

Abstract
A Disintegrin and Metalloproteinase (ADAM) and A Disintegrin and Metalloproteinase with Thrombospondin Motifs (ADAMTS) are two closely related families of proteolytic enzymes. ADAMs are largely membrane-bound enzymes that act as molecular scissors or sheddases of membrane-bound proteins, growth factors, cytokines, receptors and ligands, whereas ADAMTS are mainly secreted enzymes. ADAMs have a pro-domain, and a metalloproteinase, disintegrin, cysteine-rich and transmembrane domain. Similarly, ADAMTS family members have a pro-domain, and a metalloproteinase, disintegrin, and cysteine-rich domain, but instead of a transmembrane domain they have thrombospondin motifs. Most ADAMs and ADAMTS are activated by pro-protein convertases, and can be regulated by G-protein coupled receptor agonists, Ca2+ ionophores and protein kinase C. Activated ADAMs and ADAMTS participate in numerous vascular processes including angiogenesis, vascular smooth muscle cell proliferation and migration, vascular cell apoptosis, cell survival, tissue repair, and wound healing. ADAMs and ADAMTS also play a role in vascular malfunction and cardiovascular diseases such as hypertension, atherosclerosis, coronary artery disease, myocardial infarction, heart failure, peripheral artery disease, and vascular aneurysm. Decreased ADAMTS13 is involved in thrombotic thrombocytopenic purpura and microangiopathies. The activity of ADAMs and ADAMTS can be regulated by endogenous tissue inhibitors of metalloproteinases and other synthetic small molecule inhibitors. ADAMs and ADAMTS can be used as diagnostic biomarkers and molecular targets in cardiovascular disease, and modulators of ADAMs and ADAMTS activity may provide potential new approaches for the management of cardiovascular disorders.
Keywords: atherosclerosis, hypertension, metalloproteases, sheddase, vascular smooth muscle
1. Introduction
A disintegrin and metalloproteinase (ADAM) and a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS) are two families of proteolytic enzymes implicated in the regulation of extracellular matrix (ECM) proteins, cell-associated proteins, growth factors and cytokines, and modulation of numerous biological processes. ADAMs and ADAMTS share a highly conserved structure and protein homology in the form of a pro-domain, and a metalloproteinase, disintegrin and cysteine-rich domain. ADAMs are largely membrane-anchored by a transmembrane domain connected to a cytoplasmic tail, although some secreted ADAMs are generated by alternative splicing or cleavage. Through binding of the disintegrin domain to integrins together with the proteolytic activity of the metalloproteinase domain, ADAMs function as molecular scissors or sheddases of membrane-bound growth factors, cytokines and receptors. In contrast, ADAMTS family members are mainly secreted enzymes that lack the transmembrane domain and cytoplasmic tail, and instead have ancillary thrombospondin motifs, which structurally resemble thrombospondin 1 or 2, homotrimeric adhesive glycoproteins involved in cell-to-cell and cell-to-matrix interactions.
Several research articles have provided details of ADAMs and ADAMTS structure and function and their role in different biological processes including the regulation of the reproductive system, embryo development, central nervous system (CNS) and cell growth (Colige, et al., 2005; Lemarchant, et al., 2013; Mead & Apte, 2018; Porter, Clark, Kevorkian, & Edwards, 2005; Seals & Courtneidge, 2003; Stone, Kroeger, & Sang, 1999; Takeda, 2016; E. P. C. van der Vorst, Weber, & Donners, 2018). ADAMs and ADAMTS are also involved in pathological conditions such as inflammation, autoimmune disease and cancer (Mead & Apte, 2018). ADAMs and ADAMTS also participate in vascular processes such as angiogenesis, vascular smooth muscle cell (VSMC) proliferation and migration, and play a role in the vascular malfunction associated with cardiovascular disease (CVD) including hypertension (HTN), atherosclerosis, coronary artery disease (CAD) and dilated cardiomyopathy (Fedak, et al., 2006; Santamaria & de Groot, 2020). Highlighting the role of ADAMs and ADAMTS in vascular malfunction should help to further understand the mechanisms of CVD.
In this review, we will use data published in PubMed and Web of Science to describe the biochemical and biological aspects of ADAMs and ADAMTS and their role in vascular malfunction and CVD. We will briefly describe ADAMs and ADAMTS structure, activators, targets, substrates, and inhibitors. We will introduce the general functions of representative ADAMs and ADAMTS and their mouse knockout phenotypes, then describe their specific roles in vascular processes such as angiogenesis, VSMC proliferation and migration, intimal hyperplasia and neointima formation, vascular cell apoptosis, and tissue repair and wound healing. We will follow with description of the role of ADAMs and ADAMTS in CVD such as HTN, atherosclerosis, CAD, myocardial infarction (MI), heart failure, ischemia-reperfusion (I/R) injury, peripheral artery disease (PAD), and vascular aneurysm. We will conclude with a perspective on how ADAMs and ADAMTS can be used as potential biomarkers and molecular targets in the diagnosis and management of CVD.
2. ADAMs and ADAMTS Structure
ADAMs, formerly known as metalloproteinase/disintegrin/cysteine-rich (MDC) proteins, are zinc (Zn2+)-dependent proteolytic enzymes (Fig. 1). ADAMs and ADAMTS are members of the adamalysin subfamily, and they share similar structure and protein sequence including a pro-domain, a metalloproteinase, disintegrin, and cysteine-rich domain (Takeda, 2016).
Fig. 1.
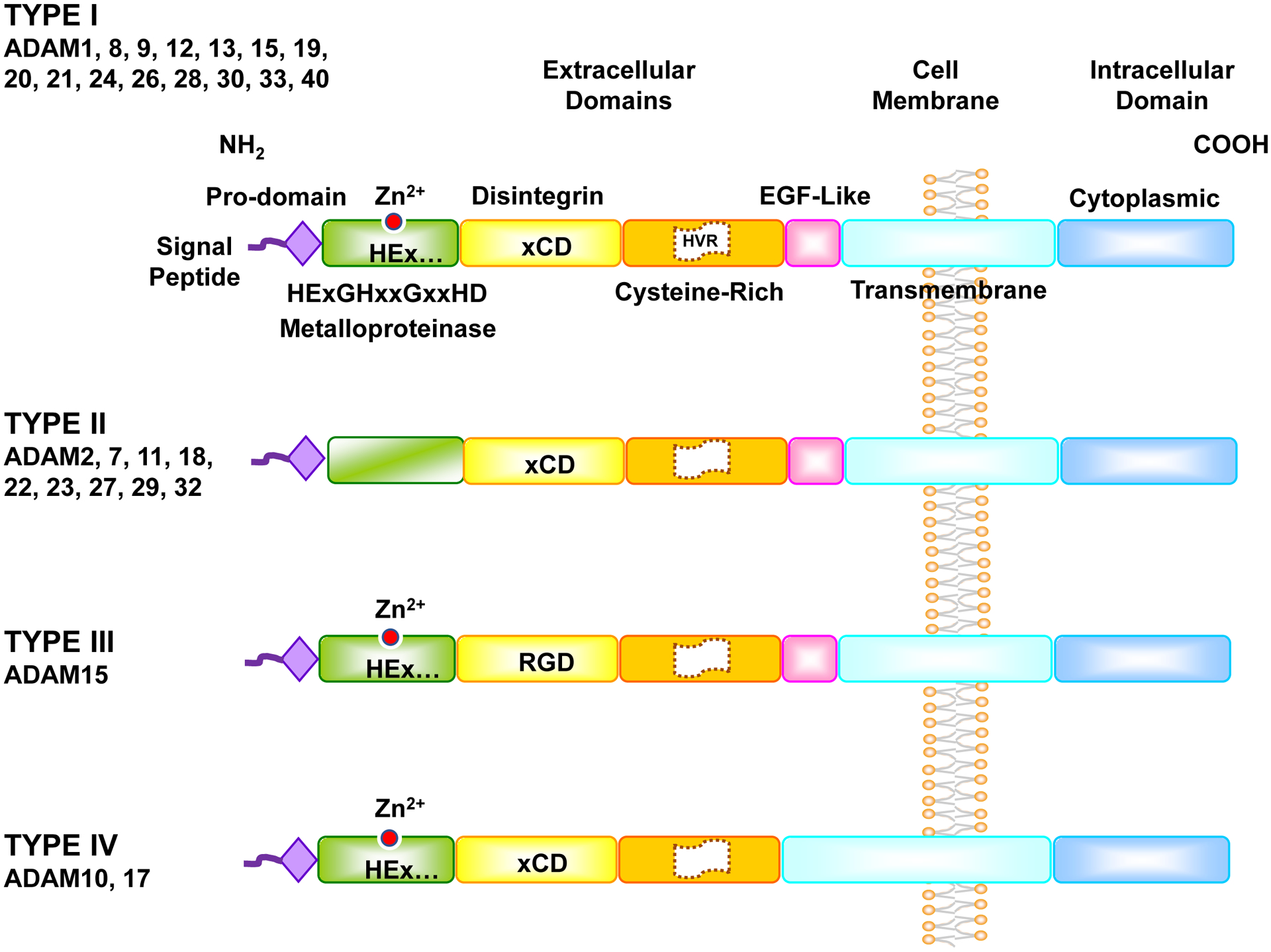
ADAMs Structure. Like MMPs, ADAMs have a signal peptide, pro-domain, and metalloproteinase domain. Also, like snake venom metalloproteinases (SVM), ADAMs have a disintegrin domain and cysteine-rich domain. Compared to SVM, ADAMs have a highly variable region in the cysteine-rich domain, and an additional epidermal growth factor (EGF)-like region, transmembrane domain and cytoplasmic tail. ADAMs have four types. Type I ADAMs have typical metalloprotease M-domain with the characteristic catalytic Zn2+ binding signature (HExGHxxGxxHD), and their disintegrin D-domain is based on xCD sequence. In Type II ADAMs the M-domain differs from that of Type I ADAMs, and lacks the catalytic-Zn2+ binding signature. In Type III ADAMs the M-domain contains the catalytic-Zn2+ binding signature, but the D-domain is based on RGD sequence rather than the xCD sequence in most ADAMs. In Type IV ADAMs the D-domain is based on xCD sequence, but they lack the EGF-like region found in Type I, II, and III.
The ADAM molecule starts with an N-terminus signal peptide which directs the enzyme to carry out its function, followed by the pro-domain which serves to correct protein folding, stabilize the protein and maintain enzyme latency via a “cysteine switch” that blocks the catalytic Zn2+ activation site. The cysteine switch in the pro-domain is different from the cysteine-rich domain (Bode, Gomis-Ruth, & Stockler, 1993; Van Wart & Birkedal-Hansen, 1990; P. Zhang, Shen, Fernandez-Patron, & Kassiri, 2016).
ADAMs have a C-shape arm that comprises a metalloproteinase (M), a disintegrin (D), and a cysteine-rich (C) domain with a Highly Variable Region (HVR) (Takeda, Igarashi, Mori, & Araki, 2006). The C-shape arm is essential for target recognition, protein interaction, and proteolytic activity.
The metalloproteinase or M-domain proteolytically interacts with various ligands, receptors and ion channels on the same cell or neighboring cell surface. The M-domain contains the catalytic Zn2+ binding signature (HExGHxxGxxHD), where H denotes histidine, E glutamic acid, G glycine, D aspartic acid, and x variable amino acid (aa). Through the M-domain, ADAMs serve as molecular scissors and sheddases of various ligands and receptors, and participate in cell signaling, proliferation, apoptosis, differentiation, tissue organization and organ function (Saha, Robev, Himanen, & Nikolov, 2019).
The disintegrin or D-domain follows the M-domain. The term “disintegrin” originated from its high similarity to the Arg-Gly-Asp (RGD)-containing sequence in snake venom proteins, which binds to integrins in snake bite victims, inhibits platelet aggregation and causes hemorrhage (Gould, et al., 1990). Oddly, only ADAM15 still conserve the RGD sequence, while other ADAMs have a Glu-Cys-Asp (ECD) or xCD sequence, hence the more accurate term “disintegrin-like domain” (Blobel, 1997). ADAMs present their disintegrin domain on the cell surface where they interact with integrins (Blobel & White, 1992).
The cysteine-rich or C-domain has cell adhesive and fusogenic properties. Along with the cell surface protein syndecan, the C-domain of ADAM12 regulates inflammatory cell adhesion and tissue distribution in an integrin-dependent manner (Iba, et al., 2000). The C-domain of ADAM13 is also involved in cell adhesion as it binds to the basement membrane proteins laminin and fibronectin (Smith, et al., 2002). ADAM10 and 17 have less sequence similarities in the C-domain with other canonical ADAMs, allowing them to have a distinct but related role in specific pathological conditions and in CVD (Takeda, et al., 2006). The highly variable region (HVR) of the C-domain also acts as a protein-protein adhesive interface (Takeda, et al., 2006).
The epidermal growth factor (EGF)-like region, transmembrane domain and cytoplasmic tail connect the extracellular and intracellular components of ADAMs together, help to fix the C-shape arm to the cell membrane, and transmit the extracellular signal into the cell in order to regulate mRNA expression and protein phosphorylation (Stone, et al., 1999).
Currently, 37 ADAMs have been identified in rats, 34 in mice, and 22 in the human genome, of which 13 proteins are proteolytically active (Edwards, Handsley, & Pennington, 2008; P. Zhang, et al., 2016). ADAMs share common domains, but have some differences shown in 4 types (Fig. 1). Type I is the most common with the classic domain structure, and it includes ADAM1, 8, 9, 12, 13, 15, 19, 20, 21, 24, 26, 28, 30, 33, 40. In type I, the M-domain has the characteristic catalytic Zn2+ binding signature (HExGHxxGxxHD), the D-domain is based on xCD sequence, and the EGF-like region is present. Type II includes ADAM2, 7, 11, 18, 22, 23, 27, 29, 32, where the M-domain does not have the catalytic-Zn2+ binding signature, and is more variable. Type III is unique for ADAM15, where the M-domain contains the catalytic-Zn2+ binding signature, but the D-domain uniquely contains RGD sequence. Type IV is unique for ADAM10 and 17, where the D-domain is based on xCD sequence, but they lack the EGF-like region. The high similarity in the structure of ADAM10 and 17 may explain the similarities in some of their functions.
ADAMTS are multidomain extracellular proteases. Like ADAMs, ADAMTS have the common signal peptide, pro-domain, metalloproteinase M-domain, disintegrin D-domain and cysteine-rich C-domain. The main difference is that ADAMTS lack the EGF-like region, transmembrane domain and cytoplasmic tail, but have thrombospondin repeats (TSR) and spacer (Fig. 2). This makes secreted ADAMTS more soluble in the circulation, and enables them to function in remote tissues.
Fig. 2.
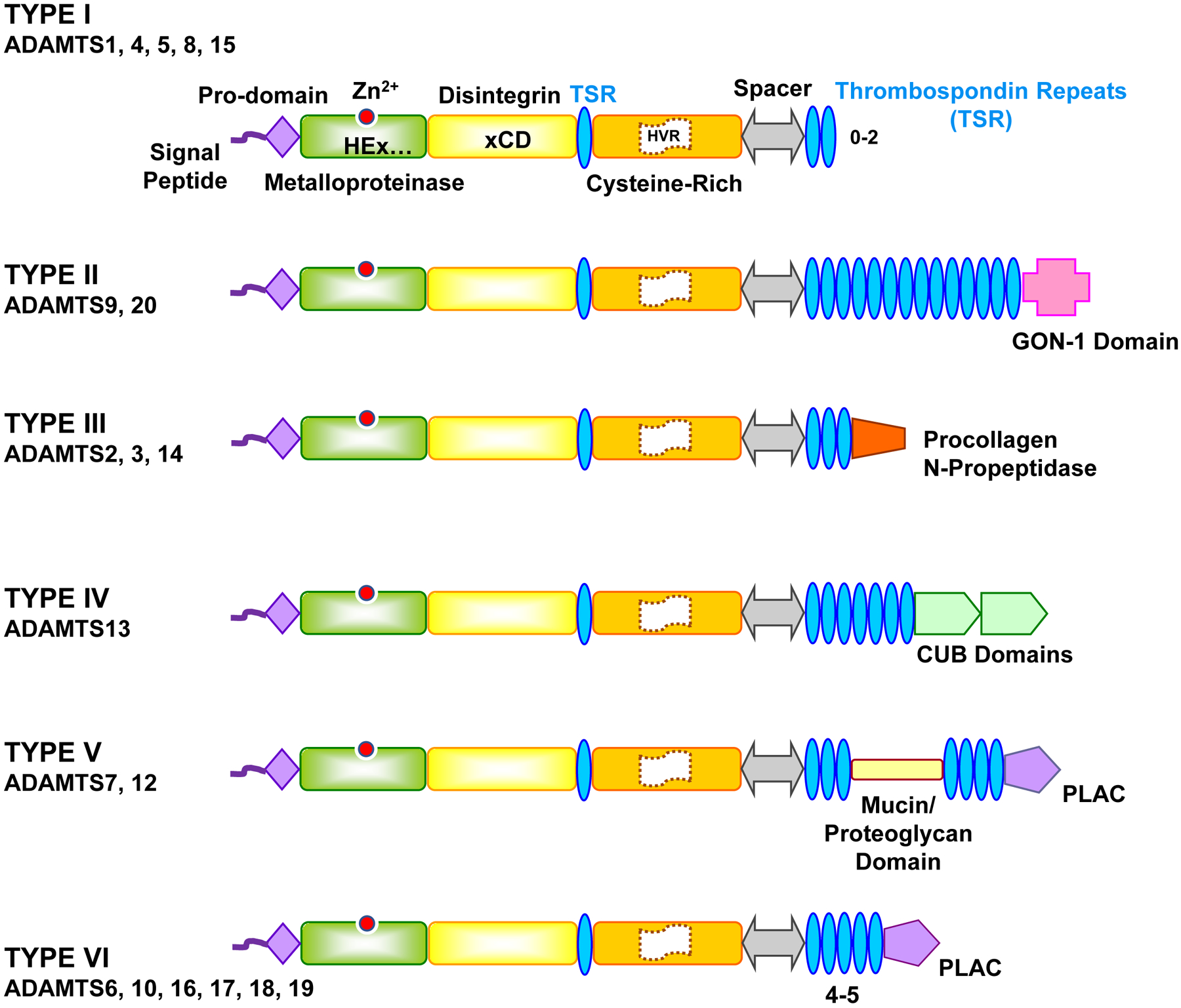
Structure of different types of ADAMTS. ADAMTS have similar structure to ADAMs, but they lack the EGF-like region, transmembrane domain and cytoplasmic tail, and instead have thrombospondin repeats (TSR) and spacer. ADAMTS members have six types. Type I includes several members of ADAMTS and the protein structure comprises the basic pro-domain, metalloproteinase, disintegrin, cysteine-rich domain, thrombospondin repeats (TSR) and spacer. Type II ADAMTS has a unique GON-1 domain, type III has a procollagen N propeptidase, Type IV has a complement C1r/C1s, Uegf, Bmp1 (CUB) domain, Type V has a protease and lacunin (PLAC) region and mucin/proteoglycan domain, and Type VI has only PLAC region. The number of TSR varies among different ADAMTS types and within type I and type VI ADAMTS.
The ADAMTS ancillary TSR are critical for interaction with ECM, substrate recognition, and regulation of the enzyme activity. ADAMTS have a 50 aa TSR highly similar to thrombospondins 1, 2 (Adams & Lawler, 2011), followed by a cysteine-rich domain of more than 100 aa, and a cysteine-free spacer of 103 to 160 aa. The spacer is followed by 0 to 14 TSR modules, which in some ADAMTS are connected to GON-1 domain (present in the ADAMTS GON-1 identified in C. elegans), CUB (complement C1r/C1s, Uegf, Bmp1) domain, or PLAC (protease and lacunin) domain (Fig. 2).
There are 19 ADAMTS genes in mammalian genomes designated 1 to 20, except ADAMTS11 which was assigned a gene previously identified for ADAMTS5. Therefore, there are 19 ADAMTS proteins in humans, divided into 6 types. Type I has the basic structure and includes several members of ADAMTS family. Type II ADAMTS has a unique GON-1 domain, type III has a procollagen N-propeptidase, Type IV has CUB domains, type V has a PLAC domain and mucin/proteoglycan domain, and type VI has only a PLAC domain.
3. Sources and Tissue Distribution of ADAM and ADAMTS Family
ADAMs are produced by different cells and have wide tissue distribution (Table 1). Many ADAMs, including ADAM1, 2, 3, 4, 5, 6, 7, 20, 21, 24 (testase 1), 25, 26, 27, 28, 29, 30, 32 and 34 are involved in reproduction processes such as spermatogenesis in the testis, sperm maturation and gain of fertilizing ability in the epididymis, and sperm migration from the uterus to the oviducts where it fuses with the oocyte (Cho, 2012). ADAM8, 9, 10, 12, 15, 17 and 19 are involved in cardiovascular development, and the abnormal expression or function of these ADAMs may lead to CVD.
Table 1.
Human ADAM Family Members, Gene Locus, Molecular Weight (MW) of Full-length Protein, Source/Tissue Distribution, Targets/Substrates, and Implications in Biology or Disease
| ADAM Other Name |
Gene Locus | MW kDa | Source/Tissue Distribution | Target/Substrate | Implications in Biology or Disease | Ref |
|---|---|---|---|---|---|---|
|
1 (a & b) Fertilin α, PH-30α |
12q24.12 | 84 | Sperm head surface, testis, epididymis, vas deferens | Integrins, ADAM3 | Spermatogenesis, sperm maturation, sperm-egg interaction | (Cho, 2012) |
|
2 Fertilin β, PH-30β, Cancer/testis antigen 15 |
8p11.22 | 82 | Sperm head, surface, testis, epididymis, vas deferens | Integrin α6β1, epididymal protein DE (CRISP-1), intracellular chaperone calnexin, oocyte CD9, ADAM3 | Sperm maturation & migration in female reproductive tract, sperm-egg interaction | (Cho, 2012) |
|
3 Cyritestin, sperm surface antigen tMDC I, CYRN |
8p11.22 | 80 | Sperm surface, testis, epididymis | Integrins, testis-expressed gene 101, epididymal protein DE (CRISP-1), chaperones calnexin & calreticulin | Sperm maturation, sperm migration, sperm-egg interaction | (Cho, 2012) |
|
6 Testis-derived tMDC IV |
14q32.33 | 60 | Testis | Integrins | Sperm maturation, sperm-egg interaction | (Cho, 2012) |
|
7 EAPI, Sperm maturation-related glycoprotein GP-83 |
8p21.2 | 86 | Sperm plasma membrane, testis, epididymis | Integrin α4β1 & α9β1, intracellular chaperone calnexin, heat shock protein 5, integral membrane protein 2B | Sperm maturation, sperm-egg interaction, melanoma progression | (Cho, 2012) |
|
8 Cell surface antigen MS2, CD156a |
10q26.3 | 90 | Lung epithelium, ECs, SMCs, immune B cells, eosinophils, neutrophils, monocytes, macrophages, dendritic cells, invasive HLA-G- trophoblasts, intervertebral discs, joints, osteoclasts, neurons | Integrin β1, neural cell adhesion molecule close homologue of L1 (CHL-1), L-selectin, P-selectin glycoprotein ligand (PSGL-1), low-affinity IgE receptor CD23, low-affinity receptor FcεRII, CD-30-ligand, amyloid precursor protein (APP), fibronectin, myelin basic protein (MBP), brevican, vitronectin, VCAM-1, ADAM17 | Angiogenesis, cell adhesion, neutrophil rolling, trans-endothelial extravasation, atherosclerosis, CAD, MI, ascending aortic dissection, ovulation, placental development, trophoblast migration, allergy, asthma, chronic obstructive pulmonary disease neurodegeneration, neuroinflammation, neoplastic meningitis, osteoclast differentiation, rheumatoid arthritis, breast, gastric, pancreatic, colorectal & brain cancer | (Hsia, et al., 2019; Kelly, et al., 2005; Klein & Bischoff, 2011; Le, et al., 2018; Levula, et al., 2011; Polverino, et al., 2018; Schick, et al., 2019; Vuohelainen, et al., 2011) |
|
9 MDC9, Meltrin-γ, γ Myeloma cell metalloprotease |
8p11.22 | 91 | Lung epithelium, muscle, brain, neutrophils, monocytes, macrophages | Pro-heparin-binding EGF-like growth factor (Pro-HB-EGF), EGF, FGFR2IIIb, VEGF, integrin αvβ3, α6β1 & α9β1, APP, fibronectin, tenascin, vitronectin, gelatin, collagen XVII, ACE, laminin, delta-like ligand 1, insulin-B chain, insulin-like growth factor binding protein-5, IL-11R | α-secretase, angiogenesis, retinal neovascularization, endocardial cushion development, myogenesis, formation of myotubes, wound healing, bone formation, fertilization, cell proliferation & /migration, cell-cell interactions, AAA, lung inflammation, chronic obstructive pulmonary disease, chondrocyte differentiation and proliferation, osteoarthritis, gastric pancreatic, colorectal and ovarian carcer. Decreased in anterior polar cataract | (Hodgkinson, et al., 2010; Horiuchi, Zhou, Kelly, Manova, & Blobel, 2005; Roychaudhuri, et al., 2014; G. Shen, et al., 2020) |
|
10 Kuzbanian protein homologue, CD156c, MDAM |
15q21.3 | 84 | Embryonic fibroblast, ECs, SMCs, cardiomyocytes, blood eosinophils, monocytes, macrophages, lung epithelium | Pro-HB-EGF, pro-TNFα, Notch, APP, cellular prion precursor, gelatin, DDR-1, β-cellulin, CD23, CD30, CD44, collagen IV & XVII, fractalkine, delta-like ligand 1, desmoglein, E-cadherin, N-cadherin, VE-cadherin, EGF, ephrin-A2 & A5, ErbB2 ligands, Fas-L, IL-6R, IL-11R, IL-23R, Klotho, LAG-3, MICA, Pcdh-γ, fibronectin, cystatin C, type I & II single membrane-spanning proteins, RAGE, JAM-A, ICAM-1 | α-secretase, Notch signaling, SMC proliferation, leukocyte migration, vascular cell apoptosis, wound healing, AAA, unstable atherosclerotic plaques, dilated cardiomyopathy, atrial fibrillation. acute lung inflammation, asthma, nervous system development, Alzheimer’s disease, chondrocyte proliferation & differentiation, osteoarthritis, colorectal cancer. Deficiency causes embryonic lethality | (Duru, et al., 2014; Hartmann, et al., 2002; Hodgkinson, et al., 2010; Hsia, et al., 2019; Mehta, et al., 2018; Pelisek, et al., 2012; Raucci, et al., 2008; Schulz, et al., 2008; G. Shen, et al., 2020; Speck, et al., 2015; C. Zhang, et al., 2010) |
|
11 MDC11 |
17q21.31 | 83 | Brain Hippocampus, cerebellum, brown fat | Integrin α4. Non-proteolytic | Neural development, myelination, synaptic transmission, epilepsy, breast cancer suppression. ADAM11−/− mice show learning deficiencies | (Hsia, et al., 2019; Takahashi, et al., 2006; L. Wang, et al., 2018) |
|
12 Meltrin-α, MLTN, MLTNA |
10q26.2 | 100 | SMCs, blood monocytes, macrophages, placenta | Insulin growth factor binding protein-3 (IGFBP-3), IGFBP-5, pro-HB-EGF, integrin α9β1, EGF, betacellulin, Delta-like1, gelatin, collagen IV, placental leucine aminopeptidase (P-LAP) | SMC proliferation, unstable atherosclerotic plaques, hypertrophic cardiomyopathy, bronchial asthma, breast, bladder & colorectal cancer, chondrocyte differentiation and proliferation, osteoarthritis, Down syndrome | (Asakura, et al., 2002; Duru, et al., 2014; Kurisaki, et al., 2003; Nyren-Erickson, et al., 2013; Pelisek, et al., 2012; Smiljanic, et al., 2011) |
|
15 Metargidin, MDC15, AD56, CR II-7 |
1q21.3 | 93 | Testis, ECs, SMCs, cardiomyocytes, blood monocytes, macrophages | Acrogranin, collagen IV, gelatin, amphiregulin, CD23, E-cadherin, HB-EGF, integrins α9β1 & αvβ3, Src family protein-tyrosine kinases, CD23, ADAM10 | Sperm-egg interaction, cell-cell adhesion, angiogenesis, retinal neovascularization, vascular cell apoptosis, neointima formation, wound healing, atrial fibrillation, dilated cardiomyopathy, atherosclerosis, ascending aortic dissection, inflammation, rheumatoid arthritis, meningioma | (Charrier-Hisamuddin, et al., 2008; Chung, et al., 2016; Hodgkinson, et al., 2010; Horiuchi, et al., 2003; Hou, et al., 2015; Levula, et al., 2011) |
|
17 TACE, CD156b, Snake venom-like protease, cSVP |
2p25.1 | 93 | Embryonic fibroblasts, SMCs, cardiomyocytes, blood monocytes, macrophages, lung epithelium | ECM, collagen, pro-TNFα, pro-TGF-α, pro-HB-EGF, EGFR ligand pro-amphiregulin, ACE2, TRANCE, pro-neuregulin-a-2C, Notch, Fas ligand, fractalkine, L-selectin, collagen XVII, TNFR I & II, IL-1R II, IL-6R, IL-11R, IL-23R, Erb-B4/HER4, macrophage colony-stimulating factor receptor I, nerve growth factor receptor (TrkA), growth hormone receptor MUC1, APP, cellular prion precursor, fibronectin, cystatin C, β1 integrin, N-cadherin, VE-cadherin, JAM-A, ICAM-1, VCAM-1, syndecan-1, α-Klotho | α-secretase, TNFα processing, angiogenesis, EC and epithelial permeability, trans-endothelial leukocyte migration, SMC proliferation, vascular cell apoptosis. Wound healing, Kawasaki disease, lung inflammation, neurite outgrowth and myelination, neuroinflammation, neoplastic meningitis, gastric and colorectal cancer. Knockout mice die perinatally with thickened and mis-shaped semilunar and atrioventricular valves, ventricular septal defect. Cardiomyocyte-specific knockout show reduced angiogenesis, post-MI left ventricle dilation & dysfunction | (Bertram, et al., 2015; Caolo, et al., 2015; Fan, et al., 2016; Hodgkinson, et al., 2010; Hsia, et al., 2019; Jackson, et al., 2003; Y. Jin, et al., 2013; Mukerjee, et al., 2019; Peng, et al., 2016; Perna, et al., 2017; Rizza, et al., 2015; Sahin, et al., 2004; M. Shen, et al., 2017; Speck, et al., 2015; R. Wang, et al., 2018) |
|
18 tMDC III, ADAM27 |
8p11.22 | 83 | Testis, sperm surface protein | Conotruncus (superior end of bulbus cordis of heart) | Endocardial cushion and heart development | (Cho, 2012) |
|
19 Adamalysin19, Meltrin-β, MADDAM, Metalloprotease and disintegrin dendritic antigen marker, FKSG34 |
5q33.3 | 105 | Heart, SMCs, lung epithelium, kidney, eosinophils, brain, peripheral nervous system, skeletal muscle, bone, testis | Pro-HB-EGF, ErbB ligands, NGR-β1 & β4, neuregulin, TNFα. Constitutive α-secretase activity that promotes processing of APP and could protect against Alzheimer’s disease | Cell adhesion, cell-cell & cell-matrix interactions, embryo implantation, cardiovascular morphogenesis and neurogenesis, endocardial cushion development, endocardial epithelial-to-mesenchymal transformation, formation of cardiac septa and valves, dendritic cell differentiation. Brain astrocytoma and glioblastoma, lung, colon and ovarian cancer, lung and kidney inflammation. ADAM19−/− mice show ventricular septum defect, immature valves, neuronal defects, and die soon after birth | (Kurisaki, Masuda, Osumi, Nabeshima, & Fujisawa-Sehara, 1998; Kurohara, et al., 2004; Qi, Newcomer, & Sang, 2009; Yagami-Hiromasa, et al., 1995; H. M. Zhou, et al., 2004) |
| 20 | 14q24.2 | 82 | Testis, monocytes | Integrins | Embryo development | (Blobel, 1997) |
|
21 ADAM31 |
14q24.2 | 81 | Testis, blood monocytes, glia | Integrins | Sperm maturation, neurogenesis | (Blobel, 1997; Blobel & White, 1992) |
|
22 MDC2 |
7q21.12 | 100 | Reproductive system, brain | Brain integrins. Non-proteolytic | Neural development, myelination, synaptic transmission, epilepsy, neointima formation | (Blobel, 1997; Hsia, et al., 2019; S. M. Zhang, et al., 2019) |
|
28 e-MDC II, MDC-L |
8p21.2 | 87 | Epididymis,brain, lymphocytes | Integrins α4β1, α4β7 & α9β1, CD23 | Sperm maturation, cancer | (Blobel, 1997) |
|
29 Cancer/testis antigen 73, svph 1 |
4q34.1 | 93 | Testis | Integrin α4 | Sperm maturation, melanoma progression | (Blobel, 1997; Smith, et al., 2002; L. Wang, et al., 2018) |
|
30 svph 4 |
1p12 | 89 | Sperm, testis | APP metabolism | Embryo development. Loss linked to amyloid-β deposition & Alzheimer | (Cho, 2012; Letronne, et al., 2016) |
| 32 | 8p11.22 | 88 | Sperm surface, testis, epididymis | Hyaluronic acid | Sperm development, egg fertilization | (Cho, 2012; Torabi, Bogle, Estanyol, Oliva, & Miller, 2017) |
| 33 | 20p13 | 88 | Testis,epididymis lung epithelium | Integrin α9β1 | Embryo development, SMC proliferation, bronchial asthma | (Cho, 2012; Kim, et al., 2017) |
ADAMTS members are widely distributed in different tissues (Table 2), and play a role in multiple physiological and pathological processes including CVD.
Table 2.
Human ADAMTS Family Members, Gene Locus, Molecular Weight (MW) of Full-length Protein, Tissue Distribution, Targets/Substrates, and Implications in Biology or Disease
| ADAMTS | Gene Locus | MW (kDa) | Tissue Distribution | Target/Substrate | Implications in Biology or Disease | Ref |
|---|---|---|---|---|---|---|
|
1 METH1 |
21q21.3 | 105 | Heart, aorta, smooth muscle, bronchial epithelial cells, fetal lung, liver, colon, kidney, adrenal gland, bladder, prostate, ovary, uterus, placenta, spinal cord, ciliary ganglion, olfactory bulb, adipocytes, breast stromal fibroblasts, myoepithelial cells | Aggrecan, versican, syndecan 4, dystroglycan, mac-2, gelatin (denatured collagen type I), amphiregulin, TGF-α, HB-EGF, VEGF, TFPI-2, semaphorin 3C, nidogen-1 & −2, desmocollin-3 | Embryogenesis. organ development, VSMC proliferation, inflammation, Anti-angiogenic, cancer cachexia, gastric cancer & lymph node metastasis, lung cancer | (J. Chen, et al., 2013; Gunther, et al., 2005; Kelwick, Desanlis, Wheeler, & Edwards, 2015; Kilic, et al., 2017; Kuno, et al., 1997; Y. Qu & Zhang, 2018; J. H. Tang, et al., 2017; Toba, et al., 2016; Vazquez, et al., 1999) |
| 2 | 5q35.3 | 135 | Aorta, smooth muscle, heart, lung, liver, kidney, bladder, skeletal muscle, tendon, bone, skin, retina, superior cervical ganglion, breast stromal fibroblasts adipocytes, uterus, placenta | Fibrillar procollagen type I, II, III & V, N-propeptide | Anti-angiogenic, cartilage matrix anabolism, procollagen N-proteinase, osteoarthritis, Ehlers-Danlos syndrome type VIIc | (Bekhouche & Colige, 2015; Colige, et al., 1997; Dubail, et al., 2010; Kelwick, Desanlis, et al., 2015) |
| 3 | 4q13.3 | 136 | CD105+ ECs, CD34+ cells, heart, lung, pineal gland, cartilage, bone, skeletal muscle, tendon, breast myoepithelial cells, testis, ovary, placenta, brain | Fibrillar procollagen type II N-propeptide, biglycan, pro-VEGF-C, reelin | Procollagen N-proteinase, angiogenesis, lymphangiogenesis, placental & brain functions, cartilage matrix anabolism, osteoarthritis | (Bekhouche & Colige, 2015; Fernandes, et al., 2001; Janssen, et al., 2016; Jeltsch, et al., 2014; Jha, et al., 2017; Kelwick, Desanlis, et al., 2015; Ogino, et al., 2017) |
|
4 Aggrecanase-1, ADMP-1 |
1q23.3 | 90 | Ovary, adrenal cortex, ciliary ganglion, trigeminal ganglion, brain, spinal cord, retina, heart, fetal lung, appendix, gall bladder, pancreas islets, skeletal muscle, uterus, breast myoepithelial cells, synovial fluid | Aggrecan, versican, neurocan, reelin, biglycan, brevican, matrilin-3, α2-macroglobulin, COMP | VSMC apoptosis, vascular inflammation, matrix degradation, sporadic aortic aneurysm and dissections, osteoarthritis | (Boerboom, et al., 2011; Dubail & Apte, 2015; Kelwick, Desanlis, et al., 2015; P. Ren, et al., 2017) |
|
5 ADAMTS11, Aggrecanase-2, ADMP-2 |
21q21.3 | 102 | Ovary, uterus, placenta, breast myoepithelial cells, bladder, adipocytes | Aggrecan, versican, reelin, biglycan, matrilin-4, brevican, α2-macroglobulin | Osteoarthritis, cancer (anti-tumorigenic, anti-angiogenic) | (Abbaszade, et al., 1999; Dubail & Apte, 2015; Hurskainen, Hirohata, Seldin, & Apte, 1999; Kelwick, Desanlis, et al., 2015) |
| 6 | 5q12.3 | 125 | Heart, appendix, gall bladder, superior cervical ganglion, trigeminal ganglion, breast myoepithelial cells, placenta | Fibrillin-1, LTBP1, syndecan 4 | Altered ventricular conduction, prolonged QRS interval | (Hurskainen, et al., 1999; Karoulias, Taye, Stanley, & Hubmacher, 2020; Kelwick, Desanlis, et al., 2015; Prins, et al., 2018) |
| 7 | 15q25.1 | 182 | Heart, smooth muscle, liver, pancreas, kidney, adrenal cortex, skeletal muscle, intervertebral disc, trigeminal ganglion, uterus, breast stromal fibroblasts | COMP, TSP-1 | Atherosclerosis, CAD, VSMC migration, neointima formation, cartilage catabolism, osteoarthritis | (Hanby & Zheng, 2013; Hurskainen, et al., 1999; Kelwick, Desanlis, et al., 2015; L. Wang, et al., 2009) |
|
8 METH2 |
11q24.3 | 96 | Skeletal muscle, heart, lung, appendix, liver, adrenal cortex, breast stromal fibroblasts and luminal epithelial cells, placenta, brain, superior cervical ganglion | Aggrecan | Anti-angiogenic | (Georgiadis, Hirohata, Seldin, & Apte, 1999; Kelwick, Desanlis, et al., 2015; Vazquez, et al., 1999) |
| 9 | 3p14.1 | 217 | Capillary ECs, heart, lung, kidney, pancreas, colon, ovary, skeletal muscle, dorsal root ganglion, breast myoepithelial cells, uterus, placenta | Aggrecan, versican | Cancer | (Desanlis, Felstead, Edwards, & Wheeler, 2018; Kelwick, Desanlis, et al., 2015; Koo, et al., 2010; Somerville, et al., 2003) |
| 10 | 19p13.2 | 121 | Heart, lung, liver, pancreas, kidney, brain, adipose tissue, CD8+ T-cells, breast stromal fibroblasts, uterus, placenta | Fibrillin-1 | Weill-Marchesani syndrome (short stature, acromelic dysplasia, disproportionate distal limb shortening) | (Hubmacher & Apte, 2015; Kelwick, Desanlis, et al., 2015; Kutz, et al., 2011; Le Goff & Cormier-Daire, 2009; Mead & Apte, 2018; Somerville, Jungers, & Apte, 2004) |
| 12 | 5p13.3 | 178 | Atrioventricular node, smooth muscle, liver, gall bladder, bone marrow, adipose tissue, intervertebral disc, breast stromal fibroblasts and myoepithelial cells | COMP | Anti-angiogenic. Cell adhesion, cancer, osteoarthritis | (Bai, Wang, Luan, Yu, & Liu, 2009; El Hour, et al., 2010; Kelwick, Desanlis, et al., 2015) |
| 13 | 9q34.2 | 154 | ECs, heart, lung, liver, hepatic stellate cells, pancreas, kidney podocytes, brain, testis, breast myoepithelial cells, placenta, CD71+ early erythroid cells, thyroid | von Willebrand factor (vWF) | Deficiency leads to thrombotic thrombocytopenic purpura (TTP) | (Cal, et al., 2002; Kelwick, Desanlis, et al., 2015; Lopez & Dong, 2004; Mead & Apte, 2018; Moake, 2004; X. Zheng, et al., 2001; X. L. Zheng, 2015) |
| 14 | 10q22.1 | 134 | Fibroblasts, lung, liver, gall bladder, prostate, retina, cerebellum, thalamus, fetal thyroid, adipocytes, bone marrow, skin, breast myoepithelial and luminal epithelial cells, placenta | Fibrillar procollagen type I N-propeptide (pNα1 and pNα2 chains) | Procollagen N-proteinase, cartilage matrix anabolism, osteoarthritis, multiple sclerosis | (Bekhouche & Colige, 2015; Cal, et al., 2002; Goertsches, Comabella, Navarro, Perkal, & Montalban, 2005; Kelwick, Desanlis, et al., 2015; Poonpet, Honsawek, Tammachote, Kanitnate, & Tammachote, 2013; Rodriguez-Lopez, et al., 2009) |
| 15 | 11q24.3 | 103 | Heart, colon, brain, musculoskeletal system, adipose tissue, breast myoepithelial cells, uterus, placenta | Aggrecan, versican | Anti-angiogenic, anti-tumorigenic | (Dancevic, et al., 2013; Kelwick, Desanlis, et al., 2015) |
| 16 | 5p15.32 | 136 | Aorta, gall bladder, brain, ovary, breast myoepithelial cells | ECM proteins, latency-associated peptide (LAP)-TGF-β complex | Hypertension, cardiac fibrosis and hypertrophy, heart failure. Loss linked to congenital undescended testes | (Cal, et al., 2002; Kelwick, Desanlis, et al., 2015; Sarila, et al., 2020; Yao, et al., 2020) |
| 17 | 15q26.3 | 121 | Ovary, breast myoepithelial cells | Fibrillin-1 & −2, fibronectin | Weill-Marchesani-like syndrome | (Cal, et al., 2002; Hubmacher & Apte, 2015; Karoulias, et al., 2020; Kelwick, Desanlis, et al., 2015; Mead & Apte, 2018) |
| 18 | 16q23.1 | 135 | Endothelium, heart, brain, ciliary ganglion, skin, prostate, breast myoepithelial cells, uterus, placenta | Basal membrane-specific proteoglycan Col18a1, fibronectin | Microcornea, myopic chorioretinal atrophy and telecanthus | (Ataca, et al., 2020; Cal, et al., 2002; Dubail & Apte, 2015; Kelwick, Desanlis, et al., 2015; Mead & Apte, 2018; R. Zhu, et al., 2018) |
| 19 | 5q23.3 | 134 | Dorsal root ganglion, breast myoepithelial cells, uterus, placenta | ECM Proteins | Loss causes non-syndromic heart valve disease | (Karoulias, et al., 2020; Kelwick, Desanlis, et al., 2015; Wunnemann, et al., 2020) |
| 20 | 12q12 | 215 | Heart, lung, appendix, liver, pancreas, brain, skeletal muscle, pituitary, trigeminal ganglion, prostate, testis, breast myoepithelial cells, ovary, placenta | Versican | Colorectal, brain and breast cancer. Cleft lip and palate | (Kelwick, Desanlis, et al., 2015; Llamazares, Cal, Quesada, & Lopez-Otin, 2003; Somerville, et al., 2003; Z. T. Wolf, et al., 2015) |
4. ADAMs and ADAMTS Activation
ADAMs can be activated by cytokines, proteinases, G-protein coupled receptor (GPCR) agonists, Ca2+ ionophores and protein kinase C activators. Compared to MMPs, whose activation occurs in ECM (Takawale, Sakamuri, & Kassiri, 2015), ADAMs activation occurs intracellularly where their pro-domain is removed by pro-protein convertases in the Golgi system during the transit process. The pro-protein convertase cleaves the Arg-Xaa-(Arg/Lys)-Arg or Rx(R/K)R motif of the pro-domain, allowing Zn2+ coordination to the metalloprotease domain so that ADAM can be catalytically activated and perform its sheddase activity and biological function (Anders, Gilbert, Garten, Postina, & Fahrenholz, 2001).
Several factors regulate ADAMs’ processing. ADAM8 processing by autocatalysis produces a sheddase form that can contribute to cell adhesion (Schlomann, et al., 2002). N-glycosylation is also important for processing, localization, stability, and activity of ADAM8 (Srinivasan, Romagnoli, Bohm, & Sonenshein, 2014). Tumor necrosis factor-α (TNFα) upregulates ADAM9 expression at the transcriptional level (Schouten, et al., 2016). Meprin-β is a membrane-bound metalloprotease that processes ADAM9, 10 and 17, and plays a role in ECM assembly and inflammation (Wichert, et al., 2019). Transforming growth factor-β (TGF-β) stimulates renal cells and causes upregulation of Adam10, 12, 17 and 19 (Ramdas, McBride, Denby, & Baker, 2013).
ADAM10 is a ubiquitous transmembrane metalloprotease that cleaves the ectodomain of transmembrane proteins such as Notch and amyloid precursor protein (APP). ADAM10 is compartmentalized into membrane microdomains by tetraspanins (Tspan), a family of 33 four-transmembrane proteins that regulate clustering and intracellular trafficking of other “partner” proteins. ADAM10 co-immunoprecipitates and interacts with the TspanC8 subgroup including Tspan5, 10, 14, 15, 17 and 33/Penumbra, all six are required for ADAM10 trafficking from the endoplasmic reticulum and its enzymatic maturation. ADAM10 could function as six different molecular scissors with different substrate specificities, depending on which of the six TspanC8s it is associated with. For example, human umbilical vein endothelial cells (HUVECs) express Tspan14, the knockdown of which reduces ADAM10 surface expression and activity (Haining, et al., 2012). ADAM10 is also implicated in leukocyte transmigration by cleaving vascular endothelial cadherin (VE-cadherin), which regulates endothelial barrier function. ADAM10 knockdown on HUVECs impairs transmigration of peripheral blood T lymphocytes. The formation of endothelial Tspan5- and Tspan17-ADAM10 complexes may regulate inflammation by maintaining VE-cadherin expression and promoting T lymphocyte transmigration (Reyat, et al., 2017). Mouse erythrocytes express mainly Tspan33, and ADAM10 expression is reduced in the absence of Tspan33. In contrast, ADAM10 expression is normal on Tspan33-deficient mouse platelets in which Tspan14 is the major TspanC8. In platelets, ADAM10-induced cleavage of the platelet collagen receptor GPVI is regulated by Tspan14, 15, and 33, leading to inhibition of GPVI receptor function and anti-thrombotic effects. Thus, TspanC8s regulate ADAM10 maturation and trafficking to the cell surface, and specific TspanC8-ADAM10 complexes provide cell- or substrate-specific ADAM10 targeting (Haining, et al., 2012).
In cultured VSMCs, angiotensin II (AngII) increases ADAM17 mRNA expression, protein levels, and promoter activity. Also, AngII infusion in mice for 2 weeks increases ADAM17 in the aorta, heart and kidney vessels through hypoxia inducible factor 1α (HIF1α)-dependent transcriptional upregulation, leading to vascular malfunction and end-organ damage (Obama, et al., 2015). MicroRNAs (miRs) are small single‐stranded noncoding molecules that regulate gene expression, directly target mRNA at posttranscriptional level, and play a role in different pathological conditions. miR-634 is upregulated in blood of patients with anti-neutrophil cytoplasmic antibody-associated vasculitis, and miR-634 mimics induce a proinflammatory phenotype in monocyte-derived macrophages with enhanced expression/release of ADAM17 and interleukin 6 (IL-6) (Bertram, et al., 2015).
Rhomboids are a conserved protein superfamily that binds membrane proteins and directs them to different cellular pathways. The iRhom1 and 2 have 7 transmembrane domains and regulate ADAM17 enzymatic maturation and trafficking to cell surface and distinct substrates. In platelets, iRhom2 directs ADAM17 to promote the shedding of the von Willebrand factor (vWF) platelet receptor glycoprotein Ib (GPIb), leading to inhibition of vWF receptor function. Targeting platelet iRhom2 could activate ADAM17 and provide anti-thrombotic therapy without the side effects of activating ADAM17 in other cells (Matthews, Noy, Reyat, & Tomlinson, 2017).
ADAM17 regulates EGF receptor (EGFR) signaling by shedding and liberating EGFR ligands from their membrane anchor. ADAM17 deficiency reduces EGFR signaling, leading to skin and intestinal barrier defects. Like Egfr−/− mice, Adam17−/− mice die perinatally with open eyes. ADAM17-dependent EGFR ligand shedding requires its transmembrane domain but not its cytoplasmic tail, suggesting that ADAM17 is regulated by other integral membrane proteins. While iRhom2 controls the maturation and function of ADAM17 in myeloid cells, iRhom2−/− mice appear normal. On the other hand, iRhom1/2−/− double knockout (KO) mice resemble Adam17−/− and Egfr−/− mice in that they die perinatally with open eyes, misshapen heart valves, and growth plate defects. The iRhom1/2−/− tissues lack mature ADAM17 and show reduced EGFR phosphorylation. iRhom1 regulates ADAM17 maturation in the brain, except in microglia, where ADAM17 is controlled by iRhom2. Thus, during mouse development, iRhoms1/2 regulate ADAM17-dependent EGFR signaling, and targeting iRhoms1/2 may be useful in ADAM17/EGFR-related pathologies (X. Li, et al., 2015).
ADAMs may differ in their activation process and its location. ADAM8 and 28 are activated in ECM. ADAM12 is stored as already active proteins, and transferred to the cell membrane when the cell is stimulated by cytokines (Sundberg, et al., 2004), suggesting that removal of the pro-domain and proteolytic processing is not necessary for ADAM12 activation (Cao, Kang, Zhao, & Zolkiewska, 2002). Other ADAMs auto-catalytically remove their pro-domain for their transformation from inactive pro-protein to active enzyme (Schlomann, et al., 2002).
The intracellular cytoplasmic tail and protein phosphorylation may contribute to ADAMs signal transmit and interaction with other proteins and receptors, but their role in ADAMs activation is unclear. For example, ADAM17 with truncated cytoplasmic tail can still function as a sheddase of TNFα, p75 TNFR and IL-1R-II. On the other hand, the ADAM17 transmembrane domain is essential for the shedding of these substrates (Reddy, et al., 2000). Also, while phosphorylated Ser819 is an important reaction site during activation of ADAM17, mutation of this site did not suppress ADAM17 activity or cleavage of its substrates transforming growth factor-α (TGFα), TNFα or TNFα receptors, suggesting that the cytoplasmic tail is not responsible for ADAM17 sheddase activity (Reddy, et al., 2000). However, the cytoplasmic tail of ADAM17 is linked to GPCR mediated EGFR signaling (Edwards, et al., 2008), where Src causes stimulation of phosphatidylinositol 3-kinase (PI3K) and activation of phosphoinositide-dependent kinase-1 (PDK1), which phosphorylates ADAM17 (Q. Zhang, et al., 2006). Also, agonist-induced phosphorylation of Thr735 of ADAM17 accounts for the transfer of ADAM17 from the endoplasmic reticulum to the cell surface, and in turn enhances cleavage of the TrkA neurotrophin receptor in cardiomyocytes (Soond, Everson, Riches, & Murphy, 2005).
Similar to ADAMs. ADAMTS activation involves cleavage of the N-terminal pro-domain next to the consensus sequence Arg-Xaa-(Arg/Lys)-Arg by the proprotein convertases furin and furin-like enzymes in the trans-Golgi or at the cell surface (Colige, et al., 2005; Koo & Apte, 2010; Somerville, Longpre, et al., 2004). As metalloproteases, ADAMTS activity requires a neutral to slightly basic pH, Zn2+ and Ca2+ (Colige, et al., 1995). Some ADAMTS may not require pro-domain cleavage for their activation. ADAMTS13 has an unusually short propeptide, and ADAMTS9 and 13 zymogens are active despite retention of the propeptide (Koo, et al., 2007; Majerus, Zheng, Tuley, & Sadler, 2003). ADAMTS2 undergoes autocatalytic cleavage within the C-terminal end and the procollagen N-propeptidase domain (Bekhouche & Colige, 2015; Colige, et al., 2005). Autocatalytic C-terminal cleavage and activation also occur with ADAMTS1, 4, 8, 9 and 12 (Porter, et al., 2005), thus impacting their bio-disponibility, substrate recognition and activity (Bekhouche & Colige, 2015).
5. ADAMs Targets, Substrates, Functions and Mouse KO Phenotype
ADAMs have multiple targets/substrates (Table 1). Similar to MMPs and SVMs, ADAMs were initially presumed as active proteases that cleave ECM substrates, but members of the ADAM family could be proteolytic or non-proteolytic. Proteolytic ADAMs including ADAM8, 9, 10 and 17 are active metalloproteases or sheddases that regulate proteolytic cleavage and ectodomain shedding of cell surface adhesion molecules, cytokines, chemokines and growth factors, resulting in the release of soluble proteins that exert agonistic or antagonistic effects. ADAM-mediated shedding could also affect a specific receptor, and in turn activate signaling pathways and cellular functions, or render the membrane protein or receptor inactive (E. P. C. van der Vorst, et al., 2018). The ADAMs’ M-domain, D-domain, and cysteine-rich C-domain with HVR are important for substrate recognition, proteolytic processing and protein degradation. During cell-cell and cell-matrix interactions, HVR guides proteolytically inactive ADAMs to recognize specific substrates, then the metalloproteinase M-domain conducts ADAM function as a proteolytic enzyme or sheddase. A specific factor may combine with the substrate before it can be recognized by HVR. Also, during inter-cellular interaction, an ADAM on one cell surface can liberate a ligand and activate a receptor on an adjacent cell surface. In contrast with proteolytic ADAMs, non-proteolytic ADAM11, 22 and 23 have no catalytic activity, and function as adhesion proteins or receptors (Hsia, et al., 2019).
ADAMs have been implicated in multiple cellular processes including degradation of ECM proteins, cell adhesion and fusion, intracellular signaling, cell proliferation, migration and invasion, fertilization, neurogenesis, and inflammation (Pelisek, et al., 2012). There are 22 ADAMs in humans (Klein & Bischoff, 2011), and 14 ADAMs including ADAM1, 2, 3, 6, 7, 18, 20, 21, 22, 28, 29, 30, 32 and 33 are mainly expressed in the testis, epididymis and uterus, and involved in embryo development. ADAM-induced shedding of membrane-associated growth factors also modulate key cell signaling pathways in cancer microenvironment. ADAM8, 9, 10, 12, 15, and 17 have a broader expression in human tissues particularly in the cardiovascular system, and may be involved in the pathogenesis of CVD, and therefore will be discussed in greater detail.
ADAM8 is highly expressed on immune cells, neutrophils, hematopoietic cells and breast cancer, ADAM8 is usually stored in granules and transported to the cell surface when the cell is stimulated by pro-inflammatory stimuli. L-selectin is a “homing receptor” for lymphocytes, and its shedding by ADAM8 is involved in regulation of neutrophil rolling and trans-endothelial extravasation. ADAM8 is implicated in inflammation, rheumatoid arthritis and asthma. The levels of ADAM8 are increased in the peri-infarct myocardium following MI and infiltration of inflammatory cells. ADAM8 also regulates ovulation and osteoclast differentiation (Vuohelainen, et al., 2011). ADAM8−/− mice develop normally without a disease phenotype, but show suppressed reaction to autoimmune arthritis (Kelly, et al., 2005).
ADAM9 (MDC9/meltrin-γ) is a widely expressed protease implicated in ectodomain cleavage of heparin-binding EGF-like growth factor (HB-EGF) and as an α-secretase for APP. During mouse development, ADAM9 is ubiquitously expressed mainly in the mesenchyme, heart and brain. ADAM9 regulates myogenesis, formation of myotubes and myocardium development. However, ADAM9−/− mice develop normally, are viable and fertile with no major pathological phenotype or defects in muscle formation, and still show HB-EGF shedding and APP α-secretase products formation (Weskamp, et al., 2002), suggesting that ADAM9 functions can be compensated by other ADAMs. Also, constitutive and stimulated ectodomain shedding of HB-EGF is comparable in embryonic fibroblasts from ADAM9−/− and WT mice, arguing against a role of ADAM9 in HB-EGF shedding in these cells. Also, the production of the APP α- and γ-secretase cleavage product 3-kDa peptide and of β- and γ-secretase cleavage product amyloid-β is not different in hippocampal neurons from ADAM9−/− and WT mice, arguing against a role as an α-secretase in mice (Weskamp, et al., 2002). Angiotensin-I converting enzyme (ACE) is a zinc-dependent peptidase that regulates vasoactive peptide metabolism and a transmembrane protein that undergoes shedding primarily from endothelial cells (ECs) by proteinases to release catalytically active sACE. The MMP/ADAM inhibitor BB-94, tissue inhibitor of metalloproteinases (TIMP), and ADAM9 knockdown by siRNA block lipopolysaccharide (LPS)-induced ACE shedding from ECs, confirming a role of ADAM9 in ACE shedding (English, Corvol, & Murphy, 2012).
ADAM10 is a crucial α-secretase involved in ECM degradation, ectodomain shedding of cell surface substrates, adhesion molecules, chemokines and growth factor receptors (E. P. van der Vorst, et al., 2015). ADAM10 is essential for embryonic development through cleavage of Notch proteins (Matthews, Noy, et al., 2017). By inducing Notch cleavage and controlling subsequent “regulated intramembrane proteolysis”, ADAM10 releases cleaved intracellular domains which transfer to the nucleus and finetune gene transcription. ADAM10 regulation of Notch signaling is an important step in the embryonic development of the cardiovascular system (Cong & Jia, 2011; Howard & Glynn, 1995). Because of the crucial role of ADAM10 in biological processes, intramembrane proteolysis, Notch/Delta-like 4 (Dll4) signaling and APP processing, ADAM10 KO mice do not survive during early embryonic development, show multiple defects in the CNS and somites, and die of severe cardiovascular defects, similar to mice deficient in Notch/Dll4 signaling (Cong & Jia, 2011; Howard & Glynn, 1995). ADAM10 also regulate the Eph/ephrin pathway and plays a role in cancer, neurodegenerative diseases, Alzheimer and prion disease, bacterial infection, inflammation, heart attacks, stroke and asthma (Matthews, Szyroka, Collier, Noy, & Tomlinson, 2017).
ADAM12 participates in myogenesis and embryonic development (Yagami-Hiromasa, et al., 1995). The two splice-variants ADAM12L and ADAM12S are proteolytically active, cleave membrane-bound proteins, and affect distant targets. ADAM12L promotes cardiomyocyte hypertrophy by inducing HB-EGF shedding and Notch signaling (Asakura, et al., 2002; Jorissen, et al., 2010; Karkkainen, Rybnikova, Pelto-Huikko, & Huovila, 2000). ADAM12 also contributes to ectodomain shedding of EGFR ligands and EGFR transactivation (Tanaka, et al., 2004), and may reduce inflammatory skin diseases and barrier defects. ADAM12−/− mice usually develop without functional deficiencies, although some studies found 30% mortality in ADAM12−/− pups within the first week (Kurisaki, et al., 2003).
ADAM15 is linked to cell adhesion, cell-cell and cell-matrix interactions, inflammation, and shedding of cell surface molecules (Oksala, et al., 2009), ADAM15 colocalizes with VE-cadherin. Accumulation of ADAM15 in cell-cell contacts is preceded by VE-cadherin-mediated EC adherens junction formation, supporting that ADAM15 is a component of adherens junctions that influence EC function (Ham, Levkau, Raines, & Herren, 2002). ADAM15 also cleaves epithelial cadherin (E-cadherin), thus compromising epithelial cell connections and tissue stability, and promoting inflammatory cell infiltration in different tissues (C. Sun, Wu, Lee, & Yuan, 2012). ADAM15 through cleaving E-cadherin, decreases EC stability, and promotes EC permeability and neutrophil trans-endothelial migration and vascular tissue infiltration (C. Sun, et al., 2010). ADAM15 also promotes angiogenesis by inducing the expression of vascular endothelial growth factor (VEGF), VEGF receptor VEGFR1 and VEGFR2 (Xie, et al., 2008). Recombinant human disintegrin domain of ADAM15 (rhddADAM15) inhibited proliferation/migration and induced partial G2/S arrest, apoptosis and caspases activity in Bel-7402 cells, and inhibited the growth and metastasis of Bel-7402 cell xenografts in zebrafish, suggesting inhibitory effects on tumor growth and metastasis that could be useful in cancer therapy (Hou, et al., 2013). ADAM15−/− mice show no overt pathological phenotype, but display reduced neovascularization in a mouse model of retinopathy of prematurity, and reduced size of tumors resulting from implanted mouse melanoma cells (Horiuchi, et al., 2003).
ADAM17 (TNFα converting enzyme, TACE) is expressed in most tissues, and is upregulated during inflammation and cancer. ADAM17 mediates ectodomain shedding of membrane-bound adhesion molecules, cytokines and their receptors (Black, et al., 1997; Moss, et al., 1997; Niu, Wang, & Li, 2015). ADAM17 is considered a first line of defense against infection and injury by releasing TNFα and promoting inflammation (Matthews, Noy, et al., 2017). ADAM17 also regulates the shedding of TNFα receptors, the growth factors TGFα, HB-EGF, VEGF, VEGFR2 and neuregulins (Blobel, 2005; Obama, et al., 2015; Sommer, et al., 2017). Shedding of membrane-anchored pro-TGFα (Peschon, et al., 1998) and pro-HB-EGF (Higashiyama & Nanba, 2005) to activate EGFR is among the critical functions of ADAM17 (Jackson, et al., 2003; D. C. Lee, et al., 2003; Sahul, et al., 2011). ADAM17 is also involved in egg fertilization, embryonic development, angiogenesis and neurogenesis (Canault, Certel, Schatzberg, Wagner, & Hynes, 2010; Peschon, et al., 1998). Mice with TACEΔZn/ΔZn null mutation die at birth and show failure of eyelid fusion, hair and skin defects, abnormal lung development, and enlarged hearts with increased myocardial trabeculation, reduced cell compaction, larger cardiomyocyte size and increased cell proliferation. The heart of TACEΔZn/ΔZn KO mouse shows reduced EGFR expression, attenuated cleavage of the receptor tyrosine-protein kinase ErbB4, and altered mitogen-activated protein kinase (MAPK) activity, supporting a role of TACE-mediated cell surface protein ectodomain shedding in cardiac development and modeling (Shi, et al., 2003). ADAM17 KO mice embryos also show thickened and mis-shaped heart valves, similar to the mice lacking HB-EGF (Jackson, et al., 2003; D. C. Lee, et al., 2003; Sahul, et al., 2011; Shi, et al., 2003). Because ADAM17 regulates TNFα processing and TNFα levels are elevated in CVD, TNFα antagonists have been tested and have shown benefits in animal models of CVD (Sato, et al., 2006; M. Sun, et al., 2007). However, anti-TNFα treatment did not show much improvement in patients with late-stage heart failure (Anker & Coats, 2002; Coletta, Clark, Banarjee, & Cleland, 2002). Also, in contrast with the perinatal death of ADAM17 KO mice, TNFα-deficient mice develop normally and are viable and fertile, indicating that ADAM17 has other essential functions (Canault, et al., 2010). Interestingly, TNFα processing is not completely inhibited in ADAM17 KO mice, as ADAM9 and 10 (Lunn, et al., 1997; Rosendahl, et al., 1997), and MMP17 (English, et al., 2000) also promote TNFα shedding. In effect, ADAM17 could have a dual effect whereby it promotes inflammation by releasing soluble TNFα to activate TNFα receptor (TNFR), and at the same time cleaving TNFRI (p55) and TNFRII (p57) and reducing their sensitivity to TNFα (McClurg, et al., 2015; Reddy, et al., 2000; Weskamp, et al., 2004; Zeinieh, Salehi, Rajkumar, & Barker, 2015).
While ADAM17 KO mice are not viable, conditional ADAM17 KO using novel gene targeting strategy generate mice with reduced ADAM17 levels in all tissues. The resulting ADAM17ex/ex mice are viable, show reduced shedding of ADAM17 cell surface substrates, and develop eye, heart, and skin defects due to failure of release of EGFR ligands and impaired EGFR signaling. Homozygous ADAM17ex/ex mice have normal intestine, but show increased susceptibility to inflammation in response to dextran sulfate sodium-induced colitis, likely due to impaired shedding of EGFR ligands and failure to activate EGFR and phosphorylate STAT3, leading to defective regeneration of epithelial cells and breakdown of the intestinal barrier. Thus, ADAM17 appears to regulate both the systemic release of the proinflammatory cytokine TNFα, and the EGFR-mediated regenerative activities during the immune response (Chalaris, et al., 2010). ADAM17 also confers defense against skin infection and injury and maintains epidermal barrier function by proteolytic release of EGFR ligands (X. Li, et al., 2017). The skin barrier is formed by cross-linking activity of transglutaminases, and ADAM17/EGFR-driven PLCγ1 and PKC pathways promote transglutaminase-1 expression during terminal keratinocyte differentiation. Epidermal deficiency of ADAM17 in mice impedes transglutaminases activity and results in postnatal skin barrier defects (C. Wolf, Qian, Brooke, Kelsell, & Franzke, 2016). Of note, EGFR participates in CVD progression, and ADAM17 may promote atherosclerosis, cardiac hypertrophy, and cardiac and renal fibrosis through the release of EGFR ligands and activation of EGFR (Oikawa, et al., 2014).
ADAM17 share aa sequence, crystal structure and function with ADAM10, and they both modulate vascular permeability by cleaving junctional adhesion molecule A (JAM-A) and VE-cadherin (Speck, et al., 2015). Other ADAM17 substrates include syndecan-1 (Bertram, et al., 2015), soluble intercellular adhesion molecule-1 (sICAM-1), soluble vascular cell adhesion molecule-1 (sVCAM-1), soluble IL-6 receptor (sIL-6R) and sTNFR1 (Rizza, et al., 2015). Individuals with the single nucleotide polymorphism (SNP) rs2228145 of IL-6R (IL-6R Asp358Ala variant) show increased sIL-6R serum levels, reduced IL-6-induced C-reactive protein, and decreased risk for CAD likely due to increased ADAM10 and 17 mediated shedding of cell surface IL-6R Asp358Ala variant versus the common IL-6R variant (Garbers, et al., 2014).
Angiotensin-converting enzyme-2 (ACE2) regulates heart function, and could act as a receptor for SARS-CoV spike protein. ACE2 is a type I transmembrane protein, with an extracellular N-terminal domain containing the active site and a short intracellular C-terminal tail. sACE2, lacking the transmembrane and cytosolic domains, could act as a decoy receptor for SARS-CoV spike protein preventing it from binding to the native ACE2 receptor (Y. Xiang, Wang, Chen, & Chen, 2021). ADAM17 promotes ACE2 shedding, and ADAM17 siRNA reduces ACE2 shedding (Lambert, et al., 2005).
Besides shedding of ADAM10, ADAM8 proteolytically releases the ADAM17 ectodomain. A study comparing the substrate spectrum of soluble ectodomains of ADAM10/17 (sADAM10/17) and their membrane-bound counterparts identified 134 protein cleavage events in total and 45 common substrates for sADAM10/17 within the secretome of murine cardiomyocytes. Further studies verified fibronectin, cystatin C, sN-cadherin, PCPE-1, and sAPP as direct substrates of sADAM10/17 (Scharfenberg, et al., 2020).
6. ADAMTS Targets, Substrates, Functions and Mouse KO Phenotype
In comparison with ADAMs, ADAMTS are secreted proteases and most of their targets/substrates are ECM proteins (Table 2) (Dubail & Apte, 2015; Mead & Apte, 2018). ADAMTS cleave proteoglycans including aggrecan, brevican, neurocan and versican, and degrade aggrecan and versican in blood vessels. ADAMTS also play a role in embryonic development, angiogenesis, coagulation, ECM proteolysis, and cartilage degradation, and ADAMTS KO mice show strong phenotypes in morphogenesis, mobility and reproduction.
ADAMTS1 (METH1) was originally cloned from a cancer cell line. ADAMTS1 is expressed in the yolk sac, placenta, heart, lung, liver, pancreas, spleen, kidney, brain, and limb bud of the developing mouse embryo and the tunica media of the aorta and pulmonary and hepatic vessels of adult mice (Thai & Iruela-Arispe, 2002). Osteotropic agents such as parathyroid hormone (PTH)-related protein (PTHrP), and prostaglandin PGE2 cause bone turnover partly via the cAMP/PKA pathway and rapid and transient increase in ADAMTS1 expression (Miles, et al., 2000). ADAMTS1 deficient mice show morphological defects in the kidney, adrenal gland, heart and aorta, and different lymphocyte and myeloid cell population in the spleen and bone marrow (Rodriguez-Baena, et al., 2018; Shindo, et al., 2000). Almost 50% of newborn Adamts1 null mice die likely due to a kidney malformation that becomes apparent at birth. Surviving female Adamts1 null mice are subfertile, while males reproduce normally (Boerboom, et al., 2011; Mittaz, et al., 2005; Mittaz, et al., 2004). LPS administration in mice induces ADAMTS1 expression in the heart and kidney, suggesting a role in inflammation. In cultured fibroblast-like COS-7 cells treated with heparin, the mature form of ADAMTS1 is detected in the culture medium and associated with ECM. Deletion mutants of ADAMTS1 revealed that the spacer region and three thrombospondin 1 (TSP1) motifs in the C-terminal region are critical for interaction with ECM (Kuno & Matsushima, 1998).
Mutations in ADAMTS2, 3, 10, 13, 17 and 20 are linked to Mendelian disorders or birth defects (Mead & Apte, 2018). ADAMTS2, 3 and 14 are procollagen N-proteinases that play a role in procollagen processing and collagen fibrils formation, maturation and assembly (Bekhouche & Colige, 2015). ADAMTS2 heterozygous mice are normal at birth, but show progressively fragile skin, and adult males are sterile (S. W. Li, et al., 2001).
ADAMTS3 is important for lymphangiogenesis and ADAMTS3 deficient mouse embryos do not survive past gestational day 15 due to lack of lymphangiogenesis and severe lymphedema (Janssen, et al., 2016).
ADAMTS5 cleaves versican and plays a role in the development of heart valves. Adamts5−/− mice fed a high-fat diet show impaired cleavage of versican in the heart and increased diastolic posterior wall thickness and left ventricle volume, but with little effect on cardiac function (Hemmeryckx, Carai, & Roger Lijnen, 2019), making it important to further assess the effects of ADAMTS5 inhibition in obesity-related heart injury. ADAMTS5 is also implicated in osteoarthritis by degrading its cartilage substrate aggrecan (Mead & Apte, 2018).
vWF mediates platelet aggregation to sites of blood vessel injury, and its ultra-large multimer functions as a substrate that is proteolytically cleaved and regulated by ADAMTS13 (Petri, et al., 2019).
ADAMTS15 is co-localized with hyaluronan in the developing heart and musculoskeletal system of the mouse embryo, and is expressed in the adult mouse colon and other systems with high versican processing capacity. In cultured HEK293T and COS-7 cells, ADAMTS15 is localized on cell surface where it undergoes propeptide processing and activation before cleaving V1 versican (Dancevic, et al., 2013).
ADAMTS16 has been linked to blood pressure regulation, osteoarthritis, cancer, kidney function and testis development. Male homozygous Adamts16 deficient mice show cryptorchidism and sterility with small testes ~10% of controls, and seminiferous tubules that do not perform spermatogenesis, are gradually depleted of germ cells, show vacuoles, and do not contain sperms. Of note, male heterozygous Adamts16 mutants on C57BL/6N background show normal testis determination during fetal development and are fertile at 3 and 6 months, suggesting a recessive trait (Livermore, et al., 2019). Adamts16 mutant rats also show renal abnormalities, suggesting that Adamts16 plays a role in maintaining normal functions of the male genitourinary system (Abdul-Majeed, Mell, Nauli, & Joe, 2014).
Heart valve defects are common disorders, making it important to determine their genetic underpinning. Studies have identified two consanguineous families, each family has two members affected with heart valve disease early in life, and whole-exome sequencing revealed homozygous alleles in ADAMTS19 in the affected four individuals. Also, Adamts19 KO mice show aortic valve dysfunction, supporting the role of ADAMTS19 gene mutation in the human heart valve disorder (Wunnemann, et al., 2020)
7. ADAMs and ADAMTS Inhibitors
The ‘cysteine-switch’ is a potent ADAM inhibitory mechanism, in which the cysteine residues in the pro-domain inhibit the protease activity (Lemjabbar & Basbaum, 2002; Ohtsu, Dempsey, & Eguchi, 2006). However, targeting the ‘cysteine-switch’ in the metalloproteinase domain is not very specific due to structural similarity between ADAMs, ADAMTS and MMPs (Nyren-Erickson, Jones, Srivastava, & Mallik, 2013). TIMPs are endogenous four-member family with a broad inhibitory effect on most MMPs (Brew & Nagase, 2010), and relatively more specific inhibitory effects on ADAMs. TIMP-1 and −3 inhibit ADAM10, 12, 15 and 17 (Pelisek, et al., 2012). In endotoxin-activated human macrophages, TIMP-3 inhibits ADAM17 only when it is bound to the cell surface, and the TIMP-3 cell surface levels are controlled by the endocytic receptor LRP1 (Schubert, Collins, Green, Nagase, & Troeberg, 2019). ADAM8, 9 and 19 are not inhibited by any TIMP (Amour, et al., 2002; Chesneau, et al., 2003). ADAM33 can be inhibited by TIMP-3 and −4, but not TIMP-1 (Zou, et al., 2004).
TIMPs inhibit MMPs and ADAMs with different efficacies. The N-terminal domain of TIMPs accounts for inhibiting MMPs, while the N-terminus of TIMP-1 or −3 or that of TIMP-2 has little effect on ADAM10 or ADAM17, respectively. Also, while TIMP-3 has a solid inhibitory effect on the isolated catalytic domain of ADAM17, when the other domains are added or some parts of the domains such as cysteine-rich domain are modified, the TIMP-3 inhibitory effect is reduced (Blobel, 2005).
Most synthetic small molecule inhibitors also target the catalytic Zn2+ and have a broad inhibitory spectrum towards ADAMs and MMPs (Table 3). Zn2+ chelators 1,10-phenanthroline and hydroxamate are potent ADAMs inhibitors (Glassey & Civetta, 2004). INCB3619 is a potent inhibitor of ADAM10 and 17, but is a less potent against ADAM8, 9 and 33 (Fridman, et al., 2007). TAPI-1, TAPI-2, and batimastat (BB-94) are potent inhibitors of ADAM17, but could affect MMPs. CGS27023, GW280264 and GI254023 exhibit specific inhibitory effect on ADAM9, 10 and 17 (Ludwig, et al., 2005). Glycosylation of a substrate such as TNFα inhibited ADAM10 activity, but increased ADAM8 and 17 activities (Minond, et al., 2012), and adopting the glycosylation strategy could allow targeting of a specific ADAM in CVD.
Table 3.
Endogenous and synthetic inhibitors of ADAMs and ADAMTS.
| Inhibitor | Target | Reference |
|---|---|---|
|
Endogenous: TIMP-1 |
ADAM10, 12, 15, 17 | (Pelisek, et al., 2012; Rapti, et al., 2008) |
| TIMP-2 | ADAM17, 33 ADAMTS1 |
(Kveiborg, et al., 2010) |
| TIMP-3 | ADAM10, 12, 15, 17, 28, 33 ADAMTS1, 2, 4, 5 |
(Bekhouche & Colige, 2015; Kashiwagi, Tortorella, Nagase, & Brew, 2001; Pelisek, et al., 2012; Rapti, et al., 2008; Schubert, et al., 2019; W. M. Wang, et al., 2006) |
| TIMP-4 | ADAM17, 28, 33 | (Heijink, et al., 2011) |
| α−2 macroglobulin | ADAMTS2, 4, 5 | (Bekhouche & Colige, 2015; Tortorella, et al., 2004; W. M. Wang, et al., 2006) |
| Papilin | ADAMTS2 | (Kramerova, et al., 2000) |
|
Synthetic: Batimastat (BB-94) |
ADAM8, 9, 17, MMPs | (Heijink, et al., 2011) |
| BK-1361 inhibitor peptide | ADAM8 | (J. Chen, et al., 2016) |
| Calcium pentosan polysulfate | ADAMTS4, 5 | (Takizawa, et al., 2008; Troeberg, et al., 2008; Vistnes, et al., 2014) |
| CGS27023 | ADAM9, 10, 17 | (Qian, et al., 2002) |
| Cis-1(S)2(R)-amino-2-indanol-based compounds | ADAMTS4, 5 | (Tortorella, et al., 2009) |
| cyclo(RLsKDK) cyclic peptide | ADAM8 | (Yim, et al., 2016) |
| Doxycycline | ADAMTS13, MMPs | (Bartoli, Kang, et al., 2015) |
| FC143, FC387 | ADAM8, 17 | (Schlomann, et al., 2019) |
| GI254023 | ADAM9, 10, 17, MMPs | (P. Zhang, et al., 2016) |
| Glycoconjugated arylsulfonamide | ADAMTS5 | (Santamaria, et al., 2021) |
| GM6001 | ADAM9, MMPs | (Maretzky, et al., 2017) |
| Granulin-epithelin precursor | ADAMTS7, 12 | (Guo, et al., 2010; Kelwick, Desanlis, et al., 2015) |
| GW280264X | ADAM8, 9, 10, 17 | (P. Zhang, et al., 2016) |
| INCB3619 | ADAM8, 9, 10, 17, 33 | (P. Zhang, et al., 2016) |
| KP457 | ADAM10, 17 | (P. Zhang, et al., 2016) |
| α-Lipolic acid | ADAM17 | (de Queiroz, et al., 2015) |
| Marimastat | ADAM9, 17, MMPs | (Maretzky, et al., 2017; Parrish, et al., 2018) |
| Propofol anesthetic | ADAM8 | (X. Yu, Shi, Wang, & Zhang, 2019) |
| TAPI-1 | ADAM17, MMPs | (Moss & Rasmussen, 2007) |
| TAPI-2 | ADAM9, 17, other ADAMs, MMPs | (Moss & Rasmussen, 2007) |
In Neuro2A cells, AngII increases ADAM17 expression and oxidative stress and these effects are attenuated by α-lipoic acid. Also, α-lipoic acid improves dysautonomia and baroreflex sensitivity, ameliorate HTN, and blunts the increase in ADAM17 and NADPH oxidase, and the decrease in ACE2 activity in the hypothalamus of deoxycorticosterone acetate (DOCA)-salt hypertensive mice. Thus, α-lipoic may preserve ACE2 compensatory activity by inhibiting ADAM17 and oxidative stress, resulting in amelioration of neurogenic HTN (de Queiroz, Xia, Filipeanu, Braga, & Lazartigues, 2015).
Cilostazol is a phosphodiesterase inhibitor that inhibits VSMC proliferation by regulating ADAM17 expression. In cultured VSMCs, cilostazol inhibits IL-1α and IL-1β-induced ADAM17 expression likely through inhibition of MAPK kinase (MEK), extracellular signal-regulated protein kinase (ERK) phosphorylation and the ERK/NF-κB pathway (Takaguri, Morimoto, Imai, & Satoh, 2016).
ADAMTS family members have different susceptibilities to endogenous and synthetic inhibitors (Murphy, 2011; Santamaria, et al., 2021; Santamaria & de Groot, 2019). As with other proteinases, α2-macroglobulin entrap ADAMTS2 after cleavage within its bait region (Bekhouche & Colige, 2015). ADAMTS1 is inhibited by TIMP-2 and 3, but not TIMP-1 and 4 (Rodriguez-Manzaneque, et al., 2002). TIMP-3 inhibits ADAMTS2, 4 and 5 (Hashimoto, Aoki, Nakamura, Tanzawa, & Okada, 2001; W. M. Wang, Ge, Lim, Nagase, & Greenspan, 2006), and aggrecan and heparan sulfate promote the inhibition by interacting with TSR and spacer regions (Troeberg, et al., 2009; Troeberg, et al., 2014). Papilin inhibits ADAMTS2 by binding both the enzyme and its substrate (Kramerova, et al., 2000). Calcium pentosan polysulfate interacts with the noncatalytic spacer domain of ADAMTS4 and the cysteine-rich domain of ADAMTS5, blocking activity against their natural substrate aggrecan (Troeberg, et al., 2008). Granulin-epithelin precursor binds to the ancillary domains of ADAMTS7 and 12 and block the degradation of cartilage oligomeric matrix protein (COMP) substrate (Guo, et al., 2010).
Many of the ADAMs and ADAMTS inhibitors have a broad spectrum and interact with other proteases such MMPs, and therefore their use has been limited to in vitro biochemical or cellar studies or in animal models in vivo. The development of new ADAMs and ADAMTS inhibitors with improved specificity would enhance their use in clinical applications.
8. ADAMs and ADAMTS in Vascular Processes and Malfunction
ADAMs and ADAMTS play a role in several vascular processes including angiogenesis, VSMC proliferation and migration, intimal hyperplasia and neointima formation, vascular cell apoptosis, EC permeability, vascular inflammation, and tissue repair and wound healing (Fig. 3).
Fig. 3.
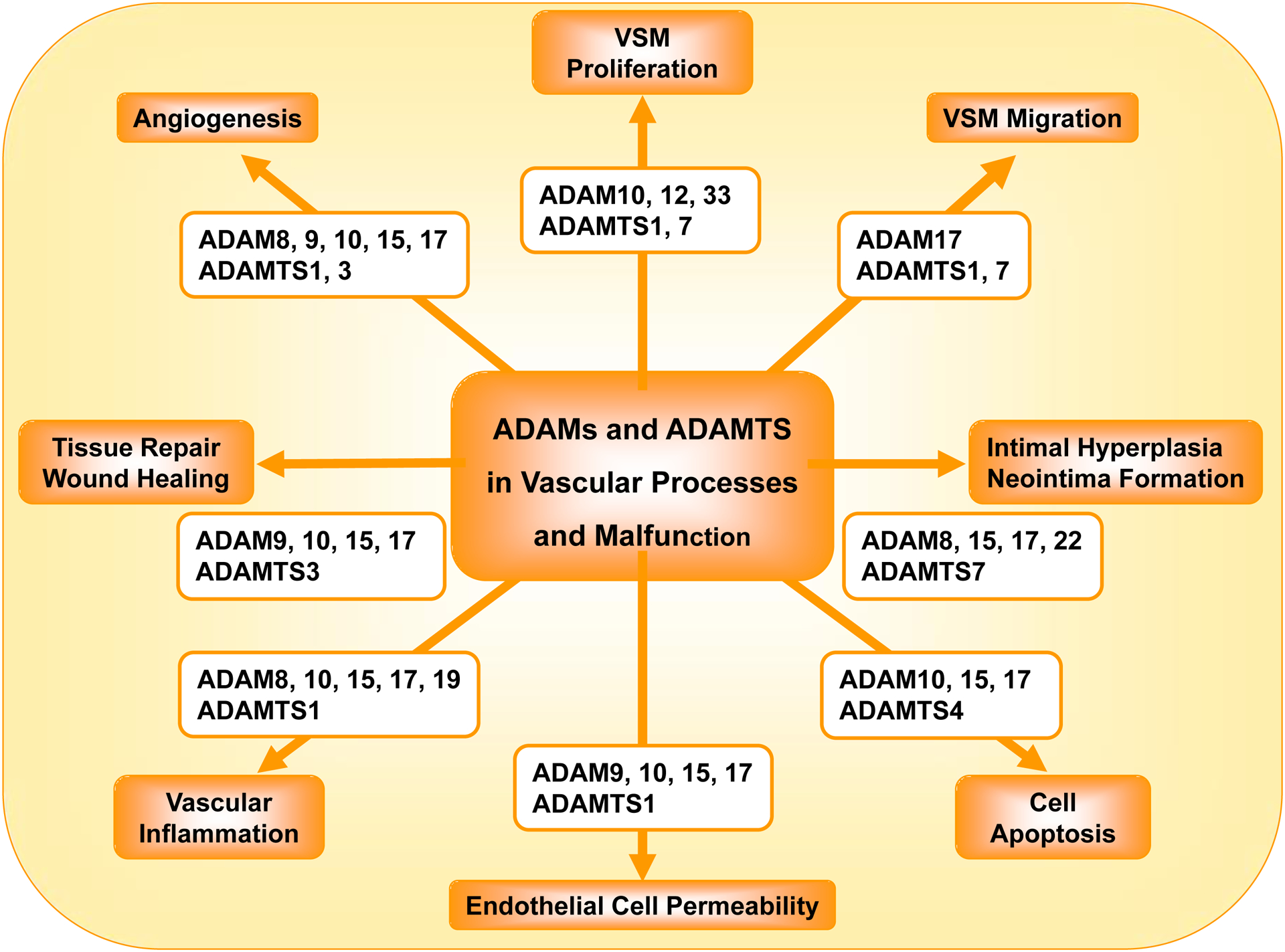
Role of ADAMs and ADAMTS in vascular processes and malfunction.
8.1. ADAMs and ADAMTS in Angiogenesis
During sprouting angiogenesis, VEGF induces tip ECs to start sprouting and express delta-like 4 (Dll4), which activates Notch in the adjacent stalk cells and limit their sprouting. Notch signaling also regulates the level of VEGFR, and in turn affects the expression of the Notch ligand Dll4, thus creating an intercellular feedback loop (Jakobsson, et al., 2010). VEGF also activates ADAMs, which promote Notch ectodomain shedding. Both ADAM10 and 17 are implicated in Notch-signaling, but they have different roles in angiogenesis. In mouse retinas, inhibition of ADAM10 induces, while inhibition of ADAM17 reduces, vascular sprouting. Retinal vessels analysis in ADAM17 hypomorphic mice (ADAM17ex/ex) confirmed the requirement of ADAM17 in angiogenesis. ADAM17 overexpression decreased the expression of a naturally occurring inhibitor of angiogenesis Thrombospondin 1 (TSP1). Genetic and pharmacological ADAM17 blockade increased TSP1 expression in mouse retina. During ADAM17 inhibition, the TSP1 inhibitor LSKL restored VEGF-induced angiogenesis and EC tube formation. Thus, ADAM10 and 17 have opposite effects on sprouting angiogenesis that may be unrelated to Notch signaling and involve differentially expressed TSP1 (Caolo, et al., 2015).
The density of preexisting collaterals varies widely and is a major determinant of severity of MI, stroke and PAD. VEGF affects collateral density in newborn and adult mice. In the mouse embryo, pial collaterals begin sprouting from existing cerebral arterioles. Global VEGF-A overexpressing mice (Vegfhi/+) form more, and Vegflo/+ form fewer, collaterals during embryogenesis. Conditional global reduction of Vegf reduces collateral formation, and the effects remain in adulthood. EC-specific reduction of Adam10 and inhibition of γ-secretase increase collateral formation, consistent with their roles in VEGF-induced Notch1 activation and suppression of sprouting signals. EC-specific Adam17 knockdown reduces collateral formation, consistent with its role in EC migration and embryonic vascular stabilization (Lucitti, et al., 2012).
EC loss occurs in the first week after spinal cord injury, and ADAMs are important for EC survival and angiogenesis. Adult C57Bl/6 mice with a spinal cord contusion show increased ADAM8 in blood vessels, ECs and spinal cord lysates. ADAM8 colocalized with platelet-EC adhesion molecule (PECAM) and with intravenously-injected isolectin B4 in a subpopulation of blood vessels, coincident with angiogenesis. ADAM8 and ADAM8-positive proliferating cells were seen at the leading end of isolectin B4-positive blood vessels, supporting a role of ADAM8 in proliferation and migration of ECs during angiogenesis following spinal cord injury (Mahoney, Benton, Maddie, Whittemore, & Hagg, 2009).
Proteolytic cleavage of VEGFR and the VEGF coreceptor neuropilin-1 are important factors in angiogenesis. Neuropilin-1 species that include the C-terminal and transmembrane domains but lack the ligand-binding A and B regions are constitutively expressed in ECs, upregulated by phorbol ester and Ca2+ ionophore, and reduced by ADAM9 knockdown using siRNA or by ADAM10 inhibitor. VEGF upregulates neuropilin-1 species in an ADAM9/ADAM10-dependent manner. Transfection of ECs with adenoviral constructs expressing neuropilin-1 C-terminal domain inhibited VEGF-induced EC motility and angiogenesis in aortic ring sprouting assays, suggesting a role of neuropilin-1 proteolysis by ADAM9 and 10 in modulating VEGF angiogenic signaling (Mehta, et al., 2018).
ADAMTS1 has been linked to angiogenesis in gastric and lung cancer (J. Chen, Zhi, Chang, Zhang, & Dai, 2013; Kilic, Aynekin, Kara, Icen, & Demircan, 2017; J. H. Tang, Zhang, Zhang, & Zhang, 2017). ADAMTS1 is abundant in rat embryonal epithelia of the intestines, nasal cavity, choroid plexus and skin, and is involved in the development of the heart, liver, adrenal glands, muscle, neuronal system and adipose tissue. At the time of birth, ADAMTS1 is reduced in most organs, but increases in the developing bone and skin. A 62 kDa ADAMTS1 fragment is detected in the heart, lung, intestines, kidneys and adrenal glands of the developing rat embryo and to a less extent in the corresponding adult rat organs. ADAMTS1 is found in principal cells of collecting ducts and renal medulla, ependymal cells of the ventricles, and some neurons. The distribution of ADAMTS1 in multiple organs during embryogenesis suggests a role in tissue remodeling and angiogenesis (Gunther, Skaftnesmo, Arnold, Bjerkvig, & Terzis, 2005).
ADAMTS3 cleaves pro-VEGF-C into active VEGF-C during embryonic lymphangiogenesis and placental angiogenesis (Janssen, et al., 2016), and regulates VEGFR-3 signaling during lymphatic EC differentiation (Jeltsch, et al., 2014). Loss of ADAMTS3 is linked to Hennekam lymphangiectasia-lymphedema syndrome (Brouillard, et al., 2017).
On the other hand, recombinant ADAMTS2 (rADAMTS2) reduces EC proliferation and branching of capillary-like structures, and inhibits vessel formation in embryoid bodies (Dubail, et al., 2010), partly through interaction of one of its C-terminal domains with nucleolin (Bekhouche & Colige, 2015; Dubail, et al., 2010). Similarly, ADAMTS12 shows antiangiogenic effects independent of its enzymatic activity, as both the WT control form and mutated inactive form similarly inhibit EC sprouting in the aortic ring assay. These ADAMTS12 antiangiogenic properties could protect against tumor progression (El Hour, et al., 2010). ADAMTS15 also reduces angiogenesis and liver cancer metastasis by modulating cell-ECM interactions (Kelwick, Wagstaff, et al., 2015).
8.2. ADAMs and ADAMTS in VSMC Proliferation and Migration
VSMC proliferation and migration are key events in atherosclerosis and intimal hyperplasia. In cultured human coronary VSMCs, the C-terminal fragment of the plasminogen activator urokinase (CTF-uPA) increases DNA synthesis, cell proliferation and EGFR phosphorylation/activation, and these effects are blocked by plasmin, EGFR inhibitors, TIMPs, and inhibitors of ADAM10, ADAM12 and HB-EGF (Duru, Fu, & Davies, 2014). The N-terminal fragment of uPA (NTF-uPA) also promotes migration, EGFR phosphorylation/activation and HB-EGF release in human coronary VSMCs, and these effects are blocked by EGFR inhibitor AG1478, MMP and HB-EGF inhibitors, ADAM inhibitors TAPI-0 and TAPI-1, and ADAM9 and 10 siRNA. Thus, uPA induces domain-dependent VSMC proliferation through transactivation of EGFR by a plasmin-mediated, ADAM-induced and HB-EGF-dependent process, and EGFR and ADAM9, 10 or 12 could be potential targets to inhibit cell migration (Bakken, Protack, Roztocil, Nicholl, & Davies, 2009).
ADAM12 is an important mediator of VSMC proliferation in carotid artery lesions (Pelisek, et al., 2012; Smiljanic, et al., 2011).
In human airway SMCs, recombinant disintegrin domain of ADAM15 (ddADAM15), which contains an RGD integrin-binding motif, adhered to SMCs in a β1-integrin dependent manner. ddADAM15 inhibited platelet-derived growth factor (PDGF)-induced airway SMC migration, and this inhibition was reversed by β1-integrin antibody, supporting a role of ddADAM15 in inhibiting airway SMC adhesion and migration through interaction with β1-integrin (Lu, et al., 2007).
ADAM17 is linked to VSMC hypertrophy and hyperplasia, possibly through activation of EGFR (M. Shen, Morton, Davidge, & Kassiri, 2017). AngII-induced HTN is suppressed in mice lacking ADAM17 in SMCs (Adam17f/f/CreSm22) during the first week of AngII infusion, but by the second week, HTN increases to levels observed in mice with intact ADAM17 (Adam17f/f). Adam17f/f/CreSm22 mice also exhibit less cardiac hypertrophy and fibrosis and renal fibrosis at 2 weeks post-AngII, but this protection is reversed by the fourth week. EGFR activation is suppressed in Adam17f/f/CreSm22-AngII arteries. AngII-induced proliferation and migration, EGFR activation and Erk1/2 signaling were suppressed in VSMCs isolated from Adam17f/f/CreSm22. Thus, Adam17-deficiency suppresses AngII-induced VSMC remodeling, and transiently protect against AngII-mediated HTN and end-organ damage (M. Shen, et al., 2017).
ADAM33, which is evolutionally related to ADAM15, has been suggested as a crucial factor in airway SMC proliferation and bronchial asthma pathology (Kim, et al., 2017)
Plasma of CAD patients shows increased ADAMTS1 and decreased miR-362–3p. In human coronary VSMCs, PDGF-BB inhibits miR-365b-3p and upregulates ADAMTS1 expression. Overexpression of miR-365b-3p downregulates ADAMTS1 and attenuates PDGF-BB-induced VSMC proliferation/migration and G1/S cell cycle transition. miR-362–3p binds to ADAMTS1 3’-untranslated region and decreases its expression. Overexpression of ADAMTS1 reverses miR-362–3p-induced inhibition of VSMC proliferation/migration and, cell cycle. Thus, miR-362–3p inhibits VSMC proliferation/migration and attenuates atherosclerosis by targeting ADAMTS1 (M. Li, et al., 2017; Y. Qu & Zhang, 2018).
ADAMTS7 could mediate VSMC migration and neointima formation (L. Wang, et al., 2009). In rat model of carotid artery balloon-injury, luminal adenoviral delivery of ADAMTS7 aggravates intimal hyperplasia and increases proliferating cell nuclear antigen-positive cells in the intima and media. Perivascular ADAMTS7 siRNA reduces intimal thickening and VSMC replication. Cultured rat VSMCs show enhanced replication with ADAMTS7 overexpression and decreased proliferation with ADAMTS7 siRNA, supporting that ADAMTS7 promotes VSMC proliferation (L. Zhang, et al., 2015). ADAMTS7 KO mice also show reduced risk of atherosclerosis likely due to loss of ADAMTS7-mediated VSM migration (Bauer, et al., 2015).
8.3. ADAMs and ADAMTS in Neointimal Hyperplasia and Vascular Restenosis
Neointimal hyperplasia and vascular restenosis are major limitations of coronary angioplasty and involve abnormal proliferation/migration of VSMCs. In response to vascular injury, adventitia stem/progenitor cells (AdSPCs) migrate into the intima, where they differentiate into SMCs and participate in neointimal hyperplasia. Macrophage-derived MMP8 promotes SMC differentiation from AdSPCs, and contributes to injury-induced neointimal SMC hyperplasia by modulating TGF-β activity and ADAM10/Notch1 signaling (Yang, et al., 2020). In support, the mouse carotid artery wire injury model shows fewer proliferating cells and smaller neointimal lesions in ApoE−/−MMP8−/− mice. Also, MMP8 KO VSMCs and WT VSMCs treated with ADAM10 inhibitor GI254023X or transfected with ADAM10 siRNA show less proliferation/migration, and contain more pro-ADAM10 and less mature ADAM10, more N-cadherin and β-catenin in the plasma membrane, less β-catenin in the nucleus, and less cyclin D1. Treatment of MMP8 KO VSMCs with rADAM10 increases cell proliferation/migration and cyclin D1 expression. Immunohistochemistry show colocalization of ADAM10 with VSMCs and N-cadherin, and nuclear accumulation of β-catenin in the neointima of ApoE−/−MMP8+/+ mice, suggesting that MMP8 promotes VSMC proliferation/migration and neointima formation via an ADAM10, N-cadherin and β-catenin-mediated pathway (Xiao, et al., 2014).
Methyl protodioscin reduces neointima formation in rat carotid artery balloon injury model, and inhibits growth and migration, arrests cell cycle at the G1 phase, and decreases ADAM15 expression in A7r5 VSMCs, suggesting a role of ADAM15 in neointima formation (Chung, et al., 2016).
In rat carotid arteries after balloon angioplasty, neointimal cells are strongly positive for ADAM17. Intimal hyperplasia and proliferating cell nuclear antigen (PCNA)- and phospho-EGFR-positive cells are reduced in a dominant-negative ADAM17 adenovirus-treated carotid artery, and enhanced by WT ADAM17 adenovirus, supporting a role of ADAM17 in EGF activation and neointimal hyperplasia after vascular injury (Takaguri, et al., 2011).
ADAM22 is upregulated in mouse carotid artery injury model and PDGF-BB-treated VSMCs. Wire injury induced neointima formation is ameliorated in ADAM22 heterozygous mice. VSMCs overexpressing ADAM22 show increased proliferation/migration and phenotypic switching, and these effects are reversed by ADAM22 siRNA or ERK inhibitors, suggesting a role of ERK phosphorylation in ADAM22-mediated neointima formation (S. M. Zhang, et al., 2019).
ADAMTS7 accumulates in the neointima of rat carotid artery after balloon injury. Luminal delivery of ADAMTS7 adenovirus to carotid arteries exacerbates, while perivascular application of ADAMTS7 siRNA attenuates, intimal thickening. TNFα and PDGF-BB enhance ADAMTS7 expression in VSMCs. ADAMTS7 overexpression accelerates, and its siRNA knockdown retards VSMC migration. ADAMTS7 promotes degradation of cartilage oligomeric matrix protein (COMP) in injured vessels, and overexpression of COMP suppresses postinjury neointima formation and VSMC migration, suggesting that ADAMTS7 facilitates intimal hyperplasia by degrading COMP (L. Wang, et al., 2009). Conversely, ADAMTS7 inhibits proliferation/migration in cultured ECs. In mouse carotid artery wire injury model, Adamts7 KO promoted re-endothelialization and ameliorates neointima formation. ADAMTS7 degraded TSP1, and the inhibitory effect of ADAMTS7 on postinjury endothelium recovery was absent in Tsp1−/− mice. Thus ADAMTS7 inhibits re-endothelialization, delays EC repair and promotes TSP1 degradation and neointima formation, and could be a potential target in postinjury intimal hyperplasia and postangioplasty restenosis (Kessler, et al., 2015).
8.4. ADAMs and ADAMTS in Vascular Cell Apoptosis
ADAM10, 15 and 17 play a role in vascular cell apoptosis (Fig. 3). ADAM10 cleaves N-cadherin and increases apoptosis in atherosclerotic lesions and the risk of vulnerable plaque (Schulz, et al., 2008). Other studies suggest that ADAM10 promotes vascular cell survival, as ionizing radiation reduces ADAM10 expression and induces neurovascular cell senescence and death (McRobb, et al., 2017). Recombinant human ADAM15 (rhADAM15) induces vascular apoptosis and inhibits tumor formation and angiogenesis (Hou, et al., 2015). ADAM17 mediates vascular apoptosis and migratory invasion of liver cancer stem cells, making ADAM17 a promising target in cancer therapy (Y. Jin, et al., 2013; R. Wang, et al., 2018). Deletion of Cdc42 increases ADAM17 activity, enhances VEGFR-2 shedding, and reduces vascular EC survival and angiogenesis, and ADAM17 inhibition reverses Cdc42 deletion-induced EC apoptosis (Y. Jin, et al., 2013).
Versican is a large proteoglycan required for development of the circulatory system and proper limb development (Timms & Maurice, 2020). ADAMTS4 degrades versican, induces VSMC apoptosis, and promotes inflammatory cell infiltration of blood vessels (P. Ren, et al., 2017). Also, ADAMTS20 mutants have reduced versican proteolysis and impaired interdigit web regression, leading to soft tissue syndactyly. Versikine, the ADAMTS20-generated versican N-terminal bioactive proteolytic fragment, restores interdigit apoptosis in ADAMTS mutant webs, and may play an essential role in interdigital web regression (Nandadasa, et al., 2021).
8.5. ADAMs and ADAMTS in Endothelial Permeability
VE-cadherin controls endothelial permeability, vascular integrity and leukocyte transmigration, and soluble VE-cadherin is elevated in atherosclerosis. In HUVECs, ADAM9 is localized at cell-cell junctions with VE-cadherin (English, Siviter, Hansen, & Murphy, 2017). Also, ADAM10 cleaves VE-cadherin ectodomain from HUVECs, and this cleavage is induced by Ca2+ influx or staurosporine, suggesting that it contributes to dissolution of adherens junctions during EC activation or apoptosis, respectively. ADAM10 contributes to thrombin-induced decrease of endothelial cell-cell adhesion and increased endothelial permeability. Also, ADAM10 siRNA inhibits T-cell transmigration in HUVECs (Schulz, et al., 2008).
The renal glomeruli filter blood through fenestrated endothelium, glomerular basement membrane and podocyte foot processes. Glomeruli of mice lacking ADAM10 in ECs (ADAM10ΔEC) show increased vascular diameter and intussusceptive or splitting angiogenesis, suggesting a role of endothelial ADAM10 and Notch signaling in glomerular vessels development (Farber, et al., 2018).
In bovine aortic ECs high shear stress modulates gene expression, promotes an anti-coagulant, anti-inflammatory, proliferative and promatrix remodeling, and upregulates the ECM processing factor ADAMTS1, metalloproteinase inhibitor TIMP-3, and the fibrinolysis factors tissue plasminogen activator (tPA) and urokinase plasminogen activator (uPA). Also, ADAMTS1 and uPA are increased in rabbit basilar arteries exposed to high flow after bilateral carotid artery ligation (Dolan, Sim, Meng, & Kolega, 2012).
Blood-brain barrier (BBB) defects and cerebrovascular dysfunction contribute to amyloid-β accumulation and Alzheimer disease. In APP and presenilin 1 (PS1) APPPS1 transgenic mice, ADAMTS13 deficiency leads to BBB breakdown, reduced vessel density, capillary perfusion and cerebral blood flow, impeded clearance of amyloid-β, increased brain plaque load and cerebral amyloid angiopathy, and further cognitive decline. Virus-induced expression of ADAMTS13 in APPPS1 mice attenuates BBB disruption, increases microvessels, capillary perfusion, cerebral blood flow and cerebral amyloid-β clearance, and improves cognitive performance, supporting that ADAMTS13 deficiency contributes to cerebrovascular dysfunction in Alzheimer disease (Cao, et al., 2019).
8.6. ADAMs and ADAMTS in Vascular Inflammation
Cell-cell and cell-matrix interactions participate in leukocyte recruitment to inflammatory sites, and ADAMs adhesive and proteolytic activities contribute to the inflammatory process. In human macrophages, meprin-β promotes inflammation and increases the production of IL-1β, IL-6 and IL-18 through activation of ADAM10-ERK1/2 pathway and phosphorylation of NF-κB (Y. J. Li, Fan, Tang, Li, & Yu, 2014).
ADAM15 through modulation of cell-cell and cell-ECM interactions and ectodomain shedding activities could mediate rheumatoid arthritis and other inflammatory disorders (Charrier-Hisamuddin, Laboisse, & Merlin, 2008).
ADAM17 regulates inflammation by shedding membrane-bound proteins, converting TNFα precursor to its mature form, and processing of TNFR1 and TNFR2 (Chemaly, et al., 2017). Plasma ADAM17 and its substrate syndecan-1 are elevated in patients with anti-neutrophil cytoplasmic antibody-associated vasculitis (Bertram, et al., 2015). TNFα and ADAM17 are also upregulated in endomyocardial biopsies from patients with myocarditis, and correlate positively with left ventricular volume and negatively with left ventricular systolic function, suggesting a role in myocarditis and cardiac dysfunction (Satoh, et al., 2000).
Kawasaki disease is a childhood disease of systemic vasculitis and coronary artery lesions. ADAM17 cleaves TGF-β receptor type-1 and reduces TGF-β signaling. In the Han Chinese population three ADAM17 SNPs are associated with Kawasaki disease risk and coronary artery lesions likely through modulation of the TGF-β/SMAD3 pathway (Peng, et al., 2016).
IL-1 induces ADAMTS1 expression in colon-26 cells, and injection of LPS in mice upregulates ADAMTS1 in the heart and kidney, supporting a role of ADAMTS1 in inflammation (Hirohata, Inagaki, & Ohtsuki, 2017; Kuno, et al., 1997).
8.7. ADAMS and ADAMTS in Tissue Repair and Wound Healing
ADAM and ADAMTS show differential expression in wound healing tissues versus normal tissues. ADAM9, 10, 15 and 17 are highly expressed, and ADAMTS3 shows differential expression in healing wounds, suggesting potential roles in tissue repair (Hodgkinson, Wang, Duncan, Edwards, & Wormstone, 2010).
ADAMTS5 could play a role in tissue repair and wound healing through regulation of versican proteolysis, pericellular matrix volume and dermal fibroblast-myofibroblast transition (Hattori, et al., 2011). In a murine model of excisional wound healing, ADAMTS5 ablation was associated with impaired contraction and dermal collagen deposition, and accumulation of cell aggregates, fibroblastic cells and full-length aggrecan enriched pericellular matrix in the dermal layer. ADAMTS5 deficiency may block dermal repair through CD44-mediated aggrecan accumulation and changes in TGF-β1 signaling (Velasco, et al., 2011)
9. ADAMs and ADAMTS in Cardiovascular Disease
ADAM8, 9, 10, 12, 13, 15, 17, 19 and 28, as well as ADAMTS1, 4 and 5 have been associated with different pathological conditions (Mochizuki & Okada, 2007). ADAM17 is involved in diabetes and inflammatory, immune and cardiovascular disorders (Chemaly, et al., 2017; Palau, Pascual, Soler, & Riera, 2019). ADAMTS13 deficiency is linked to thrombotic microangiopathy (Akyol, Akyol, & Chen, 2016). ADAMs and ADAMTS have also been implicated in HTN, atherosclerosis, CAD, MI, heart failure, ischemic stroke, PAD and vascular aneurysm (Nyren-Erickson, et al., 2013; Obama, et al., 2015; Palau, et al., 2019; Santamaria & de Groot, 2020) (Fig. 4).
Fig. 4.
Role of ADAMs and ADAMTS in cardiovascular disease.
9.1. ADAMs and ADAMTS in Hypertension
Neurogenic, renal and vascular factors contribute to different forms of hypertension (HTN), and ADAMs and ADAMTS may be involved. Receptors-for-Advanced-Glycation-End-products (RAGE) activate inflammatory processes during cell stress. ADAM10 cleaves RAGE and releases sRAGE, which neutralizes RAGE ligands and reduces cardiovascular effects of increased sympathetic activity in HTN and diabetes (Raucci, et al., 2008). Myocardial fibrosis and RAGE are increased, sRAGE is decreased, and ADAM10 myocardial and serum levels are elevated in obese spontaneously hypertensive rats (SHR), and these effects are reversed by renal denervation. Isoproterenol induces β1-adrenergic receptor-mediated RAGE expression in splenocytes and β2-adrenergic receptor-mediated suppression of ADAM10 activity and decreased sRAGE secretion from splenocytes and cardiac fibroblasts. Thus, sympathetic activity suppresses ADAM10 activity and sRAGE/RAGE balance, and sympathetic inhibition or renal denervation reverse these effects and prevent RAGE-induced cardiac damage in HTN and metabolic syndrome (Selejan, et al., 2018).
ADAM12 has been implicated in HTN (Nyren-Erickson, et al., 2013). GPCR agonists upregulate ADAM12 and other mediators to increase BP and cardiac hypertrophy (Berry, Bosonea, Wang, & Fernandez-Patron, 2013). Acute HTN induced by catecholamines, AngII or nitric oxide synthase inhibitor requires transcription of vascular MMP-7 and ADAM12. Myocardial ADAM12 is upregulated in mice with AngII-induced HTN and cardiac hypertrophy, and MMP-7 knockdown attenuates HTN, ADAM12 overexpression and cardiac hypertrophy, making the MMP-7/ADAM12 pathway a potential therapeutic target in HTN (X. Wang, F. L. Chow, et al., 2009).
ADAM17 is overexpressed in HTN (Obama, et al., 2015; Palau, et al., 2019). C57Bl/6 mice infused with AngII for 2 weeks show cardiac and vascular medial hypertrophy, perivascular fibrosis, increased vascular ADAM17, EGFR activation, and endoplasmic reticulum stress. These phenotypes are diminished in AngII-infused vascular ADAM17-deficient mice or C57Bl/6 mice treated with ADAM17 antibody independent of BP changes, suggesting a role of vascular ADAM17 in mediating AngII-induced cardiovascular remodeling via EGFR activation (Takayanagi, et al., 2016). In other studies, AngII-infused mice show cardiac hypertrophy and fibrosis, and increased myocardial hypertrophy markers (α-actin, β-myosin heavy chain and brain natriuretic peptide), fibrosis markers (collagen types I and III and fibronectin), MMP-2 and ADAM12, and these effects are prevented by ADAM17 siRNA (X. Wang, T. Oka, et al., 2009). Also, loss of ADAM17 specifically in SMCs suppressed AngII-induced HTN, cardiac hypertrophy and fibrosis, and renal fibrosis (M. Shen, et al., 2017). Thus, ADAM17, likely through transcriptional regulation of MMP-2 and ADAM12, may play a role in AngII-induced cardiac hypertrophy and fibrosis (X. Wang, T. Oka, et al., 2009).
Neurogenic HTN is characterized by increased sympathetic activity and drug resistance. Chronic activation of the brain renin angiotensin system (RAS) causes HTN by altering autonomic balance. ACE2 cleaves AngII to Ang(1–7) peptide, which counteracts the adverse effects of AngII. ACE2 activity is increased in the cerebrospinal fluid and correlates with systolic BP in patients with HTN. ADAM17 is also upregulated in the brain of patients with HTN as TNFα levels are increased in the cerebrospinal fluid. ADAM17 is also upregulated in neurons of DOCA-salt hypertensive mice, but not in mice lacking AngII receptor (AT1R) in neurons. Thus, neuronal AT1R, likely through increasing reactive oxygen species and ERK, causes ADAM17 activation, increased ADAM17-mediated ACE2 shedding, and reduced ACE2 compensatory activity in neurogenic HTN (J. Xu, et al., 2017).
ADAM17 is robustly expressed and promotes RAS activation in the brain. DOCA-salt increases BP, and cardiac and vascular sympathetic activity in WT mice, and these effects are reduced in mice lacking ADAM17 in glutamatergic neurons. Within the paraventricular nucleus, AngII-induced activation of kidney-related presympathetic glutamatergic neurons is reduced in ADAM17 knockdown mice, confirming the role of neuronal ADAM17 in increasing presympathetic neuronal activity and salt-sensitive HTN (Xu, et al., 2019). In cultured cortical neurons, glutamate increases ADAM17 activity and reduces ACE2 activity, and these effects are reversed by blockers of oxidative stress, supporting a relationship between exaggerated glutamate stimulation, ADAM17-mediated impairment in ACE2 activity, oxidative stress and neurogenic HTN (Xu, Sriramula, & Lazartigues, 2018). Interestingly, α-lipoic acid reduces neurogenic HTN by blunting oxidative stress and the increase in ADAM17 (de Queiroz, et al., 2015).
Neuronal ACE2 deletion reduces inhibitory inputs to presympathetic neurons involved in BP regulation. In primary neuron cultures, ACE2 is expressed on GABAergic neurons synapsing onto excitatory neurons within the hypothalamus. ADAM17 colocalizes with AT1R on Sim1 neurons. ADAM17 knockdown in Sim1 neurons reduces FosB gene expression, increases vagal tone, and prevents the acute pressor response to centrally administered AngII. Sim1 neurons ADAM17 deficient mice show blunted BP and preserved ACE2 activity during DOCA salt-sensitive HTN, supporting contrasting effects of ACE2 and ADAM17 on neuronal excitability of presympathetic neurons within the paraventricular nucleus, and on neurogenic HTN (Mukerjee, et al., 2019).
The renal RAS regulates salt and water excretion, plasma volume and BP. ADAM17 is highly expressed in distal renal tubules and in the diabetic kidney. In addition to shedding ACE2, ADAM17 promotes inflammation and fibrosis in chronic kidney disease (Palau, et al., 2019).
HTN-in-pregnancy and preeclampsia (PE) affect 5–8% of pregnancies in the United States. Maternal serum level of sADAM12, a marker of fetal aneuploidy and growth, is altered in PE. In the first‐trimester, serum sADAM12 level is reduced in women who developed PE and further reduced in those delivering prior to 35 weeks. Combining sADAM12 with pregnancy-associated plasma protein A (PAPP‐A) results in a further 1% increase in detecting women who developed PE. Combining sADAM12 with uterine artery Doppler Pulsatility Index increases PE detection rate to 66%. In the second trimester at 22–24 weeks, serum sADAM12 levels are increased in women developing PE (Spencer, Cowans, & Stamatopoulou, 2008). Other studies have shown that in normal pregnant women ADAM12 levels increase with fetal crown rump length, decrease with maternal weight, are higher in African-American than white women, and show an association with PAPP-A levels. ADAM12 levels are reduced in pregnant women with small for gestational age neonates, and are not markedly different in PE pregnancies, gestational HTN, or spontaneous preterm delivery (Poon, Chelemen, Granvillano, Pandeva, & Nicolaides, 2008). Also, studies examining the efficiency of ADAM12, PAPP-A, uterine artery Doppler and maternal characteristics in predicting PE in the first-trimester showed that ADAM12 serum levels were lower in women who developed PE than those who did not. ADAM12, PAPP-A, or uterine artery Doppler pulsatility indices each individually in combination with maternal characteristics identified ~50% of women who developed PE. Combining these first-trimester parameters did not improve their predictive efficiency (Goetzinger, et al., 2013). Other studies showed an association between decreased first trimester ADAM12s levels and gestational HTN, but combined ADAM12s and placental protein 13 (PP13) measurements did not further predict PE, gestational HTN and small for gestational age fetuses (Deurloo, et al., 2013), making it important to more thoroughly examine the role of ADAM12 in PE.
α-Klotho is an anti-aging protein produced by the kidney, brain and placenta. Because of mRNA splicing, α-klotho is present as both a full-length membrane-bound form and a truncated circulating soluble protein. Membrane-bound α-klotho is an obligate co-receptor for fibroblast growth factor 23 (FGF23) and its action on FGF receptor (FGFR). ADAM17 causes proteolytic shedding of α-Klotho from the cell membrane, a process that could affect kidney function and the course of chronic kidney disease (Perna, et al., 2017). Maternal plasma levels of α-Klotho are elevated in PE, and patients with the highest levels have less placental accelerated villous maturation. α-Klotho, ADAM17 and FGFR are expressed in syncytiotrophoblasts and cytotrophoblasts. Between 32 and 40 weeks gestation, placental levels of α-Klotho, ADAM17 and FGFR are decreased in normal pregnant, but not PE women, suggesting their involvement in PE (Loichinger, Towner, Thompson, Ahn, & Bryant-Greenwood, 2016).
Pulmonary arterial HTN (PAH) is characterized by pulmonary vascular remodeling, SMC proliferation, endothelial dysfunction, and excess ECM. Collagen accumulation plays a role in hypoxic pulmonary HTN, and baicalin prevents bleomycin-induced pulmonary fibrosis. ADAMTS1 degrades ECM substrates associated with vascular remodeling. In rats exposed to hypoxia for 4 weeks, baicalin treatment reduced pulmonary arterial pressure, increased ADAMTS1 expression, attenuated pulmonary artery remodeling, and decreased synthesis of type I collagen (Liu, et al., 2015).
In old mice, AngII infusion for 3 days is associated with HTN, kidney damage and increased ADAMTS7 mRNA expression and protein levels, suggesting that ADAMTS7 may contribute to AngII-mediated early inflammatory kidney damage and age-related HTN (Y. X. Gao, et al., 2013).
ADAMTS8 is upregulated in pulmonary artery SMCs (PASMCs) and the lung of mice with hypoxia-induced PAH. Hypoxia-induced PAH is attenuated in SMC-specific ADAMTS8 KO (ADAMTS8ΔSM22) mice. ADAMTS8 overexpression increases PASMC proliferation and downregulates AMP-activated protein kinase (AMPK), while ADAMTS8 deletion causes decreased PASMC proliferation and AMPK upregulation. ADAMTS8 deletion reduces mitochondrial fragmentation in hypoxic conditions. rADAMTS8 induces endothelial dysfunction and MMP activation. Cardiomyocyte-specific ADAMTS8 KO (ADAMTS8ΔαMHC) mice show ameliorated right ventricle failure in response to chronic hypoxia, enhanced angiogenesis and reduced right ventricle ischemia and fibrosis. Mebendazole reduces ADAMTS8 expression and PAH-PASMC proliferation, and ameliorates HTN and right ventricular failure in PAH models, suggesting that ADAMTS8 could be a potential therapeutic target in PAH (Omura, et al., 2019).
Non-cirrhotic intrahepatic portal HTN (NCIPH) is characterized by thrombotic microangiopathy of the portal venous system. Plasma vWF/ADAMTS13 balance is altered in NCIPH. Initial sequencing of ADAMTS13 CUB domain revealed one NCIPH patient with a rare missense variant at position c.3829C >T (rs14045669) associated with severe ADAMTS13 deficiency, high vWF, and other missense variants in ADAMTS13, vWF and complement genes. The patient liver biopsy revealed globules of ADAMTS13 in stellate cells. Missense variants in ADAMTS13, vWF and complement genes, and decreased ADAMTS13 activity may play a role in NCIPH (Goel, et al., 2017).
ADAMTS16 has been linked to systolic and diastolic BP in the Quebec Family Study. Multiple variants including an Ala to Pro variant in codon 90 (rs2086310) of human ADAMTS16 have been associated with resting systolic BP. Also, a BP quantitative trait locus on rat chromosome 1 was isolated in a short congenic segment spanning 804.6 kb, and contained two genes, LOC306664 and LOC306665. LOC306664 is predicted to translate into Adamts16. Similar to human ADAMTS16, the rat Adamts16 transcript was detected in multiple tissues including the kidney. Also, several genes related to BP were differentially expressed between congenic and Dahl Salt-sensitive (S) rat, and in kidney cell lines with or without Adamts16 knockdown (Joe, et al., 2009). Systolic BP is lower and aortic pulse wave velocity and vascular media thickness are less in homozygous Adamts16 mutant rats than S rats. The mechanosensory cilia of ECs are longer and glomerular capillaries show splitting, thickening and longer survival rate in Adamts16 mutant than S rats, suggesting a link between Adamts16, the vascular system and BP regulation (Gopalakrishnan, et al., 2012).
9.2. ADAMs and ADAMTS in Atherosclerosis
Atherosclerosis is characterized by accumulation of lipoproteins and inflammatory cytokines, and formation of lipid-rich plaques on the arterial inner walls. Macrophages play a role in all stages of atherosclerosis including foam cell formation, ROS production, inflammation, and plaque stability. Apoptosis in the fibrous cap, which is rich in VSMCs and macrophages, and its subsequent weakening and erosion affect plaque stability (Musumeci, et al., 2014). In the unstable plaque, perturbed ECs secrete vWF, which interacts with platelet GPIbα and enables platelet adherence to the damaged endothelium. Following plaque rupture, vWF and platelets contact subendothelial collagen, which further supports platelet aggregation, leading to thrombus formation and ischemic events (Howes, Knauper, Malcor, & Farndale, 2020).
ADAM8 is implicated in inflammatory processes, atherosclerosis and MI by regulating immune cell response. In patients undergoing coronary artery bypass graft (CABG), serum levels of sADAM8 are correlated with sCXCL16 levels. ADAM8 expression is increased in left internal thoracic (mammary) arterial graft from coronary bypass patients, suggesting an association with atherosclerosis and CAD. ADAM8 is also upregulated in ECs, circulating neutrophils and in macrophages of high fat diet-fed ApoE−/− and Ldlr−/− mice models of atherosclerosis and mouse model of coronary artery ligation and MI, Human mononuclear cells and neutrophils, and cultured human umbilical artery ECs and arterial SMCs under basal and inflammatory conditions show correlation of ADAM8 with the vascular disease markers ICAM-1, VCAM-1, TNFα, IL-6 and CCL-2 (Schick, et al., 2019). ADAM8 deficient mouse macrophages show reduced inflammatory mediators. Although whole-body or hematopoietic-specific ADAM8 deficiency does not influence atherosclerotic lesion size and plaque development in mice, ADAM8 expression is increased in vulnerable atherosclerotic lesions and might still distinguish between stable and unstable lesions (Theodorou, et al., 2017).
In atherosclerotic plaques from patients undergoing carotid endarterectomy, ADAM8, 9, 10, 12, 15 and 17 are expressed in both early and advanced lesions, with the highest expression for ADAM15 then ADAM8. ADAM10 and 12 are upregulated in unstable plaques, suggesting their contribution to atherosclerosis progression (Pelisek, et al., 2012). ADAM9, 15, and 17 are upregulated and co-localize with CD68-positive cells of monocytic origin in advanced human atherosclerotic lesions from carotid, aortic and femoral territories. Cells expressing ADAMs co-localize with SMCs in the carotid territory, and with CD31-positive ECs in femoral territory, supporting vascular bed-specific ADAM distribution (Oksala, et al., 2009). ADAM9 and 15 and αvβ3 and α5β1 integrins are upregulated and colocalized in human atherosclerotic arteries. PDGF increases ADAM9 and 15 and αvβ3 and α5β1 in human VSMCs. ADAM9 and 15 could regulate integrin-ECM interactions and αvβ3 and α5β1 promotion of SMC proliferation/migration during neointima progression (Al-Fakhri, et al., 2003).
ADAM10, 15 and 17 regulate inflammatory cell recruitment and macrophage activity by cleaving TNFα, TNFR1, TNFR2 and the scavenger receptor CD163, and promote neovessel formation and atherosclerosis by shedding of VEGFR2 and the angiopoietin growth factor receptor Tie-2 (E. P. van der Vorst, Keijbeck, de Winther, & Donners, 2012). In low density lipoprotein receptor Ldlr−/− mice on atherogenic diet, myeloid Adam10 deficiency increases plaque collagen content. Adam10-deficient macrophages show impaired MMP-2, 9 and 13 expression/activity, reduced migration toward monocyte chemoattractant protein-1 (MCP-1) and transmigration through collagen, increased anti-inflammatory IL-10, and reduced release of TNFα, IL-12 and nitric oxide in response to LPS, suggesting a role of myeloid ADAM10 in leukocyte recruitment, cytokine production, ECM degradation, and plaque progression/stability (E. P. van der Vorst, et al., 2015). Atherosclerotic plaques and THP-1 macrophage-derived foam cells show upregulated ADAM10 and downregulated cyclin-dependent kinase inhibitor 2B antisense RNA-1 (CDKN2B-AS1). CDKN2B-AS1 overexpression or ADAM10 silencing reduces lipid accumulation and increases cholesterol efflux. Nuclear CDKN2B-AS1 may bind DNA methyltransferase, increase methylation of ADAM10 promoter, and inhibit ADAM10 transcription, leading to suppressed atherosclerotic inflammatory response and increased cholesterol efflux (H. Li, et al., 2019). Atherosclerotic plaques from internal carotid artery of symptomatic patients show increased caspase-3 and ADAM10, and decreased N-cadherin in the fibrous cap, suggesting a role of apoptotic events triggered by ADAM10-induced N-cadherin cleavage in plaque vulnerability (Musumeci, et al., 2014). VEGF upregulates ADAM10 in ECs, induces ADAM10-mediated cleavage of VE-cadherin, increases vascular permeability and facilitates EC migration, while ADAM10 inhibition reduces EC migration, supporting a role of ADAM10 in atherosclerosis, neovascularization and VEGF-induced EC migration (Donners, et al., 2010).
Endothelium injury is an initiating event in atherosclerosis, and ADAM15 regulates endothelial permeability. In ApoE−/− mice, ADAM15 ablation decreases aortic atherosclerotic lesion size, plaque macrophage infiltration and SMC accumulation. EC-derived ADAM15 causes endothelium barrier dysfunction, dissociation of endothelial adherens junctions VE-cadherin/γ-catenin and monocyte transmigration across mouse aortic ECs and HUVECs (C. Sun, et al., 2012). Platelet adhesion to immobilized rADAM15 and ADAM15-overexpressing HUVECs is enhanced under both static and high shear rate conditions, and is reduced by blocking GPIIb-IIIa using neutralizing anti-αIIbβ3 antibody. sADAM15 binds to activated GPIIb-IIIa. Platelets adherent to ADAM15 attract more platelets under high shear rates indicating platelet-ADAM15 interactions during thrombus formation. Endothelial ADAM15, through GPIIb-IIIa binding, may promote platelet adhesion and activation and thrombus formation, making ADAM15 a potential target for antithrombotic therapies (Langer, May, Bultmann, & Gawaz, 2005).
ADAM17 is upregulated in atherosclerosis (Obama, et al., 2015; Palau, et al., 2019). In advanced atherosclerosis, vulnerable plaques show defective efferocytosis and reduced removal of apoptotic cells by phagocytic cells. In macrophages, AngII decreases surface expression of Mer tyrosine kinase (MerTK) through ADAM17-mediated shedding of sMerTK, leading to efferocytosis suppression. AngII-activated ADAM17 requires ROS and p38 MAPK phosphorylation. AT1R blockers suppress ROS production, and ROS scavengers prevent p38 MAPK phosphorylation. Mutant MERTKΔ483−488 resists AngII-induced shedding and efferocytosis suppression. Thus AngII-activated AT1R/ROS/MAPK/ADAM17 pathway and MerTK shedding in macrophages leads to defective efferocytosis and atherosclerosis progression (Y. Zhang, et al., 2019). Among patients from the Vascular Diabetes study the risk for nonfatal MI, nonfatal strokes, peripheral artery procedures and cardiovascular death is increased in individuals with high ADAM17 circulating substrates sICAM-1, sVCAM-1, sIL-6R and sTNFR1. ADAM17 score increased the prediction accuracy of the Framingham Recurring-Coronary-Heart-Disease-Score, and improved the discrimination and correct reclassification of events and non-events, supporting potential usefulness of measuring ADAM17 activity in predicting cardiovascular events in individuals with atherosclerosis (Rizza, et al., 2015). Fish oil exerts anti-atherogenic effects by decreasing ADAM10 and 17 activity. In Ldlr−/− mice fed a high fat diet, fish oil decreased early atherosclerotic lesions, and reduced circulating EC adhesion molecules JAM-A, ICAM-1 and VE-cadherin, indicating decreased ADAM activity in ECs. The reduced release of adhesion molecules from ECs should improve endothelial barrier function and prevent intimal lipoprotein insudation and macrophage accumulation (Speck, et al., 2015). Some studies suggest an opposite role of ADAM17 in atherosclerosis. Adam17 deficient mice (Adam17ex/ex) crossed onto Ldlr−/−, develop larger atherosclerotic lesions with more macrophages and SMCs than control Adam17wt/wt.Ldlr−/− mice. Reduced Adam17-mediated shedding leads to increased membrane-resident TNFα and TNFR2 activation. Cultured Adam17-deficient macrophages and SMCs show proatherosclerotic potential, increased proliferation, reduced apoptosis, and increased adhesion to ECs, and these effects are prevented by Tnfr2 siRNA. Thus, ADAM17 may exert atheroprotective effects mediated by cleaving membrane-bound TNFα and TNFR2, and preventing overactivation of TNFR2 (Nicolaou, et al., 2017).
Oxidized low-density lipoprotein (ox-LDL)-induced oxidative stress and apoptosis play a role in atherosclerosis partly through ADAM22 activation. In macrophages, ox-LDL suppresses miR-221–3p expression, transfection of miR-221–3p mimic or silencing ADAM22 reduces, and a miR-221–3p inhibitor increases ox-LDL-induced foam cell formation and cell apoptosis. Thus, miR-221–3p may inhibit ox-LDL-induced foam cell formation and apoptosis by targeting ADAM22 (Zhuang, et al., 2019).
ADAM33 is upregulated in atheromas’ SMCs and inflammatory cells. ADAM33 gene variants are associated with the extent of atherosclerosis in patients with CAD. ADAM33 neutralizing antibody increases SMC migration through a reconstituted basement membrane, suggesting an inhibitory effect of ADAM33 on SMC migration that could affect the course of atherosclerosis (Holloway, et al., 2010).
ADAMTS members also participate in atherosclerosis (Santamaria & de Groot, 2020). ADAMTS1, 4, 5 and 13 are expressed in carotid plaques, especially in SMCs and macrophages, with higher expression of ADAMTS1, little increase in ADAMTS4 and 5, and reduced TIMP-1 in unstable versus stable plaques (Pelisek, et al., 2017). Also, while ADAMTS4 or 5 immunoreactive areas are similar, ADAMTS1 immunoreactive area is greater and overlap with those positive for CD68 and versican cleavage products in coronary atherectomy specimens from patients with acute MI versus stable angina, supporting a role of ADMATS1 in the fibrous cap weakening and plaque instability (C. W. Lee, et al., 2011). Shear stress on ECs promotes atherogenesis, fibrous cap thinning and rupture. Pericytes in the sub-endothelial space have vasoprotective effects, but are subjected to shear stress when EC integrity is disrupted. In cultured ECs and pericytes subjected to high shear stress ECs upregulate ADAMTS1 while pericytes upregulate the ADAMTS1 inhibitor TIMP-3, and ECM proteins are differentially expressed in ECs and pericytes co-cultures. Thus during atherosclerosis development, pericytes sense direct flow and counter-regulate EC ADAMTS1 by protective TIMP-3 expression to prevent ECM degradation and maintain vascular stability (Schrimpf, et al., 2017). ADAMTS1, 4 and 5 are upregulated during differentiation of monocytes into macrophages. TGF-β induced ADAMTS1 and 5 and downregulated ADAMTS4. IFN-γ suppressed ADAMTS1 expression. TL-1A or IL-17A alone did not affect ADAMTS1, 4 and 5 expression, but induced their expression synergistically when present together, suggesting differential cytokine-induced regulation of ADAMTS proteases in macrophages which would affect atherosclerotic lesion progression and plaque stability (Ashlin, Kwan, & Ramji, 2013).
ADAMTS1 is upregulated in atherosclerotic plaques and in proliferating/migrating aortic SMCs. ADAMTS1 cleaves versican and other proteoglycans purified from cultured human aortic SMCs. Also, the carotid artery no-flow mouse model shows increased intimal hyperplasia in ADAMTS1 transgenic/ApoE−/− versus ApoE−/− mice, supporting that ADAMTS1 promotes atherogenesis by cleaving versican and facilitating SMC migration (Jonsson-Rylander, et al., 2005). In THP-1 macrophages, lauric fatty acid reduces PI3K and JNK activity and downregulates ADAMTS1, and these effects are reversed by JNK1 siRNA, suggesting that lauric acid stabilizes atherosclerotic plaques and prevents thrombosis by interfering with PI3K/JNK pathways and ADAMTS1 expression (Ong, Wong, Tengku-Muhammad, Choo, & Chew, 2019).
ADAMTS4 regulates versican turnover in the arterial wall. In patients undergoing carotid endarterectomy, ADAMTS4 expression in the atherosclerotic plaques and its serum level are higher, versican levels are lower, and symptomatic cerebral ischemic events are higher in the vulnerable than stable group, suggesting that ADAMTS4 is an independent risk factor and a potential biomarker for plaque vulnerability (Dong, et al., 2018). ADAMTS4 is expressed in macrophages of atherosclerotic lesions, and TGF-β likely through Smads, p38 MAPK and c-Jun pathways downregulates ADAMTS4 in human macrophages and in turn promotes anti-atherogenic and plaque stabilization actions (Salter, et al., 2011). In ApoE−/− mice fed high-fat diet, ADAMTS4 expression is increased in plaques as atherosclerosis progresses. ApoE−/−Adamts4−/− showed reduced plaque burden, vulnerability index, lipid content and macrophages, more stable plaques, increased SMCs, collagen deposition and fibrotic cap thickness, and altered plasma inflammatory cytokines, further supporting a role of ADAMTS4 in atherosclerosis progression and plaque instability (Y. C. Chen, et al., 2013; Kumar, et al., 2016).
Biglycan and versican are important lipoprotein-binding proteoglycans. ADAMTS5 releases versican and aggrecan fragments, biglycan and link protein from the aortic wall. ADAMTS5 is reduced while biglycan and versican are increased in atherosclerotic aortas of ApoE−/− mice. Aortas of ADAMTS5-deficient mice show no detectable versican fragments and increased biglycan levels, highlighting the role of ADAMTS5 in degrading vascular proteoglycans reducing their LDL binding ability, and releasing LDL from aortic lesions, thus reducing lipoprotein retention in atherosclerosis (Didangelos, Mayr, Monaco, & Mayr, 2012). ADAMTS5 also degrade the ECM protein matrilin-2. Calcified human aortic valves show reduced ADAMTS5 and increased matrilin-2 and α-smooth muscle actin (α-SMA). Treatment of normal valvular interstitial cells with soluble matrilin-2 increases α-SMA, and these effects are enhanced by ADAMTS5 knockdown. α-SMA knockdown reduces, while α-SMA overexpression increases pro-osteogenic factors and calcium deposits in valvular interstitial cells. Thus, ADAMTS5 deficiency and matrilin-2 accumulation in valvular interstitial cells promote myofibroblastic transition and pro-osteogenic activity, which may contribute to calcific aortic valve disease (F. Li, et al., 2017).
Large-scale genome-wide association studies (GWAS) have identified SNPs in ADAMTS7 gene with associations to CAD. ADAMTS7 levels were higher in carotid plaques from patients with cerebrovascular symptoms than asymptomatic patients, and correlated with intense CD68-staining and high lipid content and low SMC and collagen content, which are characteristics of a vulnerable plaque. ADAMTS7 levels above median were associated with increased risk for postoperative cardiovascular events, supporting an association of ADAMTS7 with a vulnerable plaque phenotype in human carotid lesions (Bengtsson, et al., 2017). COMP is a matricellular protein expressed in cartilage and VSMCs, and maintains VSMC contractile phenotype. COMP is also secreted by platelets and reduces hemostasis and thrombosis. COMP mediates its effects through COMP binding proteins such as integrin α7β1, integrin β3, thrombin, and bone morphogenetic protein 2. COMP deficient mice show increased VSMC migration, atherosclerosis and dilated cardiomyopathy. ADAMTS7 degrades COMP, and ADAMTS7 expression correlates with atherosclerosis and vascular calcification in both human GWAS and animal models (Fu & Kong, 2017). In rat carotid artery balloon injury model, ADAMTS7 is accumulated in the neointima mainly in SMCs. Adenovirus-induced ADAMTS7 overexpression increases VSMC proliferation/migration and neointima thickening, and ADAMTS7 siRNA produces opposite effects. Thus, ADAMTS7 facilitates VSMC migration by degrading COMP, and may be a target in atherosclerosis and postangioplasty restenosis (L. Wang, Wang, & Kong, 2010).
ADAMTS4 and 8 are expressed in macrophage-rich areas of human atherosclerotic carotid plaques and coronary unstable plaques. ADAMTS4 is upregulated in atherosclerotic aortas of Ldlr−/−ApoB100/100 mice. ADAMTS4 and 8 are induced during monocyte to macrophage differentiation, and IFN-γ and TNFα enhance ADAMTS4 and 8 expression in macrophages, supporting a role of ADAMTS4 and 8 in the regulation of macrophage-rich areas of atherosclerotic plaques (Wagsater, et al., 2008). Also, cardiovascular risk score is increased and show positive correlation with ADAMTS4 and 9 levels in patients with primary hyperparathyroidism and atherosclerosis (Karakose, et al., 2018).
ADAMTS13 is released by hepatic stellate cells and ECs, and modulates early atherosclerosis through proteolytic cleavage of ultra-large vWF multimers. vWF is synthesized by ECs and megakaryocytes. Endothelium-derived vWF contributes to atherosclerosis by promoting platelet adhesion and vascular inflammation, whereas platelet-derived vWF, even in the absence of ADAMTS13, is not sufficient to promote atherosclerosis (Doddapattar, et al., 2018). Low plasma ADAMTS13 activity is associated with cardiovascular events, MI and stroke. ADAMTS13 co-localizes with vWF in coronary thrombi from patients with acute MI. ADAMTS13 cleaves vWF and reduces inflammation, plaque formation and thrombosis during early atherosclerosis in mice. In kidney podocytes, simvastatin increases while IL-6 decreases ADAMTS13 expression, and simvastatin reverses IL-6 induced inhibition of ADAMTS13 expression, which contributes to its anti-atherosclerotic and anti-thrombotic properties (L. Shen, Lu, Dong, Ma, & Ruan, 2013). In ApoE−/− and Adamts13−/−ApoE−/− mice fed a high-fat diet, plasma cholesterol levels are similar, but Adamts13−/−ApoE−/− mice show more atherosclerotic lesions in the aorta, increased ratio of plasma high- to low-molecular-weight vWF multimers, reduced leukocyte rolling velocities, and increased number of leukocyte rolling and macrophage infiltration into the atherosclerotic lesions (S. Y. Jin, et al., 2012). Also, an antibody to ADAMTS13 disintegrin domain augments the average surface covered by platelet adhesion and the long axes of platelet thrombi, reduces the ability of plasma ADAMTS13 to cleave ultra-large vWF multimers in type I collagen-coated flow chamber, and augments thrombus formation on injured neointima of rabbit femoral artery, supporting a role of ADAMTS13 in attenuating thrombus growth on injured arteries (Moriguchi-Goto, et al., 2009).
9.3. ADAMTS13 Deficiency and Thrombotic Thrombocytopenic Purpura (TTP)
vWF is a multimeric hemostatic plasma glycoprotein that acts as a bridging molecule at sites of vascular injury for normal platelet adhesion, and platelet aggregation during high shear. Plasma ADAMTS13 recognizes and cleaves ultra-large vWF multimer into smaller less coagulant forms, and thereby counterbalances its platelet adhesion and prothrombogenic effects (X. L. Zheng, 2015). ADAMTS13-mediated proteolysis is determined by rheological shear stress-induced conformational changes in vWF that unfold the A2 domain cleavage site, and its own conformational activation. ADAMTS13 circulates in a closed conformation maintained by a CUB-spacer domain binding, and is conformationally activated on demand through interaction with vWF (South, et al., 2014). The ADAMTS13 cysteine-rich and spacer domain exosites bring the enzyme and substrate into proximity to each other, then binding of ADAMTS13 disintegrin domain to vWF allosterically activates the adjacent metalloprotease domain to facilitate proteolysis, such that vWF functions as both a substrate and activating cofactor for ADAMTS13 (Petri, et al., 2019). ADAMTS13 cleaves vWF at the Tyr1605-Met1606 (P1-P1’) bond in vWF A2 domain (Y. Xiang, de Groot, Crawley, & Lane, 2011). Truncated ADAMTS13 variants suggest the importance of the disintegrin domain for enzyme activity. Targeted mutagenesis of nonconserved regions in the disintegrin domain identified 3 ADAMTS13 mutants (R349A, L350G, V352G) with reduced proteolytic activity toward vWF115 (vWF residues 1554–1668). These residues form a predicted exposed exosite on the disintegrin domain ~26 Å from the active site. Kinetic analysis of vWF115 carrying the D1614A mutation suggested that Arg349 in ADAMTS13 disintegrin domain interacts directly with Asp1614 in vWF A2. This interaction positions the scissile bond within the active site of ADAMTS13 and in turn determines the cleavage parameters (Km and kcat), while the binding affinity (Kd) of ADAMTS13 for vWF is determined by the spacer domain (de Groot, Bardhan, Ramroop, Lane, & Crawley, 2009).
ADAMTS13 deficiency (inherited or acquired) increases ultra-large vWF multimers and leads to platelet clumping and a microvascular thrombotic disorder termed thrombotic thrombocytopenic purpura (TTP, Moschcowitz disease). Mutations in ADAMTS13 gene cause chronic relapsing TTP, and ADAMTS13 autoantibodies cause sporadic TTP (X. Zheng, Majerus, & Sadler, 2002). TTP is characterized by severe thrombocytopenia, microangiopathic hemolytic anemia and ischemic damage in the brain, heart and kidneys. In the microvasculature high shear environment, hyperreactive ultra-large vWF strings remain uncleaved after EC secretion and anchorage, bind to platelets, cause platelet aggregation and form microthrombi, leading to the clinical manifestations of TTP.
Congenital TTP (Upshaw-Schulman syndrome) is a rare autosomal recessive thrombomicroangiopathy caused by inherited ADAMTS13 deficiency. Data from the United Kingdom over 15 years showed 73 cases diagnosed with congenital TTP and confirmed by genetic analysis, with presentation peaks in childhood (age ~3.5 years) and adulthood, typically related to pregnancy (age ~31 years). Genetic mutations differed by age of onset with prespacer mutations more likely associated with childhood onset (Alwan, et al., 2019). Congenital TTP results from homozygous or compound heterozygous mutations in ADAMTS13, whereas acquired TTP is an autoimmune disorder caused by circulating ADAMTS13 autoantibodies. TTP is diagnosed by a combination of symptoms, severe deficiency in plasma ADAMTS13 activity, and detection of ADAMTS13 antibodies (Crawley & Scully, 2013), with some cases showing open ADAMTS13 conformation and exposed cryptic epitope in the spacer domain (Roose, et al., 2018).
Current treatment of acute TTP includes plasma exchange to provide functional ADAMTS13 and remove inhibitory antibodies. In patients with congenital TTP in the United Kingdom, fresh-frozen plasma and factor VIII concentrate were used, and 88% of patients showed normal blood counts and symptoms resolution. The 3-weekly regimen of fresh-frozen plasma was insufficient for 70% of patients, and weekly or fortnightly infusions were required. Stroke incidence was reduced with prophylactic therapy, but long-term the risk of end-organ damage was seen in 75% of patients with late diagnosis of congenital TTP (Alwan, et al., 2019). Immunosuppressive drugs such as corticosteroids and rituximab (RTX) supplement plasma exchange (PEX) therapy in patients with acquired TTP (Kremer Hovinga, et al., 2017). Following PEX/RTX therapy, patients remain in remission for extended periods, with continuing reduction in ADAMTS13 antibodies (Becerra, et al., 2015). Anti-CD20, rADAMTS13, hyperreactive ADAMTS13 variant and anti-vWF therapy are under development (Crawley & Scully, 2013). Prophylactic ADAMTS13 replacement may decrease the risk of end-organ damage and ischemic stroke (Alwan, et al., 2019). In patients with severe congenital ADAMTS13 deficiency and plasma activity <6% rADAMTS13 (BAX 930) increased vWF cleavage products and reduced vWF multimeric size, and was well tolerated with no serious adverse events or detection of anti-ADAMTS13 antibodies. (Scully, et al., 2017).
Acute MI may occur secondary to TTP. A report described an 80-year-old male with acute MI, who was treated by percutaneous coronary intervention, but then developed thrombocytopenia, acute kidney injury and hemolytic anemia. Acquired TTP was diagnosed based on decreased ADAMTS13 levels and the presence of ADAMTS13 autoantibodies, and the patient eventually died of multiorgan failure despite repeated plasma exchanges and immunosuppressive therapies with corticosteroid and RTX, thus alerting that TTP could be an underlying cause of acute MI (Takimoto, et al., 2016).
TTP can also present in pregnancy with typical microangiopathy and thrombocytopenia, and risk of in utero fetal loss. ADAMTS13 activity < 10% is consistent with late-onset congenital TTP or acquired TTP; the former will require plasma therapy and the latter immunosuppression (Scully, 2016).
ADAMTS13−/− mice receiving rh-ultra-large vWF multimers show TTP-like features, thrombocytopenia, schistocytosis (increased fragmented red blood cells), decreased hematocrit, elevated serum lactate dehydrogenase, platelet aggregation in the ventricles and myocardial necrosis, and rhADAMTS13 reduces the incidence and severity of TTP in ADAMTS13−/− mice (Schiviz, et al., 2012).
Of note, complete ADAMTS13 deficiency does not lead to continuous microangiopathy, Plasminogen binds to vWF A1 domain. In the absence of ADAMTS13, plasminogen activation by uPA or streptokinase causes rapid degradation of platelet-vWF complexes on ECs. Plasminogen activation and plasma levels of plasmin-α2-antiplasmin complexes increase with the extent of thrombocytopenia in acute TTP, independent of ADAMTS13 activity. Also, streptokinase shows benefits in Adamts13−/− mouse model of TTP. Thus, plasminogen activation on ECs may be a natural backup for ADAMTS13 to degrade platelet-vWF complexes, and thrombolytic agents may bypass ADAMTS13 autoantibodies and have benefits in microangiopathies (Tersteeg, et al., 2014).
Bleeding events are frequent complications of left ventricular assist device, due to shear stress-induced unfolding of large vWF multimers and its accelerated proteolysis by ADAMTS13 (Proudfoot, Davidson, & Strueber, 2017). Blood of patients with a left ventricular assist device shows reduced large vWF multimers and increased 10/11 vWF degradation fragments. Also, in normal human blood exposed to supraphysiologic shear stress in a laboratory vortexer ADAMTS13 cleaves large vWF multimers and generates 11/11 vWF degradation fragments, suggesting that ADAMTS13 could be targeted to reduce vWF degradation and bleeding complications of left ventricular assist device (Bartoli, Restle, Zhang, Acker, & Atluri, 2015).
9.4. ADAMs and ADAMTS in Coronary Artery Disease
Coronary artery disease (CAD) results from blockage of the coronary arteries by plaque buildup. In HUVECs, IL-6 and sIL-6R induce inflammation, increase glycoprotein130 and decrease soluble glycoprotein130 levels, and these effects are reversed by estradiol treatment. Estradiol-through activation of estrogen α/β receptor and increased release of ADAM10 and 17 could alter the inflammatory pathways and the course of inflammation in atherosclerosis and CAD (M. Zhou, Dai, Cui, & Li, 2020).
Decreased endothelial ADAM10 activity is linked to dysregulated Notch signaling and defective coronary arterial cell differentiation. Endothelium-specific ADAM10 deficient mice (ADAM10ΔEC) show reduced coronary endothelial EGFR signaling, enlarged hearts, abnormal myocardial compaction, and immature endothelium markers, implicating ADAM10/Notch signaling in coronary arterial cell specification and heart development, and suggesting a role of endothelial ADAM10 in EGFR signaling and the regenerative capacity and maturation of the coronary vasculature (Farber, et al., 2019).
Circulating ADAM17 substrates sICAM-1, sVCAM-1, sIL-6R and sTNFR1 have been linked to recurring cardiovascular events in patients with atherosclerosis, making ADAM17 an attractive biomarker and therapeutic target in CAD (Rizza, et al., 2015). ADAM17 also regulate angiogenesis and myocardial cell repair post-MI in patients with CAD (Fan, et al., 2015; E. P. van der Vorst, et al., 2012).
ADAMTS aggrecanases regulate vascular proteoglycans and ECM remodeling during in-stent restenosis. Immunostaining shows more aggrecan and its fragments in human stented coronary arteries at the contacts of the stent struts with the artery, in human arteries than veins, and on grafting a vein into the arterial circulation, suggesting aggrecan contribution to vascular plasticity. In pig coronary arteries implanted with overstretched drug-eluting stents, the neointima shows increases in calcification proteins, and the media shows changes in inflammatory and thrombotic factors, followed by changes in ECM proteins. By day 28 post-implantation, basement membrane proteins are less in drug-eluting than bare-metal stents, and aggrecan is increased with a shift from ADAMTS1 and 5 to ADAMTS4 gene expression, implicating aggrecan and aggrecanases in the vascular injury response after stenting. Mice lacking ADAMTS5 show aggrecan accumulation and dilation of thoracic aorta, supporting a role of aggrecanases in vascular remodeling (Suna, et al., 2018).
Plasma ADAMTS4 level is increased in acute coronary syndromes, and further elevated with progression from stable angina to unstable angina, to non-ST-segment elevation MI (NSTEMI), to ST-segment elevation MI (STEMI), and serial measurements of plasma ADAMTS4 may be a marker of plaque destabilization in acute coronary syndromes (Zha, et al., 2010). Also, in patients undergoing elective coronary angiography, ADAMTS4 levels are higher in patients with than without CAD, are increased with the number of diseased vessels, are associated with the severity of stenosis score, are correlated with the presence/severity of CAD, and are lower in statin-treated than non-treated patients, suggesting that serum ADAMTS4 levels may be an independent predictor of CAD (L. Chen, Yang, Zha, & Cui, 2011).
In patients with stable obstructive CAD, plasma ADAMTS7 levels are positively correlated with the Syntax score CAD severity tertiles (low, moderate, high), and are greater in the high than the low Syntax score group. Event-free survival rate is less in the high than low plasma ADAMTS7 group, supporting that ADAMTS7 contributes to severity of CAD (J. Yu, et al., 2016). ADAMTS7 accumulates in VSMCs in coronary atherosclerotic plaques. In a population-based study, an inverse association was observed between atherosclerosis prevalence and rs3825807, a nonsynonymous SNP (A to G) leading to a Ser-to-Pro substitution in ADAMTS7 pro-domain. The rs3825807 variant-mediated protection from atherosclerosis and CAD may involve reduced ADAMTS7 maturation and its prodomain cleavage, decreased cleavage of thrombospondin-5, and reduced VSMC migration (Pu, et al., 2013). CABG is a beneficial approach in CAD, but is prone to vein graft restenosis. Upregulation of ADAMTS7 contributes to neointima formation and graft restenosis by promoting VSMC proliferation/migration and inhibiting EC proliferation/migration. Aberrant miR-423 levels in plasma of CAD patients prior to and following CABG suggest that it may affect vein graft patency. Dual-luciferase reporter gene assay indicates that miR-423 interacts with ADAMTS7. Overexpression of miR-423 suppresses ADAMTS7, decreases proliferation/migration of human umbilical vein SMCs, increases proliferation/migration of HUVECs, enhances re-endothelialization and decreases neointima formation in a rat vein graft model, suggesting that upregulation of miR-423 and downregulation of ADAMTS7 could reduce autologous vein graft restenosis in patients undergoing CABG (W. Ren, et al., 2020). Rat model of carotid artery balloon injury show neointima formation and increased Adamts7 expression. Adamts7-deficient mice show reduced neointima formation following vascular injury, supporting that ADAMTS7 could be a target for intervention in CAD (Muller, Kessler, Schunkert, Erdmann, & Tennstedt, 2016).
Studies examined the association of genetic variants of ADAMTS13 (P475S, Q448E, rs2073932, P618A, A900V, S903L, rs652600, rs4962153) and vWF (V1565L, Y1584C), as well as ADAMTS13 activity, vWF antigen and vWF activity with CAD risk in Thai CAD and non-CAD patients. None of ADAMTS13 polymorphisms or haplotypes was associated with ADAMTS13 activity. vWF V1565L polymorphism was associated with increased ADAMTS13 activity, and the QAGA or H4 haplotype of ADAMTS13 gene had an independent protective effect on CAD (Lasom, et al., 2017).
9.5. ADAMs and ADAMTS in Myocardial Infarction
Genotyping of samples from the Finnish cardiovascular study showed increased MI risk and higher sADAM8 serum levels for carriers of the rs2995300 C allele and rs2275725 A allele, suggesting a prognostic potential of ADAM8 in MI (Raitoharju, et al., 2011). In rats undergoing cardiac transplantation after cardiac arrest to induce I/R injury and rats undergoing left anterior coronary artery ligation to cause MI, edematous intramyocardial artery nuclei and periadventitial inflammation were more prominent in MI than I/R injury and control rats. In MI rats, ADAM8 expression was increased and localized to the vicinity of the infarcted area and to remote intramyocardial arteries (Vuohelainen, et al., 2011).
In Wistar rats subjected to MI by ligation of left anterior descending coronary artery, ADAM15 expression initially increases then gradually decreases. ADAM15 is localized at cardiac myocytes in the border area of MI and at macrophages in the border and infarcted areas, suggesting a role in the inflammatory response and cardiac remodeling associated with MI (J. K. Li, Du, Jiang, & Tian, 2009).
Compared with sham-controls, Wistar rats with ligated coronary artery show MI, worse systolic function, greater left ventricular weight index, increased ADAM17 expression, upregulated TNFα, and decreased TIMP-3. In MI rats, ADAM17 expression correlates positively with increased left ventricular end-diastolic and end-systolic diameters and negatively with left ventricular ejection fraction, suggesting a link between ADAM17 expression, increased TNFα, and post-MI cardiac remodeling (D. Y. Zheng, Zhao, Yang, Wang, & Zhang, 2016). Other studies suggest a protective role of ADAM17 in ischemic cardiomyopathy. Cardiomyocyte-specific ADAM17 knockdown mice (ADAM17flox/flox/α-MHC-Cre) are more vulnerable to coronary artery ligation-induced MI than control mice (ADAM17flox/flox), and show unfavorable survival, higher rates of cardiac rupture, more severe ventricular dilation and decreased ejection fraction, and the infarcted areas show reduced myocardial vascular density, lectin perfusion, CD31 staining, NF-κB activity and VEGFR2 expression. Thus ADAM17 may promote post-MI recovery by regulating VEGFR2 transcription and angiogenesis, and limiting left ventricular dysfunction (Fan, et al., 2015).
ADAMTS1 shows diffuse distribution in the myocardium of individuals who died of MI or trauma. The MI group show no ADAMTS1 staining around fibrotic areas, and slight ADAMTS1 staining and similar localization of fragmented versican in coagulative and necrotic zones (Pehlivan, et al., 2016). In rat MI model, ADAMTS1 is upregulated in the endothelium and myocardium of infarcted heart 3 hours post-MI, making it one of the early genes expressed in the ischemic heart (K. Nakamura, et al., 2004).
In patients with STEMI and NSTEMI, plasma ADAMTS7 levels are higher in patients with left ventricular ejection fraction (LVEF) ≤35% than those with LVEF >35%, correlate positively with brain natriuretic peptide, left ventricular mass index, and left ventricular end-diastolic and end-systolic diameters, and correlate negatively with the 6-min walk test of aerobic capacity and endurance, suggesting a role of ADAMTS7 in ventricular remodeling after acute MI (Wu, et al., 2015).
Culprit atherosclerotic plaques from patients with acute MI show a smaller area for smooth muscle α-actin, and a greater area for the EC marker CD31 or macrophage marker CD68 than plaques from patients with stable angina. Also, the immunoreactive areas for ADAMTS2, 3, and 13 overlap with CD31 or CD68, and are larger in acute MI (C. W. Lee, et al., 2012). On the other hand, the Glasgow MI Study showed no correlation between ADAMTS13 and vWF levels in MI cases. ADAMTS13 levels correlated positively with serum cholesterol and triglycerides and body mass index, and negatively with high-density lipoprotein-cholesterol. vWF levels correlated with age, fibrinogen and C-reactive protein. vWF correlated positively and ADAMTS13 correlated negatively with risk of MI (Crawley, Lane, Woodward, Rumley, & Lowe, 2008). Another study showed elevated vWF activity and vWF antigen and decreased ADAMTS13 activity following percutaneous coronary intervention in STEMI patients with intramyocardial hemorrhage versus those without intramyocardial hemorrhage. However, vWF and ADAMTS13 activities were not correlated with infarct size. Also, intracoronary administration of rADAMTS13 did not decrease infarct size or intramyocardial hemorrhage in a porcine model of myocardial I/R, suggesting that vWF/ADAMTS13 imbalance may not be the cause of no reflow (Eerenberg, et al., 2016). Nevertheless, vWF/ADAMTS13 imbalance should be monitored in patients with MI. A study reported an old male patient who was receiving the purinergic P2Y12 receptor antagonist ticagrelor for STEMI, and presented with severe thrombocytopenia, anemia, renal and liver dysfunction, heart failure and fever. Blood samples showed schistocytosis and low ADAMTS13 activity, consistent with TTP. Cessation of ticagrelor and plasma exchange improved the patient condition, but re-administration of ticagrelor aggravated TTP and led to the patient’s death, highlighting the risk of purinergic receptor antagonist-induced low ADAMTS13 and TTP in patients with MI (X. Wang, et al., 2018).
9.6. ADAMs and ADAMTS in Cardiac Hypertrophy and Heart Failure
Patients with dilated cardiomyopathy show increased myocardial ADAM10 and 15 expression and integrin-β1D cleavage. ADAM10 expression correlates with chamber dilatation and systolic dysfunction. Hemodynamic unloading reduced ADAM10 and 12 and increased integrin-β1D expression. ADAM12 and integrin-β1D are increased in hypertrophic obstructive cardiomyopathy, and ADAM17 is increased in dilated cardiomyopathy and hypertrophic obstructive cardiomyopathy, suggesting different roles of ADAMs in myocardial structural remodeling and integrin shedding (Fedak, et al., 2006).
Cardiac ADAM12 levels are increased in a mouse model of heart failure induced by aorta-to-vena cava shunt (Mishra, Tyagi, Sen, Givvimani, & Tyagi, 2010), and hydrogen sulfide (H2S) donors ameliorate heart failure by decreasing ADAM12 (Donnarumma, Trivedi, & Lefer, 2017; Mishra, et al., 2010; Salloum, 2015). In cardiomyocytes, GPCR agonists cause hypertrophy and HB-EGF shedding through metalloproteinase and EGFR transactivation. Also, the ADAM12 inhibitor KB-R7785 blocks HB-EGF shedding and ameliorates cardiac hypertrophy in mouse transaortic constriction model (Asakura, et al., 2002). In mice with transverse aortic constriction, atorvastatin causes reduction of heart/body weight, decreased left ventricular end-diastolic pressure, increased fractional shortening, downregulation of ADAM12, ADAM17 and HB-EGF, and reduced EGFR and ERK activity. In neonatal rat cardiomyocytes, atorvastatin inhibits phenylephrine-induced protein synthesis, EGFR phosphorylation and ERK activity (Liao, et al., 2008). However, 4 weeks after transaortic constriction ADAM12 KO mice show advanced cardiac hypertrophy, higher mortality rates, and more cardiac fibrosis and collagen-related gene expression in failing hearts than WT mice. ADAM12 deficient mice also show enhanced focal adhesion- and fibrosis-related signaling pathways, increased integrin β1 and TGF-β receptor I and III, and phosphorylation of focal adhesion kinase, Akt, mammalian target of rapamycin, ERK, and Smad2/3 in the heart, resulting in cardiac dysfunction (Y. Nakamura, et al., 2020). These discrepant findings make it important to further examine the role of ADAM12 in heart failure.
ADAM17 and TNFα levels are higher in mononuclear cells from patients with severe congestive heart failure (CHF) than mild CHF patients and controls (M. Satoh, et al., 2004). The peroxisome proliferator-activated receptor-α (PPAR-α) agonist fenofibrate reduces heart/body weight, left ventricular wall thickness and ADAM17 expression In aortic constriction-induced hypertensive rats, and reduces AngII-induced hypertrophy and ADAM17 expression in cardiomyocytes, suggesting that PPAR-α agonists ameliorate cardiac hypertrophy by downregulating ADAM17 (Zeng, Lu, Yan, & Zou, 2018). ACE2 cleaves AngII to Ang-(1–7) and protects against cardiac hypertrophy, while ADAM17 cleaves ACE2 and contributes to AngII-induced heart damage. In AngII-infused mice Dickkopf-3 overexpression is associated with less cardiac hypertrophy and fibrosis, decreased expression of the hypertrophic genes atrial natriuretic peptide and β-myosin heavy chain and the fibrotic genes collagen I and III, reduced ADAM17 phosphorylation, and increased ACE2 expression and AngII degradation (C. G. Zhai, et al., 2018). Rats with aortic coarctation-induced myocardial hypertrophy show increased nicotinamide adenine dinucleotide phosphate oxidase 4 (Nox4) and ADAM17. Nox4 and ROS mediate AngII-induced hypertrophy of cardiac myocytes partly through upregulation of ADAM17 and increased release of mature HB-EGF, and ADAM17 siRNA decreases phospho-EGFR/EGFR ratio and EGFR activation in hypertrophic cardiomyocytes (Zeng, et al., 2013). However, in mice with cardiac pressure-overload induced by transverse aortic constriction, cardiomyocyte-specific ADAM17 knockdown mice (ADAM17f/f/Cre) show enhanced myocardial hypertrophy, fibrosis, left ventricular dilation, and systolic dysfunction, and greater upregulation of integrin β1 and activation of the focal adhesion kinase pathway versus mice with intact ADAM17 (ADAM17f/f). ADAM17 knockdown does not affect AngII infusion-induced myocardial hypertrophy, which does not require integrin β1-mediated pathway. ADAM17 knockdown also enhances hypertrophy induced by cyclic mechanical stretching in neonatal rat cardiomyocytes. Thus, ADAM17 could be cardioprotective in pressure overload but not agonist-induced cardiac hypertrophy likely through cleavage of cardiac integrin β1 (Fan, et al., 2016).
ADAM22 expression is increased in patients with hypertrophic cardiomyopathy. In mice with transaortic constriction, cardiac-specific ADAM22 KO deteriorates, while ADAM22 overexpression mitigates, cardiac hypertrophy. Cardiac-specific ADAM22 KO is associated with enhanced Akt activity in transaortic constriction-induced mice and AngII-stimulated neonatal rat cardiomyocytes, and Akt inhibitors attenuate transaortic constriction-induced cardiac hypertrophy, suggesting that ADAM22 reduces Akt activity and cardiac hypertrophy (L. Ren, et al., 2018).
ADAM23 expression is decreased in failing human heart and hypertrophic mouse heart. Cardiac-specific ADAM23 KO mice show increased cardiac hypertrophy, fibrosis, and dysfunction, and mice overexpressing ADAM23 in the heart show reduced pressure overload-induced cardiac hypertrophy. Similar findings are observed in AngII-induced neonatal rat cardiomyocyte hypertrophy. The focal adhesion kinase inhibitor PF-562271 reverses the detrimental effects in ADAM23 KO mice subjected to aortic banding, supporting that ADAM23 protects against cardiac hypertrophy through inhibition of the focal adhesion kinase-Akt signaling pathway (M. Xiang, et al., 2018).
ADAMTS2 is upregulated in failing human heart and hypertrophic mouse heart. GPCR agonists induce cardiac hypertrophy, and Adamts2 gene could drive isoproterenol-induced cardiac hypertrophy in mice (Rau, et al., 2017). Other studies suggest that ADAMTS2 cleaves the N-propeptides from procollagens type I, II, III and V, thus allowing the formation and assembly of collagen fibrils (Bekhouche & Colige, 2015). Mice lacking ADAMTS2 show increased pressure overload-induced cardiac hypertrophy, because their collagen fibrils have inadequate mechanical strength to counteract pressure overload, while mice with cardiac-specific overexpression of ADAMTS2 show alleviated cardiac hypertrophy. Also, loss or gain of function experiments in neonatal rat cardiomyocytes show that ADAMTS2 decreases AngII-induced cardiomyocyte hypertrophy. Blocking the phosphoinositide 3-kinase (PI3K)/Akt pathway ameliorates the hypertrophic response to the loss of ADAMTS2, supporting that ADAMTS2 attenuates cardiac hypertrophy through inhibition of PI3K/Akt signaling (X. Wang, et al., 2017).
Myocardial ADAMTS1, 4 and 8, and versican and aggrecan levels are increased in rat aortic banding model, and TNFα and IL-1β increase ADAMTS4 and versican in neonatal cardiomyocytes. Pentosan polysulfate was used to inhibit ADAMTS4 and was found to improve left ventricular systolic function and fractional shortening after aortic banding, and reduce extracellular versican fragments, supporting a role of increased versicanase activity in hypertrophic cardiomyopathy, and that ADAMTS4 inhibition could improve systolic function in heart failure (Vistnes, et al., 2014).
In mice with pressure overload hypertrophic cardiomyopathy, treatment with rhADAMTS13 is associated with less endothelium-associated vWF, platelet aggregates and profibrotic TGF-β1 levels, preservation of cardiac function and less fibrotic remodeling (Witsch, et al., 2018).
9.7. ADAMs and ADAMTS in Ischemic Stroke
Ischemic stroke is linked to Alzheimer’s disease and increased amyloid-β and APP. ADAM10 is upregulated in patients with atherosclerotic cerebral infarction carrying the rs653765 C > T mutation (Y. Li, et al., 2013). In neuronal SH-SY5Y cells, exposure to hypoxia decreases ADAM10 levels and reduces APP processing by the neuroprotective ADAM10 α-secretase pathway (Webster, Green, Settle, Peers, & Vaughan, 2004).
The frequencies of ADAM17 SNPs rs11684747, rs11689958, rs12692386, rs55790676 and rs1524668 or ADAM17 haplotypes do not differ between ischemic stroke patients and healthy subjects. However, ADAM17-rs1524668 A>G polymorphism shows higher ADAM17 expression and an association with the partial anterior circulation infarcts subtype of ischemic stroke (Y. Li, et al., 2014). ADAM17-induced ectodomain shedding of platelet GPIbα is important in hemostasis and thrombosis. Patients with acute atherosclerotic ischemic stroke show increased plasma glycocalicin and ADAM17 levels and decreased platelet GPIbα, suggesting ADAM17 as a risk factor for acute ischemic stroke, and GPIbα expression as a measure for stroke severity (Ling, et al., 2013).
ADAMTS1, 4, 5 and 9 proteoglycanases play a role in ECM turnover in the CNS and their expression is dysregulated in cerebral ischemic stroke and acute spinal cord injury (Lemarchant, et al., 2013). Studies of ADAMTS1 gene polymorphism suggest that the frequencies of the rs402007 GC+CC genotype and the C allele are different and may represent susceptibility factors in ischemic stroke patients versus controls (Lyu, et al., 2015). Rat models of focal cerebral ischemia induced by transient middle cerebral artery occlusion show increased ADAMTS1 and 4, IL-1β, IL-1 receptor antagonist and TNFα in the occluded hemisphere. Also, in human astrocytes, TNFα upregulates ADAMTS4, suggesting that TNFα-induced ADAMTS1 and 4 facilitate ECM breakdown, inflammatory cell infiltration and brain injury following stroke (Cross, et al., 2006). Other studies confirmed an increase in ADAMTS4 expression in the ischemic brain hemisphere of stroke patients and mice models of cerebral ischemia. However, in primary microglia or astrocytes, pretreatment with rhADAMTS4 decreased while transfection with ADAMTS4 siRNA increased LPS-induced production of NO and the inflammatory cytokines MCP-1, TNFα, IL-1β and MMP-9. In a mouse model of neuroinflammation induced by middle cerebral artery occlusion, ADAMTS4 decreased astrogliosis and macrophage infiltration, and increased microglia expressing arginase-1, a marker of reduced inflammation, and IL-10 production in the peri-ischemic area, suggesting ADAMTS4 anti-inflammatory and neuro-regenerative effects that could be useful in ischemic stroke and traumatic brain or spinal cord injury (Lemarchant, et al., 2016).
Studies of SNPs of ADAMTS13 gene showed an association of rs2285467 with stroke. Also, ADAMTS13 mRNA is elevated in blood cells of systemic lupus erythematosus patients with history of stroke (Lopez De Padilla, et al., 2013). In a prospective study, patients with ADAMTS13 activity in the lowest quartile had a higher risk of ischemic stroke than those in the highest quartile. Adding ADAMTS13 to the predictive model improved the accuracy of risk prediction of ischemic stroke (Sonneveld, et al., 2015). Patients with cerebral infarction also show higher vWF antigen, and lower ADAMTS13, vWF collagen binding activity/vWF antigen and ADAMTS13/vWF antigen, supporting that increased vWF levels and reduced ADAMTS13 activity may contribute to the pathogenesis of cerebral infarction (L. Qu, et al., 2016). vWF/ADAMTS13 imbalance is also a risk factor of neonatal thrombosis, pediatric stroke and secondary microangiopathies (Katneni, Ibla, Hunt, Schiller, & Kimchi-Sarfaty, 2019). In pediatric patients with ischemic stroke, 22% of patients showed decreased ADAMTS13 levels (Lambers, et al., 2013). Also, among pediatric stroke patients 241 variants (SNPs or insertions/deletions) in the ADAMTS13 gene have been identified, 10 SNPs were associated with pediatric stroke, and rs2285489 and rs28793911 were associated with ADAMTS13 levels. A protective haplotype H1.1. was associated with increased ADAMTS13 levels, supporting a link between decreased ADAMTS13 levels and stroke susceptibility (Stoll, et al., 2016).
Inherited TTP may present with ischemic stroke in adults. A young male with recurrent strokes, who relapsed despite anticoagulant therapy showed plasma ADAMTS13 activity <5% with no anti-ADAMTS13 IgG, and sequencing of the ADAMTS13 gene led to the diagnosis of Upschaw-Schulman syndrome. The patient received fresh frozen plasma every 2 weeks, and one year after diagnosis was free of neurological symptoms (Beauvais, et al., 2019). ADAMTS13 may promote brain angiogenesis, neovascularization and ischemic brain repair. Studies in patients undergoing endovascular treatment of ischemic stroke showed that low ADAMTS13 activity and missing of statin therapy were independently associated with poor clinical outcome to recanalization therapies (Schuppner, et al., 2018). While thrombolysis with tissue plasminogen activator (tPA) is effective in acute ischemic stroke, ADAMTS13 by virtue of cleaving vWF could be an alternative therapy if tPA is not available or in tPA resistant thrombi (X. Chen, Cheng, Zhang, & Wu, 2019). Notably, in acute ischemic stroke cases with arterial occlusion, patients who achieved intravenous tPA-induced recanalization had higher baseline ADAMTS13 activity compared to futile recanalization in patients treated with mechanical thrombectomy. Absence of early ischemic signs and ADAMTS13 activity >75% were independent predictors of recanalization, and reduced ADAMTS13 level was an independent predictor of futile recanalization (Bustamante, et al., 2018).
In mouse model of ischemic stroke, Adamts13−/− mice show reduced neovascularization, brain capillary perfusion, pericyte and SMC coverage on microvessels and expression of tight junction proteins, and accelerated BBB breakdown and extravascular deposits of serum proteins in the peri-infarct cortex, as well as decreased angiopoietin-2 and galectin-3 in brain microvessels, and these effects were reversed in vWF deficient mice, by treatment with anti-vWF antibody, or by adenovirus-mediated overexpression of angiopoietin-2 or galectin-3 (H. Xu, et al., 2017). Also, in mouse model of focal cerebral ischemia, vWF deficiency reduced infarct volume, while ADAMTS13 deficiency resulted in larger infarctions. Infusion of rhADAMTS13 in WT mice reduced infarct volume, increased neovascularization and vascular repair, and improved functional outcome, supporting potential benefits of ADAMTS13 in ischemic stroke (Zhao, et al., 2009). Although tPA is approved for treatment of acute ischemic stroke, it increases the risk of cerebral hemorrhage. In normal mice, rADAMTS13 blocked tPA- and vWF-induced BBB permeability. In mouse stroke model, intravenous tPA exacerbated BBB disruption and cerebral hemorrhage. rADAMTS13 attenuated these effects through inhibition of tPA-mediated Akt phosphorylation, RhoA activation and upregulation of VEGF, and rvWF reversed the antihemorrhagic effect of rADAMTS13 (L. Wang, et al., 2013). Also, in a mouse model of cerebral hemorrhage induced by intracerebral infusion of autologous blood, rADAMTS13 reduced chemokines and cytokines levels, neutrophil recruitment, myeloperoxidase activity and microglia activation. rADAMTS13 also reduced ICAM-1 levels and MMP-9 activity, attenuated BBB disruption, cerebral edema and hemorrhagic lesion volume, increased pericyte coverage of brain microvessels, and improved neurological functions. Also, in brain ECs, rADAMTS13 decreased LPS-induced IL-6 expression, and rvWF reversed this effect, supporting potential benefits of rADAMTS13 against inflammation and BBB dysfunction following intracerebral hemorrhage (Cai, et al., 2015).
ADAMTS18 secretion by ECs enhances platelet fragmentation and thrombus dissolution. A C-terminal 385-aa fragment of ADAMTS18 induces platelet fragmentation and destroys platelet aggregates induced ex vivo by ADP or collagen and fibrinogen. Also, the C-terminal ADAMTS18 fragment with a glutathione S-transferase tag (named rADAMTS-351) promotes platelet fragmentation (Dang, et al., 2012), facilitates platelet thrombus dissolution, and protects against FeCI3-induced carotid artery thrombosis and cerebral infarction in a mouse model of ischemic stroke (Z. Li, et al., 2009). In support, anti-ADAMTS18 antibody shortens the tail vein bleeding time (Z. Li, et al., 2009). Also, Adamts18−/− mice display accelerated FeCl3-induced carotid artery thrombosis and aggravated postischemic cerebral infarction, due to lack of ADAMTS18-mediated-platelet fragmentation, (Dang, et al., 2018).
9.8. ADAMs and ADAMTS in Ischemia-Reperfusion Injury
I/R injury causes structural and functional changes in the heart, liver, kidney and brain. I/R injury interferes with blood flow restoration to ischemic myocardium, and causes oxidative stress and apoptosis. In mouse models of I/R injury, all-trans retinoic acid promotes anti-apoptotic effects, improves cardiac function, decreases the infarcted area, ROS production and phosphorylation of MAPKs p38, JNK and ERK, increases ADAM10 expression and RAGE cleavage, and decreases RAGE, and the cardiac protective effects are not observed in RAGE KO mice (Z. Zhu, et al., 2015).
In isolated rabbit heart, ischemic preconditioning (induced by two cycles of 5 min ischemia/5 min reperfusion) or pretreatment with AngII reduced infarction size (induced by 30 min ischemia/2 hour reperfusion), increased TNFα release into the coronary effluent, and augmented tyrosine phosphorylation of target proteins running at ~60 and 90 kDa in gel electrophoresis of cardiac tissue samples, and these effects were abolished by the tyrosine kinase inhibitor genistein or ADAM17 inhibitor KB-R7785. Pretreatment with TNFα decreased infarct size and induced tyrosine phosphorylation of 60 kDa protein, suggesting that ADAM17 regulates TNFα shedding, and promotes ischemic preconditioning through a tyrosine kinase pathway (Ichikawa, et al., 2004).
The endothelial glycocalyx could be damaged during hypoxia, I/R injury, stress-related sympathoadrenal activation, and inflammation. In HUVECs, epinephrine or epinephrine+hydrogen peroxide (H2O2) increased ADAM17 and MMP-9 activity, glycocalyx degradation and shedding of syndecan-1, hyaluronic acid and TNFα, and these effects were prevented by the serine protease inhibitor tranexamic acid or MMP inhibitor doxycycline, supporting the clinical practice of early tranexamic acid treatment in severely injured patients (Diebel, Martin, Liberati, & Diebel, 2018).
Hepatic I/R injury could cause serious liver damage and affect the outcome of liver surgery or transplantation. TNFα and its receptors play a role in the inflammatory response after hepatic I/R injury. ADAM17 levels are low in normal rat liver. Rats subjected to hepatic I/R show increases in ADAM17 expression and upregulation of TNFα, TNFR1 and IL-6. ADAM17 inhibition by TIMP-3 decreased circulating TNFα and alanine transferase and improved hepatic I/R injuries (Z. Y. Tang, Loss, Carmody, & Cohen, 2006).
In rats subjected to bilateral renal I/R injury, renal VEGF and VEGFR1 (flt-1) were reduced, plasma soluble flt-1 was elevated, and the expression of ADAMTS1, which inhibits VEGF, was increased in proximal tubules. ADAMTS1-mediated inhibition of the VEGF pathway during early renal I/R injury may reduce renal microvascular density and lead to chronic renal damage (Basile, Fredrich, Chelladurai, Leonard, & Parrish, 2008).
ADAMTS13 deficiency causes thrombotic microangiopathy and multi-organ damage during I/R of the heart, liver, kidney and brain. In mice with MI produced by transient ligation of left anterior descending coronary artery, mortality and infarct size was in ADAMTS13−/− > WT > WT+ADAMTS13 mice, and microvascular reflow in the risk area was in ADAMTS13−/− < WT < WT+ADAMTS13 mice. Platelet glycoprotein GPIbα and vWF imaging signals were higher in the postischemic risk area of ADAMTS13−/− than WT mice, and were abolished in WT mice treated with ADAMTS13. (Ozawa, et al., 2018).
Children with biliary cirrhosis due to congenital biliary atresia sometimes show thrombotic microangiopathy and decreased ADAMTS13 activity. In these children, disappearance of clinical TTP after liver transplantation confirms that hepatic stellate cells are major sources of plasma ADAMTS13. Also, acute rejection in liver transplant recipients and patients who underwent hepatectomy for liver tumors often show decreased ADAMTS13 levels and the presence of ultra-large vWF, and the ADAMTS13/vWF imbalance in the hepatic sinusoids may represent a localized TTP-like microcirculatory disturbance leading to liver damage following liver surgery (Ko, et al., 2015). Studies supported an association between plasma ADAMTS13 activity, vWF antigen and ultra-large vWF multimers, and adverse events in liver transplant recipients, and measurements of ADAMTS13 and vWF could be good indicators of graft dysfunction in the early stage after liver transplantation (Ko, et al., 2006). In mice with hepatic I/R injury, vWF KO mice showed improved hepatic blood flow, and reduced alanine aminotransferase levels, hepatocellular necrosis and neutrophil infiltration of vessel wall. Also, in WT mice, treatment with rhADAMTS13, improved alanine aminotransferase levels and hepatic blood flow and decreased neutrophil infiltration and hepatic I/R injury (Urisono, et al., 2018).
Mice with bilateral renal I/R injury show increased plasma vWF and decreased ADAMTS13 levels, and treatment with rhADAMTS13 recovers renal function, reduces tubular cell apoptosis, renal inflammation, p38/ERK phosphorylation and cyclooxygenase-2 expression, and improves eNOS phosphorylation, afferent arteriolar dilation and microvascular endothelial function (S. Zhou, et al., 2019).
During brain I/R, vWF multimer promotes platelet aggregation and microvascular plugging. ADAMTS13 cleaves vWF multimer, and ADAMTS13 deficiency amplifies post-ischemic cerebral hypoperfusion. In mouse model of middle cerebral artery occlusion and reperfusion, ADAMTS13−/− mice show larger brain infarcts and less regional cerebral blood flow, suggesting protective effects of ADAMTS13 against brain I/R injury by reducing vWF-mediated microvascular plugging (Fujioka, et al., 2012).
9.9. ADAMs and ADAMTS in Peripheral Artery Disease
Peripheral artery disease (PAD) presents as intermittent claudication and critical limb ischemia. ADAM12 may modify the severity of PAD possibly through preserving endothelial and skeletal muscle cells (Okeke & Dokun, 2018). In mice with experimental PAD, ADAM12 showed less expression in Balb/c mice which exhibited poor outcomes than in C57Bl/6 mice which had favorable outcomes. ADAM12 overexpression improved outcomes in Balb/c mice, and ADAM12 knockdown worsened outcomes in C57Bl/6 mice. Also, ADAM12 expression modulated EC proliferation and survival through activation of tyrosine-protein kinase receptor (Tie-2) (Dokun, et al., 2015).
ADAMTS7 mRNA levels are elevated in peripheral blood mononuclear cells of Turkish patients with PAD. The frequency of rs1994016 and rs3825807 SNPs was not different in PAD patients versus controls, but in PAD patients ADAMTS7 mRNA levels were increased in the CC genotype of rs1994016 and TT genotype of rs3825807, suggesting a role of ADAMTS7 in PAD (Bayoglu, et al., 2018).
PAD patients that experienced cardiovascular events may not show statistically higher vWF levels and vWF/ADAMTS13 ratio than PAD controls, but in patients with complete bimonthly data a trend toward an increase in vWF/ADAMTS13 was observed, supporting the value of measuring vWF/ADAMTS13 ratio in monitoring PAD patients for ischemic cardiovascular events (Green, et al., 2017).
9.10. ADAMs and ADAMTS in Vascular Aneurysm
Abdominal aortic aneurysm (AAA) involves ECM degradation and aortic wall inflammation. ADAM9 promotes inflammation, macrophage infiltration and cell apoptosis and is overexpressed in murine models of AAA and in AngII treated human aortic ECs and SMCs. Expression of miR-126 was decreased and inversely correlated with ADAM9 expression in AngII-treated ECs and SMCs. Also, miR-126 decreased ADAM9 gene expression, inflammatory cytokines and aneurysm formation, and improved cell survival in murine AAA, supporting that ADAM9 plays a role in AAA, and that miR-126 controls AAA development by modulating ADAM9 expression (G. Shen, et al., 2020).
Dilation of the ascending aorta involves ECM modifications and inflammation, and acute aortic dissection involves progressive degeneration of the aortic media. In patients undergoing aortic repair, B-cells in the entire aortic wall and intimal plasma cells were more abundant, and ADAM8 and 15 expression in the aortic media was greater in aortic dissection than aortic dilation. Also, two of the patients with aortic dilation and increased ADAM8 and 15 developed aortic dissection (Levula, et al., 2011). However, a causal relationship between ADAM8 and 15 and aortic dissection needs to be further tested.
ADAM10 and 17 have been linked to AAA (Folkesson, et al., 2015). TNFα and ADAM17 mRNA is greater in the transition zone between non-dilated aorta and proximal aspect of AAA than the mid-portion of AAA, and in AAA than control infrarenal aorta. TNFα mRNA expression correlated with ADAM17 mRNA, and both TNFα and ADAM17 were localized in CD68 macrophages in the media and adventitia of the transition zone of AAA (H. Satoh, et al., 2004). In WT mice treated with AngII and the lysyl oxidase inhibitor β-aminopropionitrile, 52.4% of mice died from aortic rupture, and the surviving mice showed increased ADAM17 expression in the AAA lesions. In contrast, all mice lacking ADAM17 in SMCs survived and showed reduced aortic diameter, and no aortic EGFR activation, IL-6 induction, endoplasmic reticulum/oxidative stress or ECM deposition. Treatment of WT mice with ADAM17 antibody prevented AAA formation, suggesting that decreasing SMC ADAM17 could prevent AAA formation by suppressing EGFR activation (Kawai, et al., 2017). Ursolic acid decreased AAA incidence, alleviated degradation of elastin fibers and inflammation, and decreased MMP2, MMP9, ADAM17 and phospho-STAT3 (signal transducer and activator of transcription 3) in AngII-infused ApoE−/− mice and in AngII- and TNFα-treated VSMCs, suggesting that ursolic acid ameliorates AAA by inhibiting ADAM17 and phospho-STAT3 expression (M. Zhai, et al., 2018). ADAM17 is increased in tunica media and intima of thoracic aortic aneurysm (TAA). In experimental elastase-induced TAA, mice lacking ADAM17 in SMCs (Adam17f/f/Sm22Cre/+) or ECs (Adam17f/f/Tie2Cre/+) showed decreased TAA dilation. ADAM17 deficiency in SMCs prevented the contractile-to-synthetic phenotypic switching, perivascular fibrosis, inflammation and adverse aortic remodeling. Loss of ADAM17 in ECs protected the intimal barrier integrity and preserved the adherens junction (VE-cadherin) and tight junctions (JAM-A and claudin). ADAM17 inhibitor PF-548 prevented TAA progression, supporting a role of ADAM17 in aortic SMC remodeling, impaired EC barrier integrity and TAA progression (M. Shen, Hu, Fedak, Oudit, & Kassiri, 2018).
ADAMTS1, 5, and 16 are increased in AAA, TAA and acute aortic dissection (Y. Gao, et al., 2016; Gunes, et al., 2016). Also, while ADAMTS1, 4, 5, 6, 8, 9, 10, 13 and 17 mRNA may show a decrease, ADAMTS1 protein levels are elevated in SMCs and macrophages of AAA (Vorkapic, et al., 2017). ADAMTS1 is also elevated in blood of acute aortic dissection patients. Aged AngII-infused mouse model of acute aortic dissection shows upregulation of ADAMTS1 in aortic media, macrophages and neutrophils, and increased degradation of versican (Y. Gao, et al., 2016). Other studies have shown decreased ADAMTS1 and elevated inducible NOS2 levels in the aorta of patients with Marfan syndrome. Adamts1-deficient mice show TAA and dissection and elevated aortic NO and NOS2 levels similar to mouse model of Marfan syndrome, and Nos2 inactivation protected both types of mice from aortic dilation and medial degeneration, suggesting a role for the ADAMTS1-NOS2 axis in TAA and dissection, and potential benefits of NOS2 inhibitors (Oller, et al., 2017).
ADAMTS2 gene variant rs11750568 has been associated with cerebral aneurysm and pediatric stroke (Arning, et al., 2012; Arning, et al., 2016). Also, ADAMTS4 is increased in aortic SMCs from patients with sporadic ascending TAA and dissection. In mice with sporadic aortic aneurysm and dissection induced by high-fat diet and AngII infusion, ADAMTS4 deficiency reduced aortic diameter, aneurysm formation, dissection and rupture. Also, Adamts4−/− aortas showed less elastic fiber destruction, versican degradation, macrophage infiltration and SMC apoptosis (P. Ren, et al., 2017).
ADAMTS5-mediated aggrecan cleavage is important during development of the aorta. Adamts5−/−Smad2+/− and Adamts5−/− mice exhibit aortic anomalies and accumulation of aggrecan at the onset of elastogenesis. Aggrecan accumulation could promote the generation of DIPEN and FFGVG aggrecan neoepitope by local MMPs. In support, Adamts5−/− aortas show aggrecan neo-DIPEN and neo-FFGVG fragments in the aortic adventitia, and increased aggrecan neo-FFGVG in the aortic media concomitant with SMC loss (Dupuis, et al., 2019). In AngII-induced aortic dilation model, mice lacking ADAMTS5 catalytic domain (Adamts5Δcat) show attenuated BP, increased ascending aorta dilation and versican, and decreased versican cleavage products (versikine) and LDL-related protein 1. Silencing LDL-related protein 1 expression in human aortic SMCs reduced ADAMTS5 expression and versikine production, suggesting a role of ADAMTS5 in TAA, partly through regulation of versican metabolism (Fava, et al., 2018).
ADAMTS7 degrades COMP, and ADAMTS7 mRNA expression is increased and COMP protein level is decreased in human aortic aneurysm (Qin, Cao, Li, Chen, & Chen, 2017). Whether the observed ADAMTS increased expression and its degradation of COMP are causally linked to atherosclerosis and aortic aneurysm need to be further tested.
While genetic ADAMTS13 variants rs2301612 and rs2285489 have been associated with the risk of cerebral aneurysm, ADAMTS12 variant rs1364044, and ADAMTS13 variants rs739469 and rs4962153 may be protective against cerebral aneurysm (Arning, et al., 2016).
9.11. ADAMs and ADAMTS in Venous Thromboembolism
Studies investigating the relationship between plasma levels of ADAMTS13 and vWF antigen and venous thromboembolism (VTE) showed that the median ADAMTS13 level was reduced and the mean vWF antigen level was elevated in VTE. Whether lower ADAMTS13 levels and higher vWF antigen levels cause VTE or are related to another pathology remain to be examined (Karakaya, Tombak, Serin, & Tiftik, 2016).
9.12. ADAMs and ADAMTS in Obesity and Diabetes-Related Vascular disease
Obesity and diabetes are major risks for CVD particularly in aging individuals. Obesity is associated with impaired endothelium-dependent dilation in coronary arterioles from aging patients undergoing open heart surgery and from aged mice. Transplantation of adipose tissue from aged obese mice increased serum TNFα levels, and impaired coronary artery dilation in young recipient mice. In aging patients and mice, obesity was associated with activation of endothelial ADAM17, partly due to its decreased inhibition by caveolin-1 resulting from reduced caveolin-1 expression with age. In bioassay cascade experiments, ADAM17-induced TNFα shedding in the upstream adipose tissue arteries of older obese patients reduced dilation in the downstream coronary arterioles. Thus, aging- and obesity-related reduction in caveolin-1 increases endothelial ADAM17 activity and TNFα release in adipose tissue, leading to coronary microvascular dysfunction (Dou, et al., 2017).
ADAM28 is upregulated in the liver of mice with metabolic syndrome. ADAM28 KO or its downregulation by siRNA is associated with reduced body weight and liver TNFα, and increased insulin sensitivity/glucose tolerance and high density lipoprotein cholesterol in diet-induced obese mouse, supporting a role of ADAM28 in metabolic syndrome and high fat-induced obesity (Herat, et al., 2017).
Increased prevalence of obesity has been linked to non-alcoholic steatohepatitis. Studies in mice on a high fat diet have suggested that ADAMTS5 may enhance visceral/gonadal adipose tissue expansion and promote liver pathology (Bauters, Scroyen, Deprez-Poulain, & Lijnen, 2016). In a high-fat diet induced obesity mouse model, ADAMTS5−/− mice showed lower liver weight and less steatohepatitis and fibrosis than wild-type mice, suggesting that ADAMTS5 deficiency preserves liver integrity in mice with diet-induced obesity, and that targeting ADAMTS5 could reduce obesity-related steatohepatitis (Bauters, Spincemaille, et al., 2016).
ADAMTS13 gene polymorphisms and the frequencies of QE genotype and E allele carrier of ADAMTS13 Q448E, AG genotype and G allele carrier of ADAMTS13 rs2073932, and AA genotype of ADAMTS13 rs652600 are higher in diabetic individuals than controls. E allele and AA genotype of Q448E and rs652600 are associated with multi-vessel coronary stenosis, and the E and G allele of Q448E and rs2073932 are associated with high CAD severity score, suggesting contribution of ADAMTS13 polymorphisms to atherosclerotic coronary stenosis in diabetes (Lasom, et al., 2018). WT mice with streptozotocin-induced diabetes show low plasma ADAMTS13 activity and high vWF levels. Adamts13−/− diabetic mice show impaired renal function, and increased albuminuria, urea, intrarenal thrombosis, plasminogen activator inhibitor-1, vWF, fibrin(ogen), and ECM deposition, and these effects are reversed by deletion of vWF, supporting that vWF/ADAMTS13 imbalance contributes to thrombotic angiopathy, intrarenal thrombosis and diabetic nephropathy (Dhanesha, et al., 2017).
Visceral adiposity poses greater risk than subcutaneous adiposity for diabetes and CVD. In mice, Adamts18 mRNA expression is abundant in visceral (gonadal) white adipose tissue during early development after birth. Adamts18−/− mice show increased body fat, larger adipocyte size in visceral white adipose tissue, early metabolic syndrome, hyperlipidemia and HTN, suggesting a role of ADAMTS18 in the regulation of visceral white adipose tissue and metabolic disorders (R. Zhu, et al., 2018).
10. Perspective
ADAMs play a role in the shedding of various membrane-bound proteins, and regulate numerous biological processes by cleaving different cytokines, growth factors and their receptions. ADAMs are also associated with different vascular processes, vascular malfunction and CVD. ADAM10 and 17 are promising biomarkers and therapeutic targets in HTN and atherosclerosis, ADAMTS family members also contribute to various biological processes and may be involved in vascular malfunction and CVD. Decreased levels of ADAMTS13 have been related to the TTP, and modulation of its activity could be useful in treatment of thrombotic microangiopathies. TIMPs are endogenous inhibitors of specific ADAMs and ADAMTS. Synthetic ADAM and ADAMTS inhibitors have also been developed, but with varying specificity and selectivity. Further research into the potential substrates, targets and functions of ADAMs and ADAMTS should help develop specific modulators with potential use in the management of CVD.
Acknowledgements
This work was supported by BRI Fund to Sustain Research Excellence from Brigham Research Institute, and grants from National Heart, Lung, and Blood Institute (HL111775, R56HL147889, and R01HL147889-A1). Dr. HaiFeng Yang was a visiting scholar from Department of Neurosurgery, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei, China. We thank Yael Day, Clara Frothingham, Annie Heuer, Jedidiah Kim, Cameron Muniz, and Vijay Sherring for their assistance in reviewing and editing the manuscript.
List of Abbreviations:
- aa
amino acid
- AAA
abdominal aortic aneurysm
- ACE
angiotensin-I converting enzyme
- ACE2
angiotensin-converting enzyme-2
- ADAM
A Disintegrin and Metalloproteinase
- ADAMTS
A Disintegrin and Metalloproteinase with Thrombospondin Motifs
- AngII
angiotensin II
- ApoE−/−
apolipoprotein E deficient
- APP
amyloid precursor protein
- CABG
coronary artery bypass graft
- CAD
coronary artery disease
- C-domain
cysteine-rich domain
- CHF
congestive heart failure
- CNS
central nervous system
- COMP
cartilage oligomeric matrix protein
- CUB
C1r/C1s Uegf Bmp1
- CVD
cardiovascular disease
- D-domain
disintegrin domain
- DOCA
deoxycorticosterone acetate
- Dll4
delta-like 4
- EC
endothelial cell
- E-cadherin
epithelial cadherin
- ECM
extracellular matrix
- EGF
epidermal growth factor
- EGFR
EGF Receptor
- ERK
extracellular signal-regulated protein kinase
- GPCR
G-protein coupled receptor
- GPIb
platelet glycoprotein Ib
- GWAS
genome-wide association studies
- HB-EGF
heparin-binding EGF-like growth factor
- HUVECs
human umbilical vein endothelial cells
- HTN
hypertension
- HVR
highly variable region
- ICAM-1
intercellular adhesion molecule-1
- IL-6
interleukin 6
- I/R
ischemia-reperfusion
- JAM-A
junctional adhesion molecule A
- KO
knockout
- M-domain
metalloproteinase domain
- LDL
low density lipoprotein
- LPS
lipopolysaccharide
- MAPK
mitogen activated protein kinase
- MCP-1
monocyte chemoattractant protein-1
- MDC
metalloproteinase/disintegrin/cysteine-rich
- MI
myocardial infarction
- MMP
matrix metalloproteinase
- NSTEMI
non-ST-segment elevation MI
- PAD
peripheral artery disease
- PAI-1
plasminogen activator inhibitor-1
- PE
preeclampsia
- PLAC
protease and lacunin
- RAGE
Receptors-for-Advanced-Glycation-End-products
- RGD
Arg-Gly-Asp
- rh
recombinant human
- SMC
smooth muscle cell
- SNP
single nucleotide polymorphism
- SVM
snake venom metalloproteinase
- STEMI
ST-segment elevation MI
- TAA
thoracic aortic aneurysm
- Tie-2
tyrosine-protein kinase receptor
- TIMP
tissue inhibitor of metalloproteinases
- TNFα
tumor necrosis factor-α
- TACE
TNFα-converting enzyme
- tPA
tissue plasminogen activator
- Tspan
tetraspanin
- TSP1
thrombospondin 1
- TSR
thrombospondin repeat
- TTP
thrombotic thrombocytopenic purpura
- uPA
urokinase plasminogen activator
- VCAM-1
vascular cell adhesion molecule-1
- VE-cadherin
vascular endothelial cadherin
- VEGF
vascular endothelial growth factor
- VEGFR
VEGF receptor
- VSM
vascular smooth muscle
- VSMC
VSM cell
- VTE
venous thromboembolism
- vWF
von Willebrand factor
- WT
wild-type
Footnotes
Conflict of Interest None
References
- Abbaszade I, Liu RQ, Yang F, Rosenfeld SA, Ross OH, Link JR, et al. (1999). Cloning and characterization of ADAMTS11, an aggrecanase from the ADAMTS family. J Biol Chem, 274(33), 23443–23450. [DOI] [PubMed] [Google Scholar]
- Abdul-Majeed S, Mell B, Nauli SM, & Joe B (2014). Cryptorchidism and infertility in rats with targeted disruption of the Adamts16 locus. PLoS One, 9(7), e100967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams JC, & Lawler J (2011). The thrombospondins. Cold Spring Harb Perspect Biol, 3(10), a009712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akyol O, Akyol S, & Chen CH (2016). Update on ADAMTS13 and VWF in cardiovascular and hematological disorders. Clin Chim Acta, 463, 109–118. [DOI] [PubMed] [Google Scholar]
- Al-Fakhri N, Wilhelm J, Hahn M, Heidt M, Hehrlein FW, Endisch AM, et al. (2003). Increased expression of disintegrin-metalloproteinases ADAM-15 and ADAM-9 following upregulation of integrins alpha5beta1 and alphavbeta3 in atherosclerosis. J Cell Biochem, 89(4), 808–823. [DOI] [PubMed] [Google Scholar]
- Alwan F, Vendramin C, Liesner R, Clark A, Lester W, Dutt T, et al. (2019). Characterization and treatment of congenital thrombotic thrombocytopenic purpura. Blood, 133(15), 1644–1651. [DOI] [PubMed] [Google Scholar]
- Amour A, Knight CG, English WR, Webster A, Slocombe PM, Knauper V, et al. (2002). The enzymatic activity of ADAM8 and ADAM9 is not regulated by TIMPs. FEBS Lett, 524(1–3), 154–158. [DOI] [PubMed] [Google Scholar]
- Anders A, Gilbert S, Garten W, Postina R, & Fahrenholz F (2001). Regulation of the alpha-secretase ADAM10 by its prodomain and proprotein convertases. FASEB J, 15(10), 1837–1839. [DOI] [PubMed] [Google Scholar]
- Anker SD, & Coats AJ (2002). How to RECOVER from RENAISSANCE? The significance of the results of RECOVER, RENAISSANCE, RENEWAL and ATTACH. Int J Cardiol, 86(2–3), 123–130. [DOI] [PubMed] [Google Scholar]
- Arning A, Hiersche M, Witten A, Kurlemann G, Kurnik K, Manner D, et al. (2012). A genome-wide association study identifies a gene network of ADAMTS genes in the predisposition to pediatric stroke. Blood, 120(26), 5231–5236. [DOI] [PubMed] [Google Scholar]
- Arning A, Jeibmann A, Kohnemann S, Brokinkel B, Ewelt C, Berger K, et al. (2016). ADAMTS genes and the risk of cerebral aneurysm. J Neurosurg, 125(2), 269–274. [DOI] [PubMed] [Google Scholar]
- Asakura M, Kitakaze M, Takashima S, Liao Y, Ishikura F, Yoshinaka T, et al. (2002). Cardiac hypertrophy is inhibited by antagonism of ADAM12 processing of HB-EGF: metalloproteinase inhibitors as a new therapy. Nat Med, 8(1), 35–40. [DOI] [PubMed] [Google Scholar]
- Ashlin TG, Kwan AP, & Ramji DP (2013). Regulation of ADAMTS-1, −4 and −5 expression in human macrophages: differential regulation by key cytokines implicated in atherosclerosis and novel synergism between TL1A and IL-17. Cytokine, 64(1), 234–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ataca D, Aouad P, Constantin C, Laszlo C, Beleut M, Shamseddin M, et al. (2020). The secreted protease Adamts18 links hormone action to activation of the mammary stem cell niche. Nat Commun, 11(1), 1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai XH, Wang DW, Luan Y, Yu XP, & Liu CJ (2009). Regulation of chondrocyte differentiation by ADAMTS-12 metalloproteinase depends on its enzymatic activity. Cell Mol Life Sci, 66(4), 667–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakken AM, Protack CD, Roztocil E, Nicholl SM, & Davies MG (2009). Cell migration in response to the amino-terminal fragment of urokinase requires epidermal growth factor receptor activation through an ADAM-mediated mechanism. J Vasc Surg, 49(5), 1296–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartoli CR, Kang J, Restle DJ, Zhang DM, Shabahang C, Acker MA, et al. (2015). Inhibition of ADAMTS-13 by Doxycycline Reduces von Willebrand Factor Degradation During Supraphysiological Shear Stress: Therapeutic Implications for Left Ventricular Assist Device-Associated Bleeding. JACC Heart Fail, 3(11), 860–869. [DOI] [PubMed] [Google Scholar]
- Bartoli CR, Restle DJ, Zhang DM, Acker MA, & Atluri P (2015). Pathologic von Willebrand factor degradation with a left ventricular assist device occurs via two distinct mechanisms: mechanical demolition and enzymatic cleavage. J Thorac Cardiovasc Surg, 149(1), 281–289. [DOI] [PubMed] [Google Scholar]
- Basile DP, Fredrich K, Chelladurai B, Leonard EC, & Parrish AR (2008). Renal ischemia reperfusion inhibits VEGF expression and induces ADAMTS-1, a novel VEGF inhibitor. Am J Physiol Renal Physiol, 294(4), F928–936. [DOI] [PubMed] [Google Scholar]
- Bauer RC, Tohyama J, Cui J, Cheng L, Yang J, Zhang X, et al. (2015). Knockout of Adamts7, a novel coronary artery disease locus in humans, reduces atherosclerosis in mice. Circulation, 131(13), 1202–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauters D, Scroyen I, Deprez-Poulain R, & Lijnen HR (2016). ADAMTS5 promotes murine adipogenesis and visceral adipose tissue expansion. Thromb Haemost, 116(4), 694–704. [DOI] [PubMed] [Google Scholar]
- Bauters D, Spincemaille P, Geys L, Cassiman D, Vermeersch P, Bedossa P, et al. (2016). ADAMTS5 deficiency protects against non-alcoholic steatohepatitis in obesity. Liver Int, 36(12), 1848–1859. [DOI] [PubMed] [Google Scholar]
- Bayoglu B, Arslan C, Tel C, Ulutin T, Dirican A, Deser SB, et al. (2018). Genetic variants rs1994016 and rs3825807 in ADAMTS7 affect its mRNA expression in atherosclerotic occlusive peripheral arterial disease. J Clin Lab Anal, 32(1):e22174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauvais D, Venditti L, Chassin O, Joly B, Ameri A, Boisseau P, et al. (2019). Inherited Thrombotic Thrombocytopenic Purpura Revealed by Recurrent Strokes in a Male Adult: Case Report and Literature Review. J Stroke Cerebrovasc Dis, 28(6), 1537–1539. [DOI] [PubMed] [Google Scholar]
- Becerra E, Scully MA, Leandro MJ, Heelas EO, Westwood JP, De La Torre I, et al. (2015). Effect of rituximab on B cell phenotype and serum B cell-activating factor levels in patients with thrombotic thrombocytopenic purpura. Clin Exp Immunol, 179(3), 414–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekhouche M, & Colige A (2015). The procollagen N-proteinases ADAMTS2, 3 and 14 in pathophysiology. Matrix Biol, 44–46, 46–53. [DOI] [PubMed] [Google Scholar]
- Bengtsson E, Hultman K, Duner P, Asciutto G, Almgren P, Orho-Melander M, et al. (2017). ADAMTS-7 is associated with a high-risk plaque phenotype in human atherosclerosis. Sci Rep, 7(1), 3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry E, Bosonea AM, Wang X, & Fernandez-Patron C (2013). Insights into the activity, differential expression, mutual regulation, and functions of matrix metalloproteinases and a disintegrin and metalloproteinases in hypertension and cardiac disease. J Vasc Res, 50(1), 52–68. [DOI] [PubMed] [Google Scholar]
- Bertram A, Lovric S, Engel A, Beese M, Wyss K, Hertel B, et al. (2015). Circulating ADAM17 Level Reflects Disease Activity in Proteinase-3 ANCA-Associated Vasculitis. J Am Soc Nephrol, 26(11), 2860–2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, et al. (1997). A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature, 385(6618), 729–733. [DOI] [PubMed] [Google Scholar]
- Blobel CP (1997). Metalloprotease-disintegrins: links to cell adhesion and cleavage of TNF alpha and Notch. Cell, 90(4), 589–592. [DOI] [PubMed] [Google Scholar]
- Blobel CP (2005). ADAMs: key components in EGFR signalling and development. Nat Rev Mol Cell Biol, 6(1), 32–43. [DOI] [PubMed] [Google Scholar]
- Blobel CP, & White JM (1992). Structure, function and evolutionary relationship of proteins containing a disintegrin domain. Curr Opin Cell Biol, 4(5), 760–765. [DOI] [PubMed] [Google Scholar]
- Bode W, Gomis-Ruth FX, & Stockler W (1993). Astacins, serralysins, snake venom and matrix metalloproteinases exhibit identical zinc-binding environments (HEXXHXXGXXH and Met-turn) and topologies and should be grouped into a common family, the ‘metzincins’. FEBS Lett, 331(1–2), 134–140. [DOI] [PubMed] [Google Scholar]
- Boerboom D, Lafond JF, Zheng X, Lapointe E, Mittaz L, Boyer A, et al. (2011). Partially redundant functions of Adamts1 and Adamts4 in the perinatal development of the renal medulla. Dev Dyn, 240(7), 1806–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brew K, & Nagase H (2010). The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim Biophys Acta, 1803(1), 55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouillard P, Dupont L, Helaers R, Coulie R, Tiller GE, Peeden J, et al. (2017). Loss of ADAMTS3 activity causes Hennekam lymphangiectasia-lymphedema syndrome 3. Hum Mol Genet, 26(21), 4095–4104. [DOI] [PubMed] [Google Scholar]
- Bustamante A, Ning M, Garcia-Berrocoso T, Penalba A, Boada C, Simats A, et al. (2018). Usefulness of ADAMTS13 to predict response to recanalization therapies in acute ischemic stroke. Neurology, 90(12), e995–e1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai P, Luo H, Xu H, Zhu X, Xu W, Dai Y, et al. (2015). Recombinant ADAMTS 13 Attenuates Brain Injury After Intracerebral Hemorrhage. Stroke, 46(9), 2647–2653. [DOI] [PubMed] [Google Scholar]
- Cal S, Obaya AJ, Llamazares M, Garabaya C, Quesada V, & Lopez-Otin C (2002). Cloning, expression analysis, and structural characterization of seven novel human ADAMTSs, a family of metalloproteinases with disintegrin and thrombospondin-1 domains. Gene, 283(1–2), 49–62. [DOI] [PubMed] [Google Scholar]
- Canault M, Certel K, Schatzberg D, Wagner DD, & Hynes RO (2010). The lack of ADAM17 activity during embryonic development causes hemorrhage and impairs vessel formation. PLoS One, 5(10), e13433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Kang Q, Zhao Z, & Zolkiewska A (2002). Intracellular processing of metalloprotease disintegrin ADAM12. J Biol Chem, 277(29), 26403–26411. [DOI] [PubMed] [Google Scholar]
- Cao Y, Xu H, Zhu Y, Shi MJ, Wei L, Zhang J, et al. (2019). ADAMTS13 maintains cerebrovascular integrity to ameliorate Alzheimer-like pathology. PLoS Biol, 17(6), e3000313. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Caolo V, Swennen G, Chalaris A, Wagenaar A, Verbruggen S, Rose-John S, et al. (2015). ADAM10 and ADAM17 have opposite roles during sprouting angiogenesis. Angiogenesis, 18(1), 13–22. [DOI] [PubMed] [Google Scholar]
- Chalaris A, Adam N, Sina C, Rosenstiel P, Lehmann-Koch J, Schirmacher P, et al. (2010). Critical role of the disintegrin metalloprotease ADAM17 for intestinal inflammation and regeneration in mice. J Exp Med, 207(8), 1617–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charrier-Hisamuddin L, Laboisse CL, & Merlin D (2008). ADAM-15: a metalloprotease that mediates inflammation. FASEB J, 22(3), 641–653. [DOI] [PubMed] [Google Scholar]
- Chemaly M, McGilligan V, Gibson M, Clauss M, Watterson S, Alexander HD, et al. (2017). Role of tumour necrosis factor alpha converting enzyme (TACE/ADAM17) and associated proteins in coronary artery disease and cardiac events. Arch Cardiovasc Dis, 110(12), 700–711. [DOI] [PubMed] [Google Scholar]
- Chen J, Deng L, Dreymuller D, Jiang X, Long J, Duan Y, et al. (2016). A novel peptide ADAM8 inhibitor attenuates bronchial hyperresponsiveness and Th2 cytokine mediated inflammation of murine asthmatic models. Sci Rep, 6, 30451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Zhi Y, Chang X, Zhang S, & Dai D (2013). Expression of ADAMTS1 and its correlation with angiogenesis in primary gastric cancer and lymph node metastasis. Dig Dis Sci, 58(2), 405–413. [DOI] [PubMed] [Google Scholar]
- Chen L, Yang L, Zha Y, & Cui L (2011). Association of serum a disintegrin and metalloproteinase with thrombospodin motif 4 levels with the presence and severity of coronary artery disease. Coron Artery Dis, 22(8), 570–576. [DOI] [PubMed] [Google Scholar]
- Chen X, Cheng X, Zhang S, & Wu D (2019). ADAMTS13: An Emerging Target in Stroke Therapy. Front Neurol, 10, 772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YC, Bui AV, Diesch J, Manasseh R, Hausding C, Rivera J, et al. (2013). A novel mouse model of atherosclerotic plaque instability for drug testing and mechanistic/therapeutic discoveries using gene and microRNA expression profiling. Circ Res, 113(3), 252–265. [DOI] [PubMed] [Google Scholar]
- Chesneau V, Becherer JD, Zheng Y, Erdjument-Bromage H, Tempst P, & Blobel CP (2003). Catalytic properties of ADAM19. J Biol Chem, 278(25), 22331–22340. [DOI] [PubMed] [Google Scholar]
- Cho C (2012). Testicular and epididymal ADAMs: expression and function during fertilization. Nat Rev Urol, 9(10), 550–560. [DOI] [PubMed] [Google Scholar]
- Chung YL, Pan CH, Wang CC, Hsu KC, Sheu MJ, Chen HF, et al. (2016). Methyl Protodioscin, a Steroidal Saponin, Inhibits Neointima Formation in Vitro and in Vivo. J Nat Prod, 79(6), 1635–1644. [DOI] [PubMed] [Google Scholar]
- Coletta AP, Clark AL, Banarjee P, & Cleland JG (2002). Clinical trials update: RENEWAL (RENAISSANCE and RECOVER) and ATTACH. Eur J Heart Fail, 4(4), 559–561. [DOI] [PubMed] [Google Scholar]
- Colige A, Beschin A, Samyn B, Goebels Y, Van Beeumen J, Nusgens BV, et al. (1995). Characterization and partial amino acid sequencing of a 107-kDa procollagen I N-proteinase purified by affinity chromatography on immobilized type XIV collagen. J Biol Chem, 270(28), 16724–16730. [DOI] [PubMed] [Google Scholar]
- Colige A, Li SW, Sieron AL, Nusgens BV, Prockop DJ, & Lapiere CM (1997). cDNA cloning and expression of bovine procollagen I N-proteinase: a new member of the superfamily of zinc-metalloproteinases with binding sites for cells and other matrix components. Proc Natl Acad Sci U S A, 94(6), 2374–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colige A, Ruggiero F, Vandenberghe I, Dubail J, Kesteloot F, Van Beeumen J, et al. (2005). Domains and maturation processes that regulate the activity of ADAMTS-2, a metalloproteinase cleaving the aminopropeptide of fibrillar procollagens types I-III and V. J Biol Chem, 280(41), 34397–34408. [DOI] [PubMed] [Google Scholar]
- Cong L, & Jia J (2011). Promoter polymorphisms which regulate ADAM9 transcription are protective against sporadic Alzheimer’s disease. Neurobiol Aging, 32(1), 54–62. [DOI] [PubMed] [Google Scholar]
- Crawley JT, Lane DA, Woodward M, Rumley A, & Lowe GD (2008). Evidence that high von Willebrand factor and low ADAMTS-13 levels independently increase the risk of a non-fatal heart attack. J Thromb Haemost, 6(4), 583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawley JT, & Scully MA (2013). Thrombotic thrombocytopenic purpura: basic pathophysiology and therapeutic strategies. Hematology Am Soc Hematol Educ Program, 2013, 292–299. [DOI] [PubMed] [Google Scholar]
- Cross AK, Haddock G, Stock CJ, Allan S, Surr J, Bunning RA, et al. (2006). ADAMTS-1 and −4 are up-regulated following transient middle cerebral artery occlusion in the rat and their expression is modulated by TNF in cultured astrocytes. Brain Res, 1088(1), 19–30. [DOI] [PubMed] [Google Scholar]
- Dancevic CM, Fraser FW, Smith AD, Stupka N, Ward AC, & McCulloch DR (2013). Biosynthesis and expression of a disintegrin-like and metalloproteinase domain with thrombospondin-1 repeats-15: a novel versican-cleaving proteoglycanase. J Biol Chem, 288(52), 37267–37276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang S, Bu D, Lu T, Wang Z, Liu J, & Zhang W (2018). Adamts18 deficiency increases arterial thrombus formation associated with vascular defects in mice. Biochem Biophys Res Commun, 496(4), 1362–1368. [DOI] [PubMed] [Google Scholar]
- Dang S, Hong T, Bu D, Tang J, Fan J, & Zhang W (2012). Optimized refolding and characterization of active C-terminal ADAMTS-18 fragment from inclusion bodies of Escherichia coli. Protein Expr Purif, 82(1), 32–36. [DOI] [PubMed] [Google Scholar]
- de Groot R, Bardhan A, Ramroop N, Lane DA, & Crawley JT (2009). Essential role of the disintegrin-like domain in ADAMTS13 function. Blood, 113(22), 5609–5616. [DOI] [PubMed] [Google Scholar]
- de Queiroz TM, Xia H, Filipeanu CM, Braga VA, & Lazartigues E (2015). alpha-Lipoic acid reduces neurogenic hypertension by blunting oxidative stress-mediated increase in ADAM17. Am J Physiol Heart Circ Physiol, 309(5), H926–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desanlis I, Felstead HL, Edwards DR, & Wheeler GN (2018). ADAMTS9, a member of the ADAMTS family, in Xenopus development. Gene Expr Patterns, 29, 72–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deurloo KL, Linskens IH, Heymans MW, Heijboer AC, Blankenstein MA, & van Vugt JM (2013). ADAM12s and PP13 as first trimester screening markers for adverse pregnancy outcome. Clin Chem Lab Med, 51(6), 1279–1284. [DOI] [PubMed] [Google Scholar]
- Dhanesha N, Doddapattar P, Chorawala MR, Nayak MK, Kokame K, Staber JM, et al. (2017). ADAMTS13 Retards Progression of Diabetic Nephropathy by Inhibiting Intrarenal Thrombosis in Mice. Arterioscler Thromb Vasc Biol, 37(7), 1332–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didangelos A, Mayr U, Monaco C, & Mayr M (2012). Novel role of ADAMTS-5 protein in proteoglycan turnover and lipoprotein retention in atherosclerosis. J Biol Chem, 287(23), 19341–19345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebel ME, Martin JV, Liberati DM, & Diebel LN (2018). The temporal response and mechanism of action of tranexamic acid in endothelial glycocalyx degradation. J Trauma Acute Care Surg, 84(1), 75–80. [DOI] [PubMed] [Google Scholar]
- Doddapattar P, Dhanesha N, Chorawala MR, Tinsman C, Jain M, Nayak MK, et al. (2018). Endothelial Cell-Derived Von Willebrand Factor, But Not Platelet-Derived, Promotes Atherosclerosis in Apolipoprotein E-Deficient Mice. Arterioscler Thromb Vasc Biol, 38(3), 520–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokun AO, Chen L, Okutsu M, Farber CR, Hazarika S, Jones WS, et al. (2015). ADAM12: a genetic modifier of preclinical peripheral arterial disease. Am J Physiol Heart Circ Physiol, 309(5), H790–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolan JM, Sim FJ, Meng H, & Kolega J (2012). Endothelial cells express a unique transcriptional profile under very high wall shear stress known to induce expansive arterial remodeling. Am J Physiol Cell Physiol, 302(8), C1109–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Du T, Premaratne S, Zhao CX, Tian Q, Li Y, et al. (2018). Relationship between ADAMTS4 and carotid atherosclerotic plaque vulnerability in humans. J Vasc Surg, 67(4), 1120–1126. [DOI] [PubMed] [Google Scholar]
- Donnarumma E, Trivedi RK, & Lefer DJ (2017). Protective Actions of H2S in Acute Myocardial Infarction and Heart Failure. Compr Physiol, 7(2), 583–602. [DOI] [PubMed] [Google Scholar]
- Donners MM, Wolfs IM, Olieslagers S, Mohammadi-Motahhari Z, Tchaikovski V, Heeneman S, et al. (2010). A disintegrin and metalloprotease 10 is a novel mediator of vascular endothelial growth factor-induced endothelial cell function in angiogenesis and is associated with atherosclerosis. Arterioscler Thromb Vasc Biol, 30(11), 2188–2195. [DOI] [PubMed] [Google Scholar]
- Dou H, Feher A, Davila AC, Romero MJ, Patel VS, Kamath VM, et al. (2017). Role of Adipose Tissue Endothelial ADAM17 in Age-Related Coronary Microvascular Dysfunction. Arterioscler Thromb Vasc Biol, 37(6), 1180–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubail J, & Apte SS (2015). Insights on ADAMTS proteases and ADAMTS-like proteins from mammalian genetics. Matrix Biol, 44–46, 24–37. [DOI] [PubMed] [Google Scholar]
- Dubail J, Kesteloot F, Deroanne C, Motte P, Lambert V, Rakic JM, et al. (2010). ADAMTS-2 functions as anti-angiogenic and anti-tumoral molecule independently of its catalytic activity. Cell Mol Life Sci, 67(24), 4213–4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuis LE, Nelson EL, Hozik B, Porto SC, Rogers-DeCotes A, Fosang A, et al. (2019). Adamts5(−/−) Mice Exhibit Altered Aggrecan Proteolytic Profiles That Correlate With Ascending Aortic Anomalies. Arterioscler Thromb Vasc Biol, 39(10), 2067–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duru EA, Fu Y, & Davies MG (2014). Protease-mediated human smooth muscle cell proliferation by urokinase requires epidermal growth factor receptor transactivation by triple membrane signaling. J Surg Res, 192(2), 254–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards DR, Handsley MM, & Pennington CJ (2008). The ADAM metalloproteinases. Mol Aspects Med, 29(5), 258–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eerenberg ES, Teunissen PF, van den Born BJ, Meijers JC, Hollander MR, Jansen M, et al. (2016). The role of ADAMTS13 in acute myocardial infarction: cause or consequence? Cardiovasc Res, 111(3), 194–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Hour M, Moncada-Pazos A, Blacher S, Masset A, Cal S, Berndt S, et al. (2010). Higher sensitivity of Adamts12-deficient mice to tumor growth and angiogenesis. Oncogene, 29(20), 3025–3032. [DOI] [PubMed] [Google Scholar]
- English WR, Corvol P, & Murphy G (2012). LPS activates ADAM9 dependent shedding of ACE from endothelial cells. Biochem Biophys Res Commun, 421(1), 70–75. [DOI] [PubMed] [Google Scholar]
- English WR, Puente XS, Freije JM, Knauper V, Amour A, Merryweather A, et al. (2000). Membrane type 4 matrix metalloproteinase (MMP17) has tumor necrosis factor-alpha convertase activity but does not activate pro-MMP2. J Biol Chem, 275(19), 14046–14055. [DOI] [PubMed] [Google Scholar]
- English WR, Siviter RJ, Hansen M, & Murphy G (2017). ADAM9 is present at endothelial cell - cell junctions and regulates monocyte - endothelial transmigration. Biochem Biophys Res Commun, 493(2), 1057–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan D, Takawale A, Shen M, Samokhvalov V, Basu R, Patel V, et al. (2016). A Disintegrin and Metalloprotease-17 Regulates Pressure Overload-Induced Myocardial Hypertrophy and Dysfunction Through Proteolytic Processing of Integrin beta1. Hypertension, 68(4), 937–948. [DOI] [PubMed] [Google Scholar]
- Fan D, Takawale A, Shen M, Wang W, Wang X, Basu R, et al. (2015). Cardiomyocyte A Disintegrin And Metalloproteinase 17 (ADAM17) Is Essential in Post-Myocardial Infarction Repair by Regulating Angiogenesis. Circ Heart Fail, 8(5), 970–979. [DOI] [PubMed] [Google Scholar]
- Farber G, Hurtado R, Loh S, Monette S, Mtui J, Kopan R, et al. (2018). Glomerular endothelial cell maturation depends on ADAM10, a key regulator of Notch signaling. Angiogenesis, 21(2), 335–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farber G, Parks MM, Lustgarten Guahmich N, Zhang Y, Monette S, Blanchard SC, et al. (2019). ADAM10 controls the differentiation of the coronary arterial endothelium. Angiogenesis, 22(2), 237–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fava M, Barallobre-Barreiro J, Mayr U, Lu R, Didangelos A, Baig F, et al. (2018). Role of ADAMTS-5 in Aortic Dilatation and Extracellular Matrix Remodeling. Arterioscler Thromb Vasc Biol, 38(7), 1537–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedak PW, Moravec CS, McCarthy PM, Altamentova SM, Wong AP, Skrtic M, et al. (2006). Altered expression of disintegrin metalloproteinases and their inhibitor in human dilated cardiomyopathy. Circulation, 113(2), 238–245. [DOI] [PubMed] [Google Scholar]
- Fernandes RJ, Hirohata S, Engle JM, Colige A, Cohn DH, Eyre DR, et al. (2001). Procollagen II amino propeptide processing by ADAMTS-3. Insights on dermatosparaxis. J Biol Chem, 276(34), 31502–31509. [DOI] [PubMed] [Google Scholar]
- Folkesson M, Li C, Frebelius S, Swedenborg J, Wagsater D, Williams KJ, et al. (2015). Proteolytically active ADAM10 and ADAM17 carried on membrane microvesicles in human abdominal aortic aneurysms. Thromb Haemost, 114(6), 1165–1174. [DOI] [PubMed] [Google Scholar]
- Fridman JS, Caulder E, Hansbury M, Liu X, Yang G, Wang Q, et al. (2007). Selective inhibition of ADAM metalloproteases as a novel approach for modulating ErbB pathways in cancer. Clin Cancer Res, 13(6), 1892–1902. [DOI] [PubMed] [Google Scholar]
- Fu Y, & Kong W (2017). Cartilage Oligomeric Matrix Protein: Matricellular and Matricrine Signaling in Cardiovascular Homeostasis and Disease. Curr Vasc Pharmacol, 15(3), 186–196. [DOI] [PubMed] [Google Scholar]
- Fujioka M, Nakano T, Hayakawa K, Irie K, Akitake Y, Sakamoto Y, et al. (2012). ADAMTS13 gene deletion enhances plasma high-mobility group box1 elevation and neuroinflammation in brain ischemia-reperfusion injury. Neurol Sci, 33(5), 1107–1115. [DOI] [PubMed] [Google Scholar]
- Gao Y, Wu W, Yu C, Zhong F, Li G, Kong W, et al. (2016). A disintegrin and metalloproteinase with thrombospondin motif 1 (ADAMTS1) expression increases in acute aortic dissection. Sci China Life Sci, 59(1), 59–67. [DOI] [PubMed] [Google Scholar]
- Gao YX, Yu CA, Lu JH, Gao HM, Li G, Kong W, et al. (2013). ADAMTS-7 expression increases in the early stage of angiotensin II-induced renal injury in elderly mice. Kidney Blood Press Res, 38(1), 121–131. [DOI] [PubMed] [Google Scholar]
- Garbers C, Monhasery N, Aparicio-Siegmund S, Lokau J, Baran P, Nowell MA, et al. (2014). The interleukin-6 receptor Asp358Ala single nucleotide polymorphism rs2228145 confers increased proteolytic conversion rates by ADAM proteases. Biochim Biophys Acta, 1842(9), 1485–1494. [DOI] [PubMed] [Google Scholar]
- Georgiadis KE, Hirohata S, Seldin MF, & Apte SS (1999). ADAM-TS8, a novel metalloprotease of the ADAM-TS family located on mouse chromosome 9 and human chromosome 11. Genomics, 62(2), 312–315. [DOI] [PubMed] [Google Scholar]
- Glassey B, & Civetta A (2004). Positive selection at reproductive ADAM genes with potential intercellular binding activity. Mol Biol Evol, 21(5), 851–859. [DOI] [PubMed] [Google Scholar]
- Goel A, Raghupathy V, Amirtharaj GJ, Chapla A, Venkatraman A, Ramakrishna B, et al. (2017). ADAMTS13 missense variants associated with defective activity and secretion of ADAMTS13 in a patient with non-cirrhotic portal hypertension. Indian J Gastroenterol, 36(5), 380–389. [DOI] [PubMed] [Google Scholar]
- Goertsches R, Comabella M, Navarro A, Perkal H, & Montalban X (2005). Genetic association between polymorphisms in the ADAMTS14 gene and multiple sclerosis. J Neuroimmunol, 164(1–2), 140–147. [DOI] [PubMed] [Google Scholar]
- Goetzinger KR, Zhong Y, Cahill AG, Odibo L, Macones GA, & Odibo AO (2013). Efficiency of first-trimester uterine artery Doppler, a-disintegrin and metalloprotease 12, pregnancy-associated plasma protein a, and maternal characteristics in the prediction of preeclampsia. J Ultrasound Med, 32(9), 1593–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalakrishnan K, Kumarasamy S, Abdul-Majeed S, Kalinoski AL, Morgan EE, Gohara AF, et al. (2012). Targeted disruption of Adamts16 gene in a rat genetic model of hypertension. Proc Natl Acad Sci U S A, 109(50), 20555–20559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould RJ, Polokoff MA, Friedman PA, Huang TF, Holt JC, Cook JJ, et al. (1990). Disintegrins: a family of integrin inhibitory proteins from viper venoms. Proc Soc Exp Biol Med, 195(2), 168–171. [DOI] [PubMed] [Google Scholar]
- Green D, Tian L, Greenland P, Liu K, Kibbe M, Tracy R, et al. (2017). Association of the von Willebrand Factor-ADAMTS13 Ratio With Incident Cardiovascular Events in Patients With Peripheral Arterial Disease. Clin Appl Thromb Hemost, 23(7), 807–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunes MF, Akpinar MB, Comertoglu I, Akyol S, Demircelik B, Gurel OM, et al. (2016). The Investigation of a Disintegrin and Metalloproteinase with ThromboSpondin Motifs (ADAMTS) 1, 5 and 16 in Thoracic Aortic Aneurysms and Dissections. Clin Lab, 62(3), 425–433. [PubMed] [Google Scholar]
- Gunther W, Skaftnesmo KO, Arnold H, Bjerkvig R, & Terzis AJ (2005). Distribution patterns of the anti-angiogenic protein ADAMTS-1 during rat development. Acta Histochem, 107(2), 121–131. [DOI] [PubMed] [Google Scholar]
- Guo F, Lai Y, Tian Q, Lin EA, Kong L, & Liu C (2010). Granulin-epithelin precursor binds directly to ADAMTS-7 and ADAMTS-12 and inhibits their degradation of cartilage oligomeric matrix protein. Arthritis Rheum, 62(7), 2023–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haining EJ, Yang J, Bailey RL, Khan K, Collier R, Tsai S, et al. (2012). The TspanC8 subgroup of tetraspanins interacts with A disintegrin and metalloprotease 10 (ADAM10) and regulates its maturation and cell surface expression. J Biol Chem, 287(47), 39753–39765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ham C, Levkau B, Raines EW, & Herren B (2002). ADAM15 is an adherens junction molecule whose surface expression can be driven by VE-cadherin. Exp Cell Res, 279(2), 239–247. [DOI] [PubMed] [Google Scholar]
- Hanby HA, & Zheng XL (2013). Biochemistry and physiological functions of ADAMTS7 metalloprotease. Adv Biochem, 1(3): 10.11648/j.ab.20130103.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann D, de Strooper B, Serneels L, Craessaerts K, Herreman A, Annaert W, et al. (2002). The disintegrin/metalloprotease ADAM 10 is essential for Notch signalling but not for alpha-secretase activity in fibroblasts. Hum Mol Genet, 11(21), 2615–2624. [DOI] [PubMed] [Google Scholar]
- Hashimoto G, Aoki T, Nakamura H, Tanzawa K, & Okada Y (2001). Inhibition of ADAMTS4 (aggrecanase-1) by tissue inhibitors of metalloproteinases (TIMP-1, 2, 3 and 4). FEBS Lett, 494(3), 192–195. [DOI] [PubMed] [Google Scholar]
- Hattori N, Carrino DA, Lauer ME, Vasanji A, Wylie JD, Nelson CM, et al. (2011). Pericellular versican regulates the fibroblast-myofibroblast transition: a role for ADAMTS5 protease-mediated proteolysis. J Biol Chem, 286(39), 34298–34310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heijink IH, Brandenburg SM, Noordhoek JA, Slebos DJ, Postma DS, & van Oosterhout AJ (2011). Role of aberrant metalloproteinase activity in the pro-inflammatory phenotype of bronchial epithelium in COPD. Respir Res, 12, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmeryckx B, Carai P, & Roger Lijnen H (2019). ADAMTS5 deficiency in mice does not affect cardiac function. Cell Biol Int, 43(6), 593–604. [DOI] [PubMed] [Google Scholar]
- Herat L, Rudnicka C, Okada Y, Mochizuki S, Schlaich M, & Matthews V (2017). The Metalloproteinase ADAM28 Promotes Metabolic Dysfunction in Mice. Int J Mol Sci, 18(4):884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashiyama S, & Nanba D (2005). ADAM-mediated ectodomain shedding of HB-EGF in receptor cross-talk. Biochim Biophys Acta, 1751(1), 110–117. [DOI] [PubMed] [Google Scholar]
- Hirohata S, Inagaki J, & Ohtsuki T (2017). Diverse Functions of a Disintegrin and Metalloproteinase with Thrombospondin Motif-1. Yakugaku Zasshi, 137(7), 811–814. [DOI] [PubMed] [Google Scholar]
- Hodgkinson LM, Wang L, Duncan G, Edwards DR, & Wormstone IM (2010). ADAM and ADAMTS gene expression in native and wound healing human lens epithelial cells. Mol Vis, 16, 2765–2776. [PMC free article] [PubMed] [Google Scholar]
- Holloway JW, Laxton RC, Rose-Zerilli MJ, Holloway JA, Andrews AL, Riaz Z, et al. (2010). ADAM33 expression in atherosclerotic lesions and relationship of ADAM33 gene variation with atherosclerosis. Atherosclerosis, 211(1), 224–230. [DOI] [PubMed] [Google Scholar]
- Horiuchi K, Weskamp G, Lum L, Hammes HP, Cai H, Brodie TA, et al. (2003). Potential role for ADAM15 in pathological neovascularization in mice. Mol Cell Biol, 23(16), 5614–5624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi K, Zhou HM, Kelly K, Manova K, & Blobel CP (2005). Evaluation of the contributions of ADAMs 9, 12, 15, 17, and 19 to heart development and ectodomain shedding of neuregulins beta1 and beta2. Dev Biol, 283(2), 459–471. [DOI] [PubMed] [Google Scholar]
- Hou Y, Chu M, Cai Y, Lei J, Chen Y, Zhu R, et al. (2015). Antitumor and anti-angiogenic activity of the recombinant human disintegrin domain of A disintegrin and metalloproteinase 15. Mol Med Rep, 12(2), 2360–2366. [DOI] [PubMed] [Google Scholar]
- Hou Y, Chu M, Du FF, Lei JY, Chen Y, Zhu RY, et al. (2013). Recombinant disintegrin domain of ADAM15 inhibits the proliferation and migration of Bel-7402 cells. Biochem Biophys Res Commun, 435(4), 640–645. [DOI] [PubMed] [Google Scholar]
- Howard L, & Glynn P (1995). Membrane-associated metalloproteinase recognized by characteristic cleavage of myelin basic protein: assay and isolation. Methods Enzymol, 248, 388–395. [DOI] [PubMed] [Google Scholar]
- Howes JM, Knauper V, Malcor JD, & Farndale RW (2020). Cleavage by MMP-13 renders VWF unable to bind to collagen but increases its platelet reactivity. J Thromb Haemost, 18(4), 942–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsia HE, Tushaus J, Brummer T, Zheng Y, Scilabra SD, & Lichtenthaler SF (2019). Functions of ‘A disintegrin and metalloproteases (ADAMs)’ in the mammalian nervous system. Cell Mol Life Sci, 76(16), 3055–3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubmacher D, & Apte SS (2015). ADAMTS proteins as modulators of microfibril formation and function. Matrix Biol, 47, 34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurskainen TL, Hirohata S, Seldin MF, & Apte SS (1999). ADAM-TS5, ADAM-TS6, and ADAM-TS7, novel members of a new family of zinc metalloproteases. General features and genomic distribution of the ADAM-TS family. J Biol Chem, 274(36), 25555–25563. [DOI] [PubMed] [Google Scholar]
- Iba K, Albrechtsen R, Gilpin B, Frohlich C, Loechel F, Zolkiewska A, et al. (2000). The cysteine-rich domain of human ADAM 12 supports cell adhesion through syndecans and triggers signaling events that lead to beta1 integrin-dependent cell spreading. J Cell Biol, 149(5), 1143–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa Y, Miura T, Nakano A, Miki T, Nakamura Y, Tsuchihashi K, et al. (2004). The role of ADAM protease in the tyrosine kinase-mediated trigger mechanism of ischemic preconditioning. Cardiovasc Res, 62(1), 167–175. [DOI] [PubMed] [Google Scholar]
- Jackson LF, Qiu TH, Sunnarborg SW, Chang A, Zhang C, Patterson C, et al. (2003). Defective valvulogenesis in HB-EGF and TACE-null mice is associated with aberrant BMP signaling. Embo j, 22(11), 2704–2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsson L, Franco CA, Bentley K, Collins RT, Ponsioen B, Aspalter IM, et al. (2010). Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat Cell Biol, 12(10), 943–953. [DOI] [PubMed] [Google Scholar]
- Janssen L, Dupont L, Bekhouche M, Noel A, Leduc C, Voz M, et al. (2016). ADAMTS3 activity is mandatory for embryonic lymphangiogenesis and regulates placental angiogenesis. Angiogenesis, 19(1), 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeltsch M, Jha SK, Tvorogov D, Anisimov A, Leppanen VM, Holopainen T, et al. (2014). CCBE1 enhances lymphangiogenesis via A disintegrin and metalloprotease with thrombospondin motifs-3-mediated vascular endothelial growth factor-C activation. Circulation, 129(19), 1962–1971. [DOI] [PubMed] [Google Scholar]
- Jha SK, Rauniyar K, Karpanen T, Leppanen VM, Brouillard P, Vikkula M, et al. (2017). Efficient activation of the lymphangiogenic growth factor VEGF-C requires the C-terminal domain of VEGF-C and the N-terminal domain of CCBE1. Sci Rep, 7(1), 4916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SY, Tohyama J, Bauer RC, Cao NN, Rader DJ, & Zheng XL (2012). Genetic ablation of Adamts13 gene dramatically accelerates the formation of early atherosclerosis in a murine model. Arterioscler Thromb Vasc Biol, 32(8), 1817–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Liu Y, Lin Q, Li J, Druso JE, Antonyak MA, et al. (2013). Deletion of Cdc42 enhances ADAM17-mediated vascular endothelial growth factor receptor 2 shedding and impairs vascular endothelial cell survival and vasculogenesis. Mol Cell Biol, 33(21), 4181–4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joe B, Saad Y, Dhindaw S, Lee NH, Frank BC, Achinike OH, et al. (2009). Positional identification of variants of Adamts16 linked to inherited hypertension. Hum Mol Genet, 18(15), 2825–2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson-Rylander AC, Nilsson T, Fritsche-Danielson R, Hammarstrom A, Behrendt M, Andersson JO, et al. (2005). Role of ADAMTS-1 in atherosclerosis: remodeling of carotid artery, immunohistochemistry, and proteolysis of versican. Arterioscler Thromb Vasc Biol, 25(1), 180–185. [DOI] [PubMed] [Google Scholar]
- Jorissen E, Prox J, Bernreuther C, Weber S, Schwanbeck R, Serneels L, et al. (2010). The disintegrin/metalloproteinase ADAM10 is essential for the establishment of the brain cortex. J Neurosci, 30(14), 4833–4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakaya B, Tombak A, Serin MS, & Tiftik N (2016). Change in plasma a disintegrin and metalloprotease with thrombospondin type-1 repeats-13 and von Willebrand factor levels in venous thromboembolic patients. Hematology, 21(5), 295–299. [DOI] [PubMed] [Google Scholar]
- Karakose M, Caliskan M, Arslan MS, Demirci T, Karakose S, Tutal E, et al. (2018). Association of ADAMTS4 and ADAMTS9 levels with cardiovascular risk in patients with primary hyperparathyroidism. Endocr Res, 43(1), 15–20. [DOI] [PubMed] [Google Scholar]
- Karkkainen I, Rybnikova E, Pelto-Huikko M, & Huovila AP (2000). Metalloprotease-disintegrin (ADAM) genes are widely and differentially expressed in the adult CNS. Mol Cell Neurosci, 15(6), 547–560. [DOI] [PubMed] [Google Scholar]
- Karoulias SZ, Taye N, Stanley S, & Hubmacher D (2020). The ADAMTS/Fibrillin Connection: Insights into the Biological Functions of ADAMTS10 and ADAMTS17 and Their Respective Sister Proteases. Biomolecules, 10(4):596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiwagi M, Tortorella M, Nagase H, & Brew K (2001). TIMP-3 is a potent inhibitor of aggrecanase 1 (ADAM-TS4) and aggrecanase 2 (ADAM-TS5). J Biol Chem, 276(16), 12501–12504. [DOI] [PubMed] [Google Scholar]
- Katneni UK, Ibla JC, Hunt R, Schiller T, & Kimchi-Sarfaty C (2019). von Willebrand factor/ADAMTS-13 interactions at birth: implications for thrombosis in the neonatal period. J Thromb Haemost, 17(3), 429–440. [DOI] [PubMed] [Google Scholar]
- Kawai T, Takayanagi T, Forrester SJ, Preston KJ, Obama T, Tsuji T, et al. (2017). Vascular ADAM17 (a Disintegrin and Metalloproteinase Domain 17) Is Required for Angiotensin II/beta-Aminopropionitrile-Induced Abdominal Aortic Aneurysm. Hypertension, 70(5), 959–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly K, Hutchinson G, Nebenius-Oosthuizen D, Smith AJ, Bartsch JW, Horiuchi K, et al. (2005). Metalloprotease-disintegrin ADAM8: expression analysis and targeted deletion in mice. Dev Dyn, 232(1), 221–231. [DOI] [PubMed] [Google Scholar]
- Kelwick R, Desanlis I, Wheeler GN, & Edwards DR (2015). The ADAMTS (A Disintegrin and Metalloproteinase with Thrombospondin motifs) family. Genome Biol, 16, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelwick R, Wagstaff L, Decock J, Roghi C, Cooley LS, Robinson SD, et al. (2015). Metalloproteinase-dependent and -independent processes contribute to inhibition of breast cancer cell migration, angiogenesis and liver metastasis by a disintegrin and metalloproteinase with thrombospondin motifs-15. Int J Cancer, 136(4), E14–26. [DOI] [PubMed] [Google Scholar]
- Kessler T, Zhang L, Liu Z, Yin X, Huang Y, Wang Y, et al. (2015). ADAMTS-7 inhibits re-endothelialization of injured arteries and promotes vascular remodeling through cleavage of thrombospondin-1. Circulation, 131(13), 1191–1201. [DOI] [PubMed] [Google Scholar]
- Kilic MO, Aynekin B, Kara A, Icen D, & Demircan K (2017). Differentially regulated ADAMTS1, 8, and 18 in gastric adenocarcinoma. Bratisl Lek Listy, 118(2), 71–76. [DOI] [PubMed] [Google Scholar]
- Kim SH, Pei QM, Jiang P, Yang M, Qian XJ, & Liu JB (2017). Effect of active vitamin D3 on VEGF-induced ADAM33 expression and proliferation in human airway smooth muscle cells: implications for asthma treatment. Respir Res, 18(1), 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein T, & Bischoff R (2011). Active metalloproteases of the A Disintegrin and Metalloprotease (ADAM) family: biological function and structure. J Proteome Res, 10(1), 17–33. [DOI] [PubMed] [Google Scholar]
- Ko S, Chisuwa H, Matsumoto M, Fujimura Y, Okano E, & Nakajima Y (2015). Relevance of ADAMTS13 to liver transplantation and surgery. World J Hepatol, 7(13), 1772–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko S, Okano E, Kanehiro H, Matsumoto M, Ishizashi H, Uemura M, et al. (2006). Plasma ADAMTS13 activity may predict early adverse events in living donor liver transplantation: observations in 3 cases. Liver Transpl, 12(5), 859–869. [DOI] [PubMed] [Google Scholar]
- Koo BH, & Apte SS (2010). Cell-surface processing of the metalloprotease pro-ADAMTS9 is influenced by the chaperone GRP94/gp96. J Biol Chem, 285(1), 197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo BH, Coe DM, Dixon LJ, Somerville RP, Nelson CM, Wang LW, et al. (2010). ADAMTS9 is a cell-autonomously acting, anti-angiogenic metalloprotease expressed by microvascular endothelial cells. Am J Pathol, 176(3), 1494–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo BH, Longpre JM, Somerville RP, Alexander JP, Leduc R, & Apte SS (2007). Regulation of ADAMTS9 secretion and enzymatic activity by its propeptide. J Biol Chem, 282(22), 16146–16154. [DOI] [PubMed] [Google Scholar]
- Kramerova IA, Kawaguchi N, Fessler LI, Nelson RE, Chen Y, Kramerov AA, et al. (2000). Papilin in development; a pericellular protein with a homology to the ADAMTS metalloproteinases. Development, 127(24), 5475–5485. [DOI] [PubMed] [Google Scholar]
- Kremer Hovinga JA, Coppo P, Lammle B, Moake JL, Miyata T, & Vanhoorelbeke K (2017). Thrombotic thrombocytopenic purpura. Nat Rev Dis Primers, 3, 17020. [DOI] [PubMed] [Google Scholar]
- Kumar S, Chen M, Li Y, Wong FH, Thiam CW, Hossain MZ, et al. (2016). Loss of ADAMTS4 reduces high fat diet-induced atherosclerosis and enhances plaque stability in ApoE(−/−) mice. Sci Rep, 6, 31130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuno K, Kanada N, Nakashima E, Fujiki F, Ichimura F, & Matsushima K (1997). Molecular cloning of a gene encoding a new type of metalloproteinase-disintegrin family protein with thrombospondin motifs as an inflammation associated gene. J Biol Chem, 272(1), 556–562. [DOI] [PubMed] [Google Scholar]
- Kuno K, & Matsushima K (1998). ADAMTS-1 protein anchors at the extracellular matrix through the thrombospondin type I motifs and its spacing region. J Biol Chem, 273(22), 13912–13917. [DOI] [PubMed] [Google Scholar]
- Kurisaki T, Masuda A, Osumi N, Nabeshima Y, & Fujisawa-Sehara A (1998). Spatially- and temporally-restricted expression of meltrin alpha (ADAM12) and beta (ADAM19) in mouse embryo. Mech Dev, 73(2), 211–215. [DOI] [PubMed] [Google Scholar]
- Kurisaki T, Masuda A, Sudo K, Sakagami J, Higashiyama S, Matsuda Y, et al. (2003). Phenotypic analysis of Meltrin alpha (ADAM12)-deficient mice: involvement of Meltrin alpha in adipogenesis and myogenesis. Mol Cell Biol, 23(1), 55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurohara K, Komatsu K, Kurisaki T, Masuda A, Irie N, Asano M, et al. (2004). Essential roles of Meltrin beta (ADAM19) in heart development. Dev Biol, 267(1), 14–28. [DOI] [PubMed] [Google Scholar]
- Kutz WE, Wang LW, Bader HL, Majors AK, Iwata K, Traboulsi EI, et al. (2011). ADAMTS10 protein interacts with fibrillin-1 and promotes its deposition in extracellular matrix of cultured fibroblasts. J Biol Chem, 286(19), 17156–17167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kveiborg M, Jacobsen J, Lee MH, Nagase H, Wewer UM, & Murphy G (2010). Selective inhibition of ADAM12 catalytic activity through engineering of tissue inhibitor of metalloproteinase 2 (TIMP-2). Biochem J, 430(1), 79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambers M, Goldenberg NA, Kenet G, Kirkham FJ, Manner D, Bernard T, et al. (2013). Role of reduced ADAMTS13 in arterial ischemic stroke: a pediatric cohort study. Ann Neurol, 73(1), 58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert DW, Yarski M, Warner FJ, Thornhill P, Parkin ET, Smith AI, et al. (2005). Tumor necrosis factor-alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J Biol Chem, 280(34), 30113–30119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer H, May AE, Bultmann A, & Gawaz M (2005). ADAM 15 is an adhesion receptor for platelet GPIIb-IIIa and induces platelet activation. Thromb Haemost, 94(3), 555–561. [DOI] [PubMed] [Google Scholar]
- Lasom S, Komanasin N, Settasatian N, Settasatian C, Kukongviriyapan U, & Intharapetch P (2018). Association of a disintegrin and metalloproteinase with a thrombospondin type 1 motif member 13 polymorphisms with severity of coronary stenosis in type 2 diabetes mellitus. J Res Med Sci, 23, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasom S, Komanasin N, Settasatian N, Settasatian C, Kukongviriyapan U, Intharapetch P, et al. (2017). Protective effect of a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 haplotype on coronary artery disease. Blood Coagul Fibrinolysis, 28(4), 286–294. [DOI] [PubMed] [Google Scholar]
- Le Goff C, & Cormier-Daire V (2009). Genetic and molecular aspects of acromelic dysplasia. Pediatr Endocrinol Rev, 6(3), 418–423. [PubMed] [Google Scholar]
- Le HT, Atif J, Mara DL, Castellana B, Treissman J, Baltayeva J, et al. (2018). ADAM8 localizes to extravillous trophoblasts within the maternal-fetal interface and potentiates trophoblast cell line migration through a beta1 integrin-mediated mechanism. Mol Hum Reprod, 24(10), 495–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CW, Hwang I, Park CS, Lee H, Park DW, Kang SJ, et al. (2011). Comparison of ADAMTS-1, −4 and −5 expression in culprit plaques between acute myocardial infarction and stable angina. J Clin Pathol, 64(5), 399–404. [DOI] [PubMed] [Google Scholar]
- Lee CW, Hwang I, Park CS, Lee H, Park DW, Kang SJ, et al. (2012). Expression of ADAMTS-2, −3, −13, and −14 in culprit coronary lesions in patients with acute myocardial infarction or stable angina. J Thromb Thrombolysis, 33(4), 362–370. [DOI] [PubMed] [Google Scholar]
- Lee DC, Sunnarborg SW, Hinkle CL, Myers TJ, Stevenson MY, Russell WE, et al. (2003). TACE/ADAM17 processing of EGFR ligands indicates a role as a physiological convertase. Ann N Y Acad Sci, 995, 22–38. [DOI] [PubMed] [Google Scholar]
- Lemarchant S, Dunghana H, Pomeshchik Y, Leinonen H, Kolosowska N, Korhonen P, et al. (2016). Anti-inflammatory effects of ADAMTS-4 in a mouse model of ischemic stroke. Glia, 64(9), 1492–1507. [DOI] [PubMed] [Google Scholar]
- Lemarchant S, Pruvost M, Montaner J, Emery E, Vivien D, Kanninen K, et al. (2013). ADAMTS proteoglycanases in the physiological and pathological central nervous system. J Neuroinflammation, 10, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemjabbar H, & Basbaum C (2002). Platelet-activating factor receptor and ADAM10 mediate responses to Staphylococcus aureus in epithelial cells. Nat Med, 8(1), 41–46. [DOI] [PubMed] [Google Scholar]
- Letronne F, Laumet G, Ayral AM, Chapuis J, Demiautte F, Laga M, et al. (2016). ADAM30 Downregulates APP-Linked Defects Through Cathepsin D Activation in Alzheimer’s Disease. EBioMedicine, 9, 278–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levula M, Paavonen T, Valo T, Pelto-Huikko M, Laaksonen R, Kahonen M, et al. (2011). A disintegrin and metalloprotease −8 and −15 and susceptibility for ascending aortic dissection. Scand J Clin Lab Invest, 71(6), 515–522. [DOI] [PubMed] [Google Scholar]
- Li F, Song R, Ao L, Reece TB, Cleveland JC Jr., Dong N, et al. (2017). ADAMTS5 Deficiency in Calcified Aortic Valves Is Associated With Elevated Pro-Osteogenic Activity in Valvular Interstitial Cells. Arterioscler Thromb Vasc Biol, 37(7), 1339–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Han S, Sun Q, Yao Y, Li S, Yuan C, et al. (2019). Long non-coding RNA CDKN2B-AS1 reduces inflammatory response and promotes cholesterol efflux in atherosclerosis by inhibiting ADAM10 expression. Aging (Albany NY), 11(6), 1695–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JK, Du WJ, Jiang SL, & Tian H (2009). Expression of ADAM-15 in rat myocardial infarction. Int J Exp Pathol, 90(3), 347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Liu Q, Lei J, Wang X, Chen X, & Ding Y (2017). MiR-362–3p inhibits the proliferation and migration of vascular smooth muscle cells in atherosclerosis by targeting ADAMTS1. Biochem Biophys Res Commun, 493(1), 270–276. [DOI] [PubMed] [Google Scholar]
- Li SW, Arita M, Fertala A, Bao Y, Kopen GC, Langsjo TK, et al. (2001). Transgenic mice with inactive alleles for procollagen N-proteinase (ADAMTS-2) develop fragile skin and male sterility. Biochem J, 355(Pt 2), 271–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Maretzky T, Perez-Aguilar JM, Monette S, Weskamp G, Le Gall S, et al. (2017). Structural modeling defines transmembrane residues in ADAM17 that are crucial for Rhbdf2-ADAM17-dependent proteolysis. J Cell Sci, 130(5), 868–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Maretzky T, Weskamp G, Monette S, Qing X, Issuree PD, et al. (2015). iRhoms 1 and 2 are essential upstream regulators of ADAM17-dependent EGFR signaling. Proc Natl Acad Sci U S A, 112(19), 6080–6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Cui LL, Li QQ, Ma GD, Cai YJ, Chen YY, et al. (2014). Association between ADAM17 promoter polymorphisms and ischemic stroke in a Chinese population. J Atheroscler Thromb, 21(8), 878–893. [DOI] [PubMed] [Google Scholar]
- Li Y, Liao F, Yin XJ, Cui LL, Ma GD, Nong XX, et al. (2013). An association study on ADAM10 promoter polymorphisms and atherosclerotic cerebral infarction in a Chinese population. CNS Neurosci Ther, 19(10), 785–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YJ, Fan YH, Tang J, Li JB, & Yu CH (2014). Meprin-beta regulates production of pro-inflammatory factors via a disintegrin and metalloproteinase-10 (ADAM-10) dependent pathway in macrophages. Int Immunopharmacol, 18(1), 77–84. [DOI] [PubMed] [Google Scholar]
- Li Z, Nardi MA, Li YS, Zhang W, Pan R, Dang S, et al. (2009). C-terminal ADAMTS-18 fragment induces oxidative platelet fragmentation, dissolves platelet aggregates, and protects against carotid artery occlusion and cerebral stroke. Blood, 113(24), 6051–6060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Zhao H, Ogai A, Kato H, Asakura M, Kim J, et al. (2008). Atorvastatin slows the progression of cardiac remodeling in mice with pressure overload and inhibits epidermal growth factor receptor activation. Hypertens Res, 31(2), 335–344. [DOI] [PubMed] [Google Scholar]
- Ling JY, Shen L, Liu Q, Xue S, Ma W, Wu H, et al. (2013). Changes in platelet GPIbalpha and ADAM17 during the acute stage of atherosclerotic ischemic stroke among Chinese. J Huazhong Univ Sci Technolog Med Sci, 33(3), 438–442. [DOI] [PubMed] [Google Scholar]
- Liu P, Yan S, Chen M, Chen A, Yao D, Xu X, et al. (2015). Effects of baicalin on collagen Iota and collagen IotaIotaIota expression in pulmonary arteries of rats with hypoxic pulmonary hypertension. Int J Mol Med, 35(4), 901–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livermore C, Warr N, Chalon N, Siggers P, Mianne J, Codner G, et al. (2019). Male mice lacking ADAMTS-16 are fertile but exhibit testes of reduced weight. Sci Rep, 9(1), 17195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llamazares M, Cal S, Quesada V, & Lopez-Otin C (2003). Identification and characterization of ADAMTS-20 defines a novel subfamily of metalloproteinases-disintegrins with multiple thrombospondin-1 repeats and a unique GON domain. J Biol Chem, 278(15), 13382–13389. [DOI] [PubMed] [Google Scholar]
- Loichinger MH, Towner D, Thompson KS, Ahn HJ, & Bryant-Greenwood GD (2016). Systemic and placental alpha-klotho: Effects of preeclampsia in the last trimester of gestation. Placenta, 41, 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez De Padilla CM, Hein MS, Crowson CS, Choo CS, Green AB, Petri M, et al. (2013). Increased expression of ADAMTS13 mRNA correlates with ischemic cerebrovascular disease in systemic lupus erythematosus patients. SAGE Open Med, 1, 2050312113514404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez JA, & Dong JF (2004). Cleavage of von Willebrand factor by ADAMTS-13 on endothelial cells. Semin Hematol, 41(1), 15–23. [DOI] [PubMed] [Google Scholar]
- Lu D, Xie S, Sukkar MB, Lu X, Scully MF, & Chung KF (2007). Inhibition of airway smooth muscle adhesion and migration by the disintegrin domain of ADAM-15. Am J Respir Cell Mol Biol, 37(4), 494–500. [DOI] [PubMed] [Google Scholar]
- Lucitti JL, Mackey JK, Morrison JC, Haigh JJ, Adams RH, & Faber JE (2012). Formation of the collateral circulation is regulated by vascular endothelial growth factor-A and a disintegrin and metalloprotease family members 10 and 17. Circ Res, 111(12), 1539–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig A, Hundhausen C, Lambert MH, Broadway N, Andrews RC, Bickett DM, et al. (2005). Metalloproteinase inhibitors for the disintegrin-like metalloproteinases ADAM10 and ADAM17 that differentially block constitutive and phorbol ester-inducible shedding of cell surface molecules. Comb Chem High Throughput Screen, 8(2), 161–171. [DOI] [PubMed] [Google Scholar]
- Lunn CA, Fan X, Dalie B, Miller K, Zavodny PJ, Narula SK, et al. (1997). Purification of ADAM 10 from bovine spleen as a TNFalpha convertase. FEBS Lett, 400(3), 333–335. [DOI] [PubMed] [Google Scholar]
- Lyu C, Chen Y, Zhu M, Jin X, Liu P, Zheng Z, et al. (2015). [Association of ADAMTS-1 gene polymorphisms with ischemic stroke caused by large artery atherosclerosis]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi, 32(6), 844–848. [DOI] [PubMed] [Google Scholar]
- Mahoney ET, Benton RL, Maddie MA, Whittemore SR, & Hagg T (2009). ADAM8 is selectively up-regulated in endothelial cells and is associated with angiogenesis after spinal cord injury in adult mice. J Comp Neurol, 512(2), 243–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majerus EM, Zheng X, Tuley EA, & Sadler JE (2003). Cleavage of the ADAMTS13 propeptide is not required for protease activity. J Biol Chem, 278(47), 46643–46648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maretzky T, Swendeman S, Mogollon E, Weskamp G, Sahin U, Reiss K, et al. (2017). Characterization of the catalytic properties of the membrane-anchored metalloproteinase ADAM9 in cell-based assays. Biochem J, 474(9), 1467–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews AL, Noy PJ, Reyat JS, & Tomlinson MG (2017). Regulation of A disintegrin and metalloproteinase (ADAM) family sheddases ADAM10 and ADAM17: The emerging role of tetraspanins and rhomboids. Platelets, 28(4), 333–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews AL, Szyroka J, Collier R, Noy PJ, & Tomlinson MG (2017). Scissor sisters: regulation of ADAM10 by the TspanC8 tetraspanins. Biochem Soc Trans, 45(3), 719–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClurg UL, Danjo K, King HO, Scott GB, Robinson PA, & Crabtree JE (2015). Epithelial cell ADAM17 activation by Helicobacter pylori: role of ADAM17 C-terminus and Threonine-735 phosphorylation. Microbes Infect, 17(3), 205–214. [DOI] [PubMed] [Google Scholar]
- McRobb LS, McKay MJ, Gamble JR, Grace M, Moutrie V, Santos ED, et al. (2017). Ionizing radiation reduces ADAM10 expression in brain microvascular endothelial cells undergoing stress-induced senescence. Aging (Albany NY), 9(4), 1248–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mead TJ, & Apte SS (2018). ADAMTS proteins in human disorders. Matrix Biol, 71–72, 225–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta V, Fields L, Evans IM, Yamaji M, Pellet-Many C, Jones T, et al. (2018). VEGF (Vascular Endothelial Growth Factor) Induces NRP1 (Neuropilin-1) Cleavage via ADAMs (a Disintegrin and Metalloproteinase) 9 and 10 to Generate Novel Carboxy-Terminal NRP1 Fragments That Regulate Angiogenic Signaling. Arterioscler Thromb Vasc Biol, 38(8), 1845–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles RR, Sluka JP, Halladay DL, Santerre RF, Hale LV, Bloem L, et al. (2000). ADAMTS-1: A cellular disintegrin and metalloprotease with thrombospondin motifs is a target for parathyroid hormone in bone. Endocrinology, 141(12), 4533–4542. [DOI] [PubMed] [Google Scholar]
- Minond D, Cudic M, Bionda N, Giulianotti M, Maida L, Houghten RA, et al. (2012). Discovery of novel inhibitors of a disintegrin and metalloprotease 17 (ADAM17) using glycosylated and non-glycosylated substrates. J Biol Chem, 287(43), 36473–36487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra PK, Tyagi N, Sen U, Givvimani S, & Tyagi SC (2010). H2S ameliorates oxidative and proteolytic stresses and protects the heart against adverse remodeling in chronic heart failure. Am J Physiol Heart Circ Physiol, 298(2), H451–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittaz L, Ricardo S, Martinez G, Kola I, Kelly DJ, Little MH, et al. (2005). Neonatal calyceal dilation and renal fibrosis resulting from loss of Adamts-1 in mouse kidney is due to a developmental dysgenesis. Nephrol Dial Transplant, 20(2), 419–423. [DOI] [PubMed] [Google Scholar]
- Mittaz L, Russell DL, Wilson T, Brasted M, Tkalcevic J, Salamonsen LA, et al. (2004). Adamts-1 is essential for the development and function of the urogenital system. Biol Reprod, 70(4), 1096–1105. [DOI] [PubMed] [Google Scholar]
- Moake JL (2004). von Willebrand factor, ADAMTS-13, and thrombotic thrombocytopenic purpura. Semin Hematol, 41(1), 4–14. [DOI] [PubMed] [Google Scholar]
- Mochizuki S, & Okada Y (2007). ADAMs in cancer cell proliferation and progression. Cancer Sci, 98(5), 621–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriguchi-Goto S, Yamashita A, Tamura N, Soejima K, Takahashi M, Nakagaki T, et al. (2009). ADAMTS-13 attenuates thrombus formation on type I collagen surface and disrupted plaques under flow conditions. Atherosclerosis, 203(2), 409–416. [DOI] [PubMed] [Google Scholar]
- Moss ML, Jin SL, Milla ME, Bickett DM, Burkhart W, Carter HL, et al. (1997). Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature, 385(6618), 733–736. [DOI] [PubMed] [Google Scholar]
- Moss ML, & Rasmussen FH (2007). Fluorescent substrates for the proteinases ADAM17, ADAM10, ADAM8, and ADAM12 useful for high-throughput inhibitor screening. Anal Biochem, 366(2), 144–148. [DOI] [PubMed] [Google Scholar]
- Mukerjee S, Gao H, Xu J, Sato R, Zsombok A, & Lazartigues E (2019). ACE2 and ADAM17 Interaction Regulates the Activity of Presympathetic Neurons. Hypertension, 74(5), 1181–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller M, Kessler T, Schunkert H, Erdmann J, & Tennstedt S (2016). Classification of ADAMTS binding sites: The first step toward selective ADAMTS7 inhibitors. Biochem Biophys Res Commun, 471(3), 380–385. [DOI] [PubMed] [Google Scholar]
- Murphy G (2011). Tissue inhibitors of metalloproteinases. Genome Biol, 12(11), 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musumeci G, Coleman R, Imbesi R, Magro G, Parenti R, Szychlinska MA, et al. (2014). ADAM-10 could mediate cleavage of N-cadherin promoting apoptosis in human atherosclerotic lesions leading to vulnerable plaque: a morphological and immunohistochemical study. Acta Histochem, 116(7), 1148–1158. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Hirohata S, Murakami T, Miyoshi T, Demircan K, Oohashi T, et al. (2004). Dynamic induction of ADAMTS1 gene in the early phase of acute myocardial infarction. J Biochem, 136(4), 439–446. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Kita S, Tanaka Y, Fukuda S, Obata Y, Okita T, et al. (2020). A disintegrin and metalloproteinase 12 prevents heart failure by regulating cardiac hypertrophy and fibrosis. Am J Physiol Heart Circ Physiol, 318(2), H238–H251. [DOI] [PubMed] [Google Scholar]
- Nandadasa S, Burin des Roziers C, Koch C, Tran-Lundmark K, Dours-Zimmermann MT, Zimmermann DR, et al. (2021). A new mouse mutant with cleavage-resistant versican and isoform-specific versican mutants demonstrate that proteolysis at the Glu(441)-Ala(442) peptide bond in the V1 isoform is essential for interdigital web regression. Matrix Biol Plus, 10, 100064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolaou A, Zhao Z, Northoff BH, Sass K, Herbst A, Kohlmaier A, et al. (2017). Adam17 Deficiency Promotes Atherosclerosis by Enhanced TNFR2 Signaling in Mice. Arterioscler Thromb Vasc Biol, 37(2), 247–257. [DOI] [PubMed] [Google Scholar]
- Niu A, Wang B, & Li YP (2015). TNFalpha shedding in mechanically stressed cardiomyocytes is mediated by Src activation of TACE. J Cell Biochem, 116(4), 559–565. [DOI] [PubMed] [Google Scholar]
- Nyren-Erickson EK, Jones JM, Srivastava DK, & Mallik S (2013). A disintegrin and metalloproteinase-12 (ADAM12): function, roles in disease progression, and clinical implications. Biochim Biophys Acta, 1830(10), 4445–4455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obama T, Takayanagi T, Kobayashi T, Bourne AM, Elliott KJ, Charbonneau M, et al. (2015). Vascular induction of a disintegrin and metalloprotease 17 by angiotensin II through hypoxia inducible factor 1alpha. Am J Hypertens, 28(1), 10–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogino H, Hisanaga A, Kohno T, Kondo Y, Okumura K, Kamei T, et al. (2017). Secreted Metalloproteinase ADAMTS-3 Inactivates Reelin. J Neurosci, 37(12), 3181–3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsu H, Dempsey PJ, & Eguchi S (2006). ADAMs as mediators of EGF receptor transactivation by G protein-coupled receptors. Am J Physiol Cell Physiol, 291(1), C1–10. [DOI] [PubMed] [Google Scholar]
- Oikawa H, Maesawa C, Tatemichi Y, Nishinari Y, Nishiya M, Mizugai H, et al. (2014). A disintegrin and metalloproteinase 17 (ADAM17) mediates epidermal growth factor receptor transactivation by angiotensin II on hepatic stellate cells. Life Sci, 97(2), 137–144. [DOI] [PubMed] [Google Scholar]
- Okeke E, & Dokun AO (2018). Role of genetics in peripheral arterial disease outcomes; significance of limb-salvage quantitative locus-1 genes. Exp Biol Med (Maywood), 243(2), 190–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksala N, Levula M, Airla N, Pelto-Huikko M, Ortiz RM, Jarvinen O, et al. (2009). ADAM-9, ADAM-15, and ADAM-17 are upregulated in macrophages in advanced human atherosclerotic plaques in aorta and carotid and femoral arteries--Tampere vascular study. Ann Med, 41(4), 279–290. [DOI] [PubMed] [Google Scholar]
- Oller J, Mendez-Barbero N, Ruiz EJ, Villahoz S, Renard M, Canelas LI, et al. (2017). Nitric oxide mediates aortic disease in mice deficient in the metalloprotease Adamts1 and in a mouse model of Marfan syndrome. Nat Med, 23(2), 200–212. [DOI] [PubMed] [Google Scholar]
- Omura J, Satoh K, Kikuchi N, Satoh T, Kurosawa R, Nogi M, et al. (2019). ADAMTS8 Promotes the Development of Pulmonary Arterial Hypertension and Right Ventricular Failure: A Possible Novel Therapeutic Target. Circ Res, 125(10), 884–906. [DOI] [PubMed] [Google Scholar]
- Ong MH, Wong HK, Tengku-Muhammad TS, Choo QC, & Chew CH (2019). Pro-atherogenic proteoglycanase ADAMTS-1 is down-regulated by lauric acid through PI3K and JNK signaling pathways in THP-1 derived macrophages. Mol Biol Rep, 46(3), 2631–2641. [DOI] [PubMed] [Google Scholar]
- Ozawa K, Packwood W, Varlamov O, Qi Y, Xie A, Wu MD, et al. (2018). Molecular Imaging of VWF (von Willebrand Factor) and Platelet Adhesion in Postischemic Impaired Microvascular Reflow. Circ Cardiovasc Imaging, 11(11), e007913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palau V, Pascual J, Soler MJ, & Riera M (2019). Role of ADAM17 in kidney disease. Am J Physiol Renal Physiol, 317(2), F333–F342. [DOI] [PubMed] [Google Scholar]
- Parrish DC, Francis Stuart SD, Olivas A, Wang L, Nykjaer A, Ripplinger CM, et al. (2018). Transient denervation of viable myocardium after myocardial infarction does not alter arrhythmia susceptibility. Am J Physiol Heart Circ Physiol, 314(3), H415–H423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pehlivan S, Gurses MS, Ural MN, Akyol S, Eren F, Turkmen Inanir N, et al. (2016). The Role of ADAMTS1 and Versican in Human Myocardial Infarction: A Postmortem Study. Lab Med, 47(3), 205–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelisek J, Deutsch L, Ansel A, Pongratz J, Stadlbauer T, Gebhard H, et al. (2017). Expression of a metalloproteinase family of ADAMTS in human vulnerable carotid lesions. J Cardiovasc Med (Hagerstown), 18(1), 10–18. [DOI] [PubMed] [Google Scholar]
- Pelisek J, Pongratz J, Deutsch L, Reeps C, Stadlbauer T, & Eckstein HH (2012). Expression and cellular localization of metalloproteases ADAMs in high graded carotid artery lesions. Scand J Clin Lab Invest, 72(8), 648–656. [DOI] [PubMed] [Google Scholar]
- Peng Q, Deng Y, Yang X, Leng X, Yang Y, & Liu H (2016). Genetic variants of ADAM17 are implicated in the pathological process of Kawasaki disease and secondary coronary artery lesions via the TGF-beta/SMAD3 signaling pathway. Eur J Pediatr, 175(5), 705–713. [DOI] [PubMed] [Google Scholar]
- Perna AF, Pizza A, Di Nunzio A, Bellantone R, Raffaelli M, Cicchella T, et al. (2017). ADAM17, a New Player in the Pathogenesis of Chronic Kidney Disease-Mineral and Bone Disorder. J Ren Nutr, 27(6), 453–457. [DOI] [PubMed] [Google Scholar]
- Peschon JJ, Slack JL, Reddy P, Stocking KL, Sunnarborg SW, Lee DC, et al. (1998). An essential role for ectodomain shedding in mammalian development. Science, 282(5392), 1281–1284. [DOI] [PubMed] [Google Scholar]
- Petri A, Kim HJ, Xu Y, de Groot R, Li C, Vandenbulcke A, et al. (2019). Crystal structure and substrate-induced activation of ADAMTS13. Nat Commun, 10(1), 3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polverino F, Rojas-Quintero J, Wang X, Petersen H, Zhang L, Gai X, et al. (2018). A Disintegrin and Metalloproteinase Domain-8: A Novel Protective Proteinase in Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med, 198(10), 1254–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon LC, Chelemen T, Granvillano O, Pandeva I, & Nicolaides KH (2008). First-trimester maternal serum a disintegrin and metalloprotease 12 (ADAM12) and adverse pregnancy outcome. Obstet Gynecol, 112(5), 1082–1090. [DOI] [PubMed] [Google Scholar]
- Poonpet T, Honsawek S, Tammachote N, Kanitnate S, & Tammachote R (2013). ADAMTS14 gene polymorphism associated with knee osteoarthritis in Thai women. Genet Mol Res, 12(4), 5301–5309. [DOI] [PubMed] [Google Scholar]
- Porter S, Clark IM, Kevorkian L, & Edwards DR (2005). The ADAMTS metalloproteinases. Biochem J, 386(Pt 1), 15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins BP, Mead TJ, Brody JA, Sveinbjornsson G, Ntalla I, Bihlmeyer NA, et al. (2018). Exome-chip meta-analysis identifies novel loci associated with cardiac conduction, including ADAMTS6. Genome Biol, 19(1), 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proudfoot AG, Davidson SJ, & Strueber M (2017). von Willebrand factor disruption and continuous-flow circulatory devices. J Heart Lung Transplant, 36(11), 1155–1163. [DOI] [PubMed] [Google Scholar]
- Pu X, Xiao Q, Kiechl S, Chan K, Ng FL, Gor S, et al. (2013). ADAMTS7 cleavage and vascular smooth muscle cell migration is affected by a coronary-artery-disease-associated variant. Am J Hum Genet, 92(3), 366–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi B, Newcomer RG, & Sang QX (2009). ADAM19/adamalysin 19 structure, function, and role as a putative target in tumors and inflammatory diseases. Curr Pharm Des, 15(20), 2336–2348. [DOI] [PubMed] [Google Scholar]
- Qian LW, Mizumoto K, Urashima T, Nagai E, Maehara N, Sato N, et al. (2002). Radiation-induced increase in invasive potential of human pancreatic cancer cells and its blockade by a matrix metalloproteinase inhibitor, CGS27023. Clin Cancer Res, 8(4), 1223–1227. [PubMed] [Google Scholar]
- Qin W, Cao Y, Li L, Chen W, & Chen X (2017). Upregulation of ADAMTS7 and downregulation of COMP are associated with aortic aneurysm. Mol Med Rep, 16(4), 5459–5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu L, Jiang M, Qiu W, Lu S, Zhao Y, Xia L, et al. (2016). Assessment of the Diagnostic Value of Plasma Levels, Activities, and Their Ratios of von Willebrand Factor and ADAMTS13 in Patients with Cerebral Infarction. Clin Appl Thromb Hemost, 22(3), 252–259. [DOI] [PubMed] [Google Scholar]
- Qu Y, & Zhang N (2018). miR-365b-3p inhibits the cell proliferation and migration of human coronary artery smooth muscle cells by directly targeting ADAMTS1 in coronary atherosclerosis. Exp Ther Med, 16(5), 4239–4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raitoharju E, Seppala I, Levula M, Kuukasjarvi P, Laurikka J, Nikus K, et al. (2011). Common variation in the ADAM8 gene affects serum sADAM8 concentrations and the risk of myocardial infarction in two independent cohorts. Atherosclerosis, 218(1), 127–133. [DOI] [PubMed] [Google Scholar]
- Ramdas V, McBride M, Denby L, & Baker AH (2013). Canonical transforming growth factor-beta signaling regulates disintegrin metalloprotease expression in experimental renal fibrosis via miR-29. Am J Pathol, 183(6), 1885–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapti M, Atkinson SJ, Lee MH, Trim A, Moss M, & Murphy G (2008). The isolated N-terminal domains of TIMP-1 and TIMP-3 are insufficient for ADAM10 inhibition. Biochem J, 411(2), 433–439. [DOI] [PubMed] [Google Scholar]
- Rau CD, Romay MC, Tuteryan M, Wang JJ, Santolini M, Ren S, et al. (2017). Systems Genetics Approach Identifies Gene Pathways and Adamts2 as Drivers of Isoproterenol-Induced Cardiac Hypertrophy and Cardiomyopathy in Mice. Cell Syst, 4(1), 121–128 e124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raucci A, Cugusi S, Antonelli A, Barabino SM, Monti L, Bierhaus A, et al. (2008). A soluble form of the receptor for advanced glycation endproducts (RAGE) is produced by proteolytic cleavage of the membrane-bound form by the sheddase a disintegrin and metalloprotease 10 (ADAM10). FASEB J, 22(10), 3716–3727. [DOI] [PubMed] [Google Scholar]
- Reddy P, Slack JL, Davis R, Cerretti DP, Kozlosky CJ, Blanton RA, et al. (2000). Functional analysis of the domain structure of tumor necrosis factor-alpha converting enzyme. J Biol Chem, 275(19), 14608–14614. [DOI] [PubMed] [Google Scholar]
- Ren L, Wu C, Yang K, Chen S, Ye P, Wu J, et al. (2018). A Disintegrin and Metalloprotease-22 Attenuates Hypertrophic Remodeling in Mice Through Inhibition of the Protein Kinase B Signaling Pathway. J Am Heart Assoc, 7(2):e005696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren P, Hughes M, Krishnamoorthy S, Zou S, Zhang L, Wu D, et al. (2017). Critical Role of ADAMTS-4 in the Development of Sporadic Aortic Aneurysm and Dissection in Mice. Sci Rep, 7(1), 12351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren W, Liang L, Li Y, Wei FY, Mu N, Zhang L, et al. (2020). Upregulation of miR423 improves autologous vein graft restenosis via targeting ADAMTS7. Int J Mol Med, 45(2), 532–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyat JS, Chimen M, Noy PJ, Szyroka J, Rainger GE, & Tomlinson MG (2017). ADAM10-Interacting Tetraspanins Tspan5 and Tspan17 Regulate VE-Cadherin Expression and Promote T Lymphocyte Transmigration. J Immunol, 199(2), 666–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizza S, Copetti M, Cardellini M, Menghini R, Pecchioli C, Luzi A, et al. (2015). A score including ADAM17 substrates correlates to recurring cardiovascular event in subjects with atherosclerosis. Atherosclerosis, 239(2), 459–464. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Baena FJ, Redondo-Garcia S, Peris-Torres C, Martino-Echarri E, Fernandez-Rodriguez R, Plaza-Calonge MDC, et al. (2018). ADAMTS1 protease is required for a balanced immune cell repertoire and tumour inflammatory response. Sci Rep, 8(1), 13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Lopez J, Pombo-Suarez M, Loughlin J, Tsezou A, Blanco FJ, Meulenbelt I, et al. (2009). Association of a nsSNP in ADAMTS14 to some osteoarthritis phenotypes. Osteoarthritis Cartilage, 17(3), 321–327. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Manzaneque JC, Westling J, Thai SN, Luque A, Knauper V, Murphy G, et al. (2002). ADAMTS1 cleaves aggrecan at multiple sites and is differentially inhibited by metalloproteinase inhibitors. Biochem Biophys Res Commun, 293(1), 501–508. [DOI] [PubMed] [Google Scholar]
- Roose E, Vidarsson G, Kangro K, Verhagen O, Mancini I, Desender L, et al. (2018). Anti-ADAMTS13 Autoantibodies against Cryptic Epitopes in Immune-Mediated Thrombotic Thrombocytopenic Purpura. Thromb Haemost, 118(10), 1729–1742. [DOI] [PubMed] [Google Scholar]
- Rosendahl MS, Ko SC, Long DL, Brewer MT, Rosenzweig B, Hedl E, et al. (1997). Identification and characterization of a pro-tumor necrosis factor-alpha-processing enzyme from the ADAM family of zinc metalloproteases. J Biol Chem, 272(39), 24588–24593. [DOI] [PubMed] [Google Scholar]
- Roychaudhuri R, Hergrueter AH, Polverino F, Laucho-Contreras ME, Gupta K, Borregaard N, et al. (2014). ADAM9 is a novel product of polymorphonuclear neutrophils: regulation of expression and contributions to extracellular matrix protein degradation during acute lung injury. J Immunol, 193(5), 2469–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha N, Robev D, Himanen JP, & Nikolov DB (2019). ADAM proteases: Emerging role and targeting of the non-catalytic domains. Cancer Lett, 467, 50–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin U, Weskamp G, Kelly K, Zhou HM, Higashiyama S, Peschon J, et al. (2004). Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J Cell Biol, 164(5), 769–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahul ZH, Mukherjee R, Song J, McAteer J, Stroud RE, Dione DP, et al. (2011). Targeted imaging of the spatial and temporal variation of matrix metalloproteinase activity in a porcine model of postinfarct remodeling: relationship to myocardial dysfunction. Circ Cardiovasc Imaging, 4(4), 381–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salloum FN (2015). Hydrogen sulfide and cardioprotection--Mechanistic insights and clinical translatability. Pharmacol Ther, 152, 11–17. [DOI] [PubMed] [Google Scholar]
- Salter RC, Arnaoutakis K, Michael DR, Singh NN, Ashlin TG, Buckley ML, et al. (2011). The expression of a disintegrin and metalloproteinase with thrombospondin motifs 4 in human macrophages is inhibited by the anti-atherogenic cytokine transforming growth factor-beta and requires Smads, p38 mitogen-activated protein kinase and c-Jun. Int J Biochem Cell Biol, 43(5), 805–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santamaria S, Cuffaro D, Nuti E, Ciccone L, Tuccinardi T, Liva F, et al. (2021). Exosite inhibition of ADAMTS-5 by a glycoconjugated arylsulfonamide. Sci Rep, 11(1), 949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santamaria S, & de Groot R (2019). Monoclonal antibodies against metzincin targets. Br J Pharmacol, 176(1), 52–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santamaria S, & de Groot R (2020). ADAMTS proteases in cardiovascular physiology and disease. Open Biol, 10(12), 200333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarila G, Bao T, Abeydeera SA, Li R, Mell B, Joe B, et al. (2020). Interplay between collagenase and undescended testes in Adamts16 knockout rats. J Pediatr Surg, 55(9):1952–1958. [DOI] [PubMed] [Google Scholar]
- Sato T, Suzuki H, Shibata M, Kusuyama T, Omori Y, Soda T, et al. (2006). Tumor-necrosis-factor-alpha-gene-deficient mice have improved cardiac function through reduction of intercellular adhesion molecule-1 in myocardial infarction. Circ J, 70(12), 1635–1642. [DOI] [PubMed] [Google Scholar]
- Satoh H, Nakamura M, Satoh M, Nakajima T, Izumoto H, Maesawa C, et al. (2004). Expression and localization of tumour necrosis factor-alpha and its converting enzyme in human abdominal aortic aneurysm. Clin Sci (Lond), 106(3), 301–306. [DOI] [PubMed] [Google Scholar]
- Satoh M, Iwasaka J, Nakamura M, Akatsu T, Shimoda Y, & Hiramori K (2004). Increased expression of tumor necrosis factor-alpha converting enzyme and tumor necrosis factor-alpha in peripheral blood mononuclear cells in patients with advanced congestive heart failure. Eur J Heart Fail, 6(7), 869–875. [DOI] [PubMed] [Google Scholar]
- Satoh M, Nakamura M, Satoh H, Saitoh H, Segawa I, & Hiramori K (2000). Expression of tumor necrosis factor-alpha--converting enzyme and tumor necrosis factor-alpha in human myocarditis. J Am Coll Cardiol, 36(4), 1288–1294. [DOI] [PubMed] [Google Scholar]
- Scharfenberg F, Helbig A, Sammel M, Benzel J, Schlomann U, Peters F, et al. (2020). Degradome of soluble ADAM10 and ADAM17 metalloproteases. Cell Mol Life Sci, 77(2), 331–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schick D, Babendreyer A, Wozniak J, Awan T, Noels H, Liehn E, et al. (2019). Elevated expression of the metalloproteinase ADAM8 associates with vascular diseases in mice and humans. Atherosclerosis, 286, 163–171. [DOI] [PubMed] [Google Scholar]
- Schiviz A, Wuersch K, Piskernik C, Dietrich B, Hoellriegl W, Rottensteiner H, et al. (2012). A new mouse model mimicking thrombotic thrombocytopenic purpura: correction of symptoms by recombinant human ADAMTS13. Blood, 119(25), 6128–6135. [DOI] [PubMed] [Google Scholar]
- Schlomann U, Dorzweiler K, Nuti E, Tuccinardi T, Rossello A, & Bartsch JW (2019). Metalloprotease inhibitor profiles of human ADAM8 in vitro and in cell-based assays. Biol Chem, 400(6), 801–810. [DOI] [PubMed] [Google Scholar]
- Schlomann U, Wildeboer D, Webster A, Antropova O, Zeuschner D, Knight CG, et al. (2002). The metalloprotease disintegrin ADAM8. Processing by autocatalysis is required for proteolytic activity and cell adhesion. J Biol Chem, 277(50), 48210–48219. [DOI] [PubMed] [Google Scholar]
- Schouten LR, Helmerhorst HJ, Wagenaar GT, Haltenhof T, Lutter R, Roelofs JJ, et al. (2016). Age-Dependent Changes in the Pulmonary Renin-Angiotensin System Are Associated With Severity of Lung Injury in a Model of Acute Lung Injury in Rats. Crit Care Med, 44(12), e1226–e1235. [DOI] [PubMed] [Google Scholar]
- Schrimpf C, Koppen T, Duffield JS, Boer U, David S, Ziegler W, et al. (2017). TIMP3 is Regulated by Pericytes upon Shear Stress Detection Leading to a Modified Endothelial Cell Response. Eur J Vasc Endovasc Surg, 54(4), 524–533. [DOI] [PubMed] [Google Scholar]
- Schubert K, Collins LE, Green P, Nagase H, & Troeberg L (2019). LRP1 Controls TNF Release via the TIMP-3/ADAM17 Axis in Endotoxin-Activated Macrophages. J Immunol, 202(5), 1501–1509. [DOI] [PubMed] [Google Scholar]
- Schulz B, Pruessmeyer J, Maretzky T, Ludwig A, Blobel CP, Saftig P, et al. (2008). ADAM10 regulates endothelial permeability and T-Cell transmigration by proteolysis of vascular endothelial cadherin. Circ Res, 102(10), 1192–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuppner R, Dirks M, Grosse GM, Bockmann M, Goetz F, Pasedag T, et al. (2018). ADAMTS-13 Activity Predicts Outcome in Acute Ischaemic Stroke Patients Undergoing Endovascular Treatment. Thromb Haemost, 118(4), 758–767. [DOI] [PubMed] [Google Scholar]
- Scully M (2016). Thrombotic Thrombocytopenic Purpura and Atypical Hemolytic Uremic Syndrome Microangiopathy in Pregnancy. Semin Thromb Hemost, 42(7), 774–779. [DOI] [PubMed] [Google Scholar]
- Scully M, Knobl P, Kentouche K, Rice L, Windyga J, Schneppenheim R, et al. (2017). Recombinant ADAMTS-13: first-in-human pharmacokinetics and safety in congenital thrombotic thrombocytopenic purpura. Blood, 130(19), 2055–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seals DF, & Courtneidge SA (2003). The ADAMs family of metalloproteases: multidomain proteins with multiple functions. Genes Dev, 17(1), 7–30. [DOI] [PubMed] [Google Scholar]
- Selejan SR, Linz D, Tatu AM, Hohl M, Speer T, Ewen S, et al. (2018). Sympathoadrenergic suppression improves heart function by upregulating the ratio of sRAGE/RAGE in hypertension with metabolic syndrome. J Mol Cell Cardiol, 122, 34–46. [DOI] [PubMed] [Google Scholar]
- Shen G, Sun Q, Yao Y, Li S, Liu G, Yuan C, et al. (2020). Role of ADAM9 and miR-126 in the development of abdominal aortic aneurysm. Atherosclerosis, 297, 47–54. [DOI] [PubMed] [Google Scholar]
- Shen L, Lu G, Dong N, Ma Z, & Ruan C (2013). Simvastatin increases ADAMTS13 expression in podocytes. Thromb Res, 132(1), 94–99. [DOI] [PubMed] [Google Scholar]
- Shen M, Hu M, Fedak PWM, Oudit GY, & Kassiri Z (2018). Cell-Specific Functions of ADAM17 Regulate the Progression of Thoracic Aortic Aneurysm. Circ Res, 123(3), 372–388. [DOI] [PubMed] [Google Scholar]
- Shen M, Morton J, Davidge ST, & Kassiri Z (2017). Loss of smooth muscle cell disintegrin and metalloproteinase 17 transiently suppresses angiotensin II-induced hypertension and end-organ damage. J Mol Cell Cardiol, 103, 11–21. [DOI] [PubMed] [Google Scholar]
- Shi W, Chen H, Sun J, Buckley S, Zhao J, Anderson KD, et al. (2003). TACE is required for fetal murine cardiac development and modeling. Dev Biol, 261(2), 371–380. [DOI] [PubMed] [Google Scholar]
- Shindo T, Kurihara H, Kuno K, Yokoyama H, Wada T, Kurihara Y, et al. (2000). ADAMTS-1: a metalloproteinase-disintegrin essential for normal growth, fertility, and organ morphology and function. J Clin Invest, 105(10), 1345–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smiljanic K, Dobutovic B, Obradovic M, Nikolic D, Marche P, & Isenovic ER (2011). Involvement of the ADAM 12 in thrombin-induced rat’s VSMCs proliferation. Curr Med Chem, 18(22), 3382–3386. [DOI] [PubMed] [Google Scholar]
- Smith KM, Gaultier A, Cousin H, Alfandari D, White JM, & DeSimone DW (2002). The cysteine-rich domain regulates ADAM protease function in vivo. J Cell Biol, 159(5), 893–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somerville RP, Jungers KA, & Apte SS (2004). Discovery and characterization of a novel, widely expressed metalloprotease, ADAMTS10, and its proteolytic activation. J Biol Chem, 279(49), 51208–51217. [DOI] [PubMed] [Google Scholar]
- Somerville RP, Longpre JM, Apel ED, Lewis RM, Wang LW, Sanes JR, et al. (2004). ADAMTS7B, the full-length product of the ADAMTS7 gene, is a chondroitin sulfate proteoglycan containing a mucin domain. J Biol Chem, 279(34), 35159–35175. [DOI] [PubMed] [Google Scholar]
- Somerville RP, Longpre JM, Jungers KA, Engle JM, Ross M, Evanko S, et al. (2003). Characterization of ADAMTS-9 and ADAMTS-20 as a distinct ADAMTS subfamily related to Caenorhabditis elegans GON-1. J Biol Chem, 278(11), 9503–9513. [DOI] [PubMed] [Google Scholar]
- Sommer A, Duppe M, Baumecker L, Kordowski F, Buch J, Chico JF, et al. (2017). Extracellular sphingomyelinase activity impairs TNF-alpha-induced endothelial cell death via ADAM17 activation and TNF receptor 1 shedding. Oncotarget, 8(42), 72584–72596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonneveld MA, de Maat MP, Portegies ML, Kavousi M, Hofman A, Turecek PL, et al. (2015). Low ADAMTS13 activity is associated with an increased risk of ischemic stroke. Blood, 126(25), 2739–2746. [DOI] [PubMed] [Google Scholar]
- Soond SM, Everson B, Riches DW, & Murphy G (2005). ERK-mediated phosphorylation of Thr735 in TNFalpha-converting enzyme and its potential role in TACE protein trafficking. J Cell Sci, 118(Pt 11), 2371–2380. [DOI] [PubMed] [Google Scholar]
- South K, Luken BM, Crawley JT, Phillips R, Thomas M, Collins RF, et al. (2014). Conformational activation of ADAMTS13. Proc Natl Acad Sci U S A, 111(52), 18578–18583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speck N, Brandsch C, Schmidt N, Yazdekhasti N, Hirche F, Lucius R, et al. (2015). The Antiatherogenic Effect of Fish Oil in Male Mice Is Associated with a Diminished Release of Endothelial ADAM17 and ADAM10 Substrates. J Nutr, 145(6), 1218–1226. [DOI] [PubMed] [Google Scholar]
- Spencer K, Cowans NJ, & Stamatopoulou A (2008). ADAM12s in maternal serum as a potential marker of pre-eclampsia. Prenat Diagn, 28(3), 212–216. [DOI] [PubMed] [Google Scholar]
- Srinivasan S, Romagnoli M, Bohm A, & Sonenshein GE (2014). N-glycosylation regulates ADAM8 processing and activation. J Biol Chem, 289(48), 33676–33688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoll M, Ruhle F, Witten A, Barysenka A, Arning A, Strauss C, et al. (2016). Rare Variants in the ADAMTS13 Von Willebrand Factor-Binding Domain Contribute to Pediatric Stroke. Circ Cardiovasc Genet, 9(4), 357–367. [DOI] [PubMed] [Google Scholar]
- Stone AL, Kroeger M, & Sang QX (1999). Structure-function analysis of the ADAM family of disintegrin-like and metalloproteinase-containing proteins (review). J Protein Chem, 18(4), 447–465. [DOI] [PubMed] [Google Scholar]
- Sun C, Wu MH, Guo M, Day ML, Lee ES, & Yuan SY (2010). ADAM15 regulates endothelial permeability and neutrophil migration via Src/ERK1/2 signalling. Cardiovasc Res, 87(2), 348–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Wu MH, Lee ES, & Yuan SY (2012). A disintegrin and metalloproteinase 15 contributes to atherosclerosis by mediating endothelial barrier dysfunction via Src family kinase activity. Arterioscler Thromb Vasc Biol, 32(10), 2444–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M, Chen M, Dawood F, Zurawska U, Li JY, Parker T, et al. (2007). Tumor necrosis factor-alpha mediates cardiac remodeling and ventricular dysfunction after pressure overload state. Circulation, 115(11), 1398–1407. [DOI] [PubMed] [Google Scholar]
- Suna G, Wojakowski W, Lynch M, Barallobre-Barreiro J, Yin X, Mayr U, et al. (2018). Extracellular Matrix Proteomics Reveals Interplay of Aggrecan and Aggrecanases in Vascular Remodeling of Stented Coronary Arteries. Circulation, 137(2), 166–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundberg C, Thodeti CK, Kveiborg M, Larsson C, Parker P, Albrechtsen R, et al. (2004). Regulation of ADAM12 cell-surface expression by protein kinase C epsilon. J Biol Chem, 279(49), 51601–51611. [DOI] [PubMed] [Google Scholar]
- Takaguri A, Kimura K, Hinoki A, Bourne AM, Autieri MV, & Eguchi S (2011). A disintegrin and metalloprotease 17 mediates neointimal hyperplasia in vasculature. Hypertension, 57(4), 841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaguri A, Morimoto M, Imai S, & Satoh K (2016). Cilostazol inhibits interleukin-1-induced ADAM17 expression through cAMP independent signaling in vascular smooth muscle cells. Cell Biol Int, 40(3), 269–276. [DOI] [PubMed] [Google Scholar]
- Takahashi E, Sagane K, Oki T, Yamazaki K, Nagasu T, & Kuromitsu J (2006). Deficits in spatial learning and motor coordination in ADAM11-deficient mice. BMC Neurosci, 7, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takawale A, Sakamuri SS, & Kassiri Z (2015). Extracellular matrix communication and turnover in cardiac physiology and pathology. Compr Physiol, 5(2), 687–719. [DOI] [PubMed] [Google Scholar]
- Takayanagi T, Forrester SJ, Kawai T, Obama T, Tsuji T, Elliott KJ, et al. (2016). Vascular ADAM17 as a Novel Therapeutic Target in Mediating Cardiovascular Hypertrophy and Perivascular Fibrosis Induced by Angiotensin II. Hypertension, 68(4), 949–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda S (2016). ADAM and ADAMTS Family Proteins and Snake Venom Metalloproteinases: A Structural Overview. Toxins (Basel), 8(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda S, Igarashi T, Mori H, & Araki S (2006). Crystal structures of VAP1 reveal ADAMs’ MDC domain architecture and its unique C-shaped scaffold. Embo j, 25(11), 2388–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takimoto T, Nakao M, Nakajo T, Chinen Y, Kuroda J, & Taniwaki M (2016). Acute myocardial infarction as the initial thrombotic event of thrombotic thrombocytopenic purpura. Blood Coagul Fibrinolysis, 27(8), 948–951. [DOI] [PubMed] [Google Scholar]
- Takizawa M, Yatabe T, Okada A, Chijiiwa M, Mochizuki S, Ghosh P, et al. (2008). Calcium pentosan polysulfate directly inhibits enzymatic activity of ADAMTS4 (aggrecanase-1) in osteoarthritic chondrocytes. FEBS Lett, 582(19), 2945–2949. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Nanba D, Mori S, Shiba F, Ishiguro H, Yoshino K, et al. (2004). ADAM binding protein Eve-1 is required for ectodomain shedding of epidermal growth factor receptor ligands. J Biol Chem, 279(40), 41950–41959. [DOI] [PubMed] [Google Scholar]
- Tang JH, Zhang HM, Zhang ZH, & Zhang XL (2017). Effect of tetramethylpyrazine combined with cisplatin on VEGF, KLF4 and ADAMTS1 in Lewis lung cancer mice. Asian Pac J Trop Med, 10(8), 813–818. [DOI] [PubMed] [Google Scholar]
- Tang ZY, Loss G, Carmody I, & Cohen AJ (2006). TIMP-3 ameliorates hepatic ischemia/reperfusion injury through inhibition of tumor necrosis factor-alpha-converting enzyme activity in rats. Transplantation, 82(11), 1518–1523. [DOI] [PubMed] [Google Scholar]
- Tersteeg C, de Maat S, De Meyer SF, Smeets MW, Barendrecht AD, Roest M, et al. (2014). Plasmin cleavage of von Willebrand factor as an emergency bypass for ADAMTS13 deficiency in thrombotic microangiopathy. Circulation, 129(12), 1320–1331. [DOI] [PubMed] [Google Scholar]
- Thai SN, & Iruela-Arispe ML (2002). Expression of ADAMTS1 during murine development. Mech Dev, 115(1–2), 181–185. [DOI] [PubMed] [Google Scholar]
- Theodorou K, van der Vorst EPC, Gijbels MJ, Wolfs IMJ, Jeurissen M, Theelen TL, et al. (2017). Whole body and hematopoietic ADAM8 deficiency does not influence advanced atherosclerotic lesion development, despite its association with human plaque progression. Sci Rep, 7(1), 11670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timms KP, & Maurice SB (2020). Context-dependent bioactivity of versican fragments. Glycobiology, 30(6), 365–373. [DOI] [PubMed] [Google Scholar]
- Toba H, de Castro Bras LE, Baicu CF, Zile MR, Lindsey ML, & Bradshaw AD (2016). Increased ADAMTS1 mediates SPARC-dependent collagen deposition in the aging myocardium. Am J Physiol Endocrinol Metab, 310(11), E1027–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torabi F, Bogle OA, Estanyol JM, Oliva R, & Miller D (2017). Zona pellucida-binding protein 2 (ZPBP2) and several proteins containing BX7B motifs in human sperm may have hyaluronic acid binding or recognition properties. Mol Hum Reprod, 23(12), 803–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tortorella MD, Arner EC, Hills R, Easton A, Korte-Sarfaty J, Fok K, et al. (2004). Alpha2-macroglobulin is a novel substrate for ADAMTS-4 and ADAMTS-5 and represents an endogenous inhibitor of these enzymes. J Biol Chem, 279(17), 17554–17561. [DOI] [PubMed] [Google Scholar]
- Tortorella MD, Tomasselli AG, Mathis KJ, Schnute ME, Woodard SS, Munie G, et al. (2009). Structural and inhibition analysis reveals the mechanism of selectivity of a series of aggrecanase inhibitors. J Biol Chem, 284(36), 24185–24191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troeberg L, Fushimi K, Khokha R, Emonard H, Ghosh P, & Nagase H (2008). Calcium pentosan polysulfate is a multifaceted exosite inhibitor of aggrecanases. FASEB J, 22(10), 3515–3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troeberg L, Fushimi K, Scilabra SD, Nakamura H, Dive V, Thogersen IB, et al. (2009). The C-terminal domains of ADAMTS-4 and ADAMTS-5 promote association with N-TIMP-3. Matrix Biol, 28(8), 463–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troeberg L, Lazenbatt C, Anower EKMF, Freeman C, Federov O, Habuchi H, et al. (2014). Sulfated glycosaminoglycans control the extracellular trafficking and the activity of the metalloprotease inhibitor TIMP-3. Chem Biol, 21(10), 1300–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urisono Y, Sakata A, Matsui H, Kasuda S, Ono S, Yoshimoto K, et al. (2018). Von Willebrand Factor Aggravates Hepatic Ischemia-Reperfusion Injury by Promoting Neutrophil Recruitment in Mice. Thromb Haemost, 118(4), 700–708. [DOI] [PubMed] [Google Scholar]
- van der Vorst EP, Jeurissen M, Wolfs IM, Keijbeck A, Theodorou K, Wijnands E, et al. (2015). Myeloid A disintegrin and metalloproteinase domain 10 deficiency modulates atherosclerotic plaque composition by shifting the balance from inflammation toward fibrosis. Am J Pathol, 185(4), 1145–1155. [DOI] [PubMed] [Google Scholar]
- van der Vorst EP, Keijbeck AA, de Winther MP, & Donners MM (2012). A disintegrin and metalloproteases: molecular scissors in angiogenesis, inflammation and atherosclerosis. Atherosclerosis, 224(2), 302–308. [DOI] [PubMed] [Google Scholar]
- van der Vorst EPC, Weber C, & Donners M (2018). A Disintegrin and Metalloproteases (ADAMs) in Cardiovascular, Metabolic and Inflammatory Diseases: Aspects for Theranostic Approaches. Thromb Haemost, 118(7), 1167–1175. [DOI] [PubMed] [Google Scholar]
- Van Wart HE, & Birkedal-Hansen H (1990). The cysteine switch: a principle of regulation of metalloproteinase activity with potential applicability to the entire matrix metalloproteinase gene family. Proc Natl Acad Sci U S A, 87(14), 5578–5582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez F, Hastings G, Ortega MA, Lane TF, Oikemus S, Lombardo M, et al. (1999). METH-1, a human ortholog of ADAMTS-1, and METH-2 are members of a new family of proteins with angio-inhibitory activity. J Biol Chem, 274(33), 23349–23357. [DOI] [PubMed] [Google Scholar]
- Velasco J, Li J, DiPietro L, Stepp MA, Sandy JD, & Plaas A (2011). Adamts5 deletion blocks murine dermal repair through CD44-mediated aggrecan accumulation and modulation of transforming growth factor beta1 (TGFbeta1) signaling. J Biol Chem, 286(29), 26016–26027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vistnes M, Aronsen JM, Lunde IG, Sjaastad I, Carlson CR, & Christensen G (2014). Pentosan polysulfate decreases myocardial expression of the extracellular matrix enzyme ADAMTS4 and improves cardiac function in vivo in rats subjected to pressure overload by aortic banding. PLoS One, 9(3), e89621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorkapic E, Folkesson M, Magnell K, Bohlooly YM, Lanne T, & Wagsater D (2017). ADAMTS-1 in abdominal aortic aneurysm. PLoS One, 12(6), e0178729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuohelainen V, Raitoharju E, Levula M, Lehtimaki T, Pelto-Huikko M, Honkanen T, et al. (2011). Myocardial infarction induces early increased remote ADAM8 expression of rat hearts after cardiac arrest. Scand J Clin Lab Invest, 71(7), 553–562. [DOI] [PubMed] [Google Scholar]
- Wagsater D, Bjork H, Zhu C, Bjorkegren J, Valen G, Hamsten A, et al. (2008). ADAMTS-4 and −8 are inflammatory regulated enzymes expressed in macrophage-rich areas of human atherosclerotic plaques. Atherosclerosis, 196(2), 514–522. [DOI] [PubMed] [Google Scholar]
- Wang L, Fan W, Cai P, Fan M, Zhu X, Dai Y, et al. (2013). Recombinant ADAMTS13 reduces tissue plasminogen activator-induced hemorrhage after stroke in mice. Ann Neurol, 73(2), 189–198. [DOI] [PubMed] [Google Scholar]
- Wang L, Hoggard JA, Korleski ED, Long GV, Ree BC, Hensley K, et al. (2018). Multiple non-catalytic ADAMs are novel integrin alpha4 ligands. Mol Cell Biochem, 442(1–2), 29–38. [DOI] [PubMed] [Google Scholar]
- Wang L, Wang X, & Kong W (2010). ADAMTS-7, a novel proteolytic culprit in vascular remodeling. Sheng Li Xue Bao, 62(4), 285–294. [PubMed] [Google Scholar]
- Wang L, Zheng J, Bai X, Liu B, Liu CJ, Xu Q, et al. (2009). ADAMTS-7 mediates vascular smooth muscle cell migration and neointima formation in balloon-injured rat arteries. Circ Res, 104(5), 688–698. [DOI] [PubMed] [Google Scholar]
- Wang R, Li Y, Tsung A, Huang H, Du Q, Yang M, et al. (2018). iNOS promotes CD24(+)CD133(+) liver cancer stem cell phenotype through a TACE/ADAM17-dependent Notch signaling pathway. Proc Natl Acad Sci U S A, 115(43), E10127–E10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WM, Ge G, Lim NH, Nagase H, & Greenspan DS (2006). TIMP-3 inhibits the procollagen N-proteinase ADAMTS-2. Biochem J, 398(3), 515–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Chen W, Zhang J, Khan A, Li L, Huang F, et al. (2017). Critical Role of ADAMTS2 (A Disintegrin and Metalloproteinase With Thrombospondin Motifs 2) in Cardiac Hypertrophy Induced by Pressure Overload. Hypertension, 69(6), 1060–1069. [DOI] [PubMed] [Google Scholar]
- Wang X, Chow FL, Oka T, Hao L, Lopez-Campistrous A, Kelly S, et al. (2009). Matrix metalloproteinase-7 and ADAM-12 (a disintegrin and metalloproteinase-12) define a signaling axis in agonist-induced hypertension and cardiac hypertrophy. Circulation, 119(18), 2480–2489. [DOI] [PubMed] [Google Scholar]
- Wang X, Oka T, Chow FL, Cooper SB, Odenbach J, Lopaschuk GD, et al. (2009). Tumor necrosis factor-alpha-converting enzyme is a key regulator of agonist-induced cardiac hypertrophy and fibrosis. Hypertension, 54(3), 575–582. [DOI] [PubMed] [Google Scholar]
- Wang X, Zhang S, Li L, Hua J, Zhu L, & Zhang G (2018). Ticagrelor-induced thrombotic thrombocytopenic purpura: A case report and review of the literature. Medicine (Baltimore), 97(26), e11206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster NJ, Green KN, Settle VJ, Peers C, & Vaughan PF (2004). Altered processing of the amyloid precursor protein and decreased expression of ADAM 10 by chronic hypoxia in SH-SY5Y: no role for the stress-activated JNK and p38 signalling pathways. Brain Res Mol Brain Res, 130(1–2), 161–169. [DOI] [PubMed] [Google Scholar]
- Weskamp G, Cai H, Brodie TA, Higashyama S, Manova K, Ludwig T, et al. (2002). Mice lacking the metalloprotease-disintegrin MDC9 (ADAM9) have no evident major abnormalities during development or adult life. Mol Cell Biol, 22(5), 1537–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weskamp G, Schlondorff J, Lum L, Becherer JD, Kim TW, Saftig P, et al. (2004). Evidence for a critical role of the tumor necrosis factor alpha convertase (TACE) in ectodomain shedding of the p75 neurotrophin receptor (p75NTR). J Biol Chem, 279(6), 4241–4249. [DOI] [PubMed] [Google Scholar]
- Wichert R, Scharfenberg F, Colmorgen C, Koudelka T, Schwarz J, Wetzel S, et al. (2019). Meprin beta induces activities of A disintegrin and metalloproteinases 9, 10, and 17 by specific prodomain cleavage. FASEB J, 33(11), 11925–11940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witsch T, Martinod K, Sorvillo N, Portier I, De Meyer SF, & Wagner DD (2018). Recombinant Human ADAMTS13 Treatment Improves Myocardial Remodeling and Functionality After Pressure Overload Injury in Mice. J Am Heart Assoc, 7(3):e007004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf C, Qian Y, Brooke MA, Kelsell DP, & Franzke CW (2016). ADAM17/EGFR axis promotes transglutaminase-dependent skin barrier formation through phospholipase C gamma1 and protein kinase C pathways. Sci Rep, 6, 39780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf ZT, Brand HA, Shaffer JR, Leslie EJ, Arzi B, Willet CE, et al. (2015). Genome-wide association studies in dogs and humans identify ADAMTS20 as a risk variant for cleft lip and palate. PLoS Genet, 11(3), e1005059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Zhou Y, Li Y, Li J, Ke Y, Wang Y, et al. (2015). Association between plasma ADAMTS-7 levels and ventricular remodeling in patients with acute myocardial infarction. Eur J Med Res, 20, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wunnemann F, Ta-Shma A, Preuss C, Leclerc S, van Vliet PP, Oneglia A, et al. (2020). Loss of ADAMTS19 causes progressive non-syndromic heart valve disease. Nat Genet, 52(1), 40–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang M, Luo H, Wu J, Ren L, Ding X, Wu C, et al. (2018). ADAM23 in Cardiomyocyte Inhibits Cardiac Hypertrophy by Targeting FAK - AKT Signaling. J Am Heart Assoc, 7(18), e008604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y, de Groot R, Crawley JT, & Lane DA (2011). Mechanism of von Willebrand factor scissile bond cleavage by a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13). Proc Natl Acad Sci U S A, 108(28), 11602–11607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y, Wang M, Chen H, & Chen L (2021). Potential therapeutic approaches for the early entry of SARS-CoV-2 by interrupting the interaction between the spike protein on SARS-CoV-2 and angiotensin-converting enzyme 2 (ACE2). Biochem Pharmacol, 192:114724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Q, Zhang F, Grassia G, Hu Y, Zhang Z, Xing Q, et al. (2014). Matrix metalloproteinase-8 promotes vascular smooth muscle cell proliferation and neointima formation. Arterioscler Thromb Vasc Biol, 34(1), 90–98. [DOI] [PubMed] [Google Scholar]
- Xie B, Shen J, Dong A, Swaim M, Hackett SF, Wyder L, et al. (2008). An Adam15 amplification loop promotes vascular endothelial growth factor-induced ocular neovascularization. FASEB J, 22(8), 2775–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Cao Y, Yang X, Cai P, Kang L, Zhu X, et al. (2017). ADAMTS13 controls vascular remodeling by modifying VWF reactivity during stroke recovery. Blood, 130(1), 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Xu J, Molinas AJR, Mukerjee S, Morgan DA, Rahmouni K, Zsombok A, et al. (2019). Activation of ADAM17 (A Disintegrin and Metalloprotease 17) on Glutamatergic Neurons Selectively Promotes Sympathoexcitation. Hypertension, 73(6), 1266–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Sriramula S, & Lazartigues E (2018). Excessive Glutamate Stimulation Impairs ACE2 Activity Through ADAM17-Mediated Shedding in Cultured Cortical Neurons. Cell Mol Neurobiol, 38(6), 1235–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Sriramula S, Xia H, Moreno-Walton L, Culicchia F, Domenig O, et al. (2017). Clinical Relevance and Role of Neuronal AT1 Receptors in ADAM17-Mediated ACE2 Shedding in Neurogenic Hypertension. Circ Res, 121(1), 43–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagami-Hiromasa T, Sato T, Kurisaki T, Kamijo K, Nabeshima Y, & Fujisawa-Sehara A (1995). A metalloprotease-disintegrin participating in myoblast fusion. Nature, 377(6550), 652–656. [DOI] [PubMed] [Google Scholar]
- Yang F, Chen Q, Yang M, Maguire EM, Yu X, He S, et al. (2020). Macrophage-derived MMP-8 determines smooth muscle cell differentiation from adventitia stem/progenitor cells and promotes neointima hyperplasia. Cardiovasc Res, 116(1), 211–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Hu C, Song Q, Li Y, Da X, Yu Y, et al. (2020). ADAMTS16 activates latent TGF-beta, accentuating fibrosis and dysfunction of the pressure-overloaded heart. Cardiovasc Res, 116(5), 956–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim V, Noisier AFM, Hung KY, Bartsch JW, Schlomann U, & Brimble MA (2016). Synthesis and biological evaluation of analogues of the potent ADAM8 inhibitor cyclo(RLsKDK) for the treatment of inflammatory diseases and cancer metastasis. Bioorg Med Chem, 24(18), 4032–4037. [DOI] [PubMed] [Google Scholar]
- Yu J, Zhou B, Yu H, Han J, Cui M, Zhang F, et al. (2016). Association between plasma ADAMTS-7 levels and severity of disease in patients with stable obstructive coronary artery disease. Medicine (Baltimore), 95(48), e5523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Shi J, Wang X, & Zhang F (2019). Propofol affects the growth and metastasis of pancreatic cancer via ADAM8. Pharmacol Rep, 72(2), 418–426. [DOI] [PubMed] [Google Scholar]
- Zeinieh M, Salehi A, Rajkumar V, & Barker PA (2015). p75NTR-dependent Rac1 activation requires receptor cleavage and activation of an NRAGE and NEDD9 signaling cascade. J Cell Sci, 128(3), 447–459. [DOI] [PubMed] [Google Scholar]
- Zeng SY, Chen X, Chen SR, Li Q, Wang YH, Zou J, et al. (2013). Upregulation of Nox4 promotes angiotensin II-induced epidermal growth factor receptor activation and subsequent cardiac hypertrophy by increasing ADAM17 expression. Can J Cardiol, 29(10), 1310–1319. [DOI] [PubMed] [Google Scholar]
- Zeng SY, Lu HQ, Yan QJ, & Zou J (2018). A Reduction in ADAM17 Expression Is Involved in the Protective Effect of the PPAR-alpha Activator Fenofibrate on Pressure Overload-Induced Cardiac Hypertrophy. PPAR Res, 2018, 7916953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha Y, Chen Y, Xu F, Li T, Zhao C, & Cui L (2010). ADAMTS4 level in patients with stable coronary artery disease and acute coronary syndromes. Biomed Pharmacother, 64(3), 160–164. [DOI] [PubMed] [Google Scholar]
- Zhai CG, Xu YY, Tie YY, Zhang Y, Chen WQ, Ji XP, et al. (2018). DKK3 overexpression attenuates cardiac hypertrophy and fibrosis in an angiotensin-perfused animal model by regulating the ADAM17/ACE2 and GSK-3beta/beta-catenin pathways. J Mol Cell Cardiol, 114, 243–252. [DOI] [PubMed] [Google Scholar]
- Zhai M, Guo J, Ma H, Shi W, Jou D, Yan D, et al. (2018). Ursolic acid prevents angiotensin II-induced abdominal aortic aneurysm in apolipoprotein E-knockout mice. Atherosclerosis, 271, 128–135. [DOI] [PubMed] [Google Scholar]
- Zhang C, Tian L, Chi C, Wu X, Yang X, Han M, et al. (2010). Adam10 is essential for early embryonic cardiovascular development. Dev Dyn, 239(10), 2594–2602. [DOI] [PubMed] [Google Scholar]
- Zhang L, Yu F, Wang L, Zheng J, Du Y, Huang Y, et al. (2015). ADAMTS-7 promotes vascular smooth muscle cells proliferation in vitro and in vivo. Sci China Life Sci, 58(7), 674–681. [DOI] [PubMed] [Google Scholar]
- Zhang P, Shen M, Fernandez-Patron C, & Kassiri Z (2016). ADAMs family and relatives in cardiovascular physiology and pathology. J Mol Cell Cardiol, 93, 186–199. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Thomas SM, Lui VW, Xi S, Siegfried JM, Fan H, et al. (2006). Phosphorylation of TNF-alpha converting enzyme by gastrin-releasing peptide induces amphiregulin release and EGF receptor activation. Proc Natl Acad Sci U S A, 103(18), 6901–6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SM, Jiang L, Zhao X, Liu JF, Liang B, Liu C, et al. (2019). A disintegrin and metalloprotease 22 accelerates neointima formation by activating ERK signaling. Atherosclerosis, 283, 92–99. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wang Y, Zhou D, Zhang LS, Deng FX, Shu S, et al. (2019). Angiotensin II deteriorates advanced atherosclerosis by promoting MerTK cleavage and impairing efferocytosis through the AT1R/ROS/p38 MAPK/ADAM17 pathway. Am J Physiol Cell Physiol, 317(4), C776–C787. [DOI] [PubMed] [Google Scholar]
- Zhao BQ, Chauhan AK, Canault M, Patten IS, Yang JJ, Dockal M, et al. (2009). von Willebrand factor-cleaving protease ADAMTS13 reduces ischemic brain injury in experimental stroke. Blood, 114(15), 3329–3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng DY, Zhao J, Yang JM, Wang M, & Zhang XT (2016). Enhanced ADAM17 expression is associated with cardiac remodeling in rats with acute myocardial infarction. Life Sci, 151, 61–69. [DOI] [PubMed] [Google Scholar]
- Zheng X, Chung D, Takayama TK, Majerus EM, Sadler JE, & Fujikawa K (2001). Structure of von Willebrand factor-cleaving protease (ADAMTS13), a metalloprotease involved in thrombotic thrombocytopenic purpura. J Biol Chem, 276(44), 41059–41063. [DOI] [PubMed] [Google Scholar]
- Zheng X, Majerus EM, & Sadler JE (2002). ADAMTS13 and TTP. Curr Opin Hematol, 9(5), 389–394. [DOI] [PubMed] [Google Scholar]
- Zheng XL (2015). ADAMTS13 and von Willebrand factor in thrombotic thrombocytopenic purpura. Annu Rev Med, 66, 211–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HM, Weskamp G, Chesneau V, Sahin U, Vortkamp A, Horiuchi K, et al. (2004). Essential role for ADAM19 in cardiovascular morphogenesis. Mol Cell Biol, 24(1), 96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Dai W, Cui Y, & Li Y (2020). Estrogen downregulates gp130 expression in HUVECs by regulating ADAM10 and ADAM17 via the estrogen receptor. Biochem Biophys Res Commun, 523(3), 753–758. [DOI] [PubMed] [Google Scholar]
- Zhou S, Jiang S, Guo J, Xu N, Wang Q, Zhang G, et al. (2019). ADAMTS13 protects mice against renal ischemia-reperfusion injury by reducing inflammation and improving endothelial function. Am J Physiol Renal Physiol, 316(1), F134–F145. [DOI] [PubMed] [Google Scholar]
- Zhu R, Cheng M, Lu T, Yang N, Ye S, Pan YH, et al. (2018). A Disintegrin and Metalloproteinase with Thrombospondin Motifs 18 Deficiency Leads to Visceral Adiposity and Associated Metabolic Syndrome in Mice. Am J Pathol, 188(2), 461–473. [DOI] [PubMed] [Google Scholar]
- Zhu Z, Zhu J, Zhao X, Yang K, Lu L, Zhang F, et al. (2015). All-Trans Retinoic Acid Ameliorates Myocardial Ischemia/Reperfusion Injury by Reducing Cardiomyocyte Apoptosis. PLoS One, 10(7), e0133414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang X, Li R, Maimaitijiang A, Liu R, Yan F, Hu H, et al. (2019). miR-221–3p inhibits oxidized low-density lipoprotein induced oxidative stress and apoptosis via targeting a disintegrin and metalloprotease-22. J Cell Biochem, 120(4), 6304–6314. [DOI] [PubMed] [Google Scholar]
- Zou J, Zhu F, Liu J, Wang W, Zhang R, Garlisi CG, et al. (2004). Catalytic activity of human ADAM33. J Biol Chem, 279(11), 9818–9830. [DOI] [PubMed] [Google Scholar]