., Thanh PN., Ito J., Parrish NF., Mori H., Kurokawa K., Nakaoka H., Inoue I.. Comprehensive discovery of CRISPR-targeted terminally redundant sequences in the human gut metagenome: Viruses, plasmids, and more. PLoS Comput.Biol. 2021; 17:e1009428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Couvin D., Bernheim A., Toffano-Nioche C., Touchon M., Michalik J., Néron B., Rocha EPC., Vergnaud G., Gautheret D., Pourcel C.. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for cas proteins. Nucleic Acids Res. 2018; 46:W246–W51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lloyd-Price J., Arze C., Ananthakrishnan AN., Schirmer M., Avila-Pacheco J., Poon TW., Andrews E., Ajami NJ., Bonham KS., Brislawn CJ.et al.. Multi-Omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature. 2019; 569:655–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., Madden TL.. BLAST+: architecture and applications. BMC Bioinformatics. 2009; 10:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. Journal. 2011; 17:10–12. [Google Scholar]
- 69. Bruchmann S., Muthukumarasamy U., Pohl S., Preusse M., Bielecka A., Nicolai T., Hamann I., Hillert R., Kola A., Gastmeier P.et al.. Deep transcriptome profiling of clinical klebsiellapneumoniae isolates reveals strain and sequence type-specific adaptation. Environ. Microbiol. 2015; 17:4690–4710. [DOI] [PubMed] [Google Scholar]
- 70. Weinberg Z., Lünse CE., Corbino KA., Ames TD., Nelson JW., Roth A., Perkins KR., Sherlock ME., Breaker RR.. Detection of 224 candidate structured RNAs by comparative analysis of specific subsets of intergenic regions. Nucleic Acids Res. 2017; 45:10811–10823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Rivas E., Clements J., Eddy SR.. Estimating the power of sequence covariation for detecting conserved RNA structure. Bioinformatics. 2020; 36:3072–3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Crooks GE., Hon G., Chandonia JM., Brenner S.E. WebLogo: a sequence logo generator. Genome Res. 2004; 14:1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Nawrocki EP., Eddy SR.. Infernal 1.1: 100-Fold faster RNA homology searches. Bioinformatics. 2013; 29:2933–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sharifi F., Ye Y.. Identification and classification of reverse transcriptases in bacterial genomes and metagenomes. Nucleic Acids Res. 2022; 50:e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Cheng R., Huang F., Wu H., Lu X., Yan Y., Yu B., Wang X., Zhu B.. A nucleotide-sensing endonuclease from the gabija bacterial defense system. Nucleic Acids Res. 2021; 49:5216–5229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Millman A., Melamed S., Amitai G., Sorek R.. Diversity and classification of cyclic-oligonucleotide-based anti-phage signalling systems. Nat. Microbiol. 2020; 5:1608–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Makarova KS., Anantharaman V., Aravind L., Koonin EV.. Live virus-free or die: coupling of antivirus immunity and programmed suicide or dormancy in prokaryotes. Biol. Direct. 2012; 7:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Makarova KS., Wolf YI., Snir S., Koonin EV.. Defense islands in bacterial and archaeal genomes and prediction of novel defense systems. J. Bacteriol. 2011; 193:6039–6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kinch LN., Ginalski K., Rychlewski L., Grishin NV.. Identification of novel restriction Endonuclease-like fold families among hypothetical proteins. Nucleic Acids Res. 2005; 33:3598–3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Bernheim A., Millman A., Ofir G., Meitav G., Avraham C., Shomar H., Rosenberg MM., Tal N., Melamed S., Amitai G.et al.. Prokaryotic viperins produce diverse antiviral molecules. Nature. 2021; 589:120–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Burroughs AM., Zhang D., Schäffer DE., Iyer LM., Aravind L.. Comparative genomic analyses reveal a vast, novel network of nucleotide-centric systems in biological conflicts, immunity and signaling. Nucleic Acids Res. 2015; 43:10633–10654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Iyer LM., Koonin EV., Leipe DD., Aravind L.. Origin and evolution of the archaeo-eukaryotic primase superfamily and related palm-domain proteins: structural insights and new members. Nucleic Acids Res. 2005; 33:3875–3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kazlauskas D., Sezonov G., Charpin N., Venclovas Č., Forterre P., Krupovic M.. Novel families of archaeo-eukaryotic primases associated with mobile genetic elements of bacteria and archaea. J. Mol. Biol. 2018; 430:737–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Makarova KS., Anantharaman V., Grishin NV., Koonin EV., Aravind L.. CARF and WYL domains: ligand-binding regulators of prokaryotic defense systems. Front Genet. 2014; 5:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Yan WX., Chong S., Zhang H., Makarova KS., Koonin EV., Cheng DR., Scott DA.. Cas13d is a compact RNA-targeting type VI CRISPR effector positively modulated by a WYL-domain-containing accessory protein. Mol. Cell. 2018; 70:327–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Castelle CJ., Banfield JF.. Major new microbial groups expand diversity and alter our understanding of the tree of life. Cell. 2018; 172:1181–1197. [DOI] [PubMed] [Google Scholar]
- 87. Rousset F., Cabezas-Caballero J., Piastra-Facon F., Fernández-Rodríguez J., Clermont O., Denamur E., Rocha EPC., Bikard D.. The impact of genetic diversity on gene essentiality within the escherichia coli species. Nat. Microbiol. 2021; 6:301–312. [DOI] [PubMed] [Google Scholar]
- 88. Waldern J.M., Smith D., Piazza CL., Bailey EJ., Schiraldi NJ., Nemati R., Fabris D., Belfort M., Novikova O.. Methylation of RRNA as a host defense against rampant group II intron retrotransposition. Mob. DNA. 2021; 12:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Yushenova I.A., Arkhipova IR.. Biochemical properties of bacterial reverse transcriptase-related (Rvt) gene products: multimerization, protein priming, and nucleotide preference. Curr. Genet. 2018; 64:1287–1301. [DOI] [PubMed] [Google Scholar]
- 90. Gladyshev E.A., Arkhipova IR.. A widespread class of reverse transcriptase-related cellular genes. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:20311–20316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Rousset F., Depardieu F., Miele S., Dowding J., Laval AL., Lieberman E., Garry D., Rocha EPC., Bernheim A., Bikard D.. Phages and their satellites encode hotspots of antiviral systems. Cell Host Microbe. 2022; 30:740–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ainsworth S., Stockdale S., Bottacini F., Mahony J., van Sinderen D.. The lactococcus lactis plasmidome: much learnt, yet still lots to discover. FEMS Microbiol. Rev. 2014; 38:1066–1088. [DOI] [PubMed] [Google Scholar]
- 93. Uchiyama I., Albritton J., Fukuyo M., Kojima KK., Yahara K., Kobayashi I.. A novel approach to helicobacter pylori pan-genome analysis for identification of genomic islands. PLoS One. 2016; 11:e0159419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Koonin E.V., Makarova KS., Wolf Y.I., Krupovic M.. Evolutionary entanglement of mobile genetic elements and host defence systems: guns for hire. Nat. Rev. Genet. 2020; 21:119–131. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.