

Abstract
The design and manufacture of an origami-based liver-on-a-chip device are presented, together with demonstrations of the chip’s effectiveness at recapitulating some of the liver’s key in vivo architecture, physical microenvironment, and functions. Laser-cut layers of polyimide tape are folded together with polycarbonate nanoporous membranes to create a stack of three adjacent flow chambers separated by the membranes. Endothelial cells are seeded in the upper and lower flow chambers to simulate sinusoids, and hepatocytes are seeded in the middle flow chamber. Nutrients and metabolites flow through the simulated sinusoids and diffuse between the vascular pathways and the hepatocyte layers, mimicking physiological microcirculation. Studies of cell viability, metabolic functions, and hepatotoxicity of pharmaceutical compounds show that the endothelialized liver-on-a-chip model is conducive to maintaining hepatocyte functions and evaluation of the hepatotoxicity of drugs. Our unique origami approach speeds chip development and optimization, effectively simplifying the laboratory-scale fabrication of on-chip models of human tissues without necessarily reducing their structural and functional sophistication.
Keywords: organ-on-a-chip, tissue modeling, biofabrication, vascularization
Graphical Abstract
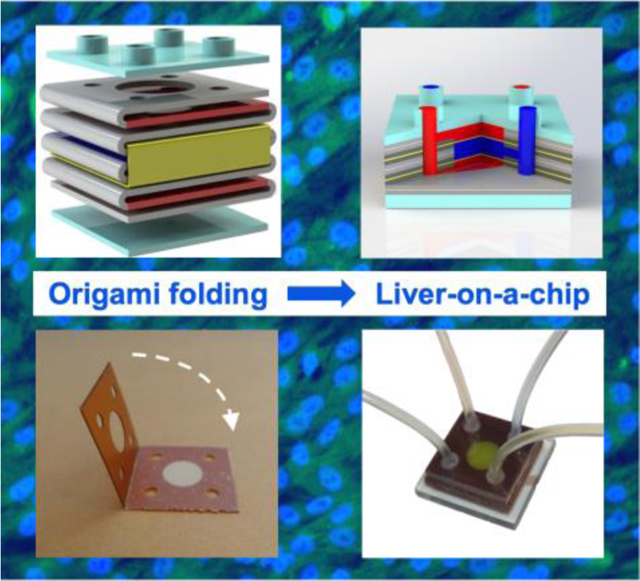
The origami-based fabrication and demonstration of a liver-on-a-chip are presented. Studies of the liver-on-a-chip demonstrate cell viability, metabolic functions, and hepatotoxicity of pharmaceuticals. The origami approach speeds chip development and optimization, simplifying the laboratory-scale fabrication of on-chip models of human tissues while retaining relevant structures and functions.
The liver is the largest metabolic organ and plays an important role in several critical functions such as glycogen storage, lipid and plasma protein biosynthesis, secretion of bile acid, drug detoxification, and endocrine signaling.[1] Hepatocytes are the primary functional cells of the liver, comprising ~80% of the liver mass and playing a central role in liver functions.[1,2] In vitro primary hepatocyte culture methods that closely emulate the in vivo microenvironment provide a potential platform for evaluating drug toxicity and screening of therapeutics, which in turn improves the success rate of drug discovery in clinical trials.
In recent years, microfabrication and microfluidic technologies have been employed to develop in vitro three-dimensional (3D) liver culture models for drug metabolism studies, including primary hepatocyte monocultures or co-cultures with non-parenchymal cells.[3–5] These 3D liver culture models include hepatic spheroids,[6,7] micropatterned[8–11] or bioprinted hepatic constructs,[12–15] and liver-on-a-chip models [16–19] that facilitate the recreation of in vivo normal liver physiological or pathophysiological conditions and the assessment of drug responses. Microfluidic systems allow the precise control of physiologically relevant parameters, such as flow rate, nutrients, and oxygen that are essential for the maintenance of hepatocyte functions in vitro. Nevertheless, the creation of in vivo-like liver-on-a-chip devices is challenging from both functional and fabrication perspectives. Functionally, conventional microfluidic chips struggle to replicate the flow along liver sinusoids in tandem with the transport of species between sinusoids and hepatocytes (Figure 1a) in a 3D architecture. In addition, the fabrication of liver-on-a-chip devices usually requires dedicated microfabrication facilities (e.g., soft lithography) and multi-step processes to realize the proper tissue chip architecture.
Figure 1.

(a) 3D architecture of liver, liver lobule, and sinusoid structure; (b) conceptual schematic cross-section of an oLOC, showing a semipermeable membrane that has been folded, seeded on opposite sides with hepatocyte and endothelial cell monolayers, and capped with top and bottom plates to define upper and lower flow channels (vascular chambers) that mimic sinusoids and a central static chamber that contains hepatocyte monolayers on the upper and lower membrane surfaces; (c) schematic diagram of the implemented oLOC, showing membrane layers (black and white speckle), flowing medium pathways (red), static medium pathway (blue), hepatocytes (purple), endothelial cells (maroon), and how it simulates liver structure with functions of flow (red arrows) and transport via diffusion (white arrows).
Origami-based design and implementation offer a potential solution to both functional and fabrication challenges. In conventional microfluidic chips, microfabricated layers of a single base material (e.g. polydimethylsiloxane (PDMS) [20] or thermoplastics such as polystyrene,[21] polycarbonate,[22] or poly(methyl methacrylate) (PMMA)[23]) are created using soft lithography or other techniques[20] and bonded together in stacks to define a network of flow channels.[24] In the origami-based approach, pre-patterned layers of multiple base materials are conveniently folded together along pre-defined fold lines into precisely aligned, multi-material stacks that can define not only a network of flow channels, but also structures that enable functions beyond simple flow.
Figure 1 shows the integration of two core modes of transport in the liver – continuous flow along sinusoids and transport between sinusoids and hepatocyte layers – into a multi-material chip comprising three adjacent folded compartments separated by two polycarbonate nanoporous membrane layers (Figure 1b). The outermost two flow channels are vascular chambers that simulate sinusoids. Each simulated sinusoid is bounded on the outer edge by a solid channel wall and on the inner edge by a nanoporous membrane. The center channel is the hepatocyte chamber and is bounded by the two nanoporous membranes. Both nanoporous membranes are seeded with an endothelial monolayer on the sinusoid side (shown in red in Figure 1c) and with a monolayer of hepatocytes on the hepatocyte side (shown in blue in Figure 1c). Similar in some respects to the space of Disse and the fenestrated endothelium in native liver tissue, the nanoporous membrane and its endothelial monolayer permit diffusion between the simulated sinusoids and the hepatocyte monolayers while preventing direct flow past the hepatocytes to protect them from shear stress.
The inlets, outlets, flow connections, perforated fold lines, and in-plane boundaries of the simulated sinusoids and hepatocyte layers are defined in patterned polyimide tape. By folding the nanoporous membranes and the nonporous polyimide tapes together into a multi-material stack, the origami approach integrates both flow channels and diffusion interfaces between cellular monolayers within a single, rapidly fabricated device. Liver functions are achieved in this flow-diffusion architecture by seeding the cellular monolayers on opposite sides of the nanoporous membrane that lies between the sinusoids and the hepatocyte chambers.
The purpose of this study was to demonstrate that a proof-of-concept origami-based liver-on-a-chip (oLOC) recapitulates some of the key in vivo architectural and physical microenvironmental parameters to support primary hepatocyte culture under physiological microcirculation. An important feature of our oLOC platform was that it employed a simple fabrication strategy to enable continuous circulation of medium through the upper and lower chambers (simulated sinusoids) in close proximity to the primary hepatocytes in the central chamber in order to refill the cellular microenvironment with nutrients while carrying away the waste products. By enabling chemical interactions between the two key cell types, the present study highlighted that the endothelialized liver model described here is conducive to maintaining cell viability and some of the key hepatocyte functions and demonstrated its potential applicability for the evaluation of the hepatotoxicity of pharmaceutical compounds. The results effectively showcase the possible utility of our unique origami method for the rapid generation of architecturally and functionally relevant organ-on-a-chip models in the future beyond those of the liver. Although similar levels of device complexity are also achievable with conventional strategies, conventional fabrication can typically be more complicated than the present origami approach.
One of the advantages of the origami platform is the ease and speed with which new design variations can be implemented in the laboratory from prepatterned materials stored on the shelf. The demonstrated per-chip time for manual fabrication from laser-cut starting materials is about 12 minutes. This rapid turnaround time from idea to implementation enables device customization and optimization to be carried out extremely quickly. A second advantage of the origami approach is that the folded features are self-aligning. The perforated fold lines and relatively stiff tape constrain the folding kinematics so that the tape can only fold one way. The precise alignment happens automatically, without the equipment required for example to align PDMS layers and without the misalignments that are inherent in conventional layer bonding. A third key advantage of the origami platform is its flexibility and ability to create different types of architectures. This flexibility comes in part from the ability to pattern different geometries into the layers prior to folding, like the rectangular structures described below and the hexagonal structures shown in supplementary Figure S1a–c. Another important source of flexibility and customizability is the ability to pick and choose which portions of the pre-patterned starting materials will be folded into the final structure. Such customizations are often more complicated with conventional chip-fabrication methods.
The flexibility to choose which portions of the pre-patterned starting materials are incorporated into a given device played a pivotal role in the design optimization of the oLOC. Three generations of oLOC devices were created, all from the same kind of starting material, and they were used for three successive rounds of testing (one round of flow testing and two rounds of testing with cells). The three generations are similar, but the precise layer sequences and assembly processes for the Generation 2 and Generation 3 devices were refined based on the results from the previous generations.
Figure 2a illustrates typical starting materials, including the strip of 240-μm-thick patterned polyimide tape (red, dark blue, and gray) and the nanoporous membrane (yellow). The polyimide tape provides the solid boundary around the channels and comprises two types of square panels connected by perforated folding joints. The flow panels (F, red, and dark blue) contain two sets of circular inlets and outlets at the corners (one for the simulated sinusoids and one for the hepatocyte chambers), a circular central chamber, and tapered flow channels that connect each central chamber to one of the inlet-outlet pairs. The F panels are included to enable through-chip flow. The chamber panels (C, gray) contain only the inlets, outlets, and central chambers, with no connecting channels. The C panels are mechanically stiffer than the F panels and are included to enable robust, leak-free assembly of the folded devices. The nanoporous membrane (M, yellow) supports the hepatocyte and endothelial monolayers and is patterned so that each square includes only the inlets and outlets at the corners. The M layers prevent leakage flow between the simulated sinusoids and hepatocyte chamber while permitting diffusion between adjacent chambers across the membrane; the inlets and outlets are defined at the corners of the M layers to allow flow into and out of the chambers. The layers are sealed together by the acrylic adhesive of the double-sided polyimide tape. Acrylic adhesive is chosen for its medical compatibility, and only its cross-section is exposed to fluid in the chip. Finally, acrylic plates (A, light blue) cap the stack at the top and bottom and provide fluid connections (top) as shown in Figure 2b.
Figure 2.

(a) Schematic diagram of the laser-patterned polyimide tape and nanoporous membrane oriented for cross-folding; (b) exploded schematic diagram of the folded structure of the oLOC (gray, red, dark blue, and yellow) and its top and bottom acrylic caps (light blue); (c) schematic and (d) cross-sectional cutaway diagrams of the completed oLOC; (e - j) photographs showing the fabrication process for the Generation 3 devices, including (e) hole punching and trimming to pattern the nanoporous membrane, (f) folding the nanoporous membrane between chamber panels to form origami sub-units, (g) layering flow panels to form additional sub-units, (h) stacking the origami sub-units on top of the solid acrylic base plate to form the device’s flow structure, (i) capping the stack with an acrylic top plate (laser-cut with four inlet/outlet ports) and adhering PVC tubing into ports, and (j) final Generation 3 device structure after assembly; (k) flow pattern of Generation 3 device shown by flowing yellow dye through the simulated sinusoids and blue dye through the hepatocyte chambers; (l) plot of concentration of trypan blue in the flow exiting the middle flow path as a function of inverse flow rate when a 0.1% solution of trypan blue in deionized water is flowed through a chip’s upper and lower flow paths and deionized water is flowed through its middle flow path with no pressure difference between flow paths.
Because the oLOC devices are built from precisely-designed panels, each patterned with flow features and connected together in strips by perforated folding joints, it is possible to store the raw material for many devices on the shelf and fold it easily, as needed, to form a variety of customized devices. For example, the Generation 1 chips (used for tests of flow and diffusion functions) have a panel sequence of A-C-F-C-M-C-F-C-M-C-F-C-A that produces three 720-μm-thick circular chambers, each of which is bounded in the plane by three folded layers of polyimide tape (Figure S2a,b). This panel sequence is identical to the sequence patterned into the polyimide tape shown in Figure 2a and represents the initial device design. Each chip contains three parallel flow paths (upper, middle, and lower) separated by two nanoporous membrane layers. The upper and lower chambers (the simulated sinusoids) connect to one inlet-outlet pair, and the middle chamber (the hepatocyte chamber) connects to the other inlet-outlet pair.
The Generation 2 chips were used for initial testing of the oLOCs with the human hepatocellular carcinoma (HepG2) cell line. They are identical to the Generation 1 chips, except that they omit the C panels adjacent to the acrylic plates at the top and bottom of the stack. This design change represents an effort to simplify the structure by omitting layers that were believed at the time to be unnecessary, and it thins the simulated sinusoid chambers at the top and bottom of the stack to 480-μm thickness while leaving the 720-μm-thick circular hepatocyte chamber unchanged. The complete panel sequence of the Generation 2 chips is A-F-C-M-C-F-C-M-C-F-A (Figure S2c,d). However, in some cases the resulting chamber heights were found to be too small for robust cell seeding.
Based on these findings, the Generation 3 chips (used for the oLOCs with primary human hepatocytes) increased the thickness of each of the three chambers by 240 μm (one additional polyimide panel). The increased chamber thicknesses minimize surface tension effects to reduce bubble formation and increase the number of cells in the chamber during cell seeding, both of which promote the formation of uniform cell layers. The panel sequence for the Generation 3 devices is A-F-F-C-M-C-F-F-C-M-C-F-F-A, yielding two 720-μm-thick circular sinusoid chambers and one 960-μm-thick circular hepatocyte chamber. To create this new panel sequence from the available starting material, the structure was folded piecewise in smaller sub-units, which were then combined to form the final oLOC chip. Folding the structure in smaller sub-units enabled the inclusion of additional panels and the increase of the chamber heights, ultimately improving the Generation 3 design. Figure 2c–d illustrates the final layer structure of the Generation 3 chips.
Figure 2e–j shows photographs of the folding process for the Generation 3 devices. After the nanoporous membrane was placed on a C panel and punched to define the inlets and outlets (Figure 2e), the C panels were folded around the membrane to form a sub-unit (Figure 2f). The second sub-unit comprises two F panels joined together (Figure 2g). Figure 2h shows the sub-units being stacked on the acrylic bottom plate; the flow panels F are visible on top of the underlying layers. After the stack was capped with the acrylic top plate, PVC tubes were adhered in the plate’s inlet and outlet ports (Figure 2i) to form the final oLOC chip (Figure 2j). The architecture of the separate flow pathways is illustrated in Figure 2k by passing yellow and blue dye through the outer and middle flow pathways of a Generation 1 oLOC chip.
The oLOC should be free of leakage, and its correct operation depends on the only transport between the sinusoid chambers and hepatocyte chambers taking place through the nanoporous membrane. Transport through the membrane is primarily diffusive, with a much smaller contribution from pressure-driven transport through the porous membrane under the slightly unequal operational pressures in the sinusoid and hepatocyte chambers. The device is designed to minimize the potential for leakage between the sinusoid and hepatocyte chambers; as long as the nanoporous membranes are intact, the only pathway for leakage flow would be along the adhesive interface to the opposite set of inlet/outlet ports.
Two types of tests confirmed that the sinusoid chambers and hepatocyte chamber are connected solely via diffusion rather than via direct leakage. In the first set of flow tests, approximately 16-μm-diameter polystyrene microspheres were suspended in ethanol, and the mixture was flowed through the hepatocyte chambers of the Generation 1 chip. Ethanol without suspended microspheres was flowed through the sinusoid chambers, and the exiting flows from hepatocyte and sinusoid chambers were examined under an optical microscope. Microspheres were observed only in the exiting hepatocyte chamber flow, indicating that there is no leakage between hepatocyte and sinusoid chambers at the 16-μm scale or above. Images of the exiting flow from the hepatocyte and simulated sinusoid chambers of the hexagonal structure of Figure S1a–c are shown in Figure S1d,e.
In the second set of leakage tests, water was flowed through the middle flow path (hepatocyte chamber) of the Generation 1 device while a solution of 0.1% trypan blue was flowed through its upper and lower flow paths (sinusoid chambers). Direct leakage and transport across the nanoporous membranes would both be reflected by an increase in trypan blue concentration in the flow exiting the middle (water) flow path. The mechanisms can be distinguished by the dependence of concentration change on flow rate. Purely diffusive transport would obey Fick’s laws of diffusion, with the concentration change being proportional to residence time (i.e. inverse flow rate). This proportionality would be expected when there is no direct leakage pathway or pressure difference across the membrane. In contrast, direct leakage would introduce an additional concentration change that is substantially independent of flow rate. Figure 2l plots the change in concentration of trypan blue in the middle flow vs. inverse flow rate through the middle path for a device in which there is no pressure difference across the nanoporous membranes; three devices were tested, and no statistically significant differences were observed among the results.[25] The concentration change is linear with inverse flow rate and passes through the origin to within the uncertainty of the measurement, indicating that the transport is diffusive rather than driven by leakage flow.
A small pressure difference develops across the nanoporous membranes during operation. To confirm that the devices remain leak tight under those conditions, the flow was driven by higher pressures across the upper and lower flow paths and by lower pressures across the middle flow path to produce a pressure difference across the membrane that was more than 50X its operational value. No leakage was observed under these conditions. Although the measured concentration change included a constant superimposed on the previously-observed proportional relationship (Figure S3), the magnitude of the offset matches Darcy’s law predictions for pressure-driven flow through the membrane to within the measurement uncertainty. The measurement was made in three devices, and no statistically significant differences were observed among the results. This result confirms that all transport takes place through the membrane (i.e. there is no leakage flow), even when a pressure difference develops between adjacent flow channels.
Hepatocytes form a critical cell layer and have a uniquely organized structure with a basal membrane facing liver sinusoidal endothelial cells.[26,27] Maintenance of hepatic phenotypes and functionality is crucial for evaluating drug-induced hepatotoxicity in vitro. To ascertain whether the oLOC sustains functional hepatocytes and the endothelial layer, primary human hepatocytes (primary hepatocytes) and human umbilical vein endothelial cells (HUVECs) were perfusion-cultured for 7 days in Generation 3 oLOCs and assessed for their viability and functions. The viability of primary hepatocytes and HUVECs within the endothelialized oLOC with initial cell seeding density of 5 × 106 cells mL−1 was determined at day 3 and day 7 using calcein AM and ethidium homodimer 1 (EthD-1) staining. Calcein AM is a non-fluorescent cell-permeable compound that is hydrolyzed into the green fluorescent anion calcein by intracellular esterase in live cells, whereas EthD-1 is a dead cell-specific red fluorescent dye that binds to DNA. Figure 3a represents the calcein AM stained live cells (green) and EthD-1 stained dead cells (red) at day 7. Further, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) (MTS) cell proliferation assay was performed for quantification of the cells’ metabolic activity in oLOCs at day 3 and 7. The NAD(P)H-dependent dehydrogenase enzymes in metabolically active cells reduce MTS tetrazolium compound to produce soluble and colored formazan product. The quantity of formazan product produced in the culture, as measured by the amount of 490-nm absorbance, is a measure of the metabolic activity of cells under given conditions and can to a good extent indicate cell viability. The quantified metabolic activity of both primary hepatocytes and HUVECs within the oLOC was found to be approximately 84.1% for primary hepatocytes and 87.0% for HUVECs at day 3 and 89.3% for primary hepatocytes and 89.9% for HUVECs at day 7 (Figure 3b). Similarly, to evaluate the applicability of the oLOC for the study of liver diseases such as hepatocellular carcinoma, HepG2 cells were cultured with HUVECs in Generation 2 oLOCs and assessed for cell viability and metabolic activity in a similar manner and duration. The quantified metabolic activity of HepG2 cells and HUVECs in the oLOC was found to be approximately 85.9% for HepG2 cells and 82.6% for HUVECs at day 3 and 89.6% for HepG2 cells and 92.6% for HUVECs at day 7 (Figure S4a,b).
Figure 3.

(a) Viability of primary hepatocytes and HUVECs assessed using calcein AM (green) and EthD-1 (red) staining on day 7 of culture; (b) quantified metabolic activities of primary hepatocytes and HUVECs at different periods of culture; (c) F-actin and nuclei staining of primary hepatocytes and HUVECs at day 7 of culture; (d) immunostaining of primary hepatocytes for CYP3A and HUVECs for CD31 and VE-cadherin at day 7 of culture.
F-actin is the most abundant cytoskeletal filamentous protein. It determines the shape, stiffness, and movement of the cell surface, and it facilitates the transduction of mechanical signals as well as generating the intracellular forces required for many cellular functions.[28] Phalloidin possesses a high binding affinity for F-actin subunits; thus fluorescent derivatives of phalloidin represent the gold standard for staining and visualizing cellular F-actin filaments.[29] Fluorescence micrographs of primary hepatocytes and HUVECs stained for F-actin and nuclei showed that these cells were homogenously distributed within their respective chambers (Figure 3c) in the oLOC. Similarly, fluorescence micrographs of HepG2 cells and HUVECs stained for F-actin and nuclei are shown in Figure S4c. Thus, the endothelialized oLOC represents the hepatic model under dynamic fluid flow through vasculature that supplies hepatic layers through diffusive transport to substantially mimic native hepatic tissue architecture.
The functions of hepatocytes were assessed by cytochrome P450 (CYP) 1A1/2 and CYP3A4 (phase I metabolic enzymes) activities, as well as albumin and urea productions.[4,30–32] Among the more than 50 CYP450 enzymes, CYP1A2, CYP3A4, CYP2C9, CYP2C19, CYP2D6, and CYP3A5 enzymes are responsible for the metabolism of 90% of drugs.[33,34] These enzymes are predominantly expressed in the liver and are involved in most of the aspects of phase I drug metabolism, including oxidation, reduction, hydrolysis, and dehydrogenation.[31,35] Similarly, human serum albumin is the most abundant plasma protein, representing 50% of circulating proteins in healthy individuals, and it is synthesized exclusively by hepatocytes.[36] Albumin has important biological functions, including molecular transport, anti-oxidation, anti-inflammation, endothelial stabilization, anti-thrombotic effects, and the adjustment of capillary permeability.[37] Reduced serum albumin concentration and impaired albumin function have been reported in a variety of liver diseases including cirrhosis.[36] Reduced serum albumin concentration is associated with decreased albumin synthesis by the hepatocytes and/or dilution due to the retention of water and sodium in the extracellular space.[38] Impaired albumin function is associated with post-translational modifications of albumin and oxidative damage resulting in a decrease in its activity.[37] Similarly, urea synthesis is an essential metabolic function of liver that plays a critical role in nitrogen homeostasis and is related to functional liver mass. A reduction in urea synthesis is associated with a decrease in total hepatic mass, decrease in the enzymes and/or substrates of the urea cycle, and alterations in portal blood flow. Decrease in urea synthesis in liver results in the accumulation of ammonia and is indicative of compromised liver functions.[39–41]
The biofunctionality of primary hepatocytes and HUVECs in the Generation 3 oLOC was confirmed by assessing the expression of CYP3A4 or CYP1A2 for primary hepatocytes and CD31 (i.e., endothelial cell adhesion molecule-1) for HUVECs via immunocytochemical analyses. At day 7, the cells in the oLOC were fixed, and immunocytochemical staining of HUVECs with CD31 and VE-cadherin antibodies and of primary hepatocytes with CYP3A4 or CYP1A2 antibody was performed. Fluorescence microscopy images of the immunostained HUVEC layer after 7 days showed the formation of a mature monolayer of spreading HUVECs with strong expression of CD31 and VE-cadherin, confirming the formation of the intercellular junctions necessary for proper functioning of the endothelium (Figure 3d). Similarly, fluorescence microscopy images of the immunostained primary hepatocytes in the oLOC exhibited strong expressions of CYP3A4 (Figure 3d). Likewise, fluorescence microscopy images of the immunostained HepG2 cells within the Generation 2 oLOC also exhibited strong expressions of both CYP3A4 and CYP1A2 (Figure S4d).
To investigate whether the oLOC can support acute hepatotoxicity testing of drugs in vitro, we evaluated the toxicity of two drugs, acetaminophen and tamoxifen, on primary hepatocytes cultured in the oLOC under conditions of dynamic flow through the vascular chambers (simulated sinusoids). In separate experiments, the toxicity of these drugs was also evaluated on HepG2 cells cultured under dynamic flow through the vascular chambers in the oLOC. Acetaminophen, also commonly known as paracetamol and N-acetyl-p-aminophenol, is an antipyretic and analgesic drug. High doses of acetaminophen (>4 g per day per adult person) induce hepatotoxicity and acute liver damage due to the CYP450-mediated oxidative metabolism of acetaminophen to the highly reactive metabolite, N-acetyl-p-benzoquinone iminine, that causes intrahepatic glutathione depletion, oxidative stress, and mitochondrial dysfunctions.[42–44] Most of the studies have used >5 mM of acetaminophen to assess the hepatotoxicity of the drug.[45–47] The acute toxicity of acetaminophen on hepatic cells, both primary hepatocytes within the Generation 3 oLOCs and HepG2 cells within the Generation 2 oLOCs, was assessed by measuring the viability and metabolic activity of hepatocytes perfused with different concentrations of acetaminophen for 48 h. Hepatotoxicity of acetaminophen on primary hepatocytes is presented in Figure 4a as live/dead staining images. Similarly, Figure S5a shows the cytotoxicity (live/dead staining) of acetaminophen on HepG2 cells. A significant acute toxic effect of acetaminophen on primary hepatocytes was observed at both 10-mM and 20-mM concentrations with metabolic activity significantly decreased to 14% and 3%, respectively (Figure 4b). In comparison, the metabolic activity of HepG2 cells was decreased to 70% at 10 mM and was significantly decreased to 10% at 20 mM (Figure S5b). Thus, it was observed that primary hepatocytes cultured in the Generation 3 oLOC were more sensitive to acetaminophen treatment at both 10 mM and 20 mM concentrations as compared to the HepG2 cells cultured in the Generation 2 oLOC. It should be noted, however, that the cell shedding upon drug treatment was more significant in the primary hepatocyte group than in the HepG2 cell group, also indicating their reduced adhesion to the surfaces when treated with acetaminophen. The hepatotoxicity of acetaminophen was further studied under normal (static) cell culture conditions for equal populations of both primary hepatocytes and HepG2 cells. The metabolic activity in the oLOCs and the metabolic activity under static culture conditions were similar for both cell types under acetaminophen treatment (Figures 4b and S5b).
Figure 4.

(a) Cytotoxicity of acetaminophen on primary hepatocytes shown via cell viability assay using calcein AM (green) and EthD-1 (red) staining after 48 h of acetaminophen treatment; (b) quantified cell metabolic activity of hepatocytes at different concentrations of acetaminophen; (c) quantified 48-h urea production by hepatocytes at different concentrations of acetaminophen; and (d) quantified 48-h albumin production by hepatocytes at different concentrations of acetaminophen. Asterisk represents significant difference between the dynamic culture group and the static culture group using two-way ANOVA (p≤0.05).
Evaluation of the specific functions (albumin and urea syntheses) of primary hepatocytes showed a total 48-h secretion of 1.5 ng of urea and 95 ng of albumin at day 7 in the absence of drug treatment. The urea and albumin secretion levels in the oLOC were compared with those for an equal population of cells under 2D static culture conditions, and it was found that the primary hepatocytes cultured in the oLOC produced higher levels of urea (Figure 4c) and albumin (Figure 4d) than those in 2D static culture. Urea and albumin secretion by HepG2 cells cultured in the oLOC also exceeded secretion by those cells in 2D static culture (Figure S5c–d). Urea and albumin syntheses were also measured at day 7 after acetaminophen treatments for 48 h under dynamic flow in the vascular chamber of the oLOC. The total amount of urea secreted by primary hepatocytes over 48 h was measured at 1.2 ng at 10-mM acetaminophen treatment and was decreased to 0.04 ng at 20-mM acetaminophen treatment, whereas total 48-h albumin secretion was decreased to 55 ng at 10 mM and to 33 ng at 20 mM of acetaminophen (Figure 4c,d). These results indicated the marked impairment of primary hepatocyte functions at higher concentrations of acetaminophen due to acute hepatotoxicity. The hepatotoxic effect of acetaminophen on urea and albumin syntheses was further assessed under static culture conditions. The toxic effect of acetaminophen on albumin synthesis by primary hepatocytes was shown to be more sensitive in the oLOC’s perfusion culture than in static culture (Figure 4d). These results indicated that the hepatocyte functions (urea and albumin syntheses) were significantly improved and maintained in the oLOC as compared to static culture.[48] Similar results were obtained for HepG2 cells under no drug treatment and under acetaminophen treatment conditions (Figure S5c,d), with a more pronounced toxic effect on urea synthesis observed for the HepG2 cells as compared with the primary hepatocytes.
In addition, the hepatoxicity of tamoxifen in the oLOC was assessed. Tamoxifen is a non-steroidal antiestrogen drug that has been used alone or as an adjuvant for the treatment and prevention of estrogen receptor-positive breast cancer at a dose of 20 mg per day.[49,50] Tamoxifen is actually a prodrug that is converted into active metabolites, 4-hydroxytamoxifen and endoxifen, in the liver by the CYP enzymes CYP2D6 and CYP3A4.[51,52] Studies have reported various side effects of tamoxifen on liver including hepatotoxicity, hepatitis, hepatic steatosis, hepatic cirrhosis, hepatic injury, and even hepatocarcinoma.[53,54] These adverse side effects of tamoxifen have been attributed to the overproduction of reactive oxygen species during metabolic activation of tamoxifen and to the impaired mitochondrial β-oxidation of fatty acids.[54–56] The toxicity of tamoxifen on hepatic cells, both primary hepatocytes cultured in the Generation 3 oLOC and HepG2 cells cultured in the Generation 2 oLOC, was evaluated by measuring their viability and metabolic activity when perfused with 10-μM and 20-μM tamoxifen for 48 h. The toxic effect of tamoxifen on HepG2 cells is presented in Figure 5a. Similarly, Figure S6a shows the toxicity of tamoxifen on primary hepatocytes. The metabolic activity of HepG2 cells was decreased by only 10% at both 10-μM and 20-μM tamoxifen (Figure 5b). Thus, tamoxifen treatment on HepG2 cells, at both 10-μM and 20-μM concentrations, did not show significant toxic effects on metabolic activity. In contrast, the metabolic activity of primary hepatocytes decreased to 56% at 10-μM tamoxifen and to 36% at 20-μM tamoxifen (Figure S6b). These results demonstrated that the metabolic activity of HepG2 cells cultured in the oLOC was less sensitive to tamoxifen than was the metabolic activity of similarly-cultured primary hepatocytes. No significant differences were observed between metabolic activity in the oLOCs and metabolic activity under static culture conditions for either cell type under tamoxifen treatment (Figures S6b and 5b).
Figure 5.

(a) Cytotoxicity of tamoxifen on HepG2 cells shown via cell viability assay using Calcein AM (green) and EthD-1 (red) staining after 48 h of tamoxifen treatment; (b) quantified cell metabolic activity of HepG2 cells at different concentrations of tamoxifen; (c) quantified 48-h urea production by HepG2 cells at different concentrations of tamoxifen; and (d) quantified 48-h albumin production by HepG2 cells at different concentrations of tamoxifen. Asterisk represents a significant difference between the dynamic culture group and the static culture group using two-way ANOVA (p≤0.05).
Urea and albumin syntheses were also measured after tamoxifen treatment for 48 h under dynamic flow in the simulated sinusoids of the oLOC. The effect of tamoxifen treatment on the synthesis of urea by HepG2 cells was not statistically significant (Figure 5c), whereas the total 48-h secretion of albumin by HepG2 cells was decreased from 1.00 ng at 0 μM to 0.18 ng at 10 μM and to 0.36 ng at 20 μM of tamoxifen at day 7 (Figure 5d). The slightly increased albumin level at 20 μM of drug treatment was likely within the biological error since no statistical significance was found between the 10-μM and 20-μM groups. For primary hepatocytes the effect of tamoxifen treatment on the synthesis of urea was also not statistically significant (Figure S6c). However, the total 48-h secretion of albumin by primary hepatocytes was decreased from 95 ng at 0 μM to 32 ng at 10 μM and to 30 ng at 20 μM of tamoxifen at day 7 (Figure S6d). These results indicated that for both HepG2 cells and primary hepatocytes, albumin synthesis was more sensitive to tamoxifen than was urea synthesis. The hepatotoxic effect of tamoxifen on urea and albumin syntheses in static culture conditions was further assessed for both HepG2 cells (Figure 5c,d) and primary hepatocytes (Figure S6c,d). No significant difference was observed between the sensitivity of urea synthesis in the oLOC and the sensitivity of urea synthesis in the static culture under tamoxifen treatment for either cell type. For primary hepatocytes, no significant difference was observed between the sensitivity of albumin synthesis in the oLOC and the sensitivity of albumin synthesis in static culture under tamoxifen treatment (Figure S6c,d).
These results show that origami-based design and creation of liver-on-a-chip can successfully reproduce a sufficient level of architectural complexity (e.g., replicating flow along sinusoids as well as the transport of species between sinusoids and hepatocytes) by reducing fabrication challenges (e.g., the need for multistep processes in dedicated microfabrication facilities). The oLOC devices were fabricated from strips of pre-patterned panels connected by perforated folding joints and stored compactly in the laboratory. The constrained kinematics of the perforated folding joints ensured correct alignment of the layers after the manual folding process. Although the materials were pre-patterned, the flexibility of the origami platform enabled the devices to be customized rapidly, simply by altering the folding sequence, ultimately yielding the successful Generation 3 oLOC devices. The Generation 3 chips demonstrated high viability of primary human hepatocytes and HUVECs in homogeneously-distributed layers in an endothelialized oLOC system that mimics native hepatic tissue architecture and functions. Urea and albumin syntheses, together with the expression of metabolic enzymes, demonstrated that the hepatocytes retain their differentiated functions to a decent degree in the oLOC environment. Hepatotoxicity testing with acetaminophen and tamoxifen in the oLOC with primary hepatocytes and HUVECs reproduces hepatotoxicity, demonstrating that the oLOC can possibly support acute hepatotoxicity evaluations. These results offer the potential opportunity for future origami-based fabrication, optimization, and practical application for pharmaceutical screening not just of liver-on-a-chip devices, but also of other origami-enabled organ-on-a-chip or multi-organ-on-a-chip systems.
Experimental Section
Materials
Double-sided Kapton® polyimide tape with 240 μm of thickness and acrylic adhesive was purchased from McMaster-Carr (IL, USA) and patterned by Micron Laser Technology (OR, USA) using a 355-nm laser tool. Acrylic sheets with 1/16” (1.59-mm) thickness, Loctite 430 Instant Bond Adhesive, Gorilla quick-set epoxy structural adhesive, and Clear Masterkleer soft PVC plastic tubing with 1/32” (0.79-mm) inner diameter (ID) and 3/32” (2.38-mm) outer diameter (OD) were purchased from McMaster-Carr (OH, USA). Nanoporous membranes (8”×12” or 200 mm×300 mm sheets of Whatman 113506 Nuclepore hydrophilic membrane with 0.2-μm pore size) were purchased from VWR (PA, USA). Polystyrene microspheres with approximately 16-μm diameter were purchased from Polysciences (PA, USA). Trypan blue 15250–061 was purchased from Gibco (Dublin, Ireland). Bovine serum albumin (BSA), fibronectin, acetaminophen, tamoxifen, triton X-100, dimethyl sulfoxide (DMSO), and urea assay kit were purchased from Sigma-Aldrich (MO, USA). Dulbecco’s modified Eagle medium (DMEM), Dulbecco’s phosphate-buffered saline (DPBS), fetal bovine serum (FBS), penicillin/streptomycin (P/S), antibiotic-antimycotic solution stabilized (Anti-Anti, 100X), DAPI, formalin (10% w/v), Live/Dead® Viability/Cytotoxicity Kit, Alexa Fluor® 594-phalloidin, mouse CYP3A4/CYP3A5 antibody, mouse anti-CYP1A2 antibody, and dialysis membrane (Mw cutoff: 12,000–14,000 Da) were obtained from Thermo Fisher Scientific (MA, USA). Primary hepatocyte medium was obtained from ScienCell Research Laboratories (CA, USA) and EGM™-2 Endothelial Cell Growth Medium-2 BulletKit™ (EGM-2) was purchased from Lonza (Basel, Switzerland). Human albumin ELISA kit, anti-CD31 [P2B1] antibody, anti-VE-cadherin antibody, and Alexa Fluor® 594- and Alexa Fluor® 488-conjugated secondary antibodies were obtained from Abcam (MA, USA). The CellTiter 96® AQueous One Solution Cell Proliferation Assay solution was obtained from Promega (WI, USA). All other chemicals used in this study were obtained from Sigma-Aldrich unless otherwise mentioned.
Fabrication of the chips
The polyimide tape was laser-patterned to define contiguous strips of 20-mm-square folding panels with flow features and perforated fold lines. Nanoporous membranes were cut to form 20-mm-wide folding strips (Generation 1 devices) or 20-mm-square panels (Generation 2 and Generation 3 devices). The acrylic top and bottom plates and the connection ports in the top plate were laser-cut from 1.59-mm-thick acrylic sheets using a 30-W Epilog Zing laser.
To fabricate the Generation 1 devices, the patterned polyimide tape was folded at 90 degrees to a strip of unpatterned nanoporous membrane (Figure 2a,b). Upon completion of folding, a 2-mm-diameter punch was inserted into the four 2-mm-diameter inlet and outlet ports at the corners of the chip, and holes were punched through the nanoporous membrane layers. To fabricate the Generation 2 and Generation 3 devices, smaller sub-units of patterned polyimide tape were folded and then stacked to create the final device. First, two C panels were folded around a nanoporous membrane, which was trimmed to the 20-mm-square size of the folded polyimide and punched to create the inlet and outlet holes (Figure 2e,f). Next, two F panels were joined to create a double layer (Figure 2g). It is necessary for this double layer to be well-enough adhered to prevent leakage, but it also needs to be able to be separated at the end of each experiment for analysis of the cell layers within the device. The pristine adhesive layers bond too strongly for device disassembly for cell characterizations, so one tape-to-tape interface within each chamber was intentionally weakened by contacting it with a glove prior to assembly. The folded subunits were then stacked (Figure 2h) to create the full device. The acrylic plates were adhered to the top and bottom of the device using the tape’s adhesive layer. Tubes with 50-mm length and 2.38-mm outer diameter were inserted into the upper acrylic plate (Figure 2i,j) and retained in place first with Loctite 430 Instant Bond Adhesive, then with a Gorilla quick-set epoxy structural adhesive later for additional strength. The oLOC chips were autoclaved prior to use; autoclaving did not appear to negatively impact the device integrity.
Characterization of inter-chamber transport
Transport between adjacent chambers was characterized by flowing deionized water through the middle flow path of the Generation 1 device as a target solution while flowing a 0.1% solution of trypan blue in deionized water through the device’s upper and lower flow paths as a source solution. In the first set of tests, the target and source solutions were dispensed at equal flow rates from 0.8 mL/min to 3.2 mL/min using a Chemyx Inc. (TX, USA) syringe pump, and samples of the target flow exiting the device were captured during steady state operation at each flow rate. In the second set of tests, the target solution was again dispensed at flow rates from 0.8 mL/min to 3.2 mL/min, while the source solution was dispensed at 1.365X higher flow rates. Three devices were characterized in each set of tests, and the experiment was repeated between 3 and 5 times at each flow rate. The light absorbance values of the exiting target flow were measured at 580 nm using a 96 well plate and a BioTek Instruments PowerWave XS plate reader spectrophotometer (VT, USA). Each well contained 200 μL of the sample, and absorbance values of each sample were measured in 3 different wells. Trypan blue concentration in the exiting target flow was calculated from the measured absorbance values and standard samples with known concentration of trypan blue (Figure S7).
Cell culture
Primary human hepatocytes were obtained from ScienCell Research Laboratories, HUVECs were obtained from Lonza, and the human hepatocellular carcinoma cell line (HepG2 cells) was obtained from American Type Culture Collection (VA, USA). Primary hepatocytes were cultured in the primary hepatocyte medium supplemented with Hepatocyte Growth Supplements, 5% (v/v) FBS, and 1% (v/v) P/S, whereas HepG2 cells were cultured in DMEM supplemented with 10% (v/v) FBS and 1% (v/v) P/S or anti-anti. Similarly, HUVECs were cultured in the EGM-2 medium. The cells were incubated at 37 °C and 5% CO2 in a 95% humidified cell incubator until 70–80% confluence. The respective culture medium was changed every 3rd day.
Cell seeding in the oLOC
Adherent primary hepatocytes, HepG2 cells, and HUVECs were trypsinized, centrifuged, and resuspended in respective medium at 5×106 viable cells mL-1. The semipermeable polycarbonate nanoporous membrane within each origami chip was pre-coated with fibronectin (200 ng mL−1) overnight at 4 °C. A volume of 100 μL of primary hepatocytes or HepG2 cell suspension was seeded in the circular liver chamber, and 100 μL of HUVECs were seeded in the vascular chamber. The oLOC device was incubated at 37 °C in a cell incubator. After 1 h of incubation at 37 °C, the oLOC was flipped upside down and the liver cells (primary hepatocytes or HepG2 cells) and HUVECs were seeded again in their respective chambers, followed by another incubation at 37 °C in a cell incubator. The oLOC was then connected to medium reservoirs, with the endothelialized chamber connected to a reservoir containing the EGM-2 medium and the hepatic chamber connected to 4 mL of primary hepatocyte medium or DMEM. The EGM-2 medium was continuously circulated at 14 μL/min using a MP2 micro peristaltic pump (Elemental Scientific, NE, USA). The oLOC was placed in a cell incubator at 37 °C for up to 7 days for downstream analyses. The culture media were replaced with fresh media every 3rd day and at the start of hepatotoxicity studies.
Cell viability and morphology assessments
The Live/Dead® Viability/Cytotoxicity Kit was utilized to evaluate cell viability at day 3 and day 7 according to manufacturer’s instructions. Briefly, cells within the chambers of the oLOC were washed with DPBS and incubated with 100 μL per well of the combined Live/Dead assay reagents (2 μM of calcein AM and 4 μM of EthD-1) for 20 min at 37 °C in a cell incubator. The cells were washed three times with DPBS. To permit direct visualization, the chips were disassembled at the interface that was prepared during device fabrication to be separable. The cells were then observed under a Zeiss Axio Observer inverted fluorescence microscope (Zeiss, NY, USA). Percentages of viable cells were determined by using the Image J software (National Institutes of Health, MD, USA). Further, the quantification of viable cells and their metabolic activities within oLOC was measured by MTS proliferation assay using the CellTiter 96® AQueous One Solution Cell Proliferation Assay kit at day 3 and day 7.
The morphology of both primary hepatocytes and the HUVECs within the oLOC was assessed by F-actin staining of the cells at day 7. The cells were washed with DPBS, fixed with 10% formalin for 15 min and permeabilized with 0.1% (v/v) Triton X-100 in DPBS for 30 min at room temperature. The cells were then blocked with 5% (v/v) goat serum in DPBS for 1 h, followed by F-actin staining by incubating the cells with either Alexa Fluor® 594-phalloidin or Alexa Fluor® 488-phalloidin (1:200 dilution in blocking buffer) for 1 h at room temperature. After washing three times with DPBS, nuclei were counter-stained with DAPI (1:1000) for 5 min at room temperature and the cells were observed under a fluorescence microscope. The images were pseudocolored to the same colors to enable visual consistency.
Functional analyses of cells in the oLOC
The functionality of the cells within the oLOC was assessed by immunostaining of the cells at day 7. The cells were washed with DPBS and fixed with 10% formalin for 30 min at room temperature. After washing with DPBS, the cells were incubated with the permeabilization buffer (0.1% (v/v) Triton X-100 in DPBS) for 1 h and blocked with 5% (v/v) goat serum in DPBS for 2 h at room temperature. The cells were then incubated with mouse cytochrome P450 3A (CYP3A4/CYP3A5 monoclonal) antibody or mouse anti-CYP1A2 antibody for primary hepatocytes and mouse anti-CD31 and mouse anti-VE-cadherin antibodies for HUVECs (1:200 dilution) overnight at 4 °C. The cells were washed three times with DPBS and incubated overnight at 4 °C with the relevant secondary antibody (either Alexa Fluor® 594-conjugated goat anti-mouse secondary antibody or Alexa Fluor® 488-conjugated goat anti-mouse secondary antibody) at 1:200 dilution. Finally, the nuclei were counterstained with DAPI after washing with DPBS and examined under a fluorescence microscope. The images were pseudocolored to the same colors to enable visual consistency.
Hepatotoxicity testing with the oLOC
Stock solutions of acetaminophen and tamoxifen at 0.5 M were prepared by dissolving the acetaminophen and tamoxifen powder in DPBS:ethanol (1:1 v/v) and DMSO, respectively. Cells were cultured in the oLOC as described above for 5 days and then treated with 0, 10, or 20 mM of acetaminophen or 0, 10 or 20 μM tamoxifen by replacing the medium in the vascular medium reservoir. After 48 h of drug treatment, the media were collected for albumin and urea production assays, and the hepatotoxicity was assessed by cell viability assay using the Live/Dead® Viability/Cytotoxicity Kit as described above and the MTS proliferation assay using the CellTiter 96® AQueous One Solution Cell Proliferation Assay kit, according to the manufacturers’ instructions. Briefly, the media were removed, and the constructs were incubated with MTS assay solution for 2 h at 37 °C in the dark. The absorbance was measured at 492 nm with a microplate reader (Tecan, Austria), and the results were expressed as percentages of the control.
Albumin and urea production assays
Albumin secreted by the hepatic cells within the oLOC was measured using the albumin ELISA kit according to the manufacturer’s protocol. Briefly, all the reagents were brought to room temperature before starting the assay. A 50-μL volume of the standard or sample was added to each well of the albumin ELISA plate and incubated for 1 h at room temperature. Each well was then washed three times with 200 μL of the wash buffer (1X). A 50-μL volume of the biotinylated albumin antibody was added to each well and incubated for 30 min. Each well was washed again three times with 200 μL of the wash buffer (1X), and 50 μL of the 1X SP conjugate was added to each well. After incubation for 30 min followed by washing, 50 μL of the chromogen substrate was added per well and incubated for 20 min. Finally, 50 μL of the stop solution was added to each well, and the absorbance was read at 450 nm on a microplate reader.
Similarly, urea produced by the hepatic cells within the oLOC was measured using a urea assay kit, according to the manufacturer’s instructions. Briefly, 0, 1, 2, 3, 4, and 5 nmol of urea standards were prepared by diluting 100-nmol urea standard solution with the urea assay buffer. A 50-μL volume of the standard or sample was added to each well of a flat-bottomed 96-well plate. Then 50 μL of the freshly prepared reaction mix was added to each well and mixed quickly by gently rocking the plate. The plate was incubated at 37 °C for 1 h, and absorbance was measured at 570 nm on the microplate reader.
Statistical analyses
All of the experiments were done in triplicates for each data point and were repeated three times, except for the characterization of inter-chamber transport, for which some experiments were repeated five times. Results were expressed as means ± standard errors of the means (SEMs) of three (or five) independent experiments, and statistical comparisons were performed using the two-way analysis of variance (ANOVA).
Supplementary Material
Acknowledgments
This research was funded by NSF and AFOSR through the EFRI-ODISSEI Program under award #1332249. In addition, support was provided by the National Institutes of Health (K99CA201603, R00CA201603, R21EB025270, R01EB028143, UG3TR003274), National Science Foundation (CBET-EBMS-1936105), the Brigham Research Institute, and the New England Anti-Vivisection Society. The authors thank Dina Atia, Daniel Scrivener, Arielle Freeman, and Jens Vittoria for their assistance with assessing flow through the microfluidic channels.
Footnotes
Potential conflict of interest: Dr. Xie and Dr. Livermore are co-founders of ApreX, Inc.; however, there is no financial involvement in any form.
Contributor Information
Xin Xie, Department of Mechanical and Industrial Engineering, Northeastern University, Boston, MA 02115, USA; Division of Engineering in Medicine, Brigham and Women’s Hospital, Harvard Medical School, Cambridge, MA 02139, USA; TransMedics, Inc., Andover, MA 01810, USA.
Sushila Maharjan, Division of Engineering in Medicine, Brigham and Women’s Hospital, Harvard Medical School, Cambridge, MA 02139, USA.
Chastity Kelly, Department of Mechanical and Industrial Engineering, Northeastern University, Boston, MA 02115, USA.
Tian Liu, Department of Mechanical and Industrial Engineering, Northeastern University, Boston, MA 02115, USA; KLA Corporation, Milpitas, CA 95035, USA.
Robert J. Lang, Lang Origami, Altadena, CA 91001
Roger Alperin, Department of Mathematics, San Jose State University, San Jose, CA 95192.
Shikha Sebastian, Division of Engineering in Medicine, Brigham and Women’s Hospital, Harvard Medical School, Cambridge, MA 02139, USA.
Diana Bonilla, Division of Engineering in Medicine, Brigham and Women’s Hospital, Harvard Medical School, Cambridge, MA 02139, USA; Escuela de Ingeniería y Ciencias, Tecnologico de Monterrey, Monterrey, NL 64849, México.
Sakura Gandolfo, Department of Mechanical and Industrial Engineering, Northeastern University, Boston, MA 02115, USA.
Yasmine Boukataya, Department of Mechanical and Industrial Engineering, Northeastern University, Boston, MA 02115, USA.
Seyed Mohammad Siadat, Department of Bioengineering, Northeastern University, Boston, MA 02115, USA.
Yu Shrike Zhang, Division of Engineering in Medicine, Brigham and Women’s Hospital, Harvard Medical School, Cambridge, MA 02139, USA.
Carol Livermore, Department of Mechanical and Industrial Engineering, Northeastern University, Boston, MA 02115, USA.
References
- [1].Kuntz E, J Hepatology Principles 2006, 31. [Google Scholar]
- [2].Guguen-Guillouzo C, Campion JP, Brissot P, Glaise D, Launois B, Bourel M, Guillouzo A, Cell biology international reports 1982, 6, 625. [DOI] [PubMed] [Google Scholar]
- [3].Godoy P, Hewitt NJ, Albrecht U, Andersen ME, Ansari N, Bhattacharya S, Bode JG, Bolleyn J, Borner C, Böttger J, Braeuning A, Budinsky RA, Burkhardt B, Cameron NR, Camussi G, Cho CS, Choi YJ, Craig Rowlands J, Dahmen U, Damm G, Dirsch O, Donato MT, Dong J, Dooley S, Drasdo D, Eakins R, Ferreira KS, Fonsato V, Fraczek J, Gebhardt R, Gibson A, Glanemann M, Goldring CE, Gómez-Lechón MJ, Groothuis GM, Gustavsson L, Guyot C, Hallifax D, Hammad S, Hayward A, Häussinger D, Hellerbrand C, Hewitt P, Hoehme S, Holzhütter HG, Houston JB, Hrach J, Ito K, Jaeschke H, Keitel V, Kelm JM, Kevin Park B, Kordes C, Kullak-Ublick GA, LeCluyse EL, Lu P, Luebke-Wheeler J, Lutz A, Maltman DJ, Matz-Soja M, McMullen P, Merfort I, Messner S, Meyer C, Mwinyi J, Naisbitt DJ, Nussler AK, Olinga P, Pampaloni F, Pi J, Pluta L, Przyborski SA, Ramachandran A, Rogiers V, Rowe C, Schelcher C, Schmich K, Schwarz M, Singh B, Stelzer EH, Stieger B, Stöber R, Sugiyama Y, Tetta C, Thasler WE, Vanhaecke T, Vinken M, Weiss TS, Widera A, Woods CG, Xu JJ, Yarborough KM, Hengstler JG, Archives of toxicology 2013, 87, 1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lee PJ, Hung PJ, Lee LP, Biotechnol Bioeng 2007, 97, 1340. [DOI] [PubMed] [Google Scholar]
- [5].Yu Y, Fisher JE, Lillegard JB, Rodysill B, Amiot B, Nyberg SL, Liver transplantation : official publication of the American Association for the Study of Liver Diseases and the International Liver Transplantation Society 2012, 18, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wang Z, Li W, Jing H, Ding M, Fu G, Yuan T, Huang W, Dai M, Tang D, Zeng M, Chen Y, Zhang H, Zhu X, Peng Y, Li Q, Yu WF, Yan HX, Zhai B, Theranostics 2019, 9, 6690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Li F, Cao L, Parikh S, Zuo R, Journal of pharmaceutical sciences 2020, 109, 1912. [DOI] [PubMed] [Google Scholar]
- [8].Trask OJ Jr., Moore A, LeCluyse EL, Assay and drug development technologies 2014, 12, 16. [DOI] [PubMed] [Google Scholar]
- [9].Chan TS, Yu H, Moore A, Khetani SR, Tweedie D, Drug metabolism and disposition: the biological fate of chemicals 2019, 47, 58. [DOI] [PubMed] [Google Scholar]
- [10].Lin C, Shi J, Moore A, Khetani SR, Drug metabolism and disposition: the biological fate of chemicals 2016, 44, 127. [DOI] [PubMed] [Google Scholar]
- [11].Moore A, Chothe PP, Tsao H, Hariparsad N, Drug metabolism and disposition: the biological fate of chemicals 2016, 44, 1910. [DOI] [PubMed] [Google Scholar]
- [12].Kizawa H, Nagao E, Shimamura M, Zhang G, Torii H, Biochemistry and biophysics reports 2017, 10, 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Grix T, Ruppelt A, Thomas A, Amler AK, Noichl BP, Lauster R, Kloke L, Genes 2018, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Knowlton S, Tasoglu S, Trends Biotechnol 2016, 34, 681. [DOI] [PubMed] [Google Scholar]
- [15].Ma X, Liu J, Zhu W, Tang M, Lawrence N, Yu C, Gou M, Chen S, Adv Drug Deliv Rev 2018, 132, 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wang Y, Wu D, Wu G, Wu J, Lu S, Lo J, He Y, Zhao C, Zhao X, Zhang H, Wang S, Theranostics 2020, 10, 300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Cong Y, Han X, Wang Y, Chen Z, Lu Y, Liu T, Wu Z, Jin Y, Luo Y, Zhang X, Micromachines 2020, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Li X, George SM, Vernetti L, Gough AH, Taylor DL, Lab Chip 2018, 18, 2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Vernetti LA, Senutovitch N, Boltz R, DeBiasio R, Shun TY, Gough A, Taylor DL, Exp Biol Med (Maywood) 2016, 241, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].and YX, Whitesides GM, 1998, 28, 153. [Google Scholar]
- [21].Young EWK, Berthier E, Guckenberger DJ, Sackmann E, Lamers C, Meyvantsson I, Huttenlocher A, Beebe DJ, Analytical chemistry 2011, 83, 1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Liu Y, Ganser D, Schneider A, Liu R, Grodzinski P, Kroutchinina N, Analytical chemistry 2001, 73, 4196. [DOI] [PubMed] [Google Scholar]
- [23].McCormick RM, Nelson RJ, Alonso-Amigo MG, Benvegnu DJ, Hooper HH, Analytical chemistry 1997, 69, 2626. [DOI] [PubMed] [Google Scholar]
- [24].Shaegh SAM, Pourmand A, Nabavinia M, Avci H, Tamayol A, Mostafalu P, Ghavifekr HB, Aghdam EN, Dokmeci MR, Khademhosseini A, Zhang YS, Sensors and Actuators B: Chemical 2018, 255, 100. [Google Scholar]
- [25].Xie X, Kelly C, Livermore C, “Origami-Enabled Microfluidics”, presented at Hilton Head Solid-State Sensors, Actuators and Microsystems Workshop (HiltonHead), Hilton Head, SC, 2018. [Google Scholar]
- [26].Gissen P, Arias IM, Journal of hepatology 2015, 63, 1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hassan S, Sebastian S, Maharjan S, Lesha A, Carpenter A-M, Liu X, Xie X, Livermore C, Zhang YS, Zarrinpar A, 2020, 71, 733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Dominguez R, Holmes KC, Annu Rev Biophys. 2011, 40, 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].De La Cruz EM, Pollard TD, Biochemistry. 1996, 35, 14054. [DOI] [PubMed] [Google Scholar]
- [30].Sivaraman A, Leach JK, Townsend S, Iida T, Hogan BJ, Stolz DB, Fry R, Samson LD, Tannenbaum SR, Griffith LG, Curr Drug Metab 2005, 6, 569. [DOI] [PubMed] [Google Scholar]
- [31].Zanger UM, Schwab M, Pharmacol Ther 2013, 138, 103. [DOI] [PubMed] [Google Scholar]
- [32].Maharjan S, Bonilla D, Sindurakar P, Li H, Li W, Duarte S, Zarrinpar A, Zhang YS, Bio-Design and Manufacturing 2021. [Google Scholar]
- [33].Lim HK, Duczak N Jr., Brougham L, Elliot M, Patel K, Chan K, Drug metabolism and disposition: the biological fate of chemicals 2005, 33, 1211. [DOI] [PubMed] [Google Scholar]
- [34].Rogers JF, Nafziger AN, Bertino JS Jr., The American journal of medicine 2002, 113, 746. [DOI] [PubMed] [Google Scholar]
- [35].Zhou SF, Clinical pharmacokinetics 2009, 48, 689. [DOI] [PubMed] [Google Scholar]
- [36].Spinella R, Sawhney R, Jalan R, Hepatology international 2016, 10, 124. [DOI] [PubMed] [Google Scholar]
- [37].Sun L, Yin H, Liu M, Xu G, Zhou X, Ge P, Yang H, Mao Y, Annals of medicine 2019, 51, 333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bernardi M, Maggioli C, Zaccherini G, Critical care (London, England) 2012, 16, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Mezey E, Annual review of nutrition 1982, 2, 21. [DOI] [PubMed] [Google Scholar]
- [40].Vilstrup H, Gut 1980, 21, 990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Rafoth RJ, Onstad GR, J Clin Invest 1975, 56, 1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hinson JA, Roberts DW, James LP, Handb Exp Pharmacol. 2010, 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Jaeschke H, McGill MR, Ramachandran A, Drug metabolism reviews 2012, 44, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Jaeschke H, McGill MR, Toxicology letters 2015, 235, 216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Amaral SS, Oliveira AG, Marques PE, Quintao JL, Pires DA, Resende RR, Sousa BR, Melgaco JG, Pinto MA, Russo RC, Gomes AK, Andrade LM, Zanin RF, Pereira RV, Bonorino C, Soriani FM, Lima CX, Cara DC, Teixeira MM, Leite MF, Menezes GB, Cell Commun Signal. 2013, 11, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].McGill MR, Yan HM, Ramachandran A, Murray GJ, Rollins DE, Jaeschke H, Hepatology. 2011, 53, 974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kennon-McGill S, McGill MR, J Clin Transl Res. 2018, 3, 297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Yu F, Deng R, Hao Tong W, Huan L, Chan Way N, IslamBadhan A, Iliescu C, Yu H, Scientific Reports 2017, 7, 14528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Jordan VC, Nature reviews. Drug discovery 2003, 2, 205. [DOI] [PubMed] [Google Scholar]
- [50].Van Hoof M, Rahier J, Horsmans Y, Annals of internal medicine 1996, 124, 855. [DOI] [PubMed] [Google Scholar]
- [51].Teft WA, Mansell SE, Kim RB, Drug metabolism and disposition: the biological fate of chemicals 2011, 39, 558. [DOI] [PubMed] [Google Scholar]
- [52].Desai PB, Nallani SC, Sane RS, Moore LB, Goodwin BJ, Buckley DJ, Buckley AR, Drug metabolism and disposition: the biological fate of chemicals 2002, 30, 608. [DOI] [PubMed] [Google Scholar]
- [53].Yang G, Nowsheen S, Aziz K, Georgakilas AG, Pharmacol Ther 2013, 139, 392. [DOI] [PubMed] [Google Scholar]
- [54].Kärki A, Mäntylä E, Hirsimäki Y, Karlsson S, Toikkanen S, Hirsimäki P, Archives of toxicology 2000, 74, 249. [DOI] [PubMed] [Google Scholar]
- [55].Ribeiro MP, Santos AE, Custódio JB, Toxicology 2014, 323, 10. [DOI] [PubMed] [Google Scholar]
- [56].Dragan YP, Fahey S, Nuwaysir E, Sattler C, Babcock K, Vaughan J, McCague R, Jordan VC, Pitot HC, Carcinogenesis 1996, 17, 585. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.