

Abstract
Acephalic spermatozoa syndrome (ASS) is one of the most severe spermatogenic failures of all infertility in men. The cognition of ASS has experienced a tortuous process. Over the past years, with the in-depth understanding of spermatogenesis and the emergence of new genetic research technologies, the unraveling of the genetic causes of spermatogenic failure has become highly active. From these advances, we established a genetic background and made significant progress in the discovery of the genetic causes of ASS. It is important to identify pathogenic genes and mutations in ASS to determine the biological reasons for the occurrence of the disease as well as provide genetic diagnosis and treatment strategies for patients with this syndrome. In this review, we enumerate various technological developments, which have made a positive contribution to the discovery of candidate genes for ASS from the past to the present. Simultaneously, we summarize the known genetic etiology of this phenotype and the clinical outcomes of treatments in the present. Furthermore, we propose perspectives for further study and application of genetic diagnosis and assisted reproductive treatment in the future.
Keywords: acephalic spermatozoa syndrome, genetic pathogenesis, male infertility
INTRODUCTION
Infertility is not only a health problem, but also a social problem, which affects approximately 15% of couples worldwide both mentally and physically.1 The rate of male factors contributing to infertility problems is approximately 50%,1 among which spermatogenic failure is significant. Spermatogenesis is completed by a series of processes in cell proliferation and differentiation, which can be divided into three crucial steps: mitotic multiplication, meiosis, and spermiogenesis. Spermatogenic failure appears, if any step is disordered. Teratospermia is often caused by failure of the third step.
Acephalic spermatozoa syndrome (ASS) is a classical type of the most serious teratozoospermia, defined as a type of abnormal sperm morphology (headless spermatozoa) predominance in the ejaculate.2 This abnormal sperm morphology was first misdescribed as “pin head spermatozoa”.3 After a series of reports paralleled to this disease2,4,5,6 and a deeper understanding of spermatogenesis, the terms “decapitated spermatozoa” and “acephalic spermatozoa” were introduced to describe a developmental disturbance of the sperm head–tail coupling apparatus (HTCA), which is the essence of the emergence of headless sperm tails in patients’ ejaculate.5 According to familial incidence, we have known the genetic origin of ASS for a long time,7 but had not identified its genetic causes in humans until 2016,8 owing to the poor understanding of spermatogenesis and the limitation of the technology.
In this review, we contextualize the contributions of different technological advances in a historical overview of the progress in genetic studies of ASS and detail the existing achievements in the study, diagnosis, and treatment of ASS (Figure 1). Additionally, we provide perspectives on future directions.
Figure 1.
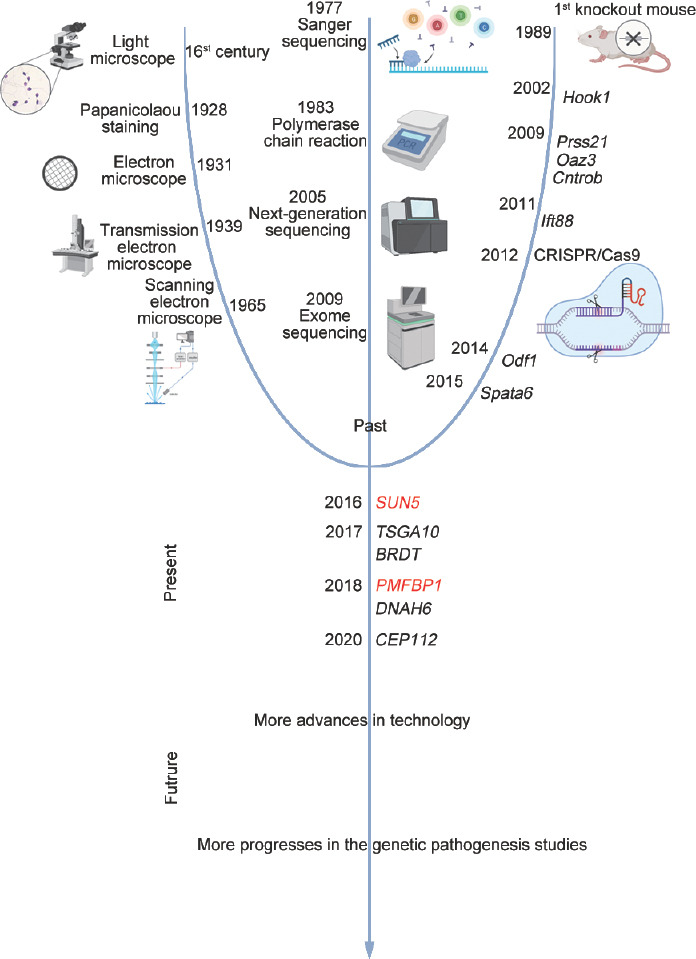
Timeline of the advancement of technologies and the discovery of key genes involved in ASS. Various technological developments were highlighted, which contributed significantly, as well as the discovery of candidate genes of mutations causing ASS according to the timeline. ASS: acephalic spermatozoa syndrome; Hook1: hook microtubule tethering protein 1; Prss21: serine protease 21; Oaz3: ornithine decarboxylase antizyme 3; Cntrob: centrobin, centriole duplication and spindle assembly protein; Ift88: intraflagellar transport 88; CRISPR/Cas9: clustered regularly interspaced short palindromic repeats/CRISPR-associated 9; Odf1: outer dense fiber of sperm tails 1; Spata6: spermatogenesis associated 6; SUN5: Sad1 and UNC84 domain containing 5; PMFBP1: polyamine-modulated factor 1 binding protein 1; TSGA10: testis-specific gene antigen 10; BDRT: bromodomain testis-specific protein; DNAH6: dynein axonemal heavy chain 6; CEP112: centrosomal protein 112.
THE PAST: THE RUDIMENTARY RECOGNITION PHASE FOR ASS
Morphologic descriptions of ASS
The cognition of the spermatozoa structure can be traced back to the 17th century, when Antonie van Leeuwenhoek used light microscopy to observe the seminal fluid of animals and men for the first time.9 The application of Papanicolaou staining allows direct and easy observation of sperm morphology, which was invented by the Greek pathologist Papanicolaou in 1928.10 Typical abnormal sperm morphological changes are hitherto the main basis of ASS diagnosis. However, because of the technical limitations of light microscopy, this diagnosis is only based on an observed alteration in the sperm shape, rather than the cellular basis of their functional incompetence responsible for male infertility. The subsequent invention of an electron microscope further uncovered the ultrastructure of the sperm. Corresponding to it, Zaneveld and Polakoski3 first referred to the acephalic sperm as “pin heads” in 1977, which was proved by ultrastructure to be a misunderstanding that the heads were in fact absent and was later corrected to “decapitated spermatozoa” and “acephalic spermatozoa”.2,5 From the careful description by the electron microscopic characteristics, we soon determined that the essence of ASS is the abnormal development of HTCA. Moreover, building on the recognition of normal spermatozoa characteristics in relatively recent times, ASS is classified into three subtypes according to the broken points in HTCA through ultrastructural observations. The normal HTCA is made up of the capitulum, proximal centriole, degenerating distal centriole, beginning of tail axoneme, and nine segmented columns around. The breakage between the two centrioles is defined as subtype I of ASS, and the fracture site of subtype II is positioned between the nucleus and the proximal centriole, whereas the broken point of subtype III is situated between the distal centriole and midpiece.
Beginning of genetics studies of ASS
As the number of similar reported cases grew, a genetic background of ASS was established and an autosomal recessive pattern of inheritance was speculated by the family clustering and homogeneity according to Mendelian's law.7 Despite our early awareness of the genetic origin of ASS, genetic studies remain at a standstill, limited by the low incidence of ASS and poor technology. In recent decades, the rapid development of next-generation sequencing (NGS) has provided the technological basis for the genetic study of ASS.
Serendipitous findings in animal models
Breeding is a ubiquitous phenomenon in biology. The advent of animal models has facilitated genetic studies on fertility problems. Limited by the extremely low incidence of ASS in humans and the lag in technologies, we first obtain serendipitous findings in animal models. Sperm with different degrees of impairment of HTCA have been reported in knockout mice, which provide valuable models for exploring the etiologies of human ASS.
Hook microtubule tethering protein 1 (Hook1)
Hook1 encodes a protein located in the microtube of sperm and belongs to the hook family. It has three crucial domains: the NH2 -domain, responsible for microtubule fixing; the central coiled-coil motif, which is easy to homodimerize; and the organelle-binding domains in the C-terminal. While studying mice with abnormal spermatozoon head shape (azh) mutation, we found decapitated sperm in mice. Further studies demonstrated that Hook1 co-localizes with the azh locus. If Hook1 does not anchor the microtubule cytoskeleton at the endocytic membrane before loss of function, the microtubule structures of the sperm will mislocate.11
Serine protease 21(Prss21)
Glycosylphosphatidylinositol-anchored Prss21 is also known as testisin. Prss21 exerts its effect as a tryptic serine protease that plays multiple roles in spermatogenesis and fertilization. In mature spermatozoa, Prss21 is located in the sperm neck region and midpiece. This could explain the appearance of decapitated spermatozoa in the spermatozoa of mice lacking Prss21, which may participate in the assembly of sperm HTCA by inserting a midpiece into the plasma membrane via glycosylphosphatidylinositol (GPI) anchors.12
Ornithine decarboxylase antizyme 3 (Oaz3)
Oaz3 is a testis-specific member of the ornithine decarboxylase antizyme gene family. Its corresponding protein product is localized in the outer dense fibrous structure and the head-tail connecting piece of sperm. A breakage between the basal plate and capitulum in the sperm from the Oaz3−/− mice was observed, similar to the phenotype of the human ASS subtype II.13,14
Centrobin, centriole duplication and spindle assembly protein (Cntrob)
Liska et al.15 designated Cntrob as a novel candidate gene for ASS. Its encoded protein, centrobin, was shown to be localized at the manchette well-known as a transporter during spermatogenesis when hypodactylous (hd) mutation was evaluated in rat. Mutant spermatids appeared decapitated, and the centrosomes broke away from the caudal nuclear region. This may be explained by disruption in the transport of proteins during HTCA formation.15
Intraflagellar transport 88 (Ift88)
Ift88 encodes a transport protein that can be detected in the trans-Golgi, the spermatid manchette, and finally in the acrosome-acroplaxome region during spermatogenesis. In Ift88−/− mice, the sperm displayed not only abnormal head shaping but also tail-lessness, which was the result of arresting in the transport of cargoes along the microtubule-containing manchette.16
Outer dense fiber of sperm tails 1 (Odf1)
Odf1 encodes a cytoskeletal protein as a structural component of the outer dense fiber surrounding the sperm tail axoneme. Odf1 assists the sperm tail to attach to the head stably, while assist mitochondrial sheaths and outer dense fibers to arrange correctly. The sperm of Odf1−/− mice also contained decapitated sperm like that in ASS patients.17,18
Spermatogenesis associated 6 (Spata6)
Spata6 is the first gene involved in the single phenotype of headless spermatozoa in mice, which is different from the genes found above. It encodes a highly conserved protein expressed in the testis, which is indispensable for the formation of segmented columns and the capitulum in HTCA. Without interaction with myosin subunits, the inactivation of SPATA6 results in the failure of the head and neck connection by disrupting myosin-based microfilament transport.19
Although the knockout of several genes in mice induced a phenotype analogous to ASS, most of their mutations were not found to be associated with ASS in humans. The genetic heterogeneity of this syndrome, combined with the different biomechanisms among species, might explained it.20 In spite of this, on account of the similarities of spermatogenesis between mice and humans, and the high conservation of exclusively expressed genes during spermatogenesis, knockout mouse models offer important and reasonable clues for decrypting the pathogenesis of ASS in humans.
THE PRESENT: SIGNIFICANT PROGRESS IN THE STUDIES OF HUMAN ASS IN GENETIC DIAGNOSIS AND TREATMENTS
Developments in sequencing technologies have made breakthroughs in the discovery of genetic causes of human ASS
Although remarkable advancements have been made in understanding and characterizing the spermiogenesis and ASS, identifying the pathogenic genes and molecular basis of ASS is still a slow process because an enormous number of genes are involved during spermatogenesis without effective means.21 Defects in any relevant genes can disturb spermatogenesis and induce ASS, particularly the genes involved in the assembly of sperm HTCA. Sequencing technology has been used as a detector for genetic defects. In the past, the application of gene sequencing was scarce because of its high price and low throughput. However, in the last decade, with the rapid development of high-throughput sequencing platforms, sequencing costs have dramatically reduced, whereas sequencing throughput has concurrently increased. The new generation of sequencing technologies, such as whole-exome sequencing (WES) and whole genome sequencing (WGS), allow an increasing number of patients to undergo a thorough inspection of their genomes, rather than only sequencing the selected and individual candidate genes. Briefly, NGS has provided a cheaper and faster genetic screening approach to discovering novel candidate genes.22 Profited by the extensive application of NGS, a series of pathogenic genes and mutations of human ASS have been revealed in recent years (Table 1). Moreover, with the huge advancements in modern morphological, biochemical, and molecular techniques, and reproductive medicine in recent years, we have demonstrated the pathogenic mechanism of these genes and mutations. The proper design of in vitro systems can assess the functional impact of the mutation by constructing individuals carrying a specific mutation. The emergence of new gene editing techniques also provides important clues for understanding the etiology of ASS by creating the mutant animal models. Certainly, the development of technologies has already and will continue to improve our ability to perform functional validation.
Table 1.
Genetic mutations discovered in infertile men with ASS
| Gene name | Gene mutation | Mutation type | Protein variant | Protein domain | ASS subtype | Study |
|---|---|---|---|---|---|---|
| SUN5 (NM_080675.4) | c.824C>T | Missense | p.Thr275Met | SUN domain | II | Zhu et al.8 |
| c.1006C>T | Missense | p.Arg356Cys | SUN domain | Zhu et al.8 | ||
| c.485T>A | Missense | p.Met162Lys | Coiled-coil domain | Zhu et al.8 | ||
| c.381delA | Frameshift | p.Val128Serfs*7 | - | Zhu et al.,8 Sha et al.,25 Zhang et al.29 | ||
| c.781G>A | Missense | p.Val261Met | SUN domain | Zhu et al.8 | ||
| c.216G>A | Nonsense | p.Trp72* | N-terminus | Zhu et al.8 | ||
| c.1043A>T | Missense | p.Asn348Ile | SUN domain | Zhu et al.8 | ||
| c.425+1G>A | Splicing mutation | - | - | Zhu et al.8 | ||
| c.851C>G | Nonsense | p.Ser284* | SUN domain | Zhu et al.8 | ||
| c.340G>A | Splicing mutation | p.Gly114Arg | Transmembrane domain | Zhu et al.8 | ||
| GRCh38-chr20:32995761_32990672delinsTGGT | Frameshift | p.Leu143Serfs*30 | - | Elkhatib et al.23 | ||
| c.475C>T | Nonsense | p.Arg159* | Coiled-coil domain | Shang et al.26 | ||
| c.829C>T | Nonsense | p.Gln277* | SUN domain | Fang et al.24 | ||
| c.1067G>A | Missense | p.Arg356His | SUN domain | Fang et al.24 | ||
| c.211+1 insGT | Frameshift | p.Ser71Cysfs11* | N-terminus | Fang et al.24 | ||
| c.772C>T | Missense | p.Arg258Cys | SUN domain | Liu et al.27 | ||
| c.211+1_211+2dup | Frameshift | p.Ser71Cysfs11* | N-terminus | Cazin et al.28 | ||
| c.675C>A | Nonsense | p.Y225* | SUN domain | Zhang et al.29 | ||
| c.88C>T | Nonsense | p.R30* | N-terminus | Zhang et al.29 | ||
| PMFBP1 (NM_031293.3) | c.1462C>T | Nonsense | p.Gln488* | Smc domain | II | Zhu et al.31 |
| c.2404C>T | Nonsense | p.Gln802* | Smc domain | Zhu et al.31 | ||
| c.2725C>T | Nonsense | p.Arg909* | C-terminus | Zhu et al.31 | ||
| c.2092delG | Frameshift | p.Ala698Profs*7 | Smc domain | Zhu et al.31 | ||
| c.2561_2562del | Frameshift | p.Lys854Argfs*5 | Smc domain | Sha et al.32 | ||
| c.327T>A | Nonsense | p.Tyr109* | N-terminus | Sha et al.32 | ||
| c.361C>T | Nonsense | p.Gln121* | N-terminus | Liu et al.27 | ||
| c.2089-1G>T | Frameshift | p.Ile687Leufs*257 | Smc domain | Liu et al.27 | ||
| c.301A>C | Missense | p.Thr101Pro | N-terminus | Lu et al.33 | ||
| TSGA10 (NM_025244.4) | c.211delG | Frameshift | p.Ala71Hisfs*12 | Phosphodiesterase | III | Sha et al.35 |
| c.1739A>C | Missense | p.Gln580Pro | COG4372 domain | Liu et al.27 | ||
| c.545dupT | Frameshift | p.Ala183Serfs*10 | Phosphodiesterase | Ye et al.36 | ||
| BRDT (NM_207189.4) | c.G2783A | Missense | p.Gly928Asp | P-TEFb binding | III | Li et al.38 |
| DNAH6 (NM_001370.2) | c.2454A>T | Missense | p.Glu818Asp | - | Unknown | Li et al.39 |
| c.7706G>A | Missense | p.Arg2569His | - | Li et al.39 | ||
| CEP112 (NM_001199165.4) | c.496C>T | Nonsense | p.Arg166* | - | Unknown | Sha et al.43 |
| c.2074C>T | Missense | p.Arg692Trp | Coiled-coil domain | Sha et al.43 | ||
| c.2104C>T | Missense | p.Arg702Cys | Coiled-coil domain | Sha et al.43 | ||
| HOOK1 (NM_015888.6) | c.848T>C | Missense | p.Gln286Arg | Central coiled-coil domain | II | Chen et al.42 |
ASS: acephalic spermatozoa syndrome; SUN5: Sad1 and UNC84 domain containing 5; PMFBP1: polyamine-modulated factor 1 binding protein 1; TSGA10: testis-specific gene antigen 10; BRDT: bromodomain testis-specific protein; DNAH6: dynein axonemal heavy chain 6; CEP112: centrosomal protein 112; HOOK1: hook microtubule tethering protein 1; -: unknown domain
Sad1 and UNC84 domain containing 5 (SUN5)
In 2016, the first identified disease-causing gene of ASS, SUN5, was discovered.8 This study also presented a feasible flowchart for genetic pathogenesis studies of ASS, which was widely used in the following genetic studies of ASS.
SUN5 encodes a transmembrane protein specifically expressed in the testis, which is composed of a nucleoplasmic domain in the N-terminus, a transmembrane domain, a coiled-coil domain, and a SUN domain in the C-terminus. SUN5 functions as a structural protein, an important component of HTCA, which participates in the assembly of the sperm neck by being involved in nuclear envelope reconstitution and nuclear migration. According to the reported cases so far, approximately 40.78% (31/76) of human ASS could impute SUN5 mutations/deletions in patient cohorts.8,23,24,25,26,27,28,29
In Sun5-knockout mice or in patients with pathogenic SUN5 mutations, it was observed that the sperm head separated from the tail because the flagellum cannot attach to the nuclear envelope.8,30
Polyamine-modulated factor 1 binding protein 1 (PMFBP1)
PMFBP1 is the second validated pathogenic gene of ASS. PMFBP1 is a testis-specific protein localized at the head-tail connecting piece of sperm. Its indirect interaction with SUN5 and SPATA6 suggests that it plays a key role in making the liaison of the sperm head to the flagella. Mutations in PMFBP1 were responsible for approximately 34.61% (9/26) of reported cases of human ASS.27,31,32,33
Male knockout mice exhibit a phenotype of acephalic spermatozoa, accompanied by disordered mitochondrial sheaths and abnormal flagellum microtubules.
Testis-specific gene antigen 10 (TSGA10)
TSGA10 encodes a protein localized in the principal piece to the midpiece of the sperm. Several studies have revealed that an important role was played by TSGA10 in the organization and position of centrioles, in the arrangement of the mitochondrial sheath, and in the development of embryo.27,34,35,36 Ultrastructural studies of ASS caused by TSGA10 mutations in sperm from infertile men demonstrated a breakage at the level of the proximal centriole. However, there was no functional validation with a knockout animal model linked to ASS, whereas, only heterozygous Tsga10 male mice were reported to present sperm motility reduction because of the mitochondrial sheath disorder.37
Bromodomain testis-specific protein (BRDT)
Li et al.38 reported a missense mutation in BRDT in a typical ASS patient whose ejaculate had 99.5% acephalic sperm diagnosed by WES in 2017. The corresponding protein encoded by BRDT functions as a transcriptional regulator and contains two standardized bromodomains, which are involved in chromatin remodeling by recognizing acetylated lysine residues. In the p.G928D mutant, 899 genes showed a remarkable change at the level of expression. Among them, the upregulated genes involved in intracellular transport may be the underlying mechanism of acephalic spermatozoa. The overproduction of a membranous vesicle system by the Golgi complex might affect the attachment of the sperm proximal centrioles to the nucleus.
Dynein axonemal heavy chain 6 (DNAH6)
DNAH6 encodes a protein as an axonemal dynein heavy chain located in the neck region of normal spermatozoa, which is critical for the sperm head-tail junction. DNAH6 mutations have been identified in a patient with 30% acephalic spermatozoa, 69% round or small headed spermatozoa, and 1% double tail or double head spermatozoa.39 However, the exact association between DNAH6 mutations and ASS needs to be further explored, because it had more to do with primary ciliary dyskinesia (PCD), azoospermia, and multiple morphological abnormalities of the sperm flagella (MMAF) according to previous studies.40,41
Hook microtubule tethering protein 1 (HOOK1)
Chen et al.42 filtered out a missense mutation in HOOK1, located at the central coiled-coil domain of the corresponding protein, in an infertile man whose semen had more than 95% acephalic spermatozoa. Consistent with the mouse model results, the incomplete and disorganized implantation fossa and the basal plate could be observed under an electron microscope in patients’ headless sperm tails without any abnormity of other structures.
Centrosomal protein 112 (CEP112)
The testis-specific gene CEP112 encodes a centrosomal protein involved in the composition of the centrosome, which is an essential component of HTCA. Recently, two ASS patients with CEP112 mutations were reported to have more than 90% decapitated sperm during ejaculation.43
In addition to an increasing number of candidate genes of ASS spring-up using WES in patients with ASS, mouse models have also exhibited novel findings. A recent study has shown that family with sequence similarity 46, member C (Fam46c), viewed as a noncanonical RNA polyadenylation polymerase, is localized in the manchette responsible for protein transport during spermiogenesis. Owing to the loss-of-function of Fam46c, the knockout mice exhibited male sterility with production of headless spermatozoa, abnormal HTCA, and incomplete segmented column.44 In addition, another study revealed that spermatogenesis and centriole associated 1-like (Spatc1l) encoded a protein located at the head-tail junction of the sperm, which is highly expressed in mouse and human testes. The Spatc1l−/− generated by clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas) 9-mediated genome engineering showed male infertility caused by acephalic spermatozoa. The study implicated that SPATC1L regulated the activity of protein kinase A (PKA) in the sperm, and that the SPATC1L-PKA complex maintained the stability of the sperm head-tail connection.45
Intracytoplasmic sperm injection (ICSI) presents a promising treatment for ASS patients
To date, because of defects in both sperm structures and functions, no spontaneous pregnancy has been reported in a couple of which the husband has primary infertility caused by ASS. Assisted reproductive treatment is the only method to obtain offspring. With the development of assisted reproductive technology, ICSI, known as the second generation of assisted reproductive techniques (ART), has been reported to be effective in patients with ASS. The use of ICSI to produce offspring in patients with ASS was first reported in 2003.46 However, ICSI outcomes are complex in the diverse types of ASS. Many studies have evaluated the impact of the type of ASS on the clinical outcomes of ICSI.
ASS subtype I
We can observe the tailless sperm head connected to the complete proximal centriole, with an intact implantation fossa and basal body; and the separated tail contained complete structures in this subtype of ASS. However, the genetic mechanisms and clinical treatments of this subtype remain unexplored.
ASS subtype II
ASS associated with mutant SUN5, PMFBP1 and HOOK1 matches the phenotypes of subtype II. The headless sperm tails from this subtype of ASS can still move in ejaculated semen; however, because of the lack of head with acrosome, acrosomal reactions cannot occur, which results in fertilization failure. Fortunately, ICSI can result in a good clinical prognosis.
ASS subtype III
ASS caused by mutant TSGA10 and BRDT belongs to subtype III. In addition to the breakage in the sperm neck, the mitochondrial sheath was also impaired. The outcomes of ICSI in this subtype of ASS were also unsatisfactory.
According to the current reports, ASS patients belonging to subtype II can obtain better ICSI outcomes than subtypes I and III, which often have a poor development of embryo and a failure of clinical pregnancy involved in the defects of the centrosome. Undoubtedly, good clinical pregnancy outcomes depend not only on the sperm but also on high-quality oocytes.
THE FUTURE: OVERCOMING EXISTING LIMITATIONS TO IMPROVE BIOLOGICAL UNDERSTANDING, CLINICAL DIAGNOSTICS AND PROGNOSTICS
Although the applications of NGS technology have revolutionized the discovery of pathogenic genes and mutations in ASS, a considerable proportion of the genetic origin underlying this disorder is not yet to be elucidated. Furthermore, genetic studies are essential to ASS, but they are not enough on their own. Advances in genetic diagnosis and assisted reproductive treatment of ASS may benefit patients more directly. Therefore, our tasks are still arduous in the future on overcoming existing limitations to improve biological understanding, clinical diagnostics, and prognostics.
Basic research
Although we understood HTCA more than before, it is still an ambiguous black box (Figure 2). In-depth analysis of the hierarchical structure model, which remains unknown, needs to be explored further. To date, our studies on HTCA have focused more on the structural proteins in the sperm head-tail junction. Many molecular changes during spermatogenesis involve not only structural proteins, but also receptors, water and ion channels and proteases. No protein can work by itself, and all proteins also need regulation by their upstream proteins and the auxiliary of their interacting proteins. Hence, in addition to studying the structure of HTCA, the genes that refer to the upstream regulation and auxiliaries of interaction proteins are also future study directions. We can pay attention to the transcriptional regulator or enzyme, similar to what we found in mouse models.
Figure 2.
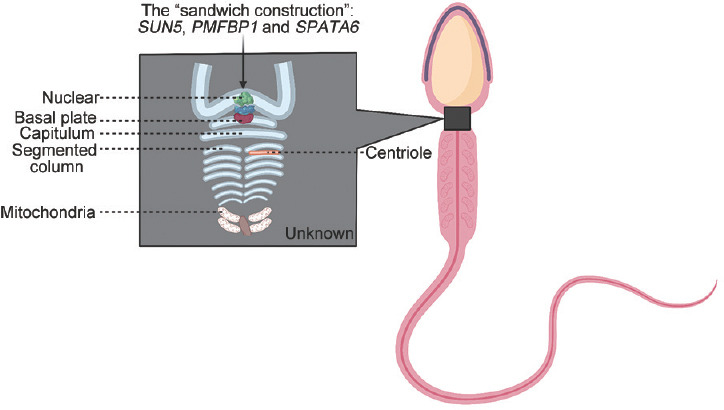
A schematic diagram of normal human spermatozoa. The black box represents the sperm HTCA, and defects in which may cause ASS. The marked parts are what we known and others need us to explore further. ASS: acephalic spermatozoa syndrome; SUN5: Sad1 and UNC84 domain containing 5; PMFBP1: polyamine-modulated factor 1 binding protein 1; SPATA6: spermatogenesis associated 6; HTCA: head–tail coupling apparatus.
In addition, the major challenge in male infertility will switch to interpretation of mutations from detection. Therefore, it is particularly important to perfect a set of relatively complete systems of functional experimental design targeting the mechanism of the occurrence of ASS.
Along with the increase in awareness of reproductive health, the phenomenon of consanguineous marriage will reduce. With the increase in sporadic genetic diseases, revealing de novo germline mutations through WES or WGS on patient-parent trios might be more important than now.47,48,49,50 Currently, increased research collaboration will drive further biological studies and understanding.
Clinical diagnosis
Although methods such as WES and WGS are widely used in laboratories to study the genetic origin of male infertility, an effective diagnostic method is still lacking in clinical diagnosis to provide patients with a rapid diagnosis and selection of treatment methods. We suggest identifying candidate biomarkers from our genetic studies for clinical assessment and establishing a unified clinical guideline for genetic diagnostics.
Treatment strategy
All deeper studies and more accurate diagnoses are aimed at providing better treatment for patients. Currently, patients diagnosed with ASS subtype I or III cannot achieve a good clinical outcome. More effective treatments are needed for the two types of patients to help them have their own offspring. Furthermore, there is still a notable question – when facilitated by ART to help patients obtain offspring, the genetic defect will pass on to the children. Therefore, the development and selection of new treatments are necessary.
CONCLUSIONS
In summary, advancements in modern technologies have remarkably improved our biological understanding of ASS by accelerating the discovery of the pathogenic genes. Nevertheless, owing to the complexity of the pathogenesis and the rarity of ASS, there were only a few pathogenic genes have been identified. Furthermore, the exact pathogenesis of some candidate genes is not fully understood. Therefore, unknown pathogenic factors and mechanisms are still the focus of future research. With more in-depth and comprehensive research on the genetics of ASS, genetic counseling, diagnosis, and fertility risk assessment for such patients will be applied to future clinical practice, and play a theoretical support and practical guidance role in the application of assisted reproduction.
AUTHOR CONTRIBUTIONS
YW reviewed the literature, collected data, wrote the manuscript, and prepared the figures. YXC and FXZ conceived, conducted the writing, and critically revised the manuscript. MFX and NZ collaborated in editing and revising the manuscript. All authors read and approved the final manuscript.
COMPETING INTERESTS
All authors declare no competing interests.
ACKNOWLEDGMENTS
We acknowledge the National Natural Science Foundation of China (No. 82071701 to FXZ), Natural Science Foundation of Anhui Province (No. 1908085J28 to FXZ), the Key R&D program of Anhui Province (No. 201904a07020050 to FXZ), the Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (No. 2019PT310002), and the Scientific Research Foundation of the Institute for Translational Medicine of Anhui Province (No. SRFITMAP 2017zhyx29 and ZHYX2020A001) supported this study.
REFERENCES
- 1.Agarwal A, Mulgund A, Hamada A, Chyatte MR. A unique view on male infertility around the globe. Reprod Biol Endocrinol. 2015;13:37. doi: 10.1186/s12958-015-0032-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perotti ME, Gioria M. Fine structure and morphogenesis of “headless” human spermatozoa associated with infertility. Cell Biol Int Rep. 1981;5:113. doi: 10.1016/0309-1651(81)90018-7. [DOI] [PubMed] [Google Scholar]
- 3.Zaneveld LJ, Polakoski KL. Collection and physical examination of the ejaculate. In: Hafez ES, editor. Techniques of Human Andrology. Amsterdam: Elsevier/North-Holland Biomedical Press; 1977. pp. 147–72. [Google Scholar]
- 4.Chemes HE, Carizza C, Scarinci F, Brugo S, Neuspiller N, et al. Lack of a head in human spermatozoa from sterile patients: a syndrome associated with impaired fertilization. Fertil Steril. 1987;47:310–6. doi: 10.1016/s0015-0282(16)50011-9. [DOI] [PubMed] [Google Scholar]
- 5.Perotti ME, Giarola A, Gioria M. Ultrastructural study of the decapitated sperm defect in an infertile man. J Reprod Fertil. 1981;63:543–9. doi: 10.1530/jrf.0.0630543. [DOI] [PubMed] [Google Scholar]
- 6.Baccetti B, Selmi MG, Soldani P. Morphogenesis of ‘decapitated’ spermatozoa in a man. J Reprod Fertil. 1984;70:395–7. doi: 10.1530/jrf.0.0700395. [DOI] [PubMed] [Google Scholar]
- 7.Chemes HE, Puigdomenech ET, Carizza C, Olmedo SB, Zanchetti F, et al. Acephalic spermatozoa and abnormal development of the head-neck attachment: a human syndrome of genetic origin. Hum Reprod. 1999;14:1811–8. doi: 10.1093/humrep/14.7.1811. [DOI] [PubMed] [Google Scholar]
- 8.Zhu F, Wang F, Yang X, Zhang J, Wu H, et al. Biallelic SUN5 mutations cause autosomal-recessive acephalic spermatozoa syndrome. Am J Hum Genet. 2016;99:942–9. doi: 10.1016/j.ajhg.2016.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gest H. Homage to Robert Hooke (1635-1703): new insights from the recently discovered Hooke Folio. Perspect Biol Med. 2009;52:392–9. doi: 10.1353/pbm.0.0096. [DOI] [PubMed] [Google Scholar]
- 10.Tan SY, Tatsumura Y. George Papanicolaou (1883-1962): discoverer of the Pap smear. Singapore Med J. 2015;56:586–7. doi: 10.11622/smedj.2015155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mendoza-Lujambio I, Burfeind P, Dixkens C, Meinhardt A, Hoyer-Fender S, et al. The Hook1 gene is non-functional in the abnormal spermatozoon head shape (azh) mutant mouse. Hum Mol Genet. 2002;11:1647–58. doi: 10.1093/hmg/11.14.1647. [DOI] [PubMed] [Google Scholar]
- 12.Netzel-Arnett S, Bugge TH, Hess RA, Carnes K, Stringer BW, et al. The glycosylphosphatidylinositol-anchored serine protease PRSS21 (testisin) imparts murine epididymal sperm cell maturation and fertilizing ability. Biol Reprod. 2009;81:921–32. doi: 10.1095/biolreprod.109.076273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tokuhiro K, Isotani A, Yokota S, Yano Y, Oshio S, et al. OAZ-t/OAZ3 is essential for rigid connection of sperm tails to heads in mouse. PLoS Genet. 2009;5:e1000712. doi: 10.1371/journal.pgen.1000712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ruan Y, Cheng M, Ou Y, Oko R, van der Hoorn FA. Ornithine decarboxylase antizyme Oaz3 modulates protein phosphatase activity. J Biol Chem. 2011;286:29417–27. doi: 10.1074/jbc.M111.274647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liska F, Gosele C, Rivkin E, Tres L, Cardoso MC, et al. Rat hd mutation reveals an essential role of centrobin in spermatid head shaping and assembly of the head-tail coupling apparatus. Biol Reprod. 2009;81:1196–205. doi: 10.1095/biolreprod.109.078980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kierszenbaum AL, Rivkin E, Tres LL, Yoder BK, Haycraft CJ, et al. GMAP210 and IFT88 are present in the spermatid Golgi apparatus and participate in the development of the acrosome-acroplaxome complex, head-tail coupling apparatus and tail. Dev Dyn. 2011;240:723–36. doi: 10.1002/dvdy.22563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang K, Meinhardt A, Zhang B, Grzmil P, Adham IM, et al. The small heat shock protein ODF1/HSPB10 is essential for tight linkage of sperm head to tail and male fertility in mice. Mol Cell Biol. 2012;32:216–25. doi: 10.1128/MCB.06158-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang K, Grzmil P, Meinhardt A, Hoyer-Fender S. Haplo-deficiency of ODF1/HSPB10 in mouse sperm causes relaxation of head-to-tail linkage. Reproduction. 2014;148:499–506. doi: 10.1530/REP-14-0370. [DOI] [PubMed] [Google Scholar]
- 19.Yuan S, Stratton CJ, Bao J, Zheng H, Bhetwal BP, et al. Spata6 is required for normal assembly of the sperm connecting piece and tight head-tail conjunction. Proc Natl Acad Sci U S A. 2015;112:E430–9. doi: 10.1073/pnas.1424648112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vogt PH. Molecular genetics of human male infertility: from genes to new therapeutic perspectives. Curr Pharm Des. 2004;10:471–500. doi: 10.2174/1381612043453261. [DOI] [PubMed] [Google Scholar]
- 21.Jan SZ, Vormer TL, Jongejan A, Roling MD, Silber SJ, et al. Unraveling transcriptome dynamics in human spermatogenesis. Development. 2017;144:3659–73. doi: 10.1242/dev.152413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fernandez-Marmiesse A, Gouveia S, Couce ML. NGS technologies as a turning point in rare disease research, diagnosis and treatment. Curr Med Chem. 2018;25:404–32. doi: 10.2174/0929867324666170718101946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elkhatib RA, Paci M, Longepied G, Saias-Magnan J, Courbiere B, et al. Homozygous deletion of SUN5 in three men with decapitated spermatozoa. Hum Mol Genet. 2017;26:3167–71. doi: 10.1093/hmg/ddx200. [DOI] [PubMed] [Google Scholar]
- 24.Fang J, Zhang J, Zhu F, Yang X, Cui Y, et al. Patients with acephalic spermatozoa syndrome linked to SUN5 mutations have a favorable pregnancy outcome from ICSI. Hum Reprod. 2018;33:372–7. doi: 10.1093/humrep/dex382. [DOI] [PubMed] [Google Scholar]
- 25.Sha YW, Xu X, Ji ZY, Lin SB, Wang X, et al. Genetic contribution of SUN5 mutations to acephalic spermatozoa in Fujian China. Gene. 2018;647:221–5. doi: 10.1016/j.gene.2018.01.035. [DOI] [PubMed] [Google Scholar]
- 26.Shang Y, Yan J, Tang W, Liu C, Xiao S, et al. Mechanistic insights into acephalic spermatozoa syndrome-associated mutations in the human SUN5 gene. J Biol Chem. 2018;293:2395–407. doi: 10.1074/jbc.RA117.000861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu G, Wang N, Zhang H, Yin S, Dai H, et al. Novel mutations in PMFBP1, TSGA10 and SUN5: expanding the spectrum of mutations that may cause acephalic spermatozoa. Clin Genet. 2020;97:938–9. doi: 10.1111/cge.13747. [DOI] [PubMed] [Google Scholar]
- 28.Cazin C, Boumerdassi Y, Martinez G, Fourati Ben Mustapha S, Whitfield M, et al. Identification and characterization of the most common genetic variant responsible for acephalic spermatozoa syndrome in men originating from North Africa. Int J Mol Sci. 2021;22:2187. doi: 10.3390/ijms22042187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang D, Huang WJ, Chen GY, Dong LH, Tang Y, et al. Pathogenesis of acephalic spermatozoa syndrome caused by SUN5 variant. Mol Hum Reprod. 2021;27:gaab028. doi: 10.1093/molehr/gaab028. [DOI] [PubMed] [Google Scholar]
- 30.Shang Y, Zhu F, Wang L, Ouyang YC, Dong MZ, et al. Essential role for SUN5 in anchoring sperm head to the tail. eLife. 2017;6:e28199. doi: 10.7554/eLife.28199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu F, Liu C, Wang F, Yang X, Zhang J, et al. Mutations in PMFBP1 cause acephalic spermatozoa syndrome. Am J Hum Genet. 2018;103:188–99. doi: 10.1016/j.ajhg.2018.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sha YW, Wang X, Xu X, Ding L, Liu WS, et al. Biallelic mutations in PMFBP1 cause acephalic spermatozoa. Clin Genet. 2019;95:277–86. doi: 10.1111/cge.13461. [DOI] [PubMed] [Google Scholar]
- 33.Lu M, Kong S, Xiang M, Wang Y, Zhang J, et al. A novel homozygous missense mutation of PMFBP1 causes acephalic spermatozoa syndrome. J Assist Reprod Genet. 2021;38:949–55. doi: 10.1007/s10815-021-02075-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Behnam B, Modarressi MH, Conti V, Taylor KE, Puliti A, et al. Expression of Tsga10 sperm tail protein in embryogenesis and neural development: from cilium to cell division. Biochem Biophys Res Commun. 2006;344:1102–10. doi: 10.1016/j.bbrc.2006.03.240. [DOI] [PubMed] [Google Scholar]
- 35.Sha YW, Sha YK, Ji ZY, Mei LB, Ding L, et al. TSGA10 is a novel candidate gene associated with acephalic spermatozoa. Clin Genet. 2018;93:776–83. doi: 10.1111/cge.13140. [DOI] [PubMed] [Google Scholar]
- 36.Ye Y, Wei X, Sha Y, Li N, Yan X, et al. Loss-of-function mutation in TSGA10 causes acephalic spermatozoa phenotype in human. Mol Genet Genomic Med. 2020;8:e1284. doi: 10.1002/mgg3.1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luo G, Hou M, Wang B, Liu Z, Liu W, et al. Tsga10 is essential for arrangement of mitochondrial sheath and male fertility in mice. Andrology. 2021;9:368–75. doi: 10.1111/andr.12889. [DOI] [PubMed] [Google Scholar]
- 38.Li L, Sha Y, Wang X, Li P, Wang J, et al. Whole-exome sequencing identified a homozygous BRDT mutation in a patient with acephalic spermatozoa. Oncotarget. 2017;8:19914–22. doi: 10.18632/oncotarget.15251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li L, Sha YW, Xu X, Mei LB, Qiu PP, et al. DNAH6 is a novel candidate gene associated with sperm head anomaly. Andrologia. 2018;50:e12953. doi: 10.1111/and.12953. [DOI] [PubMed] [Google Scholar]
- 40.Li Y, Yagi H, Onuoha EO, Damerla RR, Francis R, et al. DNAH6 and its interactions with PCD genes in heterotaxy and primary ciliary dyskinesia. PLoS Genet. 2016;12:e1005821. doi: 10.1371/journal.pgen.1005821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tu C, Nie H, Meng L, Yuan S, He W, et al. Identification of DNAH6 mutations in infertile men with multiple morphological abnormalities of the sperm flagella. Sci Rep. 2019;9:15864. doi: 10.1038/s41598-019-52436-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen H, Zhu Y, Zhu Z, Zhi E, Lu K, et al. Detection of heterozygous mutation in hook microtubule-tethering protein 1 in three patients with decapitated and decaudated spermatozoa syndrome. J Med Genet. 2018;55:150–7. doi: 10.1136/jmedgenet-2016-104404. [DOI] [PubMed] [Google Scholar]
- 43.Sha Y, Wang X, Yuan J, Zhu X, Su Z, et al. Loss-of-function mutations in centrosomal protein 112 is associated with human acephalic spermatozoa phenotype. Clin Genet. 2020;97:321–8. doi: 10.1111/cge.13662. [DOI] [PubMed] [Google Scholar]
- 44.Zheng C, Ouyang YC, Jiang B, Lin X, Chen J, et al. Non-canonical RNA polyadenylation polymerase FAM46C is essential for fastening sperm head and flagellum in mice. Biol Reprod. 2019;100:1673–85. doi: 10.1093/biolre/ioz083. [DOI] [PubMed] [Google Scholar]
- 45.Kim J, Kwon JT, Jeong J, Kim J, Hong SH, et al. SPATC1L maintains the integrity of the sperm head-tail junction. EMBO Rep. 2018;19:e45991. doi: 10.15252/embr.201845991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Porcu G, Mercier G, Boyer P, Achard V, Banet J, et al. Pregnancies after ICSI using sperm with abnormal head-tail junction from two brothers: case report. Hum Reprod. 2003;18:562–7. doi: 10.1093/humrep/deg121. [DOI] [PubMed] [Google Scholar]
- 47.Vissers LE, de Ligt J, Gilissen C, Janssen I, Steehouwer M, et al. A de novo paradigm for mental retardation. Nat Genet. 2010;42:1109–12. doi: 10.1038/ng.712. [DOI] [PubMed] [Google Scholar]
- 48.Vissers LE, Gilissen C, Veltman JA. Genetic studies in intellectual disability and related disorders. Nat Rev Genet. 2016;17:9–18. doi: 10.1038/nrg3999. [DOI] [PubMed] [Google Scholar]
- 49.Veltman JA, Brunner HG. De novo mutations in human genetic disease. Nat Rev Genet. 2012;13:565–75. doi: 10.1038/nrg3241. [DOI] [PubMed] [Google Scholar]
- 50.Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature. 2017;542:433–8. doi: 10.1038/nature21062. [DOI] [PMC free article] [PubMed] [Google Scholar]