

Abstract
The marine bacterium Pseudoalteromonas sp. strain A28 was able to kill the diatom Skeletonema costatum strain NIES-324. The culture supernatant of strain A28 showed potent algicidal activity when it was applied to a paper disk placed on a lawn of S. costatum NIES-324. The condensed supernatant, which was prepared by subjecting the A28 culture supernatant to ultrafiltration with a 10,000-Mw-cutoff membrane, showed algicidal activity, suggesting that strain A28 produced extracellular substances capable of killing S. costatum cells. The condensed supernatant was then found to have protease and DNase activities. Two Pseudoalteromonas mutants lacking algicidal activity, designated NH1 and NH2, were selected after N-methyl-N′-nitrosoguanidine mutagenesis. The culture supernatants of NH1 and NH2 showed less than 15% of the protease activity detected with the parental strain, A28. The protease was purified to homogeneity from A28 culture supernatants by using ion-exchange chromatography followed by preparative gel electrophoresis. Paper-disk assays revealed that the purified protease had potent algicidal activity. The purified protease had a molecular mass for 50 kDa, and the N-terminal amino acid sequence was determined to be Ala-Thr-Pro-Asn-Asp-Pro. The optimum pH and temperature of the protease were found to be 8.8 and 30°C, respectively, by using succinyl-Ala-Ala-Pro-Phe-p-nitroanilide as a substrate. The protease activity was strongly inhibited by phenylmethylsulfonyl fluoride, diisopropyl fluorophosphate, antipain, chymostatin, and leupeptin. No significant inhibition was detected with EDTA, EGTA, phenanthroline or tetraethylenepentamine. These results suggest that Pseudoalteromonas sp. strain A28 produced an extracellular serine protease which was responsible for the algicidal activity of this marine bacterium.
There have recently been discussions concerning the roles of marine bacteria in algal bloom dynamics (7, 15, 22). Marine bacteria may both promote and regulate algal blooms (6, 9). The fact that marine bacteria selectively promote bloom formation by algal species has recently been reported (10). On the basis of laboratory experiments, it has also been reported that some bacteria are able to inhibit the growth of red-tide algae (12). In general, bacteria that inhibit algal growth are effective through direct or indirect attack (2, 17). For example, the gliding bacterium Cytophaga sp. strain J18/M01 effectively kills diatoms and raphidophytes when it is added to algal cultures but not when filtrate alone is added (direct attack) (12). Indirect attacks are thought to be chemically mediated (17). Recent studies have demonstrated the presence of bacteria that lyse algal cells by producing extracellular substances (2, 8). Alga-lytic bacteria have also been found in coastal environments where harmful algal blooms often occur (2, 8, 12–14, 17, 19, 23). It is therefore possible that bacteria having algicidal effects are involved in the termination and decomposition of algal blooms. However, virtually nothing is known about the mechanisms underlying algicidal effects at the molecular level.
A marine bacterium, Pseudoalteromonas (formerly named Alteromonas) sp. strain A28, which had potent algicidal effects on the diatom Skeletonema costatum was previously isolated (14). This organism was also able to kill the diatoms Thalassiosira and Eucampia zodiacs and the raphidophyte Chattonella antiqua. As the first step to investigate the algicidal activity of strain A28 at the molecular level, we developed a genetic transformation system by constructing a shuttle vector that replicates in both Escherichia coli and Pseudoalteromonas sp. strain A28. In the present paper, we describe genetic and biochemical evidence that an extracellular serine protease is responsible for the algicidal activity of strain A28. The protease, which was a monomeric protein having a molecular mass of about 50 kDa, showed high killing activity against S. costatum when it was purified to homogeneity.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Pseudoalteromonas sp. strain A28 is an algicidal bacterium isolated from the Ariake Sea of Japan (14). Pseudoalteromonas cells were grown at 28°C with shaking in ASWM medium, which was a modified SWM-III medium (4) supplemented with 0.1% Casitone (Difco) and 0.05% yeast extract (Difco). ASWM agar and soft agar were prepared by adding 1.5 and 0.8% agar (Difco) to ASWM medium, respectively.
Algal cultures.
The diatom S. costatum NIES-324 was obtained from The National Institute for Environmental Studies, Tsukuba, Japan. Clonal axenic cultures were routinely maintained on modified SWM-III medium made with filtered seawater as the base. The seawater was filtered through a 0.2-μm-pore-size Nuclepore filter and stored at 4°C in darkness. Cultures were grown at 20°C under an illumination of 35 microeinsteins m−2 s−1 on a 12-h light–12-h darkness regimen.
Isolation of mutants lacking algicidal activity.
Bacterial cells grown overnight in ASWM medium were inoculated into fresh ASWM medium (a 1% inoculum), and the cultures were incubated at 28°C for 4 h with shaking. Cells were then harvested by centrifugation (4,000 × g, 10 min, 25°C). Pelleted cells were resuspended in modified SWM-III medium, washed twice with the same medium, and resuspended in 0.3 of the original volume of modified SWM-III medium. Bacterial cells were mutagenized with 50 μg of N-methyl-N′-nitro-N-nitrosoguanidine (NTG) per ml at 28°C for 40 min. The cells were washed twice with modified SWM-III medium, resuspended in ASWM medium, and then incubated overnight with shaking at 28°C. Axenic cultures of S. costatum NIES-324 were grown in modified SWM-III medium for 1 week, and 1 ml of the S. costatum culture was mixed with 2.5 ml of molten ASWM soft agar (equilibrated to 47°C). The mixture was immediately poured onto an ASWM agar plate. After the agar solidified, bacteria mutagenized with NTG were transferred onto the agar plates with toothpicks, and the plates were incubated at 20°C under an illumination of 35 microeinsteins m−2 s−1 on a 12-h light–12-h darkness regimen. Bacterial colonies which failed to produce clear zones on lawns of S. costatum were picked, purified, and maintained on ASWM agar plates.
Enzyme purification.
A28 cultures were grown for 5 h at 28°C in ASWM medium, and the cells were harvested by centrifugation at 6,500 × g for 15 min. The culture supernatant was filtered through a 0.45-μm-pore-size nitrocellulose membrane filter (Advantec Inc., Tokyo, Japan) to remove any remaining bacteria. A 3-liter filtrate was concentrated to approximately 100 ml by using a stirred ultrafiltration cell equipped with a 10,000-Mw-cutoff membrane (Advantec Inc.). The concentrated sample was then dialyzed against Tris buffer (10 mM, pH 8.0). The dialyzed sample was applied to an anion-exchange column (Poros HQ/M, 4.6 by 100 mm; PerSeptive Biosystems Inc., Framingham, Mass.). The column was washed with Tris buffer (10 mM, pH 8.0), and proteins were eluted with a linear NaCl gradient of 0 to 1 M in Tris buffer (10 mM, pH 8.0). Fractions with high protease activities were pooled and stored at −80°C until they were used for preparative native-protein gel electrophoresis. Preparative native-protein gel electrophoresis was performed using a minipreparative cell (Bio-Rad). The lower gel (4 cm) contained 8% polyacrylamide and 376 mM Tris-HCl (pH 8.8), while the upper one (1 cm) contained 4% polyacrylamide and 124 mM Tris-HCl (pH 6.8). The electrode and elution buffers contained 25 mM Tris and 192 mM glycine (pH 8.3). The sample buffer contained 62 mM Tris-HCl (pH 6.8). A 500-μl sample was mixed with 500 μl of 25% (wt/vol) glycerol and 0.012% bromophenol blue stacking dye in 62.5 mM Tris-HCl (pH 6.8) before being applied to the gel column. Electrophoresis was conducted at 400 V and 3 mA. The elution of protease was complete after 5 h at a flow rate of 0.1 ml min−1. The protein concentration was determined by the bicinchoninic acid method (26). The purity of the protease was determined by electrophoresis on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels (16). Protein bands were visualized by using the silver stain method (20). SDS-PAGE was performed with a molecular standard which included rabbit muscle phosphorylase b (97 kDa), bovine serum albumin (67 kDa), rabbit muscle aldolase (42 kDa), bovine erythrocyte carbonic anhydrase (30 kDa), soybean trypsin inhibitor (20 kDa), and egg white lysozyme (14 kDa). The native-protein molecular mass was measured by gel filtration (Superdex 200 column, l.0 by 30 cm; Pharmacia) with a flow rate of 0.5 ml min−1 (with buffer containing 25 mM potassium phosphate–0.1 M NaCl [pH 7.0]). The molecular standard used for gel filtration chromatography included bovine serum albumin (67 kDa), hen egg ovalbumin (43 kDa), bovine pancreas chymotrypsinogen (25 kDa), and bovine pancreas RNase A (14 kDa). To determine the N-terminal amino acid sequence, the purified protease was subjected to SDS–12.5% PAGE and then electroblotted onto an Immobilon-P membrane (Millipore Corporation, Bedford, Mass.) according to the manufacturer's instructions. The N-terminal amino acid sequence was determined with a Procise protein sequencing system (Applied Biosystems, Foster City, Calif). An amino acid sequence similarity search was done with the FASTA program (21) with the Protein Identification Resource amino acid sequence database.
Enzyme assays.
The protease activity during purification was measured as azocasein (Sigma) hydrolytic activity. A reaction mixture (0.5 ml) containing 10 mg of azocasein and enzyme solution appropriately diluted in 250 mM Tris-HCl buffer (pH 7.8) was incubated at 30°C for 30 min. The reaction was stopped by adding 0.5 ml of cold 10% trichloroacetic acid to the reaction mixture. The precipitate was removed by centrifugation at 10,000 × g for 5 min, and the absorbance of the supernatant at 400 nm was measured. One unit of protease activity was defined as the amount of enzyme that caused an incremental change of one absorbance unit per hour. The protease activity was also measured by using succinyl-Ala-Ala-Pro-Phe-p-nitroanilide (Sigma) as a substrate. A standard assay mixture contained 2.5 mM succinyl-Ala-Ala-Pro-Phe-p-nitroanilide, 50 mM Tris-HCl (pH 7.8), and 2% dimethylformamide. After 10 min of incubation, the absorbance of the reaction mixture was measured at 410 nm. One unit of the enzyme was expressed as the enzymatic activity giving an absorbance of 1.0 under the above-described conditions. The pH dependence of activity was determined by using 50 mM acetate buffer at pHs of 4.2 and 5.0, 50 mM phosphate buffer at pHs of 6.0 and 7.0, and 50 mM Tris buffer at pHs of 7.8, 8.8, and 9.3. Protease activity was measured at 30°C.
Cellulase and amylase activities were determined by measuring the release of reducing sugars by the dinitrosalicylic acid method (18). The reaction mixture contained 0.1 ml of bacterial culture supernatant and 0.9 ml of 50 mM Tris buffer (pH 7.8) supplemented with either 2.5% carboxymethyl cellulose or 2.5% soluble starch. The reaction mixture was incubated at 30°C for 1 h, and the reaction was stopped by boiling the mixture for 10 min. DNase activity was determined by the methods of Blaschek and Klacik (3). To determine the DNase activity, 75 μl of a 1-mg/ml DNA (type XIV; Sigma) stock solution and 725 μl of DNase buffer (20 mM Tris-HCl [pH 8.0], 50 mM NaCl, 10 mM MgCl2, 5 mM β-mercaptoethanol) were added to a 1.5-ml microcentrifugation tube. After prewarming of the solution at 30°C for 5 min, 0.1 ml of culture supernatant was added and mixed. The polymerized DNA present at time zero and remaining after 60 min at 30°C was precipitated with 0.1 ml of 5 N HCl. Turbidity was developed at 37°C for 10 min and was estimated by reading the absorbance at 600 nm. DNase activity (1 U) was defined as the amount of enzyme depolymerizing 1 μg of DNA per min at 30°C.
To assess the algicidal activity of the protease, S. costatum cells were first grown for 4 days on modified SWM-III agar plates to form algal lawns. Enzyme solutions, which were sterilized by being filtered through a 0.2-μm-pore-size polyethersulfone membrane filter (Kurabo, Osaka, Japan), were applied to 8-mm-diameter paper disks (Advantec Inc.) on the S. costatum lawns. The plates were incubated overnight at 20°C under illumination. Algicidal activity was assessed by the presence of clear zones around the paper disks.
Inhibitor studies.
Protease inhibitors tested in the present study were phenylmethylsulfonyl fluoride (PMSF; Sigma), diisopropyl fluorophosphate (DFP; Katayama, Osaka, Japan), leupeptin (Nacalai Tesque, Inc., Kyoto, Japan), antipain (Nacalai Tesque, Inc.), chymostatin (Sigma), pepstatin (Nacalai Tesque, Inc.), 1,10-phenanthroline (Sigma), tetraethylenepentamine (Sigma), EDTA (Sigma), and EGTA (Sigma). The mixture of each protease inhibitor and enzyme solution appropriately diluted in 50 mM Tris-HCl (pH 7.8) was incubated at room temperature for 30 min before succinyl-Ala-Ala-Pro-Phe-p-nitroanilide was added. Protease activity was measured at 30°C by the method described above.
RESULTS
Algicidal activity of the culture supernatant of strain A28.
The culture supernatant of strain A28 showed potent algicidal activity. When the A28 culture supernatant was applied to a paper disk placed on the lawn of S. costatum NIES-324 cells, clear zones were detected around the paper disk (Fig. 1A, panel a). No clear zone was detected with fresh ASWM medium (Fig. 1A, panel b). The algicidal activity of the A28 culture supernatant was labile to heating at 100°C for 15 min (Fig. 1A, panel c). The culture supernatant was then subjected to ultrafiltration with a 10,000-Mw-cutoff membrane, and the filtrate and concentrated supernatant were examined for the ability to kill S. costatum NIES-324 by using the paper disk assay technique. The concentrated supernatant showed algicidal activity (Fig. 1A, panel d), whereas the filtrate failed to form clear zones around paper disks (Fig. 1A, panel e). These results suggest that Pseudoalteromonas sp. strain A28 produced extracellular substances having algicidal activities.
FIG. 1.

Detection of algicidal activity. Algicidal activity was detected as described in Materials and Methods. Zones of clearing around paper disks indicate the lysis of the diatom S. costatum strain NIES-324. (A) Algicidal activity in A28 culture supernatants and samples from each of the purification steps. a, A28 culture supernatant (20 μl [0.16 U of protease activity]); b, fresh ASWM medium (20 μl); c, heated culture supernatant (20 μl); d, culture supernatant concentrated by ultrafiltration and reconstituted with fresh ASWM medium (0.16 U of protease activity); e, ultrafiltrate of culture supernatant (20 μl); f, protease I-rich fraction from a Poros HQ/M anion-exchange chromatography (0.16 U of protease activity); g, protease II-rich fraction from a Poros HQ/M anion-exchange chromatography (0.16 U of protease activity); h, protease I-rich fraction from preparative native-protein gel electrophoresis (0.16 U of protease activity). (B) Algicidal activity with Pseudoalteromonas strains A28, NH1, and NH2.
To identify the extracellular substances, the concentrated supernatants were further examined for their activities of various enzymes, including protease, DNase, cellulase, and amylase. The concentrated A28 supernatants showed protease and DNase activities (Table 1), whereas cellulase and amylase activities were not detected (data not shown). In addition, the agar plate assay convincingly showed that A28 cells had DNase activity (data not shown).
TABLE 1.
Enzymatic activities of the culture supernatants of Pseudoalteromonas sp. strain A28 and two mutants lacking algicidal activity
| Strain | Enzymatic activity (U ml of supernatant−1)
|
|
|---|---|---|
| Proteasea | DNaseb | |
| A28 | 8.2 | 9.4 |
| NH1 | 0.9 | 8.8 |
| NH2 | 1.1 | 9.0 |
Protease activity was measured by using azocasein as a substrate. One unit of protease activity was defined as the amount of enzyme that caused an incremental change of one absorbance unit per hour.
One unit of DNase activity was defined as the amount of enzyme depolymerizing 1 μg of DNA per min at 30°C.
Isolation of mutants lacking algicidal activity.
To investigate whether the enzymatic activities detected with the A28 culture supernatant are required to kill S. costatum NIES-324, we first isolated mutants lacking algicidal activity after NTG mutagenesis. A total of approximately 3,000 clones were examined for the ability to kill S. costatum NIES-324, and two mutants, designated NH1 and NH2, were unable to form detectable plaques on the S. costatum NIES-324 lawns (Fig. 1B). It was also confirmed that neither NH1 nor NH2 killed S. costatum NIES-324 in mixed algal-bacterial cultures (Fig. 2). The culture supernatant of either NH1 or NH2 showed at most about 13% of the protease activity detected with the parental strain, A28 (Table 1). Both NH1 and NH2 had DNase activities comparable to that of the parental strain (Table 1). These results suggest that the extracellular protease of strain A28 is responsible for the algicidal effects.
FIG. 2.

Influence of Pseudoalteromonas sp. strains A28, NH1, and NH2 on the growth of S. costatum strain NIES-324. S. costatum was grown in modified SWM-III medium in the presence of A28 (A), NH1 (B), or NH2 (C). Bacterial cells of A28, NH1, or NH2 were added to the S. costatum culture 4 days after the start of cultivation, as indicated by the arrows. Symbols: ●, algal cells; ○, bacterial cells. The S. costatum NIES-324 and bacterial cells were counted as described previously (14).
Protease purification.
Protease was purified to homogeneity from the concentrated culture supernatant of strain A28 by ion-exchange chromatography, followed by preparative gel electrophoresis. The results of a typical enzyme purification procedure are summarized in Table 2. Chromatography of the concentrated culture supernatant on a Poros HQ/M anion-exchange column resolved two peaks of protease activity (Fig. 3). The two peaks of activity were eluted with approximately 300 and 400 mM NaCl, respectively. However, paper disk assays revealed that only the first peak fraction had algicidal activity (Fig. 1A, panels f and g). The proteases which were detected in the first and second peak fractions were designated protease I and protease II, respectively. Protease I, which showed algicidal activity, was further purified by using preparative native-protein gel electrophoresis. After preparative native-protein gel electrophoresis, SDS-PAGE analysis showed a single protein band (Fig. 4). When protease I was applied to a paper disk placed on the lawn of S. costatum NIES-324 cells, clear zones were detected around the paper disk (Fig. 1A, panel h). Agar blocks in the clear zones were then cut out and inoculated into fresh modified SWM-III medium. No growth or S. costatum was observed (data not shown), confirming that protease I is algicidal and not algistatic (data not shown).
TABLE 2.
Purification of protease I from the culture supernatant of Pseudoalteromonas sp. strain A28
| Sample analyzed | Total protein (mg) | Total activity (U) | Sp act (U mg of protein−1) | Purificationa (fold) | Yield (%) |
|---|---|---|---|---|---|
| Culture supernatant | 613 | 1,701 | 3 | 1 | 100 |
| Concentrated supernatantb | 41.6 | 1,586 | 38 | 13 | 93 |
| Poros HQ/M column peak I | 3.0 | 496 | 165 | 55 | 29 |
| Preparative electrophoresis peak | 0.9 | 184 | 204 | 68 | 11 |
Calculated from the specific activities.
Concentrated by ultrafiltration with a 10,000-Mw-cutoff membrane.
FIG. 3.

Elution profiles of the protease activities of concentrated culture supernatants of Pseudoalteromonas sp. strain A28 from a Poros HQ/M anion-exchange column. The column was developed with a linear NaCl gradient of 0 to 1 M in Tris buffer (10 mM, pH 8.0). Protease activity was detected using the azocasein assay as described in Materials and Methods.
FIG. 4.
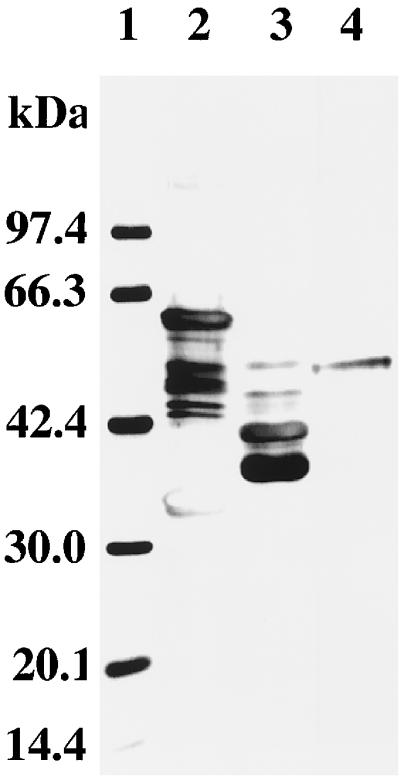
SDS-PAGE analysis of protease I-enriched fractions obtained during enzyme purification. The pooled samples from each of the purification steps were subjected to electrophoresis and silver stained. Lane 1, molecular mass markers; lane 2, concentrated culture supernatant (20 μg); lane 3, protease I-rich fraction from anion-exchange chromatography (10 μg); and lane 4, protease I-rich fraction from preparative native-protein gel electrophoresis (3 μg).
Enzyme properties.
By means of SDS-PAGE, the molecular mass of protease I was estimated to be 50 kDa. Since the molecular mass of protease I was also estimated to be 50 kDa by gel filtration, protease I should be a monomer (data not shown). Protease I was able to cleave succinyl-Ala-Ala-Pro-Phe-p-nitroanilide. However, neither succinyl-Ala-Ala-Val-Ala-p-nitroanilide nor tosyl-Gly-Pro-Lys-p-nitroanilide was cleaved by protease I. The optimum temperature for succinyl-Ala-Ala-Pro-Phe-p-nitroanilide hydrolysis activity was 30°C. The enzyme had 58, 87, and 55% of the optimum activity at 50, 37, and 22°C, respectively. The optimum pH was 8.8, and about 4, 5, 13, 81, 86, and 61% of the enzyme activity at pH 8.8 were detected at pHs 4.2, 5.0, 6.0, 7.0, 7.8, and 9.3, respectively. The protease activity was abolished by incubation at 68°C for 1 h or 100°C for 15 min (data not shown).
Ten enzyme inhibitors were tested for the ability to block the hydrolysis of succinyl-Ala-Ala-Pro-Phe-p-nitroanilide. PMSF (1 mM), DFP (1 mM), and chymostatin (0.1 mM), which are inhibitors of serine proteases, completely inhibited the activity of protease I (data not shown). Antipain (0.1 mM) and leupeptin (1 mM), which inhibit both serine and cysteine proteases, also caused complete inhibition of protease I activity (data not shown). No significant inhibition was detected with the metal protease inhibitors, including EDTA (1 mM), EGTA (1 mM), 1,10-phenanthroline (1 mM), and tetraethylenepentamine (1 mM). Pepstatin (1 mM), an inhibitor of aspartic protease, did not inhibit the activity of protease I.
The N-terminal amino acid sequence of purified protease I was determined to be Ala-Thr-Pro-Asn-Asp-Pro. Only six N-terminal amino acids could be determined because cleavage of the peptide bonds after Pro proceeded very slowly (Procise protein sequencing system user's manual, Applied Biosystems). A computer-assisted similarity search revealed that the N-terminal amino acid sequence of protease I was identical to that of the mature alkaline serine protease of Alteromonas sp. strain O-7 (29).
DISCUSSION
Several strains of marine bacteria have been found to be lethal to harmful algal bloom species (2, 8, 12–14, 17, 19, 23). They include Cytophaga sp. strain A5Y (19), Cytophaga sp. strain J18/M01 (12), Saprospira sp. strain SS90-1 (23), Alteromonas sp. strains D, K, R, and S (13), Pseudoalteromonas sp. strain A28 (14), Pseudoalteromonas sp. strain Y (17), Pseudomonas sp. strain T827/2B (2), and Flavobacterium sp. strain 5N-3 (9). These bacteria appear to be effective through direct or indirect attack (2, 17). Direct attacks require cell-to-cell contact between bacteria and algae. For example, Cytophaga sp. strain A5Y had algicidal effects on the diatoms S. costatum, Ditylum brightwellii, and Thalassiosira, as well as the raphidophyte C. antiqua, when it was added to algal cultures but not when filtrate alone was added (19). Similarly, Alteromonas sp. strains R and S also showed algicidal effects through direct attacks (13). In contrast, Alteromonas sp. strains K and D, Pseudoalteromonas sp. strain Y, and Flavobacterium sp. strain 5N-3 are known to exhibit indirect attacks (8, 13, 17). While algicidal effects were detected with the culture supernatants of these bacteria, their physical contact with algal cells was not required (8, 13, 17). Fukami et al. (8) reported that Flavobacterium sp. strain 5N-3 produced a basic compound with a molecular mass of less than 500 Da to kill the dinoflagellate Gymnodinium nagasakiense. It has also been reported that Pseudomonas sp. strain T827/2B killed the diatom Thalassiosira pseudonana by excreting a heat-labile compound having a relatively high molecular weight (2). However, none of these substances have been identified, and the mechanisms for the algicidal effects are still unclear.
It was previously reported that Pseudoalteromonas sp. strain A28 (formerly named Alteromonas sp. strain A28) was fatal to the diatoms Thalassiosira and E. zodiacs as well as the raphidophycean flagellate C. antiqua (14). The present data convincingly showed that a serine protease, designated protease I, was responsible for the algicidal effects. The algicidal effects of the culture supernatants were excluded by ultrafiltration with a 10,000-Mw-cutoff membrane. Therefore, our strain is unlikely to excrete low-molecular-weight substances capable of killing S. costatum.
To our knowledge, this is the first report to demonstrate that an extracellular protease is responsible for the algicidal effects of a marine bacterium. Inhibition studies revealed that DFP and PMSF abolished protease I activity. Both DFP and PMSF are known to be serine protease inhibitors which irreversibly react with the active-site serine residues (5, 11). Antipain and leupeptin also caused complete inhibition of protease I activity. Although antipain and leupeptin are cysteine protease inhibitors, they are also known to inhibit the serine protease trypsin (1, 27). These results suggest that protease I is a serine protease. The N-terminal amino acid sequence of protease I was identical to that of the alkaline serine protease (AprI, class I subtilase) of Alteromonas sp. strain O-7 (25, 29). Alteromonas sp. strain O-7 was isolated from a sediment sample at the Sagami Bay of Japan as a chitin-degrading bacterium (28). The molecular mass of the mature AprI (35 kDa) is smaller than that of protease I (50 kDa). It is not known whether AprI has potent algicidal activity.
Strain A28 produced two proteases, protease I and protease II. Protease II was sensitive to PMSF, indicating that it was also a serine protease (data not shown). SDS-PAGE of purified protease II revealed two protein bands corresponding to molecular masses of 48 and 33 kDa (data not shown). However, nondenaturing PAGE of protease II showed a single band at 75 kDa. These results indicate that protease II is a heterodimer. Unlike protease I, protease II did not show any algicidal activity. This may suggest that protease II had substrate specificities different from those of protease I. When bovine serum albumin was degraded by protease I and protease II, the patterns of cutting were not identical (data not shown). Alternatively, they may have different affinities for S. costatum cells. The affinity of the protease for algal cells should be of importance for causing the algicidal effects. It has been reported that Rarobacter faecitabidus produced an extracellular protease with yeast-lytic activity (24). The R. faecitabidus protease was a chimera of a serine protease on the NH2-terminal side and a mannose-binding domain with a lectin-like affinity for mannose on the COOH-terminal side. When the mannose-binding domain was truncated, the mutant protease showed normal protease activity but failed to lyse yeast cells. Some commercially available proteases, including trypsin, pepsin, subtilisin, and pronase, were examined for their algicidal activities, but none of them showed algicidal activity (data not shown).
ACKNOWLEDGMENTS
This work was supported in part by a grant from the Fisheries Agency of Japan.
We thank K. Nakashima and T. Inoue for technical assistance.
REFERENCES
- 1.Aoyagi T, Takeuchi T, Matsuzaki A, Kawamura K, Kondo S, Hamada M, Maeda K, Umezawa H. Leupeptins, new protease inhibitors from Actinomycetes. J Antibiot (Tokyo) 1969;22:283–286. doi: 10.7164/antibiotics.22.283. [DOI] [PubMed] [Google Scholar]
- 2.Baker K H, Herson D S. Interactions between the diatom Thallasiosira pseudonanna [sic] and an associated pseudomonad in a mariculture system. Appl Environ Microbiol. 1978;35:791–796. doi: 10.1128/aem.35.4.791-796.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blaschek H P, Klacik M A. Role of DNase in recovery of plasmid DNA from Clostridium perfringens. Appl Environ Microbiol. 1984;48:178–181. doi: 10.1128/aem.48.1.178-181.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen L C M, Edelstein T, MacLachlin J. Bonnemaisonia hamifera Hariot in nature and in culture. J Phycol. 1969;5:211–220. doi: 10.1111/j.1529-8817.1969.tb02605.x. [DOI] [PubMed] [Google Scholar]
- 5.Dixon G H, Kaufman D L, Neurath H. Amino acid sequence in the region of diisopropylphosphoryl binding in diisopropylphosphoryl-trypsin. J Biol Chem. 1958;233:1373–1381. [PubMed] [Google Scholar]
- 6.Doucette G J. Interactions between bacteria and harmful algae: a review. Nat Toxins. 1995;3:65–74. doi: 10.1002/nt.2620030202. [DOI] [PubMed] [Google Scholar]
- 7.Doucette G J, Kodama M, Franca S, Gallacher S. Bacterial interactions with harmful algal bloom species: bloom ecology, toxigenesis and cytology. NATO ASI Ser. 1998;41:619–647. [Google Scholar]
- 8.Fukami K, Yuzawa A, Nishijima T, Hata Y. Isolation and properties of a bacterium inhibiting the growth of Gymnodinium nagasakiense. Nippon Suisan Gakkaishi. 1992;58:1073–1077. [Google Scholar]
- 9.Fukami K, Sakaguchi K, Kanou M, Nishijima T. Effects of bacterial assemblages on the succession of blooming phytoplankton from Skeletonema costatum to Heterosigma akashiwo. In: Yasumoto T, Oshima Y, Fukuyo Y, editors. Harmful and toxic algal blooms. Paris, France: Intergovernmental Oceanographic Commission of UNESCO; 1996. pp. 335–338. [Google Scholar]
- 10.Furuki M, Kobayashi M. Interaction between Chattonella and bacteria and prevention of this red tide—EMECS '90. Mar Pollut Bull. 1991;23:189–193. [Google Scholar]
- 11.Gold A, Fahrney D. Sulfonyl fluorides as inhibitors of esterase. II. Formation and reactions of phenylmethylsulfonyl α-chymotrypsin. Biochemistry. 1964;3:783–791. doi: 10.1021/bi00894a009. [DOI] [PubMed] [Google Scholar]
- 12.Imai I, Ishida Y, Hata Y. Killing of marine phytoplankton by a gliding bacterium Cytophaga sp., isolated from the coastal sea of Japan. Mar Biol. 1993;116:527–532. [Google Scholar]
- 13.Imai I, Ishida Y, Sakaguchi K, Hata Y. Algicidal marine bacteria isolated from northern Hiroshima bay, Japan. Fish Sci. 1995;61:628–636. [Google Scholar]
- 14.Kato J, Amie J, Murata Y, Kuroda A, Mitsutani A, Ohtake H. Development of a genetic transformation system for an alga-lysing bacterium. Appl Environ Microbiol. 1998;64:2061–2064. doi: 10.1128/aem.64.6.2061-2064.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kayser H. Growth interaction between marine dinoflagellates in multispecies culture experiments. Mar Biol. 1979;52:357–369. [Google Scholar]
- 16.Laemmli U K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 17.Lovejoy C, Bowman J P, Hallegraeff G M. Algicidal effects of a novel marine Pseudoalteromonas isolate (class Proteobacteria, gamma subdivision) on harmful algal bloom species of the genera Chattonella, Gymnodinium, and Heterosigma. Appl Environ Microbiol. 1998;64:2806–2813. doi: 10.1128/aem.64.8.2806-2813.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller G L. Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal Chem. 1959;31:426–428. [Google Scholar]
- 19.Mitsutani A, Takesue K, Kirita M, Ishida Y. Lysis of Skeletonema costatum by Cytophaga sp. isolated from the coastal water of the Ariake Sea. Nippon Suisan Gakkaishi. 1992;58:2158–2167. [Google Scholar]
- 20.Ohsawa K, Ebata N. Silver stain for detecting 10-femtogram quantities of protein after polyacrylamide gel electrophoresis. Anal Biochem. 1983;135:409–415. doi: 10.1016/0003-2697(83)90703-0. [DOI] [PubMed] [Google Scholar]
- 21.Pearson W R, Lipman D J. Improved tools for biological sequence analysis. Proc Natl Acad Sci USA. 1988;85:2444–2448. doi: 10.1073/pnas.85.8.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Romalde J L, Toranzo A E, Barja J L. Changes in bacterial populations during red tides caused by Mesodinium rubrum and Gymnodinium catenatum in Northwest coast of Spain. J Appl Bacteriol. 1990;68:123–132. doi: 10.1111/j.1365-2672.1990.tb02556.x. [DOI] [PubMed] [Google Scholar]
- 23.Sakata T. Occurrence of marine Saprospira sp. possessing algicidal activity for diatoms. Nippon Suisan Gakkaishi. 1990;56:1165. [Google Scholar]
- 24.Shiomoi H, Iimura Y, Obata T, Tadenuma M. Molecular structure of Rarobacter faecitabidus protease I: a yeast-lytic serine protease having mannose-binding activity. J Biol Chem. 1992;267:25189–25195. [PubMed] [Google Scholar]
- 25.Siezen R J, de Vos W M, Leunissen J A, Dijkstra B W. Homology modelling and protein engineering strategy of subtilases, the family of subtilisin-like serine proteinases. Protein Eng. 1991;4:719–737. doi: 10.1093/protein/4.7.719. [DOI] [PubMed] [Google Scholar]
- 26.Smith P K, Krohn I, Hermanson G T, Mallia A K, Gartner F H, Provenzano M D, Fujimoto E K, Goeke N M, Olson B J, Klenk D C. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 27.Suda H, Aoyagi T, Hamada M, Takeuchi T, Umezawa H. Antipain, a new protease inhibitor isolated from Actinomycetes. J Antibiot (Tokyo) 1972;25:263–266. doi: 10.7164/antibiotics.25.263. [DOI] [PubMed] [Google Scholar]
- 28.Tsujibo H, Toshida Y, Imada C, Okami Y, Miyamoto K, Inamori Y. Isolation and characterization of a chitin degrading marine bacterium belonging to the genus Alteromonas. Nippon Suisan Gakkaishi. 1991;57:2127–2131. [Google Scholar]
- 29.Tsujibo H, Miyamoto K, Tanaka K, Kaidzu Y, Imada C, Okami Y, Inamori Y. Cloning and sequence analysis of a protease-encoding gene from the marine bacterium Alteromonas sp. strain O-7. Biosci Biotechnol Biochem. 1996;60:1284–1288. doi: 10.1271/bbb.60.1284. [DOI] [PubMed] [Google Scholar]