

Abstract
Heart failure with preserved ejection fraction (HFpEF) is increasing in prevalence worldwide, already accounting for at least half of all heart failure (HF). As most patients with HFpEF are obese with metabolic syndrome, metabolic stress has been implicated in syndrome pathogenesis. Recently, compelling evidence for bidirectional crosstalk between metabolic stress and chronic inflammation has emerged, and alterations in systemic and cardiac immune responses are held to participate in HFpEF pathophysiology. Indeed, based on both preclinical and clinical evidence, comorbidity-driven systemic inflammation, coupled with metabolic stress, have been implicated together in HFpEF pathogenesis. As metabolic alterations impact immune function(s) in HFpEF, major changes in immune cell metabolism are also recognized in HFpEF and in HFpEF-predisposing conditions. Both arms of immunity – innate and adaptive – are implicated in the cardiomyocyte response in HFpEF. Indeed, we submit that crosstalk among adipose tissue, the immune system, and the heart represents a critical component of HFpEF pathobiology. Here, we review recent evidence in support of immunometabolic mechanisms as drivers of HFpEF pathogenesis, discuss pivotal biological mechanisms underlying the syndrome, and highlight questions requiring additional inquiry.
Keywords: HFpEF, immune system, metabolism
Introduction
Recent decades have been marked by robust success in reducing the acutely lethal, typically atherothrombotic, manifestations of cardiovascular disease1,2. Clinically impactful therapies and interventions, coupled with public health efforts focusing on primary prevention, have culminated in meaningful improvements in outcomes. Owing to successes seen in much of the world, people are surviving their myocardial infarctions and ventricular tachyarrhythmias, returning to family and society with one of the major manifestations of chronic cardiovascular disease, HF.
Concomitant with these advances, modern society has witnessed dramatic increases in obesity, metabolic dysfunction, diabetes, and hypertension3. For example, it is estimated that over 40% of the US population is obese3,4 with an increase in obesity prevalence from 30.5% to 42.4% in just the last 8 years. And other parts of the world are not far behind. Strikingly, the incidences of obesity and diabetes stratified by age, sex, or ethnicity are each projected to continue to increase in the next decade3,5. It is hard to underestimate the effects of these global changes impacting all manner of cardiovascular health and disease.
As a consequence of these major changes, the clinical syndrome of HF has emerged as an important and growing public health challenge. Prevalence of HF is estimated at >60 million individuals worldwide, including >6 million in the United States alone, contributing in 2017 to 1 in 8 deaths6–8.
Two major phenotypes of HF are recognized: HF with reduced ejection fraction (HFrEF) and HF with preserved ejection fraction (HFpEF)9,10. Importantly, changes in the incidence and prevalence for these two types of HF have differed in recent decades; HFpEF has risen by 10% relative to HFrEF, and this gap is slated to continue to increase in coming years owing to progressive aging of the population and the growing prevalence of conditions predisposing to development of HFpEF, particularly obesity, metabolic syndrome, and diabetes11,12. In summary, HFpEF, a syndrome with a 35% two-year rate of hospitalization and 14% two-year mortality, is presently the most common form of HF, and one that is rising progressively, already accounting for the majority of HF worldwide.
Despite similar clinical presentations, increasing evidence supports a model in which HFpEF and HFrEF are mechanistically distinct13. Furthermore, the natural histories of HFpEF and HFrEF are dissimilar, as transitioning from HFpEF to HFrEF is rare11,14. In support of these observations is the fact that cornerstone neurohumoral therapies effective in HFrEF have failed when repurposed for HFpEF15,16. Recent results from EMPEROR-Preserved with empagliflozin, a sodium–glucose cotransporter 2 inhibitor (SGLT2i) that impacts cardiac and global metabolic profiles, are encouraging17.
Heterogeneity of the clinical manifestations of HFpEF, coupled with the complexities of its pathophysiological mechanisms, stem from the fact that HFpEF is not a disease but rather a clinical syndrome triggered by a variety of diseases; multiple comorbidities differentially contribute to the overall clinical presentation. Indeed, it is possible to distinguish phenotypes within the syndrome of HFpEF that emerge from different predisposing conditions with unique responses to therapy12,14,18. These include “cardiometabolic HFpEF”, arguably the most prevalent form of HFpEF and the subject of this review, as well as HFpEF related to auto-immune or inflammatory disease, cardio-renal HFpEF, and more (Figure 1). [It is important to note that other disorders mimic HFpEF, and likely have been included inadvertently in some HFpEF clinical trials; these include cardiac amyloidosis and hypertrophic cardiomyopathy.] As noted above, the global spread of obesity and metabolic syndrome have defined the HFpEF syndrome, with obesity and metabolic syndrome, and often type 2 diabetes, being present in most patients with HFpEF19,20. These conditions are now understood to be major drivers of HFpEF pathophysiology shaping the phenotype of what is called cardiometabolic HFpEF.
Figure 1.
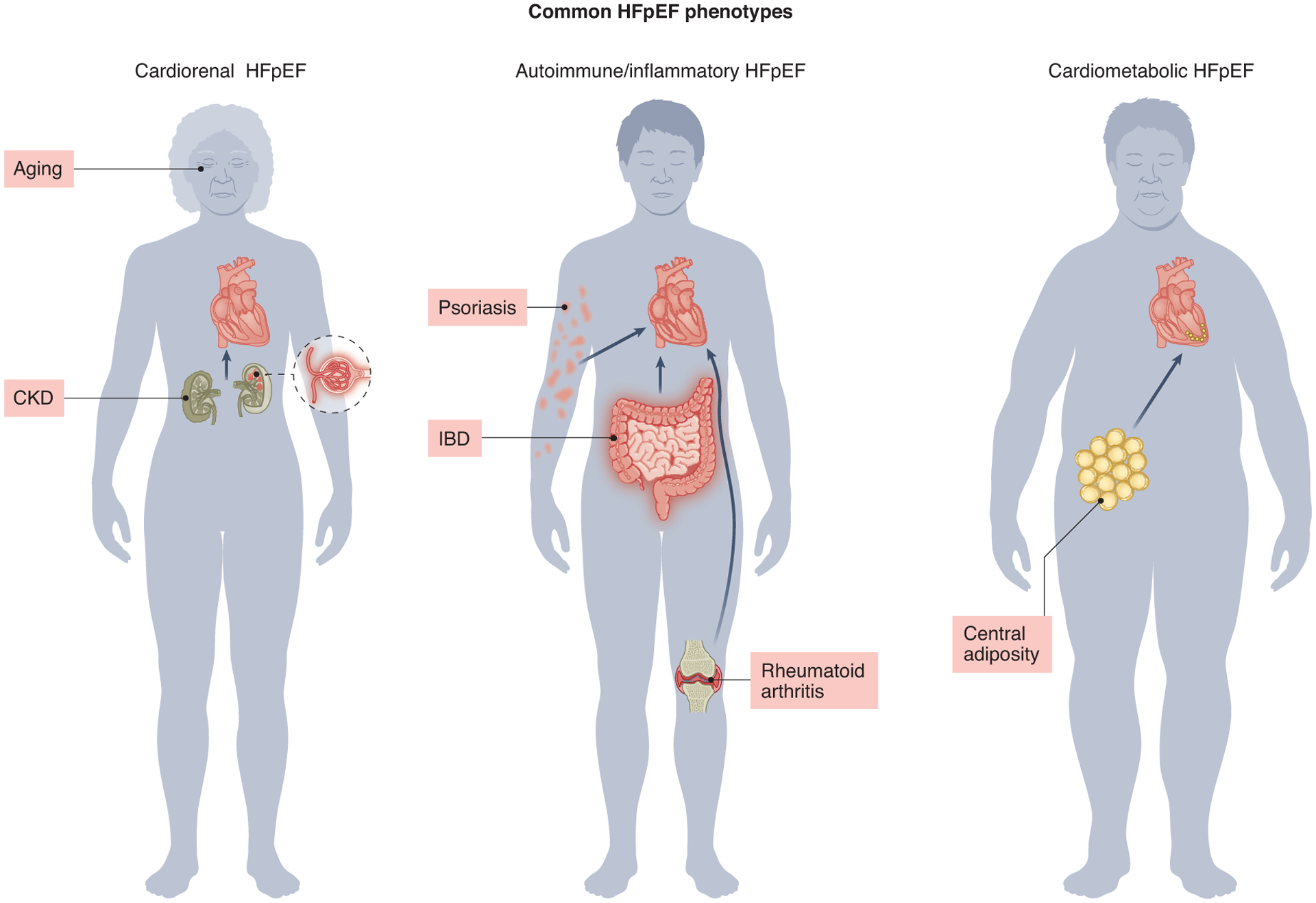
Schematic overview of common HFpEF phenotypes.
CKD: chronic kidney disease
IBD: inflammatory bowel disease
Heterogeneity in the clinical presentations of HFpEF is reflected in the complexity of preclinical modeling of the syndrome designed to unravel fundamental mechanisms21. As elucidation of pathophysiological mechanisms underlying any disease or syndrome relies on the reliability of animal and cellular models, existence of multiple phenotypes of clinical HFpEF requires highly integrated experimental approaches. Here, we discuss basic and translational data focusing on the most common form of HFpEF, cardiometabolic HFpEF. In particular, we emphasize the existence of rapidly emerging evidence pointing to bidirectional crosstalk between metabolic dysregulation triggering immune events – and vice versa – in syndrome pathogenesis.
Cardiometabolic stress and HFpEF
Cardiometabolic stress stemming from obesity is a mechanism contributing to multiple cardiovascular disorders, including both HFrEF and HFpEF11,22. And in addition to predisposing to HFpEF more than to HFrEF, obesity in HFpEF is associated with worse clinical outcomes, increased mechanical strain on the heart, insulin resistance, type 2 diabetes, and hypertension. Beyond just the heart, cardiometabolic alterations in HFpEF contribute to dysfunction of the vasculature, skeletal muscle, and other organ systems. As such, HFpEF is a systemic condition13.
Recent results from the EMPEROR-Preserved trial reveal improvement in HF hospitalization, but not mortality benefit, in HFpEF patients treated with the SGLT2i empagliflozin. Of note, this benefit was greatest in subjects with an EF 40–49%, less in subjects with an EF 50–60%, and absent in those with an EF >60%17. Whereas one could interpret these data as lack of benefit of empagliflozin in HFpEF subjects, no significant statistical interaction between the trial primary endpoint (a composite of cardiovascular death and HF hospitalization) and ejection fraction (EF) was found, suggesting that empagliflozin improved the primary endpoint independent of baseline EF. However, a recent examination of the major trial endpoints across the values of EF revealed no protective effects of empagliflozin for EF >62.5% for all the endpoints considered23. In aggregate, whereas the results of EMPEROR-Preserved support the use of SGLT2i in HFpEF patients, the cardiovascular benefit of these drugs in patients with EF >50/60% will require further confirmation.
Mechanisms of SGLT2i-afforded cardioprotection remain elusive. Yet, these agents improve metabolic parameters, lending further credence to the notion of cardiometabolic drivers of HFpEF24. Together with EMPEROR-Preserved, another recent US-only trial with the SGLT2i, dapagliflozin, demonstrated improvement in functional capacity of obese HFpEF patients25. Interestingly, this is in contrast with other trials in less obese HFpEF populations in which a similar benefit was not observed26. In summary, multiple lines of clinical and epidemiological evidence support a model in which cardiometabolic stress functions as a major driver of HFpEF. As such, targeting cardiometabolic stress may emerge as a viable therapeutic objective in the syndrome22,27.
Obesity, excess total body adipose tissue, commonly tracked as increased body mass index (BMI), is a strong – but modifiable – risk factor for HFpEF. Visceral (abdominal) adipose tissue accumulation (VAT), measured as waist circumference or waist-to-hip ratio, has the strongest correlation to HFpEF development, hospitalization and mortality28. This so-called central obesity is the strongest predictor of increased insulin resistance leading to diabetes, of decreased arterial compliance causing arterial hypertension, and of systemic endothelial dysfunction and inflammation28, all conditions considered associated with and predisposing to HFpEF. VAT expansion and related metabolic disturbances induce cardiac hypertrophy, fibrosis, and diastolic dysfunction, as VAT is associated with decreased cardiopulmonary performance and impaired left ventricular compliance28.
VAT is greater in men than women. However, recent data suggest that for a given increase in VAT, there is greater risk for development of cardiometabolic disorders in women compared with men29 suggesting that the cardiometabolic impact of VAT might exhibit a sex-dependent effect, in line with the slight predilection for women to develop HFpEF30. In women with HFpEF, a greater than 30% increase in VAT area has been identified compared with control women with the same BMI. In addition, women with increased VAT manifested a significant reduction in exercise performance with increases in estimated cardiac filling pressures compared with women with normal VAT. Intriguingly, this did not occur in men with or without excess VAT suggesting that accumulation of excess VAT plays a distinct and important role in the pathophysiology of HFpEF preferentially in women31.
VAT and epicardial adipose tissue (EAT) accumulation can each induce both systemic and local inflammation contributing to oxidative stress, microvascular injury, cardiomyocyte hypertrophy, and myocardial fibrosis. Several adipokines promote microvascular endothelial dysfunction and reduce vascular compliance in obese HFpEF patients32. These pro-inflammatory cytokines elicit the infiltration of macrophages, may cause regression (destruction) of microvascular structures, and induce pro-fibrotic pathways32. Dysfunctional adipose tissue also triggers secretion of leptin that regulates energy balance and hunger. Leptin also stimulates the secretion of aldosterone and angiotensin II33 and increases the activity of neprilysin34, together increasing sodium retention and volume expansion.
For all the reasons stated above, targeting excess body fat might represent a valid therapeutic option in HFpEF. Indeed, multiple trials with anti-diabetic drugs, such as GLP-1 (glucagon-like peptide 1) agonists, with a robust effect on body weight reduction are underway in HFpEF (e.g., STEP-HFpEF, STEP-HFpEF DM, SUMMIT).
Immunometabolic mechanisms of HFpEF
Obesity and metabolic stress elicit a low-grade, systemic inflammatory state, and dysregulation of inflammatory and immune responses are now recognized as culprit mechanisms in HFpEF pathophysiology. Indeed, concomitant with rapidly emerging evidence implicating metabolic stress in HFpEF pathogenesis, a pivotal role of immune mechanisms is also emerging. Indeed, longstanding evidence points to bidirectional crosstalk between metabolic stress and inflammation; adipose tissue, a metabolically active tissue, influences cardiac metabolism and immune activation, all to the detriment of multiple different tissues (Figure 2).
Figure 2.
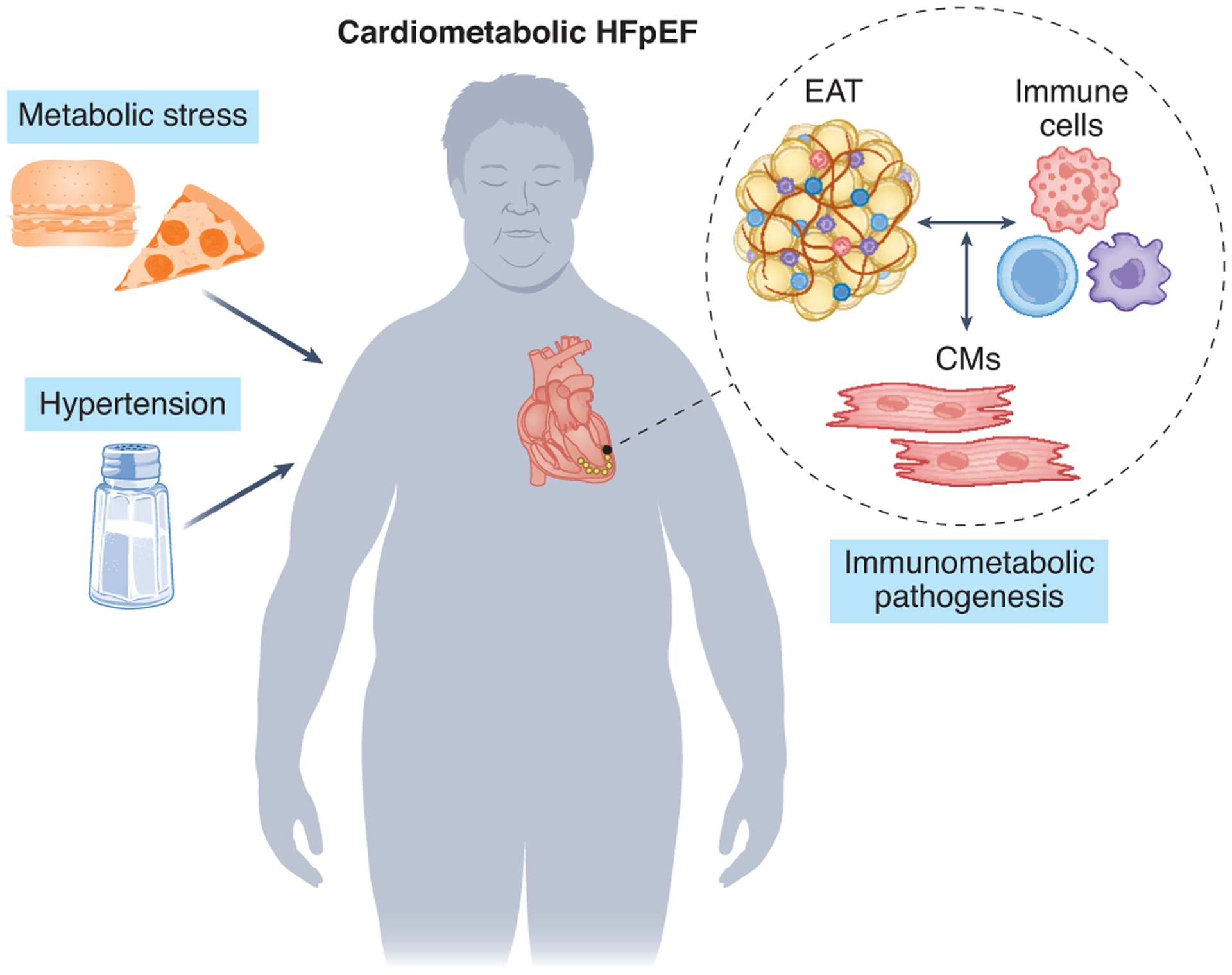
Immunometabolic mechanisms involving crosstalk between inflammatory and metabolic events contribute to the pathogenesis of cardiometabolic HFpEF.
EAT: epicardial adipose tissue
CMs: cardiomyocytes
We and others have demonstrated that oxidative and nitrosative stress drive HFpEF35–39. Furthermore, inflammatory cells have been detected in endomyocardial biopsies from HFpEF patients40. Of note, metabolic alterations promote a pro-inflammatory state as a condition termed metabolic inflammation, or meta-inflammation22,41,42. Furthermore, expansion of adipose tissue triggers release of chemokines that initiates recruitment of immune cells43, and lipids can act as inflammatory molecules, participating in the recruitment of immune cells to HF myocardium44.
Local cardiac adipose tissue can also contribute to myocardial inflammation. Secretion of cytokines from EAT has been proposed as contributing to meta-inflammation in HFpEF32, yet mechanisms of EAT-induced myocardial dysfunction in HFpEF remain unknown. The inflammatory state of VAT or EAT induces pro-inflammatory macrophage polarization and recruitment into the heart45. Similarly, other myeloid cells, mast cells46 and neutrophils are also present in obese organs, contributing to tissue damage through elastase secretion, thus promoting macrophage recruitment47. Obesity also promotes CD8+ and CD4+ T lymphocyte infiltration together with effector B cells, heightening release of pro-inflammatory factors48. In addition, VAT harbors a unique regulatory T cell (Treg) population that specializes in maintaining adipose tissue immune homeostasis and insulin sensitivity49. In mouse models of obesity, the number of Treg cells is dramatically reduced in VAT, whereas Treg cell abundances in subcutaneous adipose tissue and spleen remain unaffected50 51. Obesity also affects the phenotype and function of VAT Treg cells51. Some of these differentially expressed genes are important for maintaining the phenotype and function of VAT Treg cells. For example, the expression of the anti-inflammatory, Treg-produced cytokine IL-10, and the IL-33 receptor ST2, required for VAT Treg cell function, are reduced in VAT Treg cells from obese mice52. Similar changes have been reported in obesity in humans53. Obesity-induced VAT Treg cell dysfunction has been proposed to contribute to development of chronic inflammation and insulin resistance in metabolic syndrome49.
In aggregate, these findings highlight HFpEF as a chronic cardiovascular inflammatory syndrome that arises in the setting of multiple pro-inflammatory comorbidities, one in which the role and extent of specific immune cells and mediators of meta-inflammatory pathways remain to be elucidated (Figure 2). Despite evidence implicating inflammation and adipose tissue, drugs that specifically target these inflammatory pathways to treat metabolic syndrome, HFpEF development, and its systemic manifestations are lacking. Future studies are required to investigate whether reduction in this VAT- and EAT-triggered systemic inflammatory dysregulation may afford novel therapeutic targets.
Role of adaptive immunity
Most research on the role of inflammation in HF has focused on HFrEF; much less is known in the context of HFpEF. Experimental models of HFpEF suggest that pro-inflammatory mediators play an important role in the development and progression of the syndrome44,54,55. Clinical trials in patients with HF targeting IL-6 or TNFα have manifested no, or even negative, effects on outcomes54–57. Therefore, immune modulation in HF remains controversial.
Coordinated innate and adaptive immune responses culminate in sequential immune cell infiltration into the heart contributing to cardiac inflammation and fibrosis. Recent advances in single-cell transcriptomic technologies (scRNA seq) have revealed the variety of immune and non-immune cells in the heart adding many levels of complexity to our understanding of cardiac cell identity58 59. With respect to adaptive immunity, T cells have been identified in the inflamed heart in diverse forms of HF in patients60. Studies in experimental models of HFrEF support the notion that different T cell subsets play distinct roles in the heart depending on the inflammation-triggering event61,62. For example, T cells are involved in the response following myocardial infarction (MI)63–65. This may occur in a bi-phasic mode, with an initially beneficial66 and subsequently a chronic, detrimental phase64,67. In age-related HF, T cells have been shown to drive pathology68,69. Specifically, cardiac aging is associated with accumulation of a particular CD4+ subpopulation – FoxP3 (forkhead box P3)+ IFN-γ+ – in heart-draining lymph nodes68. Importantly, cardiotropic T cells participate in age-related cardiac inflammation and functional decline suggesting that, at least in part, cardiac aging is mediated by immunological mechanisms68. Similar T cell dysregulation occurs in pressure overload-induced HF, in which inhibition or ablation of T cells limits pathology60,70–72. Whereas the T cell immune response is dominated by dysfunctional Treg cells that elicit TNF-α in chronic ischemic HF67, pathogenic T cells polarized toward a type 1 response are central to non-ischemic HF60,73.
T cell – B cell crosstalk
Meaningful insights have been gleaned regarding the interplay between T cells and B cells in models of HFrEF, but little is known in the context of HFpEF. T cells, in most pathophysiological contexts, are mirrored in function by B cells. Thus, it is not surprising that B cells have been found to contribute to the pathophysiological mechanisms of MI in mice, and antibodies are present in human hearts post-MI74. Myocardial B cells express chemokine receptors that could allow them to form tertiary lymph nodes (formations within which adaptive responses initiate) in mice with pressure overload or MI59,75. Intriguingly, even though B cell clusters have been observed in human epicardium of coronary artery disease (CAD) patients76, canonical tertiary lymph node structures have yet to be observed in mammalian heart. T cells, in most of the conditions in which they are found to be activated, are driven by triggering of their antigen receptor. Indeed, T cell-specific activation by cardiac antigens has been reported in experimental HFrEF77,78. More specifically, in viral myocarditis, reactivity to viral antigens that mimic cardiac antigens appears to drive disease79. Molecular mimicry between microbe-derived antigens and cardiac antigens also promotes myocarditis after a response to a specific commensal bacterial strain80. In pressure overload-induced HF, the T cell response is known to involve antigen-specific reactivity72. Most recently, the driving antigens in pressure overload were found to include those modified by reactive oxygen species generated by the stress that drives the disease in the first place77 whereas alpha myosin heavy chain is a dominant cardiac antigen triggering T cell activation in mice post-myocardial infarction.
Ample evidence indicates that T cell recruitment/retention in the heart depends on several factors including differences in T cell responsiveness to specific chemokines in the myocardial environment, as well as differences in the expression of adhesion molecules in the intramyocardial vasculature61,81. These, in turn, regulate T cell-driven cardiac inflammation with consequences in cardiac remodeling and function62 (Figure 3). Thus, a specific B cell and T cell immune response induced in different types of HFrEF may combine with activation of lymphocyte subsets expressing specific chemokine receptors and adhesion molecules, promoting a unique local cardiac environment with a defined pattern of chemokine ligands and endothelial adhesion molecules to enable recruitment to the heart. As an example, CXCR3 and CCR4 define T cell cardiotropism in certain conditions that induce the cardiac CXCR3 ligands CXCL9 and CXCL10, and c-Met, a hepatocyte growth factor that can be produced in the myocardium73,81. Yet, the roles of T cell immunity and cardiotropism in HFpEF remain largely unknown.
Figure 3.
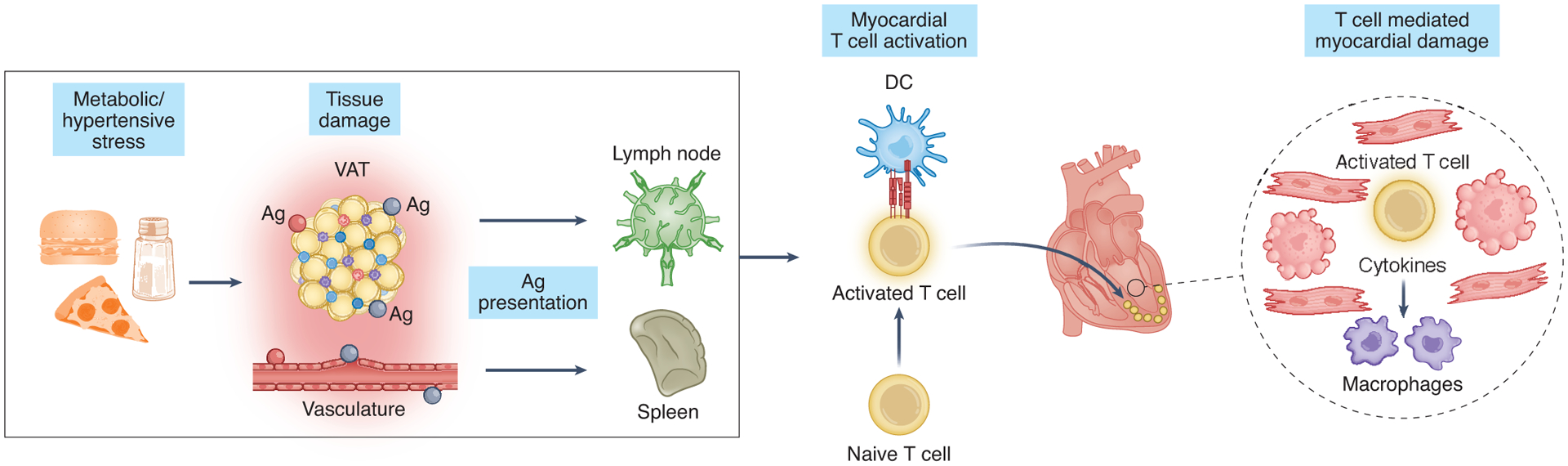
Cardiometabolic stress triggers alterations in adaptive immunity in HFpEF. Metabolic stress activates T cells in peripheral organs that, in turn, promote recruitment of other immune cells to the heart and consequent myocardial damage.
VAT: visceral adipose tissue
Ag: antigen
In HFrEF, a direct insult to the heart initiates an immune response that promotes cardiac repair or remodeling in an “inside-out” (from the heart to the periphery) manner. In contrast, HFpEF arises from systemic perturbations that may ultimately impact the heart, representing an “outside-in” mechanism of disease that implicates systemic forms of inflammation and metabolic stress that activate T cell immune responses and vascular endothelium systemically. Indeed, the latter is known to involve T cell-mediated effects51. Whereas this remains speculative, it is likely that T cells participate in HFpEF pathogenesis, potentially not only as a result of the cardiac stress driving diastolic dysfunction, but also upstream of the cardiac phenotype.
Endomyocardial biopsy data from HFpEF patients reveal a significant increase in cardiac CD3+ cells compared with control patients82. Additionally, HFpEF patients have significantly higher levels of circulating T-helper 17 (Th17) cells and significantly lower levels of circulating Treg cells compared with healthy controls83. In addition to these data that point to expansion of T cells in HFpEF, there are several lines of evidence suggesting that T cells contribute to adverse cardiac remodeling and systemic inflammation in HFpEF. First, T cells have well-characterized roles in the pathophysiology of several comorbidities that commonly present in HFpEF patients, such as hypertension, obesity, and aging68,69,84,85. Second, as noted above, endothelial cell activation and inflammation in the heart are widely recognized hallmarks of HFpEF86. Endomyocardial biopsy samples from HFpEF patients reveal significant increases in the expression of VCAM-1 (vascular cell adhesion molecule-1), ICAM-1 (intercellular adhesion molecule-1), and E-selectin82,87,88. Expression of these adhesion molecules is critical for extravasation of T cells, consistent with the premise that myocardial T cell infiltration may be a critical element in the pathophysiology of HFpEF. Preclinical data in multiple animal models of HFpEF corroborate these findings, as ZSF1 rats (obese Zucker diabetic fatty/spontaneously hypertensive hybrids) harbor similar increases in myocardial expression of ICAM and E-selectin87. Further studies, particularly using animal models that combine several comorbidities to induce HFpEF, may reveal how the intersection of risk factors affects adaptive immunity and the degrees to which T cells contribute to cardiac pathology in HFpEF.
Conditions predisposing to dysregulation of adaptive immunity
The notion that dysregulated adaptive immunity contributes to HFpEF pathogenesis is underpinned by emerging observations demonstrating that adaptive immunity is profoundly affected by systemic dysmetabolism. For example, dendritic cells (DCs) are instrumental in the initiation of the adaptive immune response and promote pro-inflammatory adaptive immunity during metabolic overload89 (Figure 3). VAT is now considered an immune site harboring an array of innate and adaptive immune cells with a direct role in immune surveillance and host defense. In homeostatic conditions, conventional DCs in VAT display a tolerogenic phenotype through upregulation of Wnt/β-catenin and the PPARγ pathways89. Upon conditions of long-term over-nutrition, however, VAT accumulates and systemic inflammation ensues. This can be attributed to adipocyte alterations reducing β-catenin and PPARγ activation89. Systemic dysmetabolism also promotes pro-inflammatory differentiation of T cells41. Specifically, obesity-induced low-grade systemic inflammation promotes recruitment of effector T cells in adipose tissue, heart, and vasculature leading to a variety of cardiovascular complications46. Metabolic stress also affects differentiation and trafficking patterns of T cells90. Memory T cells primed by overfeeding migrate preferentially to non-lymphoid, inflammatory sites due to biased T cell differentiation into effector-like T cells. A similar phenotype skew was observed in obese subjects in a general population cohort90. Mechanistically, this effect is mediated by direct exposure of CD4+ T cells to palmitate, leading to increased activation of a PI3K p110δ-Akt-dependent pathway upon priming90,91.
What about the other cardinal sign of HFpEF, viz. hypertension? Since the seminal paper by Guzik et al92 reporting that T cell-deficient mice do not develop hypertension, T cells have been known to play a major role not only in the pathogenesis of, but also the progression of, hypertensive HF. Compelling recent evidence indicates that hemodynamic overload-induced HF also involves immune dysregulation as a major etiological factor60,71,72,93. In summary, the presence of systemic DC activation, such as significantly increased CD80 (required for T-cell co-stimulation), in HF promotes the triggering of systemic T cells77,93. Under certain conditions, these activated T cells may target the heart in an autoimmune response-type manner, promoting development of hypertrophy, cardiac fibrosis, remodeling, and failure, as has been proposed recently56. We could speculate, by analogy to the generation of antigen-specific T cell responses in HFrEF, that the multi-system stress that HFpEF drivers impose on adipose tissue and the vasculature, among other tissues, could generate or modify antigens recognized by T cells, activating the T cells and thus contributing to disease (Figure 3). In aggregate, the presence of T cell alterations in humans with HFpEF justifies detailed mechanistic investigation of adaptive T cell immune responses with basic and translational studies in preclinical models that allow for elucidation of the effects of T cells in diastolic dysfunction and specific aspects of cardiac remodeling in HFpEF.
Contributions of myeloid cells
Inflammatory myeloid cells and innate macrophages have been implicated in development of diastolic dysfunction94, yet our understanding of how risk factors that predispose to HFpEF regulate myeloid mobilization and function pales, yet again, in comparison to our understanding of macrophages in HFrEF. This includes our limited understanding of how HFpEF risk factors affect the activation and differentiation of macrophage precursors such as peripheral monocytes. For example, previous studies have shown that metabolic stress can promote a “priming” of monocytes that in turn enhances monocyte adhesion and chemotaxis95. In addition, monocyte lipid metabolism is distinct from that of macrophages and is critical in the differentiation of monocytes into phagocytic macrophages96.
Tissue macrophages are conventionally associated with homeostatic clearance of dying cells, immunosurveillance, and tissue repair. Interestingly, macrophages have been linked more recently to diastolic dysfunction, such as that occurring in the context of increased salt consumption, unilateral nephrectomy, or aldosterone infusion94. However, the role of cardiac macrophages in HFpEF-associated diastolic dysfunction with an integrated metabolic contribution (i.e., obesity) remains unclear, as reviewed97. This is important given the common association of metabolic syndrome with HFpEF11. Multiple studies have examined the individual effects of hyperlipidemia on macrophage function98, yet little is known regarding how combinatorial “hits” rewire functional macrophage proinflammatory functions, including in the heart (Figure 4).
Figure 4.
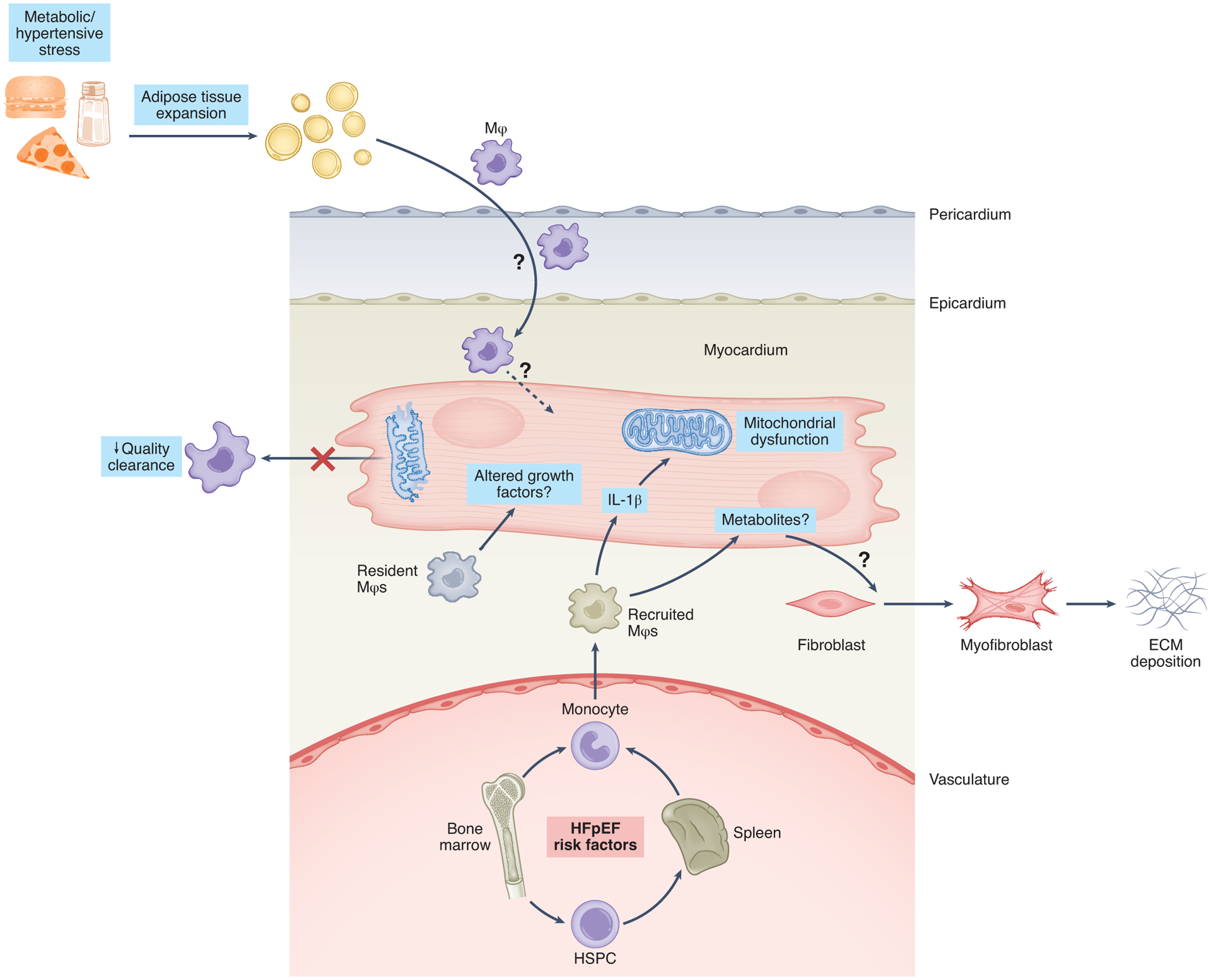
Potential contributions of innate immunity to the pathogenesis of cardiometabolic HFpEF. Depicted is a working model in which cardiometabolic risk factors, including visceral adiposity and hypertension, fuel immunometabolic mobilization of innate immune cell subsets. This immune cell mobilization, in turn, activates intercellular crosstalk that also regulates myocardial metabolic pathways.
HSPC: hematopoietic stem/progenitor cell
Mϕ: macrophages
IL-1β: interleukin 1β
As mentioned above, in contrast to myocardial “damage from within,” which often initiates HFrEF, HFpEF develops from peripheral “damage from without”86. This injury from the periphery may be fueled by the bone marrow or extramedullary myelopoiesis99. Interestingly, recent studies highlight the impact of dyslipidemia on pro-inflammatory monocyte production by the bone marrow in humans100. Specifically, CD34+ bone marrow hematopoietic stem and progenitor cells (HSPCs) of patients with familial hypercholesterolemia manifest increased gene expression in pathways involved in HSPC migration and myelomonocytic skewing. These findings are consistent with prior studies in animals demonstrating that cholesterol augments proinflammatory monocyte production101. Increased proliferation of hematopoietic stem cell clones – clonal hematopoiesis – has been associated with increased cardiovascular risk102. In addition, a causal relationship between the increase in hematopoietic stem cell division and atherosclerosis has been reported recently103. In summary, studies of cardiac myeloid-cardiomyocyte crosstalk may lead to novel insights regarding the interplay between myocardial metabolism and cardiac function (Figure 4).
As noted, the HFpEF population is characterized by multiple, usually interrelated pro-inflammatory co-morbidities such as advanced age, obesity, type 2 diabetes mellitus, kidney disease, chronic obstructive pulmonary disease, and autoimmune diseases104. Chronic cardiac and systemic inflammation is associated with capillary regression and endothelial dysfunction, cardiomyocyte hypertrophy and interstitial fibrosis. Importantly, the presence and severity of these co-morbidities correlate with poor outcome in HFpEF, with greater impact on clinical outcome than parameters of left ventricular diastolic function or brain natriuretic peptide levels105.
Whereas it is generally accepted that inflammation plays a role in HFpEF initiation and progression, the prevailing paradigm is that systemic inflammation leads to left ventricular diastolic dysfunction and myocardial hypertrophy via coronary endothelial microvascular inflammation86 that results in myocardial infiltration by activated macrophages87 and elicitation of interstitial fibrosis82; this model, however, ignores the possible role of resident macrophages. Activation of the resident macrophage population occurs much earlier than systemic inflammation and therefore may represent a crucial first step aiming to protect the heart against these inflammatory co-morbidities (Figure 4). Furthermore, within activated macrophages, remodelling of the tricarboxylic acid cycle can generate metabolites that shift the balance between inflammation activation versus inflammation resolution. For example, exogenous administration of itaconate, a metabolite significantly induced in activated macrophages, has been shown to limit cardiac inflammation and injury106, yet its role or therapeutic potential in HFpEF is currently unknown.
Studies in mice and recently in humans107 have provided evidence that resident macrophages represent a heterogeneous population of cells, serving to protect the heart against pathological stimuli such as diabetes, hypertension, and inflammation, common co-morbidities and drivers of HFpEF. These resident cardiac macrophages derive from different embryonic lineages, are long-lived, and persist independent of blood monocyte input108. Also, their behaviour is different from that of blood-derived macrophages or mononuclear cells isolated from spleen and brain, suggesting a unique phenotype of cardiac macrophages109. Recent data indicate that those resident macrophages mainly proliferate in response to external pathological stimuli to promote cardiomyocyte survival and physiological hypertrophy, prevent adverse monocyte recruitment, and stimulate vascular expansion107,110. As such, those resident macrophages may serve to protect the heart from the metabolic stress of diabetes and hypertension. Invading cardiac monocytes, on the other hand, are required to “heal” the diseased myocardium, but they also promote pathology by stimulating fibrosis, pathological hypertrophy, and vessel regression, thereby contributing to HF. Whereas evidence exists for a role of resident macrophages in preventing cardiac systolic failure upon ischemic injury, a possible role of resident macrophages in HFpEF is unclear. Importantly, myocyte injury111 and microvascular dysfunction112 113 with the capacity to contribute to myocardial ischemia and injury, have been implicated in HFpEF.
Future studies are required to determine whether stimulation of resident macrophages – tissue resident CCR2-negative versus invading CCR2-positive cardiac macrophages – antagonizes progression to HFpEF by reducing pathological hypertrophy, capillary regression and fibrosis, in the setting of diabetes, obesity, hypertension and ageing.
Immunometabolic crosstalk
Whereas direct damage to the myocardium mediated by infiltrating effector immune cells has been investigated extensively, metabolic crosstalk between inflammatory cells and the cardiac muscle itself is less well understood. The systemic increase of circulating cytokines in metabolic syndrome, a major HFpEF comorbidity, induces production of chemokines and expression of adhesion molecules by cardiomyocytes, fibroblasts, and endothelial cells leading to myocardial recruitment and retention of immune cells such as monocytes and lymphocytes114. Whereas the impact of systemic inflammation on cardiac function and remodeling in HFrEF has been well documented114,115, few studies have examined the impact of pro-inflammatory stimuli on cardiac metabolism. Even though the mutual influence of immune/parenchymal cell metabolic crosstalk in HFpEF hearts, and consequent impact on adverse remodeling, remain to be resolved, it is tempting to propose hypotheses based on parallel analysis of metabolic changes in failing (mostly HFrEF) hearts and inflammatory cell infiltrates.
Quiescent T cells rely predominantly on oxidative metabolism and consume small amounts of glucose, amino acids, or fatty acids to meet basic energetic demands. T cell activation by T cell receptor (TCR) triggering and concomitant co-stimulation via the CD28 receptor induce a dramatic shift to aerobic glycolysis to support rapid growth and differentiation into effector T cells (Teff)116. Teff cells include cytolytic T cells, secreting granzyme B, perforin, interferon-γ (IFN-γ), and helper T cells (Th) such as type-1 (Th1), type-2 (Th2), and type-17 (Th17) producing characteristic cytokines, and finally regulatory T cells (Treg)117. Each T cell subset is characterized by signaling pathways and metabolic signatures that define its fate and function118. Once activated, Teff cells infiltrate inflamed tissue, adapting to the local microenvironment (oxygen tension, acidification, and the presence of metabolites) by undergoing further metabolic reprogramming. For example, immune cells respond quickly to a decrease in oxygen availability – typical of sites of inflammation – by stabilizing the transcription factor HIF1α (hypoxia-inducible factor 1α) which, in turn, induces transcription of anabolic genes for glycolysis and mitochondrial metabolism in T cells119 and macrophages120.
Cardiac macrophages also play important roles in maintaining myocardial homeostasis. A recent study demonstrated that macrophages within the heart engulf decaying mitochondria released from cardiomyocytes121. This process requires cardiomyocyte autophagy. Furthermore, depletion of cardiac macrophages, or deficiency of macrophage phagocytic receptors, such as MerTK, lead to increases in functionally impaired mitochondria in cardiomyocytes and, in turn, reduced production of ATP. These hearts also exhibit impaired cardiac filling similar to that seen in HFpEF. Given that the extracellular domain of MerTK can be cleaved from cardiac macrophages and released into the serum as a biomarker122, it may be of interest to determine whether solubilized MerTK is a biomarker of clinical HFpEF. Mononuclear phagocytes specialize in sampling the tissue microenvironment and mediating crosstalk with neighboring cells, thus bridging innate and adaptive immunity. After stimulation, DCs undergo a burst of oxidative phosphorylation which is rapidly replaced by full engagement of aerobic glycolysis123. Macrophages undergo similar metabolic reprogramming after activation. Inflammatory macrophages manifest reduced TCA cycle activity which allows for accumulation of succinate that promotes inflammation124 via mitochondrial ROS production, HIF1α stabilization, and persistent activation of glycolysis. In contrast, anti-inflammatory M2-like macrophages rely on the TCA cycle to meet their metabolic demands125.
We propose that direct and indirect crosstalk between metabolic events and inflammatory immune cells, occurring via both nutrient and oxygen competition as well as direct signals from cytokines and metabolites, contribute to the development of HFpEF (Figures 3, 4). If correct, this hypothesis would justify therapeutic targeting of aberrant metabolic pathways in cardiac inflammatory disease. Reviews of recent trials targeting inflammation in CAD have argued that immunometabolic correction with statins is superior and less prone to severe side effects as seen with direct immunomodulation126. We have also proposed that modulation of systemic and cellular metabolism might be an attractive strategy to reduce organ inflammation42.
Cardio-immune metabolites
Under normoxic conditions, >95% of ATP generated in the heart is derived from oxidative phosphorylation in mitochondria, mainly fueled by free fatty acid oxidation. The remaining 5% derives largely from glycolysis127. There is general agreement that development of overt cardiac dysfunction is accompanied by reduced FFA oxidation127 128. HF is also characterized by alterations in glucose metabolism. Specifically, whereas glucose uptake and glycolytic rates are increased, this is not accompanied by a concomitant rise in glucose oxidation128. The profound defect in oxidative phosphorylation in the failing heart is reflected by overt mitochondrial dysfunction, with altered size and number of mitochondria, disorganized cristae, reduced density, membrane disruption, and aggregation44. Excessive ROS production from dysfunctional mitochondrial electron transport chain ATP synthesis also contributes to oxidative damage and ultimately to cardiomyocyte loss44.
Overall, both immune cells and cardiac parenchyma contribute to shaping the microenvironment in cardiac inflammation. Next, we focus on two key metabolites likely to participate in metabolic crosstalk in the failing heart.
Lactate.
Lactate is produced by highly glycolytic, activated immune cells129. Extracellular lactate, when enriched in the cytosol following uptake by lactate transporters, can signal directly to immune cells themselves and tissue parenchymal cells via lactate receptors as well as by affecting metabolic pathways.
In immune cells, lactate mainly signals via the surface-expressed G-protein-coupled receptor GPR81130, eliciting signals that are strong inhibitors of immune effector functions41. However, extracellular sodium lactate and lactic acid inhibit the motility of CD4+ and CD8+ T cells, respectively, entrapping T cells at the inflammatory site and preventing the resolution of inflammation131. Impairment of T cell motility is mediated by uptake via subtype-specific transporters (Slc5a12 and Slc16a1) expressed by CD4+ and CD8+ cells, respectively, by interference with glycolysis triggered by chemokine receptors. Importantly, sodium lactate also induces a switch toward the Th17 cell subset, promoting robust biosynthesis of the proinflammatory cytokine IL-17, enhancing fatty acid synthesis132.
Lactate accounts for a minimal component of energy production in the healthy heart at rest133, but this fraction can increase substantially during exercise or in the setting of various pathophysiological conditions. Lactate production in the cardiomyocyte cytosol is balanced by oxidation of pyruvate in mitochondria (reviewed in134). Of note, down-regulation of the mitochondrial pyruvate carrier (MCP), which transports pyruvate generated in the cytosol in the failing human heart, has been recently reported135,136. Excess extracellular lactate resulting from protracted inflammation has been linked to cardiomyocyte apoptosis in human end-stage HF137. Under inflammatory conditions, elevated concentrations of intracellular lactate promote ROS generation132. High levels of ROS, in turn, can trigger mitochondrial damage and activation of mitochondria-dependent apoptosis. Accordingly, a significant association has been identified between the lactate signaling cascade and HF and other conditions132,138,139.
Excess extracellular lactate in the heart is a metabolic indicator of ischemia. A key feature of HFpEF is endothelial dysfunction, which may contribute to chronic oxygen deficiency in the failing myocardium. Lactate has been linked to cardiomyocyte apoptosis in several experimental models of cardiovascular disease, including end-stage HF137. Despite these associations, a recent study comparing metabolomics of blood from artery, coronary sinus, and femoral vein in patients with or without HF (including patients with EF>50%) reported that the failing heart almost doubles lactate consumption133. Therefore, the overall contribution of lactate to HFpEF remains to be established.
Succinate.
Succinate accumulation is also a hallmark of the ischemic heart, where it fuels ROS production140. In addition, elevation of blood succinate has been reported in rodent models of hypertension and metabolic syndrome141. Extracellular succinate is a powerful pro-inflammatory stimulus. In the context of metabolic syndrome, exposure of adipocytes to hypoxia and hyperglycemia (such as during obesity) induces succinate release from adipose tissue in mice142 leading to macrophage infiltration and inflammation142. In DCs, exposure to succinate increases TNFα and IL-1β expression143 and the capacity of DCs to initiate adaptive immunity. In macrophages, TLR engagement by lipopolysaccharide increases intracellular succinate levels and cell surface expression of the succinate receptor SUCNR1144. Released succinate can act in an autocrine and paracrine manner to enhance pro-inflammatory cytokine production by the same cells or by nearby SUCNR1-expressing cells. Production of IL-1β further enhances SUCNR1 expression fueling this pro-inflammatory cycle. Collectively, these observations point to succinate as a likely candidate in the induction and maintenance of inflammation and adverse remodeling in HFpEF.
Therapeutic approaches in cardiometabolic HFpEF
We propose that targeting metabolic and inflammatory pathways in HFpEF is a therapeutic strategy with promise. However, targeting inflammation directly has been a longstanding challenge in cardiovascular medicine. Canonical anti-inflammatory therapies, such as anti-TNFα therapeutics, to treat HFrEF have been abandoned27. However, results from the CANTOS trial provided the first evidence that targeted anti-inflammatory approaches in cardiovascular disease have merit145. In that trial, inhibition of interleukin-1 (IL-1ß) resulted in reduced rates of recurrent cardiovascular events independent of lipid lowering145. More recently, the COLCOT trial reported that colchicine, an inexpensive, orally administered, potent, anti-inflammatory drug, led to a significantly lower risk of ischemic cardiovascular events than placebo in patients with a recent myocardial infarction146. In aggregate, the results of recent clinical trials aiming to reduce the inflammatory burden in cardiovascular disease have shown promising results, setting the stage for renewed interest in inflammation-targeting strategies in cardiovascular disease. Of note, it is important to recognize that the clinical trials mentioned above targeted conditions predisposing more often to HFrEF (e.g. atherosclerosis, CAD) than HFpEF. Hence, the validity of these therapeutic strategies in HFpEF remains to be tested.
We have reported activation of the inflammatory molecule iNOS (inducible nitric oxide synthase) in both preclinical and clinical HFpEF contributing importantly to nitrosative stress22. Furthermore, we reported that pharmacological inhibition or genetic silencing of iNOS ameliorated the HFpEF phenotype. Based on this, we suggest that the availability of clinically approved iNOS inhibitors heralds therapeutic promise in HFpEF22. Going forward, additional investigation into molecular mechanisms of immune/inflammatory activation in HFpEF will likely reveal additional targets with potential clinical efficacy.
Given the long-established crosstalk between metabolic and inflammatory mechanisms, it is conceivable that a two-pronged attack on HFpEF, combining strategies that target both metabolism and inflammation, is warranted. We and others have shown that a key metabolic alteration observed in HFpEF (and HF in general) is reduced bioavailability of NAD+ (nicotinamide adenine dinucleotide), a cofactor required for cellular respiration and sirtuin activity. We reported that reduced mitochondrial fatty acid oxidation in HFpEF is dependent, at least in part, on hyperacetylation of key mitochondrial enzymes stemming from NAD+ deficiency-derived suppression of sirtuin activity147,148. Separately, NAD+ precursors can enhance anti-inflammatory innate immune functions, including potentially in the heart149. As oral supplementation with the NAD+ precursor NR (nicotinamide riboside) increases tissue NAD+ levels in humans150, and ameliorates the HFpEF phenotype in rodents147, the prospect of pharmacologically boosting cardiac NAD+ levels emerges with the potential for rapid translation to patients.
Conclusions and perspectives
Emerging evidence implicates bidirectional crosstalk between metabolic stress and inflammation in the pathogenesis of cardiometabolic HFpEF; inflammation rewires cellular metabolism, and systemic and local metabolic perturbations dictate immune cell behavior. Given the well-established heterogeneity among HFpEF phenotypes, coupled with the robust complexity and intertwined interactions between metabolic events and inflammation, a comprehensive program of investigation will be required. That HFpEF is a systemic disorder, not simply a cardiac disorder, amplifies this complexity yet further. Nevertheless, work to unravel meta-inflammatory mechanisms contributing to HFpEF pathophysiology holds the potential to benefit millions of individuals around the globe.
Acknowledgements
This work was supported by grants from the DZHK (German Centre for Cardiovascular Research) (GGS); the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) - SFB-1470 - A02 (GGS); from IMI2-CARDIATEAM (N° 821508), and the Netherlands Cardiovascular Research Initiative, Dutch Cardiovascular Alliance CVON2016-Early HFPEF, 2015-10, CVON She-PREDICTS, grant 2017-21 (SH); US National Institutes of Health: HL144477 (PA), HL122309 (EBT), HL126012 (JAH), HL128215 (JAH), HL120732 (JAH), HL147933 (JAH), HL155765 (JAH) and American Heart Association 19TPA34910006 (JAH).
We regret that we were unable to recognize all pertinent research in this field and contributions from all investigators owing to journal space limitations.
Figures created with BioRender.com licensed to G.G.S.
Footnotes
Competing interests
The authors declare no competing interests in relation of this manuscript.
References
- 1.Mensah GA, Roth GA & Fuster V The Global Burden of Cardiovascular Diseases and Risk Factors: 2020 and Beyond. J Am Coll Cardiol 74, 2529–2532, doi: 10.1016/j.jacc.2019.10.009 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Collaborators, G. B. D. C. o. D. Global, regional, and national age-sex specific mortality for 264 causes of death, 1980–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet 390, 1151–1210, doi: 10.1016/S0140-6736(17)32152-9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Finkelstein EA et al. Obesity and severe obesity forecasts through 2030. Am J Prev Med 42, 563–570, doi: 10.1016/j.amepre.2011.10.026 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Virani SS et al. Heart Disease and Stroke Statistics-2021 Update: A Report From the American Heart Association. Circulation 143, e254–e743, doi: 10.1161/CIR.0000000000000950 (2021). [DOI] [PubMed] [Google Scholar]
- 5.Boyle JP, Thompson TJ, Gregg EW, Barker LE & Williamson DF Projection of the year 2050 burden of diabetes in the US adult population: dynamic modeling of incidence, mortality, and prediabetes prevalence. Popul Health Metr 8, 29, doi: 10.1186/1478-7954-8-29 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Timmis A et al. European Society of Cardiology: Cardiovascular Disease Statistics 2017. Eur Heart J 39, 508–579, doi: 10.1093/eurheartj/ehx628 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Bragazzi NL et al. Burden of heart failure and underlying causes in 195 countries and territories from 1990 to 2017. Eur J Prev Cardiol, doi: 10.1093/eurjpc/zwaa147 (2021). [DOI] [PubMed] [Google Scholar]
- 8.Benjamin EJ et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 139, e56–e528, doi: 10.1161/CIR.0000000000000659 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Ponikowski P et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J 37, 2129–2200, doi: 10.1093/eurheartj/ehw128 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Yancy CW et al. 2013 ACCF/AHA guideline for the management of heart failure: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 128, 1810–1852, doi: 10.1161/CIR.0b013e31829e8807 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Dunlay SM, Roger VL & Redfield MM Epidemiology of heart failure with preserved ejection fraction. Nature reviews. Cardiology 14, 591–602, doi: 10.1038/nrcardio.2017.65 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Shah SJ et al. Phenotype-Specific Treatment of Heart Failure With Preserved Ejection Fraction: A Multiorgan Roadmap. Circulation 134, 73–90, doi: 10.1161/CIRCULATIONAHA.116.021884 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mishra S & Kass DA Cellular and molecular pathobiology of heart failure with preserved ejection fraction. Nat Rev Cardiol 18:400–423, doi: 10.1038/s41569-020-00480-6 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Borlaug BA & Redfield MM Diastolic and systolic heart failure are distinct phenotypes within the heart failure spectrum. Circulation 123, 2006–2013; discussion 2014, doi: 10.1161/CIRCULATIONAHA.110.954388 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Figtree GA et al. Effects of Canagliflozin on Heart Failure Outcomes Associated With Preserved and Reduced Ejection Fraction in Type 2 Diabetes Mellitus. Circulation 139, 2591–2593, doi: 10.1161/CIRCULATIONAHA.119.040057 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Shah SJ et al. Research Priorities for Heart Failure With Preserved Ejection Fraction: National Heart, Lung, and Blood Institute Working Group Summary. Circulation 141, 1001–1026, doi: 10.1161/CIRCULATIONAHA.119.041886 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anker SD et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. The New England journal of medicine 385, 1451–1461, doi: 10.1056/NEJMoa2107038 (2021). [DOI] [PubMed] [Google Scholar]; EMPEROR-Preserved was the first large-scale randomized clinical trial to meet its primary endpoint in patients with HFpEF
- 18.Borlaug BA Evaluation and management of heart failure with preserved ejection fraction. Nat Rev Cardiol 17, 559–573, doi: 10.1038/s41569-020-0363-2 (2020). [DOI] [PubMed] [Google Scholar]
- 19.Kitzman DW & Shah SJ The HFpEF Obesity Phenotype: The Elephant in the Room. J Am Coll Cardiol 68, 200–203, doi: 10.1016/j.jacc.2016.05.019 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Obokata M, Reddy YNV, Pislaru SV, Melenovsky V & Borlaug BA Evidence Supporting the Existence of a Distinct Obese Phenotype of Heart Failure With Preserved Ejection Fraction. Circulation 136, 6–19, doi: 10.1161/CIRCULATIONAHA.116.026807 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Withaar C, Lam CSP, Schiattarella GG, de Boer RA & Meems LMG Heart failure with preserved ejection fraction in humans and mice: embracing clinical complexity in mouse models. Eur Heart J 42, 4420–4430, doi: 10.1093/eurheartj/ehab389 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schiattarella GG, Rodolico D & Hill JA Metabolic Inflammation in Heart Failure with Preserved Ejection Fraction. Cardiovascular research 117, 423–434, doi: 10.1093/cvr/cvaa217 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Packer M, Zannad F & Anker SD Heart Failure and a Preserved Ejection Fraction: A Side-by-Side Examination of the PARAGON-HF and EMPEROR-Preserved Trials. Circulation 144, 1193–1195, doi: 10.1161/CIRCULATIONAHA.121.056657 (2021). [DOI] [PubMed] [Google Scholar]
- 24.Kato ET et al. Effect of Dapagliflozin on Heart Failure and Mortality in Type 2 Diabetes Mellitus. Circulation 139, 2528–2536, doi: 10.1161/CIRCULATIONAHA.119.040130 (2019). [DOI] [PubMed] [Google Scholar]
- 25.Nassif ME et al. The SGLT2 inhibitor dapagliflozin in heart failure with preserved ejection fraction: a multicenter randomized trial. Nat Med 27, 1954–1960, doi: 10.1038/s41591-021-01536-x (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; The PRESERVED-HF randomized clinical trial showed that the SGLT2-inhibitor dapagliflozin unequivocally improved health status and exercise capacity in patients with HFpEF who fit a cardiometabolic, congested phenotype that is typically associated with high levels of systemic inflammation.
- 26.Abraham WT et al. Effect of empagliflozin on exercise ability and symptoms in heart failure patients with reduced and preserved ejection fraction, with and without type 2 diabetes. Eur Heart J 42, 700–710, doi: 10.1093/eurheartj/ehaa943 (2021). [DOI] [PubMed] [Google Scholar]
- 27.Schiattarella GG, Sequeira V & Ameri P Distinctive patterns of inflammation across the heart failure syndrome. Heart Fail Rev 26, 1333–1344, doi: 10.1007/s10741-020-09949-5 (2020). [DOI] [PubMed] [Google Scholar]
- 28.Rao VN, Fudim M, Mentz RJ, Michos ED & Felker GM Regional adiposity and heart failure with preserved ejection fraction. Eur J Heart Fail 22, 1540–1550, doi: 10.1002/ejhf.1956 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karlsson T et al. Contribution of genetics to visceral adiposity and its relation to cardiovascular and metabolic disease. Nat Med 25, 1390–1395, doi: 10.1038/s41591-019-0563-7 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Tibrewala A & Yancy CW Heart Failure with Preserved Ejection Fraction in Women. Heart Fail Clin 15, 9–18, doi: 10.1016/j.hfc.2018.08.002 (2019). [DOI] [PubMed] [Google Scholar]
- 31.Sorimachi H et al. Pathophysiologic importance of visceral adipose tissue in women with heart failure and preserved ejection fraction. Eur Heart J 42, 1595–1605, doi: 10.1093/eurheartj/ehaa823 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Packer M Epicardial Adipose Tissue May Mediate Deleterious Effects of Obesity and Inflammation on the Myocardium. J Am Coll Cardiol 71, 2360–2372, doi: 10.1016/j.jacc.2018.03.509 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Xue B et al. Leptin Mediates High-Fat Diet Sensitization of Angiotensin II-Elicited Hypertension by Upregulating the Brain Renin-Angiotensin System and Inflammation. Hypertension 67, 970–976, doi: 10.1161/HYPERTENSIONAHA.115.06736 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Packer M Leptin-Aldosterone-Neprilysin Axis: Identification of Its Distinctive Role in the Pathogenesis of the Three Phenotypes of Heart Failure in People With Obesity. Circulation 137, 1614–1631, doi: 10.1161/CIRCULATIONAHA.117.032474 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Paulus WJ Unfolding Discoveries in Heart Failure. N Engl J Med 382, 679–682, doi: 10.1056/NEJMcibr1913825 (2020). [DOI] [PubMed] [Google Scholar]
- 36.Yoon S et al. S-Nitrosylation of Histone Deacetylase 2 by Neuronal Nitric Oxide Synthase as a Mechanism of Diastolic Dysfunction. Circulation 143, 1912–1925, doi: 10.1161/CIRCULATIONAHA.119.043578 (2021). [DOI] [PubMed] [Google Scholar]
- 37.Schiattarella GG et al. Xbp1s-FoxO1 axis governs lipid accumulation and contractile performance in heart failure with preserved ejection fraction. Nat Commun 12, 1684, doi: 10.1038/s41467-021-21931-9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schiattarella GG et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature 568, 351–356, doi: 10.1038/s41586-019-1100-z (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study developed a novel, clinically meaningful, animal model of cardiometabolic HFpEF identifying metabolic inflammation (metainflammation) as a key driver of HFpEF pathogenesis.
- 39.Tong D et al. Female Sex Is Protective in a Preclinical Model of Heart Failure With Preserved Ejection Fraction. Circulation 140, 1769–1771, doi: 10.1161/CIRCULATIONAHA.119.042267 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hahn VS et al. Endomyocardial Biopsy Characterization of Heart Failure With Preserved Ejection Fraction and Prevalence of Cardiac Amyloidosis. JACC Heart Fail 8, 712–724, doi: 10.1016/j.jchf.2020.04.007 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This large human study of HFpEF patients who underwent endomyocardial biopsy showed that monocyte infiltration was greater in patients with HFpEF versus controls, suggesting that immune dysfunction may play an important role in driving HFpEF pathogenesis.
- 41.Marelli-Berg FM & Aksentijevic D Immunometabolic cross-talk in the inflamed heart. Cell Stress 3, 240–266, doi: 10.15698/cst2019.08.194 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Norata GD et al. The Cellular and Molecular Basis of Translational Immunometabolism. Immunity 43, 421–434, doi: 10.1016/j.immuni.2015.08.023 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Mouton AJ, Li X, Hall ME & Hall JE Obesity, Hypertension, and Cardiac Dysfunction: Novel Roles of Immunometabolism in Macrophage Activation and Inflammation. Circ Res 126, 789–806, doi: 10.1161/CIRCRESAHA.119.312321 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nishida K & Otsu K Inflammation and metabolic cardiomyopathy. Cardiovasc Res 113, 389–398, doi: 10.1093/cvr/cvx012 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Vyas V et al. Obesity and diabetes are major risk factors for epicardial adipose tissue inflammation. JCI Insight 6, doi: 10.1172/jci.insight.145495 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guzik TJ, Skiba DS, Touyz RM & Harrison DG The role of infiltrating immune cells in dysfunctional adipose tissue. Cardiovasc Res 113, 1009–1023, doi: 10.1093/cvr/cvx108 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Talukdar S et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med 18, 1407–1412, doi: 10.1038/nm.2885 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Winer DA et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med 17, 610–617, doi: 10.1038/nm.2353 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zeng Q et al. A Unique Population: Adipose-Resident Regulatory T Cells. Front Immunol 9, 2075, doi: 10.3389/fimmu.2018.02075 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Feuerer M et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med 15, 930–939, doi: 10.1038/nm.2002 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cipolletta D, Cohen P, Spiegelman BM, Benoist C & Mathis D Appearance and disappearance of the mRNA signature characteristic of Treg cells in visceral adipose tissue: age, diet, and PPARgamma effects. Proc Natl Acad Sci U S A 112, 482–487, doi: 10.1073/pnas.1423486112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vasanthakumar A et al. The transcriptional regulators IRF4, BATF and IL-33 orchestrate development and maintenance of adipose tissue-resident regulatory T cells. Nat Immunol 16, 276–285, doi: 10.1038/ni.3085 (2015). [DOI] [PubMed] [Google Scholar]
- 53.Wu D et al. Characterization of regulatory T cells in obese omental adipose tissue in humans. Eur J Immunol 49, 336–347, doi: 10.1002/eji.201847570 (2019). [DOI] [PubMed] [Google Scholar]
- 54.Deswal A et al. Cytokines and cytokine receptors in advanced heart failure: an analysis of the cytokine database from the Vesnarinone trial (VEST). Circulation 103, 2055–2059, doi: 10.1161/01.cir.103.16.2055 (2001). [DOI] [PubMed] [Google Scholar]
- 55.Briasoulis A, Androulakis E, Christophides T & Tousoulis D The role of inflammation and cell death in the pathogenesis, progression and treatment of heart failure. Heart Fail Rev 21, 169–176, doi: 10.1007/s10741-016-9533-z (2016). [DOI] [PubMed] [Google Scholar]
- 56.Rai A et al. Adaptive immune disorders in hypertension and heart failure: focusing on T-cell subset activation and clinical implications. J Hypertens 38, 1878–1889, doi: 10.1097/HJH.0000000000002456 (2020). [DOI] [PubMed] [Google Scholar]
- 57.Chung ES et al. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: results of the anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation 107, 3133–3140, doi: 10.1161/01.CIR.0000077913.60364.D2 (2003). [DOI] [PubMed] [Google Scholar]
- 58.Litvinukova M et al. Cells of the adult human heart. Nature 588, 466–472, doi: 10.1038/s41586-020-2797-4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study provided a picture of human heart at single-cell resolution which will be useful for the study of intercellular crosstalk.
- 59.Martini E et al. Single-Cell Sequencing of Mouse Heart Immune Infiltrate in Pressure Overload-Driven Heart Failure Reveals Extent of Immune Activation. Circulation 140, 2089–2107, doi: 10.1161/CIRCULATIONAHA.119.041694 (2019). [DOI] [PubMed] [Google Scholar]
- 60.Nevers T et al. Left Ventricular T-Cell Recruitment Contributes to the Pathogenesis of Heart Failure. Circulation. Heart failure 8, 776–787, doi: 10.1161/CIRCHEARTFAILURE.115.002225 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrated that T cells are necessary for the progression of pressure overload-induced heart failure.
- 61.Carrillo-Salinas FJ, Ngwenyama N, Anastasiou M, Kaur K & Alcaide P Heart Inflammation: Immune Cell Roles and Roads to the Heart. Am J Pathol 189, 1482–1494, doi: 10.1016/j.ajpath.2019.04.009 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Blanton RM, Carrillo-Salinas FJ & Alcaide P T-cell recruitment to the heart: friendly guests or unwelcome visitors? Am J Physiol Heart Circ Physiol 317, H124–H140, doi: 10.1152/ajpheart.00028.2019 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Borg N et al. CD73 on T Cells Orchestrates Cardiac Wound Healing After Myocardial Infarction by Purinergic Metabolic Reprogramming. Circulation 136, 297–313, doi: 10.1161/CIRCULATIONAHA.116.023365 (2017). [DOI] [PubMed] [Google Scholar]
- 64.Bansal SS et al. Activated T Lymphocytes are Essential Drivers of Pathological Remodeling in Ischemic Heart Failure. Circ Heart Fail 10, e003688, doi: 10.1161/CIRCHEARTFAILURE.116.003688 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; Study demonstrating the role of T cells in myocardial infarction-induced heart failure.
- 65.Forte E et al. Type 2 MI induced by a single high dose of isoproterenol in C57BL/6J mice triggers a persistent adaptive immune response against the heart. J Cell Mol Med 25, 229–243, doi: 10.1111/jcmm.15937 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hofmann U et al. Activation of CD4+ T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice. Circulation 125, 1652–1663, doi: 10.1161/CIRCULATIONAHA.111.044164 (2012). [DOI] [PubMed] [Google Scholar]
- 67.Bansal SS et al. Dysfunctional and Proinflammatory Regulatory T-Lymphocytes Are Essential for Adverse Cardiac Remodeling in Ischemic Cardiomyopathy. Circulation 139, 206–221, doi: 10.1161/CIRCULATIONAHA.118.036065 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ramos GC et al. Myocardial aging as a T-cell-mediated phenomenon. Proc Natl Acad Sci U S A 114, E2420–E2429, doi: 10.1073/pnas.1621047114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; Study exploring the dynamics of T cells in cardiac ageing, establishing causality between T cell behaviour and myocardial ageing.
- 69.Martini E et al. T Cell Costimulation Blockade Blunts Age-Related Heart Failure. Circulation research 127, 1115–1117, doi: 10.1161/CIRCRESAHA.119.316530 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; Study providing experimental evidence demonstrating the role T cell-targeting therapy in age-related heart failure.
- 70.Laroumanie F et al. CD4+ T cells promote the transition from hypertrophy to heart failure during chronic pressure overload. Circulation 129, 2111–2124, doi: 10.1161/CIRCULATIONAHA.113.007101 (2014). [DOI] [PubMed] [Google Scholar]; Seminal study pointing to the role of T cells in cardiac hypertrophy and heart failure development.
- 71.Kallikourdis M et al. T cell costimulation blockade blunts pressure overload-induced heart failure. Nat Commun 8, 14680, doi: 10.1038/ncomms14680 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; Study demonstrating therapeutic feasibility of targeting T cells in heart failure models.
- 72.Groschel C et al. T helper cells with specificity for an antigen in cardiomyocytes promote pressure overload-induced progression from hypertrophy to heart failure. Sci Rep 7, 15998, doi: 10.1038/s41598-017-16147-1 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ngwenyama N et al. CXCR3 regulates CD4+ T cell cardiotropism in pressure overload-induced cardiac dysfunction. JCI Insight 4, doi: 10.1172/jci.insight.125527 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sintou A et al. Mediastinal Lymphadenopathy, Class-Switched Auto-Antibodies and Myocardial Immune-Complexes During Heart Failure in Rodents and Humans. Front Cell Dev Biol 8, 695, doi: 10.3389/fcell.2020.00695 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Heinrichs M et al. The healing myocardium mobilizes a distinct B-cell subset through a CXCL13-CXCR5-dependent mechanism. Cardiovasc Res 117, 2664–2676, doi: 10.1093/cvr/cvab181 (2021). [DOI] [PubMed] [Google Scholar]
- 76.Horckmans M et al. Pericardial Adipose Tissue Regulates Granulopoiesis, Fibrosis, and Cardiac Function After Myocardial Infarction. Circulation 137, 948–960, doi: 10.1161/CIRCULATIONAHA.117.028833 (2018). [DOI] [PubMed] [Google Scholar]
- 77.Ngwenyama N et al. Isolevuglandin-Modified Cardiac Proteins Drive CD4+ T-Cell Activation in the Heart and Promote Cardiac Dysfunction. Circulation 143, 1242–1255, doi: 10.1161/CIRCULATIONAHA.120.051889 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rieckmann M et al. Myocardial infarction triggers cardioprotective antigen-specific T helper cell responses. J Clin Invest 129, 4922–4936, doi: 10.1172/JCI123859 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Basavalingappa RH et al. Viral myocarditis involves the generation of autoreactive T cells with multiple antigen specificities that localize in lymphoid and non-lymphoid organs in the mouse model of CVB3 infection. Mol Immunol 124, 218–228, doi: 10.1016/j.molimm.2020.06.017 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gil-Cruz C et al. Microbiota-derived peptide mimics drive lethal inflammatory cardiomyopathy. Science 366, 881–886, doi: 10.1126/science.aav3487 (2019). [DOI] [PubMed] [Google Scholar]
- 81.Komarowska I et al. Hepatocyte Growth Factor Receptor c-Met Instructs T Cell Cardiotropism and Promotes T Cell Migration to the Heart via Autocrine Chemokine Release. Immunity 42, 1087–1099, doi: 10.1016/j.immuni.2015.05.014 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Westermann D et al. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ Heart Fail 4, 44–52, doi: 10.1161/CIRCHEARTFAILURE.109.931451 (2011). [DOI] [PubMed] [Google Scholar]
- 83.Li N et al. The Th17/Treg imbalance exists in patients with heart failure with normal ejection fraction and heart failure with reduced ejection fraction. Clin Chim Acta 411, 1963–1968, doi: 10.1016/j.cca.2010.08.013 (2010). [DOI] [PubMed] [Google Scholar]
- 84.Lu X & Crowley SD Inflammation in Salt-Sensitive Hypertension and Renal Damage. Current Hypertension Reports 20, doi: 10.1007/s11906-018-0903-x (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liu R & Nikolajczyk BS Tissue Immune Cells Fuel Obesity-Associated Inflammation in Adipose Tissue and Beyond. Frontiers in Immunology 10, doi: 10.3389/fimmu.2019.01587 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Paulus WJ & Tschope C A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol 62, 263–271, doi: 10.1016/j.jacc.2013.02.092 (2013). [DOI] [PubMed] [Google Scholar]
- 87.Franssen C et al. Myocardial Microvascular Inflammatory Endothelial Activation in Heart Failure With Preserved Ejection Fraction. JACC Heart Fail 4, 312–324, doi: 10.1016/j.jchf.2015.10.007 (2016). [DOI] [PubMed] [Google Scholar]
- 88.van Heerebeek L et al. Diastolic stiffness of the failing diabetic heart: importance of fibrosis, advanced glycation end products, and myocyte resting tension. Circulation 117, 43–51, doi: 10.1161/CIRCULATIONAHA.107.728550 (2008). [DOI] [PubMed] [Google Scholar]
- 89.Macdougall CE et al. Visceral Adipose Tissue Immune Homeostasis Is Regulated by the Crosstalk between Adipocytes and Dendritic Cell Subsets. Cell Metab 27, 588–601 e584, doi: 10.1016/j.cmet.2018.02.007 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mauro C et al. Obesity-Induced Metabolic Stress Leads to Biased Effector Memory CD4(+) T Cell Differentiation via PI3K p110delta-Akt-Mediated Signals. Cell Metab 25, 593–609, doi: 10.1016/j.cmet.2017.01.008 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhou T et al. Upregulation of SLAMF3 on human T cells is induced by palmitic acid through the STAT5-PI3K/Akt pathway and features the chronic inflammatory profiles of type 2 diabetes. Cell Death Dis 10, 559, doi: 10.1038/s41419-019-1791-y (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Guzik TJ et al. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med 204, 2449–2460, doi: 10.1084/jem.20070657 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study pinpoints immune cells as mediators of hypertensive effects on vasculature.
- 93.Ismahil MA et al. Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure: critical importance of the cardiosplenic axis. Circ Res 114, 266–282, doi: 10.1161/CIRCRESAHA.113.301720 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hulsmans M et al. Cardiac macrophages promote diastolic dysfunction. The Journal of experimental medicine 215, 423–440, doi: 10.1084/jem.20171274 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; Data reported in this study implicate cardiac macrophages as therapeutic targets for cardiac fibrosis leading to diastolic dysfunction.
- 95.Short JD et al. Dyslipidemic Diet-Induced Monocyte “Priming” and Dysfunction in Non-Human Primates Is Triggered by Elevated Plasma Cholesterol and Accompanied by Altered Histone Acetylation. Front Immunol 8, 958, doi: 10.3389/fimmu.2017.00958 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ecker J et al. Induction of fatty acid synthesis is a key requirement for phagocytic differentiation of human monocytes. Proc Natl Acad Sci U S A 107, 7817–7822, doi: 10.1073/pnas.0912059107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.DeBerge M, Shah SJ, Wilsbacher L & Thorp EB Macrophages in Heart Failure with Reduced versus Preserved Ejection Fraction. Trends in molecular medicine 4, 328–340, doi: 10.1016/j.molmed.2019.01.002 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tabas I & Bornfeldt KE Macrophage Phenotype and Function in Different Stages of Atherosclerosis. Circ Res 118, 653–667, doi: 10.1161/CIRCRESAHA.115.306256 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sager HB et al. Proliferation and Recruitment Contribute to Myocardial Macrophage Expansion in Chronic Heart Failure. Circ Res 119, 853–864, doi: 10.1161/circresaha.116.309001 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Stiekema LCA et al. Impact of cholesterol on proinflammatory monocyte production by the bone marrow. Eur Heart J 42, 4309–4320, doi: 10.1093/eurheartj/ehab465 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Swirski FK et al. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest 117, 195–205, doi: 10.1172/jci29950 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jaiswal S & Libby P Clonal haematopoiesis: connecting ageing and inflammation in cardiovascular disease. Nat Rev Cardiol 17, 137–144, doi: 10.1038/s41569-019-0247-5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Heyde A et al. Increased stem cell proliferation in atherosclerosis accelerates clonal hematopoiesis. Cell 184, 1348–1361 e1322, doi: 10.1016/j.cell.2021.01.049 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lam CS, Donal E, Kraigher-Krainer E & Vasan RS Epidemiology and clinical course of heart failure with preserved ejection fraction. Eur J Heart Fail 13, 18–28, doi: 10.1093/eurjhf/hfq121 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Marechaux S et al. Prognostic importance of comorbidities in heart failure with preserved left ventricular ejection fraction. Heart Vessels 26, 313–320, doi: 10.1007/s00380-010-0057-5 (2011). [DOI] [PubMed] [Google Scholar]
- 106.Lampropoulou V et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab 24, 158–166, doi: 10.1016/j.cmet.2016.06.004 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bajpai G et al. Tissue Resident CCR2- and CCR2+ Cardiac Macrophages Differentially Orchestrate Monocyte Recruitment and Fate Specification Following Myocardial Injury. Circ Res 124, 263–278, doi: 10.1161/CIRCRESAHA.118.314028 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Epelman S et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 40, 91–104, doi: 10.1016/j.immuni.2013.11.019 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pinto AR et al. An abundant tissue macrophage population in the adult murine heart with a distinct alternatively-activated macrophage profile. PLoS One 7, e36814, doi: 10.1371/journal.pone.0036814 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bajpai G et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat Med 24, 1234–1245, doi: 10.1038/s41591-018-0059-x (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Obokata M et al. Myocardial Injury and Cardiac Reserve in Patients With Heart Failure and Preserved Ejection Fraction. J Am Coll Cardiol 72, 29–40, doi: 10.1016/j.jacc.2018.04.039 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Shah SJ et al. Prevalence and correlates of coronary microvascular dysfunction in heart failure with preserved ejection fraction: PROMIS-HFpEF. European heart journal 39, 3439–3450, doi: 10.1093/eurheartj/ehy531 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yang JH et al. Endothelium-dependent and independent coronary microvascular dysfunction in patients with heart failure with preserved ejection fraction. Eur J Heart Fail 22, 432–441, doi: 10.1002/ejhf.1671 (2020). [DOI] [PubMed] [Google Scholar]
- 114.Mann DL Innate immunity and the failing heart: the cytokine hypothesis revisited. Circulation research 116, 1254–1268, doi: 10.1161/CIRCRESAHA.116.302317 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Dick SA & Epelman S Chronic Heart Failure and Inflammation: What Do We Really Know? Circ Res 119, 159–176, doi: 10.1161/CIRCRESAHA.116.308030 (2016). [DOI] [PubMed] [Google Scholar]
- 116.Sukumar M et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest 123, 4479–4488, doi: 10.1172/JCI69589 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Petrova G, Ferrante A & Gorski J Cross-reactivity of T cells and its role in the immune system. Crit Rev Immunol 32, 349–372, doi: 10.1615/critrevimmunol.v32.i4.50 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Marelli-Berg FM, Fu H & Mauro C Molecular mechanisms of metabolic reprogramming in proliferating cells: implications for T-cell-mediated immunity. Immunology 136, 363–369, doi: 10.1111/j.1365-2567.2012.03583.x (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Cho SH et al. Hypoxia-inducible factors in CD4(+) T cells promote metabolism, switch cytokine secretion, and T cell help in humoral immunity. Proc Natl Acad Sci U S A 116, 8975–8984, doi: 10.1073/pnas.1811702116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Huang H, Long L, Zhou P, Chapman NM & Chi H mTOR signaling at the crossroads of environmental signals and T-cell fate decisions. Immunol Rev 295, 15–38, doi: 10.1111/imr.12845 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nicolás-Ávila JA et al. A Network of Macrophages Supports Mitochondrial Homeostasis in the Heart. Cell, doi: 10.1016/j.cell.2020.08.031 (2020). [DOI] [PubMed] [Google Scholar]; This study shows that cardiac macrophages are responsible for the elimination of defective mitochondria in cardiomyocytes, preventing metabolic alterations and maintaining ventricular function.
- 122.DeBerge M et al. MerTK Cleavage on Resident Cardiac Macrophages Compromises Repair After Myocardial Ischemia Reperfusion Injury. Circ Res 121, 930–940, doi: 10.1161/circresaha.117.311327 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.van Teijlingen Bakker N & Pearce EJ Cell-intrinsic metabolic regulation of mononuclear phagocyte activation: Findings from the tip of the iceberg. Immunol Rev 295, 54–67, doi: 10.1111/imr.12848 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mills EL et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 167, 457–470 e413, doi: 10.1016/j.cell.2016.08.064 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Vats D et al. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab 4, 13–24, doi: 10.1016/j.cmet.2006.05.011 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Reklou A et al. Reduction of Vascular Inflammation, LDL-C, or Both for the Protection from Cardiovascular Events? The open cardiovascular medicine journal 12, 29–40, doi: 10.2174/1874192401812010029 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Doenst T, Nguyen TD & Abel ED Cardiac metabolism in heart failure: implications beyond ATP production. Circ Res 113, 709–724, doi: 10.1161/CIRCRESAHA.113.300376 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kolwicz SC Jr., Purohit S & Tian R Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circulation research 113, 603–616, doi: 10.1161/CIRCRESAHA.113.302095 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Palsson-McDermott EM & O’Neill LA The Warburg effect then and now: from cancer to inflammatory diseases. Bioessays 35, 965–973, doi: 10.1002/bies.201300084 (2013). [DOI] [PubMed] [Google Scholar]
- 130.Hoque R, Farooq A, Ghani A, Gorelick F & Mehal WZ Lactate reduces liver and pancreatic injury in Toll-like receptor- and inflammasome-mediated inflammation via GPR81-mediated suppression of innate immunity. Gastroenterology 146, 1763–1774, doi: 10.1053/j.gastro.2014.03.014 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Haas R et al. Lactate Regulates Metabolic and Pro-inflammatory Circuits in Control of T Cell Migration and Effector Functions. PLoS Biol 13, e1002202, doi: 10.1371/journal.pbio.1002202 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Pucino V et al. Lactate Buildup at the Site of Chronic Inflammation Promotes Disease by Inducing CD4(+) T Cell Metabolic Rewiring. Cell Metab 30, 1055–1074 e1058, doi: 10.1016/j.cmet.2019.10.004 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Murashige D et al. Comprehensive quantification of fuel use by the failing and nonfailing human heart. Science 370, 364–368, doi: 10.1126/science.abc8861 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Fernandez-Caggiano M & Eaton P Heart failure-emerging roles for the mitochondrial pyruvate carrier. Cell Death Differ 28, 1149–1158, doi: 10.1038/s41418-020-00729-0 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Fernandez-Caggiano M et al. Mitochondrial pyruvate carrier abundance mediates pathological cardiac hypertrophy. Nat Metab 2, 1223–1231, doi: 10.1038/s42255-020-00276-5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.McCommis KS et al. Nutritional modulation of heart failure in mitochondrial pyruvate carrier-deficient mice. Nat Metab 2, 1232–1247, doi: 10.1038/s42255-020-00296-1 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Narula J et al. Apoptosis in myocytes in end-stage heart failure. N Engl J Med 335, 1182–1189, doi: 10.1056/NEJM199610173351603 (1996). [DOI] [PubMed] [Google Scholar]
- 138.Halestrap AP, Wang X, Poole RC, Jackson VN & Price NT Lactate transport in heart in relation to myocardial ischemia. Am J Cardiol 80, 17A–25A (1997). [DOI] [PubMed] [Google Scholar]
- 139.Xu J et al. Intracellular lactate signaling cascade in atrial remodeling of mitral valvular patients with atrial fibrillation. J Cardiothorac Surg 8, 34, doi: 10.1186/1749-8090-8-34 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chouchani ET et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515, 431–435, doi: 10.1038/nature13909 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; Study demonstrating a critical role of succinate as mediator of myocardial ischemia/reperfusion injury.
- 141.Sadagopan N et al. Circulating succinate is elevated in rodent models of hypertension and metabolic disease. Am J Hypertens 20, 1209–1215, doi: 10.1016/j.amjhyper.2007.05.010 (2007). [DOI] [PubMed] [Google Scholar]
- 142.van Diepen JA et al. SUCNR1-mediated chemotaxis of macrophages aggravates obesity-induced inflammation and diabetes. Diabetologia 60, 1304–1313, doi: 10.1007/s00125-017-4261-z (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Rubic T et al. Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat Immunol 9, 1261–1269, doi: 10.1038/ni.1657 (2008). [DOI] [PubMed] [Google Scholar]
- 144.Littlewood-Evans A et al. GPR91 senses extracellular succinate released from inflammatory macrophages and exacerbates rheumatoid arthritis. J Exp Med 213, 1655–1662, doi: 10.1084/jem.20160061 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ridker PM et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. The New England journal of medicine 377, 1119–1131, doi: 10.1056/NEJMoa1707914 (2017). [DOI] [PubMed] [Google Scholar]
- 146.Tardif JC et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. The New England journal of medicine 381, 2497–2505, doi: 10.1056/NEJMoa1912388 (2019). [DOI] [PubMed] [Google Scholar]
- 147.Tong D et al. NAD(+) Repletion Reverses Heart Failure With Preserved Ejection Fraction. Circulation research 128, 1629–1641, doi: 10.1161/CIRCRESAHA.120.317046 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Abdellatif M et al. Nicotinamide for the treatment of heart failure with preserved ejection fraction. Sci Transl Med 13, doi: 10.1126/scitranslmed.abd7064 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zhang S et al. Efferocytosis Fuels Requirements of Fatty Acid Oxidation and the Electron Transport Chain to Polarize Macrophages for Tissue Repair. Cell Metab 29, 443–456 e445, doi: 10.1016/j.cmet.2018.12.004 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Martens CR et al. Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD(+) in healthy middle-aged and older adults. Nat Commun 9, 1286, doi: 10.1038/s41467-018-03421-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]