

Abstract
Cilia are antenna-like organelles associated with a spectrum of human disease states. The functions of cilia remain incompletely understood, particularly in humans where cellular models do not recapitulate complex disease. Here we show that human pluripotent stem cells lacking the kinesin-2 subunits KIF3A or KIF3B, generated by CRISPR gene editing, establish a general tool for the study of ciliary function in tissue morphogenesis. Both KIF3A−/− and KIF3B−/− hPSCs lack cilia, yet remain self-renewing and pluripotent, enabling efficient derivation of diverse somatic cells. Derived tissues and organoids express multi-lineage phenotypes modeling the ciliopathy spectrum, including neuronal and nephron differentiation defects, and polycystic kidney disease. Modular culture conditions and biochemical analysis reveal that human cilia mediate a critical switch in hedgehog signaling during organoid differentiation, and constitutively release extracellular vesicles containing signaling molecules associated with these phenotypes. KIF3A−/− and KIF3B−/− hPSCs thus establish a human genetic system linking complex ciliary phenotypes with endogenous mechanisms, for therapeutics discovery and regenerative applications.
Structurally, eukaryotic cilia comprise a bundle of parallel, axonemal microtubules anchored to a basal body and encased in a sheath of plasma membrane, and may be present individually (primary cilia) or in multi-ciliated arrays 1–3. A conserved pathway called intraflagellar transport (IFT) facilitates the ciliary import and export of specific molecular cargoes along the axonemal microtubules 4, 5. Kinesin-2 is a family of motor proteins such as KIF3A and KIF3B that play important roles in IFT and ciliogenesis 6. Mutations in over fifty genes associated with ciliary structure, transport, or function cause a spectrum of ciliopathy syndromes, such as nephronophthisis (NPHP), Joubert syndrome, and PKD 7–9. Cilia are absent in mitotic cells and many tumors, and may act as a brake on cell division 10–12. The presence or absence of cilia can also affect the development of medulloblastomas and basal cell carcinomas 13, 14. Cilia have therefore emerged as an important therapeutic target for diverse disease states.
A major barrier to understanding ciliary function is the absence of cellular models that accurately reconstitute complex human phenotypes and disease mechanisms. In mice, genetic disruption of IFT results in complete loss of cilia and early embryonic lethality, precluding further analysis 15–17. Conditional or hypomorphic mutations can produce tissue-specific phenotypes, such as retinal degeneration and cystic kidney disease, but cilia are only partially ablated in these models, which do not fully genocopy or phenocopy humans 18–22. At the other end of the spectrum, primary or immortalized cells with ciliopathy mutations are rare, lineage-restricted, heterogenous, and incapable of recapitulating complex tissue phenotypes 8, 23, 24. In contrast, human pluripotent stem cells (hPSCs), including embryonic stem cells and induced pluripotent stem cells, possess an extensive capacity for complex tissue differentiation 25, 26. Undifferentiated hPSCs possess primary cilia and can exhibit defects in ciliary trafficking, number, or length linked to disease 27, 28. Gene editing methods, such as the Cas9/CRISPR (clustered regularly interspaced short palindromic repeats) system, further enable elegant genetic models featuring mutant and control hPSCs of identical genetic background 29–31. Here, we generate gene-edited hPSCs lacking KIF3A or KIF3B (kinesin-2 knockout hPSCs), and use these to recapitulate hallmark features of the ciliopathies and reveal molecular functions of these critical organelles.
RESULTS
Generation of stable hPSCs without cilia
To establish a general tool for studying the function of human cilia, we employed a genetic strategy to completely ablate these organelles in hPSCs. By applying the CRISPR-Cas9 gene-editing system to hPSCs, we introduced nonsense mutations in the KIF3A or KIF3B genes 6. Three distinct guide RNAs were applied to male or female hPSCs to generate a collection of fourteen different mutant cell lines, plus twelve isogenic control cell lines of matching passage and experimental history (Fig. 1a and Supplementary Fig. 1a). Each cell line was subjected to allele-specific sequencing to determine every knockout mutation (Fig. 1b and Supplementary Fig. 1b–e). KIF3A and KIF3B proteins were expressed in unmodified controls, but the targeted protein was undetectable in each of the gene-edited hPSC lines (Fig. 1c and Supplementary Fig. 2a–e). Whereas ~ 50 % of control hPSCs formed ciliary axonemes (ARL13B+ and acetylated α-tubulin+), cilia were completely undetectable in the gene-edited hPSCs of both targeted genes in both genetic backgrounds (Fig. 1d–e and Supplementary Fig. 3a) 27, 28. We referred to this collection of mutants as kinesin-2 knockout hPSCs.
Fig. 1. A CRISPR gene editing strategy produces stable hPSCs completely lacking cilia.
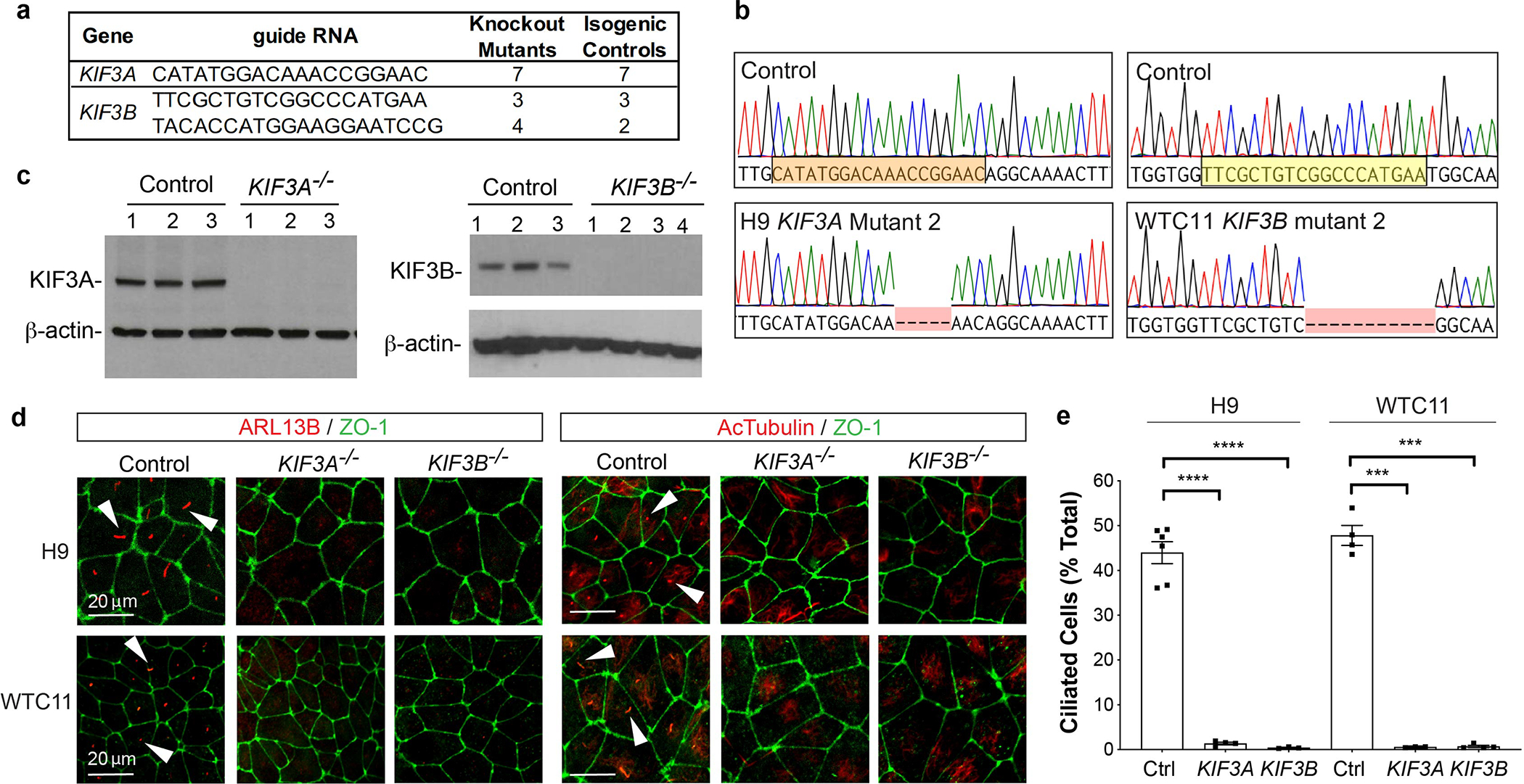
(a) Summary of new hPSC lines. (b) Representative KIF3A and KIF3B chromatograms. Two genetic backgrounds (H9 and WTC11) were used to generate isogenic pairs. The sgRNA sequence used for CRISPR targeting is highlighted. (c) KIF3A and KIF3B immunoblots showing multiple distinct control, KIF3A−/−, and KIF3B−/− hPSC lines. (d) Confocal immunofluorescence images of ARL13B and acetylated α-tubulin (AcTub) with ZO-1 counterstain in representative fields of undifferentiated control and kinesin-2 knockout hPSCs, on two distinct genetic backgrounds (H9 ES cells or WTC11 iPS cells). Representative ciliary axonemes are indicated with arrowheads. (e) Cilia counts in undifferentiated control and kinesin-2 hPSCs on two distinct genetic backgrounds (mean ± s.e.m., n ≥ 3 independent biological replicates per condition from a total of 24 distinct cell lines; ≥ 180 cells counted per cell line). Scale bars, 20 μm. ****, p < 0.0001; ***, p < 0.001.
Both KIF3A−/− and KIF3B−/− hPSCs formed flat, smooth-edged colonies with characteristic hPSC morphology, which remained stable and self-renewing with no signs of aberrant differentiation or senescence in over fifty passages in vitro (Fig. 2a). Growth rates were indistinguishable from isogenic controls (p = 0.69), and karyotyping detected no significant increase in chromosomal abnormalities (Fig. 2b–d). When cultured in three-dimensional matrix, kinesin-2 knockout hPSCs formed pluripotent epiblast spheroids with hollow, amniotic cavities of normal size and polarity, despite the loss of cilia from the luminal surface (Fig. 2e and Supplementary Fig. 3b–e) 31. Thus, cilia were dispensable in undifferentiated hPSCs.
Fig. 2. KIF3A−/− and KIF3B−/− hPSCs establish a renewable source of diverse human cell types lacking cilia.
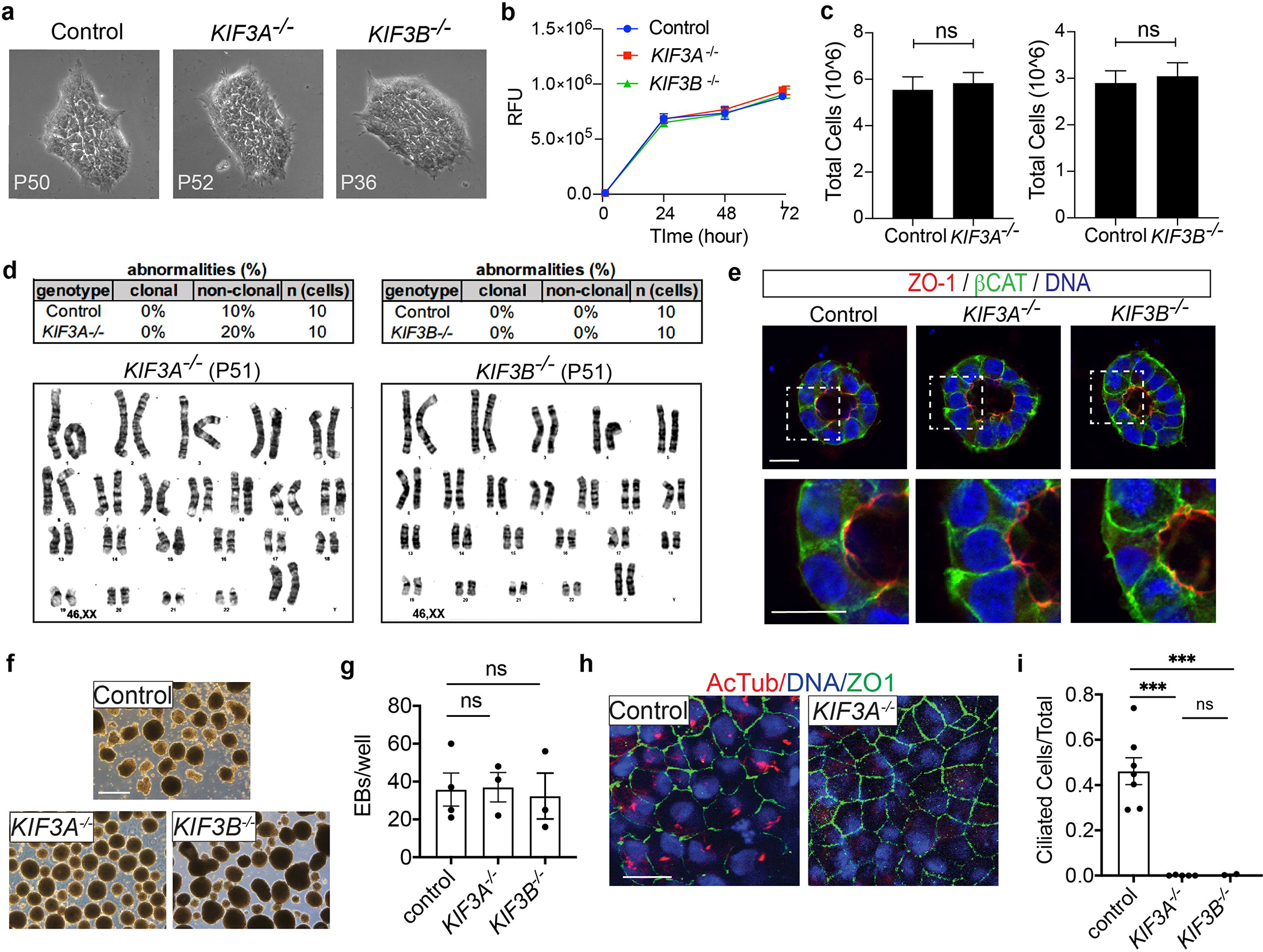
(a) hPSC colony morphology at passages > 30. No abnormalities were observed in earlier or later passages. (b) Cell viability assay (alamarBlue). Identical numbers of KIF3A−/− or KIF3B−/− hPSCs or isogenic controls were plated on day 0 and relative fluorescence units (RFU) were quantified at the indicated times points (mean ± s.e.m., n = 3 independent biological replicates per condition from a total of 9 distinct cell lines). (c) Average cell number at passage, after plating identical numbers of cells (mean ± s.e.m., n ≥ 7 biological replicates per condition from a total of 11 distinct cell lines). (d) Summary of clonal abnormalities and representative karyotypes from KIF3A−/− or KIF3B−/− hPSCs, compared to isogenic control (mean of n = 10 cells from two distinct cell lines per condition). (e) Confocal sections showing immunofluorescence for pluripotency, polarity, and ciliary markers in hPSC epiblast spheroids. Scale bars, 20 μm. (f) Representative images and (g) quantification of EB formation in control and KIF3A−/− or KIF3B−/− knockout hPSCs (n ≥ 3 biological replicates per condition from a total of 8 distinct cell lines). Scale bars, 500 μm. (h) Confocal images and (i) quantification of cilia formation in EB epithelial cells expressing the tight junction marker zonula occludens 1 (ZO-1). Scale bars, 20 μm (n ≥ 2 independent biological replicates from a total of 9 distinct cell lines. ***, p < 0.001).
Kinesin-2 knockout hPSCs produce diverse cell types lacking cilia
When switched to differentiating culture conditions, both kinesin-2 knockout hPSCs and controls differentiated efficiently into embryoid bodies (EBs) containing cores of ectoderm (neuroepithelium) bordering migratory outgrowths of endoderm (α-fetoprotein, AFP) and mesoderm (smooth muscle α-actin, SMA) (Fig. 2f–g and Supplementary Fig. 4a). Primary cilia were detected in control EBs, but not in KIF3A−/− or KIF3B−/− EBs, indicating that these lineages formed without cilia (Fig. 2h–i and Supplementary Fig. 4a). In prolonged cultures, both control and kinesin-2 knockout EBs formed patches of pigmented epithelium (Supplementary Fig. 4b). No significant difference was observed in the ability of kinesin-2 knockout hPSCs to form EBs (p = 0.92) and pigmented cells (p = 0.68), nor in their dose-dependent upregulation of brachyury (BRY) in response to CHIR99021 (Fig. 2g and Supplementary Fig. 4c–e) 32–34. These mutant hPSCs were therefore pluripotent and provided a stable and renewable source of diverse cell types.
EB culture involves complex media containing high concentrations of proteins and growth factors. To provide a more physiological environment for differentiation, hPSCs were implanted into immunodeficient animals in vivo. Both kinesin-2 knockout hPSCs and isogenic controls rapidly formed large tumors under these conditions (Supplementary Fig. 5a–b) 25. Growths from kinesin-2 knockout hPSCs were of similar size and weight to controls (p = 0.75) and contained a diversity of complex and well-differentiated tissues typical of teratomas, including pigmented epithelia and cartilage (Fig. 3a–b) 25. However, secretory epithelia were reduced > 10-fold in growths derived from kinesin-2 knockout hPSCs, while undifferentiated blasts, which stained primarily with hematoxylin rather than eosin, were ~ 5-fold more prevalent (Fig. 3c–d). These differences were statistically significant (p < 0.01). Quantities of pigmented epithelium, cartilage, and neuroepithelium derived from kinesin-2 knockout hPSCs were less than or equal to controls (Supplementary Fig. 5c–e).
Fig. 3. Cilia are critical for terminal differentiation in vivo.
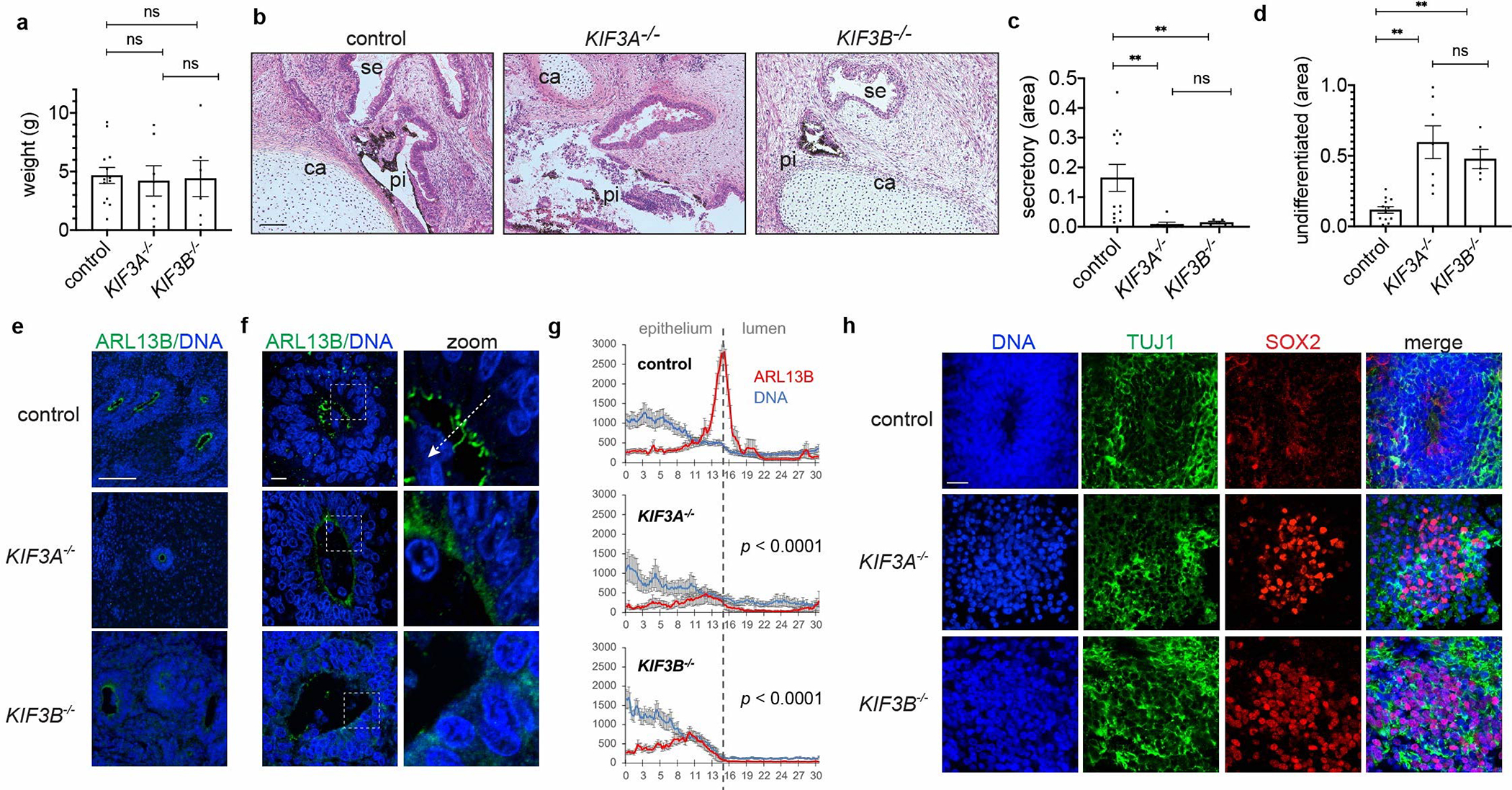
(a) Weights of growths removed from animals (mean ± s.e.m., n ≥ 6 independent biological replicates per condition from a total of 14 distinct cell lines). (b) Representative images and (c-d) quantification of tissue types in growth sections stained with hematoxylin and eosin. Labels indicate pigmented epithelium (pi), secretory epithelium (se), and cartilage (ca) regions. (mean ± s.e.m., n ≥ 5 independent biological replicates per condition from a total of 14 distinct cell lines; **, p < 0.01.) (e) Representative wide-field and (f) confocal microscope images showing ARL13B, with (g) averaged line scans drawn from neuroepithelium into rosette lumens (mean ± s.e.m., n ≥ 9 independent biological replicates per condition from a total of 8 distinct cell lines). White dashed arrow illustrates how line scans were drawn. (h) Representative confocal images showing neuronal marker immunofluorescence in teratomas. Scale bar, 100 μm (b,e) or 10 μm (f,h).
We further examined ADP-ribosylation factor-like GTPase 13b (ARL13B), which is associated with Joubert syndrome in humans and ventricular organization defects in animals 35, 36. In controls, ARL13B localized brightly to multi-ciliated arrays at the lumens of neuroepithelial rosettes, whereas in rosettes derived from KIF3A−/− or KIF3B−/− hPSCs, multi-cilia were absent, and ARL13B was restricted to a dim halo around the lumen, where its intensity was significantly reduced (Fig. 3e–g). In patches of TUJ1+ neurons, increased quantities of SOX2+ neuronal stem cells were detected in growths from kinesin-2 knockout hPSCs, compared to isogenic controls (Fig. 3h and Supplementary Fig. 5f). These experiments revealed a critical role for cilia in tissue differentiation in vivo 6.
Kidney organoid differentiation depends upon kinesin-2
Teratomas are naturally heterogenous and difficult to perturb experimentally. To elicit tissue-like phenotypes in vitro, we applied to hPSCs a kidney organoid differentiation protocol with defined, minimal media components and no FGF, which produces neuronal clusters alongside nephron-like organoids 31. Compared to isogenic controls, both KIF3A−/− and KIF3B−/− cultures showed a significant decrease in TUJ1+ neurons, despite the presence of abundant SOX2+ progenitors (Fig. 4a–c and Supplementary Fig. 6a).
Fig. 4. KIF3A−/− and KIF3B−/− kidney organoids express defects in neuronal and nephron differentiation.
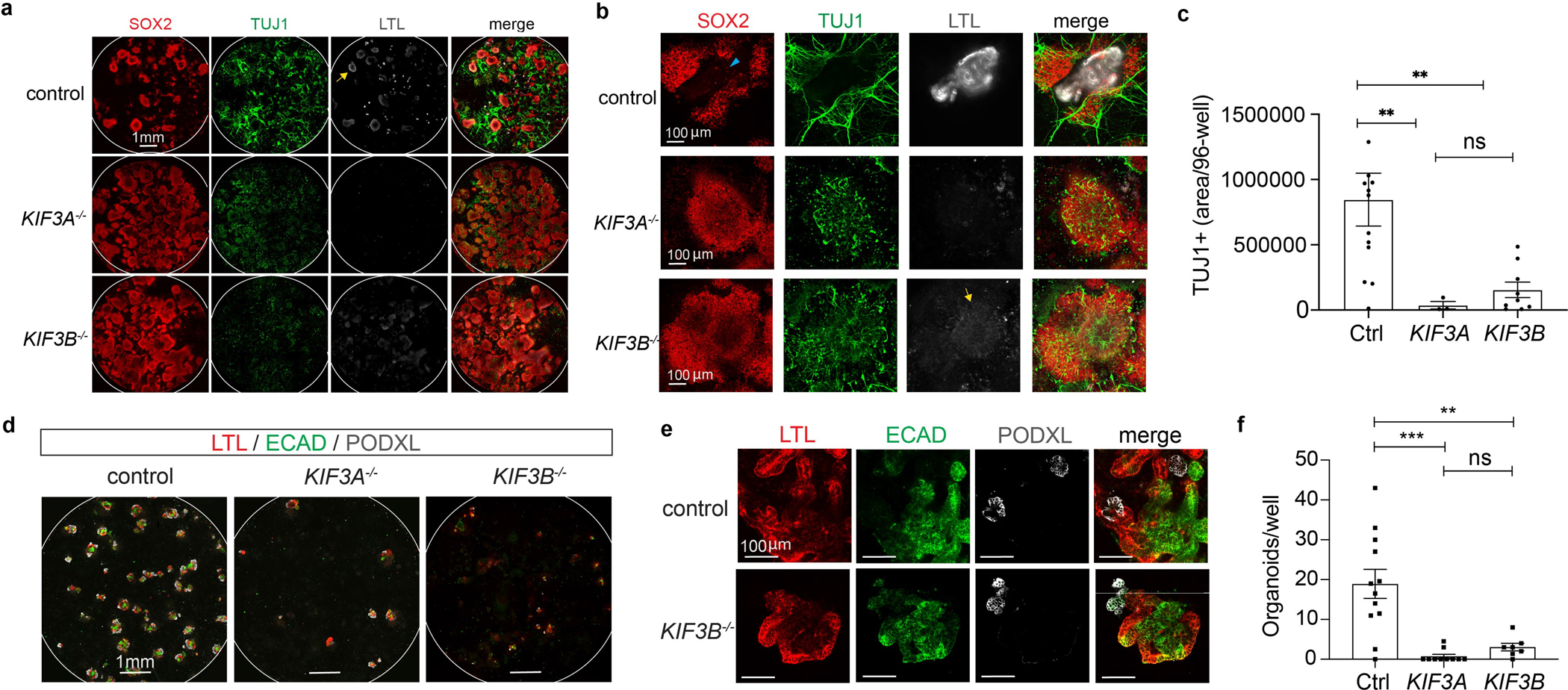
(a) Representative confocal immunofluorescence for markers of neurons and proximal tubules in control, KIF3A−/−, and KIF3B−/− kidney organoid cultures, showing whole 96-wells or (b) higher-magnification fields. A small minority of SOX2+ cells are found in proximal tubules (cyan arrowhead). In addition to tubules, fainter LTL staining is observed in neuroepithelial structures (yellow arrows). (c) Quantification of TUJ1+ neuron area in these 96-wells (mean ± s.e.m., n ≥ 3 independent biological replicates per condition from a total of 11 distinct cell lines. **, p < 0.01). (d) Confocal immunofluorescence images of nephron markers in kidney organoid differentiations in representative 96-well plates and (e) higher magnification. Each kidney organoid contains distal tubular (ECAD+), proximal tubular (LTL+), and podocyte (PODXL+) epithelial cells. (f) Quantification of kidney organoid differentiation in these lines (mean ± s.e.m., n ≥ 7 independent biological replicates per condition from a total of 13 distinct cell lines. **, p < 0.01; ***, p < 0.001).
Kidney disease is a common ciliopathy phenotype, manifesting in forms ranging from multicystic dysplastic kidney disease (MCDK) to PKD 18, 37. Kidney organoids contain distal tubules (ECAD+), proximal tubules (LTL+), and podocytes (PODXL+, NPHS1+) in continuous segments 31, 38, 39. In 96-well plates, control hPSCs formed ~ 17 organoids per well, whereas kinesin-2 knockout hPSCs produced ~ 2 organoids, indicating a requirement for cilia in the induction of nephron cells (Fig. 4d–f and Supplementary Fig. 6b).
A hedgehog signaling switch promotes organoid differentiation
Unbiased RNA-seq analysis identified several differentially expressed gene modules in kinesin-2 knockout hPSCs, including increased activation of hedgehog signaling (Fig. 5a and Supplementary Fig. 6c). As this pathway is associated with primary cilia, we conducted immunoblot analysis of effector GLI proteins and transcriptional targets of hedgehog signaling during the time course of differentiation (Fig. 5b). The hedgehog effector protein GLI3 can be expressed in full-length forms associated with pathway activation (GLI3F), as well as a shorter, partially processed forms associated with pathway repression (GLI3R) 40, 41. Immunoblots of undifferentiated hPSCs detected two major GLI3F bands of molecular weights ~ 190 and ~ 150 kDa, and a minor GLI3R band of molecular weight ~ 80 kDa (Fig. 5c and Supplementary Fig. 6d). When quantified by band intensity analysis, the GLI1R:GLI1F ratio was slightly decreased in both KIF3A−/− and KIF3B−/−hPSCs, compared to isogenic controls (Supplementary Fig. 6e).
Fig. 5. Modulation of hedgehog pathway activation via kinesin-2 promotes kidney organoid differentiation.
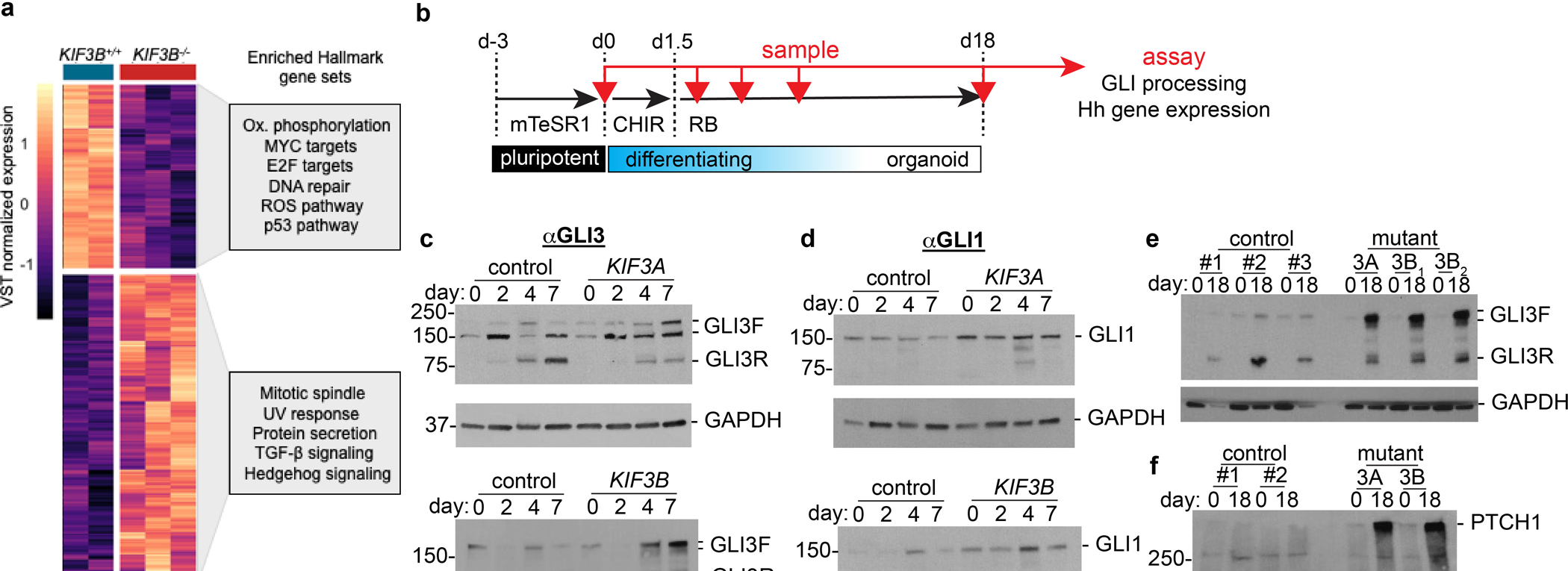
(a) Heatmap of the expression level of genes differentially expressed as a function KIF3B−/− loss in hPSCs (FDR < 0.1). Boxes to the right: Enriched gene sets from geneset analysis using the MSigDB Hallmarks gene set collection (hypergeometric test, FDR < 0.1). (b) Schematic of organoid differentiation time course, with sampling intervals indicated (red arrows). (c) Representative GLI3 and (d) GLI1 immunoblots of control versus KIF3A−/− (top) and KIF3B−/− (bottom) organoid lysates on days 0–7 of differentiation. (e) Representative GLI3 and (f) PTCH1 immunoblots of undifferentiated hPSCs (day 0) and terminally differentiated organoids (day 18) in KIF3A−/− and KIF3B−/− genotypes (3A, and 3B; subscripts indicate gRNAs 1 or 2, respectively), compared to paired isogenic controls (#1-#3).
In control cells, on days 4–7 of differentiation, GLI3F levels decreased and GLI3R levels increased relative to the undifferentiated state (Fig. 5c). In both KIF3A−/− and KIF3B−/− mutants, however, this shift in GLI3 processing was partially impaired, resulting in lower levels of GLI3R and increased levels of GLI3F, compared to isogenic controls (Fig. 5c). We further examined GLI1, a marker of hedgehog pathway activation. GLI1 was expressed in undifferentiated hPSCs, increased transiently on day 4 of differentiation, and subsequently decreased on day 7 (Fig. 5d). Compared to controls, both KIF3A−/− and KIF3B−/− mutants expressed higher levels of GLI1, peaking on day 4 with the appearance of lower molecular weight isoforms (Fig. 5d). Increased hedgehog pathway activation was thus detected in kinesin-2 mutants at early stages of differentiation.
We further examined hedgehog signaling at later stages of differentiation. Day 18 is a time point at which kidney structures in organoids express features of terminal nephron differentiation, such as podocytes 42. In control cell lines on day 18, the GLI3R:GLI3F ratio was increased ~6-fold, relative to its value in undifferentiated cells (Fig. 5e). In contrast, both KIF3A−/− and KIF3B−/− cells expressed higher levels of GLI3F, resulting in a GLI3R:GLIF ratio that was only slightly increased on day 18 compared to undifferentiated hPSCs (Fig. 5e and Supplementary Fig. 6f). Similar GLI3 profiles were observed at intermediate stages in the differentiation process (Supplementary Fig. 6g). Using a microdissection technique that we have previously established to purify nephron structures 43, 44, we found similar defects in both kidney (epithelial) and stromal subcompartments of these organoid cultures (Supplementary Fig. 6h). Thus, the observed defect in GLI3 processing in the mutants was a stable change that was not specific to a particular cell type. Despite sustained reduction in the GLI3R:GLI3F ratio in our kinesin-2 knockout mutants, GLI1 levels in these mutants on day 18 were similar to controls, being equivalent or slightly reduced compared to day 0 (Supplementary Fig. 6i). We hypothesized that this might reflect induction of inhibitors of hedgehog signal transduction such as PTCH1, which is a downstream target of hedgehog signaling 45–47. Indeed, PTCH1 was substantially upregulated on day 18 in kinesin-2 knockout mutants, but not in isogenic controls (Fig. 5f). Collectively, these data revealed that loss of kinesin-2 had long-term effects on hedgehog signaling in these cultures.
Standard kidney organoid differentiation protocols do not incorporate treatment with small molecules that target hedgehog signaling. To further test hedgehog’s role, SAG (smoothened agonist) was added to the culture media, starting on day 1.5 and for the remainder of the differentiation until day 18 (Fig. 6a). By day 4 of differentiation, control cultures treated with SAG expressed reduced levels of GLI3R and increased levels of GLI1, which resembled KIF3A−/− and KIF3B−/− mutants qualitatively, although these changes were more dramatic in SAG-treated control cells than in KIF3A−/− or KIF3B−/− mutants (Fig. 6b–c). Unlike controls, SAG had no detectable effect on GLI3 or GLI1 levels in KIF3A−/− and KIF3B−/− mutants (6b-c). By day 18, the prolonged 16.5 day treatment with SAG strongly downregulated GLI3 and upregulated GLI1 in control cultures, whereas treatment with the smoothened antagonist cyclopamine modestly increased GLI3R over the same period (Fig. 6d and Supplementary Fig. 7a–e). Despite prolonged treatment, KIF3A−/− and KIF3B−/− mutants failed to respond to SAG or cyclopamine in either of these ways (Fig. 6d and Supplementary Fig. 7a–e).
Fig. 6. Smoothened agonist perturbs organoid differentiation.
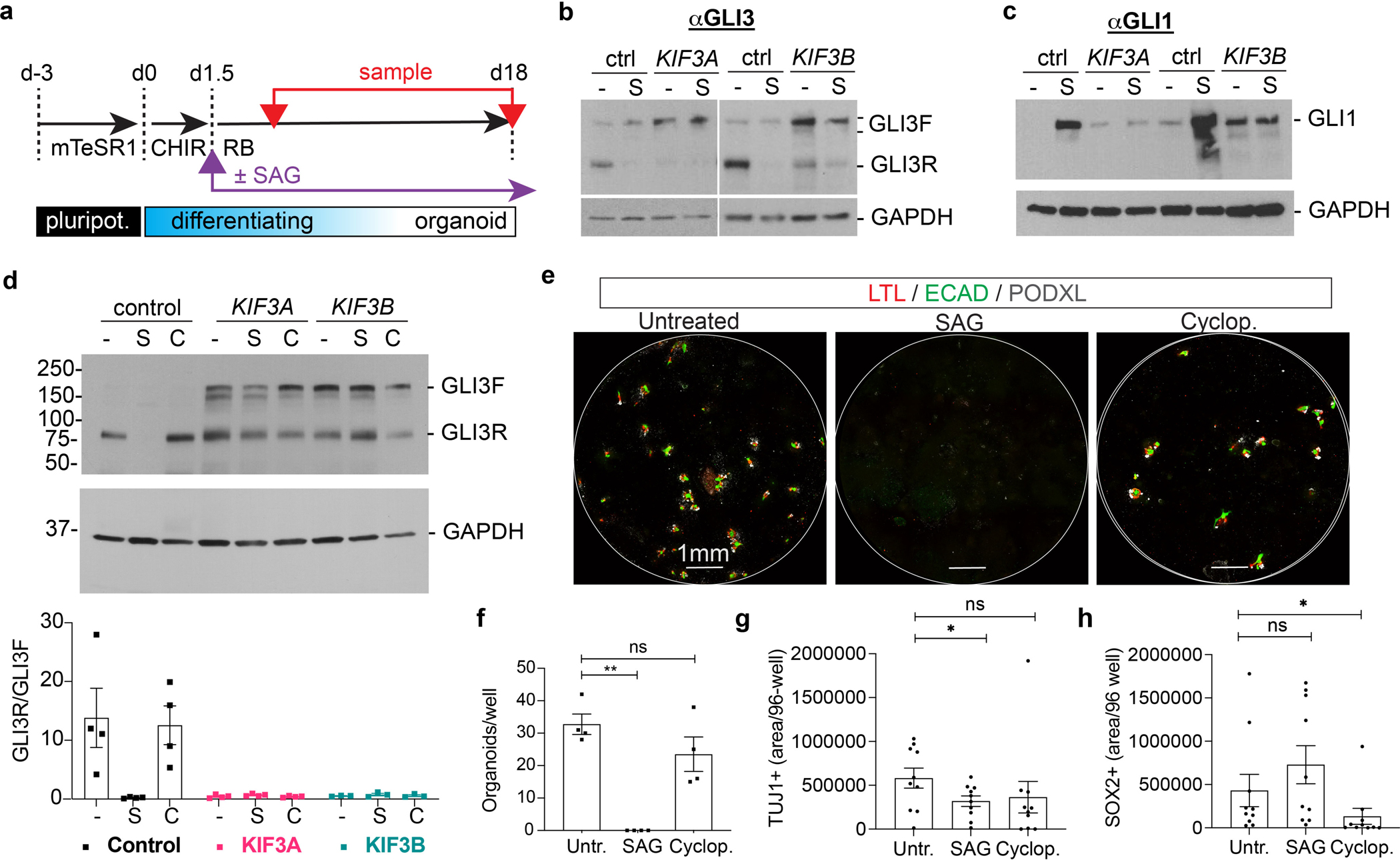
(a) Schematic of organoid differentiation time course, indicating initiation of SAG treatment (purple arrow) and sampling time points (red arrows). (b) Representative GLI3 and (c) GLI1 immunoblots of cultures on day 7 differentiation, under standard conditions (−) or supplemented with 1 μM SAG (S) starting on day 1.5 of differentiation. (d) Representative GLI3 immunoblot of control, KIF3A−/−, and KIF3B−/− organoid lysates on day 18 of differentiation. For each genotype, conditions were untreated (−), + 1 μM SAG (S), or + 10 μM cyclopamine (C) starting on day 1.5 of differentiation. Graph below the blot shows quantification of GLI3 immunoblot band intensities (mean ± s.e.m., n ≥ 3 independent biological replicates per condition from a total of 8 distinct cell lines). (e) Representative whole 96-well confocal immunofluorescence images of kidney organoid cultures derived from control hPSCs, showing nephron marker expression in these treatment conditions. (f) Quantification of nephron organoids per well in control hPSCs with hedgehog treatments (mean ± s.e.m., n = 4 independent biological replicates per condition from a total of 4 distinct cell lines; **, p < 0.01). (g) Quantification of TUJ1+ and (h) SOX2+ area in control 96 wells ± SAG (mean ± s.e.m., n = 10 independent biological replicates per condition from a total of 4 distinct cell lines. *, p < 0.05).
We examined the effects of hedgehog manipulation with these small molecules on organoid lineage differentiation in the control cultures responsive to these molecules at the molecular level. Kidney organoid differentiation was impaired by SAG, but not cyclopamine (Fig. 6e–f). Neuron differentiation (TUJ1+) was significantly suppressed by SAG, but also by cyclopamine, although cyclopamine’s effects did not reach statistical significance (Fig. 6g). To understand how SAG and cyclopamine could have similar effects, we examined SOX2+ neuronal progenitor cells. SAG increased SOX2+ neuronal progenitor cells, whereas cyclopamine had a strong inhibitory effect (Fig. 6g–h). These findings supported the conclusion that modulation of hedgehog signaling was required for proper differentiation in these organoid cultures.
Kinesin-2 mutants form nephron organoid cysts
Polycystic kidney disease (PKD) is a common ciliopathy phenotype. Previously, we have generated kidney organoids from hPSCs with PKD mutations, which form large cysts from tubular epithelial cells, recapitulating the phenotypic hallmark of PKD in vitro 31, 43. We tested whether kinesin-2 knockout organoids might also exhibit a PKD phenotype. Although differentiation was inefficient in kinesin-2 knockout lines, we succeeded in obtaining a small number of kidney organoids from mutant hPSCs, which were then transferred into suspension cultures, so that equal numbers of organoids from controls and mutants could be compared (Fig. 7a). Under these conditions, ~ 50 % of kinesin-2 knockout organoids formed large, translucent cysts after two weeks, whereas isogenic controls rarely formed cysts (Fig. 7b–c). Kinesin-2 knockout cysts arose spontaneously, without any forskolin addition. These cysts grew dramatically in size over several months, reaching diameters of > 1 cm, whereas isogenic controls were limited to mm diameters (Fig. 7d). In extended cultures, cysts could reach several centimeters in diameter and needed to be transferred into 200 mL flasks to accommodate their large size (Fig. 7e, Supplementary Fig. 8a, and Movie 1). Cysts stained positive for the proximal tubular marker LTL in patches and contained a thin layer of pH3+ cells surrounding a hollow lumen, indicating they comprised proliferating kidney tubular epithelial cells (Fig. 7f and Movie 2) 31. LTL+ cyst-lining epithelial cells co-expressed the distal tubular marker ECAD, similar to PKD1−/− organoid cysts (Fig. 7g and Supplementary Fig. 8b). PKD1−/− cysts, however, elaborated abundant primary cilia, which were absent in kinesin-2 knockout cysts (Fig. 7g). Conversely, no substantive difference was detected in the levels of the PKD1 protein product, polycystin-1 (PC1), or the PKD2 protein product, polycystin-2 (PC2), in whole cell lysates from kinesin-2 knockout hPSCs (Supplementary Fig. 8c–h). This is distinct from the PC1 and PC2 knockout lines we have described previously, in which PC1 expression is lost even in PKD2−/− cells 31.
Fig. 7. Kinesin-2 knockout organoids express the PKD cystic phenotype.
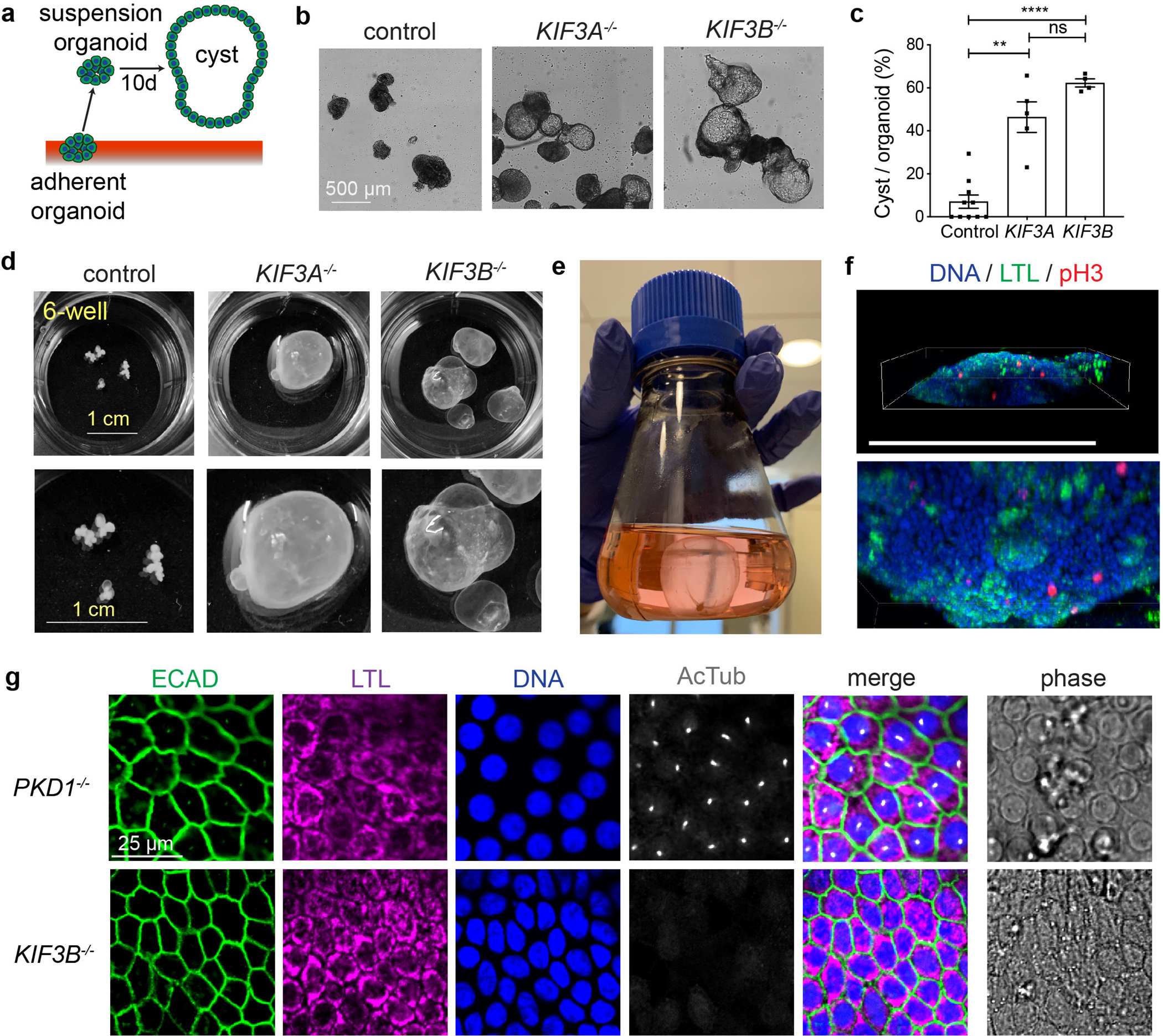
(a) Schematic of cystogenesis assay. (b) Representative phase-contrast images of control, KIF3A−/−, and KIF3B−/− organoids after 10 days in suspension culture. Scale bars, 500 μm. (c) Percentage of cystic organoids in suspension cultures (mean ± s.e.m., n ≥ 4 biological replicates per condition from a total of ten distinct cell lines. **, p < 0.01; ****, p < 0.0001). (d) Representative photographs of whole 6-wells containing organoids grown for several months in suspension culture, with zoom below. Scale bars, 1 cm. (e) 250 ml flask containing a large KIF3B−/− cystic organoid that outgrew the 6-well plates, as shown in Movie 1. (f) Scenes from Movie 2 showing 3-D confocal reconstruction of the lower edge of a cyst labeled with phospho-histone H3 (pH3) for mitotic cells and LTL for proximal tubular epithelia. Scale bar, 500 μm. (g) Confocal immunofluorescence of cilia (AcTub) and kidney epithelial cell markers in cyst-lining epithelial cells. Scale bar, 25 μm.
Movie 1.
KIF3B−/− cyst grown for several months in suspension culture and transferred into a 250 mL flask.
Movie 2.
Kinesin-2 knockout cysts contain proliferating kidney tubular epithelial cells. Volumetric reconstruction of the lower portion of a cyst derived from KIF3A−/− kidney organoids, showing proximal tubules (LTL, green) and dividing cells (pH3, red) with all nuclei labeled in blue (DAPI). A subset of dividing cells is observed just inside the lumen of the cyst (luminal mitoses).
Collectively, these experiments revealed two distinct phenotypes in kinesin-2 knockout kidney organoids: decreased nephrogenesis, and increased cystogenesis among the nephron structures that do form. This differs from the pure PKD phenotype, in which cysts form but nephrogenesis occurs normally 31. Nephrogenesis and cystogenesis may be regulated, respectively, by the hedgehog and PKD pathways, both of which likely require IFT for signal transduction and would be disrupted simultaneously in kinesin-2 knockout hPSCs 48–51. Reduced nephrogenesis would be expected to have a dominant effect in the context of PKD, which might explain the smaller size of kidneys in Kif3a-Pkd2 double-mutant mice compared to Pkd2 single mutants 19, 37.
Human cilia constitutively release extracellular vesicles containing disease-associated proteins
To further determine the mechanistic consequences of kinesin-2 loss, we developed a direct biochemical assay to measure the expression of ciliopathy-linked proteins in both whole cell lysates and extracellular vesicles (EV) (Fig. 8a). Cilia have been suggested to release EV, but these have been difficult to detect in immortalized mammalian cell lines without starvation, and even under optimized conditions do not appear to contain detectable quantities of many key membrane proteins 52, 53. Using our method, EV protein amounted to only ~ 6 % of total protein (Supplementary Fig. 9a). Per mg protein, however, the relatively small EV fraction was enriched ~ 5-fold for IFT88, an intracellular protein involved in ciliary transport, relative to whole cell lysates (Supplementary Fig. 9b–c).
Fig. 8. Kinesin-2 knockout hPSCs and organoids reveal secretion defects in disease-relevant signaling pathways.
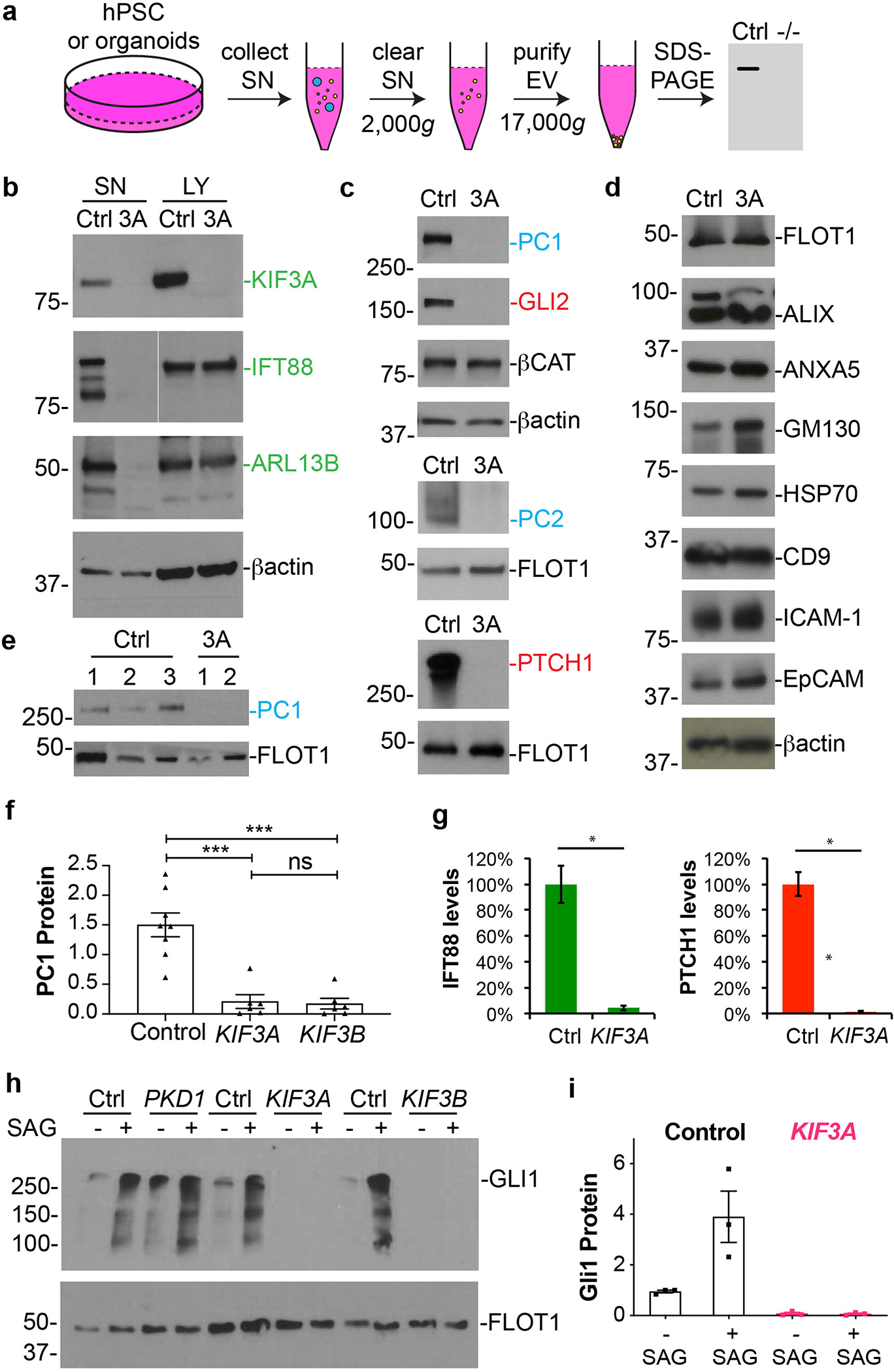
(a) Schematic of EV purification and analysis experiment. (b) Representative immunoblots of endogenous ciliary proteins (green type) in supernatants (SN) and lysates (LY) from control and KIF3A−/− hPSCs. β-actin is shown as a loading control. (c) Immunoblots of supernatants showing key components of the PKD (blue type) and hedgehog (red type) pathways in these supernatants. β-actin, β-catenin, and flotillin (FLOT1) are shown as loading controls. (d) A panel of non-ciliary EV components in supernatants. (e) PC1 expression in the supernatants of kidney organoids in suspension. Three control and two KIF3A−/− lines are shown. (f) Quantification of PC1 in EVs from control and mutant hPSCs (mean ± s.e.m., n ≥ 6 biological replicates per condition, total of 14 distinct cell lines. ***, p < 0.001). (g) Quantification of IFT88, PC1, and PTCH1 in EVs from control and mutant hPSCs (n = 4 biological replicates, p < 0.01). (h) Representative immunoblots of EV from control, PKD1−/−, and kinesin-2 knockout hPSCs, ± SAG treatment, with (i) quantification (mean ± s.e.m., n = 3 biological replicates per condition, total of 6 distinct cell lines).
In kinesin-2 knockout hPSCs, as expected, the kinesin-2 subunit targeted by genome editing (KIF3A or KIF3B) was absent in both lysates and supernatants as a control (Fig. 8b). In contrast, components of the IFT (ARL13B, IFT88), hedgehog (PTCH1, GLI2), and PKD/polycystin (PC1, PC2) pathways, as well as the non-targeted kinesin-2 subunit, were expressed at normal levels in whole cell lysates of kinesin-2 knockout hPSCs, but were dramatically reduced in EV of kinesin-2 knockout hPSCs (Fig. 8b–c and Supplementary Fig. 9d–f). The magnitude of PC1 and PC2 loss from EV in kinesin-2 knockout hPSCs was similar to that observed in PKD1−/− or PKD2−/− hPSCs (Supplementary Fig. 10a–b) 31. Compared to PC2, the proportion of cellular PC1 was highly enriched in EV, indicating a stronger propensity of PC1 to exit the cell (Supplementary Fig. 10a–b). This is significant, because PC1 is difficult to detect intracellularly, whereas PC2 contains an endoplasmic reticulum retention signal and is more readily detected inside cells 28.
The inability of kinesin-2 knockout hPSCs to release these proteins did not reflect a general inability to release EV, as non-IFT cargoes, including β-actin, HSP70, and flotillin, were efficiently released by kinesin-2 knockout hPSCs, and no differences in total protein level or banding pattern were detected (Fig. 8b–d and Supplementary Fig. 10c). Loss of extracellular PC1 was also observed in EV of kidney organoids derived from kinesin-2 knockout hPSCs (Fig. 8e). Quantification indicated an 8-fold reduction in PC1 levels, and a 20-fold reduction in ARL13B and PTCH1 levels, in kinesin-2 knockout supernatants, compared to controls (Fig. 8f–g, Supplementary Fig. 9d–f, and Supplementary Fig. 10d). In kinesin-2 knockouts, GLI1 was undetectable in EV, even after SAG stimulation, which contrasted with control or PKD1−/− hPSCs, in which GLI1 was detected in EV at steady-state and increased after SAG stimulation (Fig. 8h–i). These findings revealed that EV release in kinesin-2 knockout hPSCs is a robust, constitutive process under non-starvation conditions, enabling the sensitive detection of membrane proteins associated with ciliopathy phenotypes.
DISCUSSION
We have established a new general tool, a collection of KIF3A−/− and KIF3B−/− hPSCs, to elucidate the developmental and disease-related functions of cilia in human somatic cell types and tissues at different stages of maturation. Despite proposed roles of cilia in cell cycle regulation and signaling, both KIF3A−/− and KIF3B−/− hPSCs are stably pluripotent and self-renewing, providing an ideal starting point for differentiation experiments into diverse lineages without the complication of embryonic lethality 27, 28. Phenotypes of defective neurogenesis and nephrogenesis, as well as cystogenesis in the nephron structures that do form, manifest in complex tissues and organoids, but are not observed in undifferentiated hPSCs or simpler EBs in vitro, suggesting that cilia play a critical role in the higher-order organization of complex tissue architecture, for instance by coordinating the migration of scattered cell types. The fact that such tissue-scale phenotypes can be reconstituted in a relatively simple organoid culture in vitro demonstrates that they are driven by cell intrinsic processes, rather than complex interactions between organ systems or systemic effects that can only occur in a living organism.
KIF3A and KIF3B form the heterodimeric motor subunits of kinesin-2. KIF3B has been studied less extensively than KIF3A, shows greater genetic variation in humans, and can under certain circumstances be replaced in kinesin-2 by KIF3C 54, 55. In our experiments studying hPSC differentiation, KIF3A−/− and KIF3B−/− hPSCs have very similar phenotypes, indicating that the effects are specific to kinesin-2, and that both members of the complex are necessary for function. This is consistent with studies of Kif3a and Kif3b knockout mice, which show very similar phenotypes to one another, dying in mid-gestation with various malformations including open neural tubes 15, 16. Further study in diverse lineages may yet reveal subtle differences between the functions of KIF3A and KIF3B, and the full extent to which each of these is required for cilia formation. In addition to its effects on cilia, kinesin-2 can also play compounding non-ciliary roles, which we have not ruled out in our experiments 54. KIF3A−/− and KIF3B−/− hPSCs can be extensively passaged and expanded, establishing a long-term resource for investigating these topics.
The ability to vary differentiation and growth conditions in KIF3A−/− and KIF3B−/− hPSCs in vitro represents a major advantage of this system for elucidating functional mechanisms. For example, both KIF3A−/− and KIF3B−/− knockout cells differentiate normally in EBs, but are impaired in teratomas and organoids. Analysis of organoid cultures during the process of differentiation reveals a critical switch in hedgehog signaling from activation to repression, which depends upon kinesin-2 for proper modulation. In these cultures, differentiation is induced by transient exposure to a kinase inhibitor immediately upon withdrawal from pluripotent stem cell maintenance media 31. Our studies reveal that this step produces a transient increase in hedgehog pathway activation, while simultaneously triggering a more permanent switch to hedgehog repression. This switch and its consequences would not be discernable in somatic cells that do not undergo differentiation, nor has it been suggested in previous analyses of kidney organoids 56–58. There is genetic evidence that hedgehog repression promotes nephrogenesis in the mouse, although this role has been largely overshadowed by studies of Pallister Hall syndrome, in which GLI3R-mediated hedgehog repression is maladaptive for kidney development, and the role of cilia in this process remains poorly understood 19, 59.
The mechanism whereby GLI1 expression is increased transiently may relate to a form of negative feedback involving PTCH1, which inhibits Smoothened but is simultaneously a downstream target of hedgehog pathway activation 45–47. Increased PTCH1 in kinesin-2 knockout mutants on day 18 of differentiation thus supports sustained hedgehog activation in these mutants, and also provides a possible explanation for the reduction of GLI1 over time. While distinct from control cells, kinesin-2 knockout mutants also exhibited clear changes in GLI3 processing and in the expression levels of downstream hedgehog target genes (GLI1 and PTCH1) during the time course of differentiation. Thus, some capacity to execute hedgehog signaling responses is retained without a cilium. Kinesin-2 knockout mutants were, however, numbed in their ability to respond to SAG or cyclopamine. Based on these findings, distinct activators or inhibitors of hedgehog signaling may have different dependencies on cilia.
Long-term supplementation of our organoid differentiation protocol with hedgehog agonist (SAG) or antagonist (cyclopamine) was detrimental to neuronal differentiation. In other neuronal differentiation protocols, hedgehog agonists have been used to promote neuronal differentiation 60, 61. We did observe an increase in neuronal progenitor cells in cultures treated with SAG, which appeared to come at the expense of differentiation into mature TUJ1+ neurons. In contrast to SAG, cyclopamine had a generally detrimental effect on the differentiation of both neuronal progenitor cells and neurons. The detrimental effect of SAG on neuronal differentiation in our protocol, compared to other protocols, may be due to its prolonged treatment in these experiments, or its use in addition to high doses of a kinase inhibitor that is already sufficient to transiently activate hedgehog signaling. A balance of hedgehog repression and activation may be necessary for optimal neuronal differentiation, as well as for renal cell fates. We note that in addition to hedgehog, our RNA-seq analysis suggests that other signaling pathways such as TGF-β may be dysregulated in hPSCs devoid of cilia, which could potentially affect differentiation or disease phenotypes in these cultures 43, 62. Although we have focused here on hedgehog signaling, other such pathways remain an interesting area for further investigation.
Beyond effects on differentiation, generation of KIF3A−/− and KIF3B−/− kidney organoids enables the deconvolution of the PKD phenotype from the nephrogenesis defect, with implications for understanding the paradoxical effects of IFT knockout in the context of PKD. In contrast to polycystin knockouts, both KIF3A−/− and KIF3B−/− hPSCs express normal levels of PC1 and PC2 in whole cell lysates, but lack cilia. Using KIF3A−/− and KIF3B−/− hPSCs, we identify a robust requirement for kinesin-2 in the release of ciliopathy-related signaling molecules from cells into the surrounding microenvironment, including PC1. This is consistent with detailed cell biology analyses demonstrating release of EV from ciliary tips containing specific cargoes, which are distinct from EV released from non-ciliary sites on the plasma membrane 52, 53. As kinesin-2 knockout organoids show defects in both GLI3 processing and EV release, we cannot yet clearly distinguish which of these mechanisms is causative for cystogenesis. Deficiencies in hedgehog signaling are not evident in polycystin knockouts, suggesting that the PKD pathway is separable and independent from hedgehog, but shares the requirement for cilia as a signaling organizing center. EV release may serve to dispatch extracellular signals packaged in EV, similar to specialized neuronal cilia of nematode worms 63, or alternatively to dispose of ciliary material in order to trim ciliary length, as suggested in studies of immortalized mouse kidney cells 52, 53.
Collectively, our data suggest a model wherein human cilia function as both a protected domain for organizing signaling functions, and constitutive secretory organelles that release EV packets containing specific combinations of signaling molecules (Fig. 9a). In mutants, loss of hedgehog responsiveness in stem cells inhibits terminal differentiation, while loss of PKD activity in differentiated cells leads to cystogenesis (Fig. 9b). Ciliary function is therefore cell type- and context-dependent, and may be increased in mammalian developmental lineages poised for embryonic differentiation.
Fig. 9. Ciliary function directs morphogenesis in complex tissues derived from hPSCs.
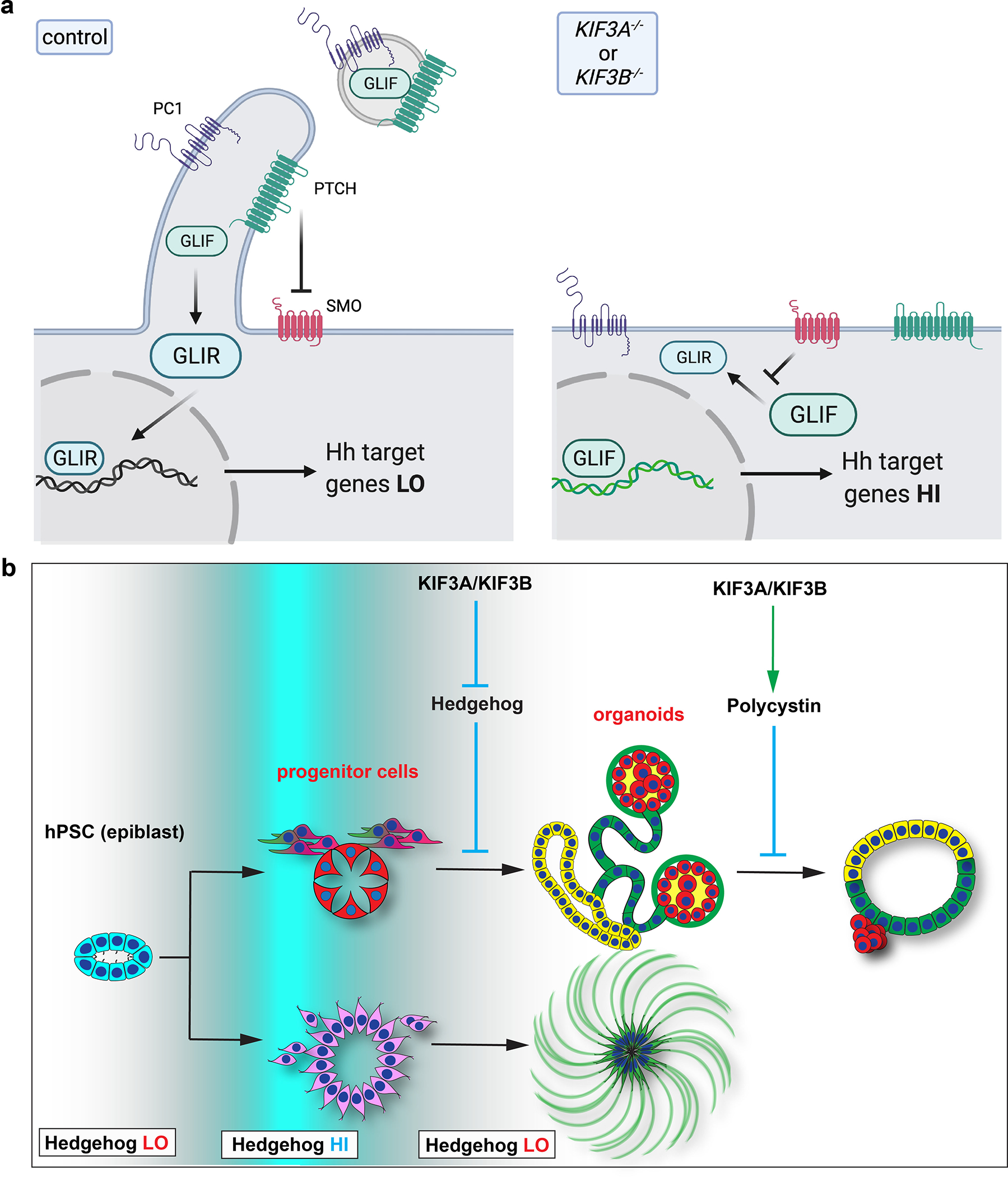
(a) Schematic of kinesin-2’s effects at the cell surface. Left: In normal cells, the cilium generated by kinesin-2 is inhabited by PTCH1 which establishes a protected zone to promote downstream GLI protein processing (relative quantity of GLIF versus GLIR), ultimately resulting in proper modulation of the hedgehog signal and levels of downstream gene expression. Hedgehog and PKD components exit the cell through ciliary EV to further modulate signaling. Right: In kinesin-2 knockout cells, absence of the cilium renders PTCH1 unable to inhibit SMO, resulting in blockade of GLI protein processing and increased activation of the hedgehog signaling pathway. Hedgehog and PKD components are unable to exit the cell. (b) Illustration depicting the role of cilia in tissues and organoids derived from hPSCs. Kinesin-2 activity is responsible for switching the hedgehog signaling between ‘HI’ and ‘LO’ states (white to blue gradient) during differentiation of the pluripotent epiblast into complex tissues (left to right). In differentiated tubular epithelia, kinesin-2 regulates PKD pathway components to restrict expansion into cysts.
In conclusion, disruption of IFT in hPSCs establishes a human genetic model of cilia function, providing critical insight into this enigmatic organelle. Using this new tool, it is now possible to perform phenotypic screens and mechanistic studies to reveal the functions of human cilia and test interventional strategies for ciliopathy syndromes or cilia-dependent cancers. This is the first time that we have been able to generate human tissues and organoids lacking cilia, and compare them to isogenic structures with cilia. A major strength of this system is its ability to reveal and de-convolve phenotypes under modular differentiation conditions, without the complication of embryonic lethality. Biochemical analysis in this system further indicates a critical role for cilia in organoid differentiation, and in the constitutive extracellular release of key signaling molecules. As these studies are performed in human stem cells at the earliest stages of embryonic development, understanding and controlling cilia may provide a path to guiding and improving regenerative therapies.
METHODS
Generation of gene-edited hPSCs
Plasmids encoding guide RNA targeting KIF3A (5’-CATATGGACAAACCGGAAC-3’) or KIF3B (5’-TTCGCTGTCGGCCCATGAA-3’ or 5’-TACACCATGGAAGGAATCCG-3’) and GFP-tagged Cas9 were co-transfected transiently into a subclone of WA09 (H9) ESCs (WiCell) or WTC11 iPSC (Coriell #25256) using Lipofectamine Stem transfection reagent (Thermo Fisher). GFP+ cells were isolated using a FACS Aria cell sorter and plated at low density in 6-well dishes coated with 1% Geltrex (Thermo Fisher). Clones were manually picked with a 10 μL pipet and transferred to a 96-well plate. DNA was extracted from the resulting clonal cell lines using QuickExtract DNA extraction solution (Biocentre), and the targeted region was PCR amplified, purified, and sequenced to detect indel mutations. At least three mutant clones and three isogenic, negative control clones (processed identically but found to be unmodified) were selected for further expansion and analysis. The primers used for amplification and sequencing analysis were the following: 5’-GAATCCAGGGGAGAAATTACATCACAG-3’ and 5’-AAAGCGGAGGGTGATACAAGGTAAAAG-3’ for KIF3A; 5’-GGCTAGCCAACAACACTGGT-3’ and 5’-AACAAGTGGTCGGAACGTCT-3’ for KIF3B gRNA 1; or 5’-AAAGGGACGGCCCATGAAAT-3’ and 5’-CGTTCATGTTGGTAGCACCG-3’ for KIF3B gRNA 2. Amplified fragments were used to clone individual alleles into the pCR™ 4 -TOPO® plasmid using a TOPO TA cloning kit for sequencing (Thermo Fisher). Plasmids were sequenced using M13 Forward primer (5’-GTAAAACGACGGCCAG-3’) and chromatograms were aligned to a sequence from control, non-edited cells to identify indel mutations.
Pluripotent stem cell culture
The pluripotent stem cell lines were maintained in feeder-free culture conditions in 5 % carbon dioxide with daily medium changes of mTesR1 (STEMCELL Technologies). For cilia detection, dissociated cells were plated into 8-well chamber slides (Lab-Tek) coated with 3% Geltrex and grown to confluency for 6 days with daily mTesR1 medium changes. To promote ciliogenesis, cells were treated with 100 μM cAMP for 48 hours and the medium was changed to DMEM/F12 for the last 24 hours of culture. For cavitated spheroid formation, hPSC were dissociated into single cells and plated at low density in 96-well plates coated with 1% Geltrex. The next morning, mTeSR1 containing 1.5% Geltrex was added to the wells. The cells were fixed with 4% paraformaldehyde 48 hours later. For monitoring cell viability, cells were plated in 96-wells and cultured 24 hours in 90 μL mTesR1 supplemented with 10 μL of alamarBlue cell viability reagent (Thermo Fisher). Fluorescence was read using a fluorescence excitation wavelength of 560 nm and an emission of 590 nm in a Perkin Elmer Envision Xcite instrument. Media was replaced with fresh mTesR1 supplemented with 10 μL of alamarBlue daily after the fluorescence measurements for the duration of the experiment.
Protein gels and immunoblotting
KIF3A−/−, KIF3B−/− and isogenic control hPSCs were lysed with RIPA buffer containing protease and phosphatase inhibitors (Roche). Protein concentration was determined using a Pierce BCA protein assay kit. 50 μg of total protein was separated in a 4–20% acrylamide gel (Bio-Rad) and analyzed with silver stain (Bio-Rad) or transferred onto a PDVF membrane using standard procedures. 5 % milk was used as a blocking agent prior to and during immunoblotting. Blots were probed with antibodies raised against PC1 (Santa Cruz sc-130554), PC2 (Santa Cruz sc-25749), KIF3A (Abcam ab11259), KIF3B (Cell Signaling 13817), β-Actin (Cell Signaling 4970), FLOT1 (Cell Signaling 18634), IFT88 (Proteintech 13967–1-AP), ARL13B (Proteintech 17711–1-AP), GLI1 (Cell Signaling 3538), GLI2 (Abcam ab26056), PTCH1 (Cell Signaling 8358), ALIX (Cell Signaling 2171), ANXA5 (Cell Signaling 8555), GM130 (Cell Signaling 12480), HPS70 (Cell Signaling 4876), CD9 (Cell Signaling 13174), ICAM-1 (Cell Signaling 4915), and EpCAM (Cell Signaling 2626).
Kidney organoid differentiation
Kidney organoids were generated as previously described 31. Briefly, dissociated hPSCs were plated into 96 or 24 well plates coated with 1% Geltrex (Thermo Fisher) in mTeSR1 (STEMCELL Technologies) supplemented with 10 μM Rho-kinase inhibitor Y27632 (Tocris Bioscience). The next morning, mTeSR1 containing 1.5% Geltrex was added to the wells, and the media was changed again to mTeSR1 24 hours later. 72 hours after plating, cells were treated with 12 μM CHIR99021 (Stemgent) in RPMI medium containing 1X Glutamax (Thermo Fisher) with 2 ng/ml recombinant BMP-4 (Peprotech) to induce kidney differentiation. 36 hours after treatment, the medium was changed to RPMI supplemented with B27 (RB medium, Thermo Fisher). RB medium changes were performed every three days. When indicated, organoid cultures were supplemented with 1 μM SAG or 10 μM Cyclopamine (both from Cayman Chemical) starting with the first addition of RB on day 1.5 (36 hours after CHIR99021 treatment on day 0) and maintained thereafter in RB media containing these compounds at all subsequent time points.
For suspension cultures, organoids in adherent cultures on day 21 of differentiation were selected for microdissection based on their three-dimensional, translucent, tubular appearance as detected by phase contrast microscopy. The organoids were gently detached from the plate using a 22 gauge needle and transferred into low adhesion plates (Corning) using a plastic transfer pipet. Organoids microdissected from cultures of mutants and isogenic controls were confirmed to be of similar size and morphology at the time of transfer. Media was replaced weekly, and cultures were maintained for one year. The 1 year old organoids are visible to the naked eyes and were photographed using a handheld digital camera. The images were opened with FIJI for quantification. The freehand selection tool was used to trace the organoid and measure the area of the organoid. For immunoblot analysis, microdissected organoids were transferred into an Eppendorf tube, allowed to settle by gravity, washed once with a full tube volume of PBS, allowed to settle, and subsequently lysed in the tube.
Generation of embryoid bodies (EBs)
To generate EBs, 400,000 dissociated hPSCs were transferred into one well of a low-adhesion 6-well plate (Corning) in mTeSR1 plus 10 μM Rho-kinase inhibitor Y27632 (Tocris Bioscience). The following day, the cells were allowed to settle and the media was changed to mTeSR1 without Y27632. Two days later, the media was exchanged to ESC media (20% Knock Out Serum Replacement, 1X nonessential amino acids, 1X Glutamax, 1X penicillin-streptomycin, and 0.1 mM β-mercaptoethanol, in DMEM/F12, all from Thermo Fisher) without FGF. Media was changed every four days for the following eight weeks, after which the EBs were transferred onto gelatin-coated 24-well plates in 10 % fetal bovine serum in DMEM and grown as outgrowths for two additional weeks. Floating EBs or EB outgrowths were subsequently fixed and processed for immunofluorescence analysis.
Teratoma formation
Animal work was performed in compliance with the strict ethical requirements and regulations of the UW IACUC under a pre-approved animal protocol. Dissociated hPSCs (400,000/well) were plated in three wells of a 6-well plate and grown to confluence in mTeSR1. Cells were dissociated, pelleted, resuspended in 500 μl of an ice-cold 1:1 mixture of DMEM/F12 (Fisher) and Matrigel (Corning). The cells were immediately injected beneath the neck scruff of immunodeficient, NOD-SCID mice (NOD.CB17-Prkdcscid/J, Jackson Labs) using a 22-gauge syringe needle. Littermate animals of equally mixed genders at 8 weeks of age were used for all experiments. Growths were harvested 15 weeks after injection, photographed, fixed in methacarn (60 % methanol, 30 % chloroform, 10 % acetic acid, all from Sigma), embedded in paraffin, sectioned, and stained with hematoxylin and eosin for histological analysis.
Immunofluorescence analysis
Cells and organoid cultures were fixed with 4% paraformaldehyde (Electron Microscopy Sciences) for 15 minutes, washed 3 times with Phosphate buffered saline (PBS, Thermo Fisher), blocked for 1 hour with blocking buffer (5% normal donkey serum and 0.3% Triton X-100 in PBS), and incubated overnight with primary antibodies including mouse acetylated α-tubulin (Sigma T7451), rabbit acetylated α-tubulin (Cell Signaling, D20G3), α-fetoprotein (AFP) (R&D MAB1368), α-smooth muscle actin (SMA) (R&D MAB1420), ZO-1 (Invitrogen, 339100), E-Cadherin (Abcam, ab11512), Nephrin (R&D, AF4269), p-Histone H3 (Santa Cruz sc-8656-R), OCT4 (Abcam ab19857), brachyury (BRY) (Santa Cruz sc-17745), SOX2 (Abcam ab97959), and β-Tubulin III (Tuj1) (Sigma-Aldrich T2200–200UL). After washing with PBS, secondary donkey antibodies conjugated with Alexa-Fluor 488, 555, or 647 (1:500, Thermo Fisher) were applied, or Fluorescein Lotus Lectin (Vector Labs, FL-1321), with DAPI to stain the nuclei. Stained cells were imaged in PBS using a Nikon A1R confocal microscope or Eclipse TE wide-field microscope.
EV Purification
1.5 × 106 control or kinesin-2 knockout hPSCs were plated onto 10 cm diameter plates and grown to confluence in mTeSR1 (10 ml/plate). Alternatively, organoids were cultured in RB media (4 ml/well) for seven days. The media was collected without cells and centrifuged at 2,000 g in a swinging bucket tabletop centrifuge in 50 ml conical tubes (Falcon) for 10 minutes to pellet any residual cells and cellular debris. The supernatant was then transferred to thick-walled polypropylene round-bottom tubes (Beckman #355642) and EV were pelleted at 17,000 g at 4 degrees Celsius for 30 minutes in a RC-6 Plus centrifuge using an HB-6 swinging bucket rotor (Sorvall). The supernatant was aspirated and the pellet was resuspended in 100 μl of RIPA buffer containing protease and phosphatase inhibitors (Roche).
Quantification and Statistics
Differentiation, processing, and analysis of mutant and control hPSCs was performed side-by-side. Multiple clones of the KIF3A−/− or KIF3B−/− genotypes were utilized in each experiment to control for any variability between clonal cell lines. Supplementary Table 1 details the use of multiple clones for each experiment. Fluorescence intensity analysis of BRY and OCT4 was performed on images processed and imaged identically, individual cells were identified automatically using Cell Profiler 2.0 and the average intensity per cell was plotted in bbp. Line scan analyses were conducted utilizing the Intensity Profile function in NIS Elements. A single line scan of equal length was performed on each structure, and subsequently these were averaged to generate the graphs, plotting pixel intensity values for each channel on the y-axis against the points along the line on the x-axis. Immunoblot band intensity levels were quantified using the ImageJ magic wand tool, normalized to the loading control, and pooled from multiple experiments to determine average and s.e.m. values. A two-tailed t-test for two samples with unequal variance was applied to determine significance. In cases where a treatment condition was applied, a paired t-test was performed. Statistical analyses were performed in GraphPad Prism 9 software.
Differential Gene Expression and Gene-Set Enrichment Analysis
To minimize batch effects, all cells were prepared in a single batch. For each condition, ~1000 cells were lysed directly in SMARTseq lysis buffer. Library was prepared from small using a combination of Clontech SMARTseq chemistry and Illumina Nextera XT kits. RNA was sequenced on an Illumina HiSeq2500 processor in one direction (single read), with a target of 5M reads per sample for 58 nucleotides (Genomics Core, Benaroya Research Institute, Seattle, WA). Differentially expression analysis was performed using the DESeq2 R package64. For all analyses, p-values were corrected for multiple hypothesis testing using the Benjamini-Hochberg procedure. For the joint analysis of KIF3A−/−, KIF3B−/− and control RNA-seq samples, differential expression due to loss of KIF3 activity was assessed using a model of “expression ~ KIF3 status”. To leverage our higher statistical power to identify signatures associated with KIF3B loss in KIF3B−/− hPSCs, we performed differential gene expression analysis on KIF3B−/− and control RNA-seq samples using a model of “expression ~ KIF3B−/− status”. For both analyses, hierarchical clustering was performed to group differentially expressed genes with similar dynamics across samples using the pheatmap R package specifying ward.D2 as the clustering method. Gene set enrichment analysis was performed on clusters of differentially expressed genes as a function of KIF3B−/− loss using the hypergeometric test implemented in the piano R package.
Code Availability
The custom algorithm for Cell Profiler analysis of BRY and OCT4 is available upon request from the corresponding author. R and DeSeq2 code is available upon request from the corresonding author.
Supplementary Material
Supplementary Figure 1. Generation of KIF3A−/− and KIF3B−/− hPSC mutants using CRISPR gene editing. (a) Table of gene edited hPSC carrying loss of function mutations in KIF3A or KIF3B generated using CRISPR gene editing. The genetic background, sgRNA used for targeting, and resulting mutation is indicated for each cell line. (b) Chromatograms of homozygote mutants showing the edited KIF3A or (c) KIF3B regions. The sgRNA sequence used for targeting is labelled in yellow in the respective control chromatograms. (d) Sanger sequencing chromatogram of compound heterozygote KIF3A or (e) KIF3B mutants and the separated allele sequences after TOPO cloning. Deletions and insertions are indicated in the chromatograms.
Supplementary Figure 2. CRISPR gene editing results in loss of KIF3A or KIF3B protein. (a) KIF3A immunoblot showing three distinct control and KIF3A−/− H9 hPSC lines at passages 4 and 17. Cropped areas used in Figure 1 are shown in bracket box. β-actin was blotted as a loading control. (b) KIF3A immunoblot for one control (C1) and two distinct KIF3A−/− WTC11 hPSC (M1 and M2). GAPDH was blotted as a loading control. (c) KIF3B and β-actin immunoblot for one H9 control (C1) and two KIF3B−/− mutant (M1, M2) hPSC. (d) KIF3B and β-actin immunoblot for three distinct control and four KIF3B−/− WTC11 mutant hPSC. Cropped areas used in Figure 1 are shown in bracket box. kDa sizes of the protein standards are indicated. (e) Band intensity quantification from immunoblots of the indicated genotype (mean ± s.e.m., n ≥ 4 independent biological replicates).
Supplementary Figure 3. Kinesin-2 is dispensable for hPSC morphology and epiblast spheroid formation. (a) Confocal immunofluorescence images of acetylated α-tubulin (AcTub) and DNA in representative fields of undifferentiated control and KIF3A−/− hPSCs. Orthogonal (top) and volume (bottom) views are shown. (b) Phase contrast images of representative control and KIF3A−/− epiblast spheroids. (c) Quantification of lumen area as percentage of the spheroid area. (d-e) Confocal sections showing immunofluorescence for pluripotency, polarity, and ciliary markers in spheroids. A midbody is seen in a KIF3A−/− spheroid (arrow), but not cilia. Scale bars, 25 μm.
Supplementary Figure 4. Kinesin-2 knockout hPSCs establish a renewable source of diverse human cell types lacking cilia. (a) Neuroepithelial (AcTub), endodermal (AFP), and mesodermal (SMA) lineages in EBs. Zoom shows magnification of dashed boxed area with AcTub and DNA intensities increased for clarity. Scale bar, 50 μm. (b) Images and (c) quantification of pigmented EBs. Scale bar, 500 μm. Knockout (KO) represent pooled data from both KIF3A−/− and KIF3B−/− EBs (mean ± s.e.m., n ≥ 9 independent biological replicates per condition from a total of 8 distinct cell lines). (d) Representative images and (e) quantification of OCT4 and BRY immunofluorescence intensities in hPSCs after treatment with increasing doses of CHIR99021 in mTeSR1 for 48 hours. Each dot represents a single cell. Data are pooled from three separate experiments. bpp, bits per pixel (raw intensity). Scale bars, 50 μm.
Supplementary Figure 5. Kinesin-2 knockout tumors exhibit differentiation defects. (a) Representative photographs of whole, unfixed growths retrieved from immunodeficient animals at the same time point. Growths are sliced through their centers to reveal the internal surface. (b) A second set of tumors, photographed intact after retrieval from the animals. (c-e) Quantification of tissue subtypes within teratomas as a fraction of total area (mean ± s.e.m., n ≥ 5 independent biological replicates per condition from a total of 14 distinct cell lines). (f) Ratio of area occupied by SOX2+ cells in TUJ1+ patches of teratoma sections (mean ± s.e.m., n ≥ 4 independent biological replicates per condition from a total of 6 distinct cell lines; *, p < 0.05).
Supplementary Figure 6. Hedgehog switching is defective in kinesin-2 knockout organoid cultures. (a) Quantification of SOX2+ area per 96-well of kidney organoid cultures (mean ± s.e.m., n ≥ 3 independent biological replicates per condition from a total of 11 distinct cell lines. **, p < 0.01). (b) Wide-field immunofluorescence images of kidney organoid differentiations in a representative 96-well plate. The three left wells contain control cell lines and the three right wells contain KIF3A−/− cell lines. Zoom of boxed regions is shown below each of the wells. Each kidney organoid contains distal tubular (ECAD), proximal tubular (LTL), and podocyte (NPHS1) epithelial cells. Arrow indicates a cluster of ECAD+ cells without proximal tubule and podocyte segments. (c) Heatmap of the expression level of genes differentially expressed as a combined function of KIF3A or KIF3B loss in hPSCs (FDR < 0.2). Gene names and labels at right refer to genes associated with enriched gene ontology processes as a function of KIF3B−/− loss in hPSCs in Fig. 5a. (d) Representative GLI3 immunoblot of control, KIF3A−/−, and KIF3B−/− undifferentiated hPSCs, with (e) band intensity quantification (mean ± s.e.m., n ≥ 3 independent biological replicates per condition from a total of 11 distinct cell lines). (f) Band intensity quantification of the blot shown in Fig. 5e, comparing cultures on day 0 to day 18. (g) Immunoblot of GLI3 in kidney organoid cultures on days 7, 11, and 18 of differentiation. 3A and 3B indicate KIF3A−/− and KIF3B−/− mutants, respectively. (h) Schematic of organoid microdissection (left), with immunoblot of GLI3 on day 18 of differentiation in lysates of whole wells (w), microdissected organoids (o), or remnant stroma (s). (i) GLI1 immunoblot of the samples shown in Fig. 5e, comparing cultures on day 0 to day 18.
Supplementary Figure 7. (a) Representative GLI3 immunoblot in organoid cultures on day 18. Blot is from independent biological replicates, compared to the blot shown in Fig. 6d. (b) Individual measurements of GLI3F and (c) GLI3R bands for the sample set quantified in ratiometric form in Fig. 6d. (d) Representative GLI1 blot of these conditions, with (e) quantification of band intensities (mean ± s.e.m., n ≥ 3 independent biological replicates per condition from a total of 6 distinct cell lines).
Supplementary Figure 8. Comparison between kinesin-2 and polycystin knockout mutants. (a) Quantification of organoid area in 12 month old suspension cultures. (b) Representative confocal optical sections showing tubular epithelial markers in cyst-lining epithelia of PKD1 versus KIF3B mutant organoids. (c-e) Representative immunoblot showing PC1 and PC2 levels in KIF3A−/− hPSCs, compared to isogenic controls. β-actin is shown as a loading control. (f) Quantification of band intensities from whole cell lysates of kinesin-2 knockout or control hPSCs (n ≥ 5 independent biological replicates per condition). (g) Immunoblot showing PC2, KIF3A, and GAPDH loading control in kinesin-2 knockout hPSCs, isogenic controls, or PKD2 knockout. (h) Quantification of PC2 band intensities from immunoblots of kinesin-2 knockout hPSCs and isogenic controls (n ≥ 3 independent biological replicates per condition).
Supplementary Figure 9. hPSCs release ciliary proteins in EV via kinesin-2. (a) Quantification of protein fraction in EV (SN), compared to whole cell lysates (LY), as a percentage of total protein (SN + LY), using BCA assay (n = four distinct hPSC lines, plotted separately). (b) Representative immunoblot showing IFT88 levels in LY vs. SN. Total protein load is indicated in μg above each lane. (c) Quantification of IFT88 band intensities per lane at increasing total protein loads in SN and LY (n = 4 independent biological replicates from a total of 4 distinct cell lines). (d-e) Representative immunoblots and (f) band intensity quantifications of key ciliary proteins in EV, in KIF3B−/− hPSCs compared to isogenic controls (mean ± s.e.m., n ≥ 2 independent biological replicates from a total of 4 distinct cell lines).
Supplementary Figure 10. Kinesin-2 knockout hPSCs exhibit secretion defects with normal cytoplasmic expression. (a) Representative immunoblots of endogenous PC1 or (b) PC2 proteins in supernatants (SN) and lysates (LY) from control, PKD1−/−, PKD2−/−, and KIF3A−/− hPSCs, with FLOT1 loading control. PC2tetra refers to the tetrameric form of PC2. (c) Silver stain of whole supernatants from two control and three KIF3A−/− hPSC lines. No consistent differences in banding pattern are observed between the two groups. (d) Uncropped immunoblots for PTCH1 and IFT88 in EV, from Fig. 8b and 8c, respectively.
Supplementary Table 1. Lines and n used for statistical analysis.
ACKNOWLEDGMENTS
The authors thank Stuart Shankland, Jonathan Himmelfarb, Charles Murry, David Beier, Dan Doherty, John Scott, and Mark Bothwell (UW) for helpful discussions. Studies were supported by Institute for Stem Cell and Regenerative Medicine Innovation Pilot Award; Lara Nowak-Macklin Research Fund; NIH Awards R01DK117914 (BSF), UG3TR000504 (JH), UG3TR002158 (JH), and U01DK127553 (BSF); the Northwest Kidney Centers, and start-up funds from the University of Washington. We acknowledge the work of many authors whose publications we were unable to cite due to space limitations.
Footnotes
COMPETING FINANCIAL INTERESTS
None of the authors have any competing financial interests to declare. B.S.F. is an inventor on patents for kidney organoid generation and disease modeling.
DATA AVAILABILITY STATEMENT
All datasets are available from the corresponding author upon reasonable request. Complete RNA-seq datasets will be deposited in NCBI as a GEO dataset at the time of publication.
REFERENCES
- 1.Sedar AW & Porter KR The fine structure of cortical components of Paramecium multimicronucleatum. J Biophys Biochem Cytol 1, 583–604 (1955). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sorokin SP Reconstructions of centriole formation and ciliogenesis in mammalian lungs. J Cell Sci 3, 207–230 (1968). [DOI] [PubMed] [Google Scholar]
- 3.McGrath J, Somlo S, Makova S, Tian X & Brueckner M Two populations of node monocilia initiate left-right asymmetry in the mouse. Cell 114, 61–73 (2003). [DOI] [PubMed] [Google Scholar]
- 4.Kozminski KG, Johnson KA, Forscher P & Rosenbaum JL A motility in the eukaryotic flagellum unrelated to flagellar beating. Proc Natl Acad Sci U S A 90, 5519–5523 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Orozco JT et al. Movement of motor and cargo along cilia. Nature 398, 674 (1999). [DOI] [PubMed] [Google Scholar]
- 6.He M, Agbu S & Anderson KV Microtubule Motors Drive Hedgehog Signaling in Primary Cilia. Trends Cell Biol 27, 110–125 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Otto EA et al. Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination. Nat Genet 34, 413–420 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nachury MV et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell 129, 1201–1213 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Ansley SJ et al. Basal body dysfunction is a likely cause of pleiotropic Bardet-Biedl syndrome. Nature 425, 628–633 (2003). [DOI] [PubMed] [Google Scholar]
- 10.Kim S et al. Nde1-mediated inhibition of ciliogenesis affects cell cycle re-entry. Nat Cell Biol 13, 351–360 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rash JE, Shay JW & Biesele JJ Cilia in cardiac differentiation. Journal of ultrastructure research 29, 470–484 (1969). [DOI] [PubMed] [Google Scholar]
- 12.Schraml P et al. Sporadic clear cell renal cell carcinoma but not the papillary type is characterized by severely reduced frequency of primary cilia. Modern pathology: an official journal of the United States and Canadian Academy of Pathology, Inc 22, 31–36 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Wong SY et al. Primary cilia can both mediate and suppress Hedgehog pathway-dependent tumorigenesis. Nat Med 15, 1055–1061 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Han YG et al. Dual and opposing roles of primary cilia in medulloblastoma development. Nat Med 15, 1062–1065 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marszalek JR, Ruiz-Lozano P, Roberts E, Chien KR & Goldstein LS Situs inversus and embryonic ciliary morphogenesis defects in mouse mutants lacking the KIF3A subunit of kinesin-II. Proc Natl Acad Sci U S A 96, 5043–5048 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nonaka S et al. Randomization of left-right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell 95, 829–837 (1998). [DOI] [PubMed] [Google Scholar]
- 17.Murcia NS et al. The Oak Ridge Polycystic Kidney (orpk) disease gene is required for left-right axis determination. Development 127, 2347–2355 (2000). [DOI] [PubMed] [Google Scholar]
- 18.Lin F et al. Kidney-specific inactivation of the KIF3A subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. Proc Natl Acad Sci U S A 100, 5286–5291 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chi L et al. Kif3a controls murine nephron number via GLI3 repressor, cell survival, and gene expression in a lineage-specific manner. PLoS One 8, e65448 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marszalek JR et al. Genetic evidence for selective transport of opsin and arrestin by kinesin-II in mammalian photoreceptors. Cell 102, 175–187 (2000). [DOI] [PubMed] [Google Scholar]
- 21.Moyer JH et al. Candidate gene associated with a mutation causing recessive polycystic kidney disease in mice. Science 264, 1329–1333 (1994). [DOI] [PubMed] [Google Scholar]
- 22.Pazour GJ et al. The intraflagellar transport protein, IFT88, is essential for vertebrate photoreceptor assembly and maintenance. J Cell Biol 157, 103–113 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tammachote R et al. Ciliary and centrosomal defects associated with mutation and depletion of the Meckel syndrome genes MKS1 and MKS3. Hum Mol Genet 18, 3311–3323 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frank V et al. Mutations in NEK8 link multiple organ dysplasia with altered Hippo signalling and increased c-MYC expression. Hum Mol Genet 22, 2177–2185 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Thomson JA et al. Embryonic stem cell lines derived from human blastocysts. Science 282, 1145–1147 (1998). [DOI] [PubMed] [Google Scholar]
- 26.Takahashi K et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Kiprilov EN et al. Human embryonic stem cells in culture possess primary cilia with hedgehog signaling machinery. J Cell Biol 180, 897–904 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Freedman BS et al. Reduced ciliary polycystin-2 in induced pluripotent stem cells from polycystic kidney disease patients with PKD1 mutations. J Am Soc Nephrol 24, 1571–1586 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jinek M et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mali P et al. RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Freedman BS et al. Modelling kidney disease with CRISPR-mutant kidney organoids derived from human pluripotent epiblast spheroids. Nat Commun 6, 8715 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davidson KC et al. Wnt/beta-catenin signaling promotes differentiation, not self-renewal, of human embryonic stem cells and is repressed by Oct4. Proc Natl Acad Sci U S A 109, 4485–4490 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lam AQ et al. Rapid and efficient differentiation of human pluripotent stem cells into intermediate mesoderm that forms tubules expressing kidney proximal tubular markers. J Am Soc Nephrol 25, 1211–1225 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sumi T, Tsuneyoshi N, Nakatsuji N & Suemori H Defining early lineage specification of human embryonic stem cells by the orchestrated balance of canonical Wnt/beta-catenin, Activin/Nodal and BMP signaling. Development 135, 2969–2979 (2008). [DOI] [PubMed] [Google Scholar]
- 35.Cantagrel V et al. Mutations in the cilia gene ARL13B lead to the classical form of Joubert syndrome. Am J Hum Genet 83, 170–179 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Higginbotham H et al. Arl13b-regulated cilia activities are essential for polarized radial glial scaffold formation. Nat Neurosci 16, 1000–1007 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma M, Tian X, Igarashi P, Pazour GJ & Somlo S Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease. Nat Genet 45, 1004–1012 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taguchi A et al. Redefining the in vivo origin of metanephric nephron progenitors enables generation of complex kidney structures from pluripotent stem cells. Cell Stem Cell 14, 53–67 (2014). [DOI] [PubMed] [Google Scholar]
- 39.Takasato M et al. Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature 526, 564–568 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Wang B, Fallon JF & Beachy PA Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell 100, 423–434 (2000). [DOI] [PubMed] [Google Scholar]
- 41.Humke EW, Dorn KV, Milenkovic L, Scott MP & Rohatgi R The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev 24, 670–682 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim YK et al. Gene-Edited Human Kidney Organoids Reveal Mechanisms of Disease in Podocyte Development. Stem Cells 35, 2366–2378 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cruz NM et al. Organoid cystogenesis reveals a critical role of microenvironment in human polycystic kidney disease. Nat Mater 16, 1112–1119 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harder JL et al. Organoid single cell profiling identifies a transcriptional signature of glomerular disease. JCI Insight 4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goodrich LV, Johnson RL, Milenkovic L, McMahon JA & Scott MP Conservation of the hedgehog/patched signaling pathway from flies to mice: induction of a mouse patched gene by Hedgehog. Genes Dev 10, 301–312 (1996). [DOI] [PubMed] [Google Scholar]
- 46.Marigo V & Tabin CJ Regulation of patched by sonic hedgehog in the developing neural tube. Proc Natl Acad Sci U S A 93, 9346–9351 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dessaud E et al. Interpretation of the sonic hedgehog morphogen gradient by a temporal adaptation mechanism. Nature 450, 717–720 (2007). [DOI] [PubMed] [Google Scholar]
- 48.Huangfu D et al. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 426, 83–87 (2003). [DOI] [PubMed] [Google Scholar]
- 49.Corbit KC et al. Vertebrate Smoothened functions at the primary cilium. Nature 437, 1018–1021 (2005). [DOI] [PubMed] [Google Scholar]
- 50.Pazour GJ et al. Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. J Cell Biol 151, 709–718 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yoder BK, Hou X & Guay-Woodford LM The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J Am Soc Nephrol 13, 2508–2516 (2002). [DOI] [PubMed] [Google Scholar]
- 52.Phua SC et al. Dynamic Remodeling of Membrane Composition Drives Cell Cycle through Primary Cilia Excision. Cell 168, 264–279 e215 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nager AR et al. An Actin Network Dispatches Ciliary GPCRs into Extracellular Vesicles to Modulate Signaling. Cell 168, 252–263 e214 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Scholey JM Kinesin-2: a family of heterotrimeric and homodimeric motors with diverse intracellular transport functions. Annu Rev Cell Dev Biol 29, 443–469 (2013). [DOI] [PubMed] [Google Scholar]
- 55.Cogne B et al. Mutations in the Kinesin-2 Motor KIF3B Cause an Autosomal-Dominant Ciliopathy. Am J Hum Genet 106, 893–904 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Glass NR et al. Multivariate patterning of human pluripotent cells under perfusion reveals critical roles of induced paracrine factors in kidney organoid development. Sci Adv 6, eaaw2746 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu H et al. Comparative Analysis and Refinement of Human PSC-Derived Kidney Organoid Differentiation with Single-Cell Transcriptomics. Cell Stem Cell 23, 869–881 e868 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Subramanian A et al. Single cell census of human kidney organoids shows reproducibility and diminished off-target cells after transplantation. Nat Commun 10, 5462 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.D’Cruz R, Stronks K, Rowan CJ & Rosenblum ND Lineage-specific roles of hedgehog-GLI signaling during mammalian kidney development. Pediatr Nephrol 35, 725–731 (2020). [DOI] [PubMed] [Google Scholar]
- 60.Mak SK et al. Small molecules greatly improve conversion of human-induced pluripotent stem cells to the neuronal lineage. Stem Cells Int 2012, 140427 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kriks S et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 480, 547–551 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Morizane R et al. Nephron organoids derived from human pluripotent stem cells model kidney development and injury. Nat Biotechnol 33, 1193–1200 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Barr MM & Sternberg PW A polycystic kidney-disease gene homologue required for male mating behaviour in C. elegans. Nature 401, 386–389 (1999). [DOI] [PubMed] [Google Scholar]
- 64.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Generation of KIF3A−/− and KIF3B−/− hPSC mutants using CRISPR gene editing. (a) Table of gene edited hPSC carrying loss of function mutations in KIF3A or KIF3B generated using CRISPR gene editing. The genetic background, sgRNA used for targeting, and resulting mutation is indicated for each cell line. (b) Chromatograms of homozygote mutants showing the edited KIF3A or (c) KIF3B regions. The sgRNA sequence used for targeting is labelled in yellow in the respective control chromatograms. (d) Sanger sequencing chromatogram of compound heterozygote KIF3A or (e) KIF3B mutants and the separated allele sequences after TOPO cloning. Deletions and insertions are indicated in the chromatograms.
Supplementary Figure 2. CRISPR gene editing results in loss of KIF3A or KIF3B protein. (a) KIF3A immunoblot showing three distinct control and KIF3A−/− H9 hPSC lines at passages 4 and 17. Cropped areas used in Figure 1 are shown in bracket box. β-actin was blotted as a loading control. (b) KIF3A immunoblot for one control (C1) and two distinct KIF3A−/− WTC11 hPSC (M1 and M2). GAPDH was blotted as a loading control. (c) KIF3B and β-actin immunoblot for one H9 control (C1) and two KIF3B−/− mutant (M1, M2) hPSC. (d) KIF3B and β-actin immunoblot for three distinct control and four KIF3B−/− WTC11 mutant hPSC. Cropped areas used in Figure 1 are shown in bracket box. kDa sizes of the protein standards are indicated. (e) Band intensity quantification from immunoblots of the indicated genotype (mean ± s.e.m., n ≥ 4 independent biological replicates).
Supplementary Figure 3. Kinesin-2 is dispensable for hPSC morphology and epiblast spheroid formation. (a) Confocal immunofluorescence images of acetylated α-tubulin (AcTub) and DNA in representative fields of undifferentiated control and KIF3A−/− hPSCs. Orthogonal (top) and volume (bottom) views are shown. (b) Phase contrast images of representative control and KIF3A−/− epiblast spheroids. (c) Quantification of lumen area as percentage of the spheroid area. (d-e) Confocal sections showing immunofluorescence for pluripotency, polarity, and ciliary markers in spheroids. A midbody is seen in a KIF3A−/− spheroid (arrow), but not cilia. Scale bars, 25 μm.
Supplementary Figure 4. Kinesin-2 knockout hPSCs establish a renewable source of diverse human cell types lacking cilia. (a) Neuroepithelial (AcTub), endodermal (AFP), and mesodermal (SMA) lineages in EBs. Zoom shows magnification of dashed boxed area with AcTub and DNA intensities increased for clarity. Scale bar, 50 μm. (b) Images and (c) quantification of pigmented EBs. Scale bar, 500 μm. Knockout (KO) represent pooled data from both KIF3A−/− and KIF3B−/− EBs (mean ± s.e.m., n ≥ 9 independent biological replicates per condition from a total of 8 distinct cell lines). (d) Representative images and (e) quantification of OCT4 and BRY immunofluorescence intensities in hPSCs after treatment with increasing doses of CHIR99021 in mTeSR1 for 48 hours. Each dot represents a single cell. Data are pooled from three separate experiments. bpp, bits per pixel (raw intensity). Scale bars, 50 μm.
Supplementary Figure 5. Kinesin-2 knockout tumors exhibit differentiation defects. (a) Representative photographs of whole, unfixed growths retrieved from immunodeficient animals at the same time point. Growths are sliced through their centers to reveal the internal surface. (b) A second set of tumors, photographed intact after retrieval from the animals. (c-e) Quantification of tissue subtypes within teratomas as a fraction of total area (mean ± s.e.m., n ≥ 5 independent biological replicates per condition from a total of 14 distinct cell lines). (f) Ratio of area occupied by SOX2+ cells in TUJ1+ patches of teratoma sections (mean ± s.e.m., n ≥ 4 independent biological replicates per condition from a total of 6 distinct cell lines; *, p < 0.05).
Supplementary Figure 6. Hedgehog switching is defective in kinesin-2 knockout organoid cultures. (a) Quantification of SOX2+ area per 96-well of kidney organoid cultures (mean ± s.e.m., n ≥ 3 independent biological replicates per condition from a total of 11 distinct cell lines. **, p < 0.01). (b) Wide-field immunofluorescence images of kidney organoid differentiations in a representative 96-well plate. The three left wells contain control cell lines and the three right wells contain KIF3A−/− cell lines. Zoom of boxed regions is shown below each of the wells. Each kidney organoid contains distal tubular (ECAD), proximal tubular (LTL), and podocyte (NPHS1) epithelial cells. Arrow indicates a cluster of ECAD+ cells without proximal tubule and podocyte segments. (c) Heatmap of the expression level of genes differentially expressed as a combined function of KIF3A or KIF3B loss in hPSCs (FDR < 0.2). Gene names and labels at right refer to genes associated with enriched gene ontology processes as a function of KIF3B−/− loss in hPSCs in Fig. 5a. (d) Representative GLI3 immunoblot of control, KIF3A−/−, and KIF3B−/− undifferentiated hPSCs, with (e) band intensity quantification (mean ± s.e.m., n ≥ 3 independent biological replicates per condition from a total of 11 distinct cell lines). (f) Band intensity quantification of the blot shown in Fig. 5e, comparing cultures on day 0 to day 18. (g) Immunoblot of GLI3 in kidney organoid cultures on days 7, 11, and 18 of differentiation. 3A and 3B indicate KIF3A−/− and KIF3B−/− mutants, respectively. (h) Schematic of organoid microdissection (left), with immunoblot of GLI3 on day 18 of differentiation in lysates of whole wells (w), microdissected organoids (o), or remnant stroma (s). (i) GLI1 immunoblot of the samples shown in Fig. 5e, comparing cultures on day 0 to day 18.
Supplementary Figure 7. (a) Representative GLI3 immunoblot in organoid cultures on day 18. Blot is from independent biological replicates, compared to the blot shown in Fig. 6d. (b) Individual measurements of GLI3F and (c) GLI3R bands for the sample set quantified in ratiometric form in Fig. 6d. (d) Representative GLI1 blot of these conditions, with (e) quantification of band intensities (mean ± s.e.m., n ≥ 3 independent biological replicates per condition from a total of 6 distinct cell lines).
Supplementary Figure 8. Comparison between kinesin-2 and polycystin knockout mutants. (a) Quantification of organoid area in 12 month old suspension cultures. (b) Representative confocal optical sections showing tubular epithelial markers in cyst-lining epithelia of PKD1 versus KIF3B mutant organoids. (c-e) Representative immunoblot showing PC1 and PC2 levels in KIF3A−/− hPSCs, compared to isogenic controls. β-actin is shown as a loading control. (f) Quantification of band intensities from whole cell lysates of kinesin-2 knockout or control hPSCs (n ≥ 5 independent biological replicates per condition). (g) Immunoblot showing PC2, KIF3A, and GAPDH loading control in kinesin-2 knockout hPSCs, isogenic controls, or PKD2 knockout. (h) Quantification of PC2 band intensities from immunoblots of kinesin-2 knockout hPSCs and isogenic controls (n ≥ 3 independent biological replicates per condition).
Supplementary Figure 9. hPSCs release ciliary proteins in EV via kinesin-2. (a) Quantification of protein fraction in EV (SN), compared to whole cell lysates (LY), as a percentage of total protein (SN + LY), using BCA assay (n = four distinct hPSC lines, plotted separately). (b) Representative immunoblot showing IFT88 levels in LY vs. SN. Total protein load is indicated in μg above each lane. (c) Quantification of IFT88 band intensities per lane at increasing total protein loads in SN and LY (n = 4 independent biological replicates from a total of 4 distinct cell lines). (d-e) Representative immunoblots and (f) band intensity quantifications of key ciliary proteins in EV, in KIF3B−/− hPSCs compared to isogenic controls (mean ± s.e.m., n ≥ 2 independent biological replicates from a total of 4 distinct cell lines).
Supplementary Figure 10. Kinesin-2 knockout hPSCs exhibit secretion defects with normal cytoplasmic expression. (a) Representative immunoblots of endogenous PC1 or (b) PC2 proteins in supernatants (SN) and lysates (LY) from control, PKD1−/−, PKD2−/−, and KIF3A−/− hPSCs, with FLOT1 loading control. PC2tetra refers to the tetrameric form of PC2. (c) Silver stain of whole supernatants from two control and three KIF3A−/− hPSC lines. No consistent differences in banding pattern are observed between the two groups. (d) Uncropped immunoblots for PTCH1 and IFT88 in EV, from Fig. 8b and 8c, respectively.
Supplementary Table 1. Lines and n used for statistical analysis.
Data Availability Statement
All datasets are available from the corresponding author upon reasonable request. Complete RNA-seq datasets will be deposited in NCBI as a GEO dataset at the time of publication.