


Abstract
NAT10 is an essential enzyme that catalyzes N4-acetylcytidine (ac4C) in eukaryotic transfer RNA and 18S ribosomal RNA. Recent studies suggested that rRNA acetylation is dependent on SNORD13, a box C/D small nucleolar RNA predicted to base-pair with 18S rRNA via two antisense elements. However, the selectivity of SNORD13-dependent cytidine acetylation and its relationship to NAT10’s essential function remain to be defined. Here, we demonstrate that SNORD13 is required for acetylation of a single cytidine of human and zebrafish 18S rRNA. In-depth characterization revealed that SNORD13-dependent ac4C is dispensable for human cell growth, ribosome biogenesis, translation and development. This loss of function analysis inspired a cross-evolutionary survey of the eukaryotic rRNA acetylation ‘machinery’ that led to the characterization of many novel metazoan SNORD13 genes. This includes an atypical SNORD13-like RNA in Drosophila melanogaster which guides ac4C to 18S rRNA helix 45 despite lacking one of the two rRNA antisense elements. Finally, we discover that Caenorhabditis elegans 18S rRNA is not acetylated despite the presence of an essential NAT10 homolog. Our findings shed light on the molecular mechanisms underlying SNORD13-mediated rRNA acetylation across eukaryotic evolution and raise new questions regarding the biological and evolutionary relevance of this highly conserved rRNA modification.
INTRODUCTION
N4-acetylcytidine (ac4C) in eukaryotic and archaeal rRNA was first documented in the 1970s, but the catalytic machinery and specific sites of this base modification have been defined only recently (1,2). In eukaryotes, ac4C is installed at two positions within small subunit (18S) rRNA (helix 34 and 45) where it is catalyzed by the nucleolar GCN5-related RNA acetyltransferase NAT10, also referred to as Kre33 or Rra1 in yeast (3–6). In addition, recent studies showed that certain archaeal species harbour >100 ac4C residues in their rRNA, including at helix 45, placing cytidine acetylation on par with prevalent nucleoside modifications such as pseudouridines and 2′-O-methylations in these organisms (7,8). Beside its involvement in rRNA modification, NAT10/Kre33 (assisted by its specific co-factor THUMPD1/Tan1) can modify C12 of serine and leucine tRNAs (5,9) and has been suggested to modify eukaryotic mRNAs (10,11). Orthogonal validation has, however, called into question the involvement of NAT10 in mRNA modification (7). In addition to modifying RNA, NAT10 may also be implicated in protein acetylation (12–14). Moreover, NAT10 is essential for pre-rRNA processing reactions leading to the synthesis of 18S rRNA (3–6), and unsurprisingly it was associated with human diseases (15,16). However, the importance of helix 45 ac4C for cell homeostasis has not been addressed.
In yeast, the ability of Kre33 to specifically acetylate 18S rRNA was demonstrated to rely on two box C/D small nucleolar RNAs (SNORD), namely snR4 and snR45, which form imperfect base pairing interactions surrounding the targeted cytidine. Indeed, knockout of snR4 and snR45 led to the specific disappearance of 18S rRNA acetylation at SSU-C1280 (helix 34) and SSU-C1773 (helix 45), respectively, while leaving tRNA acetylation unaffected (17). SNORDs are associated with four core proteins (SNU13, NOP56, NOP58, and Fibrillarin) and form nucleolar ribonucleoprotein (RNP) complexes known to mediate site-specific ribose methylation of rRNA, U6 spliceosomal snRNA and tRNAs; some of them are also involved in early nucleolar pre-rRNA processing (18). The possible contribution of snoRNA-associated proteins in rRNA acetylation remains undefined. Of note, SNORD13 (formerly U13) displays two rRNA complementarity tracts reminiscent of those present on snR45 (Figure 1), strongly implicating it as a human counterpart (5,19,20). Consistent with this notion, inhibiting SNORD13 expression in a cancer cell line using antisense oligonucleotides causes a ∼50% decrease in levels of ac4C in 18S rRNA (5). However, the nucleotide-specificity of this inhibition was not determined and the residual level of acetylation did not enable this modification's impact on cell growth or ribosome biogenesis. Finally, three SNORD13-like RNA species were also reported in Arabidopsis thaliana (snoR105, snoR108 and snoR146) but their roles in rRNA acetylation still await investigation (21).
Figure 1.

Schematic of SNORD13-dependent ribosomal RNA acetylation. (A) Through an incompletely defined antisense-mediated mechanism, human SNORD13 assists the RNA cytidine acetyltransferase NAT10 in modifying C1842 in the universally conserved helix 45 at the 3′ extremity of 18S rRNA. (B) Helix 45 is enriched with post-transcriptional RNA modifications. RNA modifying enzymes and their modified nucleotides (lollipops) are indicated: SSU-ac4C1842 (yellow), SSU-m6A1832 (green) and SSU-m26A1850 and SSU-m26A1851 (blue) are introduced by NAT10, METTL5 and DIMT1, respectively. (C) Human SNORD13 base-pairs with 18S rRNA sequences on each side of the cytidine to be acetylated. Note that SNORD13-rRNA duplexes tolerate G-U wobble base-pairs, mismatches or even bulged nucleotides and the length of the intervening loops, from either rRNA or SNORD side, also differ from one organism to another (Supplementary Figure S1).
Outside their potential to form two imperfect base-pairing interactions with 18S rRNA, vertebrate SNORD13 and its Saccharomyces cerevisiae and A. thaliana homologs are not highly conserved. This explains why, although SNORD13 was discovered thirty years ago (19,22), its relationship with snR45 was only recognized recently (17). This suggests two distinct evolutionary hypotheses: either SNORD-mediated rRNA acetylation emerged early in a common ancestor of plants, fungi and animals and diverged thereafter, or alternatively, SNORD13 arose independently in these different lineages via convergent evolution. Irrespective of the scenario, the possibility also exists that SNORD13 has been lost in some lineages and, accordingly, helix 45 acetylation may rely on a stand-alone enzyme, or no longer be present in rRNA in certain species. How conserved SNORD-guided cytidine acetylation is across the eukaryotic tree of life, as well as what cross-organismal commonalities in nucleic acid acetylation machinery govern this process, remain unknown.
Here, we use CRISPR-Cas9 genome editing to delete SNORD13 in human cells, confirming the hypothesis that this snoRNA is specifically required for acetylation of a single cytidine residue in small subunit ribosomal RNAs (SSU-ac4C1842 located in rRNA helix45). Using this cell line and related model systems, we further establish that loss of SSU-ac4C1842 does not lead to obvious defects in terms of cell growth, protein synthesis, ribosome biogenesis, or development of metazoan model organisms (zebrafish, flies). Finally, considering that Kre33/NAT10 is essential for pre-rRNA processing (5) while ac4C in helix 45 appears dispensable, we surveyed the conservation of the ribosomal RNA acetylation machinery across the eukaryotic tree of life. This cross-evolutionary analysis revealed that SNORD-mediated rRNA acetylation is widely conserved in Metazoans, but defined Caenorhabditis elegans as a multicellular organism in which rRNA acetylation has been lost. Our findings illuminate the molecular mechanisms underlying SNORD13-mediated rRNA acetylation across evolution and raise new questions regarding the role of highly conserved SSU-ac4C1842.
MATERIALS AND METHODS
Unless otherwise noted, all techniques for cloning and manipulating nucleic acids were performed according to standard protocols.
Generation and characterization of SNORD13-deficient HAP1 cells
HAP1 cells were grown at 37°C with 5% CO2 in IMDM (Gibco; 4.5 g/l glucose) supplemented with 10% fetal bovine serum (PAN biotech), 1 mM sodium pyruvate (Gibco) and 1% penicillin–streptomycin (Sigma-Aldrich). The human SNORD13 gene was disrupted via CRISPR/Cas9-mediated deletion. Two sgRNAs selected using the CRISPR design tool at https://zlab.bio/guide-design-resources were cloned into pX459 V2.0 (Addgene #62988) plasmid. HAP1 cells seeded in six-well dishes (70–80% confluency) were co-transfected using lipofectamine 2000 (Thermo Fisher Scientific) with pX459_sgRNA1 and pX459_sgRNA2 (500 ng each). 48 h post-transfection, cells were transiently treated with puromycin (2 μg/ml; ∼36 h) before being subjected to clonal selection by limiting dilution. Deletion events were validated by PCR (primer sequences are listed in Supplementary Data S3) and three clones per genotype were randomly chosen for further analyses. The rate of cell growth was assayed using a Beckman Coulter Z1 particle counter. Cells were seeded into a 24-well plate (5000 cells) and counted in quadruplicate at each time point. Six independent proliferation assays were performed. Apoptosis was detected using Annexin V-FITC/propidium iodide staining according to the manufacturer's instructions (BioLegend). Cells were analyzed using FacsVerse (Becton Dickinson) with the acquisition of 20 000 total events (BD FacsSuite). Two independent experiments were performed.
RNA extraction and rRNA analyses
Total RNA was prepared using TRI reagent (MRC) according to the manufacturer's instructions and treated with RQ1 RNAse-free DNase (Promega) and proteinase K (Sigma). For Northern blot analyses of low molecular-weight species, total RNA (10 μg) was fractionated by electrophoresis on a 6% acrylamide, 7 M urea denaturing gel and electrotransferred onto a Hybond N membrane (Amersham GE Healthcare) as described previously (23). For northern blot analyses of higher molecular-weight species, 4 μl of total RNA (2.5 μg/μl) were added to 20 μl of ‘glyoxal mix’ (60% DMSO, BTPE 1.2×, 5% glycerol, 8% glyoxal solution (Sigma) and 40 μg/ml Ethidium bromide), heat-denatured at 55°C for 60 min, chilled on ice for 10 min and separated by electrophoresis on a 1.2% agarose gel (Pipes 10 mM, Bis–Tris 300 mM, EDTA 10 mM). Capillarity transfer was performed onto a Hybond N+ membrane. In all cases, transfer was followed by UV light irradiation and membranes were pre-hybridized for 1 h in 5× SSC, 0.1% SDS, 5× Denhardt's, 150 mg/ml yeast tRNA. Hybridization was carried out overnight at 50°C with 5′-[32P]-labeled-DNA oligonucleotide probes (100 000 cpm/ml). Membranes were washed twice with 0.1× SSC, 0.1% SDS at room temperature (2 × 10 min) before autoradiography. Pulse chase experiments were performed as described in (24). The day before, 12-well plates are seeded with 500 000 cells. Cells are then incubated in methionine-free DMEM (Invitrogen) for 30 min at 37°C, labeled for exactly 15 min with l-methyl 3H methionine (50mCi/ml) and rinsed twice with IMDM containing unlabeled methionine. They were then incubated for 0 min (rinsed immediately twice with cold PBS), 30 min, 1 h, 1.5 h, 2 h, 2.5 h in regular IMDM. After the corresponding chase time, cells were rinsed twice with cold PBS and lysed with Tri-Reagent (MRC). RNA samples were glyoxal denatured and separated on a 1.2% agarose gel and passively transferred to a nylon membrane as described above. The membrane was exposed 5 weeks to Biomax KODAK MS films with a KODAK BioMax Transcreen LE. RNAse H mapping was performed as described in (24). A water solution containing 4 μg of total RNA was heat-denatured (95°C for 3 min) in the presence of an antisense oligonucleotide matching the 3′-end of 18S rRNA (RNase H_2, 100 μM). After allowing annealing by cooling down to room temperature, the RNA/oligonucleotide mixture was diluted to a 30 μl final volume with a reaction mix (1× RNase H reaction buffer, 65 μM DTT, 0.5U/μl RNasin (Promega), 50 U RNase H (New England Biolabs) and incubated at 37°C for 30 min. The reaction was stopped by adding 0.3 M sodium acetate pH 5.2/0.2 mM EDTA. RNA was phenol extracted, ethanol precipitated, loaded onto a 15% acrylamide gel and analyzed by northern blot (32P-labeled 3′18S oligo-probe) as described above. For primer extension, 2 μg of total RNA extracted from adult flies were mixed with a 5′-32P-labeled oligo (∼100 000 cpm) and cDNA synthesis, generated according to the manufacturer's instructions, were resolved onto a 12% denaturing acrylamide gel.
Sucrose gradient sedimentation and Sunset assay
Exponentially growing WT and SNORD13-KO cells (∼20 million per genotype) were treated with 100 μg/ml of cycloheximide (CHX, SIGMA) for 10 min at 37°C, rinsed twice with cold PBS/CHX (100μg/ml), collected by centrifugation (400 g, 5 min) and resuspended in 5 ml of cold Buffer A (10 mM HEPES, 1.5 mM MgCl2, 100 mM KCl, CHX 100 μg/ml, 1 mM DTT). Cells were then centrifuged (400 g, 10 min at 4°C), suspended in 700 μl of Buffer A and incubated for 10 min on ice. Cells were then mechanically disrupted with a cold Dounce homogenizer (B) and centrifuged for 10 min (1000 g at 4°C). The cytoplasmic fraction (supernatant) was collected and centrifuged twice at 10 000 g (10 min, 4°C). About 1.5 mg of protein was loaded on a 10–50% sucrose gradient and centrifuged for 2h45min at 39 000 rpm in an Optima L-100XP Ultracentrifuge (Beckman–Coulter) using a SW41 rotor (Beckman). Gradients were then monitored at 260 nm and fractions were collected from the top using a Foxy Jr. fraction collector (Teledyne ISCO). Sunset assay was performed as described in (25). Exponentially growing cells were treated with 1 μg/ml of puromycin for 20 min. As a control, cycloheximide (100 μg/ml) was also added 15 min before puromycin treatment. Total cell extracts were then processed for western blot using an anti-puromycin antibody (Millipore, clone 12D10 #MABE343) as well as an Anti-CDK9 (C12F7) mAb (cellsignal.com) as a gel loading control.
Immunoprecipitations
30 μl of rabbit R1131 anti-trimethylguanosine (3 mg/ml, a gift of Dr R. Luhrmann) were incubated with gentle agitation for 120 min at 4°C with 80 μl of Protein A Sepharose® 4B, Fast Flow from S. aureus (PAS, Sigma #P9424) in 1 ml of NET-150 buffer (50 mM Tris–HCl (pH 7.4), 150 mM NaCl, 0.5% Igepal® CA-630, SIGMA). Mouse IgG (Jackson Immunoresearch #315-005-008) was used as negative controls. PAS-Ig pellets were washed 3 times with 1 ml of NET-150 buffer. 30 μg of total RNAs were then added to PAS-R1131 (or PAS-IgG) in 0.5 ml of NET-150 and incubated with gentle agitation for 60 min at 4°C. Pellets were then collected by centrifugation and washed 7 times in 1 ml of NET-150. RNAs from pellet and supernatant were extracted by SDS/phenol extraction and analyzed by Northern blot as described above.
Searching for SNORD13-like and 18S rRNA sequences in eukaryotic genomes
Using vertebrate SNORD13 sequences as queries, iterative BLAST searches with low stringency algorithm parameters, together with manual inspection of some hits, were conducted using eukaryotic genome databases found at https://blast.ncbi.nlm.nih.gov/Blast.cgi, https://rnacentral.org/, https://www.echinobase.org/entry/, http://mgbase.qnlm.ac/home, http://www.insect-genome.com/, https://bipaa.genouest.org/is/, https://i5k.nal.usda.gov/ webapp/blast/, https://spaces.facsci.ualberta.ca/ephybase/. Eukaryotic 18S rRNA sequences were retrieved either at https://www.arb-silva.de/ or https://www.ncbi.nlm.nih.gov/nucleotide/.
Mung bean nuclease protection assay and RP-HPLC
Mung bean nuclease (MBN) protection assay was performed exactly as described before (26). 500 pmol of the synthetic deoxy oligonucleotide (5′-TAATGATCCTTCCGCAGGTTCACCTACGGAAACCTTGTTACGACTTTTAC-3′) was incubated with 50 pmol of 18S rRNA and 5% of DMSO in 0.3 volume of hybridization buffer (250 mM HEPES, 500 mM KCl at pH 7). The mixture was incubated at 90°C for 5 min and then slowly cooled down to 45°C over 2 h. After hybridization, 35 units of mung bean nuclease (New England Bio Labs (NEB)) and 0.02 mg/ml RNase A (Sigma-Aldrich) along with an appropriate amount of 10× MBN buffer (NEB) were added to start digestion. The digestion was carried out at 35°C for 1 h. The protected fragment (RNA–DNA hybrid) was extracted from the reaction mixture by phenol/chloroform, followed by overnight ethanol precipitation. The protected rRNA fragment was separated from the complementary DNA oligonucleotides on a denaturing 7 M urea 15% PAGE gel. Bands were visualized by ethidium bromide staining and the rRNA band was excised and electro-eluted using the D-Tube TM Dialyzers according to the manufacturer's protocol (Novagen). Eluted rRNA fragment was digested to nucleosides using P1 nuclease and alkaline phosphatase as described before (26). Nucleosides were then analyzed by RP-HPLC on a Supelcosil LC-18-S HPLC column (25 cm × 4.6 mm, 5 μm) equipped with a pre-column (4.6 × 20 mm) at 30°C on an Agilent 1200 HPLC system as described previously in (17).
Targeted ac4C sequencing
Nucleotide resolution sequencing of ac4C in 18S rRNA (helix 45 and helix 34) was performed as previously described (7). RNA samples were treated with sodium cyanoborohydride (100 mM in H2O) or vehicle (H2O) in a final reaction volume of 100 μl. Reactions were initiated by addition of 1 M HCl to a final concentration of 100 mM and incubated for 20 min at room temperature. Reactions were stopped by neutralizing the pH by the addition of 30 μl 1 M Tris-HCl pH 8.0. After incubation, reactions were adjusted to 200 μl with H2O, ethanol precipitated, desalted with 70% ice-cold ethanol, briefly dried on Speedvac, resuspended in H2O, and quantified by using a Nanodrop 2000 spectrophotometer. Cellular total RNA from individual reactions (200 pg) was incubated with specific h45/h34 rev primer (4.0 pmol) in a final volume of 20 μl. Individual reactions were heated to 65°C for 5 min and transferred to ice for 3 min to facilitate annealing in SuperScript III reaction buffer (Invitrogen). After annealing, reverse transcriptions were performed by adding 5 mM DTT, 200 units SuperScript III RT, 500 μM dNTPs (use 250 μM dGTP) and incubating for 60 min at 55°C. Reaction was quenched by increasing the temperature to 70°C for 15 min and store at 4°C. cDNA (2 μl) was used as template in 50 μl PCR reaction with Phusion Hot start flex (New England Biolabs). Reaction conditions: 1× supplied HF buffer, 2.5 pmol each forward and reverse primer, 200 μM each dNTPs, 2 units Phusion hot start enzyme, 2 μl cDNA template. PCR products were run on a 2% agarose gel, stained with SYBR safe and visualized on UV transilluminator at 302 nm. Bands of the desired size were excised from the gel and DNA extracted using QIA-quick gel extraction kit from Qiagen and submitted for Sanger sequencing (GeneWiz) using the forward PCR primer. Processed sequencing traces were viewed using 4Peaks software. Peak height for each base was measured and the percent misincorporation was determined using the equation: percent misincorporation = (peak intensity of T)/(sum of C and T base peaks)×100%.
Dual-luciferase reporter assay
100 000 WT and SNORD13-KO cells were seeded into a 24-well plate for 24 h. Cells were transfected using lipofectamine 2000 (Invitrogen) with 0.06 ng Renilla vector (internal control) and 500 ng of reporter Firefly vectors. Twenty-four hours post-transfection, cells were harvested, lysed and luciferase activity was monitored using the Dual-Luciferase Reporter Assay kit (Promega) according to the manufacturer's recommendations. Luciferase detection was assayed on Centro LB 960 Microplate Luminometer (Mikrowin 2000 Software). Five to six independent transfections were performed with each measurement made in triplicate.
Yeast cells and media conditions
S. cerevisiae deletion mutants of snR4 and snR45 or wild-type cells were grown at 30°C in standard YPD medium (1% yeast extract, 2% Bacto Peptone and 2% dextrose). For liquid growth assay, yeast cells were inoculated from a fresh YPD-agar plate into liquid YPD and grown for 48–72 h at 30°C or 37°C. Optical density at 600 nm (OD600) of cells was then measured and used to dilute cells into a 96-well plate (Cat # 167008, Thermo Scientific) at an equal OD of ∼0.02 (n = 3 or 12 for 30°C and 37°C samples, respectively). Cells were then grown for 90 h in an Epoch 2 microplate spectrophotometer (BID EPOCH2 Microplate Spectrometer from BioTek) at 30°C and 37°C and OD was measured every 30 min. Measured OD values were analyzed in R2. For presentation of growth curves, a regression line representing the mean of biological replicates was calculated using a generalized additive model. For the spot assay, yeast cells were inoculated from a fresh YPD-agar plate into liquid YPD and grown for 72 h at 30°C and 37°C. Optical density at 600 nm (OD600) was subsequently measured and cells were serially diluted in 2% YPD, such that the OD of cells varied from 0.02 OD to 0.00016 OD (5-fold dilutions). 3 μl of each diluted sample were used to spot cells on a YPD-agar plate, which was transferred to 30°C and 37°C. After 48 h of incubation pictures were taken of the colonies. Each strain was grown in duplicates.
Ethics statement and zebrafish care
Fish were handled in a facility certified by the French Ministry of Agriculture (approval number A3155510). The project has received an agreement number APAFIS#7124-20161 00517263944 v3. Anesthesia and euthanasia procedures were performed in tricaine methanesulfonate (MS222) solutions as recommended for zebrafish (0.16 mg/ml for anesthesia, 0.30 mg/ml for euthanasia). All efforts were made to minimize the number of animals used and their suffering, in accordance with the guidelines from the European directive on the protection of animals used for scientific purposes (2010/63/UE) and the guiding principles from the French Decret 2013-118. Embryos were raised and staged according to standard protocols and the Recommended Guidelines for Zebrafish Husbandry Conditions (27,28)
Generation of zebrafish snord13 mutants
The two guide RNAs (gRNA) were designed using CHOPCHOP CRISPR Design website (http://chopchop.cbu.uib.no). The designed oligos were annealed and ligated into the gRNA plasmid pX459 digested by BbsI (Thermo Scientific). The gRNAs were generated using the MEGAshortscript T7 transcription kit (Ambion) with PCR fragments containing the T7 promoter. Transcripts were purified by phenol–chloroform extraction and Ethanol precipitation. 1 nl of a solution containing 10 μM EnGen Cas9 NLS (NEB) and 100 ng/μl of gRNAs was injected at the one-cell stage. WT, heterozygous, and homozygous SNORD13 animals were then identified by PCR. Sequence primers can be found in (Supplementary Data S3).
RNA extraction, reverse transcription and real-time PCR
Total RNAs from 15 wild type and 15 mutant zebrafish embryos were extracted using TRI reagent (MRC) and treated with RQ1-RNase free DNase I (Promega) and Proteinase K (Sigma) as recommended by manufacturer's instructions. Total RNAs were converted into cDNA using GoScript RTase (Promega) random hexamer primers for 60 min at 42°C according to manufacturer's instructions. cDNAs were then diluted 20-fold and quantified by qPCR using SYBER green (Bio-Rad) and specific primers. Data were acquired on CFX96 Real-Time PCR detection System (Bio-Rad). Samples were analysed in triplicates and the expression level was calculated relative to zebrafish housekeeping gene EF1α. Sequence primers can be found in (Supplementary Data S3).
Immunostaining and in situ hybridization
Embryos were fixed overnight at 4°C in BT-FIX, after which they were immediately processed or dehydrated and stored at −20°C until use. After fixation or rehydration, embryos were washed twice with Phosphate Buffered Saline/1% Triton X-100 (PBST), permeabilized with PBST/0.5% trypsin for 30 s and washed twice again with PBST. After blocking with PBST/10% fetal calf serum (FCS)/1% bovine serum albumin (BSA) (hereafter termed ‘blocking solution’) for at least 1 h, embryos were incubated with antibodies directed against either cleaved Caspase-3 (Asp175) (Cell Signaling Technology), or HuC/D (Molecular Probes), in blocking solution overnight at 4°C followed by five washing steps with PBST. Embryos were then incubated with the appropriate Alexa Fluor-conjugated secondary antibodies (Molecular Probes) for at least 2 h at room temperature and washed three times. Fluorescent in situ hybridization was carried out as previously described (29). fli1 riboprobe preparation has been described elsewhere (30). Embryos were dissected, flat-mounted in glycerol and images were recorded on the confocal microscope TCS SP8 (Leica Microsystems) with an L 25× /0.95 W FLUOSTAR VIZIR objective (zoom ×1.25) using the scanner resonant mode.
Comparison of ribosome 3D structures
Atomic models of ribosomes from the following species: Homo sapiens, PDB: 6EK0 (31), Plasmodium falciparum, pdb: 6OKK and 3J79 (32); Toxoplasma gondii, PDB: 5XXU (33); Trichomonas vaginalis, PDB: 5XYI (33), Leishmania donovani, PDB: 6AZ1, 6AZ3 (34); Euglena gracilis, PDB: 6ZJ3 (35) were displayed and aligned in UCSF Chimera (36) using the ‘Matchmaker’ option, with the human 18S rRNA chain as reference. Prior to this structural alignment, cryo-EM maps of P. falciparum and T. gondii (EMD-2660 and EMD-6780, respectively) were visually inspected. Using UCSF Chimera and Coot (37), unattributed densities above C2065 in P. falciparum and C1764 in T. gondii cryo-EM map allowed to unambiguously replace unmodified residues by their ac4C counterparts in the corresponding atomic models.
RESULTS
Genetic deletion of human SNORD13 gene causes loss of SSU-ac4C1842
As a first step to probe directly the functional role of SNORD13 in rRNA modification, the human SNORD13 gene was inactivated in the haploid human chronic myelogenous leukemia-derived HAP1 cell line (38). In order to avoid potential transcriptional interference with the adjacent TTI2 gene, the 3′-end region of SNORD13 was targeted for CRISPR-Cas9-mediated deletion (Figure 2A). PCR analysis identified three independent SNORD13-deficient clones (#12/#20/#30) harboring the expected deletion (not shown), which rendered SNORD13 undetectable by northern blot (Figure 2B). Three additional clones (#13/#19/#22) were identified that were exposed to CRISPR/Cas9 without any deletion at SNORD13, providing a set of matched control cell lines subjected to identical clonal selection procedures and/or putative off-target effects. Unless otherwise specified, in subsequent experiments SNORD13-KO and WT are used to refer to a stoichiometric mixture of these three SNORD13-deficient and SNORD13-expressing clones, respectively.
Figure 2.

The specific lack of SNORD13-mediated acetylation in helix 45 does not alter cell growth or translation. (A) Schematic representation of SNORD13 locus at human chromosome 8. The relative location of the two DNA sequences targeted by sgRNAs is shown (PAM sequence is underlined). (B) Northern blot showing that SNORD13 is no longer detected in clones bearing deletions. * indicates truncated SNORD13 form which is routinely detected and corresponding to RNA species whose 5′-end is positioned 5–6 nucleotides upstream of the C-box (not shown). (C) RP-HPLC chromatograms of nucleosides obtained from helix 45 of WT (black) and SNORD13-KO (red) cells. Peak corresponding to SSU-ac4C1842 is indicated. (D) Sanger DNA sequencing of RT-PCR products obtained after borohydride treatments of total RNA extracted from WT and SNORD13-KO cells. Percentage of misincorporation (C-to-U) at SSU-C1842 (helix 45) and SSU-C1337 (helix 34) is indicated above each electropherogram. (E) Growth curves of WT and SNORD13-KO cells as judged by cell counting (n = 6 for each genotype). (F) Apoptosis levels determined by flow cytometry using propidium and Annexin V staining of WT and SNORD13-KO cells (n = 2 for each genotype). (G) SUnSET assay. Incorporation of puromycin into the elongating peptide was assayed in WT and SNORD13-KO cells (western blotting with anti-puromycin antibodies). Ponceau S staining of the membrane and detection of CDK9 (western blotting) were used as gel loading controls. Cells treated with the translation inhibitor cycloheximide (CHX) were also used as negative controls. (H) Luciferase reporter gene assays. WT and SNORD13-KO cells were transiently co-transfected by two plasmids expressing Renilla luciferase gene (LucR; internal control) or Firefly luciferase gene (lucF; translation reporter) carrying either an in-frame stop-codons (UAA, UAG), a detrimental mutation (R-to-S) or an IRES from FGF1A, FGF2 and CMV. Histograms show the normalized luciferase activity (lucF-to-lucR ratio). Data are expressed as mean ± s.e.m. and represent 5–6 independent experiments with triplicate measurements.
To define the site-specific activity of SNORD13, we took a dual approach to analyze our mutant and WT cell lines. First, we directly measured ac4C levels in helix 45 using a mung bean nuclease protection assay coupled to reversed phase high performance liquid chromatography (RP-HPLC, (5)). Briefly, total RNA extracted from SNORD13-KO and WT cells were hybridized with an antisense DNA oligonucleotide overlapping helix 45 and its flanking sequences and were treated with mung bean nuclease. Protected rRNA fragments were then gel-purified and nucleoside hydrolysates obtained after nuclease P1 digestions were subjected to RP-HPLC analysis. As shown in Figure 2C, cytosine acetylation at helix 45 was completely abolished in SNORD13-KO cells as compared to WT. As orthogonal validation, RNA isolated from SNORD13-KO and WT cells was analyzed using ac4C sequencing chemistry (39). In this approach, total RNA is treated with sodium cyanoborohydride under acidic conditions, causing chemical reduction of the acetylated residue. This reduced nucleobase causes misincorporation of non-cognate NTPs during reverse transcription (RT), enabling ac4C sites to be detected as C-to-T mutations upon cDNA sequencing. Applying this approach to the two cell lines, we detected SNORD13-dependent misincorporation at SSU-ac4C1842 but not at SSU-ac4C1337 (Figure 2D). These experiments validated our knockout strategy and unambiguously defined SNORD13 as a human counterpart of yeast snR45 required for guiding formation of ac4C at the terminal stem-loop of human 18S rRNA.
SNORD13 deletion does not disrupt cell growth, pre-rRNA processing or protein synthesis in human cells
Next, we assessed the phenotypic consequences of SNORD13 deletion and/or SSU-ac4C1842 loss. Analysis of proliferation by cell counting revealed that loss of SNORD13 did not impact cell growth significantly (Figure 2E) and that cell death rates and modalities were within normal range (Figure 2F). Moreover, SNORD13-KO cells mounted an apparent normal stress response when exposed to oxidative stress, nucleolar/ribotoxic stress, DNA damage, endoplasmic reticulum stress, chemically-induced hypoxia, serum withdrawal, altered translation or low (34°C) and high (40.5°C) temperatures (Supplementary Figure S2). In addition, we also examined the long-term fate of SNORD13-KO cells when mixed with WT cells (one passage every 3 days for 30 days). As shown in Supplementary Figure S3, this competition assay, performed in three different ratios of WT and mutant cells, did not reveal any selective advantage to WT cells. This supports the conclusion that growth of this immortalized cell line is not highly dependent on SSU-ac4C1842, including when subjected to a wide range of external stresses.
We next examined the levels of pre-rRNA precursors and mature rRNAs in SNORD13-KO cells by Northern blot (Supplementary Figure S4A-B), RNase H mapping (Supplementary Figure S4C) and pulse chase experiments (Supplementary Figure S4D). These analyses revealed that loss of SNORD13 and/or SSU-ac4C1842 does not interfere with normal pre-rRNA processing. An involvement of SNORD13 and/or SSU-ac4C1842 on ribosomal subunit assembly and global translation was also investigated by performing polysomal analysis. In this assay, whole cytoplasmic extracts from SNORD13-KO and WT cells were subjected to sucrose density gradient fractionation (10–50%). As shown in Supplementary Figure S4E, the distribution and amplitude of peaks corresponding to 40S, 60S, 80S monosomes and polysomes were virtually identical in SNORD13-KO and WT cells. Moreover, detection of nascent protein production by puromycin labelling (SUnSET assay) revealed identical, translation-dependent incorporation patterns in SNORD13-KO and WT cells, again indicating that overall protein synthesis is unlikely to be severely impaired in SNORD13-deficient cells (Figure 2G). To test whether SSU-ac4C1842 plays a more subtle role in fine-tuning aspects of ribosome function such as translational fidelity, we transfected WT and SNORD13-KO cells with firefly luciferase reporter genes bearing in-frame stop codons (UAA, UAG) or point mutations (R218S) that abrogate enzymatic luciferase activity. These reporters enable any decrease in translational accuracy to be detected via a corresponding increase in firefly luciferase activity. Our data indicate that stop codon read-through and amino acid misincorporation in SNORD13-KO cells was similar between WT and KO cells (Figure 2H, left panel). Finally, when using a set of Renilla-IRES-Firefly bicistronic vectors, we also showed that the efficiency of internal ribosome entry site (IRES)-mediated translation was apparently unaffected in SNORD13-KO cells (Figure 2H, right panel). Overall, these findings suggest that SNORD13 and SSU-ac4C1842 are not essential for ribosome biogenesis or function; at least in standard laboratory growth conditions.
SNORD13-deficient zebrafish embryos develop normally
In order to understand the biological relevance of SNORD13 at the whole organism level, a 189 bp-long, CRISPR-Cas9 mediated deletion overlapping the Danio rerio (zebrafish) snord13 gene locus on chromosome 8 was generated (Figure 3A). Crossing of individual heterozygotes led to the generation of homozygous adult snord13 knockout fish of normal appearance (not shown). To exclude any maternal contribution, three different homozygous females were crossed with heterozygous males. As expected, snord13 and ac4C in helix 45 were no longer detectable in RNA isolated from homozygous knockout fish, as judged by Northern blotting (Figure 3B) and ac4C sequencing (Figure 3C), respectively. Homozygous snord13 individuals in the progeny did not present any apparent morphological defects at 30 h post fertilization (30 hpf) and developed normally until adulthood (Supplementary Figure S5A). To explore the possible occurrence of more subtle defects, such as activation of the nucleolar stress pathway in response to impaired ribosome biogenesis (40), we performed immunostaining against activated Caspase-3. No increase in apoptotic cells was observed in the developing brain (Figure 3D, E) or spinal cord (Figure 3F-G) of snord13-deficient embryos, as compared to WT. Accordingly, the level of expression of the pro-apoptotic bcl2a, baxa and bbc3 genes, as judged by RT-qPCR, was not significantly altered in snord13 mutant embryos (Supplementary Figure S3B). Finally, we performed immunostaining in WT and mutant embryos for the pan-neural marker HuC/D which labels newly differentiated neurons in different structures including olfactory epithelium, pineal gland or optic tectum and the spinal cord. Again, no obvious difference could be detected between WT and mutant embryos (Figure 3D-G). Beside central nervous system development, we also sought to explore whether snord13 mutant displayed defects in another process such as vasculogenesis. We found that the vasculature of the trunk which is composed of the dorsal aorta (DA), the cardinal vein (CV) and intersegmental vessels was also normal in 30 hpf snord13-deficient embryos, as revealed by in situ hybridization against the key vascular transcription factor fli1 (Figures 3H-I). Similarly, the expression levels of various markers of the neural system (lhx1a, pax6a and otx5) and of the dorsal aorta trunk vasculature (cdh5 and fli1) were unchanged between WT and snord13-KO embryos, validating the lack of phenotypes observed (Supplementary Figure S5B). In conclusion, neither SNORD13 nor 18S rRNA ac4C in helix 45 appear to play any essential roles in embryonic zebrafish development.
Figure 3.

Phenotypic analyses of SNORD13-deficient zebrafish embryos. (A) Schematic representation of the zebrafish snord13 locus. The relative location of the two DNA sequences targeted by sgRNAs is shown (PAM sequence is underlined). (B) Northern blotting showing that SNORD13 is no longer detected in embryos bearing deletion events. *: truncated SNORD13 form which is routinely detected. SNORD118 was used as a gel loading control. (C) Histograms show percentage of misincorporation (C-to-U ratio) measured in borohydride-treated RNA samples prepared from WT and SNORD13-KO embryos (n = 3). (D–G) Confocal projections of WT (D, F) and SNORD13-KO (E, G) embryos after immunostaining against cleaved Caspase-3 (red) and the pan-neural marker HuC/D (green). (D, E) Dorsal view of the zebrafish brain at 30 hpf, neurons from the olfactory epithelium (oe), tectum (t), pineal gland (pg), optic tectum (ot) and trigeminal ganglion (tg) can be detected. (F, G) Lateral view of the spinal cord (sc) at 30 hpf. The white arrow indicates an apoptotic cell stained by activated caspase-3. (H, I) Confocal projections of WT (H) and SNORD13-KO (I) embryos after in situ RNA hybridization using antisense riboprobes directed against fli1 transcripts. Note that in the absence of antibodies to FLI1 that give reliable signals, fli1 mRNA expression was used as a proxy for monitoring vessel development. It does not imply that fli1 expression per se may be altered in SNORD13-KO zebrafish, whether at the mRNA or protein levels. White brackets indicate the dorsal aorta (DA) and the cardinal vein (CV) and the white arrows point to intersegmental vessels. Pictures are representative of three independent experiments (with at least 6 embryos imaged per genotype).
A cross-evolutionary survey of eukaryotic ribosomal RNA acetylation machinery
To more fully apprehend the evolutionary origin of SNORD-mediated rRNA acetylation, we revisited the phylogenetic distribution of SNORD13 throughout eukaryotes, focusing primarily on Metazoan (animals) and Archaeplastida (organisms capable of photosynthesis). Using BLAST searches with low stringency algorithm parameters together with manual inspection, we identified many SNORD13 novel candidate homologs outside the vertebrate lineage, including Urochordata, Cephalochordata, Echinodermata, Arthropoda, Mollusca, Brachiopoda, Cnidaria and even Porifera (Figure 4A, B). As depicted in Figure 4B, many of the newly-identified SNORD13 genes are located within introns, both in the sense or antisense orientation with respect to transcription of their host-gene transcript. In Archaeplastida, SNORD13 was also uncovered in phylogenetically distant land plants, including in the freshwater green alga C. braunii (Figure 4C). A full list of newly-identified SNORD13 and proposed snoRNA:rRNA duplexes is provided in Supplementary Data 1 and Supplementary Figure S1, respectively. These sequence analyses were accompanied by experimental profiling of ac4C at helix 45 in representative eukaryotic species. As listed in Figure 4A, acetylated cytidine was readily detected in the vast majority of organisms we examined, including unicellular eukaryotes such as slime molds (P. polycephalum), ciliates (P. tetraurelia, P. falciparum) or microalgaes (P. tricornutum). Hence, our findings indicate that SNORD13 is much more broadly distributed than initially thought and SNORD-mediated RNA acetylation is a widespread principle in eukaryotes. Consistent with this conclusion, NAT10 homologs were found in almost all organisms we queried, with the notable exception of E. gracilis (Figure 4A, Supplementary Data 2).
Figure 4.

Cross-evolutionary survey of eukaryotic rRNA acetylation machinery. (A) This table summarizes the conservation of the 5′-CCG-3′ motif, the acetylation status of helix 45 and the detection of SNORD13 and NAT10 genes in commonly used models or organisms for which we experimentally assayed the presence of ac4C at helix 45. n.d.: not determined. n/a: not applicable (genome is not available) (56,57). (B) Simplified phylogenetic tree of Metazoan. The genomic organization (intronic vs intergenic) of newly-identified SNORD13 genes (red arrows) is shown. Note that intronic SNORD13 can be positioned in the sense or antisense orientation with respect to their host-genes (black arrow). (C) Simplified phylogenetic tree of Archaeplastida. The acetylation status of helix 45 and the presence of SNORD13 in the genome of representative species are indicated. Note that while rRNA in algal species is acetylated, we failed to identify obvious SNORD13 counterparts. A full list of newly-identified SNORD13 sequences can be found in Supplementary data S1.
Atypical SNORD13-like homologs in Diptera
SNORD13-like genes are present in many groups of Arthropoda (Figure 5A, Supplementary Data 1), including D. melanogaster and B. mori where they had previously been identified as SNORDs of unknown functions (named Or-CD1 and Bm-10, respectively; (41,42)). In Diptera for which a genome annotation is available, Or-CD1 can be found either in large introns positioned in the 5′-UTR of two transcript isoforms generated from two alternative transcription sites (e.g. Swim, Tinagl1_2), or within short introns embedded in the open reading frame of the host-gene (e.g. Zfp15, Rps11) (Figure 5B). To our surprise, the published sequence of Or-CD1, as well as that of all newly-identified SNORD13-like in Diptera, lack conserved antisense rRNA sequence at their 5′-end (Figure 5C). One potential explanation would be that the sequence upstream of the C-box is removed by exonucleolytic trimming of the spliced-out host-intron, very likely due to the absence of a 5′-capped structure which normally plays a protective role against degradation (43). Although plausible for small introns, such as those in Zfp15 or Rps11 genes, this hypothesis cannot account for all observations. For example, many Or-CD1-like in Arthropoda are located in introns (Figure 4B), yet they still retain a conserved rRNA complementarity in their 5′-end segment (Figure 5C, Supplementary Figure S1). As an alternative, we reasoned that being intronic may not necessarily rule out independent transcription from distinct promoter regions. This is supported by the fact that SNORD13 can be found positioned either in the sense or antisense orientation relative to transcription of their host-gene (Figures 4B and 5B), a scenario that is not compatible with post-transcriptional processing from introns.
Figure 5.

Atypical SNORD13-related RNA guides rRNA acetylation in D. melanogaster. (A) Simplified phylogenetic tree of Arthropoda. Representative species in which SNORD13-like were identified are indicated. (B) Simplified phylogenetic tree of Diptera. The genomic arrangements of newly-identified SNORD13 genes (blue arrow) are shown. (C) Multiple sequence alignment of representative eukaryotic SNORD13 sequences. The small red arrow indicates the relative position of the mature 5′-end of Or-CD1 in Diptera (see also panel F) while the two horizontal black arrows in opposite orientation depict the terminal 5′-3′ stem structure. Note that SNORD13 sequences in Diptera - but not in other species - lack one of the two conserved antisense rRNA elements (denoted by horizontal blue bars). Vertical black, grey and white bars highlight Diptera, other Arthropoda and non-Arthropoda species, respectively. (D) Immunoprecipitation by anti-trimethyl cap (R1131) antibodies. OR-CD1 in various insects (as indicated below the panels) was detected by Northern blots. The same membrane was hybridized with an antisense oligo probe that recognized uncapped 5.8S rRNA used as negative controls. Input RNA (I); RNA recovered from the pellet (P). RNA recovered from the supernatant (S). The theoretical size of SNORD13 (nt) in each of the species studied is indicated in parentheses. (E) Schematic representation of two distinct modes of expression: independent transcription gives rise to short (top) or long (bottom) forms of 5′ capped SNORD13. (F) A mutant KO fly strain harboring an inserted P-element in the Or-CD1 gene (Dmel\P{EP}snoRNA:Or-CD1G9117) does not express OR-CD1 as assayed by primer extension. Note that the 5′-end of the cDNA product confirms that fly Or-CD1 lacks one 18S rRNA complementarity. The expected size (nt) of OR-CD1 with ∼4–5 nucleotides upstream of the C-box (as shown on the panel) is indicated in parentheses. 32P labeled primer (P). (G) Misincorporation (C-to-U) at SSU-C1968 (helix 45) as judged by Sanger DNA sequencing of RT-PCR products obtained after borohydride treatments of total RNA extracted from WT and Or-CD1-KO adult flies.
To probe the mode of expression of Or-CD1 in Arthropoda, total RNAs extracted from D. melanogaster, L. humile (ants) and A. mellifera (bees) were subjected to immunoprecipitation with R1131 antibodies that recognize the 2,2,7-trimethylguanosine (TMG) cap structure, enabling its use as a proxy for independent transcription. Consistent with the expected size for small RNAs lacking 5′-end extension in D. melanogaster, Northern blot analyses detected a radioactive signal migrating at about 110 nucleotides (Figure 5D, see also Figure 5F). These RNA species were specifically immunoprecipitated with the R1131 antibodies indicating that, despite being located within an intron, Or-CD1 gene is independently transcribed. This conclusion is in line with prior studies showing that Or-CD1 represents one of the very few SNORD genes that recruit RNA polymerase II and the Little Elongation Complex (LEC; (44)). In agreement with phylogenetic comparison that identifies two conserved antisense rRNA elements (Figure 5C), Or-CD1 displays a 5′-capped extension in both L. humile and A. mellifera (Figure 5D), as evidenced by the sizes of RNAs recovered in the immunoprecipitation pellet fraction. This therefore identifies a second expression strategy by which independent transcription from introns can also yield ‘regular’ extended forms of Or-CD1 (Figure 5E).
Thus, Or-CD1 in Diptera - but not in Arthropoda - displays only one antisense rRNA sequence (Figure 5C). In order to test if this remarkable fly SNORD13 was capable of guiding rRNA acetylation, a strain bearing a P-element inserted in the Or-CD1 gene was retrieved from the Bloomington Drosophila Stock Center (Dmel\PsnoRNA:Or-CD1G9117). After demonstrating that Or-CD1 was no longer detected in the mutant adult flies (Figure 5F), we showed that acetylation in helix45 was abolished (Figure 5G). Thus, SSU-C1968 in D. melanogaster can be targeted for acetylation by a SNORD13 homolog displaying a single antisense element. Of note, Or-CD1 adult knockout flies are viable, fertile and do not manifest gross abnormalities (not shown), thus strengthening the notion that N4-acetylcytidine at helix 45 is largely dispensable for viability, at least under standard laboratory conditions. Based on our findings, we propose to rename Or-CD1 as SNORD13.
Life without 18S rRNA acetylation: the cases of C. elegans and genetically modified ΔsnR4ΔsnR45 mutant yeast
A key role in ribosome biology of 18S rRNA helix 45, which in mature small ribosomal subunits lies directly adjacent to the decoding site, is implied by its extreme sequence conservation in many phylogenetically-distant organisms belonging to all known eukaryotic supergroups. Furthermore, when mutations in helix 45 arise, they are often accompanied by compensatory base changes (Supplementary Figure S6A). The 5′-CCG-3′ motif - wherein the second C is targeted for acetylation - was established as a key specificity sequence determinant for NAT10 (3,7). Remarkably, a few well-studied organisms lack this conserved motif or even the targeted substrate cytidine, including flagellated protozoan parasites (T. vaginalis, T. brucei, L. major), protozoan algae (E. gracilis), ciliate protozoa (T. thermophila), slime mold (P. polycephalum), corn smut fungus (U. maydis) or even the nematode (C. elegans) (Figure 4A). Other examples can be found in Supplementary Figure S6B. Only in the slime mold P. polycephalum did our ac4C sequencing reaction provide evidence for helix 45 acetylation within a non-consensus 5′-UCG-3′ motif, suggesting that NAT10 may have a distinct biochemical specificity in this organism (Supplementary Figure S7). Of note, helix 34 of P. polycephalum is apparently not acetylated despite the presence of a similar 5′-UCG-3′. Given its importance as animal model and the prior determination that it carries an essential NAT10 gene (named nath-10) (45), we next focused on C. elegans. As shown in Figure 6A, helix 45 of C. elegans 18S rRNA harbors a 5′-CUG-3′ motif also found in other nematodes, notably those belonging to the Clades IV and V. By definition, this motif variant that lacks a middle cytidine cannot be acetylated canonically. Nonetheless, since NAT10 is present in the worm genome, we verified the absence of any other ac4C residue within rRNA helix 45 prepared from adult C. elegans strain N2 (Figure 6B, not shown). More surprisingly, we found that the 5′-CCG-3′ motif in helix 34 was also devoid of acetylation (Figure 6B). Absence of ac4C in helix 45 and helix 34 was also true in embryos and larval stages (Supplementary Figure S7). Finally, the lack of rRNA acetylation in adult C. elegans elsewhere than in helices 34 and 45 was further examined using anti-ac4C immuno-northern blotting, in which signals for acetylated RNA were found to be limited to tRNAs (Figure 6C). It is important to note that the absence of ac4C on 18S rRNA in nematodes may concern only one of the two helices known to be modified. Indeed, in S. carpocapsae, an entomopathogenic nematode of the Steinernematidae family, only helix 34 is not acetylated (Supplementary Figure S7). The evolutionary forces that drove disappearance of rRNA acetylation in C. elegans remain unknown. Nonetheless, our results indicate that the changes in robustness of vulval cell-fate specification observed when the essential nath10 gene (45,46) was mutated did not reflect loss of rRNA modification but more likely the involvement of nath10 in pre-rRNA processing and/or tRNA acetylation. A similar conclusion was reached recently when it was shown that the involvement of Fibrillarin in neural crest cell maturation of Xenopus laevis depends on its role in pre-rRNA processing rather than in methylation (47). This is similar to prior observations in S. cerevisiae where Kre33 was shown to be essential at optimal (30°C) temperature but not snR45, neither snR4, or even double mutants ((17); Figures 6D-E). Normal growth for ΔsnR4-ΔsnR45 mutants at 30°C was also previously reported (32). In agreement with observations made with human SNORD13-KO cells, we also found that ac4C at helix 45 or helix 34 did not confer any obvious selective advantage in a growth competition assay between WT and ΔsnR45 (or ΔsnR4) mutant strains (Supplementary Figure S8A). Although loss of an individual rRNA modification in yeasts (e.g. 2′-O-methylations) can result in subtle changes in growth in the presence of antibiotics that target ribosomes (47), the sensitivity of ΔsnR45 or ΔsnR4 strains to hygromycin, cycloheximide, anisomycin and paromomycin also remains seemingly unchanged under the conditions tested (Supplementary Figure S8B). Finally, it is worth mentioning that the re-introduction of a catalytic-dead form of Kre33 (H545A) in Δkre33 mutants supports growth at 30°C and 19°C but not at 37°C while, as expected, the WT form rescued the growth of Δkre33 at the three tested temperatures (Figure 6D, left panels). Thus, lack of rRNA or tRNA acetylation at low (19°C) temperatures has no apparent detrimental growth defects. The growth of ΔsnR4 and ΔsnR45 (Figure 6D-right panels), and that of ΔsnR4-ΔsnR45 mutants as assayed in liquid or solid media (Figure 6E), were undistinguishable from that of WT at 37°C. Because Δtan1 mutants displays thermo-sensitivity at 37°C (Figure 6D, right panels), we infer that loss of tRNA acetylation is responsible for impaired growth at 37°C. This statement is in line with previous findings suggesting that decreased stability of unmodified serine tRNAs most likely accounts for growth defects of tan1 mutants at high (39°C) temperature (33). Altogether, we conclude that the essential function of Kre33 lies with its modification-independent, activity which is crucial for pre-rRNA processing and likely involves its helicase function (5), and also probably for tRNA acetylation under suboptimal conditions (high temperature) (33).
Figure 6.

Life without rRNA acetylation (A) Simplified phylogenetic tree of Nematoda. Sequence contexts of the acetylated cytidine in representative species of each clade are indicated. (B) Sanger DNA sequencing of RT-PCR products obtained after borohydride treatments of total RNA extracted from C. elegans. (C) Ac4C content in total RNA extracted from adult C. elegans was visualized by Northern blotting-based assay using anti-Ac4C antibodies. Ethidium bromide (etBr) staining indicates the relative position of rRNA and tRNA species. (D) A ten-fold serial dilution of cell suspensions of the different yeast strains (as indicated on the side of the panels) was spotted on a YPD-agar plate and incubated for 48 h at low (19°C), regular (30°C) and high (37°C) temperatures. (E) Top-panels: WT and ΔsnR4ΔsnR45 yeast strains were grown at 30°C (left) or 37°C (right) in liquid YPD. Optical density at 600 nm (OD600) was measured every 30 min. Growth curves of individual biological samples (thin lines; n = 3 or 12 for 30°C and 37°C samples, respectively) and the mean of replicates (thick lines) are presented along with a confidence interval (gray area). Bottom-panels: WT and ΔsnR4ΔsnR45 yeast strains were spotted on a YPD-agar plate and incubated for 48 h at 30°C or 37°C. Cells were grown in duplicates with a 5-fold dilution between distinct concentrations.
DISCUSSION
In order to interrogate the function of a single SNORD13-dependent cytidine acetylation in helix 45 of 18S rRNA, we have combined a cross-evolutionary survey of rRNA acetylation machinery across the eukaryotic tree of life and loss-of-function studies in four distinct models: budding yeasts, human cells, zebrafish and fly. This study represents one of the very few examples addressing the physiological roles of a SNORD in multicellular organisms bearing a germline-derived knockout (48–52). Although this peculiar rRNA ac4C is highly conserved, its loss surprisingly does not appear to grossly impact ribosome biogenesis or function, or even embryonic development. Consistent with this observation, we identify C. elegans as an evolutionary example of dispensable metazoan rRNA acetylation. Finally, the discovery of a 5′ truncated form of SNORD13 in D. melanogaster suggests that the mechanism by which SNORDs assist NAT10 in catalyzing rRNA ac4C is likely more diverse than previously anticipated and in some case may rely on a single antisense element.
An important question raised by our study concerns the specific role of ac4C in 18S rRNA helix 45. In humans, the ribosomal protein eL41 (formerly RPL41) is positioned at the interface between the 40S and 60S subunits and directly contacts ac4C1842 (31,53). This led to the proposal that acetylation may mediate functionally important crosstalk between the two subunits (54). However, our experiments failed to uncover any obvious defects in ribosomal subunit assembly or protein synthesis in human SNORD13-KO cells. This is perhaps not surprising since in yeast the knockout of eL41 does not affect translation unless the formation of another inter-subunit contact is compromised (55). Also arguing against a prominent role of ac4C in eL41 assembly and/or RNA::protein interaction is the finding that eL41 is correctly incorporated into ribosomes of T. vaginalis (33), E. gracilis (35), L. donovani (34) and T. brucei (34), all of which lack acetylation in helix 45 (Supplementary Figure S9). Furthermore, the relative positioning of eL41 in the ribosome and the 3-D architecture of helix 45 in the SSU of the archaea T. kodakarensis are not affected when TkNAT10 gene is deleted (7). Altogether, these observations point to a subtle molecular role for ac4C, possibly in fine tuning ribosome structure and/or function in sub-optimal environmental contexts and/or sensitized genetic backgrounds. Unravelling the precise physiological relevance of 18S rRNA acetylation in multicellular organisms, if any, represents a conceptually and technically formidable, but important, task for future studies.
Our bioinformatics search has considerably expanded the inventory of SNORD13 sequences in metazoans, identifying homologs well beyond vertebrates where they were previously thought to be restricted. However, we were unable to identify SNORD13 counterparts in many major phyla including Hemichordata, Annelida, Nematoda, Fungi (other than budding yeasts), algae or unicellular eukaryotes. Although the most parsimonious hypothesis would be that rapidly-evolving SNORD13 sequences make them difficult to detect, one cannot exclude the possibility that SNORD13 may have been lost in these lineages, which would imply that in these organisms rRNA acetylation may be catalyzed by NAT10 alone. More sophisticated structural and biochemical analyses will be critical to answer this question. An additional related question is whether acetylation of helix 34 is also strictly dependent on an antisense SNORD, as shown only in S. cerevisiae to date (17). The characterization of RNA-guided rRNA acetylation systems in Archaea, should they exist, represents an additional avenue that may lend invaluable evolutionary and mechanistic insights into this process (7,8).
Cross-linking analyses in yeast have previously described how SNORDs can guide the acetyltransferase to its rRNA substrate by use of two short base-pairing interactions on each side of the substrate cytosine, forming a ‘modification pocket’ akin to H/ACA snoRNAs (SNORA) acting in pseudouridylation (17). During the course of this study, we unexpectedly identified an independently-transcribed SNORD13 gene in D. melanogaster that produces RNA species lacking 5′-sequence complementary to rRNA (Figure 7). A similar strategy may be used by other Diptera for which SNORD13 is embedded within a very short intron (e.g. L. sericata, M. domestica). In such a scenario we envision that exonucleolytic processing of the host-intron produces uncapped RNAs with the C- and D-boxes brought together by a short terminal 5′-3′ terminal stem, as commonly occurring for other intron-located SNORDs (43). The genomic constraints and selection pressure that led to these different SNORD13 expression strategies across evolution are unknown, yet they have inherent mechanistic repercussions since the deposition of ac4C in helix 45 in D. melanogaster does not strictly require two antisense elements flanking the rRNA ac4C site. This finding contrasts with conclusions drawn from mutagenesis of yeast snR45 where the 5′ antisense element appears absolutely essential for acetylation (17). This implies an alternative mode of action of SNORD13 in D. melanogaster and, more broadly, in Diptera. Intriguingly, the overall structure of SNORD13 in Diptera is indistinguishable from that of the canonical intron-located SNORDs which mediate rRNA ribose methylation, raising the question as to whether other intronic SNORDs in these organisms, particularly those of uncharacterized function, might mediate acetylation of novel RNA targets.
Figure 7.
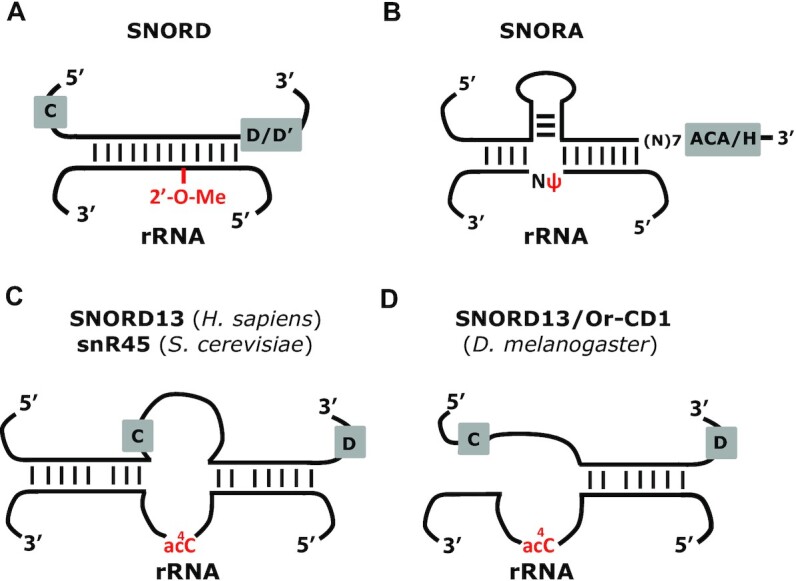
Comparative schematic representation of rRNA modification systems. (A) Through the formation of a perfect (or near perfect) RNA duplex (∼10–20 bp), SNORDs guide Fibrillarin/Nop1 to their target rRNA nucleotides, i.e. the nucleotide to be 2′-O-methylated (2′-O-Me) is paired to the fifth position upstream of the conserved D (or D’) box. (B) Through the formation of two perfect (or near perfect) RNA duplexes (∼4–10 bp each), SNORAs guide Dyskerin/Cbf5 to their target rRNA nucleotides, i.e. the uridine to be isomerized into pseudouridine (ψ) remains unpaired and it is located at ∼14–16 nt from the conserved H (or ACA) box. (C) snR45, snR4 and SNORD13 contain two imperfect antisense elements matching either side of the cytidine to be acetylated (ac4C) and, through still unknown mechanisms, guide cytosine acetylation by NAT10/Kre33. (D) In D. melanogaster, SNORD13 targets rRNA acetylation at helix 45 through the use of a single antisense rRNA sequence.
Altogether, our data shed light on interlaced questions regarding SNORD13 sequence diversity across evolution, the molecular mechanisms underlying site-specific rRNA acetylation, and the physiological importance of ac4C in rRNA in cell homeostasis, development and physiology. We propose further application of the evolutionary perspective our studies offer is warranted since—paradoxically not to mention ironically—our insights from the widely-studied model organisms C. elegans and D. melanogaster depart considerably from prior observation made in vertebrates and budding yeast.
DATA AVAILABILITY
The data that support the findings of this study are available from the corresponding author [J.C.] upon reasonable request.
Supplementary Material
ACKNOWLEDGEMENTS
This paper is dedicated to the discovery of the SNORD13/18S RNA duplex by Jean-Pierre Bachellerie in 1995. We thank Isahak Saidi for his technical assistance in the very early stages of the project. We thank the many colleagues who kindly provided us with biological samples, particularly EMBRC-France (Nathalie Turque, Régis Lasbleiz, Axel Duchene), the Botanical garden of Toulouse, Insectosphere, Botanic-Blagnac, Anna Mattout, Sylvie Tournier, Isabelle Massou, Guillaume Canal, Raphaël Jeanson, Luisa Di Stefano, Philippe Valenti, Audrey Dussutour, Mireille Bétermier, Joana Santos, Kazufumi Mochizuki, Guillaume Allorent and Pierre-Marc Delaux. We are also grateful to Aurore Laire for excellent zebrafish care and Peter Watzinger for his help with competition assay in yeasts.
Notes
Present address: Aldema Sas-Chen, The Shmunis School of Biomedicine and Cancer Research, The George S. Wise Faculty of Life Sciences, Tel Aviv University, Tel Aviv, Israel.
Present address: Sunny Sharma, Department of Cell Biology and Neurosciences, Rutgers, The State University of New Jersey, Piscataway, NJ 0885, USA.
Present address: Laurence Vandel. Université Clermont Auvergne, CNRS, INSERM, iGReD, F-63000 Clermont-Ferrand, France.
Contributor Information
Marie-Line Bortolin-Cavaillé, Molecular, Cellular and Developmental Biology (MCD), UMR5077, Centre de Biologie Intégrative (CBI), Université de Toulouse, CNRS, UPS, F-31062 Toulouse, France.
Aurélie Quillien, Molecular, Cellular and Developmental Biology (MCD), UMR5077, Centre de Biologie Intégrative (CBI), Université de Toulouse, CNRS, UPS, F-31062 Toulouse, France.
Supuni Thalalla Gamage, Chemical Biology Laboratory, National Cancer Institute, Frederick, MD 21702, USA.
Justin M Thomas, Chemical Biology Laboratory, National Cancer Institute, Frederick, MD 21702, USA.
Aldema Sas-Chen, Department of Molecular Genetics, Weizmann Institute of Science, Rehovot, Israel.
Sunny Sharma, RNA Molecular Biology, Fonds de la Recherche Scientifique (F.R.S./FNRS), Université libre de Bruxelles (ULB), Biopark campus, B-6041 Gosselies, Belgium.
Célia Plisson-Chastang, Molecular, Cellular and Developmental Biology (MCD), UMR5077, Centre de Biologie Intégrative (CBI), Université de Toulouse, CNRS, UPS, F-31062 Toulouse, France.
Laurence Vandel, Molecular, Cellular and Developmental Biology (MCD), UMR5077, Centre de Biologie Intégrative (CBI), Université de Toulouse, CNRS, UPS, F-31062 Toulouse, France.
Patrick Blader, Molecular, Cellular and Developmental Biology (MCD), UMR5077, Centre de Biologie Intégrative (CBI), Université de Toulouse, CNRS, UPS, F-31062 Toulouse, France.
Denis L J Lafontaine, RNA Molecular Biology, Fonds de la Recherche Scientifique (F.R.S./FNRS), Université libre de Bruxelles (ULB), Biopark campus, B-6041 Gosselies, Belgium.
Schraga Schwartz, Department of Molecular Genetics, Weizmann Institute of Science, Rehovot, Israel.
Jordan L Meier, Chemical Biology Laboratory, National Cancer Institute, Frederick, MD 21702, USA.
Jérôme Cavaillé, Molecular, Cellular and Developmental Biology (MCD), UMR5077, Centre de Biologie Intégrative (CBI), Université de Toulouse, CNRS, UPS, F-31062 Toulouse, France.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Agence Nationale de la Recherche [ANR-18- CE12-0008-01]; research in the lab of J.L.M. was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research [ZIA-BC011488]; research in the lab of D.L.J.L. is supported by the Belgian Fonds de la Recherche Scientifique (F.R.S./FNRS); Université Libre de Bruxelles (ULB); European Joint Programme on Rare Diseases (EJP-RD ‘RiboEurope’ and ‘DBAGencure’); Région Wallonne (SPW EER ‘RIBOcancer’); Internationale Brachet Stiftüng; Epitran COST action [CA16120]. Funding for open access charge: Agence Nationale de la Recherche [ANR-18- CE12-0008-01].
Conflict of interest statement. None declared.
REFERENCES
- 1. Bruenger E., Kowalak J.A., Kuchino Y., McCloskey J.A., Mizushima H., Stetter K.O., Crain P.F.. 5S rRNA modification in the hyperthermophilic archaea Sulfolobus solfataricus and Pyrodictium occultum. FASEB J. 1993; 7:196–200. [DOI] [PubMed] [Google Scholar]
- 2. Thomas G., Gordon J., Rogg H.. N4-Acetylcytidine. A previously unidentified labile component of the small subunit of eukaryotic ribosomes. J. Biol. Chem. 1978; 253:1101–1105. [PubMed] [Google Scholar]
- 3. Ito S., Akamatsu Y., Noma A., Kimura S., Miyauchi K., Ikeuchi Y., Suzuki T.. A single acetylation of 18 s rRNA is essential for biogenesis of the small ribosomal subunit in Saccharomyces cerevisiae. J. Biol. Chem. 2014; 289:26201–26212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ito S., Horikawa S., Suzuki T., Kawauchi H., Tanaka Y.. Human NAT10 is an ATP-dependent RNA acetyltransferase responsible for N4-acetylcytidine formation in 18 S ribosomal RNA (rRNA). J. Biol. Chem. 2014; 289:35724–35730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sharma S., Langhendries J.L., Watzinger P., Kotter P., Entian K.D., Lafontaine D.L.. Yeast Kre33 and human NAT10 are conserved 18S rRNA cytosine acetyltransferases that modify tRNAs assisted by the adaptor Tan1/THUMPD1. Nucleic Acids Res. 2015; 43:2242–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Taoka M., Ishikawa D., Nobe Y., Ishikawa H., Yamauchi Y., Terukina G., Nakayama H., Hirota K., Takahashi N., Isobe T.. RNA cytidine acetyltransferase of small-subunit ribosomal RNA: identification of acetylation sites and the responsible acetyltransferase in fission yeast, Schizosaccharomyces pombe. PLoS One. 2014; 9:e112156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sas-Chen A., Thomas J.M., Matzov D., Taoka M., Nance K.D., Nir R., Bryson K.M., Shachar R., Liman G.L.S., Burkhart B.W.et al.. Dynamic RNA acetylation revealed by quantitative cross-evolutionary mapping. Nature. 2020; 583:638–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Coureux P.D., Lazennec-Schurdevin C., Bourcier S., Mechulam Y., Schmitt E.. Cryo-EM study of an archaeal 30S initiation complex gives insights into evolution of translation initiation. Commun Biol. 2020; 3:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Johansson M.J., Bystrom A.S.. The Saccharomyces cerevisiae TAN1 gene is required for N4-acetylcytidine formation in tRNA. RNA. 2004; 10:712–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Arango D., Sturgill D., Alhusaini N., Dillman A.A., Sweet T.J., Hanson G., Hosogane M., Sinclair W.R., Nanan K.K., Mandler M.D.et al.. Acetylation of Cytidine in mRNA promotes translation efficiency. Cell. 2018; 175:1872–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tardu M., Jones J.D., Kennedy R.T., Lin Q., Koutmou K.S.. Identification and quantification of modified nucleosides in Saccharomyces cerevisiae mRNAs. ACS Chem. Biol. 2019; 14:1403–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu H.Y., Liu Y.Y., Yang F., Zhang L., Zhang F.L., Hu X., Shao Z.M., Li D.Q.. Acetylation of MORC2 by NAT10 regulates cell-cycle checkpoint control and resistance to DNA-damaging chemotherapy and radiotherapy in breast cancer. Nucleic Acids Res. 2020; 48:3638–3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu X., Tan Y., Zhang C., Zhang Y., Zhang L., Ren P., Deng H., Luo J., Ke Y., Du X.. NAT10 regulates p53 activation through acetylating p53 at K120 and ubiquitinating Mdm2. EMBO Rep. 2016; 17:349–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shen Q., Zheng X., McNutt M.A., Guang L., Sun Y., Wang J., Gong Y., Hou L., Zhang B.. NAT10, a nucleolar protein, localizes to the midbody and regulates cytokinesis and acetylation of microtubules. Exp. Cell. Res. 2009; 315:1653–1667. [DOI] [PubMed] [Google Scholar]
- 15. Balmus G., Larrieu D., Barros A.C., Collins C., Abrudan M., Demir M., Geisler N.J., Lelliott C.J., White J.K., Karp N.A.et al.. Targeting of NAT10 enhances healthspan in a mouse model of human accelerated aging syndrome. Nat. Commun. 2018; 9:1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Larrieu D., Britton S., Demir M., Rodriguez R., Jackson S.P.. Chemical inhibition of NAT10 corrects defects of laminopathic cells. Science. 2014; 344:527–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sharma S., Yang J., van Nues R., Watzinger P., Kotter P., Lafontaine D.L.J., Granneman S., Entian K.D.. Specialized box C/D snoRNPs act as antisense guides to target RNA base acetylation. PLoS Genet. 2017; 13:e1006804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Watkins N.J., Bohnsack M.T.. The box C/D and H/ACA snoRNPs: key players in the modification, processing and the dynamic folding of ribosomal RNA. Wiley Interdiscip. Rev. RNA. 2012; 3:397–414. [DOI] [PubMed] [Google Scholar]
- 19. Bachellerie J.P., Michot B., Nicoloso M., Balakin A., Ni J., Fournier M.J.. Antisense snoRNAs: a family of nucleolar RNAs with long complementarities to rRNA. Trends Biochem. Sci. 1995; 20:261–264. [DOI] [PubMed] [Google Scholar]
- 20. Cavaille J., Hadjiolov A.A., Bachellerie J.P.. Processing of mammalian rRNA precursors at the 3′ end of 18S rRNA. Identification of cis-acting signals suggests the involvement of U13 small nucleolar RNA. Eur. J. Biochem. 1996; 242:206–213. [DOI] [PubMed] [Google Scholar]
- 21. Kim S.H., Spensley M., Choi S.K., Calixto C.P., Pendle A.F., Koroleva O., Shaw P.J., Brown J.W.. Plant U13 orthologues and orphan snoRNAs identified by RNomics of RNA from Arabidopsis nucleoli. Nucleic Acids Res. 2010; 38:3054–3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tyc K., Steitz J.A.. U3, U8 and U13 comprise a new class of mammalian snRNPs localized in the cell nucleolus. EMBO J. 1989; 8:3113–3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cavaille J., Bachellerie J.P.. SnoRNA-guided ribose methylation of rRNA: structural features of the guide RNA duplex influencing the extent of the reaction. Nucleic Acids Res. 1998; 26:1576–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Montellese C., Montel-Lehry N., Henras A.K., Kutay U., Gleizes P.E., O’Donohue M.F. Poly(A)-specific ribonuclease is a nuclear ribosome biogenesis factor involved in human 18S rRNA maturation. Nucleic Acids Res. 2017; 45:6822–6836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Martineau Y., Azar R., Muller D., Lasfargues C., El Khawand S., Anesia R., Pelletier J., Bousquet C., Pyronnet S.. Pancreatic tumours escape from translational control through 4E-BP1 loss. Oncogene. 2014; 33:1367–1374. [DOI] [PubMed] [Google Scholar]
- 26. Yang J., Sharma S., Watzinger P., Hartmann J.D., Kotter P., Entian K.D.. Mapping of complete set of ribose and base modifications of yeast rRNA by RP-HPLC and mung bean nuclease assay. PLoS One. 2016; 11:e0168873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Alestrom P., D’Angelo L., Midtlyng P.J., Schorderet D.F., Schulte-Merker S., Sohm F., Warner S.. Zebrafish: housing and husbandry recommendations. Lab. Anim. 2020; 54:213–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kimmel C.B., Ballard W.W., Kimmel S.R., Ullmann B., Schilling T.F.. Stages of embryonic development of the zebrafish. Dev. Dyn. 1995; 203:253–310. [DOI] [PubMed] [Google Scholar]
- 29. Quillien A., Moore J.C., Shin M., Siekmann A.F., Smith T., Pan L., Moens C.B., Parsons M.J., Lawson N.D. Distinct notch signaling outputs pattern the developing arterial system. Development. 2014; 141:1544–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lawson N.D., Scheer N., Pham V.N., Kim C.H., Chitnis A.B., Campos-Ortega J.A., Weinstein B.M.. Notch signaling is required for arterial-venous differentiation during embryonic vascular development. Development. 2001; 128:3675–3683. [DOI] [PubMed] [Google Scholar]
- 31. Natchiar S.K., Myasnikov A.G., Kratzat H., Hazemann I., Klaholz B.P.. Visualization of chemical modifications in the human 80S ribosome structure. Nature. 2017; 551:472–477. [DOI] [PubMed] [Google Scholar]
- 32. Dudnakova T., Dunn-Davies H., Peters R., Tollervey D.. Mapping targets for small nucleolar RNAs in yeast. Wellcome Open Res. 2018; 3:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dewe J.M., Whipple J.M., Chernyakov I., Jaramillo L.N., Phizicky E.M.. The yeast rapid tRNA decay pathway competes with elongation factor 1A for substrate tRNAs and acts on tRNAs lacking one or more of several modifications. RNA. 2012; 18:1886–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shalev-Benami M., Zhang Y., Rozenberg H., Nobe Y., Taoka M., Matzov D., Zimmerman E., Bashan A., Isobe T., Jaffe C.L.et al.. Atomic resolution snapshot of Leishmania ribosome inhibition by the aminoglycoside paromomycin. Nat. Commun. 2017; 8:1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Matzov D., Taoka M., Nobe Y., Yamauchi Y., Halfon Y., Asis N., Zimermann E., Rozenberg H., Bashan A., Bhushan S.et al.. Cryo-EM structure of the highly atypical cytoplasmic ribosome of Euglena gracilis. Nucleic Acids Res. 2020; 48:11750–11761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E.. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004; 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 37. Emsley P., Lohkamp B., Scott W.G., Cowtan K.. Features and development of Coot. Acta Crystallogr. D, Biol. Crystallogr. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Essletzbichler P., Konopka T., Santoro F., Chen D., Gapp B.V., Kralovics R., Brummelkamp T.R., Nijman S.M., Burckstummer T.. Megabase-scale deletion using CRISPR/Cas9 to generate a fully haploid human cell line. Genome Res. 2014; 24:2059–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Thomas J.M., Briney C.A., Nance K.D., Lopez J.E., Thorpe A.L., Fox S.D., Bortolin-Cavaille M.L., Sas-Chen A., Arango D., Oberdoerffer S.et al.. A chemical signature for cytidine acetylation in RNA. J. Am. Chem. Soc. 2018; 140:12667–12670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Langhendries J.L., Nicolas E., Doumont G., Goldman S., Lafontaine D.L.. The human box C/D snoRNAs U3 and U8 are required for pre-rRNA processing and tumorigenesis. Oncotarget. 2016; 7:59519–59534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Huang Z.P., Zhou H., He H.L., Chen C.L., Liang D., Qu L.H.. Genome-wide analyses of two families of snoRNA genes from Drosophila melanogaster, demonstrating the extensive utilization of introns for coding of snoRNAs. RNA. 2005; 11:1303–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li D., Wang Y., Zhang K., Jiao Z., Zhu X., Skogerboe G., Guo X., Chinnusamy V., Bi L., Huang Y.et al.. Experimental RNomics and genomic comparative analysis reveal a large group of species-specific small non-message RNAs in the silkworm Bombyx mori. Nucleic Acids Res. 2011; 39:3792–3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kiss T., Filipowicz W.. Exonucleolytic processing of small nucleolar RNAs from pre-mRNA introns. Genes Dev. 1995; 9:1411–1424. [DOI] [PubMed] [Google Scholar]
- 44. Smith E.R., Lin C., Garrett A.S., Thornton J., Mohaghegh N., Hu D., Jackson J., Saraf A., Swanson S.K., Seidel C.et al.. The little elongation complex regulates small nuclear RNA transcription. Mol. Cell. 2011; 44:954–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Duveau F., Felix M.A.. Role of pleiotropy in the evolution of a cryptic developmental variation in Caenorhabditis elegans. PLoS Biol. 2012; 10:e1001230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hintze M., Katsanos D., Shahrezaei V., Barkoulas M.. Phenotypic robustness of epidermal stem cell number in c. elegans is modulated by the activity of the conserved N-acetyltransferase nath-10/NAT10. Front. Cell Dev. Biol. 2021; 9:640856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Esguerra J., Warringer J., Blomberg A.. Functional importance of individual rRNA 2'-O-ribose methylations revealed by high-resolution phenotyping. RNA. 2008; 14:649–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ding F., Li H.H., Zhang S., Solomon N.M., Camper S.A., Cohen P., Francke U.. SnoRNA Snord116 (Pwcr1/MBII-85) deletion causes growth deficiency and hyperphagia in mice. PLoS One. 2008; 3:e1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hebras J., Marty V., Personnaz J., Mercier P., Krogh N., Nielsen H., Aguirrebengoa M., Seitz H., Pradere J.P., Guiard B.P.et al.. Reassessment of the involvement of Snord115 in the serotonin 2c receptor pathway in a genetically relevant mouse model. Elife. 2020; 9:e60862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lee J., Harris A.N., Holley C.L., Mahadevan J., Pyles K.D., Lavagnino Z., Scherrer D.E., Fujiwara H., Sidhu R., Zhang J.et al.. Rpl13a small nucleolar RNAs regulate systemic glucose metabolism. J. Clin. Invest. 2016; 126:4616–4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Skryabin B.V., Gubar L.V., Seeger B., Pfeiffer J., Handel S., Robeck T., Karpova E., Rozhdestvensky T.S., Brosius J.. Deletion of the MBII-85 snoRNA gene cluster in mice results in postnatal growth retardation. PLoS Genet. 2007; 3:e235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Soeno Y., Fujita K., Kudo T., Asagiri M., Kakuta S., Taya Y., Shimazu Y., Sato K., Tanaka-Fujita R., Kubo S.et al.. Generation of a mouse model with down-regulated U50 snoRNA (SNORD50) expression and its organ-specific phenotypic modulation. PLoS One. 2013; 8:e72105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ben-Shem A., Garreau de Loubresse N., Melnikov S., Jenner L., Yusupova G., Yusupov M.. The structure of the eukaryotic ribosome at 3.0 a resolution. Science. 2011; 334:1524–1529. [DOI] [PubMed] [Google Scholar]
- 54. Sharma S., Lafontaine D.L.J.. View from a bridge’: a new perspective on eukaryotic rRNA base modification. Trends Biochem. Sci. 2015; 40:560–575. [DOI] [PubMed] [Google Scholar]
- 55. Tamm T., Kisly I., Remme J.. Functional interactions of ribosomal intersubunit bridges in Saccharomyces cerevisiae. Genetics. 2019; 213:1329–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Makarova J.A., Kramerov D.A.. SNOntology: Myriads of novel snoRNAs or just a mirage?. BMC Genom. 2011; 12:543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. McCarroll R., Olsen G.J., Stahl Y.D., Woese C.R., Sogin M.L.. Nucleotide Sequence of the Dictyostelium discoideum Small-Subunit Ribosomal Ribonucleic Acid Inferred from the Gene Sequence: Evolutionary Implications. Biochemistry. 1983; 22:5858–5868. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author [J.C.] upon reasonable request.