

Abstract
Recruiting endogenous adenosine deaminases using exogenous guide RNAs to edit cellular RNAs is a promising therapeutic strategy, but editing efficiency and durability remains low using current guide RNA designs. We engineered circular ADAR recruiting guide RNAs (cadRNAs) to enable more efficient programmable A-to-I RNA editing without requiring co-delivery of any exogenous proteins. Using these cadRNAs we observed robust and durable RNA editing across multiple sites and cell lines, in both untranslated and coding regions of RNAs, and high transcriptome-wide specificity. Additionally, we increased transcript-level specificity for the target adenosine by incorporating interspersed loops in the antisense domains, reducing bystander editing. In vivo delivery of cadRNAs via adeno-associated viruses enabled 53% RNA editing of the mPCSK9 transcript in C57BL/6J mice livers, and 12% UAG-to-UGG RNA correction of the amber nonsense mutation in the IDUA-W392X mouse model of mucopolysaccharidosis type I-Hurler (MPS I-H) syndrome. cadRNAs enable efficient programmable RNA editing in vivo with diverse protein modulation and gene therapeutic applications,
Adenosine to inosine (A-to-I) RNA editing is a common post-transcriptional modification catalyzed by adenosine deaminases acting on RNA (ADAR) enzymes1–7. ADARs edit double stranded RNA (dsRNA) predominantly in non-coding regions such as Alu repetitive elements in a promiscuous fashion, while also editing a handful of sites in coding regions with high specificity8–12. The structural similarity between inosine and guanosine results in the translation and splicing machinery recognizing the edited base as guanosine, thereby making ADARs attractive tools for recoding protein sequences13. To this end, several studies have recently repurposed the ADAR system for programmable RNA editing both in vitro14–22 and in vivo20,23 by engineering recruitment of ADARs to a target RNA sequence using ADAR recruiting guide RNAs (adRNAs). Although ADARs, and in particular ADAR1, are widely expressed throughout the body, most of these studies relied on exogenously delivered ADAR enzymes and their variants to achieve robust RNA editing efficiencies. However, as ADAR-dsRNA interactions primarily rely on structure rather than sequence dependency, a major limitation of relying on enzyme overexpression is the propensity to introduce a plethora of off-target A-to-I edits across the transcriptome18,20,24,25. Additionally, as ADARs are native to and thus not orthogonal to most mammalian systems, their overexpression can result in altered protein interactions that might impact cellular physiology. Furthermore, as this approach relies on two components, a guide RNA and the ADAR protein, it can limit delivery modalities, in particular for in vivo applications.
A solution to this is to engineer adRNAs to enable recruitment of endogenous ADARs. Towards this, we recently demonstrated that it is possible to recruit endogenous ADARs using simple long antisense RNA of length >60 bp20. This strategy is exciting since akin to short-hairpin RNAs (shRNAs) and antisense oligonucleotides (ASOs) which efficaciously recruit endogenous cellular machinery such as Argonaute26 and RNase H27,28 to enable targeted RNA knockdown, just delivery of guide RNAs alone can now enable programmable A-to-I RNA editing without requiring co-delivery of any exogenous proteins. However, the efficiency of RNA editing via this approach is typically lower than seen with enzyme overexpression, thus limiting its utility in biotechnology and therapeutic applications. Conjecturing this was due in part to the short half-life and target residence times of guide RNAs, here we engineer highly stable circular ADAR recruiting guide RNAs (cadRNAs). These vastly improve the efficiency and durability of RNA editing. We demonstrate too that targeting via cadRNAs is highly specific at the transcriptome-wide level, and via further engineering to reduce bystander editing, also highly specific at the transcript level. Furthermore, we show cadRNAs can be delivered genetically encoded via DNA, and as well via in vitro transcribed RNA at a fraction of the cost of chemically synthesized ASOs. Additionally, these enable highly robust RNA editing in both untranslated and coding regions of mRNAs, and across multiple RNA targets and cell lines. Importantly, using cadRNAs, we also demonstrate for the first time robust in vivo RNA editing via endogenous ADAR recruitment, including in the IDUA-W392X mouse model of mucopolysaccharidosis type I-Hurler (MPS I-H) syndrome.
RESULTS
Using our long antisense guide RNA design20 that can recruit endogenous ADARs as a base format, we explored two guide RNA engineering strategies to enhance RNA editing efficiencies (Figure 1a): one, we coupled recruiting domains that are derived from native RNAs sites known to be heavily edited by ADARs; and two, we coupled domains that stabilize and confer increased half-life of the guide RNAs (Supplementary Table 1).
Figure 1: Engineering circular ADAR recruiting guide RNAs (cadRNAs).
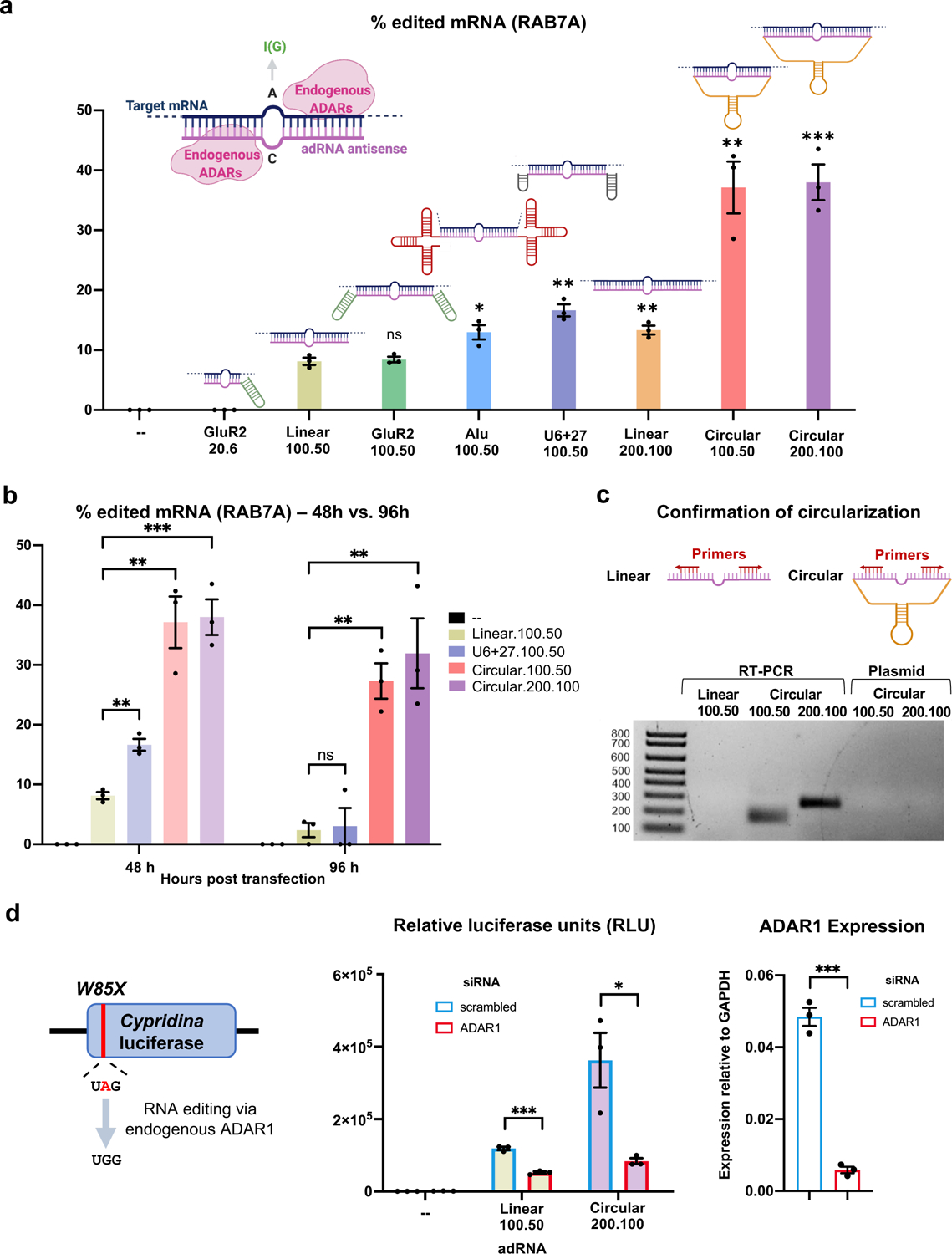
(a) A comparison of the RNA editing efficiencies in the 3’ UTR of the RAB7A transcript via various adRNA designs. Values represent mean +/− SEM (n=3; with respect to the linear.100.50, left-to-right, p=0.7289, p=0.0226, p=0.0019, p=0.0055, p=0.0027, and p=0.0006; unpaired t-test, two-tailed). In the schematics, the pink strand represents the antisense domain of the adRNA while the target mRNA is in blue. The bulge indicates the A-C mismatch between the target mRNA and adRNA. The adRNAs are labelled using the following convention: (domain name).(antisense length).(position of A-C mismatch from 5’ end of the antisense). (b) RNA editing efficiencies achieved 48 hours and 96 hours post transfection of various adRNA designs. Values represent mean +/− SEM (n=3; left-to-right, p=0.0019, p=0.0027, p=0.0006 and p=0.8488, p=0.0014, p=0.0077; unpaired t-test, two-tailed). The 48 hour panel data is reproduced from Figure 1a. (c) RT-PCR based confirmation of adRNA circularization in cells. (d) The ability of adRNAs to effect RNA editing of the cluc transcript was assessed in the presence of an siRNA targeting ADAR1. Values represent mean +/− SEM (n=3; left-to-right, p=0.0002, p=0.0216 and p=0.0001; unpaired t-test, two-tailed). All experiments were carried out in HEK293FT cells.
Towards the former we evaluated recruiting domains from the naturally occurring ADAR2 substrate GluR2 pre-messenger RNA16,17, and Alu elements which are known substrates for ADAR129. The Alu adRNAs were created by positioning the antisense domain within the Alu consensus sequence and eliminating any poly U stretches. We screened these modified guide RNAs by assaying editing at an adenosine in the 3’UTR of the RAB7A transcript in HEK293FT cells. Consistent with our previous observations20, the GluR2 domain coupled to a short antisense of length 20 bp with the A-C mismatch located 6 bp from the 5’ end of the antisense domain (GluR2.20.6) was unable to recruit endogenous ADARs resulting in no detectable RNA editing, while, as we previously demonstrated, long antisense RNAs with a centrally located A-C mismatch (linear.100.50) resulted in modest ~10% RNA editing. Coupling the GluR2 domains to the long antisense version (GluR2.100.50) did not further enhance RNA editing yields, but we observed that the addition of Alu domains (Alu.100.50) marginally enhanced the efficiency of RNA editing (1.5-fold). While significant, these designs had only a modest improvement over the base format of simple long antisense guide RNAs.
We thus focused next on evaluating the impact of persistence of guide RNAs, as this in turn could also impact target RNA search as well as their net target residence times. In particular, genetically encoded adRNAs are typically expressed via the polymerase III promoter, and thus transcribed guides lack a 5’ cap and a 3’ poly A tail and correspondingly have very short half-lives. To improve guide RNA persistence we evaluated: 1) increasing the length of the guide RNAs (linear.200.100); 2) coupling a U6+27 cassette (U6+27.100.50) which has been shown to improve stability of siRNA30; and 3) engineering circularized versions (circular.100.50 and circular.200.100) as these would be intrinsically resistant to cellular exonucleases. Specifically, leveraging an elegant methodology recently developed by Litke and colleagues31, we engineered circular ADAR recruiting guide RNAs (cadRNAs) by flanking the linear adRNAs by twister ribozymes, which upon autocatalytic cleavage leave termini that are ligated by the ubiquitous endogenous RNA ligase RtcB to yield circularized guide RNAs. Comparing the three different guide designs we observed that both the increase of adRNA length and the addition of U6+27 to the long antisense adRNA led to a 1.5-fold and 2-fold respective improvement in editing of the RAB7A transcript over the linear.100.50 designs (Figure 1a). Notably, using circular adRNA with antisense lengths 100 bp and 200 bp (i.e. circular.100.50 and circular.200.100), resulted in an even more robust 3.5-fold improvement in efficiency over the linear.100.50 designs and a 2-fold improvement over the Alu.100.50 and U6+27.100.50 designs (Figure 1a). Excitingly, we observed persistence of significant levels in RNA editing at both 48 hours and 96 hours post transfection via these, while editing via linear guide RNAs was almost undetectable by the 96 hour time point (Figure 1b). We confirmed that U6 transcribed ribozyme flanked adRNAs were covalently circularized in cells, forming cadRNAs, which were detected via RT-PCR by designing outward facing primers that selectively amplified only the circularized structure (Figure 1c).
To confirm that circularization was indeed essential for boosting RNA editing (Figures 1a, 1b), we flanked the antisense sequence with catalytically inactive mutants of the twister ribozymes (ribozyme.mutant.200.100). This led to a significant decrease in RNA editing at both 48 and 96 hours post transfections with observed RNA editing levels similar to the linear versions (Extended Data Figure 1a). qPCR analysis confirmed the absence of circular adRNAs in cells transfected with ribozyme.mutant.200.100 (Extended Data Figure 1b). Additionally, in cells transfected with circular.200.100 plasmid, a significant fraction of the U6 transcribed adRNA was present in the circular form (Extended Data Figure 1b). To further ascertain that the long half-lives of the cadRNAs were responsible for persistent RNA editing observed, we treated cells transfected with circular.200.100 and ribozyme.mutant.200.100 plasmids with actinomycin D, a transcription inhibitor. Within 6 hours post-treatment we observed a significant reduction in the amounts of the ribozyme.mutant.200.100 adRNA while the levels of circular.200.100 adRNA remained constant (Extended Data Figure 1c). We also evaluated the intracellular localization of cadRNAs and detected them at high levels both in the nucleus and the cytoplasm (Extended Data Figure 1d).
Importantly, we confirmed that RNA editing via the circular guide RNAs, similar to the linear guide RNAs, was mediated by endogenous ADAR1 recruitment. Towards this, we performed a luciferase based reporter assay, where we assayed the guide RNAs for their ability to repair a premature stop codon (UAG) in the cypridina luciferase (cluc) transcript18 in the presence of scrambled and ADAR1 specific siRNAs. We observed a significant drop in luciferase activity in the presence of ADAR1 siRNA, confirming that RNA editing via long antisense adRNAs and circular adRNAs was dependent on endogenous ADAR1 levels (Figure 1d).
We next sought to evaluate the specificity profile of cadRNAs at both the transcriptome-wide and target transcript levels. Towards the former, a circular.100.50 and a circular.200.100 sample along with an untransfected HEK293FT sample were analyzed by deep RNA-seq. Notably, in contrast with enzyme overexpression where we routinely observed 103-104 transcriptome-wide off-targets20, we noted 2–3 orders of magnitude lower off-target editing via the cadRNAs and at levels similar to the linear long antisense guide RNAs (Figure 2a). Notably, over 80% of the adenosines detected as off-targets in these analyses were located in the RAB7A transcript itself which is indicative of bystander editing via cadRNA that we also confirmed via Sanger sequencing (Extended Data Figure 2). This is attributable to the long and perfectly paired dsRNA stretch created upon adRNA-target binding. By creating a G-mismatch32 opposite all non-target adenosines (cadRNA.bulges) we could eliminate this bystander editing, however this also led to a significant drop in the on-target editing efficiency to about 50% of the unmodified circular.200.100 version (Figures 2b, 2c, 2d). To address this, we engineered the antisense region to more closely mimic dsRNA structures of natural ADAR substrates. As demonstrated previously by Lehmann and colleagues, loops of 6 bp or more help to dictate selectivity of ADAR enzymes within its dsRNA substrate33 and so we engineered 8 bp loops positioned both 5 bp upstream and 30 bp downstream of the target adenosine (cadRNA.loops). This design led to a significant reduction in bystander editing within the 36 bp region between the bulges, with the on-target editing being double that achieved by simply placing opposing G mismatches (Figures 2b, 2c, 2d). However, we still observed significant bystander editing in the adenosines flanking the 36 bp region. We hypothesized that it might be possible to eliminate these via positioning of 8 bp loops all along the antisense domain at intervals of 15 bp flanking the 36 bp central region that carries the target adenosine (cadRNA.loops.interspersed). Indeed, this design substantially reduced bystander editing in the 200 bp dsRNA stretch formed between the target mRNA and the antisense domain, while maintaining on-target editing levels similar to the unmodified circular.200.100 construct (Figures 2b, 2c, 2d, Extended Data Figure 2). Taken together, a combination of appropriately positioned 8–12 bp loops to create breaks within the long stretch of dsRNA, along with certain A-specific bulges can thus be utilized to eliminate bystander editing in a target specific manner (Figures 2b, 2c, 2d, Extended Data Figure 2).
Figure 2: Transcriptome-wide and target transcript-level specificity profiles of cadRNAs.
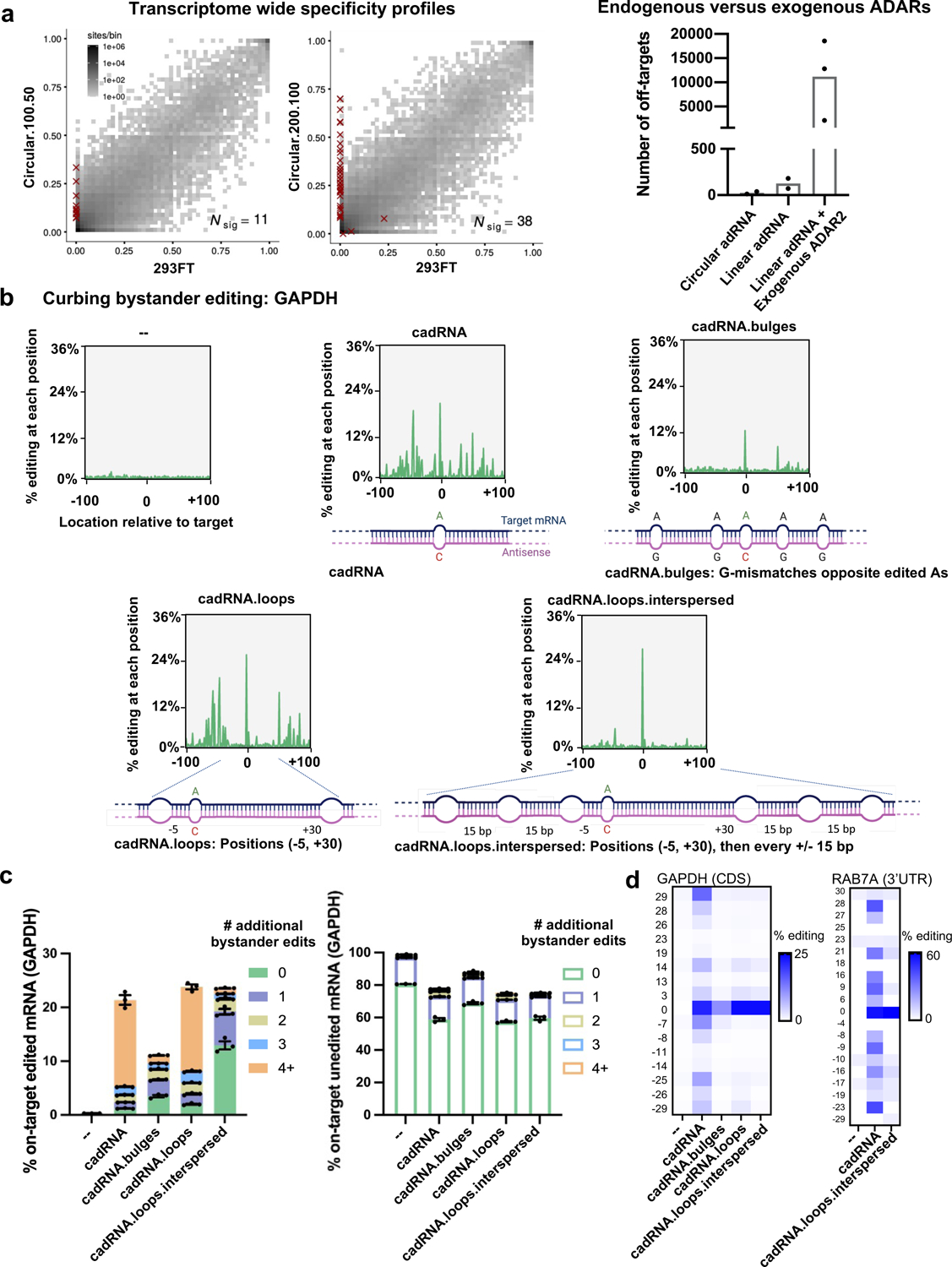
(a) (left-panel) 2D histograms comparing the transcriptome-wide A-to-G editing yields observed with a cadRNA construct (y-axis) to the yields observed with the control sample (x-axis). Each histogram represents the same set of reference sites, where read coverage was at least 10 and at least one putative editing event was detected in at least one sample. Nsig is the number of sites with significant changes in editing yield. Points corresponding to such sites are shown with red crosses. The on-target editing values obtained via Sanger sequencing for the samples are HEK293FT: 0%, circular.100.50: 40.47% and circular.200.100: 43.54% respectively. (right-panel) A comparison of the number of off-targets induced by delivery of circular adRNAs, linear adRNAs, and linear adRNAs with co-delivered ADAR220. (b) Engineered cadRNA designs for reducing bystander editing. Design 1 (cadRNA): Unmodified circular.200.100 antisense. Design 2 (cadRNA.bulges): Antisense bulges created by positioning guanosines opposite bystander edited adenosines. Design 3 (cadRNA.loops): Loops of size 8 bp created at position −5 and +30 relative to the target adenosine. Design 4 (cadRNA.loops.interspersed): Loops of size 8 bp created at position −5 and +30 relative to the target adenosine and additional 8 bp loops added at 15 bp intervals all along the antisense strand. Plots depicting the location and extent of all substitutions in the 200 bp dsRNA stretch (n=1 representative plot shown for each construct, analyzed via CRISPResso236). (c) Plots depict % of on-target edited or unedited reads with and without further A-to-G hyperedits in the 200 bp dsRNA stretch formed between the cadRNA and target RNA as observed with the various designs. Substitutions other than A-to-G were not considered for this analysis. Values represent mean % +/− SEM on-target editing in 200 bp long amplicons as quantified by NGS (n=3). (d) Heatmaps of percent editing within a 60 bp window around the target adenosine in the GAPDH and RAB7A transcripts. The positions of adenosines relative to the target adenosine (0) are listed to the left of the heatmap. Values represent mean (n=2). All experiments were carried out in HEK293FT cells.
Next, we confirmed the robustness and generalizability of the cadRNA format via their ability of to successfully edit adenosines in the 3’ UTR and coding sequence (CDS) of seven additional transcripts – GAPDH, ALDOA, DAXX, FANCC, CTNNB1, SMAD4 and TARDBP in HEK293FT cells (Figure 3a). Furthermore, in addition to delivery via a genetically encoded format in plasmids, we also explored if in vitro transcribed (IVT) circular adRNA would similarly be functional. The ribozymes flanking the antisense domain were rapidly cleaved upon transcription and these cleaved products were then delivered to cells where they underwent in situ circularization in the cells (Figure 3b, Extended Data Figure 3). 24 hours post transfection, we observed robust editing of the RAB7A and GAPDH transcripts using IVT adRNAs in HEK293FTs (Figure 3a) and also confirmed circularization of the IVT adRNAs via qPCR. Additionally, the plasmid and IVT adRNAs based editing of RAB7A in K562 cells using electroporation was similarly robust at 90% and 70% RNA editing yields respectively (Figures 3a, 3b). Notably, we confirmed that for a majority of the tested loci we did not observe significant knockdown of the targeted transcripts via the cadRNAs (Figure 3a).
Figure 3: In vitro activity of cadRNAs.
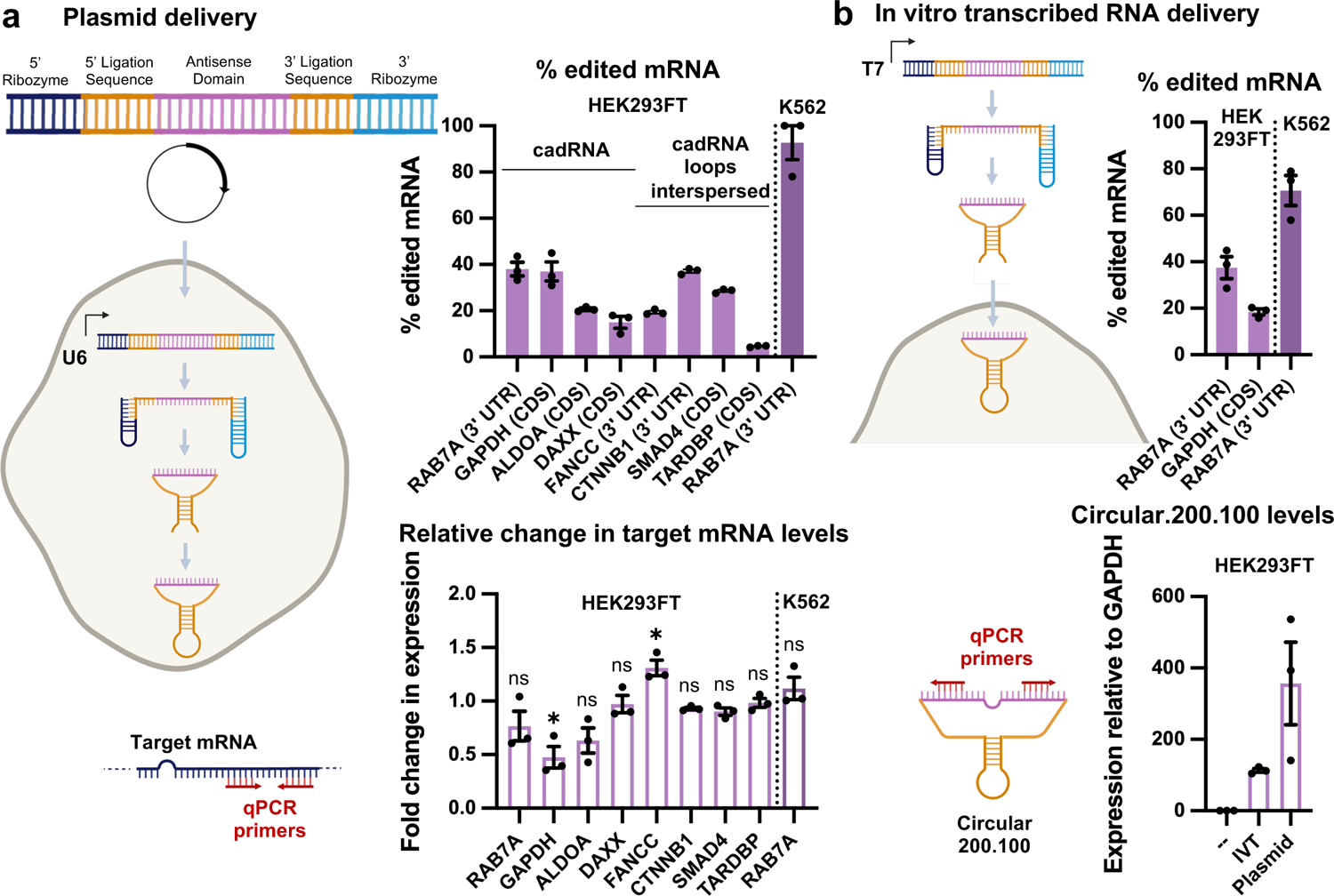
(a) Plasmid delivered in situ cadRNA generation: RNA editing efficiencies across various transcripts observed in HEK293FT and K562 cells via plasmid delivered circular.200.100 adRNA, 48 hours post transfections are shown. Values represent mean +/− SEM (n=3). These experiments were carried out using either cadRNA or cadRNA.loops.interspersed from Figure 2b. Associated changes in expression levels of target transcripts as compared to levels seen in untransfected controls is also shown, 48 hours post transfections (p=0.2599, p=0.0135, p=0.1982, p=0.7871, p=0.0144, p=0.2674, p=0.1168, p=0.7852, p=0.5145; unpaired t-test, two-tailed). (b) In vitro transcribed (IVT) circular adRNA generation: Linear forms of twister ribozyme flanked circular adRNAs were transcribed in vitro using a T7 polymerase, purified using LiCl, and transfected into cells, where they circularize in situ by the endogenous RNA ligase RtcB. RNA editing efficiencies across various transcripts observed in HEK293FT and K562 cells via IVT circular adRNA, 24 hours post transfections are shown. Values represent mean +/− SEM (n=3). Associated levels of IVT and plasmid delivered circular.200.100 adRNA targeting RAB7A measured in transfected HEK293FT cells 24 hours post transfections are also shown. Values represent mean +/− SEM (n=3).
Given the vastly improved efficiency and durability of RNA editing via cadRNAs, we next wondered if these could enable in vivo RNA editing. Since no co-delivery of proteins is required, successful demonstration here could enable a powerful gene therapy approach. Additionally, for the cadRNAs, one could leverage the already established delivery modalities and accruing knowledge from the field of shRNAs and ASOs that similarly only require delivery of nucleic acids to target tissues. To explore this, we first targeted an adenosine in the 3’ UTR of the mPCSK9 transcript via AAV8 mediated delivery of adRNAs to the mouse liver. We systematically compared RNA editing yields via linear.U6+27.100.50, one copy of circular.200.100, and two copies of circular.200.100 guide RNAs (Figure 4a). 2 weeks post injections, we harvested mice livers and did not detect any editing in the PBS injected mice, in mice injected with AAV8-mCherry, and notably in the mice injected with AAV8-linear.U6+27.100.50 guide RNAs too we did not measure detectable RNA editing (Figure 4b). Excitingly, we observed highly efficient 11% and 38% on-target editing via the AAV8 delivered single copy (1x) and two copy (2x) circular.200.100 guide RNAs respectively. Additionally, editing via AAV8–2x.circular.200.100 was persistent, with mPCSK9 editing levels of 53% observed 8 weeks post injections. We confirmed robust expression of the cadRNAs via qPCR, and noted that addition of a second copy of the circular.200.100 led to a 3-fold increase in expression levels, together suggesting that persistent and robust guide RNA expression was key to enabling efficient in vivo RNA editing (Figure 3c). Importantly, we also confirmed that cadRNAs delivered via AAVs did not alter the expression levels of the mPCSK9 transcript in mice livers (Figure 3d).
Figure 4: In vivo activity of cadRNAs.
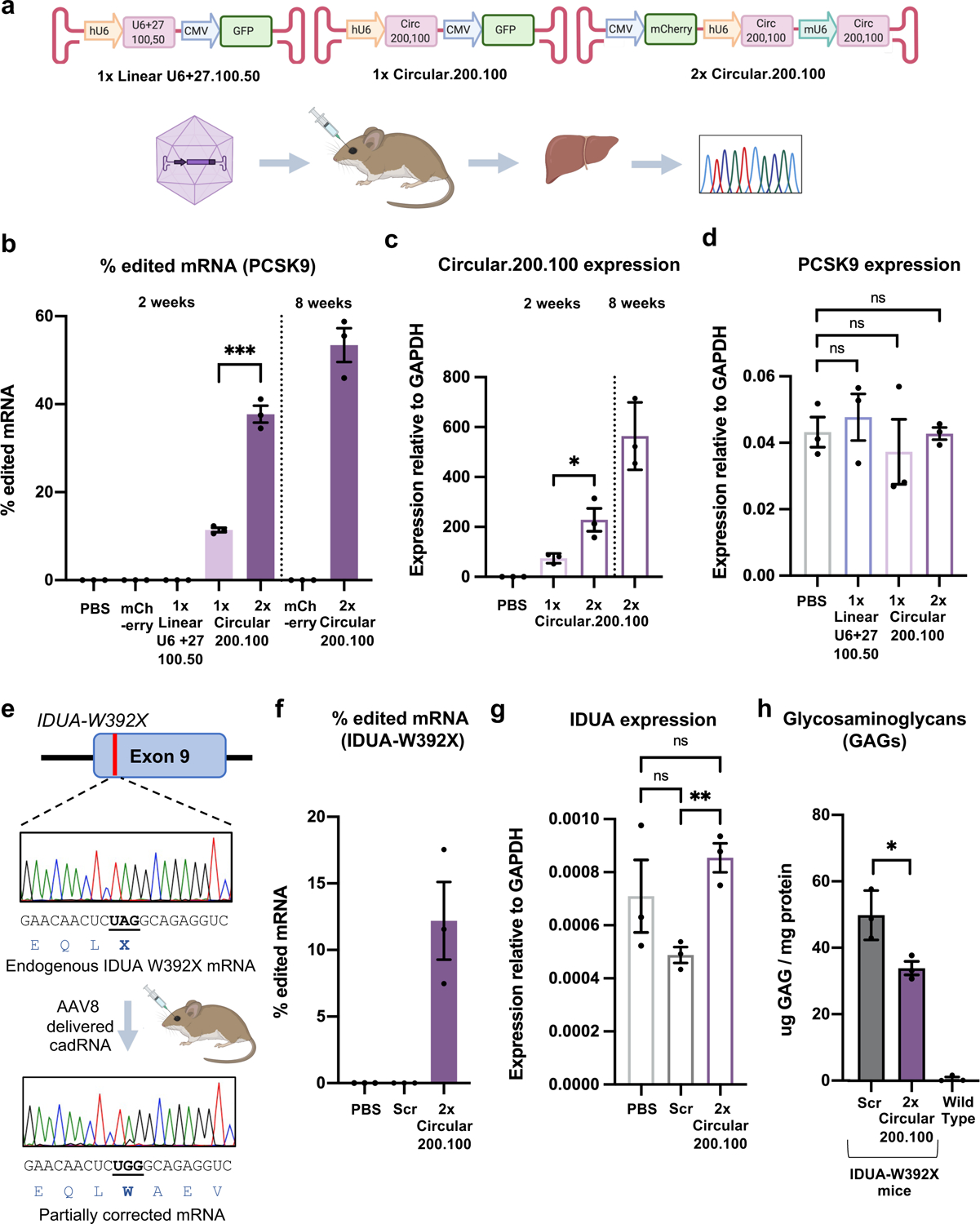
(a) (i) AAV vectors used for adRNA delivery. (ii) Schematic of the in vivo experiment. (b) In vivo RNA editing efficiencies of the mPCSK9 transcript in mice livers via systemic delivery of U6 transcribed linear (U6+27) and genetically encoded circular adRNAs packaged in AAV8. Values represent mean +/− SEM (n=3; p=0.0002; unpaired t-test, two-tailed). (c) Relative expression levels of circular adRNAs. Values represent mean +/− SEM (n=3; p=0.0305; unpaired t-test, two-tailed). (d) mPCSK9 transcript levels relative to GAPDH. Values represent mean +/− SEM (n=3; p=0.6179, p=0.6125, p=0.9323; unpaired t-test, two-tailed). (e) Schematic of the IDUA-W392X mRNA, and RNA editing experiment. (f) In vivo UAG-to-UGG RNA editing efficiencies of the IDUA transcript in mice livers via systemic delivery of genetically encoded circular adRNAs packaged in AAV8. Values represent mean +/− SEM (n=3). (g) IDUA transcript levels relative to GAPDH. Values represent mean +/− SEM (n=3; p=0.1185, p=0.3815, p=0.0042; unpaired t-test, two-tailed). (h) GAG content in mice livers of AAV8-scrambled.2x.circular.200.100 and AAV8-IDUA.2x.circular.200.100 injected IDUA-W392X mice. Wild type C57BL/6J mice were included as controls. Values represent mean +/− SEM (n=3; p=0.0285; unpaired t-test, two-tailed).
To evaluate the specificity profiles of the cadRNAs in vivo and also systematically study their effects on gene expression, we carried out RNA seq on 4 C57BL6/J litter-mates, 2 injected with AAV8-mCherry and 2 with AAV8–2x.circular.200.100, 2 weeks post injections. We observed precise transcript-specific editing of the PCSK9 mRNA in these mice (Extended Data Figure 4). Furthermore, we carried out qPCRs on several IFN-stimulated genes, especially those involved in sensing dsRNA such as RIG-I, MDA5, OAS1A, OSL, OASL2, PKR34. In the short term experiments, we did not observe significant changes in the levels of many of these genes, but observed that there is an increase in the levels of MDA5 and PKR in the mice injected with AAV8–2x.circular.200.100 as compared to the AAV8-mCherry control group. However, in the long term experiments we did not observe significant changes in the levels of any of these genes when compared to the AAV control group (Extended Data Figure 5a). Additionally, we also confirmed that presence of the cadRNAs did not significantly alter the expression of ADAR1-p110, ADAR1-p150 and ADAR2 as compared to the AAV control group (Extended Data Figure 5b). Differential expression analyses also confirmed no alterations in gene groups involved in sensing foreign RNA (Extended Data Figure 5c).
Building on these results, we next targeted a mouse model of Hurler syndrome. Hurler syndrome is a form of mucopolysaccharidosis type 1 (MPS1), a rare genetic disorder that results in the buildup of large sugar molecules called glycosaminoglycans (GAGs) in lysosomes. This occurs due to a lack of the enzyme alpha-L-iduronidase which is encoded by the IDUA gene. W402X is a commonly occurring mutation in the IDUA gene in Hurler syndrome patients and there exists a corresponding mouse model bearing the IDUA-W392X mutation35 (Figure 3e). With a goal to repair the IDUA-W392X premature stop codon, we packaged 2 copies of IDUA.circular.200.100 guide RNA into AAV8 and injected these into IDUA-W392X mice systemically. As a control we included a AAV8–2x.scrambled.circular.200.100. Two weeks post injection, we harvested mice livers and observed robust 7–17% correction of the premature stop codon in the mice injected with the AAV8–2x.IDUA.circular.200.100 adRNA (Figure 3e, 3f). We confirmed that expression of the circular.200.100 adRNA did not alter the expression levels of the IDUA transcript (Figure 3g). We also measured GAG levels in these mice, and observed about 33% less GAG accumulation in the treated animals over the 2 week period as compared to the scrambled control mice, indicating successful partial restoration of alpha-L-iduronidase activity (Figure 3h).
DISCUSSION
Use of endogenous ADARs for correction of G-to-A point mutations and premature stop codons carries immense therapeutic potential. However, the relatively short half-life of the guide RNAs limits efficacy. In this study we engineered circular guide RNAs (cadRNAs) for recruitment of endogenous ADARs, that vastly improve the efficiency and durability of programmable RNA editing. This method is highly specific at the transcriptome-level, and engineering of interspersed loops in the antisense domain also enabled high specificity at the transcript-level with significantly reduced bystander adenosine editing. Via AAV-delivered cadRNAs we also demonstrated for the first time robust, persistent, and highly transcript-specific in vivo RNA editing via endogenous ADAR recruitment, including in the IDUA-W392X mouse model of mucopolysaccharidosis type I-Hurler (MPS I-H) syndrome. While cadRNAs provide an exciting format for RNA editing, there are several areas that merit further investigation. Specifically: 1) While the circular.200.100 adRNAs provide a general framework to achieve robust and persistent RNA editing, we do observe variations in editing yields across targets. Further target specific optimizations while considering local sequence and structural contexts (such as pre-straining, or secondary structure modulation of the cadRNA, for instance, if the antisense domain is part of a stable duplex and is unavailable to bind its target) will be important to further improve cadRNA editing yields; 2) Additionally, coupling additional ADAR recruitment domains onto the cadRNA might further help boost editing yields; 3) For the IVT formats, we anticipate introduction of modified RNA bases such as pseudouridines or completion of circularization prior to delivery might be critical for enhancing cadRNA efficacy; 4) Also, as noted both in this and our prior work20, while a majority of targets maintained expression levels, for some targets clear RNAi effects are observed via both long-antisense adRNAs and cadRNAs, and correspondingly, modifying those guide designs will be critical to enable efficacious editing; 6) Additionally, impact on protein translation upon binding of the long antisense domains to the target mRNA needs further assessment. 7) Finally, on the in vivo studies front, while the two week long experiments analyzed via RNA-seq did not reveal enrichment of any gene groups involved in sensing foreign RNA, the effects of cadRNA accumulation will need to be carefully monitored over longer periods of time.
Taken together, as cadRNAs do not require the need for co-delivery of any effector proteins, and as a targeting moiety also have enhanced persistence in cells, they have the potential for broad utility in programmable RNA editing mediated transient protein modulation, and correction of G-to-A point mutations and premature stop codons for therapeutic applications. Moving beyond, we anticipate circularization of guide RNAs might also have utility in other transcriptome and genome engineering modalities, such as RNAi, ASOs, and guide RNAs in CRISPR-Cas.
ONLINE METHODS
Transfections:
Unless otherwise stated, experiments were carried out in HEK293FT cells (ThermoFisher: R70007) which were grown in DMEM supplemented with 10% FBS and 1% Antibiotic-Antimycotic (Thermo Fisher) in an incubator at 37 °C and 5% CO2 atmosphere. HEK293FT cells were seeded in 24 well plates and transfected using 1000 ng adRNA plasmid or 48 pmol of IVT RNA and 2ul of commercial transfection reagent Lipofectamine 2000 (Thermo Fisher). Cells were transfected at 25–30% confluence. Plasmid transfection experiments were harvested 48 hours post transfections while IVT RNA experiments were harvested 24 hours post transfections. For 96 hour long experiments, cells were passaged at a 1:4 ratio, 48 hours post transfections. Cells after plasmid electroporation were harvested at 48 hours, while IVT RNA experiments were harvested 24 hours post electroporation.
Electroporation:
K562 cells (ATCC: CCL-243) were grown in RPMI supplemented with 10% FBS and 1% Antibiotic-Antimycotic (Thermo Fisher) in an incubator at 37 °C and 5% CO2 atmosphere. 200,000 cells were electroporated with 1000 ng adRNA plasmid or 48 pmol of IVT RNA using the Amaxa SF cell Line 4D-Nucleofector X kit (Lonza) as per the manufacturer’s instructions.
In vitro transcription:
Sense RNA fragments and circular adRNA were made by in vitro transcription using the HiScribe T7 Quick High Yield RNA Synthesis Kit (NEB) as per the manufacturer’s protocol. DNA templates for the IVT reaction carried the T7 promoter sequence at the 5’ end and were created by PCR amplification of the desired sequence from plasmids or cDNA. PCR products were purified using a PCR Purification Kit (Qiagen) and then used for IVT.
Luciferase assay:
HEK293FT cells were grown in DMEM supplemented with 10% FBS and 1% Antibiotic-Antimycotic (Thermo Fisher) in an incubator at 37 °C and 5% CO2 atmosphere. All in vitro luciferase experiments were carried out in HEK293FT cells seeded in 96 well plates, at 25–30% confluency, using 200 ng total plasmid and 0.4 μl of commercial transfection reagent Lipofectamine 2000 (Thermo Fisher). Specifically, every well received 100 ng each of the Cluc-W85X (TAG) reporter and the adRNA plasmids. At the same time, every well also received 25 pmol siRNA. 48 hours post transfections, 20 μl of supernatant from cells was added to a Costar black 96 well plate (Corning). For the readout, 50 μl of Cypridina Glow Assay buffer was mixed with 0.5 μl Vargulin substrate (Thermo Fisher) and added to the 96 well plate in the dark. The luminescence was read within 10 minutes on Spectramax i3x or iD3 plate readers (Molecular Devices) with the following settings: 5 s mix before read, 5 s integration time, 1 mm read height.
Actinomycin D treatment:
24 hours post transfections, media with actinomycin D (5 μg/ml) was added to cells for the indicated duration of time.
Production of AAV vectors:
AAV8 particles were produced using HEK293FT cells via the triple-transfection method and purified via an iodixanol gradient. Confluency at transfection was about 50%. Two hours before transfection, cell medium was exchanged with Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 100X Antibiotic-Antimycotic (Gibco). All viruses were produced in 5×15 cm plates, where each plate was transfected with 10 μg of pXR-8, 10 μg of recombinant transfer vector and 10 μg of pHelper vector using polyethylenimine (PEI) (1 μg/μl linear PEI in ultrapure water, pH 7, using hydrochloric acid) at a PEI:DNA mass ratio of 4:1. The mixture was incubated for 10 minutes at room temperature and subsequently applied dropwise onto the cell media. The virus was harvested after 72 hours and purified using an iodixanol density gradient ultracentrifugation method. The virus was then dialyzed with 1× phosphate buffered saline (pH 7.2) supplemented with 50 mM sodium chloride and 0.0001% Pluronic F68 (Thermo Fisher) using 50 kDA filters (Millipore), to a final volume of ~1 ml, and quantified by quantitative PCR using primers specific to the ITR region, against a standard (ATCC VR-1616): AAV-ITR-F, 5′-CGGCCTCAGTGAGCGA-3′; AAV-ITR-R, 5′-GGAACCCCTAGTGATGGAGTT-3′.
Animal experiments:
All animal procedures were performed in accordance with protocol S16003 approved by the Institutional Animal Care and Use Committee of the University of California, San Diego. All mice were acquired from Jackson Labs. Mice were housed at a temperature of ~70 °F, with ~55 % humidity and a 12 hour light / 12 hour dark cycle. AAVs were injected retro-orbitally into both C57BL/6J mice and IDUA-W392X mice (B6.129S-Iduatm1.1Kmke/J), males, 6–8 weeks of age, at a dose of 1.0E13 vector genomes per mouse. At least 3 mice were injected per experimental condition. Mice were monitored three times a week for the duration of the experiment (2 weeks or 8 weeks).
GAG assay:
The GAG assay was performed following the protocol described in40. Briefly, harvested mouse tissues were homogenized in 1 ml PBS with a syringe and 16 gauge (1.6 mm) needle. Tissue homogenates were then incubated on ice for 20 min with Triton X-100 added to a final concentration of 1%. Protein concentration in the supernatant clarified via centrifugation was estimated using the Bradford assay. Supernatants were digested in 1 mg/ml Proteinase K (Qiagen) for 12 h at 55 °C then boiled for 10 min to inactivate the enzyme. Nucleic acids were digested using Benzonase nuclease (Sigma) at 37 °C for 1 h followed by 10 min boiling to inactivate the enzyme. Total amount of GAG in each sample was measured using the Blyscan GAG assay kit (Biocolor).
RNA extraction and quantification of editing:
RNA from cells was extracted using the RNeasy Mini Kit (Qiagen) while extraction from tissues was carried out using QIAzol Lysis Reagent and purified using RNeasy Plus Universal Mini Kit (Qiagen), according to the manufacturer’s protocol. 500–1000 ng RNA was incubated with 1 μl of 5 μM of a target specific sense RNA (synthesized via IVT) at 95 °C for 3 minutes followed by 4 °C for 5 minutes. This step was carried out to capture the circular adRNA which if tightly bound to the target mRNA would block reverse transcription. cDNA was then synthesized using the Protoscript II First Strand cDNA synthesis Kit (NEB). 1 μl of cDNA was amplified by PCR with primers that amplify about 300–600 bp surrounding the sites of interest (outside the length of the antisense domain) using OneTaq PCR Mix (NEB). The numbers of cycles were tested to ensure that they fell within the linear phase of amplification. PCR products were purified using a PCR Purification Kit (Qiagen) and sent out for Sanger sequencing. The RNA editing efficiency was quantified using the ratio of peak heights G/(A+G). Data was plotted using GraphPad Prism. RNA-seq libraries were prepared from 250 ng of RNA, using the NEBNext Poly(A) mRNA magnetic isolation module and NEBNext Ultra II Directional RNA Library Prep Kit for Illumina. Samples were pooled and loaded on an Illumina Novaseq 6000 (100 bp paired-end run) to obtain 40–45 million reads per sample.
qPCRs:
1 μl of 1:4 diluted cDNA was used to set up a 10 μl qPCR reaction using iTaq Universal SYBR Supermix (Biorad). Primers were designed to keep the amplicon length within 300 bp. 2 technical replicates were carried out for each sample.
Extraction of nuclear and cytoplasmic RNA:
48 hours post transfections, cells were harvested and nuclear and cytoplasmic RNA fractions were extracted using the PARIS kit (Thermo Fisher) as per the manufacturer’s protocol. The extracted RNA was treated with DNase and 100 ng was converted to cDNA using the Protoscript II First Strand cDNA synthesis Kit (NEB).
Mapping of RNA-seq reads:
Sequence read pairs from stranded RNA-seq libraries were mapped to the reference human genome hg38 by running STAR aligner version 2.7.3a41 with the following command line options: --clip3pAdapterSeq AGATCGGAAGAGCACACGTCTGAACTCCAGTCA AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT (to trim Illumina adapter sequences from the 3′ ends of the reads in each pair), --quantMode GeneCounts (to collect read counts for each gene), --alignSJDBoverhangMin 1 (following ENCODE standard practice), --peOverlapNbasesMin=10 --peOverlapMMp=0.05 (to correctly align pairs of overlapping reads), --outSAMmultNmax 1 (to limit output of multimapping reads), --alignEndsType EndToEnd (to avoid soft-clipping of reads), --outFilterMismatchNmax −1 --outFilterMismatchNoverReadLmax 0.2 --outFilterMultimapNmax 1 (to increase the likelihood of successful alignment for reads containing A-to-I editing events). The genome index for STAR aligner was built using transcript annotations from Gencode42 release 32 for the human genome assembly GRCh38. Each aligned read was retained for downstream analysis even when the corresponding mate in the pair could not be successfully aligned. Samtools version 1.1043 was used to sort the aligned reads by genomic coordinate and to mark duplicated single or paired reads. The file ReadsPerGene.out.tab generated by STAR aligner contains three types of read counts for each gene: counts collected without considering read strands, counts based on the first strand of each read pair, and counts based on the second strand. The counts based on the first strand were found to be zero for most genes, while the counts based on the second strand were comparable to the unstranded counts, thus confirming that the sequence of first (second) read in each pair of the stranded RNA-seq libraries had the same orientation as the first (second) cDNA strand, as expected from the NEBNext Ultra II Directional RNA Library Prep Kit. The RNA-seq reads obtained from mice were processed as above, except for the following differences: the version of STAR aligner was 2.7.7a; the transcript annotations were from Gencode release M27 for the mouse genome assembly GRCm39; the version of samtools was 1.11.
Analysis of differential gene expression:
RNA-seq libraries from mice were analyzed for differential gene expression using the Bioconductor package DESeq2 version 1.28.144. The per-gene counts of aligned read for each of four samples were collected by STAR aligner version 2.7.7a into a corresponding ReadsPerGene.out.tab file. The read counts corresponding to “the 2nd read strand aligned with RNA” were loaded for all samples into a DESeq2::DESeqDataSet object. Genes with less than 10 read counts in all samples were discarded. The counts for the remaining genes were processed using R function DESeq2::DESeq with default parameters. This function estimates size factors that account for differences in RNA-seq library size between the samples, estimates the dispersion parameters of the negative binomial distributions assumed for the read counts, fits generalized linear models (GLMs) to such counts, and calculates Wald statistics. The comparison between untreated and treated mice was carried out using R function DESeq2::results with default parameters, except that the significance cutoff for independent filtering optimization was set to 0.01. Shrinkage of effect sizes was carried out using R function DESeq2::lfcShrink with default parameters, thus employing the method of Approximate Posterior Estimator for GLM45.
Quantification of changes in RNA editing:
To quantify significant changes in RNA editing, the BAM files containing reads aligned to the reference genome were processed as follows. Reads marked as duplicates were ignored. To minimize the bias of library size on statistical comparisons between different samples, the remaining reads from each sample were down-sampled, using samtools view with options, to the smallest number of such reads available for any sample. The down-sampling fraction used for each sample was calculated by dividing the smallest number of uniquely aligned reads among all samples by the number of uniquely aligned reads available for the sample being down-sampled. However, reads for the control sample, which was used for all comparisons, were not down-sampled.
The first step to quantify A-to-I editing events is to count the actual bases occurring on RNA transcripts at positions that, according to the reference genome, are expected to harbor an adenine base. Thus, for transcripts oriented as the forward (reverse) reference strand, base counts must be collected at reference A-sites (T-sites). As noted above, the first (second) read in each pair of the stranded RNA-seq libraries has the same orientation as the first (second) cDNA strand, i.e., the opposite (same) orientation as the transcript from which each cDNA molecule is synthesized. Also, the Illumina sequencing technology yields a pair of reads from opposite strands of the sequenced DNA molecule. Therefore, to handle transcripts oriented as the forward reference strand, base counts were collected at reference A-sites using the second (first) read in a pair, if that read was mapped to the forward (reverse) reference strand. Conversely, to handle transcripts oriented as the reverse reference strand, base counts were collected at reference T-sites using the first (second) read in a pair, if that read was mapped to the forward (reverse) reference strand.
The C library htslib (github.com/samtools/htslib), version 1.12 was used to enumerate the aligned reads that overlapped each base position in the reference genome. Reference sites covered by less than ten reads were ignored. The value of the SAM tag MD, “String for mismatching positions”, was recorded by samtools calmd, version 1.11, in each alignment record, and was used to determine the reference base at each position of an aligned sequence read. Base deletions and insertions relative to the reference genome were ignored. Sequenced bases with a Phred quality score less than 13 were ignored. For each sample, an initial list of base counts from reads overlapping each selected reference A- and T-site was generated.
The initial lists of base counts from all samples were then used to generate a final list of reference A- and T-sites where such base counts were available for all samples, and where at least one sample had a non-zero count of G (C) at reference A-sites (T-sites). The total number of reference sites in the final list was 1600217 and 1455241 for human and mice samples respectively.
At each selected reference site in the final list, a pairwise comparison between the base counts for each treatment sample and those for the control sample was carried out using Fisher’s exact test, as implemented in R function fisher.test, with a 2-by-2 contingency table containing the counts of G (C) at reference A-sites (T-sites) in the first row, the counts of all other bases at those sites in the second row, the base counts for the control sample in the first column, and the base counts for the compared treatment sample in the second column. The resulting p-values were adjusted for multiple comparisons using the method of Benjamini and Hochberg46, as implemented in R function p.adjust. The proportion of the number of G (C) bases relative to the number of all bases was also calculated at each A-site (T-site). Reference A-sites (T-sites) with a significant change in such base proportion for at least one comparison between a treatment sample and the control sample were selected by requiring an adjusted p-value less than 0.01 and a fold change greater than 1.1 in either direction. To visually compare each treatment sample with the control sample, 2D histograms of the observed base proportions at all reference A- and T-sites in the final list were generated using ggplot247. Note: The on-target editing efficiency values obtained in the RNA seq are highly inflated due to a large number of reads coming from the cadRNAs mapping onto the target and thus were omitted from the 2D histograms. Long-read deep sequencing or Sanger sequencing was instead utilized to measure on-target editing.
Extended Data
Extended Data Fig. 1. Characterization of genetically encoded cadRNAs.
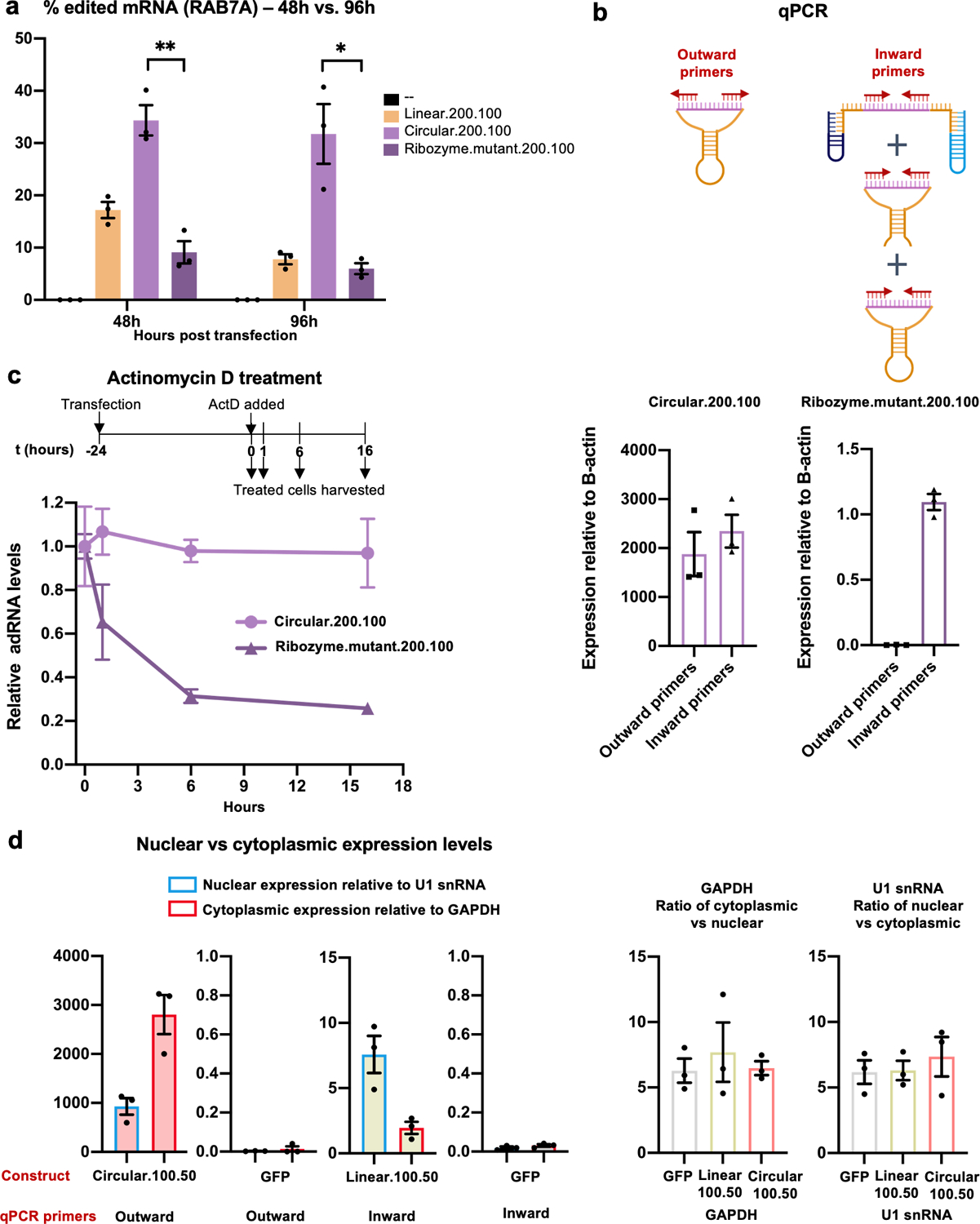
(a) RNA editing efficiencies achieved 48 hours and 96 hours post transfection of circular.200.100 and ribozyme.mutant.200.100 plasmids. Ribozyme.mutant.200.100 was created by substituting two key residues in both twister ribozymes (P3 ribozyme: residue 15 G to U and residue 16 U to G; P1 ribozyme: residue 22 A to G and residue 26 C to U) of the construct circular.200.10037,38. Values represent mean +/− SEM (n=3; p=0.0021, p=0.0112; unpaired t-test, two-tailed). (b) Schematic representation of various products detected by inward and outward binding primers used for quantification. The outward binding primers selectively amplify the cadRNA. The inward binding primers amplify uncleaved and cleaved-unligated fractions in addition to cadRNA. Values represent mean +/− SEM (n=3). (c) Cells transfected with circular.200.100 and ribozyme.mutant.200.100 plasmids were treated with actinomycin D for 1, 6 and 16 hours starting at 24 hours post transfections. qPCRs were carried out using inward binding primers from panel (b) and expression levels were normalized to untreated samples. (d) Levels of circular.100.50 and linear.100.50 adRNA were measured in the nucleus and cytoplasm. GFP transfected cells were included as controls. U1 snRNA and GAPDH were used to normalize for the nuclear and cytoplasmic compartments respectively. Relative U1 snRNA and GAPDH levels seen in the nuclear vs cytoplasmic fractions were consistent with other published work39. Values represent mean +/− SEM (n=3). All experiments were carried out in HEK293FT cells.
Extended Data Fig. 2. Curbing bystander editing of the RAB7A transcript.
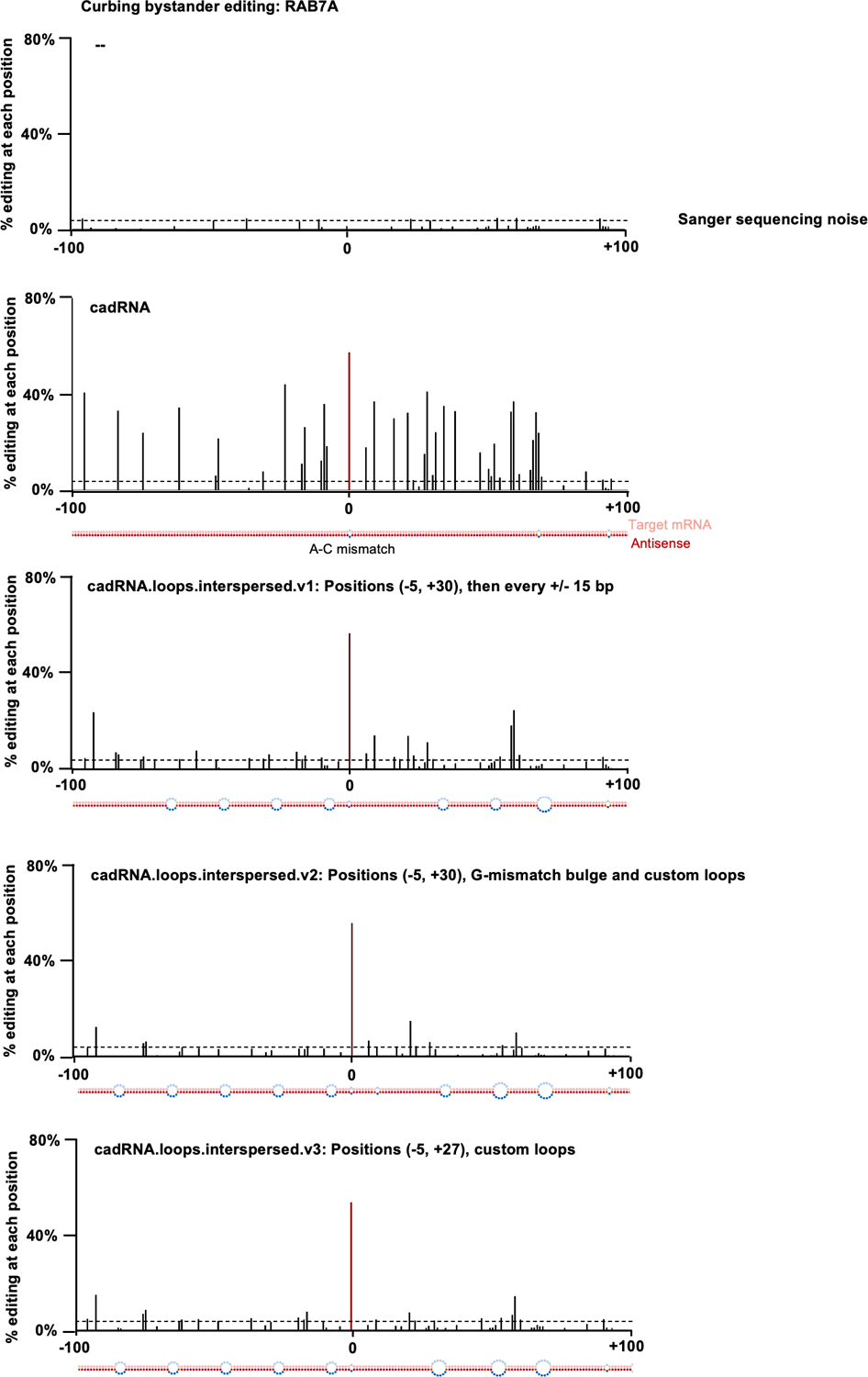
Histograms of percent A-to-G editing within a 200 bp window around the target adenosine in the RAB7A transcript as quantified by Sanger sequencing. The target adenosine is located at position 0. The dsRNA stretch formed between the antisense and the target are shown below each histogram. Design 1 (cadRNA): Unmodified circular.200.100 antisense, in addition to the A-C mismatch at position 0, two mismatches are seen at positions +66 and +91 that were created to avoid a stretch of poly Us to allow for transcription from a U6 promoter. Design 2 (cadRNA.loops.interspersed.v1): Loops of size 8 bp created at position −5 and +30 relative to the target adenosine and additional 8 bp loops added at 15 bp intervals along the antisense strand. Design 3 (cadRNA.loops.interspersed.v2): As compared to v1, a G-mismatch was positioned opposite a highly edited A (at position +9), an additional 8 bp loop was added at position −81 and the loop at position +49 was changed to a 12 bp loop. Design 4 (cadRNA.loops.interspersed.v3): As compared to v1, the 8 bp loop at +30 was changed to a 12 bp loop starting at position +27, one additional 8 bp loop was added at position −81 and the loop at position +49 was changed to a 12 bp loop. Values represent mean % editing (n=2). All experiments were carried out in HEK293FT cells.
Extended Data Fig. 3. Characterization of IVT synthesized cadRNAs.
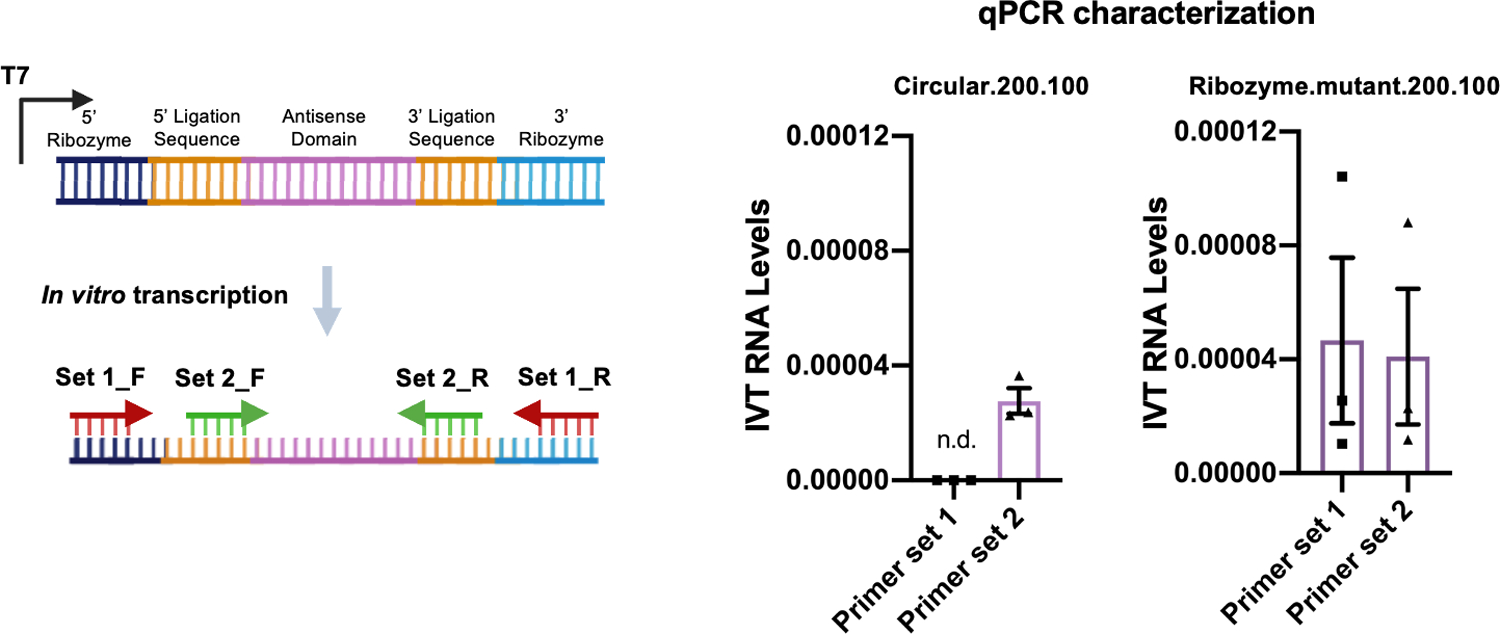
qPCRs were carried out on cDNA synthesized from IVT-circular.200.100 adRNA and IVT-ribozyme.mutant.200.100 adRNA using primers binding to the ligation stem and ribozyme sequence. n.d.: not detected. Values represent mean +/− SEM (n=3).
Extended Data Fig. 4. In vivo specificity of cadRNAs.
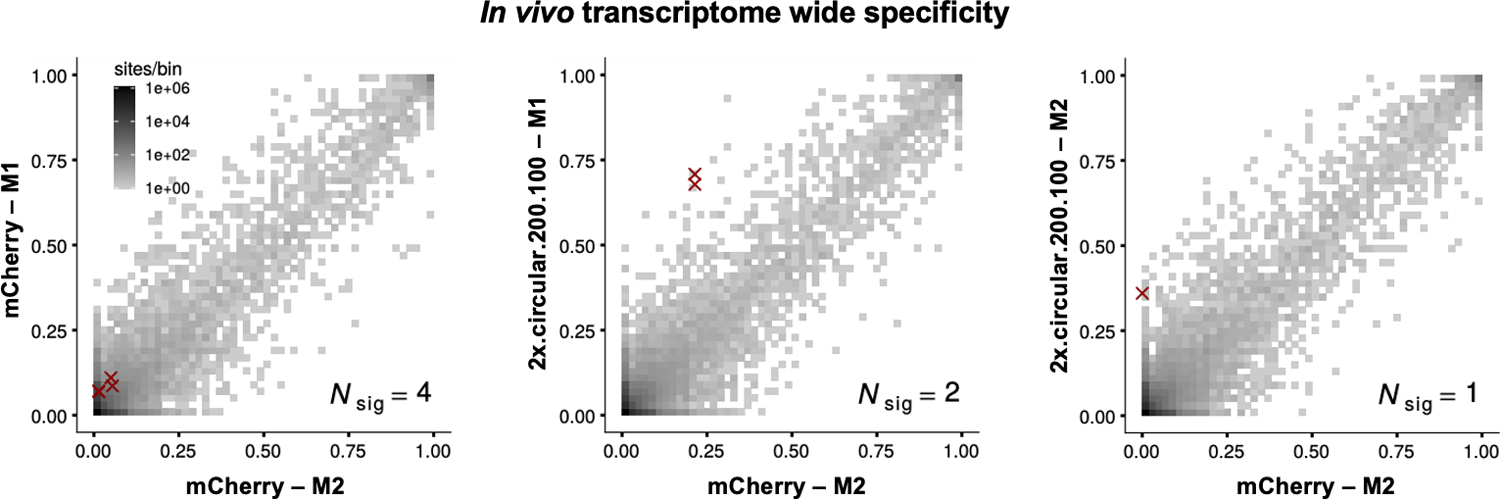
2D histograms comparing the transcriptome-wide A-to-G editing yields observed with an AAV delivered construct (y-axis) to the yields observed with the control AAV construct (x-axis). Each histogram represents the same set of reference sites, where read coverage was at least 10 and at least one putative editing event was detected in at least one sample. Nsig is the number of sites with significant changes in editing yield. Points corresponding to such sites are shown with red crosses. The on-target editing efficiency values obtained in the RNA seq are highly inflated due to a large number of reads coming from the cadRNAs mapping onto the target and thus have been omitted from the 2D histograms. The on-target editing values obtained via Sanger sequencing for the four samples analyzed by RNA seq were mCherry-M1: 0%, mCherry-M2: 0%, 2x.circular.200.100-M1: 42.94% and 2x.circular.200.100-M2: 41.32% respectively. M1 and M2 refer to injected mouse 1 and 2.
Extended Data Fig. 5. Transcriptomic changes associated with in vivo cadRNA expression.
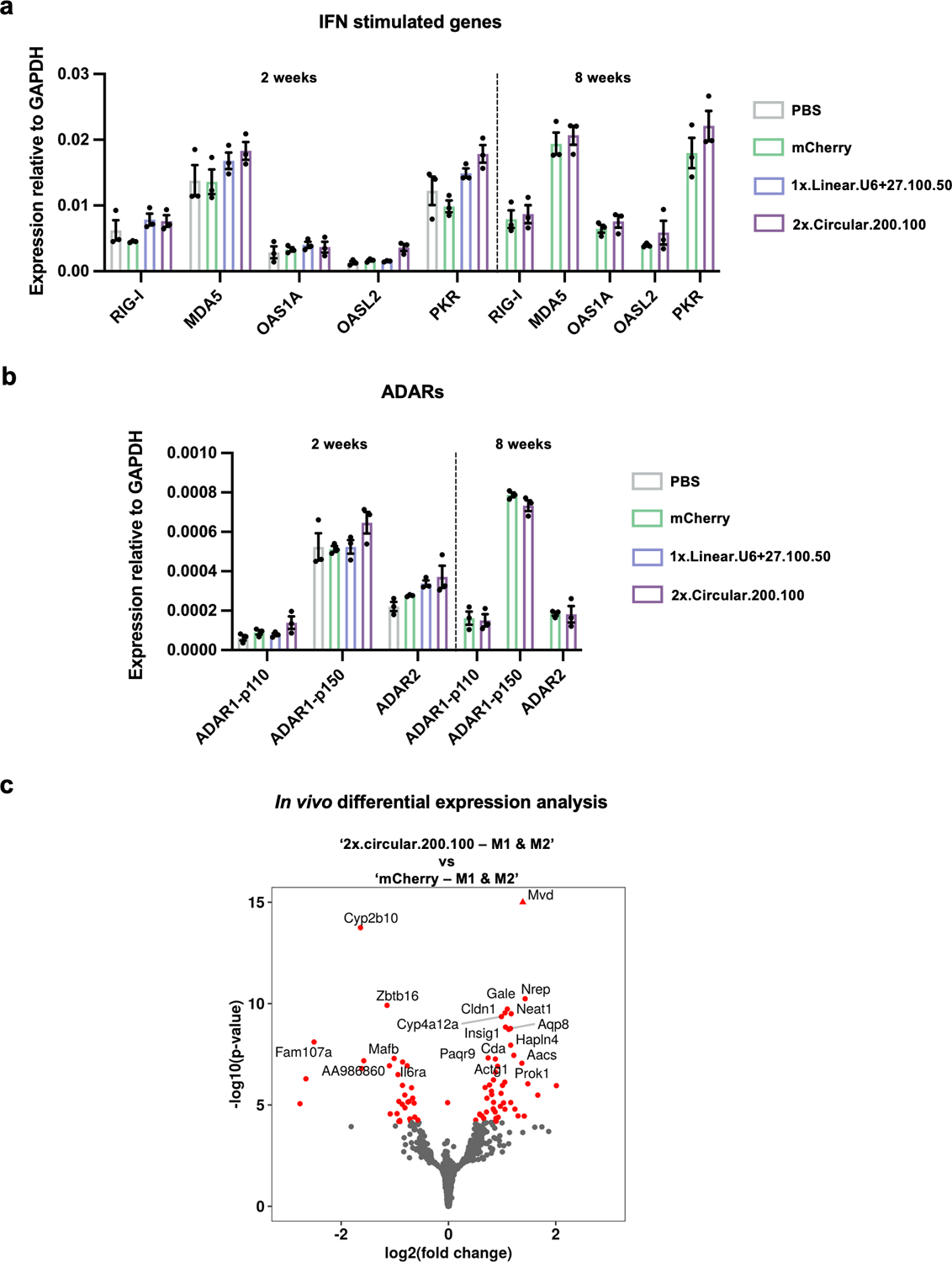
(a) qPCRs were carried out on IFN-inducible genes involved in sensing of dsRNA 2 weeks and 8 weeks post AAV injections. Values represent mean +/− SEM (n=3; p-values for 2 week long experiment, 2x.circular.200.100 vs mCherry, for genes from left to right p=0.0721, p=0.0353, p=0.8082, p=0.0748, p=0.0303; p-values for 8 week long experiment, 2x.circular.200.100 vs mCherry, for genes from left to right p=0.7276, p=0.6020, p=0.3838, p=0.3491, p=0.2746; unpaired t-test, two-tailed). (b) qPCRs were carried out on ADAR variants 2 weeks and 8 weeks post AAV injections. Values represent mean +/− SEM (n=3; p-values for 2-week long experiment, 2x.circular.200.100 vs. mCherry, for ADAR variants from left to right p=0.3165, p=0.1885, p=0.2815; p-values for 8 week long experiment, 2x.circular.200.100 vs. mCherry, for genes from left to right p=0.8150, p=0.1440, p=0.9532; unpaired t-test, two-tailed). (c) Transcriptome-wide differentially expressed genes in the two groups: 2x.circular.200.100 vs. mCherry are highlighted in red.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Genghao Chen, Lauren Hodge, Udit Parekh, Roberto Perales and other members of the Mali lab for discussions, advice and help with experiments. This work was generously supported by UCSD Institutional Funds and NIH grants (R01HG009285 (P.M.), RO1CA222826 (P.M.), RO1GM123313 (P.M.), 1K01DK119687 (D.M.)). This publication includes data generated at the UC San Diego IGM Genomics Center utilizing an Illumina NovaSeq 6000 that was purchased with funding from a National Institutes of Health SIG grant (#S10 OD026929). Schematics were created using BioRender.
Footnotes
COMPETING FINANCIAL INTERESTS
D.K. and P.M. have filed patents based on this work. P.M. is a scientific co-founder of Shape Therapeutics, Boundless Biosciences, Seven Therapeutics, Navega Therapeutics, and Engine Biosciences. The terms of these arrangements have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies. Y.S. is an employee of Shape Therapeutics. D.K. is now an employee of Shape Therapeutics. The remaining authors declare no competing interests.
CODE AVAILABILITY
Code is available from the corresponding author upon request.
PLASMID AND SEQUENCE AVAILABILITY
Plasmids used in this study are available via Addgene. All guide RNA and primer sequences are listed in Supplementary Table 1.
DATA AVAILABILITY
RNAseq data for Figure 2a and SI Figures 4, 5c is accessible via NCBI GEO under accession GSE164956. Any other data can be obtained from the corresponding author upon request. Publicly available datasets used in this study:
GRCh38, release 32, https://www.gencodegenes.org/human/release_32.html
GRCm39, release M27, https://www.gencodegenes.org/mouse/release_M27.html
REFERENCES
- 1.Melcher T et al. A mammalian RNA editing enzyme. Nature 379, 460–464 (1996). [DOI] [PubMed] [Google Scholar]
- 2.Bass BL & Weintraub H An unwinding activity that covalently modifies its double-stranded RNA substrate. Cell 55, 1089–1098 (1988). [DOI] [PubMed] [Google Scholar]
- 3.Bass BL & Weintraub H A developmentally regulated activity that unwinds RNA duplexes. Cell 48, 607–613 (1987). [DOI] [PubMed] [Google Scholar]
- 4.Mannion NM et al. The RNA-Editing Enzyme ADAR1 Controls Innate Immune Responses to RNA. Cell Rep. 9, 1482–1494 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tomaselli S et al. Modulation of microRNA editing, expression and processing by ADAR2 deaminase in glioblastoma. Genome Biol. 16, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schoft VK, Schopoff S & Jantsch MF Regulation of glutamate receptor B pre-mRNA splicing by RNA editing. Nucleic Acids Res. 35, 3723–3732 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wagner RW, Smith JE, Cooperman BS & Nishikura K A double-stranded RNA unwinding activity introduces structural alterations by means of adenosine to inosine conversions in mammalian cells and Xenopus eggs. Proc. Natl. Acad. Sci. U. S. A 86, 2647–2651 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nishikura K A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol 17, 83–96 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peng Z et al. Comprehensive analysis of RNA-Seq data reveals extensive RNA editing in a human transcriptome. Nat. Biotechnol 30, 253–260 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Eggington JM, Greene T & Bass BL Predicting sites of ADAR editing in double-stranded RNA. Nat. Commun 2, 319 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tan MH et al. Dynamic landscape and regulation of RNA editing in mammals. Nature (2017) doi: 10.1038/nature24041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levanon EY et al. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat. Biotechnol 22, 1001–1005 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Woolf TM, Chase JM & Stinchcomb DT Toward the therapeutic editing of mutated RNA sequences. Biochemistry 92, 8298–8302 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stafforst T & Schneider MF An RNA-Deaminase Conjugate Selectively Repairs Point Mutations. Angew. Chem. Int. Ed 51, 11166–11169 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Montiel-Gonzalez MF, Vallecillo-Viejo I, Yudowski GA & Rosenthal JJC Correction of mutations within the cystic fibrosis transmembrane conductance regulator by site-directed RNA editing. Proc. Natl. Acad. Sci. U. S. A 110, 18285–18290 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wettengel J, Reautschnig P, Geisler S, Kahle PJ & Stafforst T Harnessing human ADAR2 for RNA repair - Recoding a PINK1 mutation rescues mitophagy. Nucleic Acids Res. 45, 2797–2808 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fukuda M et al. Construction of a guide-RNA for site-directed RNA mutagenesis utilising intracellular A-to-I RNA editing. Sci. Rep 7, 41478 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cox DBT et al. RNA editing with CRISPR-Cas13. Science 358, 1019–1027 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Merkle T et al. Precise RNA editing by recruiting endogenous ADARs with antisense oligonucleotides. Nat. Biotechnol 37, 133–138 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Katrekar D et al. In vivo RNA editing of point mutations via RNA-guided adenosine deaminases. Nat. Methods 16, 239–242 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qu L et al. Programmable RNA editing by recruiting endogenous ADAR using engineered RNAs. Nat. Biotechnol 37, 1059–1069 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Monteleone LR et al. A Bump-Hole Approach for Directed RNA Editing. Cell Chemical Biology 26, 269–277.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sinnamon JR et al. In Vivo Repair of a Protein Underlying a Neurological Disorder by Programmable RNA Editing. Cell Rep. 32, 107878 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vallecillo-Viejo IC, Liscovitch-Brauer N, Montiel-Gonzalez MF, Eisenberg E & Rosenthal JJC Abundant off-target edits from site-directed RNA editing can be reduced by nuclear localization of the editing enzyme. RNA Biol. 15, 104–114 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vogel P et al. Efficient and precise editing of endogenous transcripts with SNAP-tagged ADARs. Nat. Methods 15, 535–538 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fire A et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391, 806–811 (1998). [DOI] [PubMed] [Google Scholar]
- 27.Zamecnik PC & Stephenson ML Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc. Natl. Acad. Sci. U. S. A 75, 280–284 (1978). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dias N & Stein CA Antisense oligonucleotides: basic concepts and mechanisms. Mol. Cancer Ther 1, 347–355 (2002). [PubMed] [Google Scholar]
- 29.Chung H et al. Human ADAR1 Prevents Endogenous RNA from Triggering Translational Shutdown. Cell 172, 811–824.e14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paul CP, Good PD, Winer I & Engelke DR Effective expression of small interfering RNA in human cells. Nat. Biotechnol 20, 505–508 (2002). [DOI] [PubMed] [Google Scholar]
- 31.Litke JL & Jaffrey SR Highly efficient expression of circular RNA aptamers in cells using autocatalytic transcripts. Nat. Biotechnol 37, 667–675 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heep M, Mach P, Reautschnig P, Wettengel J & Stafforst T Applying Human ADAR1p110 and ADAR1p150 for Site-Directed RNA Editing—G/C Substitution Stabilizes GuideRNAs against Editing. Genes 8, 34 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lehmann KA & Bass BL The importance of internal loops within RNA substrates of ADAR1. J. Mol. Biol 291, 1–13 (1999). [DOI] [PubMed] [Google Scholar]
- 34.Chen YG et al. Sensing Self and Foreign Circular RNAs by Intron Identity. Mol. Cell 67, 228–238.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang D et al. Characterization of an MPS I-H knock-in mouse that carries a nonsense mutation analogous to the human IDUA-W402X mutation. Mol. Genet. Metab 99, 62–71 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clement K et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol 37, 224–226 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Y, Wilson TJ, McPhee SA & Lilley DMJ Crystal structure and mechanistic investigation of the twister ribozyme. Nat. Chem. Biol 10, 739–744 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Felletti M, Stifel J, Wurmthaler LA, Geiger S & Hartig JS Twister ribozymes as highly versatile expression platforms for artificial riboswitches. Nat. Commun 7, 12834 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miao H et al. A long noncoding RNA distributed in both nucleus and cytoplasm operates in the PYCARD-regulated apoptosis by coordinating the epigenetic and translational regulation. PLoS Genet. 15, e1008144 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garcia-Rivera MF et al. Characterization of an immunodeficient mouse model of mucopolysaccharidosis type I suitable for preclinical testing of human stem cell and gene therapy. Brain Res. Bull 74, 429–438 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frankish A et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 47, D766–D773 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhu A, Ibrahim JG & Love MI Heavy-tailed prior distributions for sequence count data: removing the noise and preserving large differences. Bioinformatics 35, 2084–2092 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Benjamini Y & Hochberg Y Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc 57, 289–300 (1995). [Google Scholar]
- 47.Wickham H ggplot2: Elegant Graphics for Data Analysis. (Springer, 2016). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNAseq data for Figure 2a and SI Figures 4, 5c is accessible via NCBI GEO under accession GSE164956. Any other data can be obtained from the corresponding author upon request. Publicly available datasets used in this study:
GRCh38, release 32, https://www.gencodegenes.org/human/release_32.html
GRCm39, release M27, https://www.gencodegenes.org/mouse/release_M27.html