

Abstract
Top dose selection for repeated dose animal studies has generally focused on identification of apical endpoints, use of the limit dose, or determination of a maximum tolerated dose (MTD). The intent is to optimize the ability of toxicity tests performed in a small number of animals to detect effects for hazard identification. An alternative approach, the kinetically derived maximum dose (KMD), has been proposed as a mechanism to integrate toxicokinetic (TK) data into the dose selection process. The approach refers to the dose above which the systemic exposures depart from being proportional to external doses. This non-linear external-internal dose relationship arises from saturation or limitation of TK process(es), such as absorption or metabolism. The importance of TK information is widely acknowledged when assessing human health risks arising from exposures to environmental chemicals, as TK determines the amount of chemical at potential sites of toxicological responses. However, there have been differing opinions and interpretations within the scientific and regulatory communities related to the validity and application of the KMD concept. A multi-stakeholder working group, led by the Health and Environmental Sciences Institute (HESI), was formed to provide an opportunity for impacted stakeholders to address commonly raised scientific and technical issues related to this topic and, more specifically, a weight of evidence approach is recommended to inform design and dose selection for repeated dose animal studies. Commonly raised challenges related to the use of TK data for dose selection are discussed, recommendations are provided, and illustrative case examples are provided to address these challenges or refute misconceptions.
Keywords: Toxicokinetics, Dose selection, Weight of evidence, KMD
1. Background and introduction
Dose-response analysis is an essential element within the risk assessment paradigm. The shape and slope of the dose-response curve, or a defined region on the dose-response relationship determined from animal toxicity studies, is used to derive health-based guidance values for human exposures. The observed dose-response relationship in animal toxicity studies results from the combined effects of toxicokinetic (TK) and toxicodynamic (TD) processes. TK processes, which include chemical absorption, distribution, metabolism, and excretion (ADME), determine target tissue exposure from a given administered dose. TD processes are interactions of a chemical with target molecules/cells or target tissues/organs and the subsequent toxicological responses. The concentration of a chemical at the target site, as a result of TK, controls the types and degrees of responses. Thus, internal dose (often characterized as systemic exposure), relative to the administered dose, allows for better prediction of the initiation and degree of toxicological responses (Creton et al., 2012; Saghir et al., 2006; Slikker et al., 2004a, 2004b). TK measurements usually consist of plasma (or whole blood) concentrations for the parent compound and/or metabolites to determine the amount of a chemical that is systemically available over time. In some cases, tissue concentrations are also measured to further understand the distribution of chemical in the body or to a target organ.
Many ADME mechanisms involve transporters or enzymes that have saturable capacity and specificity for their substrates. For example, chemicals may demonstrate capacity-limited transport through the gut wall, saturable intestinal or hepatic metabolism, or saturable plasma protein or tissue binding. Physicochemical transitions can also lead to nonlinear TK, such as the case where limitation of solubility or dissolution can affect the extent of absorption. In other cases, TD can alter TK; for example, a chemical that causes nephrotoxicity or obstruction of the bile duct can reduce the excretion of that chemical. These and many other factors can lead to nonlinearity between administered dose and systemic exposure for a parent and metabolite(s); thus, interpretation of the dose-response data needs to be done with substantial caution and should not ignore the potential impact from nonlinear TK. For example, thioacetamide displays a less than proportional increase in liver toxicity at higher doses (Mangipudy et al., 1998; Ramaiah et al., 1998) which can be explained by the saturation of thioacetamide bioactivation to a reactive necrosis-inducing toxic metabolite (Chilakapati et al., 2005).
Several hazard/risk assessment guidance documents recommend the use of TK data to inform dose selection and interpretation of study results from repeated dose animal toxicity studies (Table 1). Some guidance documents specifically state that when systemic exposure plateaus despite increasing administered dose, it indicates dosing above absorption saturation and the maximum systemic exposure has been reached. Administration of a higher dose not only adds little value, but also is not ethically justifiable (ICH, 2020; USEPA, 2005). Alternately, when the systemic exposure steeply and disproportionally increases with increasing administered dose, which indicates saturation of clearance, appropriate adjustments for TK become necessary when interpreting dose-response findings (ICH, 1994/2008; ICH, 1995b; ICH, 1997). When systemic exposure becomes extremely high due to saturation of clearance, particularly at doses that are orders of magnitude higher than the maximum possible human exposure, observed toxicity in animals likely does not accurately reflect the toxicological properties of a chemical at doses relevant to the exposed human population.
Table 1.
Summary of regulatory guidance on the requirements for and use of toxicokinetic data for study design and interpretation.
| Region/authority | Regulation/guideline/guidance | Summary of guidance on toxicokinetics (TK) |
|---|---|---|
|
| ||
| OECD | TG 417: Toxicokinetics (OECD, 2010) | • Guidance on how to generate TK data with maximum utility for test guideline studies, including ADME, linearity vs. nonlinearity, parent, and metabolite exposure • Detailed guidance on use of TK data to inform study design, such as setting dose levels to avoid nonlinearity and excessive toxicity, potential for bioaccumulation, and route of exposure • Guidance on how TK data provide context for interpretation for human hazard/risk assessment, such as contribution to mode/mechanism of toxicity, parent and metabolite exposure, accumulation, potential for biotransformation. |
| GD 116: Conduct and Design of Chronic Toxicity and Carcinogenicity Studies, Supporting Test Guidelines 451, 452, and 453 (OECD, 2012/2014) TG 451: Carcinogenicity Studies (OECD, 2018b) TG 453: Combined Chronic Toxicity/Carcinogenicity Studies (OECD, 2018c) |
• For study design, TK data should be used to inform top dose selection to avoid TK saturation of ADME mechanisms and excessive toxicity, aligning with the need for applicability to human exposure levels. • For study design, the TK data from shorter-term repeated dose/range finding studies should be considered to avoid suspected nonlinearities in the dose response indicative of metabolic induction, saturation, and changes in the external-internal dose relationship and increase interpretability in the context of human exposure levels. |
|
| Guidance Document Supporting OECD Test Guideline 443 on the Extended One-Generation Reproductive Toxicity Test, Series on Testing and Assessment, No. 151 (EOGRTS) (OECD, 2013) | • For study design, the use of TK data to avoid saturation of ADME processes is “strongly recommended” in the selection of multiple study parameters, including dose setting, route of administration, choice of vehicle, and duration of premating exposure to achieve steady state. • For study interpretation, TK data offer insight for the extent of prenatal exposure via placental transfer and postnatal exposure via breastmilk from the conduct of TG 443. |
|
| TG 443: Extended One-Generation Reproductive Toxicity Study (EOGRTS) (OECD, 2018a) | • In addition to the use of TK data described above (OECD, 2013), it is especially useful to: • Verify exposure of developing fetuses and pups to the test compound. • Provide an estimate of internal dosimetry. • Evaluate for potential dose-dependent saturation of kinetic processes. |
|
| TG 426: Developmental Neurotoxicity Study (OECD, 2007) | • For study design, TK data should be considered to provide evidence of adequate exposure to offspring and aid in decisionmaking for direct dosing of pups. • For study interpretation, TK data are incorporated as part of a weight of evidence approach to understand neurotoxic effects. |
|
| TG 409: Repeated Dose 90-Day Oral Toxicity Study in Non-Rodents (OECD, 1998) | • This study is used when previous TK studies indicate that a specific nonrodent study would more relevant than a rodent study. • It is recommended that existing TK data be considered in dose setting to avoid excessive toxicity. |
|
| ICH | ICH Topic S3A Toxicokinetics: A Guidance for Assessing Systemic Exposure in Toxicology Studies (ICH, 1995b) | • Provides guidance for integration of TK approaches for assessing systemic exposure in toxicity testing, including details on methods, quantification, timepoints, dose setting, routes of administration, and metabolites across study types (single dose, repeat dose, genotoxicity, carcinogenicity, and reproductive). |
| ICH Detection of Reproductive and Developmental Toxicity for Human Pharmaceuticals S5(R3) (ICH, 2020) | • All available TK data should be used for study design and evaluation, to avoid dosing above saturation of absorption and to set doses reflective of potential TK differences during pregnancy. | |
| ICH Dose Selection for Carcinogenicity Studies S1C(R2) (ICH, 1994/2008) ICH Guideline on the Need for Carcinogenicity Studies of Pharmaceuticals S1A (ICH, 1995a) ICH Testing for Carcinogenicity of Pharmaceuticals S1B (ICH, 1997) |
• In study design, dose setting should take into account pharmacokinetic linearity as it relates to saturation of metabolic pathways, to avoid exposures so high as to decrease the relevance to human risk. | |
| EU | REACH: Guidance on Information Requirements and Chemical Safety Assessment, Chapter R.7c: Endpoint Specific Guidance (ECHA, 2017) | • Recommends following guidance outlined in OECD TG 417 Toxicokinetics (see above) for the generation and use of TK data in human health risk assessment. • Encourages the consideration of potential for nonlinear kinetics to support dose setting decisions for repeat-dose studies and reiterates that toxicity is a function of target tissue dose, ADME processes, bioaccumulation, and elimination halflives. |
| Regulation (EC) No 1107/2009 (European Union, 2013) | • TK data should support the design of and be included in short- and long-term studies. • Dose selection should consider TK data, such as systemic exposure levels indicative of saturation of absorption. • TK data considered in comparative animal and human in vitro metabolism studies. |
|
| USEPA | Guidelines for Carcinogen Risk Assessment (USEPA, 2005) | • TK should be considered to set top dose, as to not compromise study outcome through inducing inappropriate TK (e.g., overwhelming absorption, detoxification mechanisms) and excessive toxicity. • Overt toxicity or qualitatively altered TK due to excessively high dose may result in tumor effects that are secondary to the toxicity rather than directly attributable to the agent. • TK of saturation of metabolic processes and potential nonlinearity of the dose-response relationship inform the mode of action. |
| Guidance Document #G2003.2: Rodent Carcinogenicity Studies: Dose Selection and Evaluation (USEPA, 2003) | • TK in consideration of a key precursor event in a mode of action being more likely in children. • In determining the adequacy of the high dose for carcinogenicity studies, TK is used as part of a weight of evidence approach and should indicate that the high dose is below saturation of absorption. |
|
Abbreviations: ADME, absorption, distribution, metabolism, excretion; ECHA, European Chemicals Agency; EOGRTS, Extended One-Generation Reproductive Toxicity Study; GD, Guidance Document; ICH, International Conference on Harmonisation; OECD, Organisation for Economic Co-operation, and Development; REACH, Registration, Evaluation, Authorisation and Restriction of Chemicals; TG, Test Guideline; TK, toxicokinetics; USEPA, U.S. Environmental Protection Agency.
Historically, top dose selection for repeated dose animal studies generally has relied on three approaches: identifying target organs and toxicity effects, using the limit dose,1 or determining a maximum tolerated dose (MTD),2 with the intention to increase the capacity of toxicity studies to detect effects given the relatively small number of animals in these studies (hazard identification). As is the case with any approach intended to evaluate complex biological processes, each of these approaches has advantages and limitations depending on the intended use of the study results, such as hazard identification or risk assessment. A fourth alternative, the kinetically derived maximum dose (KMD), has been proposed by stakeholders in various sectors to integrate TK data into the dose selection process (Carmichael et al., 2006; HESI, 2001). A proposed definition for the KMD is an administered dose that is slightly above the onset of nonlinear TK as informed by administered dose versus systemic exposure data (Saghir, 2015; Saghir et al., 2013). This approach is proposed to avoid selecting a top dose that induces toxicity well outside of the expected human exposure range or is above the saturation of absorption, while still health protective and supportive of animal welfare benefits.
While variations of the KMD approach have been applied in several studies (Bartels et al., 2020a, 2020b; Borghoff et al., 2016; Loccisano et al., 2020; Saghir et al., 2012; Van Cott et al., 2018), there is no consensus on how a KMD should be estimated or when such an approach is appropriate. The lack of consensus is partly due to the vague definition of a KMD, which refers to an inflection point that signals the onset of nonlinear TK behavior (McFadden et al., 2012; Saghir, 2015; Saghir et al., 2013). However, such an inflection point does not exist in the hyperbolic relationship between administered dose and systemic exposure when one or more ADME mechanisms are nonlinear, so there are no agreed-upon statistical approaches to estimate a KMD. Furthermore, there are different perceptions as to what the “KMD approach” means and encompasses. Some users equate the KMD approach with the administered dose at which systemic exposure becomes disproportional to administered dose, while others refer to integrating nonlinear TK information with other available data as part of a weight of evidence approach. These different interpretations likely contribute to the confusion and debates surrounding the use of the KMD concept for top dose selection. Since regulatory agencies often receive submissions of studies that use the KMD concept, a multi-stakeholder working group, led by the Health and Environmental Sciences Institute (HESI), has formed to provide an opportunity for impacted stakeholders to address commonly raised scientific and technical issues related to the KMD topic. The group is comprised of leading PBPK experts from academia, government, industry, and NGOs, who are committed to addressing key scientific needs PBPK modeling practices and applications that could facilitate their use for chemical risk assessment. As the first step to engage the stakeholders and subject experts in the KMD discussions, a virtual symposium on “Opportunities and Challenges in Using the Kinetically Derived Maximum Dose Concept to Refine Risk Assessment”3 was held in September 2020. One of the key messages from the symposium is that nonlinear TK is an indication of limits or transitions of a biological process but, by itself, does not overtly suggest an adverse effect. In other words, a KMD based on the current definition, which solely relies on a disproportional relationship between administered dose and systemic exposure, is not sufficient to set a top dose or to interpret dose-response data. Rather, the design of repeated dose animal studies, including dose selection, should involve weighing available data from shorter-term in vivo and in vitro studies that inform both TK and TD of a chemical, such as potency, target organ, toxic moiety, metabolism, and mode of action (OECD TG 116) (OECD, 2012/2014). This weight of evidence approach should also include an understanding of human exposures, such as routes and scenarios, and how these exposures drive internal or systemic exposure. This integrative approach offers better insight into how a chemical elicits its toxicity following repeated exposure. It also provides a better opportunity for these longer-term animal studies to provide data that appropriately characterize the nature of specific toxic responses, describe interpretable and human-relevant dose-response relationships, and elucidate the roles of TK and TD in toxicity pathways (OECD TG 116) (OECD, 2012/2014). The experts involved in the symposium recommended developing a framework to guide the use of a weight of evidence approach, accounting for various degrees of data availability, to inform dose selection in repeated dose animal studies. They also suggested that a separate framework would be valuable to guide the collection and possible uses of TK data at various phases of the chemical safety testing program. In this article, recommendations made during the symposium are summarized to address key challenges identified regarding the KMD, or nonlinear TK information, to set top doses (Heringa et al., 2020; Woutersen et al., 2020). When available, case studies are provided to illustrate specific recommendations. Open discussions and additional analyses will continue on these topics to support an improved process of hazard characterization and reduce uncertainty that hinders confidence in human health risk assessment.
2. Challenges and recommendations
Differing interpretations of the KMD definition and divergent perceptions related to its application have likely contributed to criticism of and lack of support for the concept (Heringa et al., 2020; Woutersen et al., 2020). These critiques have led to robust discussions and case studies to help better understand when and how TK data can be used, with other pieces of evidence, to design repeated dose animal studies or to interpret dose-response results. This section briefly summarizes five commonly raised challenges related to the KMD concept, or more general use of TK data, for dose selection. Recommendations are provided, with case studies when available, to address these challenges or refute misconceptions. Table 2 provides an overview of the main discussion points and recommendations.
Table 2.
An overview of the main challenges related to KMD and TK saturation data, examples from the text, and recommendations to address these challenges.
| Challenges | Case studies/examples | Recommendations |
|---|---|---|
|
| ||
| Inflection point: No inflection point in the hyperbolic relationship between administered dose and systemic exposure of a chemical that exhibits nonlinear TK behavior. | • Computational modeling to simulate administered dose versus systemic exposure relationships • Collecting TK data using in vitro approaches • See Section 2.1 |
• The current definition of a KMD that uses the term “inflection point” or “onset of nonlinear behavior” is not appropriate. • Use in vitro and in vivo TK data, along with available hazard data and human exposure information, in a weight of evidence approach to achieve a scientifically defensible decision on dose selection or response interpretation. • Develop guidance regarding the use of statistical methods to analyze proportionality between administered dose and systemic exposure, given a criterion determined from the weight of evidence assessment. • Use TK modeling throughout the chemical safety testing program to organize and integrate available data, test hypotheses on the mode of action, and recommend targeted studies to inform the role of TK on toxicity pathways. |
| Exposure: Human exposure levels are hard to predict, making it difficult to ensure that a top dose selected using TK information is higher than human exposures by a sufficient margin. | • Occupational worker dermal and inhalation exposures to pesticides • Dermal and inhalation exposures to a wide range of environmental chemicals • Screening level exposure estimates for 448 chemicals • See Section 2.2 |
• Many valid and robust exposure data resources and approaches are available to estimate human exposure levels, and uncertainty analysis has been an integral part of exposure assessment. • Due to the conservative nature of exposure assessment approaches, there is more uncertainty in the lower end of possible exposure prediction, rather than in the higher end. In most cases, all dose levels used in repeated dose animal toxicity studies are likely to be higher than human exposure levels by a sufficient margin. |
| 3Rs: Obtaining TK data requires additional animals, which does not meet the 3Rs principles. | • Microsampling within the main study • In vitro methods to obtain ADME parameters + TK modeling (rifampicin) • See Section 2.3 |
• Dose selection is best supported by a scientific weight of evidence approach that collectively considers the available body of data • Leverage alternative methods (such as in vitro bioassays), novel techniques (such as microsampling), and computational modeling to reliably collect and integrate TK data without additional satellite animal groups. |
| Limitations in TK studies: Animal TK studies are not always reflective of human exposure scenarios; TK differences between animals and humans cannot be determined; TK endpoints from animal studies may not be an appropriate internal dose metric for the toxic moiety. | • MPP+ (selective concentration of the toxic moiety) • Dichloropropane (glutathione depletion in the lungs) • See Section 2.4 |
• To improve the relevance and predictability of animal models to human hazard identification and risk assessment, we recommend designing fit-for-purpose animal studies and using in vitro approaches and computational modeling (such as PBK analysis) to understand TK and TD differences between test species and humans. • Use the internal dose metric, or an appropriate surrogate, that is most closely related to toxicity responses to understand the impact of nonlinear TK on the dose-response relationship. |
| Conflicts with some regulatory requirements: Lower dose studies may not meet requirements for some regulatory purposes, such as classification and labeling or endocrine disruption assessment. | • See Section 2.5 | • Use a weight of evidence approach to optimize the study design of repeated dose animal studies. • TK data may have a different weight in the dose selection process across regulatory programs, but these data can still be critical for other risk assessment objectives. |
| Insufficiency of TK information for top dose setting: TK data are not sufficient on their own to inform study design. | • Saturation of absorption (florpyrauxifen-benzyl, fenpicoxamid) • Saturation of absorption and clearance (sulfoxaflor) • See Section 3 |
• A weight of evidence approach that incorporates available data, such as hazard, TK, mechanistic, and exposure information, should be used to inform dose setting. |
Abbreviations: 3Rs, reduction, replacement, and refinement; ADME, absorption, distribution, metabolism, excretion; TK, toxicokinetics.
2.1. Inflection point
2.1.1. Challenge
Current definitions of the KMD refers to an inflection point at the onset of nonlinear TK behavior, or a maximum administered dose at which systemic exposure is not proportional to administered dose (Saghir, 2015; Saghir et al., 2013). However, there is no inflection point in the hyperbolic relationship between administered dose and systemic exposure of a chemical that exhibits nonlinear TK behaviors. In addition, systemic exposure can start to deviate from proportionality at very low administered dose when one or more ADME process(es) are nonlinear.
2.1.2. Recommendations
The current definition of a KMD that uses the term “inflection point” or “onset of nonlinear behavior” is not appropriate.
Use in vitro and in vivo TK data, along with available information, in a weight of evidence approach to achieve a scientifically defensible decision on dose selection or response interpretation.
Develop guidance regarding the use of statistical methods to analyze degree of proportionality between administered dose and systemic exposure, given a criterion determined from the weight of evidence assessment.
Use TK modeling throughout the chemical safety testing program to organize and integrate available TK and dose-response data, test hypotheses on the mode of action, and recommend targeted studies to inform the role of TK in toxicity pathways.
One of the main objections to the KMD concept is related to the use of terms “inflection point,” “point of nonlinearity,” or “slightly above the threshold of TK saturation” to define a KMD (ECHA, 2017; McFadden et al., 2012; Saghir, 2015; Saghir et al., 2013). We acknowledge that using the terms “inflection point” or “threshold” can lead to confusion, and a KMD estimated based on these definitions in not appropriate for top dose selection or interpretation of a dose-response relationship. However, the lack of an inflection point in nonlinear TK behavior does not preclude TK data from being useful for dose selection and dose-response interpretation. On the contrary, a greater understanding of how administered dose relates to systemic exposure or delivered dose available to elicit a toxicological response in tissues supports optimal design of toxicity testing and characterization of uncertainty in hazard and risk assessments. Understandably, the challenges in conducting these evaluations would be dependent on the available data, the nature of the effect, and the overall toxicity profile. For instance, using TK data to interpret subtle effects may be more difficult than for severe effects.
To illustrate how nonlinear TK can lead to a substantially higher or lower than expected systemic exposure based on proportionality to the administered dose, a case study is presented. This example compares simulated systemic exposures in rats, given first order/saturable metabolism or first-order/saturable oral absorption. A three-compartment physiologically based kinetic (PBK) model was constructed to simulate, at pseudo steady state, the plasma area under the curve (AUC) for both the parent and a metabolite in rats from repeated oral bolus dosing to a hypothetical chemical over a wide range of administered doses. (Fig. 1). This model is implemented in Berkeley Madonna (v.10) and the code is available upon request. The rate of oral absorption and the rate of parent metabolism is either first order or saturable, which is described as Michaelis-Menten kinetics. In the case of saturable metabolism, the simulated AUCs of the parent shows a hyperbolic relationship with the administered dose; except at very low doses (in the range where rate of metabolism is approaching first order) or very high doses (in the range of zero-order rate of metabolism), AUCs are proportional to administered doses (Fig. 1A). In this example (Vmax = 6 μmol/h; Km = 2.5 μmol/L), when exposed to the limit dose of 1000 mg/kg/day, the parent AUC is more than 500-fold higher when metabolism is saturable compared with linear metabolism. On the other hand, the increase in simulated AUCs of the metabolite is lower than proportional with increasing administered dose until a plateau is reached after parent metabolism is saturated (Fig. 1A).
Fig. 1.
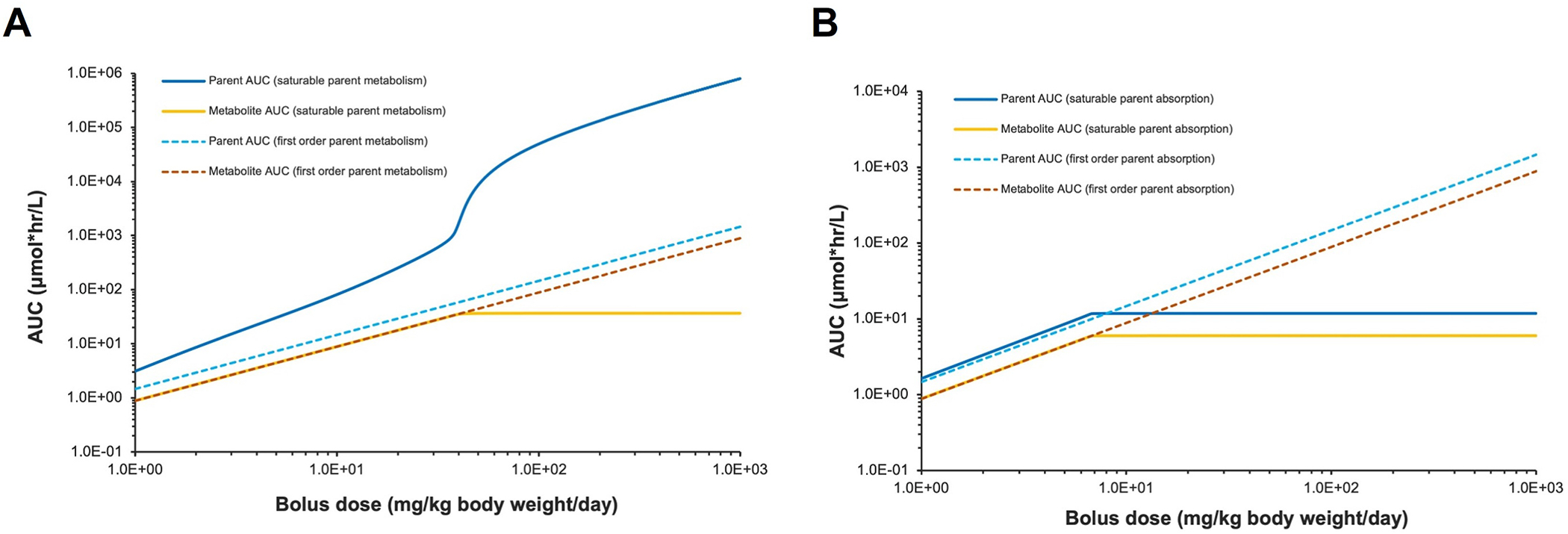
Simulated administered dose versus area under the curve (AUC) of plasma concentration of a hypothetical chemical and its metabolite, assuming (A) either first-order or saturable metabolism of the hypothetical chemical or (B) either first-order or saturable oral absorption of the hypothetical chemical.
In the case of saturable oral absorption, both the parent and metabolite AUCs became disproportionally lower with increasing administered dose and approach a plateau after absorption is saturated (Fig. 1B). While this case study was based on the most common exposure route (oral bolus), test species (rats), and dose metric (plasma AUC), the PBK modeling approach can be used to simulate other exposure routes (such as dietary, inhalation, dermal) for other test species (such as mice, dogs) and humans and for other dose metrics (such as peak concentration). Several lessons can be learned from this case study. Saturation or limitation of ADME process(es) can result in different nonlinear relationships between the administered dose and the internal/systemic exposure for a parent and a metabolite (Fig. 1A). These nonlinear relationships can then lead to systemic exposure that is substantially higher or lower than expected based on a proportional relationship to the administered dose, making it difficult to interpret dose-response relationship at the administered dose level. Thus, nonlinear TK data should be included in a weight of evidence assessment to inform better design of longer-term toxicity studies.
This simulation study also brings to light an issue regarding common approaches used to estimate a KMD by evaluating the degree of disproportionality of systemic exposure to administered dose. These approaches include visual inspection of the administered dose versus systemic exposure data, comparison of fold changes in administered doses and systemic exposures (Loccisano et al., 2020; Saghir et al., 2013; Terry et al., 2014), and statistical methods to determine if any of the data points deviate from the linear assumption (Bartels et al., 2020a, 2020b; McFadden et al., 2012; Saghir et al., 2012). All these approaches are based on the premise that a KMD is the onset (or an inflection point) of nonlinear TK, but as shown in our case study, systemic exposure can deviate from proportionality starting at very low administered dose (Fig. 1). This issue further reiterates the need to redefine the KMD and that TK data should not be used in isolation for dose selection or response interpretation. Furthermore, we recommend developing guidance on how to use statistical methods to objectively analyze proportionality between administered dose and systemic exposure and quantitatively address uncertainty. The weight of evidence assessment can inform a specific criterion for statistical analysis, such as the observed systemic exposure being more than a certain percentage above/below the expected proportional level.
Finally, this simulation study demonstrates the potential use of PBK modeling, as part of a weight of evidence approach, to explore how one or more nonlinearities in ADME processes influence the relationship between administered dose and systemic exposure/internal dose, or between administered dose and toxicological response. A PBK model can be used to integrate data collected from shorter-term studies (such as dose-response, toxic moiety, TK) to optimize the design of longer-term studies, including appropriate dose selection and placement. A PBK model can also be valuable during earlier stages of the safety testing program to test hypotheses on mode of action or suggest targeted studies to fill data gaps. For example, these models can be used to calculate alternative measures of internal dose (such as peak concentration in target tissue, average concentration in plasma, rate of metabolism) to identify the approach that best correlates with the observed toxic effect (USEPA, 2006). These models can also be used to characterize the relationships between potential dose metrics and early biochemical responses to suggest or confirm possible mode of action. As more in vitro and in silico tools are available to estimate enzyme and transporter functions in tissues, it becomes easier to construct provisional PBK models with limited resources. These provisional models can continuously be refined throughout the safety testing program when more data become available and to address different science questions.
2.2. Human exposure predictions
2.2.1. Challenge
Human exposure levels are hard to predict and are associated with large uncertainties, making it difficult to ensure that a top dose selected using TK information is higher than human exposures by a sufficient margin.
2.2.2. Recommendation
Many valid and robust exposure data resources and approaches are available to estimate human exposure levels, and uncertainty analysis has been an integral part of exposure assessment. In addition, due to the conservative nature of exposure assessment approaches, there is more uncertainty in the lower end of possible exposure prediction, rather than in the higher end. In most cases, all dose levels used in repeated dose animal toxicity studies are likely to be higher than human exposure levels by a sufficient margin.
Exposure assessment is, and has been, an integral part of the risk assessment process. It is one of the four major steps in risk assessment, along with hazard identification, dose-response assessment, and risk characterization (National Research Council, 1983). Exposure assessments can provide important information for toxicity study dose setting, including the magnitude, frequency, duration, and route(s) of entry. Exposure assessment tools are available for various chemistries and use scenarios to conservatively predict human exposure levels. Many of these exposure resources/tools have been extensively peer reviewed and are used routinely to estimate occupational and consumer exposures. Exposure assessment tools are fit-for-purpose and tailored to meet specific requirements of risk assessment while accounting for data and resource availability. The ability to estimate exposure levels expected under typical use conditions using these available resources allows for confidence in the use of TK data in a weight of evidence approach for dose selection. Since the proposed uses of TK data under discussion here apply to repeated dose animal studies, exposure scenarios such as those related to acute toxicity, calamities, accidents, or intentional misuse are outside the scope of relevant design considerations.
To address the concern that human exposure levels are hard to predict and are associated with large uncertainties, several case studies were conducted to estimate human exposures for both pesticides and industrial chemicals with different degrees of data availability, from data-rich to data-poor chemicals, including those with very limited use information. The details of these case studies are provided in the companion paper, “Exposure Considerations Related to the Use of Toxicokinetic Data to Inform Top Dose Selection in Repeated-Dose Toxicity Studies” (Lowe, 2021). In this article, a short summary of some of the case studies are presented below. Also, to address the concern that it is difficult to ensure a top dose selected using TK information is higher than human exposures by a sufficient margin, when available, the estimated human exposure levels were compared to points of departure (PODs) reported from repeated dose animal studies, such as no-observed-adverse-effect levels (NOAELs) or lowest-observed-adverse-effect levels (LOAELs). Animal PODs are used as proxies for top doses in these case studies because they are much easier to obtain from literature or databases. And, regardless how a top dose is selected (such as MTD or based on TK consideration), animal PODs are likely to be one to two orders of magnitude lower than the top dose, unless a chemical exhibited no toxicity, and the NOAEL is equal to the top dose.
In one case study, occupational worker dermal and inhalation exposures to pesticides were estimated based on the U.S. Environmental Protection Agency (USEPA) scenario-based exposure assessment methodology for nondietary exposures to pesticides (USEPA, 2021b; USEPA, 2021c). A scenario represents a specific job task that involves a combination of application equipment (such as groundboom tractors, handheld sprays), formulation type (such as liquids, granules), and job function (such as mixer/loaders, applicators). The analysis was conducted for common occupational exposure scenarios (803 total). Assumptions were made to provide higher end exposure estimates, such as typical work clothing was worn with no additional personal protective equipment (PPE), and exposures were combined across both the dermal and inhalation routes. The estimated exposure levels for all 803 scenarios had an average combined exposure of 0.74 mg/kg body weight/day (with a standard deviation of 3.4 mg/kg bw/day). The maximum estimated combined dermal/inhalation exposure was 67 mg/kg bw/day, with dermal exposures being much higher (maximum of 66 mg/kg bw/day) compared with inhalation exposures (maximum of 0.84 mg/kg bw/day). In this case study, dermal NOAELs/LOAELs for all conventional pesticides obtained from submitted subchronic exposure duration dermal administration toxicity studies (except for 2 out of 227 studies) were used for comparison to the estimated exposures. The estimated maximum exposure levels were lower than the average dermal NOAEL (130 mg/kg bw/day; range: 0.05 to 1000 mg/kg bw/day; SD 208 mg/kg bw/day; N = 217) and dermal LOAEL (384 mg/kg bw/day; range: 0.2 to 4000 mg/kg bw/day; SD 527 mg/kg bw/day; N = 204) identified for conventional pesticides. The average combined exposure of 0.74 mg/kg bw/day was ~175 × lower than the average dermal NOAEL and ~500 × lower than the average dermal LOAEL.
Another case study used existing occupational exposure models from the U.S. EPA Office of Pollution Prevention and Toxic’s Chemical Screening Tool for Exposures and Environmental Releases (ChemSTEER) (with an Office of Research and Development [ORD] developed interface) to estimate exposure levels in the workplace through both chemical-specific and chemical-agnostic models contained within the tool (USEPA, 2021a). In this case study, the weight fraction of a chemical in the media of interest (particulate matter or air for inhalation; liquid, solid, or container residue for skin contact) was varied from 0.1 to 1 in the chemical-agnostic models. Even at the weight fraction of 1, all estimated average daily doses were <0.5 mg/kg bw/day.
In another case study, one of the two chemical-specific models in ChemSTEER, the user-defined inhalation model, was used to predict exposure levels using chemical-specific inputs obtained from the U.S. Occupational Safety and Health Administration’s Chemical Exposure Health Data (CEHD). CEHD reports both dermal residues and air concentrations across various U.S. workplaces and industries. In this case study, CEHD inhalation data collected from 1984 to 2018 for more than 1000 chemicals were aggregated to provide a high-end exposure for airborne chemical concentrations across industrial sectors and subsectors. Ninety-nine percent of the data points in this dataset had average daily doses that were <1 mg/kg bw/day, with most being much lower; the median value was 9.09 × 10−5 mg/kg bw/day.
Exposure levels can also be predicted based on physicochemical properties of a chemical and minimal information, such as their functional role in formulated products (Dellarco et al., 2017; Wambaugh et al., 2019). As an example, the ExpoCast Systematic Empirical Evaluations of Models (SEEM) framework can use chemical structure to first predict relevant exposure pathways, and then use calibrated exposure models to make predictions of daily intake rate (in mg/kg bw/day). In one other case study, version 2 of the SEEM framework (SEEM2) was used to predict screening level exposures for 448 chemicals (Paul Friedman et al., 2020). Using the 95th percentile of the predicted exposure ranges for each chemical, 431 out of 448 chemicals (96%) had predicted exposure levels <1 mg/kg bw/day.
The above case studies, and others provided in the companion paper (Lowe, 2021), address several of the concerns that have been noted regarding the uncertainty related to how human exposure levels compared to top doses used in animal toxicity studies. The case studies demonstrate that human exposure levels can be easily estimated in both data-rich and data-poor situations using readily available exposure assessment tools and resources. The approaches for exposure estimation typically involve conservative assumptions and address all routes of potential exposure to ensure that the resulting exposure levels are not underestimated. For the purposes of this paper, the pesticide case study only presented nondietary exposure estimates; however, risk assessments for pesticides routinely consider aggregate exposure (i.e., exposure to a single chemical from multiple sources; nondietary, food, and water) for the general population. For pesticides, aggregate exposure is assessed under a risk-only statute (i.e., without consideration of the benefits of the pesticide), where human exposures are compared to adjusted PODs, which takes into consideration uncertainty factors. Human exposure levels are usually several orders of magnitude lower than the dose where effects were observed in animal studies. Biomonitoring data, such as those collected by the Centers for Disease Control and Prevention under the National Health and Nutrition Examination Survey (NHANES), can provide additional information related to aggregate and cumulative exposure (exposure to multiple chemicals) for a number of chemicals. NHANES data can be used to determine populations with increased body burdens, identify exposure levels in populations of concern, and assess trends over time.
Another concern that was raised, that is not directly addressed by the case studies presented here or in the companion paper (Lowe, 2021), is related specifically to cumulative exposure; that is, people can be exposed to several substances at the same time and that when these substances have a similar mode of action, it is the accumulated exposure, not the exposure to a single substance, that should be compared to the top dose in a toxicity study. In a recent paper (Price, 2020), an analysis was conducted to investigate a common observance, following the Pareto principle,4 that risks posed by combined exposures from multiple chemicals are typically driven by exposures from a few chemicals/sources and also that individuals with the highest exposures were driven by a single chemical from a single source. The paper identifies characteristics of those cases where cumulative exposure is driven by a limited set of chemicals/sources and proposes that it may be possible to develop criteria to predict if risks from combined exposures would exceed the risks from separate exposures to individual chemicals. Rather than conducting comprehensive cumulative assessments, which can be resource intensive, this predictive approach would provide another exposure assessment tool that could be used to address the challenge related with cumulative exposure.
As noted above, exposure assessment approaches can involve chemical-specific data or can be chemical agnostic, allowing for estimates of exposure for many possible use scenarios with limited chemical information. A number of exposure data resources and approaches are available to assess the potential exposure from chemicals with various degrees of information available. Generally, these approaches have received public scrutiny and peer review to ensure their rigor. Based on our analysis, human exposures to a wide range of environmental chemicals can be easily predicted and are generally lower than animal PODs (in this case, NOAEL/LOAELs). While NOAEL/LOAEL values have been used in these analyses as a proxy for the top doses in animal studies, as stated earlier, the NOAEL/LOAEL values are likely to be one to two orders of magnitude lower than the top dose. As a result, the human exposure estimates are expected to be orders of magnitude lower than the actual top doses tested in the animal studies.
2.3. The 3Rs
The 3Rs refers to the principle of reduction, replacement, and refinement of animal studies. The objective of reduction is to decrease the number of animals used for research and regulatory purposes. The replacement approach includes in vitro or in silico methods. Refinement involves reducing, eliminating, or relieving animals’ pain or distress, and thereby improving their well-being.
2.3.1. Challenge
Obtaining TK data requires additional animals, which does not meet the 3Rs principles.
2.3.2. Recommendation
Dose selection is best supported by a scientific weight of evidence approach that collectively considers the available body of data, such as apical toxicity, TK data, and human exposure information. We recommend leveraging alternative methods (such as in vitro bioassays), novel techniques (such as microsampling), and computational modeling to reliably collect and integrate TK data without additional satellite animal groups.
Selecting a top dose that is too high or too low, especially in a longer-term repeated dose toxicity study, can have a markedly negative impact on the 3Rs (reduction, replacement, and refinement of animal testing). Dosing at a level that is too high has significant potential to induce unjustifiable animal suffering and generate data of questionable scientific validity. For example, the use of excessively high doses can result in the need to terminate dose groups early, increasing the likelihood of the need for repeat studies. It is also possible that maximum systemic exposures have been achieved at higher doses. In both cases, data generated from the high dose groups are essentially of no value from a human health hazard and risk assessment perspective. In other cases, additional animal studies may be triggered to further explore whether toxicities observed at high doses are relevant to realistic human exposures, or to investigate the underlying biological processes leading to the effect(s) for differentiating chemical-specific responses from nonspecific toxic effects. Conversely, dosing at a level that is deemed too low may also result in replication of a study at a higher dose level to demonstrate overt toxicity, using additional animals. Thus, it is critical to ensure the most scientifically appropriate and human-relevant doses are selected, and to reduce animal use and suffering by using only the necessary number of animals to achieve impactful results (Sewell et al., 2020).
Dose level selection should be guided by a scientific weight of evidence approach that considers available data from shorter-term repeated dose or range finding studies (OECD, 2012/2014) (OECD TG 116). These shorter-term studies provide valuable dose-response information, such as adaptive versus toxic effects or progression of toxic effects at different dose levels. It cannot be stressed enough that available TK data should always be considered as it provides information on how the conditions of the earlier studies (e.g., administration methods, the range of doses tested) influence the relationship between administered dose and systemic exposure/internal dose. Besides data on apical toxicity, dose response, and TK, other available mechanistic and human exposure information should also be evaluated to decide whether dose levels used in shorter-term repeated dose studies should be increased or decreased in longer-term studies, or if additional dose groups are warranted. There are many examples where it is appropriate for apical toxicity data to drive the dose selection, and there are cases where other data, such as TK data, provide the best scientific basis for selecting adequate dose groups to avoid producing false negative or false positive results.
Early in the safety testing program, TK data are collected to satisfy regulatory (Table 1) and scientific needs. These data may help to identify which circulating moieties are responsible for toxicity, evaluate bioavailability and the potential for chemical accumulation in tissues, and aid in understanding the mechanism of toxicity (OECD TG417) (OECD, 2010). In probe ADME studies, blood/plasma and urine samples may be collected at various time points to determine appropriate biomarkers (parent and major metabolites) to be monitored in future repeated dose studies. Single dose TK studies using radiolabeled or non-radiolabeled chemicals may provide information on mass balance, metabolism, accumulation and/or persistence, and changes in TK such as enzyme inhibition or ADME saturation. In shorter-term toxicity studies, without the addition of satellite animals, TK data may be generated to understand steady-state systemic exposure to the test material at various administered dose levels. From these data, several TK parameters can be calculated and analyzed (such as AUC, peak concentration, time to peak concentration, biological half-lives, and dose proportionality) to understand the TK influence on dose-response relationship, identify toxic moiety, or propose mode of action hypothesis. Furthermore, in vitro or in silico TK analyses, such as comparative metabolism study or PBK modeling, can also be conducted to understand TK differences in species, routes of exposure, and life stages.
Concerns have been raised that animal use might increase as a result of adding satellite groups into repeated dose toxicity studies to obtain samples for TK assessments, or even conducting additional standalone TK studies to investigate dose proportionality. Generating TK data does not necessarily require the use of additional animals. We recommend, when appropriate, replacing or complementing in vivo studies with novel techniques and alternative methods to collect TK information in the future. For example, over the past decade, the use of microsampling has gained acceptance as the standard blood sampling method for measuring chemical concentrations in rodent blood/plasma. The use of these smaller samples (typically 50 μl, compared to conventional samples of 200 μl) enables TK profiles to be obtained from animals in the main study, with no detrimental effect on other parameters such as clinical pathology (Chapman et al., 2014). The microsampling method negates additional animal use, improves animal welfare (for example, less time in the warming chamber; quicker, less-invasive technique; less blood loss), has the scientific benefit of allowing direct comparisons between toxicity and systemic exposures in the same animal, and has financial benefits related to fewer animals required per study (for example, less compound used, quicker procedures, fewer cages for husbandry) (Caron et al., 2015; Chapman et al., 2014; Powles-Glover et al., 2014). Currently, microsampling has been predominantly used within the pharmaceutical industry and is supported by specific regulatory guidance (EMA, 2017; EMA, 2020). The wider adoption of this technique in the field of chemical risk assessment would facilitate more acquisition of TK data while applying the 3Rs.
Many in vitro methods are available to estimate ADME parameters or investigate if ADME processes may be nonlinear over the range of possible administered doses used in animal studies (OECD et al., 2021). Parameters that are commonly measured with in vitro methods are fractional plasma protein binding (Rotroff et al., 2010; Wetmore et al., 2012), oral and dermal absorption rates (Hansch et al., 2004; Lehman et al., 2011; Potts and Guy, 1992; Zhao et al., 2003), tissue/plasma partitioning (Smith and Waters, 2018), and metabolism by whole cells (such as hepatocytes), key proteins in cellular membranes (such as microsomes), or individual enzyme (such as CYPs) (Franzosa et al., 2021; Obach et al., 1997; Rotroff et al., 2010; Wetmore et al., 2012). Some of these parameters can also be predicted in silico based on chemical structure and physiochemical properties (Dawson et al., 2021; Ingle et al., 2016; Obach et al., 2008; Poulin and Krishnan, 1996a, 1996b; Poulin and Theil, 2000; Sarigiannis et al., 2017; Schmitt, 2008). In addition, a read-across approach may be used to fill some data gaps when similar ADME-relevant properties can be identified and characterized for both the target chemical and its analogues (Ball et al., 2020; Laroche et al., 2018; Paini et al., 2021).
PBK models can be used to integrate these in vitro or in silico estimated ADME parameters for predicting systemic exposure/internal dose across a wide range of administered dose to evaluate the impact of any nonlinear ADME processes. For example, an antibiotic, rifampicin, demonstrates saturable biliary excretion (Acocella, 1978; Acocella et al., 1971), dose-dependent bioavailability (Chirehwa et al., 2016), and dose-dependent metabolism via autoinduction (Chirehwa et al., 2016). A recent study used a population-based TK model to incorporate these nonlinear mechanisms to facilitate optimization of rifampicin dosing in tuberculosis treatment (Svensson et al., 2018). The resulting model can also be used to explore future clinical trial designs by simulating different dosing and sampling regimes to evaluate exposure response, regarding both safety and efficacy.
2.4. Limitations in TK studies
2.4.1. Challenge
Animal TK studies are not always reflective of human exposure scenarios (routes, formulations, durations). Since human TK data cannot be generated in vivo, TK differences between animals and humans cannot be determined to allow for using animal TK data for dose selection. Additionally, the TK endpoints from animal studies, most frequently AUC of parent in plasma, may not be an appropriate internal dose metric for the toxic moiety.
2.4.2. Recommendations
To improve the relevance and predictability of animal models to human hazard identification and risk assessment, we recommend designing fit-for-purpose animal studies and using in vitro approaches and computational modeling (such as PBK analysis) to understand TK and TD differences between test species and humans.
We recommend using the internal dose metric, or an appropriate surrogate, that is most closely related to toxicity responses to understand the impact of nonlinear TK on the dose-response relationship.
The limitations in animal TK studies and the challenges associated with biological differences between species are not unique to TK studies; rather, these are general limitations when using animal models to derive human risk assessment endpoints. Methods have been developed and are commonly used in risk assessment to address the need to extrapolate and apply animal data to estimate human health risks in different contexts, including exposure scenarios or routes, life-stage sensitivities, and both TK and TD differences between animal models and humans. Collection of TK information is needed to improve the relevance and predictability of animal studies to human health risks and can also be used to inform dose setting. Optimizing the design of repeated dose studies involves scientific questions that can only be answered by using multiple pieces of evidence. Thus, we recommend following the OECD guidance (TG 116) (OECD, 2012) to consider using existing toxicological and TK data (Barton et al., 2006) from earlier studies to inform dose selection.
TK data from the species that will be tested in a longer-term study are the most relevant and directly applicable. Even for the purpose of identifying and quantifying interspecies differences in TK, many in vitro approaches using species-relevant biomaterials have been appropriately validated in relation to in vivo phenomena and are available to investigate TK between laboratory animals and humans. Furthermore, the increased availability of computing power and in vitro/in silico generated ADME data, as well as progressively sophisticated modeling techniques, readily allow for PBK models to be developed for predicting, in an increasingly accurate way, TK behaviors in test species and human populations. These capabilities are particularly useful for evaluating the relationship between administered dose/exposure levels and systemic exposure/internal dose under different dosing/exposure scenarios (such as routes, durations, frequencies), and for expected range of human exposure levels and appropriate dose range used in animal studies.
Analyses that examine dose proportionality and other TK behaviors are not restricted only to measurements based on plasma AUC; and indeed, the mathematical and kinetic basis of these analyses allows them to relate administered dose to any measures of the biologically active form of a chemical (such as parent or metabolites) in the appropriate biological medium (such as blood, target or surrogate tissues) for the duration of interest (such as lifetime or during a specific development period) and metric that reflect the toxic effect of interest (such as peak or average concentration). In situations where there is a lack of a quantifiable relationship between free plasma concentration and adverse effect at the target site, it is more appropriate to use other dose metrics, such as tissue or even cell concentrations of the toxicologically critical penultimate moiety(moieties). For example, 1-methyl-4-phenylpyridinium (MPP+), the toxic metabolite of 1-methyl-4-phenyl-1,2,3,6-tetrahy-dropyridine (MPTP), selectively targets dopaminergic cells of the substantia nigra where the compound has been shown to accumulate in primates, despite being eliminated in other parts of the brain (Irwin and Langston, 1985). This selective concentration of the toxic moiety illustrates the importance and feasibility of selecting a dose metric that is most relevant for the observed toxicity; in this example, the toxic moiety is metabolite substantia nigra concentration rather than blood AUC of the parent. Alternatively, surrogate measures, such as a molecular initiating event or keystone key event in an adverse outcome pathway (AOP) or AOP network, can be collected in addition to measures of systemic exposure. For example, a recent study demonstrated that higher exposure concentrations of 1,3-dichloropropane result in the depletion of glutathione in the lung, and that the depletion of glutathione is associated with an increased susceptibility of the tissue to oxidative stress (Bartels et al., 2020b). The data collected in this study on the concentration response for glutathione depletion in the lungs of the exposed animals could be used to estimate a mode of action related dose-dependent transition. In another commonly known example with acetaminophen-induce hepatotoxicity, 80%–90% glutathione depletion in hepatocytes is found to be necessary for the development of necrosis (Henderson et al., 2000; Jollow et al., 1974). In the future, combining in vitro studies on the concentration response for key events, such as glutathione depletion or genomic indicators of glutathione homeostasis, with TK modeling could allow for predicting the shape of the dose-response curve prior to conducting in vivo studies. Effects in liver may be better related to portal or inlet concentrations following oral exposure estimated from TK data, as has been shown for drug-drug interactions impacting liver metabolism (Ito et al., 2002). We recommend taking advantage of alternative methods to facilitate identification of mode of action, and to link internal dose levels to precursor key events for more informed design of animal studies.
2.5. Conflicts with some regulatory requirements
2.5.1. Challenge
If a top dose is much lower than the limit dose, it cannot provide information on a chemical’s potential to cause adverse effects at higher doses, which is required for some regulatory purposes such as classification and labeling or endocrine disruption assessment.
2.5.2. Recommendation
We recommend using a weight of evidence approach to optimize the design of repeated dose animal studies. We acknowledge that TK data may have a different weight in the dose selection process across regulatory programs, but these data can still be critical for other risk assessment objectives.
In a weight of evidence approach, pieces of evidence that are potentially pertinent are collectively integrated and weighed to identify a decision. We acknowledge that TK data are not considered pertinent when toxicity information at very high dose levels is required by specific regulatory authorities, such as the case where the hazards of a chemical or mixture need to be identified by assigning a certain hazard class and category to provide warning information for accidental high exposure. In some jurisdictions, requirements to identify potential endocrine disrupting substances is another instance where high doses may be required by regulators. The European Union, for instance, has introduced statutory requirements intended to phase out endocrine disruptors in water, industrial chemicals, plant protection products, and biocides. In EU Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH) program, endocrine disrupting chemicals can be considered as substances of very high concern alongside chemicals known to cause cancer, mutations, and reproduction toxicity (European Commission, 2021). The European Union Regulation (EC) No. 1107/2009 states that.
An active substance, safener or synergist shall only be approved if … it is not considered to have endocrine disrupting properties that may cause adverse effect in humans unless the exposure of humans to that active substance, safener or synergist in a plant protection product, under realistic proposed conditions of use, is negligible, that is, the product is used in closed systems or in other conditions excluding contact with humans and where residues of the active substance, safener or synergist concerned on food and feed do not exceed the default value set in accordance with point (b) of Article 18(1) of Regulation (EC) No 396/2005 (European Union, 2009).
In contrast to the European Union, U.S. regulations emphasize a risk-based approach in which exposure and hazard considerations, in concert, drive the regulatory and risk management decisions. These decisions may range from limiting conditions of use (such as decreasing application rates, use sites, frequency of use) to cancelling or banning a chemical. Repeat dose TK data also may not be relevant under the circumstances associated with acute exposure (a single instance or multiple instances over a period of <24 h), massive overdose/massive exposure, suicidal ideation-associated ingestion or off-label uses, and rare accidental events in industrial or catastrophic situations. However, classification for the above situations is based on acute toxicology studies, which is outside of the scope of study design for repeated dose studies.
TK data are not needed for chemicals that do not exert systemic toxicity, or when portal of entry effects (e.g., irritation, sensitization) or localized effects (Thompson et al., 2016) are more sensitive toxic endpoints. We recommend, when appropriate, considering TK as one piece of evidence in a whole suite of techniques and knowledge to optimize the design of repeated dose studies that are more interpretable and relevant to human risk assessment.
3. A weight of evidence approach
One of the general principles for dose selection in longer-term repeated dose animal studies is to consider any existing toxicological and TK data available for the chemical or related materials, such as data collected from shorter-term studies or range finding studies (Foran, 1997; OECD, 2012; Rhomberg et al., 2007). Such a weight of evidence approach is critical for achieving a scientifically defensible, consistent, and transparent conclusion (EFSA Scientific Committee et al., 2017). In some cases, the impact of TK on the expected dose-response relationship is more direct and easier to interpret, such as the case where systemic exposures of the known toxic moiety remain at a plateau, despite an increasing administered dose. In fact, saturation of absorption measured by systemic availability of a substance and/or metabolite is one of the high dose selection criteria recommended by several agencies (ICH Regulation No 283/2013) (ICH, 1994/2008). Even when absorption is not completely saturated, the nonlinear relationship between the administered dose and systemic exposure can still inform dose selection and dose spacing decisions. For example, in a population-based meta-analysis study that compared the two dosing regimens for the antibiotic roxithromycin (Dolton and D’Argenio, 2017), it was found that the 300 mg once-daily dose regimen achieves 37% and 53% less total and free AUC, respectively, in comparison to the 150 mg twice-daily dose regimen. The lower than proportional AUC at the higher dose is due to saturation of absorption and protein binding to α-1-glycoprotein (Andrews et al., 1987; Lassman et al., 1988; Tremblay et al., 1988; Zini et al., 1988). Based on this result, a twice-daily regimen can be recommended as a preferable option for pathogens less susceptible to roxithromycin (Dolton and D’Argenio, 2017).
In other cases, the relevance and impact of TK should be weighed with toxicity data from dose range finding and shorter-term bioassays, other relevant data from in vitro and in silico studies, human exposure predictions, and additional mechanistic evidence that suggests a dose-dependent transition in the mode of action (Slikker et al., 2004a, 2004b), for dose selection and response interpretation.
3.1. Shorter-term repeated dose animal studies
Short or subchronic studies—typically ranging in duration from a week to 90 days—have historically been used to inform the dose selection process for chronic/long-term toxicity studies. These studies collect data on a variety of endpoints including, but not limited to, body weights, clinical signs, hematology, clinical chemistry, organ weights, and histopathology. These data, in turn, help identify target organs/systems and critical effects elicited by the compound under evaluation. Based on the effects seen in these studies and using a weight of evidence approach, inferences regarding appropriate top doses for the chronic studies may be made. As an example, clinical observations are conducted daily. If clinical signs of toxicity worsen during the study duration (increased incidence and/or severity), progression of the effect(s) has been established and dose selection for a chronic study would/should take into account these data along with any available additional information, including ADME data. Likewise, if effects (such as hyperplasia or hypertrophy), based on underlying biological processes may be reasonably expected to worsen with longer exposures, are noted during the histopathological examination in a short-term toxicity study, these observations would help guide/inform top dose selection for longer-term studies. Rather than designing long-term studies based solely on these shorter-term studies, use of these studies in a weight of evidence approach provides an opportunity to utilize the results in a more impactful way.
3.2. Dose-dependent transition in the mode of action
Dose-dependent transition in the mode of action may be determined by using genomic studies with the target tissue cell or by performing benchmark dose analysis to estimate the onset of key changes, such as glutathione depletion (Bartels et al., 2020b), receptor activation (McMullen et al., 2020), oxidative stress/inflammation/proliferative signaling, or DNA damage response (Clewell et al., 2014; Klapacz et al., 2016; Paini et al., 2012). To be useful, these in vitro studies would need to be conducted over a wide range of concentrations; the appropriate in vitro concentrations could be selected based on in vivo blood concentrations associated with potential in vivo exposures in the human and experimental animals using PBK (Louisse et al., 2017; Yoon et al., 2012). Preferably, in vitro cell/tissue culture studies from both the experimental animal species and humans would be conducted in parallel. The in vitro concentration at which there is evidence, in the experimental animal, of a dose-dependent transition in the mode of action could then be converted to an administered dose, using quantitative in vitro to in vivo extrapolation (Louisse et al., 2017; Rotroff et al., 2010; Yoon et al., 2012) to inform dose selection in the in vivo animal studies. The same approach can also be applied to estimating human equivalent doses, using human cells and human PK or PBK models (Wetmore, 2015; Yoon et al., 2015). With the move toward AOP-based testing in the field of toxicology and the commitment to support the 3Rs principles, the quantitative relationship that links administered dose, systemic exposure, molecular initiating events, key event responses, and adverse outcomes is paramount to appropriate dose selection or interpretation of repeated dose animal studies.
The following examples demonstrate how TK data can be used to inform study design, including dose selection, when coupled with an understanding of the underlying processes associated with observed nonlinearity.
3.3. Top dose selection considering saturation of absorption
In a 90-day rat study for florpyrauxifen-benzyl at dose levels of 0, 100, 300, and 1000 mg/kg bw/day, no toxicity was observed at dose levels up to the limit dose, 1000 mg/kg bw/day (Murphy et al., 2018). Blood samples were collected toward the end of the 90-day study. The blood concentrations of the acid metabolite were used to calculate the AUC, which was a systemic exposure dose metric relevant to florpyrauxifen-benzyl toxicity. A plot of the log-transformed AUC versus the log-transformed administered dose showed the blood AUCs (last 24 h) did not increase proportionally at administered doses >100 mg/kg bw/day, suggesting saturation of absorption (Fig. 2). Thus, 300 mg/kg bw/day was considered the most relevant top dose for the rat 2-year dietary oncogenicity study, based on no toxicity observed at the limit dose, and absorption was saturated at doses >100 mg/kg bw/day. Dose levels selected for the rat 2-year chronic/oncogenicity study were 0, 10, 50 and 300 mg/kg/day. Also in this study, the acid metabolite was measured in blood at 6 and 12 months, and the AUC versus administered dose indicated saturation of absorption at 300 mg/kg/day and 50 mg/kg/day, respectively.
Fig. 2.
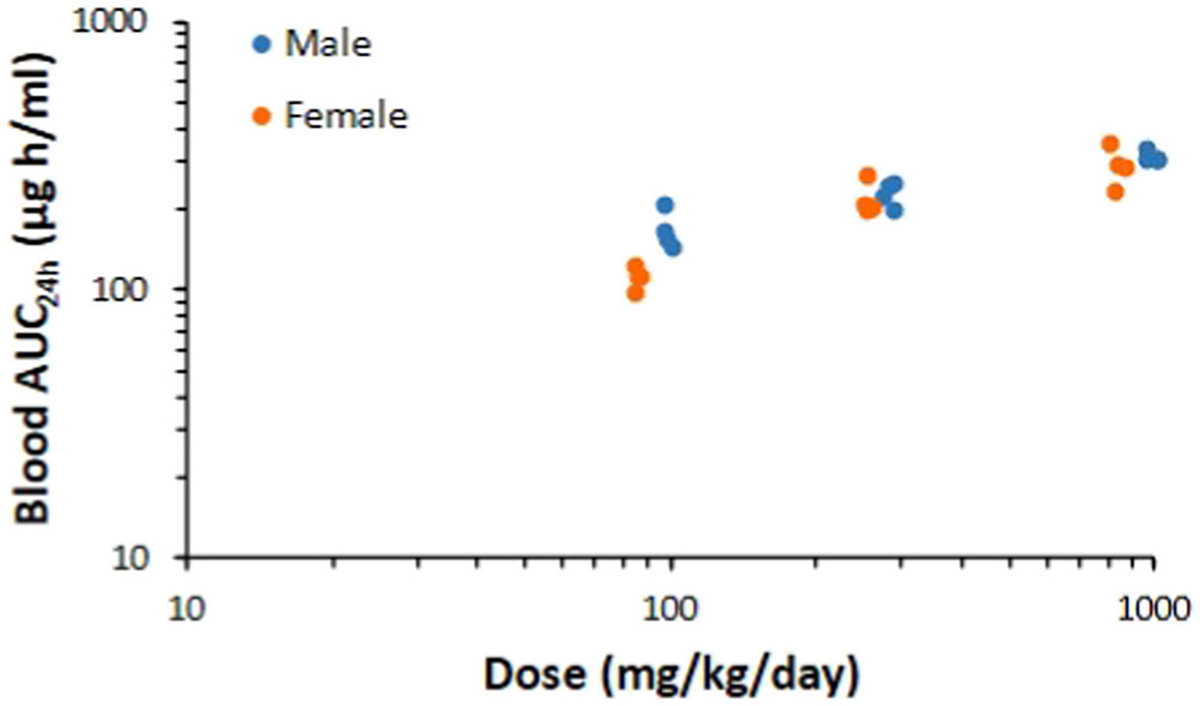
Blood area under the curve (AUC) for a florpyrauxifen-benzyl acid metabolite vs. florpyrauxifen-benzyl intake from a 90-day study in rats. Individual AUC data plotted on a log-log scale for males and females. Data indicate saturation of absorption above 100 mg/kg/day.
If the limit dose had been selected as the top dose for the 2-year study, then dose level spacing would have resulted in all dose levels being above saturation of absorption. This result translates to animals in all dose levels receiving the same systemic dose, which is of no added value in understanding the dose-response relationship and animals would have been wasted. In addition, by selecting the top dose based on the TK saturation in this example, a resultant lower value for the POD at 100 mg/kg/day, rather than 1000 mg/kg/day, was determined for risk assessment purposes (Murphy et al., 2018).
In another example, a 90-day dietary study, male and female mice were exposed to fenpicoxamid in the diet at 0, 300, 1,500, 3,000, 6000 (females only), and 9000 (males only) ppm. Fenpicoxamid is extensively metabolized with three measurable metabolites. Liver effects observed in both males and females consisted of hepatocellular hypertrophy (males at 1500 ppm, and females at 3000 ppm) with very slight necrosis at 1500 ppm observed in males only. Blood samples were collected during the last week of study, and blood levels for one of the metabolites indicated saturation of absorption in both males and females (Fig. 3). The 90-day NOAEL was 300 ppm for both males and females. A weight of evidence approach considering apical endpoints and TK was used to set the top dose level in the 18-month mouse study, with 1500 ppm in the diet as the top dose for males and 3000 ppm in females. Dietary concentrations in the 18-month mouse study were 0, 50, 300, and 1500 ppm in males and 0, 50, 300, and 3000 ppm for females. During the 18-month study, blood AUCs of metabolites exhibited sublinear behavior with saturation of absorption at 300 and 3000 ppm. There were no treatment-related neoplasms in the 18-month mouse study, even though there were treatment-related adverse liver effects with a resultant NOAEL of 300 ppm for male and female mice.
Fig. 3.

Blood concentration of a fenpicoxamid metabolite vs. fenpicoxamid intake from a 90-day study in mice. Individual animal data plotted on a log-log scale. Data indicate saturation of absorption with increasing dose administered. Doses converted from ppm to mg/kg/day with dose group averages for males: 33, 161, 361, and 1000 mg/kg/day; and females: 46, 275, 470, and 995 mg/kg/day.
3.4. Top dose selection considering saturable of elimination and saturable of absorption
A 90-day mouse study was conducted at dietary concentrations of sulfoxaflor at 0, 100, 750, and 1250 ppm in males and 0, 100, 1,500, and 3000 ppm in females. Sulfoxaflor is not metabolized; therefore, parent is the measured biomarker. These selected dietary concentrations for the 90-day study were based on a previous 28-day dietary mouse study. With sulfoxaflor, there was apparent saturation of elimination observed above the mid-dose of 750 ppm (92.29 mg/kg/day) in males and apparent saturation of absorption was observed above the mid-dose of 1500 ppm (226.54 mg/kg/day) in females (Terry et al., 2016, 2020) (Fig. 4). In the 90-day study, treatment-related effects were observed at the mid and high dose levels (1250 ppm in males and 3000 ppm for females), such as increased liver weights, hepatocyte hypertrophy, and lower kidney weights in males only. Signs of toxicity were observed while systemic exposure was in the linear range. These toxicity findings and TK data were used in a weight of evidence approach to set the top dose for the subsequent 18-month mouse carcinogenicity study (Terry et al., 2016). The dietary concentrations selected for the 18-month study were 0, 25, 100, and 750 ppm in males and 0, 25, 250, and 1250 ppm in females. During the 18-month study, systemic exposure was proportional to administered dose. Even though TK data contributed to the dose selection process for the 18-month study, hepatocellular adenomas and carcinomas were observed in males and females at 750 and 1250 ppm, respectively. In other words, the doses selected based on the weight of evidence approach were still high enough to result in toxicity in the 18-month study. The resultant NOEL was 100 ppm in males and 25 ppm in female mice. The NOAEL in female mice was 250 ppm. Findings of liver adenomas and carcinomas from the 18-month mouse study were followed with mode of action studies to demonstrated that the mouse tumors were not relevant to humans. Indeed, constitutive androstane receptor/pregnane X receptor activation was responsible for liver effects in the mouse, and this is not a relevant mode of action in humans.
Fig. 4.

Plasma concentration (ug/g) vs. sulfoxaflor intake in mg/kg bw/day from a 90-day dietary study in mice. Individual animal data plotted on a log-log scale for males and females. Data indicate saturation of clearance with increasing dose administered in males and saturation of absorption with increasing dose administered in females.
For each of the three examples above, the top dose selected for the longer-term study is 4–5 orders of magnitude higher than the estimate human exposures (Fig. 5). Noteworthy is that the top dose selected for the long-term study was only 2- to 3-fold different from the top dose in the shorter-term study. The above examples demonstrate that a weight of evidence approach is not in conflict with the conventional MTD or limit dose-based testing. In some cases, a MTD or the limit dose may be the most appropriate top dose. In other cases, considering TK nonlinearity with apical endpoints may provide a scientifically defensible biological basis for selecting a lower top dose than a MTD or the limit dose, thus reducing unnecessary animal use and suffering, while still being health protective and better predictive of human health risks.
Fig. 5.

The high dose in mg/kg/bw (mkd) used in a 90-day study, the high dose selected in the chronic/oncogenicity studies with the NOAELs as compared to the acceptable daily intake (ADI) and estimated human dietary exposures for florpyrauxifen-benzyl, fenpicoxamid, and sulfoxaflor. ADI and estimated human exposure data form EFSA conclusions (EFSA, 2014; EFSA, 2018a; EFSA, 2018b).
We recommend using a weight of evidence approach to inform dose selection in repeated dose animal toxicity studies by collectively evaluating data obtained from exposure assessment, in vitro and in silico studies, TK studies and TK modeling, and short-term in vivo studies. We also recommend not using the term “KMD” to represent the consideration of TK nonlinearity in a weight of evidence approach. In addition, the term “KMD” implies an administered dose determined based on data on TK nonlinearity alone, whether it is due to saturation of absorption or saturation of clearance. Such definition contradicts our recommendation of a weight of evidence approach, so we recommend to not use the term “KMD.”
4. Conclusions
Based on the findings presented here, recommendations are as follows.
First, the observed dose-response relationship in animal toxicity studies results from the combined effects of TK and TD processes. Characterizing interspecies differences in TK and TD provides a stronger biological basis for conducting extrapolations and comparing responses across studies, species, exposure routes, and dose levels.
Second, saturation of ADME processes can profoundly affect the design and interpretation of animal toxicity tests in two quite different ways. The first is that saturation of absorption, through capacity-limited absorption or nonlinear bioaccessibility, will result in plateauing of systemic exposure regardless of increasing administered dose above a certain level (Fig. 2). A similar effect can occur when toxicity is due to formation of a metabolite, and formation is capacity-limited within the experimental dose range (Fig. 1). The consequence is that no additional information on the effect of dosing is obtained above saturation, as systemic/target tissue exposure no longer increases with increasing administered dose. Not only is no useful information obtained from these dose groups, but their use is not ethically justifiable. The second situation is saturation of the clearance of the active moiety. This process may lead to dose-dependent transitions of mode of action, which are of no relevance at the much lower levels of exposure occurring in exposed populations. The result may be substantial overprediction of risk. However, the data may be relevant if saturation of clearance can occur at human exposure levels or if the mode of action is the same at all doses, and hence a weight of evidence approach is necessary, before discounting such information.
Third, the lack of consensus on how a KMD should be estimated or when such an approach is appropriate may be due to different interpretations of the KMD definition and applications. The term “KMD” implies an administered dose that is determined based on TK nonlinearity alone, whether it is due to saturation of absorption or saturation of clearance.
Fourth, rather than redefining the KMD, we recommend using a weight of evidence approach, which considers data demonstrating saturation or limitation of TK mechanism(s) in combination with hazard data from shorter term and range finding studies and human exposure information, to inform dose selection for repeated dose animal toxicity studies or interpreting dose-response findings from these studies.
Fifth, a weight of evidence approach may result in the conventional MTD or the limit dose as a top dose, or a top dose lower than a MTD or the limit dose. A good practice is to justify, in the study report, the dose selection process and rationale. A scientifically defensible top dose will have animal welfare benefits, be health protective, and be better predictive of human health risks.
Sixth, regardless of which dose selection method is used, a top dose in a repeated dose animal toxicity study is typically several orders of magnitude higher than anticipated human exposure levels. Many exposure data resources and approaches are available to estimate human exposure levels, which is an additional line of evidence to be considered when designing repeated dose animal toxicity studies.
Seventh, TK data can be reliably collected without additional satellite animal groups. During dose range finding and shorter-term toxicity studies, in vivo TK data may be collected to increase interpretability of results from these studies. In addition, advances in computational modeling and in vitro approaches can aid in identifying doses where one or more TK mechanisms saturate, or where there is a dose-dependent transition in the mode of action. A follow-up manuscript specifically focused on these in vitro methods and their application to inform study design is being developed by this working group.
Eighth, the internal dose metric, or its surrogate, that is most closely related to toxicity responses should be used to understand the impact of TK nonlinearity on dose-response relationship.
Finally, it is acknowledged that knowledge related to TK nonlinearity is important for some, but not all, risk assessment purposes. Investigators should consider the regulatory requirements for the data being generated and how these data are used to meet different risk assessment purposes across regulatory organizations.
Funding
This work was supported in part by the Health and Environmental Sciences Institute PBPK Committee. It is recognized via a Memorandum of Understanding between the USEPA and HESI that outlines joint commitments to a multisector and multidisciplinary HESI working group on PBPK modeling. We acknowledge the committee members for their support and helpful feedback on development of this document.
Abbreviations
- 3Rs
reduction, replacement, and refinement
- ADME
absorption, distribution, metabolism, and excretion
- AOP
adverse outcome pathway
- AUC
area under the curve
- CEHD
Chemical Exposure Health Data
- ChemSTEER
Chemical Screening Tool for Exposures and Environmental Releases
- HESI
Health and Environmental Sciences Institute
- ICH
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use
- KMD
kinetically derived maximum dose
- LOAEL
lowest-observed-adverse-effect level
- MTD
maximum tolerated dose
- NHANES
National Health and Nutrition Examination Survey
- NOAEL
no-observed-adverse-effect level
- OECD
Organisation for Economic Co-Operation and Development
- ORD
Office of Research and Development
- PBK
physiologically based kinetic
- POD
point of departure
- PPE
personal protective equipment
- REACH
Registration, Evaluation, Authorisation and Restriction of Chemicals
- SEEM2
Systematic Empirical Evaluations of Models, version 2
- TD
toxicodynamics
- TK
toxicokinetics
- USEPA
U.S. Environmental Protection Agency
Footnotes
Declaration of competing interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: Three of the case studies presented in this paper are based on chemicals manufactured by Corteva, with whom Jean Domoradzki is employed. The other authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
Yu-Mei Tan: Conceptualization, Methodology, Writing – original draft, Writing – review & editing, Formal analysis. Hugh A. Barton: Conceptualization, Methodology, Writing – original draft, Writing – review & editing. Alan Boobis: Conceptualization, Writing – original draft, Writing – review & editing. Rachel Brunner: Investigation, Writing – original draft, Visualization. Harvey Clewell: Conceptualization, Methodology, Writing – original draft, Writing – review & editing. Rhian Cope: Conceptualization, Writing – review & editing. Jeffrey Dawson: Methodology, Formal analysis, Investigation, Data curation, Writing – original draft, Writing – review & editing. Jeanne Domoradzki: Conceptualization, Methodology, Writing – original draft, Writing – review & editing, Formal analysis. Peter Egeghy: Validation, Writing – review & editing. Pankaj Gulati: Writing – review & editing. Brandall Ingle: Investigation, Writing – original draft. Nicole Kleinstreuer: Conceptualization, Methodology, Writing – review & editing. Kelly Lowe: Methodology, Formal analysis, Investigation, Data curation, Writing – original draft, Writing – review & editing, Visualization, Project administration. Anna Lowit: Conceptualization, Methodology, Writing – review & editing. Elizabeth Mendez: Conceptualization, Writing – original draft, Writing – review & editing. David Miller: Methodology, Writing – review & editing. Jeffrey Minucci: Methodology, Formal analysis, Investigation, Writing – original draft, Writing – review & editing. James Nguyen: Methodology, Formal analysis, Investigation, Writing – original draft. Alicia Paini: Conceptualization, Methodology, Writing – original draft, Writing – review & editing, Formal analysis. Monique Perron: Conceptualization, Writing – review & editing. Katherine Phillips: Methodology, Formal analysis, Investigation, Writing – original draft, Writing – review & editing, Visualization. Hua Qian: Conceptualization, Writing – review & editing. Tharacad Ramanarayanan: Conceptualization, Writing – review & editing. Fiona Sewell: Conceptualization, Writing – original draft, Writing – review & editing. Philip Villanueva: Methodology, Writing – original draft, Writing – review & editing. John Wambaugh: Methodology, Investigation, Writing – original draft, Writing – review & editing. Michelle Embry: Conceptualization, Writing – original draft, Writing – review & editing, Supervision, Project administration.
An oral dose of 1000 mg/kg body weight/day as a default maximum dose in repeated dose systemic toxicity studies (OECD Guideline 407, 1995) (OECD, 1995/2008). For acute oral studies, the limit dose may be 2000 mg/kg or 5000 mg/kg (OECD Guideline 425, 2001) (OECD, 2001/2008). The focus of this article is only on repeated dose studies.
OECD Guidance Notes on Chronic/Carcinogenicity Testing No. 35: the maximum tolerated dose is defined as the highest dose to produce toxic effects without causing death and to decrease body weight by no more than 10% relative to controls (OECD, 2002. Guidance Notes for Analysis and Evaluation of Chronic Toxicity and Carcinogenicity Studies.).
A recording of the symposium, presenters’ slides and abstracts, and some Q&A are available at https://ntp.niehs.nih.gov/go/kmd-2020.
Vilfredo Pareto (1848–1923) observed that 20% of the income in Italy was received by 80% of the Italian population; and that 20% of the population owned 80% of the property. Joseph Juran (1904–2008) called the ‘80–20 rule’ the Pareto principle. The principle states that roughly 80% of consequences come from 20% of causes.
References
- Acocella G, 1978. Clinical pharmacokinetics of rifampicin. Clin. Pharmacokinet 3, 108–127. [DOI] [PubMed] [Google Scholar]
- Acocella G, et al. , 1971. Kinetic studies on rifampicin. Chemotherapy 16, 356–370. [DOI] [PubMed] [Google Scholar]
- Andrews JM, et al. , 1987. Factors affecting the in-vitro activity of roxithromycin. J. Antimicrob. Chemother 20 (Suppl. B), 31–37. [DOI] [PubMed] [Google Scholar]
- Ball N, et al. , 2020. Key read across framework components and biology based improvements. Mutat. Res. Genet. Toxicol. Environ. Mutagen 853, 503172. [DOI] [PubMed] [Google Scholar]
- Bartels M, et al. , 2020a. Review of the pharmacokinetics and metabolism of triclopyr herbicide in mammals: impact on safety assessments. Regul. Toxicol. Pharmacol 116, 104714. [DOI] [PubMed] [Google Scholar]
- Bartels MJ, et al. , 2020b. Metabolic basis for nonlinearity in 1,3-dichloropropene toxicokinetics and use in setting a kinetically-derived maximum inhalation exposure concentration in mice. Toxicol. Sci 174, 16–24. [DOI] [PubMed] [Google Scholar]
- Barton HA, et al. , 2006. The acquisition and application of absorption, distribution, metabolism, and excretion (ADME) data in agricultural chemical safety assessments. Crit. Rev. Toxicol 36, 9–35. [DOI] [PubMed] [Google Scholar]
- Borghoff SJ, et al. , 2016. Dose- and time-dependent changes in tissue levels of tetrabromobisphenol A (TBBPA) and its sulfate and glucuronide conjugates following repeated administration to female Wistar Han Rats. Toxicology Reports 3, 190–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmichael NG, et al. , 2006. Agricultural chemical safety assessment: a multisector approach to the modernization of human safety requirements. Crit. Rev. Toxicol 36, 1–7. [DOI] [PubMed] [Google Scholar]
- Caron A, et al. , 2015. Clinical and anatomic pathology effects of serial blood sampling in rat toxicology studies, using conventional or microsampling methods. Regul. Toxicol. Pharmacol 72, 429–439. [DOI] [PubMed] [Google Scholar]
- Chapman K, et al. , 2014. Reducing pre-clinical blood volumes for toxicokinetics: toxicologists, pathologists and bioanalysts unite. Bioanalysis 6, 2965–2968. [DOI] [PubMed] [Google Scholar]
- Chilakapati J, et al. , 2005. Saturation toxicokinetics of thioacetamide: role in initiation of liver injury. Drug Metab. Dispos 33, 1877–1885. [DOI] [PubMed] [Google Scholar]
- Chirehwa MT, et al. , 2016. Model-based evaluation of higher doses of rifampin using a semimechanistic model incorporating autoinduction and saturation of hepatic extraction. Antimicrob. Agents Chemother 60, 487–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clewell RA, et al. , 2014. Profiling dose-dependent activation of p53-mediated signaling pathways by chemicals with distinct mechanisms of DNA damage. Toxicol. Sci 142, 56–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creton S, et al. , 2012. Use of toxicokinetics to support chemical evaluation: informing high dose selection and study interpretation. Regul. Toxicol. Pharmacol 62, 241–247. [DOI] [PubMed] [Google Scholar]
- Dawson DE, et al. , 2021. Designing QSARs for parameters of high-throughput toxicokinetic models using open-source descriptors. Environ. Sci. Technol 55, 6505–6517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dellarco M, et al. , 2017. Using exposure bands for rapid decision making in the RISK21 tiered exposure assessment. Crit. Rev. Toxicol 47, 317–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolton MJ, D’Argenio DZ, 2017. Population-based meta-analysis of roxithromycin pharmacokinetics: dosing implications of saturable absorption and protein binding. J. Antimicrob. Chemother 72, 1129–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ECHA, 2017. Guidance on Information Requirements and Chemical Safety Assessment, Chapter R.7c: Endpoint Specific Guidance. European Chemicals Agency. [Google Scholar]
- EFSA, 2014. Conclusion on the peer review of the pesticide risk assessment of the active substance sulfoxaflor. EFSA Journal 12. [Google Scholar]
- EFSA, 2018a. Conclusion on the peer review of the pesticide risk assessment of the active substance fenpicoxamid (XDE-777). EFSA Journal 16, 5146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA, 2018b. Conclusion on the peer review of the pesticide risk assessment of the active substance florpyrauxifen (variant assessed florpyrauxifen-benzyl). EFSA Journal 16, 5378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee, 2017. Guidance on the use of the weight of evidence approach in scientific assessments. EFSA J 15, e04971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EMA, 2017. ICH Guideline S3A: Note for Guidance on Toxicokinetics: the Assessment of Systemic Exposure in Toxicity Studies - Questions and Answers. Committee for Human Medicinal Products. EMA/CHMP/ICH/320985/2016, London, UK. [Google Scholar]
- EMA, 2020. ICH Guideline S11 on Nonclinical Safety Testing in Support of Development of Paediatric Pharmaceuticals. EMA/CHMP/ICH/616110/2018, Amsterdam, NL. [Google Scholar]
- European Commission, 2021. Endocrine Disruptors.
- European Union, 2009. Regulation (EC) No 1107/2009 of the European parliament and of the council of 21 October 2009 concerning the placing of plant protection products on the market and repealing council directives 79/117/EEC and 91/414/EEC. Off. J. Eur. Union 52, 1–50. [Google Scholar]
- European Union, 2013. Commission Regulation (EU) No 283/2013 of 1 March 2013: setting out the data requirements for active substances. In: Accordance with Regulation (EC) No 1107/2009 of the European Parliament and of the Council Concerning the Placing of Plant Protection Products on the Market. Off. J. Eur. Union. OJ L 93. [Google Scholar]
- Foran JA, 1997. Principles for the Selection of Doses in Chronic Rodent Bioassays. ILSI Risk Science Working Group on Dose Selection, vol. 105. Environmental health perspectives, pp. 18–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzosa JA, et al. , 2021. High-throughput toxicogenomic screening of chemicals in the environment using metabolically competent hepatic cell cultures. NPJ Syst Biol Appl 7, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansch C, et al. , 2004. QSAR and ADME. Bioorg. Med. Chem 12, 3391–3400. [DOI] [PubMed] [Google Scholar]
- Henderson CJ, et al. , 2000. Increased resistance to acetaminophen hepatotoxicity in mice lacking glutathione S-transferase Pi. Proc. Natl. Acad. Sci. U. S. A 97, 12741–12745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heringa MB, et al. , 2020. Use of the kinetically-derived maximum dose concept in selection of top doses for toxicity studies hampers proper hazard assessment and risk management. Regul. Toxicol. Pharmacol 114, 104659. [DOI] [PubMed] [Google Scholar]
- HESI, 2001. Developing Strategies for Agricultural Chemical Safety Evaluation: a Report from April 22–23, 2001 Workshop. ILSI HESI, Washington, DC. [Google Scholar]
- ICH, 1994/2008. Dose Selection for Carcinogenicity Studies S1C(R2).
- ICH, 1995a. Guideline on the Need for Carcinogenicity Studies of Pharmaceuticals S1A.
- ICH, 1995b. ICH Topic S3A Toxicokinetics: A Guidance for Assessing Systemic Exposure in Toxicology Studies.
- ICH, 1997. ICH Guideline: Testing for Carcinogenicity of Pharmaceuticals S1B. [Google Scholar]
- ICH, 2020. ICH Harmonised Guideline: Detection of Reproductive and Developmental Toxicity for Human Pharmaceuticals. S5(R3).
- Ingle B, et al. , 2016. Toxicity Challenges in Environmental Chemicals: Prediction of Human Plasma Protein Binding through Quantitative Structure-Activity Relationship (QSAR) Models. IVIE Workshop RTP, Research Triangle Park, NC, USA. [Google Scholar]
- Irwin I, Langston JW, 1985. Selective accumulation of MPP+ in the substantia nigra: a key to neurotoxicity? Life Sci. 36, 207–212. [DOI] [PubMed] [Google Scholar]
- Ito K, et al. , 2002. Which concentration of the inhibitor should be used to predict in vivo drug interactions from in vitro data? AAPS PharmSci 4, E25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jollow DJ, et al. , 1974. Acetaminophen-induced hepatic necrosis. VI. Metabolic disposition of toxic and nontoxic doses of acetaminophen. Pharmacology 12, 251–271. [DOI] [PubMed] [Google Scholar]
- Klapacz J, et al. , 2016. Contributions of DNA repair and damage response pathways to the non-linear genotoxic responses of alkylating agents. Mutat. Res. Rev. Mutat. Res 767, 77–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laroche C, et al. , 2018. Finding synergies for 3Rs – toxicokinetics and read-across: report from an EPAA partners’ forum. Regul. Toxicol. Pharmacol 99, 5–21. [DOI] [PubMed] [Google Scholar]
- Lassman HB, et al. , 1988. Pharmacokinetics of roxithromycin (RU 965). J. Clin. Pharmacol 28, 141–152. [DOI] [PubMed] [Google Scholar]
- Lehman PA, et al. , 2011. Percutaneous absorption in man: in vitro-in vivo correlation. Skin Pharmacol. Physiol 24, 224–230. [DOI] [PubMed] [Google Scholar]
- Loccisano AE, et al. , 2020. Use of toxicokinetic data for afidopyropen to determine the dose levels in developmental toxicity studies. Regul. Toxicol. Pharmacol 113, 104644. [DOI] [PubMed] [Google Scholar]
- Louisse J, et al. , 2017. Use of physiologically based kinetic modeling-based reverse dosimetry to predict in vivo toxicity from in vitro data. Chem. Res. Toxicol 30, 114–125. [DOI] [PubMed] [Google Scholar]
- Lowe K, et al. , 2021. Exposure Considerations Related to the Use of Toxicokinetic Data to Inform Top Dose Selection in Repeated-Dose Toxicity Studies. Regulatory Toxicology & Pharmacology. In press. [Google Scholar]
- Mangipudy RS, et al. , 1998. Dose-dependent modulation of cell death: apoptosis versus necrosis in thioacetamide hepatotoxicity. Int. J. Toxicol 17, 193–211. [Google Scholar]
- McFadden LG, et al. , 2012. Statistical methodology to determine kinetically derived maximum tolerated dose in repeat dose toxicity studies. Regul. Toxicol. Pharmacol 63, 344–351. [DOI] [PubMed] [Google Scholar]
- McMullen PD, et al. , 2020. Identifying qualitative differences in PPARα signaling networks in human and rat hepatocytes and their significance for next generation chemical risk assessment methods. Toxicol. Vitro 64, 104463. [DOI] [PubMed] [Google Scholar]
- Murphy LA, et al. , 2018. The Future of Agrochemical Risk Assessment: Benefits, Challenges, and the Evolution of Integrated Endpoints for the Rat 90-Day Toxicity Study. The Toxicologist, vol. 162. Oxford Press, San Antonio, TX, USA. [Google Scholar]
- National Research Council, 1983. Risk Assessment in the Federal Government: Managing the Process. National Academies Press, Washington, DC. [PubMed] [Google Scholar]
- Obach RS, et al. , 1997. The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J. Pharmacol. Exp. Therapeut 283, 46–58. [PubMed] [Google Scholar]
- Obach RS, et al. , 2008. Trend analysis of a database of intravenous pharmacokinetic parameters in humans for 670 drug compounds. Drug Metab. Dispos 36, 1385–1405. [DOI] [PubMed] [Google Scholar]
- OECD, 1998. Test No. 409: Repeated Dose 90-Day Oral Toxicity Study in Non-rodents.
- OECD, 2002. Guidance Notes for Analysis and Evaluation of Chronic Toxicity and Carcinogenicity Studies.
- OECD, 2007. Test No. 426. Developmental Neurotoxicity Study.
- OECD, 2010. Test No. 417. Toxicokinetics.
- OECD, 2012. Guidance Document 116 on the Conduct and Design of Chronic Toxicity and Carcinogenicity Studies, Support Test Guidelines 451, 452 and 453, second ed. OECD Environmental, Health and Safety Publications, Series on Testing and Assessment.
- OECD, 2012/2014. Guidance document 116 on the conduct and design of chronic toxicity and carcinogenicity studies. Supporting Test Guidelines 451, 452–453 second ed.
- OECD, 2013. Guidance Document Supporting OECD Test Guideline 443 on the Extended One-Generation Reproductive Toxicity Test. Series on Testing and Assessment. No. 151.
- OECD, 2018a. Extended One-Generation Reproductive Toxicity Study (EOGRTS) (OECD TG 443). OECD Publishing, Paris. [Google Scholar]
- OECD, 2018b. Test No. 451: Carcinogenicity Studies.
- OECD, 2018c. Test No. 453: Combined Chronic Toxicity/Carcinogenicity Studies.
- OECD, 2021. In: Environment, H.a.S., Environment Directorate (Eds.), Guidance Document on the Characterization, Validation, and Reporting of Physiologically Based Kinetic (PBK) Models for Regulatory Purposes. Paris, France. [Google Scholar]
- Paini A, et al. , 2012. In vivo validation of DNA adduct formation by estragole in rats predicted by physiologically based biodynamic modelling. Mutagenesis 27, 653–663. [DOI] [PubMed] [Google Scholar]
- Paini A, et al. , 2021. Assessment of the predictive capacity of a physiologically based kinetic model using a read-across approach. Computational Toxicology 18, 100159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul Friedman K, et al. , 2020. Utility of in vitro bioactivity as a lower bound estimate of in vivo adverse effect levels and in risk-based prioritization. Toxicol. Sci 173, 202–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potts RO, Guy RH, 1992. Predicting skin permeability. Pharmaceut. Res 9, 663–669. [DOI] [PubMed] [Google Scholar]
- Poulin P, Krishnan K, 1996a. A mechanistic algorithm for predicting blood:air partition coefficients of organic chemicals with the consideration of reversible binding in hemoglobin. Toxicol. Appl. Pharmacol 136, 131–137. [DOI] [PubMed] [Google Scholar]
- Poulin P, Krishnan K, 1996b. A tissue composition-based algorithm for predicting tissue:air partition coefficients of organic chemicals. Toxicol. Appl. Pharmacol 136, 126–130. [DOI] [PubMed] [Google Scholar]
- Poulin P, Theil FP, 2000. A priori prediction of tissue:plasma partition coefficients of drugs to facilitate the use of physiologically-based pharmacokinetic models in drug discovery. J. Pharmacol. Sci 89, 16–35. [DOI] [PubMed] [Google Scholar]
- Powles-Glover N, et al. , 2014. Assessment of haematological and clinical pathology effects of blood microsampling in suckling and weaned juvenile rats. Regul. Toxicol. Pharmacol 69, 425–433. [DOI] [PubMed] [Google Scholar]
- Price P, 2020. Interindividual variation in source-specific doses is a determinant of health impacts of combined chemical exposures. Risk Anal. 40, 2572–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaiah SK, et al. , 1998. Diet restriction enhances compensatory liver tissue repair and survival following administration of lethal dose of thioacetamide. Toxicol. Appl. Pharmacol. 150, 12–21. [DOI] [PubMed] [Google Scholar]
- Rhomberg LR, et al. , 2007. Issues in the design and interpretation of chronic toxicity and carcinogenicity studies in rodents: approaches to dose selection. Crit. Rev. Toxicol 37, 729–837. [DOI] [PubMed] [Google Scholar]
- Rotroff DM, et al. , 2010. Incorporating human dosimetry and exposure into high-throughput in vitro toxicity screening. Toxicol. Sci 117, 348–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saghir SA, 2015. Rethinking guideline toxicity testing. Regul. Toxicol. Pharmacol 72, 423–428. [DOI] [PubMed] [Google Scholar]
- Saghir SA, et al. , 2012. Assessment of diurnal systemic dose of agrochemicals in regulatory toxicity testing–an integrated approach without additional animal use. Regul. Toxicol. Pharmacol 63, 321–332. [DOI] [PubMed] [Google Scholar]
- Saghir SA, et al. , 2013. Life-stage-, sex-, and dose-dependent dietary toxicokinetics and relationship to toxicity of 2,4-dichlorophenoxyacetic acid (2,4-D) in rats: implications for toxicity test dose selection, design, and interpretation. Toxicol. Sci 136, 294–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saghir SA, et al. , 2006. Strategies to assess systemic exposure of chemicals in subchronic/chronic diet and drinking water studies. Toxicol. Appl. Pharmacol 211, 245–260. [DOI] [PubMed] [Google Scholar]
- Sarigiannis D, et al. , 2017. Development of QSARs for parameterizing physiology based ToxicoKinetic models. Food Chem. Toxicol 106, 114–124. [DOI] [PubMed] [Google Scholar]
- Schmitt W, 2008. General approach for the calculation of tissue to plasma partition coefficients. Toxicol. Vitro 22, 457–467. [DOI] [PubMed] [Google Scholar]
- Sewell F, et al. , 2020. Use of the kinetically-derived maximum dose: opportunities for delivering 3Rs benefits. Regul. Toxicol. Pharmacol 116, 104734. [DOI] [PubMed] [Google Scholar]
- Slikker W Jr., et al. , 2004a. Dose-dependent transitions in mechanisms of toxicity. Toxicol. Appl. Pharmacol 201, 203–225. [DOI] [PubMed] [Google Scholar]
- Slikker W Jr., et al. , 2004b. Dose-dependent transitions in mechanisms of toxicity: case studies. Toxicol. Appl. Pharmacol 201, 226–294. [DOI] [PubMed] [Google Scholar]
- Smith SA, Waters NJ, 2018. Pharmacokinetic and pharmacodynamic considerations for drugs binding to alpha-1-acid glycoprotein. Pharm. Res. (N. Y.) 36, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson RJ, et al. , 2018. A population pharmacokinetic model incorporating saturable pharmacokinetics and autoinduction for high rifampicin doses. Clin. Pharmacol. Ther 103, 674–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry C, et al. , 2020. Letter to the editor regarding Heringa et al. (2020) paper entitled “Use of the Kinetically-derived Maximum Dose concept in selection of top doses for toxicity studies hampers proper hazard assessment and risk management. Regul. Toxicol. Pharmacol 117, 104765. [DOI] [PubMed] [Google Scholar]
- Terry C, et al. , 2016. Implementing a framework for integrating toxicokinetics into human health risk assessment for agrochemicals. Regul. Toxicol. Pharmacol 75, 89–104. [DOI] [PubMed] [Google Scholar]
- Terry C, et al. , 2014. Application of a novel integrated toxicity testing strategy incorporating “3R” principles of animal research to evaluate the safety of a new agrochemical sulfoxaflor. Crit. Rev. Toxicol 44 (Suppl. 2), 1–14. [DOI] [PubMed] [Google Scholar]
- Thompson RA, et al. , 2016. Reactive metabolites: current and emerging risk and hazard assessments. Chem. Res. Toxicol 29, 505–533. [DOI] [PubMed] [Google Scholar]
- Tremblay D, et al. , 1988. Pharmacokinetics of three single doses (150, 300, 450 mg) of roxithromycin in young volunteers. Br. J. Clin. Pract 42, 49–50.3052558 [Google Scholar]
- USEPA, 2003. Interim Guidance Document #G2003.02: Rodent Carcinogenicity Studies: Dose Selection and Evaluation. U.S. Environmental Protection Agency, Washington, DC. [Google Scholar]
- USEPA, 2005. Guidelines for Carcinogen Risk Assessment. EPA/630/P-03/001F.
- USEPA, 2006. Approaches for the Application of Physiologically Based Pharmacokinetic (PBPK) Models and Supporting Data in Risk Assessment (Final Report). U.S. Environmental Protection Agency, Washington, D.C.. EPA/600/R-05/043F, 2006. U. S. Environmental Protection Agency, Washington, DC. [Google Scholar]
- USEPA, 2021a. ChemSTEER: Chemical Screening Tool for Exposures and Environmental Releases.
- USEPA, 2021b. Occupational Pesticide Handler Exposure Data.
- USEPA, 2021c. Standard Operating Procedures for Residential Pesticide Exposure Assessment.
- Van Cott A, et al. , 2018. Uterine adenocarcinoma in the rat induced by afidopyropen. An analysis of the lesion’s induction, progression and its relevance to humans. Regul. Toxicol. Pharmacol. 95, 29–51. [DOI] [PubMed] [Google Scholar]
- Wambaugh JF, et al. , 2019. New approach methodologies for exposure science. Curr. Opin. Toxicol 15, 76–92. [Google Scholar]
- Wetmore BA, 2015. Quantitative in vitro-to-in vivo extrapolation in a high-throughput environment. Toxicology 332, 94–101. [DOI] [PubMed] [Google Scholar]
- Wetmore BA, et al. , 2012. Integration of dosimetry, exposure, and high-throughput screening data in chemical toxicity assessment. Toxicol. Sci 125, 157–174. [DOI] [PubMed] [Google Scholar]
- Woutersen M, et al. , 2020. Regulating human safety: how dose selection in toxicity studies impacts human health hazard assessment and subsequent risk management options. Regul. Toxicol. Pharmacol 114, 104660. [DOI] [PubMed] [Google Scholar]
- Yoon M, et al. , 2012. Quantitative in vitro to in vivo extrapolation of cell-based toxicity assay results. Crit. Rev. Toxicol 42, 633–652. [DOI] [PubMed] [Google Scholar]
- Yoon M, et al. , 2015. Use of in vitro data in developing a physiologically based pharmacokinetic model: carbaryl as a case study. Toxicology 332, 52–66. [DOI] [PubMed] [Google Scholar]
- Zhao YH, et al. , 2003. Evaluation of rat intestinal absorption data and correlation with human intestinal absorption. Eur. J. Med. Chem 38, 233–243. [DOI] [PubMed] [Google Scholar]
- Zini R, et al. , 1988. In vitro study of roxithromycin binding to serum proteins and erythrocytes in humans. Br. J. Clin. Pract 42, 54. [Google Scholar]