

ABSTRACT
Manganese neurotoxicity is a hallmark of hypermanganesemia with dystonia 2, an inherited manganese transporter defect caused by mutations in SLC39A14. To identify novel potential targets of manganese neurotoxicity, we performed transcriptome analysis of slc39a14−/− mutant zebrafish that were exposed to MnCl2. Differentially expressed genes mapped to the central nervous system and eye, and pathway analysis suggested that Ca2+ dyshomeostasis and activation of the unfolded protein response are key features of manganese neurotoxicity. Consistent with this interpretation, MnCl2 exposure led to decreased whole-animal Ca2+ levels, locomotor defects and changes in neuronal activity within the telencephalon and optic tectum. In accordance with reduced tectal activity, slc39a14−/− zebrafish showed changes in visual phototransduction gene expression, absence of visual background adaptation and a diminished optokinetic reflex. Finally, numerous differentially expressed genes in mutant larvae normalised upon MnCl2 treatment indicating that, in addition to neurotoxicity, manganese deficiency is present either subcellularly or in specific cells or tissues. Overall, we assembled a comprehensive set of genes that mediate manganese-systemic responses and found a highly correlated and modulated network associated with Ca2+ dyshomeostasis and cellular stress.
This article has an associated First Person interview with the first author of the paper.
KEY WORDS: Zebrafish, Slc39a14, Manganese, Calcium, Transcriptome
Summary: Transcriptome analysis of zebrafish slc39a14−/− mutants demonstrates that loss of slc39a14 leads to concurrent manganese neurotoxicity and deficiency, both of which are associated with Ca2+ dyshomeostasis.
INTRODUCTION
SLC39A14 is a manganese (Mn) uptake transporter that is essential for the maintenance of Mn homeostasis (Thompson and Wessling-Resnick, 2019). Mutations in SLC39A14 impair cellular Mn uptake and result in systemic Mn overload characterised by hypermanganesemia and neurodegeneration (Tuschl et al., 2016; Juneja et al., 2018; Marti-Sanchez et al., 2018; Rodan et al., 2018; Zeglam et al., 2018). In patients, subsequent accumulation of Mn in the globus pallidus, part of the basal ganglia involved in motor control, leads to rapidly progressive dystonia-parkinsonism with onset in early childhood, a condition known as hypermanganesemia with dystonia 2 (HMNDYT2, OMIM 617013). In a small number of patients, treatment has been attempted using intravenous disodium calcium edetate (Na2CaEDTA) for Mn chelation (Tuschl et al., 2016; Rodan et al., 2018; Lee and Shin, 2022), similar to a protocol established for HMNDYT1 (OMIM 613280), which is caused by mutations in SLC30A10, a Mn exporter required for biliary excretion of Mn (Tuschl et al., 2012a,b). Brain magnetic resonance imaging (MRI) appearances of patients with either disorder are indistinguishable for the hyperintensity of both the basal ganglia, particularly the globus pallidus, and the white matter on T1-weighted imaging (Tuschl et al., 2012b, 2016). Although patients with HMNDYT1 show significant improvement of neurological symptoms upon treatment initiation (Tuschl et al., 2008, 2012b), individuals with HMNDYT2 have variable treatment responses, with some patients experiencing a worsening of their movement disorder (Tuschl et al., 2016; Marti-Sanchez et al., 2018). The reasons for the difference in treatment response are poorly understood.
Although an essential trace metal, excess Mn acts as a neurotoxicant. Environmental Mn overexposure leads to preferential Mn accumulation in the globus pallidus, similar to that observed in inherited Mn transporter defects, and causes manganism, a Parkinsonian movement disorder characterised by bradykinesia, akinetic rigidity and dystonia, accompanied by psychiatric disturbances (Blanc, 2018; Chen et al., 2018). Despite the recognised role of Mn in neurodegenerative disease processes, the mechanisms related to Mn neurotoxicity remain poorly understood. The clinical similarities between manganism and Parkinson's disease (PD) suggest that dopaminergic signalling is impaired upon Mn toxicity. However, in manganism, dopaminergic neurons within the substantia nigra are intact and the response to L-3,4-dihydroxyphenylalanine (L-DOPA), the mainstay of treatment in PD, is poor (Koller et al., 2004). Glutamatergic excitotoxicity as well as altered gamma-aminobutyric acid (GABA) signalling have also been proposed to underlie Mn-associated neurodegeneration (Caito and Aschner, 2015). Indeed, Mn toxicity is likely mediated by a number of processes including oxidative stress, impaired mitochondrial function, protein misfolding and aggregation and neuroinflammation (Martinez-Finley et al., 2013; Tjalkens et al., 2017).
We have recently established and characterised a zebrafish loss-of-function mutant slc39a14U801/U801 (herein referred to as slc39a14−/−) that closely resembles the human phenotype with systemic accumulation of Mn, particularly in the brain (Tuschl et al., 2016). Homozygous mutants develop increased susceptibility to Mn toxicity and impaired locomotor behaviour upon Mn exposure. Mn levels can be lowered through chelation with Na2CaEDTA similar to what is observed in human patients (Tuschl et al., 2016).
In this study, we performed RNA sequencing on individual scl39a14−/− larvae and their unaffected siblings to identify novel potential targets of Mn toxicity. Furthermore, we determined the transcriptional signature elicited in response to MnCl2 treatment in scl39a14−/− and their unaffected sibling larvae. Our results provide evidence that, in addition to Mn neurotoxicity, partial Mn deficiency that was corrected upon Mn treatment is a prominent feature of slc39a14 loss-of-function. We determined that Ca2+ dyshomeostasis is a likely key event in both Mn deficiency and overload. Mn neurotoxicity is further associated with activation of the unfolded protein response (UPR), oxidative stress, mitochondrial dysfunction, apoptosis, autophagy and disruption of proteostasis. These changes accompany impaired neuronal activity within the telencephalon and optic tectum, as well as associated behaviours, of slc39a14−/− zebrafish.
RESULTS
Transcriptome analysis of slc39a14−/− mutants identifies increased sensitivity to Mn toxicity as well as Mn deficiency effects
To investigate the transcriptional profiles of slc39a14−/− mutants in the absence and presence of Mn treatment, embryos from a heterozygous incross were split into two groups and either raised under standard conditions (subsequently referred to as unexposed), or treated with 50 µM MnCl2 from 2 to 5 days post fertilisation (dpf) (Fig. 1A). We have previously shown that this concentration elicits a pronounced locomotor phenotype in homozygous mutant larvae compared to their siblings (Tuschl et al., 2016). We then carried out transcriptional profiling of individual 5 dpf larvae using 3′ tag sequencing (differential expression transcript counting technique, DeTCT) (Collins et al., 2015). Principal component analysis (PCA) showed an effect of homozygosity and treatment status, but no difference between heterozygous and wild-type individuals (Fig. 1B, Table S1). We therefore pooled the wild-type and heterozygous embryos in the analysis for better statistical confidence and simplicity.
Fig. 1.

DeTCT analysis identifies three groups of differentially expressed genes. (A) Diagram of the experiment. Embryos from a slc39a14+/− incross were either left unexposed or exposed to 50 µM MnCl2 from 2 to 5 dpf. (B) Principal component analysis of the samples. Principal component (PC) 1 is plotted on the x-axis and PC2 on the y-axis. Samples belonging to the same condition are grouped together. Circles represent wild-type embryos, diamonds represent heterozygotes and squares represent homozygote mutants. Unexposed sibling embryos are indicated in light blue and MnCl2-exposed embryos are in dark blue. Unexposed mutants are coloured light red and exposed mutants are dark red. (C) Group 1 (Mn toxicity) genes were defined as those with a significant difference between exposed and unexposed siblings (red bar with asterisk). An example plot of normalised counts for the soul5 gene is shown. The colour scheme for C-E is the same as in B. (D) Group 2 (Increased sensitivity) genes were defined as those with a significant difference between exposed mutants and unexposed siblings (red bar with asterisk), but without significant differences between either unexposed mutants or exposed siblings when compared to unexposed siblings (black bars labelled NS, not significant). An example plot of normalised counts for the opn1mw2 gene is shown. (E) Group3 (Mutant effect) was defined as genes with a significant difference between unexposed mutants and unexposed siblings (red bar with asterisk). Example plot of normalised counts for the cdh24b gene. *P<0.05 determined using the Wald test (see Materials and Methods).
Analysis of differentially expressed genes between the four conditions produced three groups of genes, each with a characteristic expression profile. The first group consisted of genes that were differentially expressed in MnCl2-exposed siblings compared with their unexposed siblings, and represent a response to an increased concentration of Mn in the embryos (Fig. 1C, Mn toxicity). The second group consisted of genes that show increased sensitivity to Mn in slc39a14−/− mutants. These are defined as genes that are differentially expressed in MnCl2-exposed mutants compared with their unexposed siblings, but not differentially expressed in unexposed mutants compared with their unexposed siblings or exposed siblings compared with their unexposed siblings (Fig. 1D, Increased sensitivity). The third group was composed of genes that were differentially expressed in unexposed mutants compared with their unexposed siblings (Fig. 1E, Mutant effect). We then considered these three groups of genes in turn (see Table 1 for examples and Tables S2 and S3 for the top ten upregulated and downregulated genes and the highest P-values from each differentially expressed gene list).
Table 1.
Differentially expressed genes grouped by function.

Mn toxicity causes genotype-independent differential gene expression
MnCl2 treatment caused differential expression of 328 genes independent of the genotype (comparing MnCl2-exposed and unexposed siblings) (Fig. 2A, Table 1; Table S1). Among them is brain-derived neurotrophic factor (bdnf), a previously reported read-out for Mn exposure (Zou et al., 2014), that also showed diminished expression in untreated mutants compared to their siblings (Fig. 2B). BDNF signalling has been linked to the maturation of Parvalbumin-positive cells, mainly GABAergic interneurons (Fairless et al., 2019). However, Parvalbumin-encoding genes were more highly expressed upon Mn exposure in mutants (pvalb1, pvalb2 and pvalb8) as well as their siblings (pvalb2 and pvalb8).
Fig. 2.

Manganese overexposure causes neurotoxicity and metabolic defects in slc39a14+/+ or slc39a14+/− embryos. (A) Heatmap of the expression of all 328 genes with a significant difference between exposed and unexposed siblings (Group 1 – Mn toxicity, Table S1). Each row represents a different gene and each column is a sample. Mutant embryos are displayed for completeness; however, the group of genes is defined by the response in siblings only. The normalised counts for each gene have been mean centred and scaled by dividing by the standard deviation. (B) Plot of the normalised counts for each sample of the gene bdnf in Group 1. Unexposed sibling embryos are indicated in light blue and MnCl2-exposed ones are in dark blue. Unexposed mutants are coloured light red and exposed mutants are dark red. FC, fold change. Wald test test was used to determine significance. (C) Enrichment of Gene Ontology (GO) terms associated with the genes in A. Diagram produced using the CytoScape ClueGO app. Nodes represent enriched GO terms and edges connect GO terms that have annotated genes in common. Different components of the network are coloured according to the categories labelled on the diagram. The sizes of the circles represent the adjusted P-values for each GO term as indicated on the right (Wald test). See Fig. S1 for GO enrichment split by up- and downregulation.
Among other brain-expressed genes affected by MnCl2 exposure were those involved in synaptic vesicle function (rims2b, stxbp1a, sv2a, sypb and syt9a) and genes encoding the Metabotropic glutamate receptor 8a (grm8a), β-Synuclein (sncb) and Ephrin-B membrane proteins (efnb1 and efnb2a), all of which had decreased expression (Table 1).
Analysis of annotations to Gene Ontology (GO) terms (Fig. 2C; Table S4; Fig. S1 for GO enrichment split by up- and downregulation) showed enrichment of terms related to lipid metabolism (driven by upregulation of, for example, apoa4b.2, apoa4a and apoea), blood cell development (upregulation of alas1, fech and soul5), translation (35 ribosomal protein-encoding genes, most of which were upregulated) and circadian rhythm (upregulation of cry1aa, cry1bb, cryba4 and per3). These findings were similar to previous reports in which links between Mn toxicity and lipid metabolism (Luo et al., 2020), circadian clock gene regulation (Li et al., 2017), haem-enzyme biogenesis (Chino et al., 2018) and protein biosynthesis (Hernandez et al., 2019) have been described.
Mn is important for connective tissue integrity and bone mineralisation as a constituent of metalloenzymes and enzyme activator (Sirri et al., 2016; Zofkova et al., 2017). Consistent with its role in connective tissue maintenance, transcriptome analysis confirmed that Mn exposure in zebrafish led to reduced expression of multiple connective tissue-related genes (col2a1b, col4a5, col9a1a, col9a2, col11a2, dcn, fbn2b and matn1).
slc39a14−/− mutants show increased sensitivity to MnCl2 treatment
Our analysis showed that 613 genes were differentially expressed in MnCl2-exposed mutants compared with their unexposed siblings, with no significant expression changes in either unexposed mutants or their exposed siblings. Therefore, these were genes that showed increased sensitivity to MnCl2 exposure in slc39a14−/− mutant larvae (Fig. 3A, Table 1). Of these genes, 15% (95/613) also had a significant genotype-treatment interaction effect, meaning that there was a synergistic effect on gene expression in mutant embryos with MnCl2 treatment – that is, the combined estimated effects of genotype and MnCl2 treatment alone were significantly less than the estimated log2 fold change for MnCl2-exposed mutants when compared with their unexposed siblings (Fig. 3B; see Table S1 for synergistic genes in bold). The remaining genes (518/613) showed expression changes consistent with additive effects of the sub-significance threshold responses to genotype and MnCl2 exposure alone (Fig. 3C). Results from the transcriptome analysis were validated by quantitative reverse transcription PCR (qRT-PCR) for a subset of six genes (bdnf, gnat2, hspa5, opn1mw2, pde6ha and prph2b) using RNA extracted from equivalent embryos in a different experiment (Fig. 3D,E; Fig. S2, Table S5). Changes in gene expression observed by qRT-PCR for all six genes were consistent with the results obtained from transcript counting (compare Fig. 3B with Fig. 3D and Fig. 3C with Fig. 3E).
Fig. 3.

Effect of Mn treatment in slc39a14−/− mutants. (A) Heatmap of the expression of all 613 genes with a significant difference between exposed mutant and unexposed sibling embryos, but without without significant effects of treatment or genotype. The heatmaps are split into genes that show either synergistic or additive effects of the individual genotype and treatment effects. Each row represents a different gene and each column is a sample. The normalised counts for each gene have been mean centred and scaled by dividing by the standard deviation. (B) Example of a gene (hspa5) with a synergistic effect of treatment and genotype. The difference between the exposed mutants and unexposed siblings cannot be explained by adding together the separate effects of Mn treatment and the slc39a14−/− mutation. Unexposed sibling embryos are indicated in light blue and MnCl2-exposed embryos are in dark blue. Unexposed mutants are coloured light red and exposed mutants are dark red. (C) Example of a gene (pde6c) that has an additive effect of treatment and genotype. The difference between exposed mutants and unexposed siblings is consistent with adding together the two sub-threshold effects of treatment and genotype produce. Colour scheme is as in B. FC, fold change. Wald test was used to determine significance in B,C. (D) qRT-PCR showed comparable gene expression changes for hspa5 as for the single-embryo sequencing dataset. The individual samples are displayed as fold change relative to the mean value for unexposed siblings, and the mean and 95% confidence intervals for each condition indicated are in orange. Compare with B. (E) Enrichment Map network of the Zebrafish Anatomy Ontology (ZFA) enrichment results. Each node represents an enriched ZFA term and the edges join nodes that have overlapping genes annotated to them. The width of each edge is proportional to the amount of overlap; nodes are coloured by −log10[adjusted P-value] (Wald test); and the size represents the number of significant genes annotated to the term. (F) ClueGO network diagram of the enrichment of GO terms. Nodes represent enriched GO terms and edges connect nodes that share annotations to the significant genes. Different components of the network are coloured according to the categories as labelled on the diagram. The sizes of the circles represent the adjusted P-values for each GO term as indicated below (Wald test).
Enrichment of zebrafish anatomy (ZFA) terms showed that genes differentially expressed upon MnCl2 exposure in slc39a14−/− mutants were disproportionately expressed in the nervous system, including the eye (Fig. 3F; Fig. S3, Table S6). This was confirmed by the enrichment of GO terms such as visual perception and phototransduction, associated with genes that were downregulated (Fig. 3F; Fig. S1). Also enriched were terms related to the ribosome, translation and the UPR, suggesting effects on protein synthesis and folding (Fig. 3F; Fig. S1, Table S4).
Increased sensitivity of slc39a14−/− mutants to MnCl2 treatment leads to Mn neurotoxicity
Enriched ZFA terms identified in MnCl2-exposed slc39a14−/− mutants that were not present in siblings showed a high number of differentially expressed genes in the nervous system (Table S6), confirming the role of increased Mn levels in neurotoxicity. We found slc1a2a, encoding the astrocytic glutamate transporter excitatory amino acid transporter (EAAT2), to be the fifth most highly and significantly downregulated gene upon MnCl2 exposure (Tables S2 and S3). A role for astrocyte-mediated Mn neurotoxicity and neuroinflammation was further suggested by increased expression of the astrocyte-related genes atf5a, atf5b and gfap. In addition, expression of the teleost-specific glutamate transporters slc1a2b (upregulated) and slc1a8a (downregulated) was altered, pointing towards involvement of the glutamate-glutamine cycle in Mn neurotoxicity. Two genes required for the regulation of ionotropic AMPA-type glutamate receptors (nsg2 and prrt1) also showed diminished expression in MnCl2-treated mutants (Table 1).
Furthermore, we observed increased expression of slc6a11b, encoding a GABA uptake transporter, as well as the Parvalbumin-encoding gene (pvalb1) present in GABAergic interneurons. Expression of the GABA-A receptor-encoding genes gabra6a and gabrb3 and the Neuronal pentraxin receptor-encoding gene nptxrb, which is expressed in Parvalbumin-positive interneurons (Kikuchihara et al., 2015), was reduced.
Despite the assumption that abnormal dopamine signalling is a major player in Mn neurotoxicity (Guilarte and Gonzales, 2015), only two genes linked to dopamine, gnb5b (downregulated) and gpr37l1b (upregulated), both of which encode proteins that interact with neurotransmission via the Dopamine D2 receptor (Octeau et al., 2014; Hertz et al., 2019), were differentially expressed.
In order to assess the effects of Mn neurotoxicity on neuronal function, we performed in situ hybridisation chain reaction to map gad1b mRNA in MnCl2-exposed wild-type and mutant siblings. gad1b was chosen because Mn preferentially accumulates in the globus pallidus, a region that is particularly rich in GABAergic projections, both in individuals with Mn overexposure and those with inherited Mn transporter defects. However, spatial gad1b expression analysis did not suggest changes in GABAergic signalling and brain structure (Fig. S4).
MnCl2 exposure alters resting-state neuronal activity and locomotor behaviour
cfos (officially known as fosab) is an immediate-early gene induced in response to neuronal activity, and thus, we performed in situ hybridisation chain reaction to map changes in cfos expression in response to MnCl2 exposure as a proxy for identifying resting-state changes in neuronal activity. We observed pronounced alterations in cfos expression in both MnCl2-treated wild-type and mutant siblings (Fig. 4A-D). Consistent with the increased sensitivity to Mn neurotoxicity suggested by RNA sequencing, homozygous mutant larvae showed more extensive changes in cfos expression compared to their siblings. Specifically, enhanced expression of cfos, reflecting increased neuronal activity, was particularly evident within the telencephalon in mutant versus wild-type larvae, whereas lower expression was observed within the optic tectum of mutants (Fig. 4D).
Fig. 4.
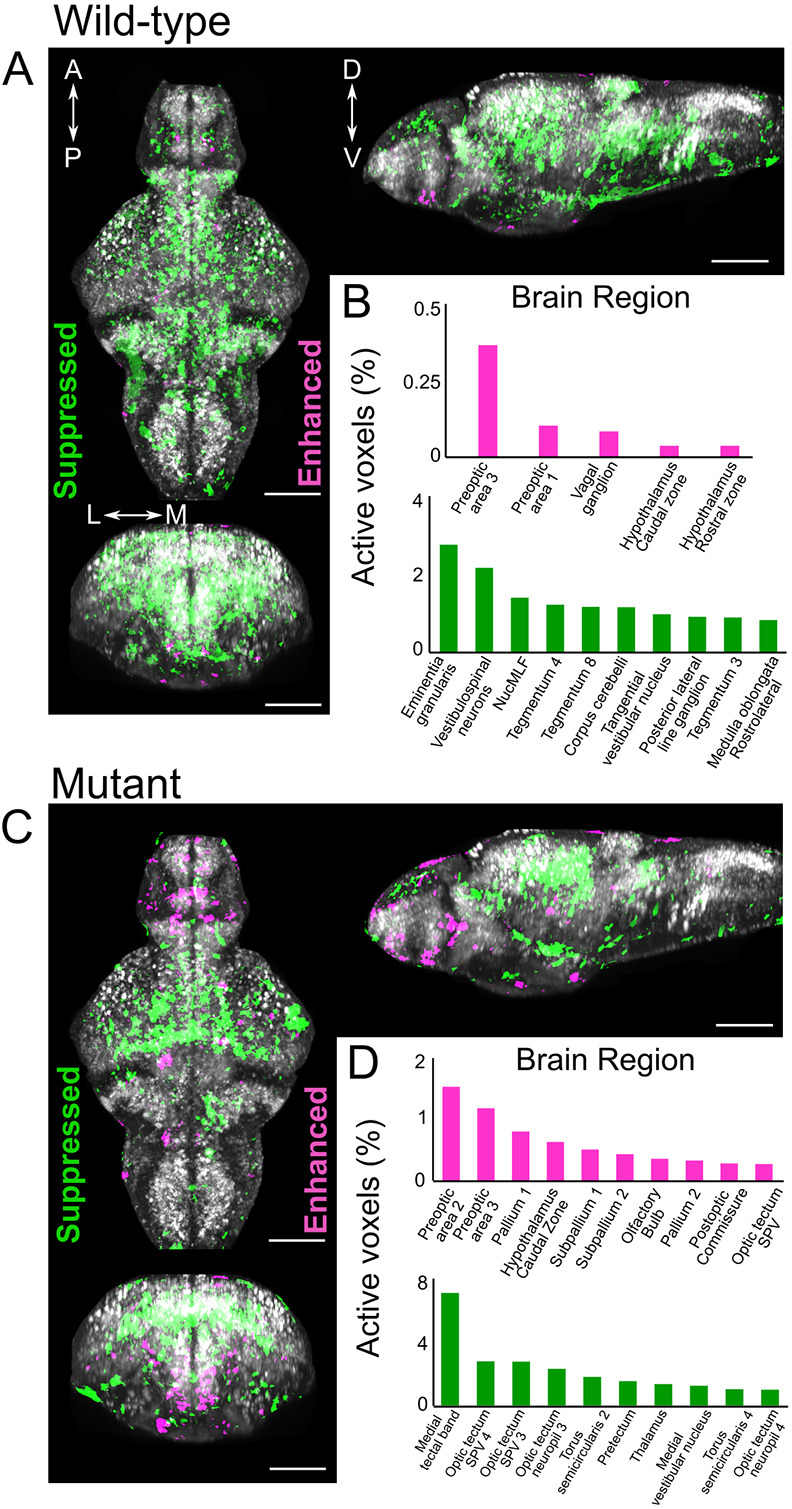
MnCl2 treatment alters neuronal activity in both wild-type and slc39a14−/− larvae. (A-D) Z-projection of cfos mRNA expression in the brain of wild-type (A) and homozygous mutant larvae (C) at 6 dpf following treatment with 50 µM MnCl2 from 2 dpf. B and D list the brain regions with enhanced (magenta) and suppressed (green) neuronal activity by genotype. A, anterior; P, posterior; D, dorsal; V, ventral; L, lateral; M, medial. Scale bars: 100 µm.
We next tracked the locomotor behaviour of unexposed and MnCl2-exposed wild-type and mutant larvae from 4 to 7 dpf on a 14:10 h light-dark cycle. Homozygous mutants showed a dose-dependent reduction in average locomotor activity during the day and increased locomotor activity during the night, whereas wild-type larvae remained unaffected by MnCl2 exposure (Fig. 5A; Table S7). Wild-type fish sharply increased their locomotor activity immediately following lights OFF and gradually, over several minutes, returned to baseline locomotor activity, a behaviour known as the visual motor response (VMR) (Burton et al., 2017). Frame-by-frame analysis of larval locomotion showed that slc39a14−/− zebrafish had a preserved VMR but showed hyperlocomotion throughout the first few hours following lights OFF, with larvae returning to baseline activity only towards the second half of the night (Fig. 5B).
Fig. 5.

MnCl2 treatment causes locomotor abnormalities and Ca2+ dyshomeostasis. (A) Average locomotor activity of wild-type (blue) and slc39a14−/− larvae (red) during the day and night in response to increasing concentrations of MnCl2. Data are presented as mean±s.e.m. (two-way ANOVA with Tukey's posthoc test; *P<0.05), n=24 larvae per group. The y-axes represent the average activity of larval zebrafish in seconds per 10 minutes. (B) Frame-by-frame analysis of the locomotor activity of wild-type and slc39a14−/− larvae at 6 dpf that were unexposed and exposed to 50 µM MnCl2. White shading representing the day (lights ON) and grey shading represents the night (lights OFF). The arrow indicates the ‘lights OFF’ switch. Summed and smoothed Δ pixels traces (Delta Px) are shown as mean±s.e.m. (bold lines and shaded surrounding areas). n=24 larvae per group. (C) Calcium, magnesium and manganese concentrations determined by ICP-MS in untreated and MnCl2 (50 µM)-treated wild-type and slc39a14−/− larvae. Data are presented as mean±s.d. (one-way ANOVA with Tukey's post hoc test; *P<0.05; **P<0.01; ***P<0.001). (D) Apoptotic cell death upon MnCl2 exposure in both wild-type (blue) and mutant (red) larvae at 5 dpf detected by TUNEL staining in the telencephalon, diencephalon and hindbrain. Data are presented as mean±s.d.
Increased sensitivity of slc39a14−/− mutants to MnCl2 treatment is associated with gene expression changes affecting Ca2+ and protein homeostasis, and the UPR
Mn toxicity is known to cause protein misfolding and aggregation (Angeli et al., 2014; Harischandra et al., 2019b) and, as previously shown for Mn overexposure in Caenorhabditis elegans (Angeli et al., 2014), multiple genes involved in the UPR had increased expression in slc39a14−/− mutants, with hspa5, atf3 and xbp1 observed to be the most highly and significantly upregulated genes upon MnCl2 treatment (Table 1; Tables S2 and S3). This is supported by transcription factor motif-enrichment analysis using Hypergeometric Optimization of Motif EnRichment analysis (HOMER) (Heinz et al., 2010), which showed that the dysregulated genes are enriched for Chop/Atf4-binding sites among others (Fig. S3, Table S8). Degradation of misfolded and aggregated proteins occurs via the ubiquitin-proteasome system within the cytosol (Tamas et al., 2014), and MnCl2-exposed slc39a14−/− mutants showed gene expression changes linked to ubiquitination (Table 1). Ca2+ homeostasis within the endoplasmic reticulum (ER) plays a major role during the UPR, and vice versa (Groenendyk et al., 2021). Potentially linked to the UPR, over a dozen Ca2+-associated/dependent genes were differentially expressed in MnCl2-treated slc39a14−/− mutants (Table 1; Table S1).
Given the observed changes in the expression of Ca2+-linked genes, we next assessed total Ca, Mg and Mn levels in both wild-type and mutant larvae by inductively coupled plasma mass spectrometry (ICP-MS). Consistent with a disturbance in Ca2+ homeostasis, we found that MnCl2 treatment in both wild-type and mutant larvae led to a marked decrease in total Ca and Mg levels (Fig. 5C). As previously observed, Mn accumulation was much greater in mutant larvae (Fig. 5C). These results confirm that Mn overload leads to Ca2+ dyshomeostasis that is associated with changes in expression of key genes responsible for Ca2+ regulation.
Activation of the UPR as well as Ca2+ dyshomeostasis can promote apoptosis and autophagy. Concordantly, genes involved in autophagy and apoptosis were differentially expressed (Table 1). In particular, faim2b, which encodes the recently identified regulator of autophagy FAIM2B (Hong et al., 2020), was the third most highly upregulated gene in MnCl2-exposed slc39a14−/− mutants (Table S2). Also, the expression of rubcn, encoding a beclin 1 interactor and responsible for autophagy initiation (Liu et al., 2019), was increased upon MnCl2 exposure.
To further explore whether increased apoptotic cell death might be responsible for the high number of downregulated genes observed upon MnCl2 exposure, we performed terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) staining on brains from unexposed and MnCl2-exposed wild-type and mutant larvae (Fig. 5D). However, there was no difference in the number of TUNEL-positive cells between unexposed and MnCl2-exposed larvae of either genotype, suggesting that functional rather than neurodegenerative changes are responsible for Mn neurotoxicity effects.
Oxidative stress and mitochondrial dysfunction are prominent features of Mn toxicity (Smith et al., 2017; Harischandra et al., 2019a). Consistent with this observation, essential genes of the thioredoxin/peroxiredoxin system (prdx1, txn and txnrd3) were activated in MnCl2-exposed slc39a14−/− mutants. Likewise, genes related to mitochondrial function showed differential expression in MnCl2-treated mutants (Table 1). Our data therefore further support a role of mitochondrial impairment in Mn-induced neurotoxicity.
Increased sensitivity of slc39a14−/− mutants to MnCl2 causes visual impairment
Consistent with the reduced cfos expression/neuronal activity observed within the optic tectum (Fig. 4), 30 genes involved in phototransduction were differentially expressed (27/30 genes were reduced) in MnCl2-exposed mutants but not in their siblings (Fig. 6A; Table S1). These included some of the most significantly upregulated genes such as pde6ha, opn1mw2, opn1mw1 and rcvrna in the increased sensitivity group (Tables S2 and S3). Hence, we further examined the vision of slc39a14−/− mutants. In zebrafish, visual background adaptation (VBA), which is the ability to aggregate and disperse melanosomes in order to adapt their body pigmentation to the environment, requires retinal input and is impaired in blind larvae (Mueller and Neuhauss, 2014).
Fig. 6.

slc39a14−/− mutants develop a visual phenotype upon MnCl2 exposure. (A) Schematic showing the process of phototransduction (Kaupp and Seifert, 2002) with the differentially expressed genes observed in MnCl2-exposed slc39a14−/− mutants indicated in italics. cGMP, cyclic guanosine monophosphate; CNG, cyclic nucleotide-gated non-selective cation channels; GC, guanylyl cyclase; GCAP, guanylate cyclase-activating protein; PDE, phosphodiesterase; GRK, G protein-coupled receptor kinase; GAP, GTPase-activating protein. (B) Dorsal views during light exposure of wild-type siblings (slc39a14+/+, left) and slc39a14−/− larvae (right) at 5 dpf that were unexposed and exposed to 50 µM MnCl2. Note the darker pigmentation of mutants exposed to MnCl2 (red asterisk). Scale bar: 500 µm. (C) Graph showing the eye velocity in response to moving stimuli of different spatial frequencies (average of both eyes) of slc39a14−/− larvae that were unexposed (dark green circles) and exposed to 50 µM MnCl2 (light green squares). Data are presented as mean±s.e.m. from five independent experiments (two-tailed unpaired Student's t-test; **P<0.01; ***P<0.001). (D) Histologic analysis of retinal sections with Richardson–Romeis staining of wild-type siblings (slc39a14+/+, top row) and slc39a14−/− larvae (bottom row) at 5 dpf exposed to 50 µM MnCl2. Scale bars: 200 µm (image of both eyes), 100 µm (image of single eye).
We observed that MnCl2-exposed slc39a14−/− mutant larvae lacked melanosome aggregation and remained dark following light exposure from 4 dpf, whereas exposed wild-type larvae and unexposed mutants demonstrated a normal VBA (Fig. 6B). Next, we analysed the optokinetic response (OKR) in homozygous slc39a14−/− larvae at 5 dpf after MnCl2 exposure. Exposed mutant larvae demonstrated a significant reduction in slow-phase eye velocity at high spatial frequencies, suggesting impaired visual acuity (Fig. 6C; Table S9). Retinal histology of mutant and MnCl2-exposed animals appeared normal, suggesting functional rather than overt structural deficits (Fig. 6D). In conclusion, the reduced expression of phototransduction genes in combination with reduced cfos expression/neuronal activity within the optic tectum, impaired VBA and OKR, as well as abnormal VMR reveal that Mn exposure in slc39a14−/− larvae leads to visual impairment.
Most differentially expressed genes in unexposed slc39a14−/− mutants are rescued by Mn treatment suggesting Mn deficiency
When compared to unexposed siblings, 266 genes showed significantly different expression due to the homozygous state alone (unexposed homozygous mutants versus unexposed unaffected siblings) (Fig. 7A; Table S1). Expression of 12% of these genes (31/266) was also significantly different between MnCl2-exposed mutants and their unexposed siblings (Fig. 7B). Seven of these genes overlapped with those differentially expressed in siblings upon MnCl2 exposure, suggesting that these genes were the most sensitive targets of Mn toxicity (alas1, atp2a1, bdnf, crim1, dio3b, dip2ca and rims2b). However, the majority (88%, 235/266) of differentially expressed genes in unexposed mutants were not significantly differentially expressed when comparing MnCl2-exposed mutants and their unexposed siblings (Fig. 7C). This suggests that the homozygous U801 mutation creates an Mn deficiency leading to gene expression changes that return to levels observed in unexposed and unaffected siblings upon MnCl2 treatment.
Fig. 7.

Exogenous Mn restores normal expression of genes that are differentially expressed in unexposed slc39a14−/− mutants. (A) Heatmap of the expression of 266 genes with a significant difference between unexposed mutants and unexposed siblings. Each row represents a different gene and each column is a sample. The normalised counts for each gene have been mean centred and scaled by dividing by the standard deviation. (B) Plot of normalised counts for the add2 gene. Gene expression is decreased in both unexposed and MnCl2-exposed mutant embryos. Unexposed sibling embryos are indicated in light blue and Mn-exposed embryos are in dark blue. Unexposed mutants are coloured light red and exposed mutants are dark red. (C) Plot of normalised counts for the pcdh7b gene. There were decreased counts in the unexposed mutant embryos that were rescued back to wild-type levels upon 50 µM MnCl2 treatment. Colour scheme is as in B. FC, fold change. Wald test was used to determine significance in B,C. (D) Enrichment Map diagram of the enrichment of ZFA terms for the genes differentially expressed in unexposed mutants that are rescued by Mn treatment. Nodes represent enriched ZFA terms and edges connect nodes that share annotations to the significant genes. The width of each edge is proportional to amount of overlap, nodes are coloured by −log10[adjusted P-value] (Wald test) and the size represents the number of significant genes annotated to the term. (E) ClueGO network diagram of the enrichment of GO terms associated with the genes that are rescued by Mn treatment. Nodes represent enriched GO terms and edges connect nodes that share annotations to the significant genes. Different components of the network are coloured according to the categories as labelled on the diagram. The sizes of the circles represent the adjusted P-values for each GO term as indicated on the right (Wald test).
Analysis of ZFA terms within this rescued set of genes demonstrated enrichment of terms related to the nervous system (Fig. 7D; Fig. S3, Table S6). For instance, brain-expressed genes that showed reduced expression upon Mn deficiency include those essential for synaptic function and vesicle formation (snap25a, sv2a, sypb, syt6a and syt9a), neurite and axonal growth (dock3, gas7a, kalrna, kalrnb and lrrc4ca) and potassium channels (kcnc1a and kcnc3a). GO term analysis linked differential gene expression to cell-cell adhesion and cell-cell interactions (Fig. 7E; Fig. S1, Table S4). Expression of seven protocadherin-encoding genes was altered with pcdh7b being the third most highly downregulated gene within this group. Protocadherins are Ca2+-dependent cell-adhesion proteins that are primarily expressed in the brain, where they regulate synapse maturation, function and plasticity (Mancini et al., 2020). In addition, the expression of several other Ca2+-associated genes returned to normal levels by Mn treatment, which were distinct from those changed due to Mn toxicity. These included genes encoding Ca2+ ATPases (atp2a1 and atp2b3b), Ca2+ channels (cacnb4b), Ca2+-activated potassium channels (kcnma1a and kcnn1a), calmodulins (calm1b and calm3a) and calmodulin-binding proteins (camta1b and strn4). These results suggest that in addition to causing a systemic increase in Mn levels, the loss of slc39a14 function might also result in local Mn deficiency with gene expression changes that can be rescued with exogenous Mn. Differentially expressed genes in both the Mn sensitivity and Mn rescue group link to Ca2+ regulation, suggesting that disturbed Mn homeostasis has significant consequences on Ca2+-dependent genes with a distinctly affected gene set for each group (Table 1).
DISCUSSION
Transcriptional profiling of slc39a14 mutant zebrafish identified distinct gene groups that are differentially expressed in physiological conditions and upon MnCl2 exposure. Consistent with the neurodegenerative phenotype observed in HMNDYT2 patients and the previously described accumulation of Mn in the brain of slc39a14−/− zebrafish mutants (Tuschl et al., 2016), the majority of differentially expressed genes map to the central nervous system (CNS) and the eye. Mn treatment leads to gene expression changes in both slc39a14−/− mutant and sibling zebrafish. However, the expression of a much greater number of genes changes in mutant larvae upon MnCl2 treatment than in treated non-mutant siblings, confirming an increased sensitivity to Mn toxicity that is consistent with previous observations (Tuschl et al., 2016). This is corroborated by the changes in brain activity, locomotor and visual behaviour observed in mutant larvae. Intriguingly, 88% (235/266) of differentially expressed genes in unexposed slc39a14−/− mutants normalised upon MnCl2 treatment. This suggests that Mn treatment in slc39a14−/− mutants rescues some of the transcriptomic changes observed in unexposed mutants, and implies that SLC39A14 loss leads to Mn deficiency, in parallel to the observed Mn accumulation.
Unexposed slc39a14−/− mutants as well as MnCl2-treated mutants and siblings show evidence of Mn neurotoxicity
The mechanisms underlying Mn neurotoxicity are heterogenous, suggesting extensive roles for Mn in brain pathobiology (Soares et al., 2020). The neuronal subtypes affected by Mn neurotoxicity remain the subject of debate. In agreement with previous reports, we observed altered expression of genes involved in glutamatergic and GABAergic neurotransmission in MnCl2-treated slc39a14−/− mutants (Marreilha Dos Santos et al., 2017). The most highly and significantly downregulated genes included slc1a2a, encoding the astrocytic glutamate reuptake transporter EAAT2. Transcriptional repression of SLC1A2 with subsequent impaired glutamate uptake and excitotoxicity has been observed in MnCl2-exposed human astrocytes (Rizor et al., 2021), suggesting that Mn neurotoxicity affects the glutamate-glutamine cycle.
In humans, Mn preferentially accumulates in the globus pallidus, a region that is particularly rich in GABAergic neurons (Sidoryk-Wegrzynowicz and Aschner, 2013; Tuschl et al., 2016). In MnCl2-treated slc39a14−/− zebrafish, the expression of genes encoding the GABA-A receptor (gabra6a and gabrb3) and the GABA reuptake transporter (slc6a11b) was reduced, similar to studies in rats where Mn exposure led to diminished GABA-A receptor mRNA expression and interfered with GABA uptake in astrocytes (Fordahl and Erikson, 2014; Ou et al., 2017). Increased expression of genes encoding Parvalbumin (pvalb1, pbalb2 and pvalb8) in slc39a14−/− mutants and their siblings upon MnCl2 treatment might further indicate that GABAergic interneurons are a target of Mn neurotoxicity (Kikuchihara et al., 2015). Parvalbumin, a Ca2+-binding protein, can also bind Mn2+ with high affinity (Nara et al., 1994). Mn might therefore interact with Parvalbumin directly or via changes in Ca2+ homeostasis, which are clearly evident in slc39a14−/− zebrafish. Despite the observed gene expression changes related to GABAergic neurotransmission, the spatial localisation of gad1b mRNA expression was unchanged in both MnCl2-exposed wild-type and slc39a14−/− larvae. However, the observed marked alterations in cfos expression suggested altered neuronal activity in slc39a14−/− compared with wild-type fish. Enhanced expression/activity was evident within preoptic, hypothalamic, pallidal and subpallidal regions.
Because manganism resembles Parkinson's disease to some extent (e.g. both cause an akinetic movement disorder, albeit with distinct clinical features), it has long been hypothesised that dopaminergic neurons are affected by Mn neurotoxicity (Ijomone et al., 2016). However, transcriptome analysis of slc39a14−/− mutants provides little evidence that Mn neurotoxicity causes primary gene expression changes related to dopaminergic signalling.
Consistent with predominant accumulation of Mn in astrocytes rather than neurons (Tjalkens et al., 2017; Gorojod et al., 2018; Popichak et al., 2018), Mn exposure in slc39a14−/− mutants leads to increased expression of the astrocyte-related genes atf5a, atf5b and gfap, as well as the astrocyte-expressed glutamate and GABA uptake transporter genes slc1a2 and slc6a11b, respectively, corroborating a role for glia in Mn neurotoxicity.
Although we observed gene expression changes linked to apoptosis, TUNEL staining did not reveal increased apoptotic cell death upon MnCl2 exposure. This suggests that Mn neurotoxicity initially and primarily causes deficits in neuronal function rather than neurodegeneration, which is in keeping with clinical observations that the neuronal phenotype of affected individuals is to some extent reversible (Tuschl et al., 2016).
Mn toxicity in slc39a14−/− mutants is associated with Ca2+ dyshomeostasis, activation of the UPR and oxidative stress
Our results clearly indicate that Mn imbalance interferes with Ca2+ homeostasis and causes changes in expression of Ca2+-associated genes coupled with altered total Ca2+ levels. It is understood that Mn2+ can replace Ca2+ in its biologically active sites (Kalbitzer et al., 1978; Song et al., 2017) and disrupt Ca2+ homeostasis at the mitochondria and the ER, thereby affecting intracellular Ca2+ concentrations (Quintanar et al., 2012). Mn overexposure has previously been shown to disrupt neurotransmitter release via interaction with the SNARE complex and subsequent activation of calpain, a Ca2+-/Mn2+-activated neutral protease (Wang et al., 2018). MnCl2 treatment in slc39a14−/− mutants indeed affects the expression of genes encoding parts of the presynaptic neurotransmitter release machinery, suggesting that Mn neurotoxicity might be mediated through impaired presynaptic exocytosis. Whether this is facilitated via direct interaction of Mn with neurotransmitter release or via Ca2+ dysregulation needs to be determined in future studies. Nevertheless, our results provide evidence that Ca2+ dysregulation is a key feature of Mn neurotoxicity. This has also been shown for other neurodegenerative disorders including Parkinson's, Alzheimer's and Huntington's diseases, in which Ca2+ dyshomeostasis occurs upstream of protein aggregation (Jadiya et al., 2021).
Ca2+ homeostasis is maintained by the ER, the key organelle for regulating proteostasis (Wang et al., 2012). Ca2+ dysregulation is closely linked to the UPR and ER stress that is evident in MnCl2-exposed slc39a14−/− mutants, with upregulation of multiple UPR-associated genes. HOMER analysis also confirms enrichment of the Chop/Atf4 motif in MnCl2-treated mutants. This is consistent with previous studies that show increased expression of ATF6 and HSPA5 as well as increased Xbp1 mRNA splicing in Mn-exposed brain slices (Xu et al., 2013).
In addition to Ca2+ dyshomeostasis, oxidative stress and mitochondrial dysfunction are shared characteristics among neurodegenerative disorders and metal toxicity (Harischandra et al., 2019a). Mn accumulates in mitochondria, where it leads to the generation of reactive oxygen species (ROS) (Rizor et al., 2021). ROS production can further exacerbate protein misfolding (Nakamura et al., 2021). Oxidative stress is highlighted in MnCl2-exposed slc39a14−/− mutants by the upregulation of the thioredoxin/thioredoxin reductase and peroxiredoxin system, similar to previous results in rats (Taka et al., 2012). ROS also cause apoptosis and autophagy via lysosomal membrane permeabilisation and cathepsin release (Gorojod et al., 2017; Wang et al., 2017; Porte Alcon et al., 2018; Zhi et al., 2019; Tinkov et al., 2021). In accordance, we observed changes in autophagy and cathepsin gene expression upon MnCl2 treatment in mutant larvae; however, we did not see alterations in the number of apoptotic cells as determined by TUNEL staining.
In summary, transcriptome analysis of slc39a14−/− zebrafish suggests that Mn overexposure affects a multitude of molecular processes. The future challenge will be the identification of the trigger event that leads to Mn-induced Ca2+ dyshomeostasis as well as mitochondrial and lysosomal dysfunction, a prerequisite for finding novel therapeutic targets for the treatment of Mn neurotoxicity.
Mn toxicity in slc39a14−/− zebrafish causes impairment in retinal function
Transcriptome analysis revealed an unsuspected Mn toxicity effect in slc39a14−/− zebrafish, with more than 30 retinal phototransduction genes being differentially expressed. Changes in expression were accompanied by impaired VBA and an altered OKR. Combined with the reduced neuronal activity observed within the optic tectum, this suggests that Mn has toxic effects on the function of the zebrafish retina. Although this has not been observed in affected patients or rodent models of Mn overload, both Mn uptake transporters SLC39A8 and SLC39A14 are highly expressed in the retinal pigment epithelium (Leung et al., 2008). Furthermore, Mn plays an essential role in retinal function, in which it is required for normal ultrastructure of the retina (Gong and Amemiya, 1996). Possible differences between the human and zebrafish phenotypes might simply be caused by the direct contact of the zebrafish eye with Mn in the water, contributing to enhanced ocular Mn uptake and toxicity. We cannot exclude a direct effect of Mn on the oculomotor system and melanophore function leading to the changes in the OKR and VBA observed, but the large number of differentially expressed phototransduction genes, as well as reduced tectal neuronal activity, make Mn-induced retinal dysfunction more likely. It is plausible that Mn also affects non-retinal photoreceptors, which might link to the expression changes observed for several circadian clock genes, as well as the altered VMR. Mn administration in rats has previously been shown to cause dysregulation of circadian clock gene expressions (Li et al., 2017). Locomotor behavioural analysis of slc39a14−/− zebrafish did indeed reveal changes in the locomotor activity pattern, with decreased activity during the day and increased activity during the night, as well as an altered VMR at light-dark transitions, a behaviour linked to the function of non-visual photoreceptors (Fernandes et al., 2012).
Loss of slc39a14 function in zebrafish causes Mn deficiency
Perhaps the most intriguing observation was that most differentially expressed genes (235/266) in unexposed slc39a14−/− mutants normalised upon MnCl2 treatment. This indicates that although SLC39A14 deficiency leads to systemic Mn accumulation, it also causes deficiency of Mn in parts of the cell or specific types of cells due to its role as an Mn uptake transporter. This partial Mn deficiency might explain why chelation therapy in patients with HMNDYT2 is less effective compared to those with HMNDYT1, with some patients deteriorating upon Mn chelation (Tuschl et al., 2016; Marti-Sanchez et al., 2018; Rodan et al., 2018).
Mn deficiency in slc39a14−/− mutants suggests that some features of HMNDYT2 might overlap with those observed in SLC39A8 deficiency, an inherited Mn transporter defect leading to systemic Mn deficiency (OMIM 616721). Affected individuals present with intellectual disability, developmental delay, hypotonia, epilepsy, strabismus, cerebellar atrophy and short stature (Boycott et al., 2015; Park et al., 2015). However, HMNDYT2 does not share these features aside from cerebellar atrophy described in some patients.
As for Mn toxicity, the majority of ‘rescued’ genes map to the CNS. Several differentially expressed genes link to Ca2+ homeostasis and binding; however, these are different to those identified upon Mn overload. Notably, the expression of protocadherins and formin-related genes is reduced in unexposed slc39a14−/− mutants. Protocadherins are mainly expressed in the CNS where they are required for normal neural circuitry activity and regulate synaptic function (Kim et al., 2011). Loss of protocadherin function in mice has previously been associated with neurodegeneration (Hasegawa et al., 2016). Formins are required for the stabilisation of E-cadherins (Rao and Zaidel-Bar, 2016), which might link the changes observed in (proto-)cadherin expression with that of formin-associated genes.
How partial Mn deficiency arises within the brain of slc39a14−/− zebrafish remains to be determined. It might stem from differences in the expression patterns of various metal transporters. In the future, single-cell RNA sequencing, spatial transcriptomics and proteomics might allow us to distinguish neurons/glial cells affected by Mn neurotoxicity from those with deficiency. Identifying the overlap between chelator-treated and mutant larvae as well as analysis of Slc39a14-deficient neuronal cultures will aid to delineate the molecular events underlying partial Mn deficiency in slc39a14−/− mutants.
In conclusion, our results demonstrate that partial Mn deficiency might be an additional feature to Mn neurotoxicity in slc39a14−/− zebrafish. Overall, the slc39a14U801 loss-of-function zebrafish mutants are an excellent disease model to study the disease pathogenesis of HMNDYT2 as well as Mn neurotoxicity.
MATERIALS AND METHODS
Zebrafish husbandry
Zebrafish were reared on a 14:10 h light-dark cycle at 28.5°C at the University College London (UCL) Zebrafish Facility. Embryos were obtained by natural spawning, and staging was performed according to standard criteria (Kimmel et al., 1995). Previously generated slc39a14U801 loss-of-function zebrafish and their siblings were used for all experiments (Tuschl et al., 2016). Ethical approval for zebrafish experiments was obtained from the Home Office UK under the Animal Scientific Procedures Act 1986.
Preparation of larvae for RNA and DNA extraction
The progeny of a single incross of slc39a14U801/+ fish were raised in 10-cm Petri dishes filled with fish water (0.3 g/l Instant Ocean, 50 embryos per dish) at 28°C. At 2 dpf, half of the larvae were exposed to MnCl2 that was added to the fish water at a concentration of 50 µM (stock solution 1 M MnCl2, made up in water). After 72 h of exposure (at 5 dpf), single larvae were collected in the wells of a 96-well plate, immediately frozen on dry ice and stored at −80°C. For sequencing, frozen embryos were lysed in 100 µl RLT buffer (Qiagen) containing 1 µl of 14.3 M β-mercaptoethanol (Sigma). The lysate was allowed to bind to 1.8 volumes of Agencourt RNAClean XP (Beckman Coulter) beads for 10 mins. The plate was then applied to a plate magnet (Invitrogen) until the solution cleared and the supernatant was removed without disturbing the beads. While still on the magnet, the beads were washed three times with 70% ethanol and total nucleic acid was eluted from the beads as per the manufacturer's instructions. Nucleic acid samples were used for genotyping of individual larvae by KASP assay (LGC Genomics) according to the manufacturer's instructions, and the following primers were used: wild-type allele, 5′-GGCACATAATAATCCTCCATGGG-3′; mutant allele, 5′-GGGCACATAATAATCCTCCATGGT-3′; and common primer, 5′-CCCTGTATGTAGGCCTTCGGGTT-3′. After DNase treatment, RNA was quantified using either Qubit RNA HS assay or Quant-iT RNA assay (Invitrogen).
Transcript counting
DeTCT libraries were generated as described previously (Collins et al., 2015). Briefly, 300 ng of RNA from each genotyped sample was fragmented and bound to streptavidin beads. The 3′ ends of the fragmented RNA were pulled down using a biotinylated polyT primer. An RNA oligo containing the partial Illumina adapter 2 was ligated to the 5′ end of the bound fragment. The RNA fragment was eluted and reverse transcribed using an anchored oligo-dT reverse-transcriptase primer containing one of the 96 unique index sequences and part of the Illumina adapter 1. The Illumina adapters were completed during a library amplification step and the libraries were quantified using either the BioPhotometer (Eppendorf) or Pherastar (BMG Labtech). This was followed by size selection for an insert size of 70-270 bases. Equal quantities of libraries for each experiment were pooled, quantified by qRT-PCR, and sequenced on either HiSeq 2000 or HiSeq 2500 (Illumina).
Sequencing data were analysed as described previously (Collins et al., 2015). Briefly, sequencing reads were processed with the DeTCT detag_fastq.pl (https://github.com/iansealy/DETCT) script and aligned to the GRCz11 zebrafish reference genome with BWA 0.5.10 (Li and Durbin, 2009). The resulting BAM files were processed using the DeTCT pipeline, which results in a list of regions (for simplicity referred to as genes in the Results) representing 3′ ends, together with a count for each sample. These counts were used for differential expression analysis with DESeq2 (Love et al., 2014). Each region was associated with Ensembl 95 (Yates et al., 2020) gene annotation based on the nearest transcript in the appropriate orientation. False positive 3′ ends representing, for example, polyA-rich regions of the genome, were filtered using the DeTCT filter_output.pl script with the ‘—strict’ option. Gene sets were analysed using the Cytoscape plugin ClueGO (Bindea et al., 2009) for GO enrichment and Ontologizer (Bauer et al., 2008) for ZFA Ontology enrichment.
qRT-PCR
RNA extraction from 30 zebrafish larvae from the same genotype (homozygous mutant or wild-type) was performed using 500 µL TRIzol reagent (Invitrogen) according to the manufacturer's protocol and purified using the RNeasy MiniKit (Qiagen). cDNA was generated using GoScript Reverse Transcriptase (Promega). qRT-PCR was performed using GoTaq qPCR Master Mix (Promega) according to the recommended protocol. All samples were run in triplicates. qRT-PCR was carried out on a CFX96 Touch Real-Time PCR Detection System (Bio-Rad). Only primer pairs with R2 values >0.99 and amplification efficiencies between 95% and 105% were used. Relative quantification of gene expression was determined using the 2−ΔΔCt method, with elongation factor 1α (ef1α, also known as eef1a1l1) as a reference gene (Livak and Schmittgen, 2001). The following primer sequences were used: ef1a forward, 5′-GTACTTCTCAGGCTGACTGTG-3′; ef1a reverse, 5′-ACGATCAGCTGTTTCACTCC-3′; bdnf forward, 5′-AGATCGGCTGGCGGTTTATA-3′; bdnf reverse 5′-CATTGTGTACACTATCTGCCCC-3′; gnat2 forward, 5′-GCTGGCAGACGTCATCAAAA-3′; gnat2 reverse, 5′-CTCGGTGGGAAGGTAGTCAG-3′; hspa5 forward, 5′-GCTGGGCTGAATGTCATGAG-3′; hspa5 reverse 5′-CAGCAGAGACACGTCAAAGG-3′; opn1mw2 forward, 5′-GCTGTCATTTCTGCGTTCCT-3′; opn1mw2 reverse, 5′-GACCATGCGTGTTACTTCCC-3′; pde6ha forward, 5′-CTCGCACCTTCAAGAGCAAG-3′; pde6ha reverse, 5′-CATGTCTCCAAACGCTTCCC-3′; prph2b forward, 5′-GCCCTGGTGTCCTACTATGG-3′; prph2b reverse, 5′-CTCTCGGGATTCTCTGGGTC-3′.
ICP-MS analysis of metal ions
ICP-MS analysis of zebrafish larvae was performed as previously described (Tuschl et al., 2016). In brief, ten larvae of the same genotype, anaesthetised with MS-222 (4% Tricaine), were pooled and washed several times with distilled water. Samples were digested in 200 µl concentrated nitric acid at 95°C until dry and resuspended in 1 mL 3% nitric acid. Further dilution with 20% nitric acid to a final volume of 2 ml was done prior to analysis. The metals (24Mg, 44Ca and 55Mn) were measured using an Agilent 7500ce ICP-MS instrument with collision cell (in He mode) and Integrated Autosampler (I-AS) using 72Ge as an internal standard. The following experimental parameters were used: (1) plasma: RF power, 1500 W; sampling depth, 8.5 mm; carrier gas, 0.8 l/min; make-up gas, 0.11 l/min; and (2) quadrupole: mass range, 1-250 amu; dwell time, 100 msec; replicates, three; integration time, 0.1 s/point. Calibration solutions were prepared for each element between 0 and 200 ng/ml using certified reference standards (Fisher Scientific, UK).
Apoptosis analysis
The TUNEL assay was used to determine apoptotic cell death. Larvae were fixed at 5 dpf overnight at 4°C in 4% paraformaldehyde (PFA) and 4% sucrose. Brains were manually dissected, transferred to methanol and stored at −20°C. After rehydration in PBS with 0.5% Triton X-100 (PBSTr), brains were permeabilised using 1× proteinase K for 15 min. Following washes in PBSTr, the samples were incubated at −20°C in pre-chilled ethanol:acetone (2:1) for 10 min, followed by washes in PBSTr. After a 1 h incubation in Apoptag equilibration buffer (Millipore), the samples were incubated in 35 µl of TdT enzyme mix [24 µl reaction buffer, 12 µl TdT enzyme (both Millipore), 1 µl 10% Triton X-100] at 37°C overnight. Following washes in PBSTr and incubation in blocking solution (for 1 ml: 100 μl normal goat serum, 10 μl of DMSO, 0.89 ml PBSTr) for 2 h at room temperature, the samples were incubated with polyclonal anti-digoxigenin-AP antibody (Roche) at a concentration of 1:2000 in blocking solution at 4°C overnight, and then developed using 4-nitro blue tetrazolium chloride and 5-bromo-4-chloro-3-indolyl-phosphate toluidine-salt (Roche). Imaging was performed in 80% methanol on a Nikon Eclipse E1000 microscope using the Openlab 4.0.2 software package.
Whole mount in situ hybridisation chain reaction (HCR)
Larvae were fixed in PFA with 4% sucrose overnight at 4°C, transferred into PBS the next morning and the brain dissected by removing skin, cartilage and eyes with forceps. For each target mRNA, custom DNA probe sets were designed and single-stranded DNA (ssDNA) oligos ordered from Life Technologies, ThermoFisher. DNA HCR amplifiers (comprising a pair of fluorophore-labelled DNA hairpins for Alexa Fluor 488 and Alexa Fluor 568) and hybridisation, wash and amplification buffers were purchased from Molecular Instruments (molecularinstruments.org). In situ HCR was performed using a published protocol (https://files.molecularinstruments.com/MI-Protocol-HCRv3-Zebrafish-Rev7.pdf) (Choi et al., 2018). For probe sets and amplifier details for each target mRNA (cfos and gad1b), see Table S10. Imaging was performed on a Zeiss Z1 Lightsheet microscope with a 10× imaging objective. Whole-brain image stacks were registered to a gad1b reference brain aligned to Zebrafish Brain Browser (Marquart et al., 2015) co-ordinates using Advanced Normalization Tools (ANTs) as reported in Marquart et al. (2017).
Following registration, we applied image analysis using ImageJ, MATLAB and custom-written scripts in Python. We first applied a 3D median filter to the image stacks, and subsequently performed permutation testing to detect changes in cfos signal between two groups as described in Randlett et al. (2015). This resulted in image stacks with cfos pixels that were enhanced or suppressed between control and treatment groups. These image stacks were then processed to determine their distribution across 168 different anatomical regions. Publicly available masks for these anatomical regions were used (Gupta et al., 2018). We used a false discovery rate (FDR) threshold of 0.05%, resulting in a 99.5% significance threshold for each active voxel. Python script for active voxel calculation in each brain mask and the ANTs script for registration can be found on Figshare: https://dx.doi.org/10.6084/m9.figshare.19550998 and https://dx.doi.org/10.6084/m9.figshare.19551007.
Locomotor behavioural analysis
The behavioural assay was conducted as described previously (Tuschl et al., 2016). In brief, zebrafish embryos and larvae were raised on a 14:10 h light/dark cycle at 28°C. Single larvae were transferred to each well of a flat-bottom, clear polystyrene 96-square-well plate (Whatman) in fish water (650 µl) at 4 dpf. Mn exposure was achieved by adding MnCl2 directly to the fish water at the desired concentration (stock solution of 1 M MnCl2 made in distilled water). The 96-well plate was maintained at a constant temperature (28.5°C) and exposed to a 14:10 h white light/dark schedule with constant infrared illumination within a custom-modified Zebrabox (Viewpoint Life Sciences). The locomotor behaviour of zebrafish larvae was tracked from 4 to 7 dpf using an automated video tracking system (Viewpoint Life Sciences). Larval movement was recorded using Videotrack Quantisation mode. The Videotrack detection parameters were empirically defined for clean detection of larval movement with minimal noise. A custom-designed MATLAB code was used to extract the average activity data of each larva as described previously (Rihel et al., 2010). Frame-by-frame analysis (25 frames per second) was performed as described by Ghosh and Rihel (2020) using the published MATLAB code.
Optokinetic response
The optokinetic response (OKR) was examined using a custom-built rig to track horizontal eye movements in response to whole-field motion stimuli. Larvae at 4 dpf were immobilised in 1.5% agarose in a 35 mm Petri dish and analysed at 5 dpf. The agarose surrounding the eyes was removed to allow normal eye movements. Sinusoidal gratings with spatial frequencies of 0.05, 0.1, 0.13 and 0.16 cycles/degree were presented on a cylindrical diffusive screen that was 25 mm from the centre of the fish's head. Gratings had a constant velocity of 10 degrees/second and changed direction and/or spatial frequency every 20 s. Eye movements were tracked under infrared illumination (720 nm) at 60 Hz using a Flea3 USB machine vision camera and custom-written software. A custom-designed MATLAB code was used to determine the eye velocity in degrees per second (available at https://bitbucket.org/biancolab/okrsuite).
Retinal histology
5 dpf larvae were fixed in 4% PFA overnight at 4°C. Dehydration was achieved by a series of increasing ethanol concentrations in PBS (50%, 70%, 80%, 90%, 95% and 100% ethanol). After dehydration, larvae were incubated in a 1:1 ethanol:Technovit 7100 solution (1% Hardener 1 in Technovit 7100 basic solution) for 1 h followed by incubation in 100% Technovit solution overnight at room temperature (Heraeus Kulzer, Germany). Larvae were than embedded in plastic moulds in Technovit 7100 polymerisation medium and dried at 37°C for 1 h. Sections of 3 μm thickness were prepared with a microtome, mounted onto glass slides and dried at 60°C. Sections were stained with Richardson (Romeis) solution (0.5% borax, 0.5% Azur II, 0.5% Methylene Blue) and slides were mounted with Entellan (Merck, Darmstadt, Germany). Images were taken in the brightfield mode of a BX61 microscope (Olympus).
Experimental design and statistical analyses
Animals were divided into four experimental groups: unexposed homozygous slc39a14−/− mutants and their siblings (wild-type and heterozygous genotypes), and MnCl2-exposed homozygous slc39a14−/− mutants and their siblings (wild-type and heterozygous genotypes). For the DeTCT data, an equal number of wild-type and heterozygous embryos were selected (see Fig. 1 for numbers of embryos for each experimental group). This was to investigate the possibility of transcriptional changes in the heterozygous embryos compared with wild-type ones. This was not the case; the PCA showed that heterozygous embryos grouped with wild-type embryos and not separately. Differential expression analysis returned only 11 genes that were statistically different between untreated heterozygotes and untreated wild-type embryos (four of these genes are on the same chromosome as slc39a14 and likely represent the effect of allele-specific expression linked to the mutation). There were only 20 genes that were differentially expressed between Mn-exposed heterozygotes and Mn-exposed wild-type embryos, with three being linked to slc39a14. These lists have been included in Table S1 for completeness. Because of the lack of effect, wild-type and heterozygous embryos were pooled as unaffected siblings for the remaining analyses.
Embryos were all derived from a single cross to minimise the amount of biological variance not caused by the experimental conditions (i.e. genotype and Mn exposure). One wild-type Mn-exposed embryo was excluded from the data after visual inspection of the PCA as it did not group with any of the other samples. DESeq2 was used for differential expression analysis with the following model: ∼genotype+treatment+genotype:treatment. This models the observed counts as a function of the genotype (homozygous versus siblings) and the treatment (Mn exposed versus unexposed) and an interaction between the two, and tests for significant parameters were performed using the Wald test with an adjusted P-value (Benjamini–Hochberg) threshold of 0.05.
The three groups of differentially expressed genes were defined as follows: (1) Mn toxicity, significant in Mn-exposed siblings versus unexposed siblings; (2) increased sensitivity, significant in Mn-exposed mutants versus unexposed siblings AND NOT significant in unexposed mutants versus unexposed siblings AND NOT significant in Mn-exposed siblings versus unexposed siblings; and (3) mutant effect, significant in unexposed mutants versus unexposed siblings. These groups were not mutually exclusive and some genes appeared in more than one group because of the way the groups were defined.
For qRT-PCR, metal and behavioural locomotor analysis, ANOVA with Tukey post hoc testing, and for OKR analysis, a two-tailed unpaired Student's t-test was used to determine statistical significance, using the GraphPad Prism software (version 5). For GO term analysis, the settings for ClueGO were as follows: a right-sided hypergeometric test (enrichment only) was used with the Bonferroni step-down (Holm–Bonferroni) correction for multiple testing, and terms with corrected P-values >0.05 were discarded. For ZFA enrichment analysis, the Ontologizer Parent-Child-Union calculation method was used with Bonferroni correction.
Transcription factor motif analysis
Transcription factor motif enrichment was performed using HOMER's findMotifs.pl tool (v4.10.3) with default settings (Heinz et al., 2010). The GRCz11 promoter set used was created with HOMER's updatePromoters.pl tool based on RefSeq genes from −2000 bp to 2000 bp relative to the transcription start site.
Supplementary Material
Acknowledgements
We thank the members of the Wilson, Rihel and Bianco labs and other zebrafish groups at University College London (UCL) for helpful discussions. We are grateful to Neha Wali and the Sanger Institute sequencing pipelines for sample processing and sequencing. We thank all supporting staff at UCL including the Fish Facility staff for fish care and husbandry.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: K.T., R.J.W., E.M.B.-N.; Methodology: K.T., R.J.W.; Validation: K.T., L.E.V., S.C.F.N.; Formal analysis: K.T., R.J.W., C.T., S.C.F.N., C.D., R.G.-M.; Investigation: S.C.F.N., J.R.; Resources: E.M.B.-N.; Data curation: K.T., R.J.W., C.T., I.H.B., C.D., I.M.S.; Writing - original draft: K.T., R.J.W.; Writing - review & editing: L.E.V., S.C.F.N., I.H.B., R.G.-M., I.M.S., S.C.F.N., C.H., J.R., S.W.W., E.M.B.-N.; Supervision: L.E.V., I.H.B., S.C.F.N., C.H., J.R., S.W.W., E.M.B.-N.; Project administration: K.T., E.M.B.-N.; Funding acquisition: S.W.W., E.M.B.-N.
Funding
K.T. was supported by Action Medical Research (GN1999), the Academy of Medical Sciences, the National Institute for Health Research (NIHR, Academic Clinical Lectureship), the Great Ormond Street Hospital Charity (V0018) and the Medical Research Council (MR/V006754/1). K.T., S.C.F.N and S.W.W. were supported by the University College London and the Neuroscience Center Zurich, University of Zurich Collaboration. L.E.V. was funded by the Fondo Nacional de Desarrollo Científico y Tecnológico (FONDECYT) grant (11160951), Comisión Nacional de Investigación Científica y Tecnológica (CONICYT) International network grants (REDI170300 and REDES170010), and Universidad Mayor FDP grant (PEP I-2019074). C.T., L.E.V. and S.W.W. were supported by Medical Research Council grants (MR/L003775/1 and MR/T020164/1) and a Wellcome Trust investigator award (104682/Z/14/Z). S.C.F.N. was supported by the Schweizerischer Nationalfonds zur Förderung der Wissenschaftlichen Forschung (Swiss National Science Foundation; 31003A_173083) J.R. was supported by the Biotechnology and Biological Sciences Research Council (BBSRC; BB/T001844/1) and Wellcome Trust (217150/Z/19/Z). E.M.B.-N. was supported by core grants to the Wellcome Sanger Institute (WT098051 and 206194). This publication presents independent research funded by the NIHR. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health and Social Care. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Open Access funding provided by the Wellcome Trust and UKRI. Deposited in PMC for immediate release.
Data availability
Sequence data can be downloaded from the European Nucleotide Archive (ENA) using the links provided in Table S11. Supplementary tables are available at Figshare (DOIs specified in the supplementary information). The processed count data are available at Figshare: https://doi.org/10.6084/m9.figshare.11808789.
References
- Angeli, S., Barhydt, T., Jacobs, R., Killilea, D. W., Lithgow, G. J. and Andersen, J. K. (2014). Manganese disturbs metal and protein homeostasis in Caenorhabditis elegans. Metallomics 6, 1816-1823. 10.1039/C4MT00168K [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer, S., Grossmann, S., Vingron, M. and Robinson, P. N. (2008). Ontologizer 2.0--a multifunctional tool for GO term enrichment analysis and data exploration. Bioinformatics 24, 1650-1651. 10.1093/bioinformatics/btn250 [DOI] [PubMed] [Google Scholar]
- Bindea, G., Mlecnik, B., Hackl, H., Charoentong, P., Tosolini, M., Kirilovsky, A., Fridman, W. H., Pages, F., Trajanoski, Z. and Galon, J. (2009). ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25, 1091-1093. 10.1093/bioinformatics/btp101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc, P. D. (2018). The early history of manganese and the recognition of its neurotoxicity, 1837-1936. Neurotoxicology 64, 5-11. 10.1016/j.neuro.2017.04.006 [DOI] [PubMed] [Google Scholar]
- Boycott, K. M., Beaulieu, C. L., Kernohan, K. D., Gebril, O. H., Mhanni, A., Chudley, A. E., Redl, D., Qin, W., Hampson, S., Küry, S.et al. (2015). Autosomal-recessive intellectual disability with cerebellar atrophy syndrome caused by mutation of the manganese and zinc transporter gene SLC39A8. Am. J. Hum. Genet. 97, 886-893. 10.1016/j.ajhg.2015.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton, C. E., Zhou, Y., Bai, Q. and Burton, E. A. (2017). Spectral properties of the zebrafish visual motor response. Neurosci. Lett. 646, 62-67. 10.1016/j.neulet.2017.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caito, S. and Aschner, M. (2015). Neurotoxicity of metals. Handb Clin Neurol 131, 169-189. 10.1016/B978-0-444-62627-1.00011-1 [DOI] [PubMed] [Google Scholar]
- Chen, P., Bornhorst, J. and Aschner, M. (2018). Manganese metabolism in humans. Front Biosci (Landmark Ed) 23, 1655-1679. 10.2741/4665 [DOI] [PubMed] [Google Scholar]
- Chino, M., Leone, L., Zambrano, G., Pirro, F., D'alonzo, D., Firpo, V., Aref, D., Lista, L., Maglio, O., Nastri, Fet al. et al. (2018). Oxidation catalysis by iron and manganese porphyrins within enzyme-like cages. Biopolymers 109, e23107. 10.1002/bip.23107 [DOI] [PubMed] [Google Scholar]
- Choi, H. M. T., Schwarzkopf, M., Fornace, M. E., Acharya, A., Artavanis, G., Stegmaier, J., Cunha, A. and Pierce, N. A. (2018). Third-generation in situ hybridization chain reaction: multiplexed, quantitative, sensitive, versatile, robust. Development 145, dev165753. 10.1242/dev.165753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins, J. E., Wali, N., Sealy, I. M., Morris, J. A., White, R. J., Leonard, S. R., Jackson, D. K., Jones, M. C., Smerdon, N. C., Zamora, J.et al. (2015). High-throughput and quantitative genome-wide messenger RNA sequencing for molecular phenotyping. BMC Genomics 16:578. 10.1186/s12864-015-1788-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairless, R., Williams, S. K. and Diem, R. (2019). Calcium-binding proteins as determinants of central nervous system neuronal vulnerability to disease. Int. J. Mol. Sci. 20. 10.3390/ijms20092146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes, A. M., Fero, K., Arrenberg, A. B., Bergeron, S. A., Driever, W. and Burgess, H. A. (2012). Deep brain photoreceptors control light-seeking behavior in zebrafish larvae. Curr. Biol. 22, 2042-2047. 10.1016/j.cub.2012.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fordahl, S. C. and Erikson, K. M. (2014). Manganese accumulation in membrane fractions of primary astrocytes is associated with decreased gamma-aminobutyric acid (GABA) uptake, and is exacerbated by oleic acid and palmitate. Environ. Toxicol. Pharmacol. 37, 1148-1156. 10.1016/j.etap.2014.03.016 [DOI] [PubMed] [Google Scholar]
- Ghosh, M. and Rihel, J. (2020). Hierarchical Compression reveals sub-second to day-long structure in larval Zebrafish behavior. eNeuro 7, ENEURO.0408-19.2020. 10.1523/ENEURO.0408-19.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong, H. and Amemiya, T. (1996). Ultrastructure of retina of manganese-deficient rats. Invest. Ophthalmol. Vis. Sci. 37, 1967-1974. [PubMed] [Google Scholar]
- Gorojod, R. M., Alaimo, A., Porte Alcon, S., Saravia, F. and Kotler, M. L. (2017). Interplay between lysosomal, mitochondrial and death receptor pathways during manganese-induced apoptosis in glial cells. Arch. Toxicol. 91:3065-3078. 10.1007/s00204-017-1936-7 [DOI] [PubMed] [Google Scholar]
- Gorojod, R. M., Alaimo, A., Porte Alcon, S., Martinez, J. H., Cortina, M. E., Vazquez, E. S. and Kotler, M. L. (2018). Heme Oxygenase-1 protects astroglia against manganese-induced oxidative injury by regulating mitochondrial quality control. Toxicol. Lett. 295:357-368. 10.1016/j.toxlet.2018.07.045 [DOI] [PubMed] [Google Scholar]
- Groenendyk, J., Agellon, L. B. and Michalak, M. (2021). Calcium signaling and endoplasmic reticulum stress. Int Rev Cell Mol Biol 363, 1-20. 10.1016/bs.ircmb.2021.03.003 [DOI] [PubMed] [Google Scholar]
- Guilarte, T. R. and Gonzales, K. K. (2015). Manganese-induced parkinsonism is not idiopathic Parkinson's disease: environmental and genetic evidence. Toxicol. Sci. 146, 204-212. 10.1093/toxsci/kfv099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta, T., Marquart, G. D., Horstick, E. J., Tabor, K. M., Pajevic, S. and Burgess, H. A. (2018). Morphometric analysis and neuroanatomical mapping of the zebrafish brain. Methods 150, 49-62. 10.1016/j.ymeth.2018.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harischandra, D. S., Ghaisas, S., Zenitsky, G., Jin, H., Kanthasamy, A., Anantharam, V. and Kanthasamy, A. G. (2019a). Manganese-Induced Neurotoxicity: New Insights Into the Triad of Protein Misfolding, Mitochondrial Impairment, and Neuroinflammation. Front Neurosci 13, 654. 10.3389/fnins.2019.00654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harischandra, D. S., Rokad, D., Neal, M. L., Ghaisas, S., Manne, S., Sarkar, S., Panicker, N., Zenitsky, G., Jin, H., Lewis, M.et al. (2019b). Manganese promotes the aggregation and prion-like cell-to-cell exosomal transmission of alpha-synuclein. Sci. Signal. 12, eaau4543. 10.1126/scisignal.aau4543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa, S., Kumagai, M., Hagihara, M., Nishimaru, H., Hirano, K., Kaneko, R., Okayama, A., Hirayama, T., Sanbo, M., Hirabayashi, M.et al. (2016). Distinct and Cooperative Functions for the Protocadherin-alpha, -beta and -gamma Clusters in Neuronal Survival and Axon Targeting. Front Mol Neurosci 9:155. 10.3389/fnmol.2016.00155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz, S., Benner, C., Spann, N., Bertolino, E., Lin, Y. C., Laslo, P., Cheng, J. X., Murre, C., Singh, H. and Glass, C. K. (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576-589. 10.1016/j.molcel.2010.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez, R. B., Moteshareie, H., Burnside, D., Mckay, B. and Golshani, A. (2019). Manganese-induced cellular disturbance in the baker's yeast, Saccharomyces cerevisiae with putative implications in neuronal dysfunction. Sci. Rep. 9, 6563. 10.1038/s41598-019-42907-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz, E., Terenius, L., Vukojevic, V. and Svenningsson, P. (2019). GPR37 and GPR37L1 differently interact with dopamine 2 receptors in live cells. Neuropharmacology 152, 51-57. 10.1016/j.neuropharm.2018.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong, C. J., Yeon, J., Yeo, B. K., Woo, H., An, H. K., Heo, W., Kim, K. and Yu, S. W. (2020). Fas-apoptotic inhibitory molecule 2 localizes to the lysosome and facilitates autophagosome-lysosome fusion through the LC3 interaction region motif-dependent interaction with LC3. FASEB J. 34, 161-179. 10.1096/fj.201901626R [DOI] [PubMed] [Google Scholar]
- Ijomone, O. M., Miah, M. R., Peres, T. V., Nwoha, P. U. and Aschner, M. (2016). Null allele mutants of trt-1, the catalytic subunit of telomerase in Caenorhabditis elegans, are less sensitive to Mn-induced toxicity and DAergic degeneration. Neurotoxicology 57, 54-60. 10.1016/j.neuro.2016.08.016 [DOI] [PubMed] [Google Scholar]
- Jadiya, P., Garbincius, J. F. and Elrod, J. W. (2021). Reappraisal of metabolic dysfunction in neurodegeneration: Focus on mitochondrial function and calcium signaling. Acta Neuropathol. Commun. 9:124. 10.1186/s40478-021-01224-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juneja, M., Shamim, U., Joshi, A., Mathur, A., Uppili, B., Sairam, S., Ambawat, S., Dixit, R. and Faruq, M. (2018). A novel mutation in SLC39A14 causing hypermanganesemia associated with infantile onset dystonia. J. Gene Med. 20, e3012. 10.1002/jgm.3012 [DOI] [PubMed] [Google Scholar]
- Kalbitzer, H. R., Stehlik, D. and Hasselbach, W. (1978). The binding of calcium and magnesium to sarcoplasmic reticulum vesicles as studied by manganese electron paramagnetic resonance. Eur. J. Biochem. 82, 245-255. 10.1111/j.1432-1033.1978.tb12017.x [DOI] [PubMed] [Google Scholar]
- Kaupp, U. B. and Seifert, R. (2002). Cyclic nucleotide-gated ion channels. Physiol. Rev. 82, 769-824. 10.1152/physrev.00008.2002 [DOI] [PubMed] [Google Scholar]
- Kikuchihara, Y., Abe, H., Tanaka, T., Kato, M., Wang, L., Ikarashi, Y., Yoshida, T. and Shibutani, M. (2015). Relationship between brain accumulation of manganese and aberration of hippocampal adult neurogenesis after oral exposure to manganese chloride in mice. Toxicology 331, 24-34. 10.1016/j.tox.2015.02.005 [DOI] [PubMed] [Google Scholar]
- Kim, S. Y., Yasuda, S., Tanaka, H., Yamagata, K. and Kim, H. (2011). Non-clustered protocadherin. Cell Adh Migr 5, 97-105. 10.4161/cam.5.2.14374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel, C. B., Ballard, W. W., Kimmel, S. R., Ullmann, B. and Schilling, T. F. (1995). Stages of embryonic development of the zebrafish. Dev. Dyn. 203, 253-310. 10.1002/aja.1002030302 [DOI] [PubMed] [Google Scholar]
- Koller, W. C., Lyons, K. E. and Truly, W. (2004). Effect of levodopa treatment for parkinsonism in welders: A double-blind study. Neurology 62, 730-733. 10.1212/01.WNL.0000113726.34734.15 [DOI] [PubMed] [Google Scholar]
- Lee, J. H. and Shin, J. H. (2022). Effect of chelation therapy on a korean patient with brain manganese deposition resulting from a compound heterozygous mutation in the SLC39A14 gene. J. Mov. Disord. 15, 171-174. 10.14802/jmd.21143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung, K. W., Liu, M., Xu, X., Seiler, M. J., Barnstable, C. J. and Tombran-Tink, J. (2008). Expression of ZnT and ZIP zinc transporters in the human RPE and their regulation by neurotrophic factors. Invest. Ophthalmol. Vis. Sci. 49, 1221-1231. 10.1167/iovs.07-0781 [DOI] [PubMed] [Google Scholar]
- Li, H. and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754-1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H., Fan, X., Luo, Y., Song, S., Liu, J. and Fan, Q. (2017). Repeated manganese administration produced abnormal expression of circadian clock genes in the hypothalamus and liver of rats. Neurotoxicology 62, 39-45. 10.1016/j.neuro.2017.05.007 [DOI] [PubMed] [Google Scholar]
- Liu K., Guo C., Lao Y., Yang J., Chen F., Zhao Y., Yang Y., Yang J. and Yi J. (2019). A fine-tuning mechanism underlying self-control for autophagy: deSUMOylation of BECN1 by SENP3. Autophagy, 6, 975-990. 10.1080/15548627.2019.1647944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak, K. J. and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402-408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- Love, M. I., Huber, W. and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, X., Liu, Z., Ge, X., Huang, S., Zhou, Y., Li, D., Li, L., Chen, X., Huang, L., Hou, Q.et al. (2020). High manganese exposure decreased the risk of high triglycerides in workers: a cross-sectional study. BMC Public Health 20, 874. 10.1186/s12889-020-09011-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancini, M., Bassani, S. and Passafaro, M. (2020). Right place at the right time: how changes in protocadherins affect synaptic connections contributing to the etiology of neurodevelopmental disorders. Cells 9. 10.3390/cells9122711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquart, G. D., Tabor, K. M., Brown, M., Strykowski, J. L., Varshney, G. K., Lafave, M. C., Mueller, T., Burgess, S. M., Higashijima, S. and Burgess, H. A. (2015). A 3D Searchable Database of Transgenic Zebrafish Gal4 and Cre Lines for Functional Neuroanatomy Studies. Front Neural Circuits 9, 78. 10.3389/fncir.2015.00078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquart, G. D., Tabor, K. M., Horstick, E. J., Brown, M., Geoca, A. K., Polys, N. F., Nogare, D. D. and Burgess, H. A. (2017). High-precision registration between zebrafish brain atlases using symmetric diffeomorphic normalization. Gigascience 6, 1-15. 10.1093/gigascience/gix056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marreilha Dos Santos, A. P., Andrade, V. and Aschner, M. (2017). Neuroprotective and Therapeutic Strategies for Manganese-Induced Neurotoxicity. Clin Pharmacol Transl Med 1:54-62. [PMC free article] [PubMed] [Google Scholar]
- Marti-Sanchez, L., Ortigoza-Escobar, J. D., Darling, A., Villaronga, M., Baide, H., Molero-Luis, M., Batllori, M., Vanegas, M. I., Muchart, J., Aquino, L.et al. (2018). Hypermanganesemia due to mutations in SLC39A14: further insights into Mn deposition in the central nervous system. Orphanet J. Rare Dis. 13, 28. 10.1186/s13023-018-0758-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Finley, E. J., Gavin, C. E., Aschner, M. and Gunter, T. E. (2013). Manganese neurotoxicity and the role of reactive oxygen species. Free Radic. Biol. Med. 62, 65-75. 10.1016/j.freeradbiomed.2013.01.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller, K. P. and Neuhauss, S. C. (2014). Sunscreen for fish: co-option of UV light protection for camouflage. PLoS One 9, e87372. 10.1371/journal.pone.0087372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura, T., Oh, C. K., Zhang, X. and Lipton, S. A. (2021). Protein S-nitrosylation and oxidation contribute to protein misfolding in neurodegeneration. Free Radic. Biol. Med. 172, 562-577. 10.1016/j.freeradbiomed.2021.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nara, M., Tasumi, M., Tanokura, M., Hiraoki, T., Yazawa, M. and Tsutsumi, A. (1994). Infrared studies of interaction between metal ions and Ca(2+)-binding proteins. Marker bands for identifying the types of coordination of the side-chain COO- groups to metal ions in pike parvalbumin (pI=4.10). FEBS Lett. 349, 84-88. 10.1016/0014-5793(94)00645-8 [DOI] [PubMed] [Google Scholar]
- Octeau, J. C., Schrader, J. M., Masuho, I., Sharma, M., Aiudi, C., Chen, C. K., Kovoor, A. and Celver, J. (2014). G protein beta 5 is targeted to D2-dopamine receptor-containing biochemical compartments and blocks dopamine-dependent receptor internalization. PLoS One 9, e105791. 10.1371/journal.pone.0105791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou, C. Y., Luo, Y. N., He, S. N., Deng, X. F., Luo, H. L., Yuan, Z. X., Meng, H. Y., Mo, Y. H., Li, S. J. and Jiang, Y. M. (2017). Sodium P-Aminosalicylic Acid Improved Manganese-Induced Learning and Memory Dysfunction via Restoring the Ultrastructural Alterations and gamma-Aminobutyric Acid Metabolism Imbalance in the Basal Ganglia. Biol. Trace Elem. Res. 176, 143-153. 10.1007/s12011-016-0802-4 [DOI] [PubMed] [Google Scholar]
- Park, J. H., Hogrebe, M., Grüneberg, M., Duchesne, I., von der Heiden, A. L., Reunert, J., Schlingmann, K. P., Boycott, K. M., Beaulieu, C. L., Mhanni, A. A.et al. (2015). SLC39A8 Deficiency: A Disorder of Manganese Transport and Glycosylation. Am. J. Hum. Genet. 97:894-903. 10.1016/j.ajhg.2015.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popichak, K. A., Afzali, M. F., Kirkley, K. S. and Tjalkens, R. B. (2018). Glial-neuronal signaling mechanisms underlying the neuroinflammatory effects of manganese. J. Neuroinflammation 15, 324. 10.1186/s12974-018-1349-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porte Alcon, S., Gorojod, R. M. and Kotler, M. L. (2018). Regulated Necrosis Orchestrates Microglial Cell Death in Manganese-Induced Toxicity. Neuroscience 393:206-225. 10.1016/j.neuroscience.2018.10.006 [DOI] [PubMed] [Google Scholar]
- Quintanar, L., Montiel, T., Marquez, M., Gonzalez, A. and Massieu, L. (2012). Calpain activation is involved in acute manganese neurotoxicity in the rat striatum in vivo. Exp. Neurol. 233, 182-192. 10.1016/j.expneurol.2011.09.032 [DOI] [PubMed] [Google Scholar]
- Randlett, O., Wee, C. L., Naumann, E. A., Nnaemeka, O., Schoppik, D., Fitzgerald, J. E., Portugues, R., Lacoste, A. M., Riegler, C., Engert, F.et al. (2015). Whole-brain activity mapping onto a zebrafish brain atlas. Nat. Methods 12, 1039-1046. 10.1038/nmeth.3581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao, M. V. and Zaidel-Bar, R. (2016). Formin-mediated actin polymerization at cell-cell junctions stabilizes E-cadherin and maintains monolayer integrity during wound repair. Mol. Biol. Cell 27, 2844-2856. 10.1091/mbc.e16-06-0429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rihel, J., Prober, D. A., Arvanites, A., Lam, K., Zimmerman, S., Jang, S., Haggarty, S. J., Kokel, D., Rubin, L. L., Peterson, R. T. et al. (2010). Zebrafish behavioral profiling links drugs to biological targets and rest/wake regulation. Science 327, 348-351. 10.1126/science.1183090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizor, A., Pajarillo, E., Nyarko-Danquah, I., Digman, A., Mooneyham, L., Son, D. S., Aschner, M. and Lee, E. (2021). Manganese-induced reactive oxygen species activate IkappaB kinase to upregulate YY1 and impair glutamate transporter EAAT2 function in human astrocytes in vitro. Neurotoxicology 86, 94-103. 10.1016/j.neuro.2021.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodan, L. H., Hauptman, M., D'gama, A. M., Qualls, A. E., Cao, S., Tuschl, K., Al-Jasmi, F., Hertecant, J., Hayflick, S. J., Wessling-Resnick, M.et al. (2018). Novel founder intronic variant in SLC39A14 in two families causing Manganism and potential treatment strategies. Mol. Genet. Metab. 124, 161-167. 10.1016/j.ymgme.2018.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidoryk-Wegrzynowicz, M. and Aschner, M. (2013). Manganese toxicity in the central nervous system: the glutamine/glutamate-gamma-aminobutyric acid cycle. J. Intern. Med. 273, 466-477. 10.1111/joim.12040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirri, F., Maiorano, G., Tavaniello, S., Chen, J., Petracci, M. and Meluzzi, A. (2016). Effect of different levels of dietary zinc, manganese, and copper from organic or inorganic sources on performance, bacterial chondronecrosis, intramuscular collagen characteristics, and occurrence of meat quality defects of broiler chickens. Poult. Sci. 95, 1813-1824. 10.3382/ps/pew064 [DOI] [PubMed] [Google Scholar]
- Smith, M. R., Fernandes, J., Go, Y. M. and Jones, D. P. (2017). Redox dynamics of manganese as a mitochondrial life-death switch. Biochem. Biophys. Res. Commun. 482, 388-398. 10.1016/j.bbrc.2016.10.126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares, A. T. G., Silva, A. C., Tinkov, A. A., Khan, H., Santamaria, A., Skalnaya, M. G., Skalny, A. V., Tsatsakis, A., Bowman, A. B., Aschner, M.et al. (2020). The impact of manganese on neurotransmitter systems. J. Trace Elem. Med. Biol. 61:126554. 10.1016/j.jtemb.2020.126554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, D., Ma, J., Chen, L., Guo, C., Zhang, Y., Chen, T., Zhang, S., Zhu, Z., Tian, L. and Niu, P. (2017). FOXO3 promoted mitophagy via nuclear retention induced by manganese chloride in SH-SY5Y cells. Metallomics 9, 1251-1259. 10.1039/C7MT00085E [DOI] [PubMed] [Google Scholar]
- Taka, E., Mazzio, E., Soliman, K. F. and Renee, R. R. (2012). Microarray genomic profile of mitochondrial and oxidant response in manganese chloride treated PC12 cells. Neurotoxicology 33, 162-168. 10.1016/j.neuro.2012.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamas, M. J., Sharma, S. K., Ibstedt, S., Jacobson, T. and Christen, P. (2014). Heavy metals and metalloids as a cause for protein misfolding and aggregation. Biomolecules 4, 252-267. 10.3390/biom4010252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, K. J. and Wessling-Resnick, M. (2019). ZIP14 is degraded in response to manganese exposure. Biometals 32, 829-843. 10.1007/s10534-019-00216-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinkov, A. A., Paoliello, M. M. B., Mazilina, A. N., Skalny, A. V., Martins, A. C., Voskresenskaya, O. N., Aaseth, J., Santamaria, A., Notova, S. V., Tsatsakis, A.et al. (2021). Molecular Targets of Manganese-Induced Neurotoxicity: A Five-Year Update. Int. J. Mol. Sci. 22. 10.3390/ijms22094646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjalkens, R. B., Popichak, K. A. and Kirkley, K. A. (2017). Inflammatory Activation of Microglia and Astrocytes in Manganese Neurotoxicity. Adv Neurobiol 18, 159-181. 10.1007/978-3-319-60189-2_8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuschl, K., Mills, P. B., Parsons, H., Malone, M., Fowler, D., Bitner-Glindzicz, M. and Clayton, P. T. (2008). Hepatic cirrhosis, dystonia, polycythaemia and hypermanganesaemia--a new metabolic disorder. J. Inherit. Metab. Dis. 31, 151-163. 10.1007/s10545-008-0813-1 [DOI] [PubMed] [Google Scholar]
- Tuschl, K., Clayton, P. T., Gospe, S. M. and Mills, P. B. (2012a). Hypermanganesemia with Dystonia 1. In GeneReviews® (ed. Adam M. P., Mirzaa G. M., Pagon R. A., Wallace S. E., Bean L. J. H., Gripp K. W. and Amemiya A.). University of Washington, Seattle. https://www.ncbi.nlm.nih.gov/books/NBK100241/ [PubMed] [Google Scholar]
- Tuschl, K., Clayton, P. T., Gospe, S. M., Jr, Gulab, S., Ibrahim, S., Singhi, P., Aulakh, R., Ribeiro, R. T., Barsottini, O. G., Zaki, M. S.et al. (2012b). Syndrome of hepatic cirrhosis, dystonia, polycythemia, and hypermanganesemia caused by mutations in SLC30A10, a manganese transporter in man. Am. J. Hum. Genet. 90:457-466. 10.1016/j.ajhg.2012.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuschl, K., Meyer, E., Valdivia, L. E., Zhao, N., Dadswell, C., Abdul-Sada, A., Hung, C. Y., Simpson, M. A., Chong, W. K., Jacques, T. S.et al. (2016). Mutations in SLC39A14 disrupt manganese homeostasis and cause childhood-onset parkinsonism-dystonia. Nat. Commun. 7:11601. 10.1038/ncomms11601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, W. A., Groenendyk, J. and Michalak, M. (2012). Calreticulin signaling in health and disease. Int. J. Biochem. Cell Biol. 44, 842-846. 10.1016/j.biocel.2012.02.009 [DOI] [PubMed] [Google Scholar]
- Wang, D., Zhang, J., Jiang, W., Cao, Z., Zhao, F., Cai, T., Aschner, M. and Luo, W. (2017). The role of NLRP3-CASP1 in inflammasome-mediated neuroinflammation and autophagy dysfunction in manganese-induced, hippocampal-dependent impairment of learning and memory ability. Autophagy 13, 914-927. 10.1080/15548627.2017.1293766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, C., Ma, Z., Yan, D. Y., Liu, C., Deng, Y., Liu, W., Xu, Z. F. and Xu, B. (2018). Alpha-Synuclein and Calpains Disrupt SNARE-Mediated Synaptic Vesicle Fusion During Manganese Exposure in SH-SY5Y Cells. Cells 7. 10.3390/cells7120258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, B., Shan, M., Wang, F., Deng, Y., Liu, W., Feng, S., Yang, T. Y. and Xu, Z. F. (2013). Endoplasmic reticulum stress signaling involvement in manganese-induced nerve cell damage in organotypic brain slice cultures. Toxicol. Lett. 222, 239-246. 10.1016/j.toxlet.2013.08.001 [DOI] [PubMed] [Google Scholar]
- Yates, A. D., Garavís, M., Gonzã¡Lez, C., Jameson, G. B., Filichev, V. V. and Hale, T. K. . (2020). Ensembl 2020. Nucleic Acids Res. 48:D682-D688. 10.1093/nar/gkz1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeglam, A., Abugrara, A. and Kabuka, M. (2018). Autosomal-recessive iron deficiency anemia, dystonia and hypermanganesemia caused by new variant mutation of the manganese transporter gene SLC39A14. Acta Neurol. Belg. 119, 379-384. 10.1007/s13760-018-1024-7 [DOI] [PubMed] [Google Scholar]
- Zhi, C. N., Lai, L. L., Dou, C. S., Wang, X. H., Zhao, P., Fu, J. L. and Yao, B. Y. (2019). [The role of lysosomes in manganese-induced toxicity in SK-N-SH cells]. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi 37, 332-336. 10.3760/cma.j.issn.1001-9391.2019.05.003 [DOI] [PubMed] [Google Scholar]
- Zofkova, I., Davis, M. and Blahos, J. (2017). Trace elements have beneficial, as well as detrimental effects on bone homeostasis. Physiol. Res. 66:391-402. 10.33549/physiolres.933454 [DOI] [PubMed] [Google Scholar]
- Zou, Y., Qing, L., Zeng, X., Shen, Y., Zhong, Y., Liu, J., Li, Q., Chen, K., Lv, Y., Huang, D.et al. (2014). Cognitive function and plasma BDNF levels among manganese-exposed smelters. Occup. Environ. Med. 71, 189-194. 10.1136/oemed-2013-101896 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.