

Abstract
Sixteen compounds (TR1–TR16) were synthesized and evaluated for their inhibitory activities against monoamine oxidase A and B (MAOs). Most of the derivatives showed potent and highly selective MAO-B inhibition. Compound TR16 was the most potent inhibitor against MAO-B with an IC50 value of 0.17 μM, followed by TR2 (IC50 = 0.27 μM). TR2 and TR16 selectivity index (SI) values for MAO-B versus MAO-A were 84.96 and higher than 235.29, respectively. Compared to the basic structures, the para-chloro substituent in TR2 and TR16 increased the inhibitory activity of MAO-B. TR2 and TR16 were reversible MAO-B inhibitors that were competitive, with Ki values of 0.230 ± 0.004 and 0.149 ± 0.016 µM, respectively. The PAMPA method indicated that compounds TR2 and TR16 had the tendency to traverse the blood–brain barrier. Docking investigations revealed that lead compounds were beneficial for MAO-B inhibition via association with key as well as selective E84 or Y326 residues, but not for MAO-A inhibition via interaction primarily driven by hydrophobic contacts. In conclusion, TR2 and TR16 are therapeutic prospects for the management of multiple neurodegenerative diseases.
Keywords: pyridazinones, monoamine oxidase-B, kinetics, reversibility, PAMPA, docking
1. Introduction
Parkinson’s disease (PD) is a neurological illness that affects 6.1 million individuals throughout the world [1]. Parkinsonism is a syndrome characterized by rigidity, bradykinesia, rest tremor, and postural instability [1,2,3]. PD is defined pathologically by the depletion of dopamine neurons in nigra pars compacta, which is followed by Lewy bodies, which are cytoplasmic inclusions within the midbrain comprising insoluble alpha-synuclein clusters. Nevertheless, PD is distinguished by more common pathology in different areas of the brain, which encompasses nondopaminergic neurons. [2]. The symptoms of disease can be treated with drugs that increase the level of dopamine, though they are not so effective.
Monoamine oxidases (MAO-A and MAO-B) are key enzymes that deaminate biogenic amines in the tissues of both peripheral and brain regions, controlling neurotransmitter levels including dopamine, norepinephrine, and epinephrine [4,5]. Selective MAO-A inhibitors have been shown to be useful in the management of depression [6]. Selective MAO-B inhibitors limit dopamine degradation in the central nervous system (CNS), blocking dopamine reduction. MAO-B inhibitors not only impede dopamine degradation, but also diminish the generation of neurotoxic byproducts of the MAO enzymatic reaction, such as hydrogen peroxide and aldehyde. L-Dopa has always remained the cornerstone of Parkinson’s therapy and continues to be the most efficacious suggestive therapeutic medication. L-Dopa is often used with selective MAO-B inhibitors. As a corollary, adopting MAO-B inhibitors in the initial phases of the disease may postpone the emergence of significant symptoms and the necessity for L-dopa. [7].
MAO inhibitors have various scaffolds with different structural frameworks such as anilide, benzothiazinone, chalcone, chromone, coumarin, enamide, hydrazone, indolalkylamine, pyrazoline, oxazolidinones, and propargylamine [8,9,10,11,12,13,14]. Pyridazinone is a six-membered cyclic hydrazine non-aromatic heterocyclic ring. The ring features in two conterminous nitrogen atoms, with one endocyclic double bond and one carbonyl functional moiety bearing from the unit. Diverse pharmacologic activity investigations on molecules with the 3(2H)-pyridazinone framework have been undertaken. Multiple bioactivities have really been documented for the compounds, including analgesic, anti-inflammatory, antihypertensive, cardiotonic, antiplatelet, anticholinesterase, anti-bacterial, antifungal, and antitumoral effects. [15,16]. In particular, pyridazinone derivatives are recognized as agents with noteworthy effects in the cardiovascular system attributed to their inhibition of platelet aggregation, antihypertensive activity, and cardiotonic qualities (Table 1).
Table 1.
Some therapeutic drugs with pyridazinone core.
| Structure | Compound | Activity |
|---|---|---|

|
Zardaverine | Cardiotonic |

|
Levosimendan | Vasodilator |

|
Pimobendan | Vasodilator |

|
Imazodan | Cardiotonic |

|
Emorfazone | Analgesic |
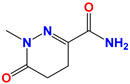
|
Medazomide | Antitussif |
In our previous reports, the synthesis of 3(2H)-pyridazinone candidates and the exploration of their MAO inhibitory properties were described. In the reports, new classes of pyridazinone core with substitution of benzalhydrazones, piperazine and morpholine analogues were designed and synthesized [17]. In addition, it was hypothesized that the addition of substituted benzalhydrazone to the second position of pyridazinone and the substitution of electron withdrawing groups in the benzalhydrazone ring caused MAO-B inhibitory activity to increase [18].
In particular, in this study, we aimed to synthesize, characterize, and determine the MAO-B inhibition activity of pyridazinone derivatives according to the hypothesis as shown below (Scheme 1).
Scheme 1.

Rational design of the new hMAO inhibitors.
2. Results and Discussion
2.1. General Chemistry of Synthetic Scheme
The targeted pyridazinone compounds (TR1-16) were synthesized in accordance with the literature, as shown in Scheme 2.
Scheme 2.

Synthesis of the compounds TR1-16.
The nucleophilic substitution reaction of 3,6-dichloropyridazine with phenylpiperazine or morpholine derivatives in ethanol was carried out to produce 3-chloro-6-substituted pyridazine. The hydrolysis of 3-chloro-6-(4-(3,4-dichlorophenyl)piperazine-1-yl)pyridazine by heating in glacial acetic acid afforded 6-(4-(3-Methoxy/3-trifluoromethylphenyl)piperazine-1-yl/morpholino)-3(2H)-pyridazinone [19]. Subsequently, the reaction of 6-(4-(3-methoxy/3-tifluoromethylphenyl)piperazine-1-yl/morpholino)-3(2H)-pyridazinone with ethyl bromoacetate in the presence of K2CO3 in acetone replaced the hydrogen atom of tertiary nitrogen atom in the pyridazinone ring and formed ethyl 6-(4-(3-methoxy/3-trifluoromethylphenyl)piperazine-1-yl/morpholino)-3(2H)-pyridazinone-2-ylacetate. The introduction of hydrazine hydrate (99%) can remove the ethyl group from the former intermediate and generate corresponding acid hydrazides [20]. Finally, nucleophilic addition reaction with various substituted aromatic aldehydes with acid hydrazides resulted in the formation of final targeted candidates (TR1-TR16). In this study, all of the title compounds were published for the first time. The compounds’ reaction yields vary from around 57% to 92%. The maximum yield (91.67%) was achieved with compound TR8, while the lowest yield was achieved with compound TR3 (57.10%). All structures were validated using 1H-NMR, 13C-NMR and mass spectral data. Table 2 lists the molecular structures and physical characterization of TR1-TR16.
Table 2.
Chemical-physical data of titled compounds.

| |||||
| Entry | R1 | R2 | Yield (%) | M.P. (°C) | Molecular Formula |
| TR1 | OCH3 | H | 79.23 | 232–235 | C24H26N6O3 |
| TR2 | OCH3 | 4-Cl | 82.49 | 205 | C24H25ClN6O3 |
| TR3 | OCH3 | 4-F | 57.10 | 172 | C24H25FN6O3 |
| TR4 | OCH3 | 4-OCH3 | 67.08 | 154 | C25H28N6O4 |
| TR5 | OCH3 | 4-CH3 | 76.73 | 201–202 | C25H28N6O3 |
| TR6 | OCH3 | 4-Br | 82.54 | 214 | C24H25BrN6O3 |
| TR7 | CF3 | H | 76.90 | 126 | C24H23F3N6O2 |
| TR8 | CF3 | 4-Cl | 91.67 | 242 | C24H22ClF3N6O2 |
| TR9 | CF3 | 4-F | 78.09 | 224 | C24H22F4N6O2 |
| TR10 | CF3 | 4-OCH3 | 86.36 | 195 | C25H25F3N6O3 |
| TR11 | CF3 | 4-CH3 | 83.43 | 142 | C25H25F3N6O2 |
| TR12 | CF3 | 4-N(CH3)2 | 80.21 | 227 | C26H28F3N7O2 |
| TR13 | CF3 | 4-Br | 82.64 | 241 | C24H22BrF3N6O2 |

| |||||
| Entry | R2 | Yield (%) | M.P. (°C) | Molecular Formula | |
| TR14 | H | 71.22 | 206 | C17H19N5O3 | |
| TR15 | 4-F | 61.17 | 119 | C17H18FN5O3 | |
| TR16 | 4-Cl | 80.73 | 152 | C17H18ClN5O3 | |
Piperazine signals in compounds TR1–13 were observed approximately between 3.2 and 3.5 ppm in 1H-NMR. Morpholine signals in compounds TR14–16 were shifted between 3.2 and 3.68 ppm due to the oxygen atom in the ring (-OCH2-). Unlike other compounds TR7–16, compounds TR1–6 with -OCH3 at the R1 position had a singlet signal at 3.80 ppm in 1H-NMR and a signal at 55 ppm in 13C-NMR. Compounds TR5 and TR11 with -CH3 at the R2 position were confirmed by the signal at 2.39 ppm in 1H-NMR and the signal at 21 ppm in 13C-NMR. TR12 with 4-N(CH3)2 at the R2 position was confirmed by the signal at 3.04 ppm (6H, integral) in 1H-NMR.
2.2. Biochemistry
2.2.1. MAO Inhibition Studies
At 10 μM, all compounds evaluated exhibited higher than 50% residual activity for MAO-A; however, only six compounds showed lower than 50% residual activity for MAO-B (Table 3). TR16 had the strongest inhibitory action against MAO-B, with an IC50 value of 0.17 μM, followed by TR2 (IC50 = 0.27 μM) (Table 3). The substituent -Cl atom at the para position of TR16 showed 235.3- and 2.5-times higher MAO-B inhibitory activity over the basic structure TR14 (IC50 = >40 μM) and -F atom at the para position of TR15 (IC50 = 0.43 μM), respectively. On the other hand, the -Cl atom at the para position of TR2 increased MAO-B inhibitory potency 148.1-fold (IC50 = 0.27 μM) compared to the basic structure TR1 (IC50 = >40 μM). When TR2 and TR16 were compared, TR16 had a 1.59-fold higher IC50 for MAO-B than TR2. Both TR2 and TR16 showed selective inhibition for MAO-B with selectivity index (SI) values of >235.29 and 84.96, respectively (Table 3). The -OCH3 substituted derivatives at the R1 position (TR1-6) showed higher MAO-B inhibitory activity than the -CF3 substituted derivatives at the same position (TR7-13), except for the derivatives with -F substituent at the para position of R2, i.e., TR3 and TR9. However, substitutions of –OCH3 and -CF3 at the R1 position did not show a significant difference in MAO-A inhibitory activity.
Table 3.
Inhibition of MAO-A and MAO-B by the TR series a.
| Compounds | Residual Activity at 10 µM (%) | IC50 (µM) | SI b | ||
|---|---|---|---|---|---|
| MAO-A | MAO-B | MAO-A | MAO-B | ||
| TR1 | 82.04 ± 1.20 | 80.54 ± 1.01 | 17.32 ± 1.20 | >40 | <0.43 |
| TR2 | 65.85 ± 0.66 | −4.37 ± 4.13 | 22.94 ± 0.24 | 0.27 ± 0.02 | 84.96 |
| TR3 | 71.66 ± 0.71 | 20.11 ± 1.15 | 19.65 ± 0.30 | 2.11 ± 0.51 | 9.31 |
| TR4 | 61.00 ± 3.54 | 88.64 ± 8.61 | 16.53 ± 1.66 | >40 | <0.41 |
| TR5 | 72.83 ± 1.02 | 45.56 ± 2.56 | 18.19 ± 0.09 | 4.42 ± 0.23 | 4.12 |
| TR6 | 51.73 ± 4.55 | 70.23 ± 3.80 | 12.33 ± 0.11 | 15.12 ± 0.18 | 0.81 |
| TR7 | 55.40 ± 0.84 | 90.66 ± 6.64 | 14.43 ± 3.55 | >40 | <0.36 |
| TR8 | 52.87 ± 0.82 | 69.09 ± 0.95 | 12.02 ± 1.75 | 20.00 ± 0.00 | 0.60 |
| TR9 | 72.28 ± 0.40 | 46.51 ± 4.93 | 17.08 ± 0.21 | 1.00 ± 0.15 | 17.08 |
| TR10 | 56.00 ± 2.26 | 91.86 ± 7.09 | 13.28 ± 1.81 | >40 | <0.33 |
| TR11 | 55.65 ± 2.28 | 83.87 ± 0.56 | 18.54 ± 5.83 | 32.00 ± 1.21 | 0.57 |
| TR12 | 52.55 ± 1.77 | 85.33 ± 1.59 | 14.72 ± 2.96 | >40 | <0.37 |
| TR13 | 74.24 ± 6.43 | 100.32 ± 5.43 | 31.00 ± 4.33 | >40 | <0.78 |
| TR14 | 97.47 ± 0.71 | 106.69 ± 2.01 | >40 | >40 | 1.00 |
| TR15 | 85.59 ± 1.04 | 12.50 ± 3.54 | >40 | 0.43 ± 0.05 | >93.02 |
| TR16 | 85.00 ± 3.08 | 13.06 ± 0.64 | >40 | 0.17 ± 0.04 | >235.29 |
| Toloxatone | 1.08 ± 0.03 | - | |||
| Lazabemide | - | 0.11 ± 0.02 | |||
| Clorgyline | 0.007 ± 0.001 | - | |||
| Pargyline | - | 0.14 ± 0.01 | |||
a The results represent the averages of duplicate or triple studies, including standard errors. b SI values are presented for MAO-B compared with MAO-A. The IC50 concentration was determined to be 40 µM as the highest concentration.
2.2.2. Kinetic Study
Five different substrate concentrations were used in the kinetics. TR2 and TR16 met a point on the y-axis in the Lineweaver–Burk plots (Figure 1A,C), and their secondary plots exhibited Ki values of 0.230 ± 0.004 and 0.149 ± 0.016 µM, respectively, in the kinetic investigations for MAO-B. (Figure 1B,D). TR2 and TR16 are competitive inhibitors that compete with the substrate and bind to the MAO-B active site.
Figure 1.


Lineweaver–Burk plots for MAO-B inhibition by TR2 and TR16 (A,C), and their respective secondary plots (B,D) of slopes versus inhibitor concentrations.
2.2.3. Reversibility Studies
TR2 and TR16 concentrations in the reversibility studies were 0.54 and 0.34 μM, respectively, whereas the respective concentrations for the references, the reversible inhibitor lazabemide and the irreversible inhibitor pargyline, were 0.22 and 0.28 µM, respectively. The relative activity of undialyzed (AU) as well as dialyzed (AD) samples were compared to establish reversibility profiles. TR2 and TR16 inhibitions of MAO-B were restored from 28.0 % (AU) to 75.0 % (AD), and from 31.8% to 73.4%, respectively (Figure 2). When compared to the reference inhibitor, the compounds recovered to the levels comparable to lazabemide, the reference reversible inhibitor against MAO-B (varying between 25.0% and 73.4%), but were distinct from pargyline, a reference irreversible inhibitor against MAO-B. (i.e., varying between 34.5% and 35.2%). These data revealed that TR2 and TR16 were reversible inhibitors of MAO-B.
Figure 2.

Recoveries of MAO-B inhibitions by TR2 and TR16 using dialysis experiments.
2.3. Parallel Artificial Membrane Permeability Assay (PAMPA)
The blood–brain barrier (BBB) permeation of the lead pyridaziones molecules (TR2 and TR16) was confirmed by PAMPA [21]. The compounds showed good CNS permeabilities, with CNS+ positive standards such as progesterone and verapamil (Table 4).
Table 4.
BBB permeation of standard and TR compounds by PAMPA.
| Compounds | Bibliography Pe (×10−6 cm/s) a |
Experimental Pe (×10−6 cm/s) |
Prediction |
|---|---|---|---|
| Dopamine | 0.2 | 0.21 ± 0.01 | CNS- |
| Lomefloxacin | 1.1 | 1.13 ± 0.01 | CNS- |
| Verapamil | 16.0 | 15.35 ± 0.33 | CNS+ |
| Progesterone | 9.3 | 9.02 ± 0.17 | CNS+ |
| TR2 | 9.33 ± 0.33 | CNS+ | |
| TR16 | 10.62 ± 0.26 | CNS+ |
CNS-: Pe (10−6 cm/s) <2, less BBB permeable; CNS+, Pe (10−6 cm/s) >4, good BBB permeable. a from [21].
2.4. Computational Studies
2.4.1. Docking Studies
Molecular modeling simulations were performed to give a sound indication of molecular interactions responsible for MAO-A and MAO-B inhibitions. Figure 3 and Figure 4 show the best poses returned from docking simulations. For completeness, the 2D interaction schemes were enclosed as Supporting Information in Figures S49 and S50.
Figure 3.

Zoomed in view at MAO-A binding site. Best poses of TR2 (A), TR15 (B), and TR16 (C) were reported as green, cyan, and magenta sticks, respectively. For completeness, docking scores returned from simulations were equal to −7.99, −8.29, and −8.16 kcal/mol for TR2, TR15, and TR16, respectively.
Figure 4.

Zoomed in view at MAO-B binding site. Best poses of TR2 (A), TR15 (B), and TR16 (C) were reported as green, cyan, and magenta sticks, respectively. Red arrows and green lines indicate hydrogen bonds and π–π contacts, respectively. For completeness, docking scores returned from simulations were equal to −10.051, −9.452 and −8.558 kcal/mol for TR2, TR15 and TR16, respectively.
Compounds TR2, TR15, and TR16 interact within the MAO-A binding site through hydrophobic contacts. Morpholine ring of TR15 and TR16 faces the aromatic rings of FAD, Y407, and Y444, a binding room not engaged by the meta-methoxy ring of TR2, due to its bulky structure.
As shown in Figure 4, compound TR2 reaches the aromatic cage formed by FAD, Y398, and Y435 through its meta-methoxy ring and establishes a hydrogen bond with the side chain of E84, a MAO-B selective residue (changed to V93 in MAO-A). Moreover, compounds TR15 and TR16 engage the same interactions with Y398 through π–π contacts and with the phenol group of MAO-B selective residue Y326 by forming a hydrogen bond with the carbonyl groups of the pyridazinone rings. Overall, molecular docking analyses suggested that compounds were not very effective at engaging the MAO-A binding pocket, due to the interaction mainly driven by hydrophobic contacts. Finally, the induced-fit docking protocol was used in order to deepen the study concerning the interactions occurring between TR16 and MAO-B (Figure 5).
Figure 5.

Best docked pose of TR16 resulted from the induced-fit docking protocol. Green lines and red arrows depict π–π contacts and hydrogen bonds, respectively. Black lines show the original conformation of MAO-B sidechains. For completeness, docking score was equal to −11.544 kcal/mol.
As reported in Figure 5, the binding mode of TR16 was consistent with previous standard docking simulation. In detail, the interactions with selective residue Y326 and with Y398 were detected, and an additional aromatic residue, F343, was recruited to engage a new π–π contact, with a little shift from its initial position.
2.4.2. Bioavailability Prediction
The bioavailability of lead molecules was predicted by SwissADME online platform. The bioavailability radar panel assesses drug candidates based on physicochemical parameters such as lipophilicity, flexibility, polarity insolubility, size, and saturation [22]. Interestingly, both TR2 and TR16 molecules had been reported to be in the pink region (optimal ranges) and to have drug-like properties (Figure 6).
Figure 6.

The bioavailability radar of TR2 and TR16.
3. Materials and Methods
3.1. Chemistry
The synthetic procedures of the titled compounds are provided in the supplementary information.
3.2. Biological Analysis
3.2.1. Enzyme Assays
Recombinant MAO-A and MAO-B were used in MAO inhibitory activity assays using kynuramine (0.06 mM) and benzylamine (0.3 mM) as substrates, respectively [23]. The MAO-A and MAO-B reactions were carried out at 25 °C and monitored for 30 min at 316 and 250 nm, respectively, in 0.5 mL mixture of 50 mM sodium phosphate (pH 7.2). Clorgyline and toloxatone were the drug references used for MAO-A, and pargyline and lazabemide were used for MAO-B as reference compounds. The IC50 values were determined by measuring the residual activity at different concentrations of the compounds and by using GraphPad Prism software 5.0. The Km value of benzylamine against MAO-B was measured to be 0.40 mM [24]. On the other hand, multitarget analysis, ChE (AChE, BChE) inhibitory activity assays were carried out at 25 °C and monitored for 15 min at 412 nm in a 0.5 mL mixture of 100 mM sodium phosphate (pH 7.5). BACE1 inhibitory activities were tested by the BACE1 activity detection kit. The assay method was described previously [25]. Recombinant human MAO-A and MAO-B, AChE from Electrophorus electricus, BChE from equine serum, BACE1, and their substrates and reference inhibitors were purchased from Sigma-Aldrich (St. Louis, MO, USA).
3.2.2. Inhibition Profile of MAOs and Kinetic Studies
TR1 and TR16 MAO inhibitory actions were evaluated at 10 μM for primary screening. For the compounds with <80% residual activities, their IC50 values were calculated. TR2 and TR16 kinetics with MAO-B were investigated at five different substrate concentrations (0.0375–0.6 μM). Lineweaver–Burk plots were used to examine the inhibition patterns, and secondary plots were used to calculate the Ki values with three inhibitor dosages [26,27,28].
3.2.3. Inhibition Reversibility of TR2 and TR16
After preincubation with the enzyme for 30 min at ~2 × IC50 (i.e., 0.54 and 0.34 μM, respectively), the reversibility of TR2 or TR16 against MAO-B inhibition was evaluated using a dialysis approach and employing a dialysis kit, as previously described [29,30]. Lazabemide (a reversible MAO-B inhibitor) and pargyline (an irreversible MAO-B inhibitor) were preincubated at 0.22 and 0.28 μM, respectively, as reference compounds. To determine reversibility patterns, the activities of dialyzed (AD) and undialyzed (AU) specimens were compared.
3.3. BBB Study by PAMPA Method
Using the PAMPA method, blood–brain barrier permeation abilities of lead compounds TR2 and TR16 were analyzed [21].
3.4. Docking Studies
X-ray crystallographic structures of MAO-A and MAO-B were taken from the Protein Data Bank by finding records 2Z5X and 2V5Z, respectively [31,32]. With the assistance of the protein preparation wizard tool, both protein structures were evaluated for the ionization states of acid/basic side chains at physiological pH, to remove nonfunctional water and to carry out energy minimization steps [33,34]. Ligands to be docked were prepared by using the Ligprep tool to sample allowed tautomers and ionization states for all the possible conformers. Standard docking protocol (SP) was carried out using GLIDE by utilizing the OPLS3 force field and setting 50,000 poses per ligand for the first phase and 4000 poses per ligand for energy minimization [35,36,37]. Best poses yielded Root Mean Square Deviation (RMSD) values of 0.39 Å and 0.76 Å for MAO-A and MAO-B cognate ligands, respectively, which was satisfactory. Eventually, the induced-fit docking protocol was employed in order to better understand the interactions between compound TR16 and MAO-B. This protocol allows the conformational changes in residues involved in the ligand binding to be considered, thus enhancing the reliability and the accuracy of computational simulations [38]
3.5. Bioavailiability Predicition
The bioavailability prediction of lead molecules TR2 and TR16 was assessed at http://www.swisstargetprediction.ch [39] (accessed on 1 March 2022).
4. Conclusions
The current work evaluated the MAO-A and MAO-B inhibitory characteristics of a series of sixteen pyridazinone derivatives that were synthesized. The majority of the chemicals inhibited MAO-B more effectively than MAO-A. The lead compounds TR2 and TR16 have been shown to be competitive and reversible MAO-B inhibitors in kinetic and reversibility experiments. The PAMPA test revealed that the lead candidates have a high level of CNS permeability. According to docking studies, the strong inhibitory efficiency of compounds TR2 and TR16 is due to the possibility of interaction with important and selective residues of E84 or Y326 in MAO-B. These findings are consistent with the experimental data. The compounds’ pharmacological similarity was also shown by the favorable bioavailability radar.
Acknowledgments
The authors would like to extend their sincere appreciation to Taif University Researchers Supporting Project number (TURSP-2020/56), Taif University, Taif, Saudi Arabia. Additionally, the authors deeply acknowledge the Researchers Supporting Program (TUMA-Project-2021-6), AlMaarefa University, Riyadh, Saudi Arabia.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules27123801/s1, Experimental procedures for the synthesis of the titled compounds. Figures S1–S48; 1H-NMR, 13C-NMR, and HRMS spectra of the compounds TR1~16. Figures S49 and S50; 2D-schemes of TR2, TR15, and TR16 interactions to MAO-B.
Author Contributions
Conceptualization: M.A.A. (Mohamed A. Abdelgawad), B.M. and H.K.; synthesis: M.A.A., Y.N.Z. and Z.Ö.; biological activity: J.M.O., H.K., M.A.A. (Mehmet Abdullah Alagöz) and I.A.N.; docking analysis: N.G. and O.N.; original draft writing: B.M., M.A.A. (Mehmet Abdullah Alagöz) and H.K.; review and editing: H.K., B.M., M.M.G. and M.A.A. (Mohamed A. Abdelgawad); supervision: H.K. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
MDPI Research Data Policies” at https://www.mdpi.com/ethics (accessed on 25 February 2022).
Conflicts of Interest
The authors have no conflict of interest to declare.
Sample Availability
Samples of the compounds TR1 to TR16 are available from the authors.
Funding Statement
This work was financially supported by the Research Foundation of İnönü University (Grant number TCD-2021-2558).
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Jankovic J., Sherer T. The future of research in Parkinson disease. JAMA Neurol. 2014;71:1351–1352. doi: 10.1001/jamaneurol.2014.1717. [DOI] [PubMed] [Google Scholar]
- 2.Simon D.K., Tanner C.M., Brundin P. Parkinson disease epidemiology, pathology, genetics, and pathophysiology. Clin. Geriatr. Med. 2020;36:1–12. doi: 10.1016/j.cger.2019.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wakabayashi K., Tanji K., Mori F., Takahashi H. The Lewy body in Parkinson’s disease: Molecules implicated in the formation and degradation of α-synuclein aggregates. Neuropathology. 2007;27:494–506. doi: 10.1111/j.1440-1789.2007.00803.x. [DOI] [PubMed] [Google Scholar]
- 4.Entzeroth M., Ratty A.K. Monoamine oxidase inhibitors-revisiting a therapeutic principle. Open J. Depress. 2017;6:31–68. doi: 10.4236/ojd.2017.62004. [DOI] [Google Scholar]
- 5.Kumar B., Sheetal Mantha A.K., Kumar V. Recent developments on the structure-activity relationship studies of MAO inhibitors and their role in different neurological disorders. RSC Adv. 2016;6:42660–42683. doi: 10.1039/C6RA00302H. [DOI] [Google Scholar]
- 6.Rehuman N.A., Mathew B., Jat R.K., Nicolotti O., Kim H. A Comprehensive review of monoamine oxidase-A inhibitors in their syntheses and potencies. Comb. Chem. High Throughput Screen. 2020;23:898–914. doi: 10.2174/1386207323666200428091306. [DOI] [PubMed] [Google Scholar]
- 7.Özdemir Z., Alagöz M.A., Bahçecioğlu Ö.F., Gök S. Monoamine oxidase-B (MAO-B) inhibitors in the treatment of Alzheimer’s and Parkinson’s disease. Curr. Med. Chem. 2021;28:6045–6065. doi: 10.2174/0929867328666210203204710. [DOI] [PubMed] [Google Scholar]
- 8.Guglielmi P., Carradori S., Ammazzalorso A., Secci D. Novel approaches to the discovery of selective human monoamine oxidase-B inhibitors: Is there room for improvement? Expert Opin. Drug Discov. 2019;14:995–1035. doi: 10.1080/17460441.2019.1637415. [DOI] [PubMed] [Google Scholar]
- 9.Tripathi A.C., Upadhyay S., Paliwal S., Saraf S.K. Privileged scaffolds as MAO inhibitors: Retrospect and prospects. Eur. J. Med. Chem. 2018;145:445–497. doi: 10.1016/j.ejmech.2018.01.003. [DOI] [PubMed] [Google Scholar]
- 10.Rangarajan T.M., Mathew B. Recent updates on pyrazoline derivatives as promising candidates for neuropsychiatric and neurodegenerative disorders. Curr. Top. Med. Chem. 2021;21:2695–2714. doi: 10.2174/1568026621999210902123132. [DOI] [PubMed] [Google Scholar]
- 11.Guglielmi P., Mathew B., Secci D., Carradori S. Chalcones: Unearthing their therapeutic possibility as monoamine oxidase B inhibitors. Eur. J. Med. Chem. 2020;205:112650. doi: 10.1016/j.ejmech.2020.112650. [DOI] [PubMed] [Google Scholar]
- 12.Koyiparambath V.P., Rajappan K.P., Rangarajan T.M., Al-Sehemi A.G., Pannipara M., Bhaskar V., Nair A.S., Sudevan S.T., Kumar S., Mathew B. Deciphering the detailed structure-activity relationship of coumarins as monoamine oxidase enzyme inhibitors—An updated review. Chem. Biol. Drug Des. 2021;98:655–673. doi: 10.1111/cbdd.13919. [DOI] [PubMed] [Google Scholar]
- 13.Mellado M., González C., Mella J., Aguilar L.F., Viña D., Uriarte E., Cuellar M., Matos M.J. Combined 3D-QSAR and docking analysis for the design and synthesis of chalcones as potent and selective monoamine oxidase B inhibitors. Bioorg. Chem. 2021;108:104689. doi: 10.1016/j.bioorg.2021.104689. [DOI] [PubMed] [Google Scholar]
- 14.Mellado M., Mella J., González C., Viña D., Uriarte E., Matos M.J. 3-Arylcoumarins as highly potent and selective monoamine oxidase B inhibitors: Which chemical features matter? Bioorg. Chem. 2020;101:103964. doi: 10.1016/j.bioorg.2020.103964. [DOI] [PubMed] [Google Scholar]
- 15.Özdemir Z., Yılmaz H., Sarı S., Karakurt A., Şenol F.S., Uysal M. Design, synthesis, and molecular modeling of new 3(2H)-pyridazinone derivatives as acetylcholinesterase/butyrylcholinesterase inhibitors. Med. Chem. Res. 2017;26:2293–2308. doi: 10.1007/s00044-017-1930-x. [DOI] [Google Scholar]
- 16.Özdemir Z., Alagöz M.A., Uslu H., Karakurt A., Erikci A., Ucar G., Uysal M. Synthesis, molecular modelling and biological activity of some pyridazinone derivatives as selective human monoamine oxidase-B inhibitors. Pharmacol. Rep. 2020;72:692–704. doi: 10.1007/s43440-020-00070-w. [DOI] [PubMed] [Google Scholar]
- 17.Çeçen M., Oh J.M., Özdemir Z., Büyüktuncel S.E., Uysal M., Abdelgawad M.A., Musa A., Gambacorta N., Nicolotti O., Mathew B., et al. Design, synthesis, and biological evaluation of pyridazinones containing the (2-fluorophenyl) piperazine moiety as selective MAO-B inhibitors. Molecules. 2020;25:5371. doi: 10.3390/molecules25225371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Özçelik A.B., Özdemir Z., Sari S., Utku S., Uysal M. A new series of pyridazinone derivatives as cholinesterases inhibitors: Synthesis, in vitro activity and molecular modeling studies. Pharmacol. Rep. 2019;71:1253–1263. doi: 10.1016/j.pharep.2019.07.006. [DOI] [PubMed] [Google Scholar]
- 19.Bozbey İ., Özdemir Z., Uslu H., Özçelik A.B., Şenol F.S., Erdoğan-Orhan İ., Uysal M. A series of new hydrazone derivatives: Synthesis, molecular docking and anticholinesterase activity studies. Mini-Rev. Med. Chem. 2020;20:1042–1060. doi: 10.2174/1389557519666191010154444. [DOI] [PubMed] [Google Scholar]
- 20.Alagöz M.A., Özdemir Z., Uysal M., Carradori S., Gallorini M., Ricci A., Zara S., Mathew B. Synthesis, cytotoxicity and anti-proliferative activity against AGS cells of new 3(2H)-pyridazinone derivatives endowed with a piperazinyl linker. Pharmaceuticals. 2021;14:183. doi: 10.3390/ph14030183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Di L., Kerns E.H., Fan K., McConnell O.J., Carter G.T. High throughput artificial membrane permeability assay for blood-brain barrier. Eur. J. Med. Chem. 2003;38:223–232. doi: 10.1016/S0223-5234(03)00012-6. [DOI] [PubMed] [Google Scholar]
- 22.Al-Sehemi A.G., Olotu F.A., Dev S., Pannipara M., Soliman M.E.S., Carradori S., Mathew B. Natural products database screening for the discovery of naturally occurring SARS-Cov-2 spike glycoprotein blockers. ChemistrySelect. 2020;5:13309–13317. doi: 10.1002/slct.202003349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Venkidath A., Oh J.M., Dev S., Amin E., Rasheed S.P., Vengamthodi A., Gambacorta N., Khames A., Abdelgawad M.A., George G., et al. Selected class of enamides bearing nitro functionality as dual-acting with highly selective monoamine oxidase-B and BACE1 inhibitors. Molecules. 2021;26:6004. doi: 10.3390/molecules26196004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee J.P., Kang M.G., Lee J.Y., Oh J.M., Baek S.C., Leem H.H., Park D., Cho M.L., Kim H. Potent inhibition of acetylcholinesterase by sargachromanol I from Sargassum siliquastrum and by selected natural compounds. Bioorg. Chem. 2019;89:103043. doi: 10.1016/j.bioorg.2019.103043. [DOI] [PubMed] [Google Scholar]
- 25.Mathew B., Oh J.M., Khames A., Abdelgawad M.A., Rangarajan T.M., Nath L.R., Agoni C., Soliman M.E.S., Mathew G.E., Kim H. Replacement of chalcone-ethers with chalcone-thioethers as potent and highly selective monoamine oxidase-B inhibitors and their protein-ligand interactions. Pharmaceuticals. 2021;14:1148. doi: 10.3390/ph14111148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heo J.H., Eom B.H., Ryu H.W., Kang M.G., Park J.E., Kim D.Y., Kim J.H., Park D., Oh S.R., Kim H. Acetylcholinesterase and butyrylcholinesterase inhibitory activities of khellactone coumarin derivatives isolated from Peucedanum japonicum Thurnberg. Sci. Rep. 2020;10:21695. doi: 10.1038/s41598-020-78782-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jeong G., Kang M.-G., Lee J., Lee S., Park D., Cho M., Kim H. Inhibition of butyrylcholinesterase and human monoamine oxidase-B by the coumarin glycyrol and liquiritigenin isolated from Glycyrrhiza uralensis. Molecules. 2020;25:3896. doi: 10.3390/molecules25173896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jeong G.S., Kang M.G., Han S.A., Noh J.I., Park J.E., Nam S.J., Park D., Yee S.T., Kim H. Selective inhibition of human monoamine oxidase B by 5-hydroxy-2-methyl-chroman-4-one isolated from an endogenous lichen fungus Daldinia fissa. J. Fungi. 2021;7:84. doi: 10.3390/jof7020084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee H.W., Ryu H.W., Kang M.G., Park D., Oh S.R., Kim H. Potent selective monoamine oxidase B inhibition by maackiain, a pterocarpan from the roots of Sophora flavescens. Bioorg. Med. Chem. Lett. 2016;26:4714–4719. doi: 10.1016/j.bmcl.2016.08.044. [DOI] [PubMed] [Google Scholar]
- 30.Baek S.C., Lee H.W., Ryu H.W., Kang M.G., Park D., Kim S.H., Cho M.L., Oh S.R., Kim H. Selective inhibition of monoamine oxidase A by hispidol. Bioorg. Med. Chem. Lett. 2018;15:584–588. doi: 10.1016/j.bmcl.2018.01.049. [DOI] [PubMed] [Google Scholar]
- 31.Son S.Y., Ma J., Kondou Y., Yoshimura M., Yamashita E., Tsukihara T. Structure of human monoamine oxidase A at 2.2-A resolution: The control of opening the entry for substrates/inhibitors. Proc. Natl. Acad. Sci. USA. 2008;105:5739–5744. doi: 10.1073/pnas.0710626105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Binda C., Wang J., Pisani L., Caccia C., Carotti A., Salvati P., Edmondson D.E., Mattevi A. Structures of human monoamine oxidase B complexes with selective noncovalent inhibitors: Safinamide and coumarin analogs. J. Med. Chem. 2007;50:5848–5852. doi: 10.1021/jm070677y. [DOI] [PubMed] [Google Scholar]
- 33.Sastry G.M., Adzhigirey M., Day T., Annabhimoju R., Sherman W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013;27:221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 34.Schrödinger . Protein Preparation Wizard. Schrödinger, L.L.C.; New York, NY, USA: 2020. [Google Scholar]
- 35.Schrödinger . LigPrep. Schrödinger, L.L.C.; New York, NY, USA: 2020. [Google Scholar]
- 36.Friesner R.A., Banks J.L., Murphy R.B., Halgren T.A., Klicic J.J., Mainz D.T., Repasky M.P., Knoll E.H., Shelley M., Perry J.K., et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 37.Harder E., Damm W., Maple J., Wu C., Reboul M., Xiang J.Y., Wang L., Lupyan D., Dahlgren M.K., Knight J.L., et al. OPLS3: A force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 2016;12:281–296. doi: 10.1021/acs.jctc.5b00864. [DOI] [PubMed] [Google Scholar]
- 38.Sherman W., Day T., Jacobson M.P., Friesner R.A., Farid R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006;49:534–553. doi: 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- 39.Daina A., Michielin O., Zoete V. SwissTargetPrediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019;47:W357–W364. doi: 10.1093/nar/gkz382. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
MDPI Research Data Policies” at https://www.mdpi.com/ethics (accessed on 25 February 2022).