

Abstract
Background
Pathogenic variants in SPTAN1 have been linked to a remarkably broad phenotypical spectrum. Clinical presentations include epileptic syndromes, intellectual disability, and hereditary motor neuropathy.
Objectives
We investigated the role of SPTAN1 variants in rare neurological disorders such as ataxia and spastic paraplegia.
Methods
We screened 10,000 NGS datasets across two international consortia and one local database, indicative of the level of international collaboration currently required to identify genes causative for rare disease. We performed in silico modelling of the identified SPTAN1 variants.
Results
We describe 22 patients from 14 families with five novel SPTAN1 variants. Of six patients with cerebellar ataxia four carry a de novo SPTAN1 variant, two showed a sporadic inheritance. In this group one variant (p.Lys2083del) is recurrent in four patients. Two patients have novel de novo missense mutations (p.Arg1098Cys, p.Arg1624Cys) associated with cerebellar ataxia, in one patient accompanied by intellectual disability and epilepsy. We furthermore report a recurrent missense mutation (p.Arg19Trp) in 15 patients with spastic paraplegia from seven families with a dominant inheritance pattern in four and a de novo origin in one case. One more patient carrying a de novo missense mutation (p.Gln2205Pro) has a complex spastic ataxic phenotype. Through protein modelling we show that mutated amino acids are located at crucial interlinking positions, interconnecting the three-helix bundle of a spectrin repeat.
Conclusions
We show that SPTAN1 is a relevant candidate gene for ataxia and spastic paraplegia. We suggest that for the mutations identified in this study, disruption of the interlinking of spectrin helices could be a key feature of the pathomechanism.
Introduction
The paradigm of “one gene - one phenotype” is increasingly challenged by the discovery of considerable pleiotropy for many genes, leading to overlapping disease spectra across many neurological diseases such as motor neuron and frontotemporal diseases, motor neuropathies, spastic paraplegia, and Charcot-Marie-Tooth disease. The widespread implementation and increasing accessibility of next generation sequencing (NGS) platforms accelerates the identification of new gene-disease-relations in rare disease (RD) patients and constantly broadens our understanding of such overlapping clinical entities.1–3
Of recent and particular interest is the wide range of Mendelian gene-disease links established for neurological diseases and mutations in the SPTAN1 gene. Initially, SPTAN1 mutations were reported to be linked with severe epileptic syndromes and intellectual disability.4 In these patients, brainstem and cerebellar atrophy, cerebral hypomyelination and microcephaly are frequently observed. We recently identified three families with nonsense mutations causing a hereditary motor neuropathy (HMN) phenotype.5 An independent report of a de novo nonsense mutation in a HMN patient confirmed the pathogenicity of SPTAN1 nonsense mutations.6 Later SPTAN1 was also implicated in a sensory-motor peripheral neuropathy phenotype accompanied by a developmental disorder.7 There are also two reports on putatively recessively inherited SPTAN1 variants in hereditary spastic paraplegia (HSP) patients.8,9 One patient with severe intellectual disability was reported with cerebellar ataxia, but pathogenicity of the SPTAN1 intronic variant could not be conclusively established due to a pathological expansion at the ATXN8 locus. Other reported symptoms include autism and migraine. Aggregation of spectrin complexes was found in cortical neurons differentiated from induced pluripotent stem cells (iPSCs) of a patient with epileptic encephalopathy, however, this aggregate formation cannot be considered as a pathogenic hallmark for all other epilepsy associated SPTAN1 mutations.4,10 In contrast, nonsense-mediated mRNA decay haploinsufficiency was proposed in the HMN cohort.5
The SPTAN1 gene encodes for α-II-spectrin, a major component of the cytoskeleton expressed in all cells, except erythrocytes, which express α-I-spectrin.11 In neurons, α-II-spectrin forms a complex with one of four β-spectrins (SPTBN1, SPTBN2, SPTBN4, SPTBN5) through heterotetramerization of two α- and two β-subunits (Figure 1). While the α-subunit is always α-II-spectrin in neuronal cells, the composition of the complex in terms of its β-spectrin can differ depending on the cell type and even its subcellular localization.12,13 The canonical function of the spectrin complex is to interlink actin rings, giving rise to the membrane-associated periodic skeleton (MPS). The α-II-spectrin protein counts 20 spectrin repeats, each composed of a three-helix bundle.14 Additionally the spectrin complex has a scaffolding function ensuring the correct localization of several ankyrins, known to be of critical importance for neuronal development and homeostasis (Figure 1).14
Figure 1: Function, structure, and subcellular compositions of spectrin.
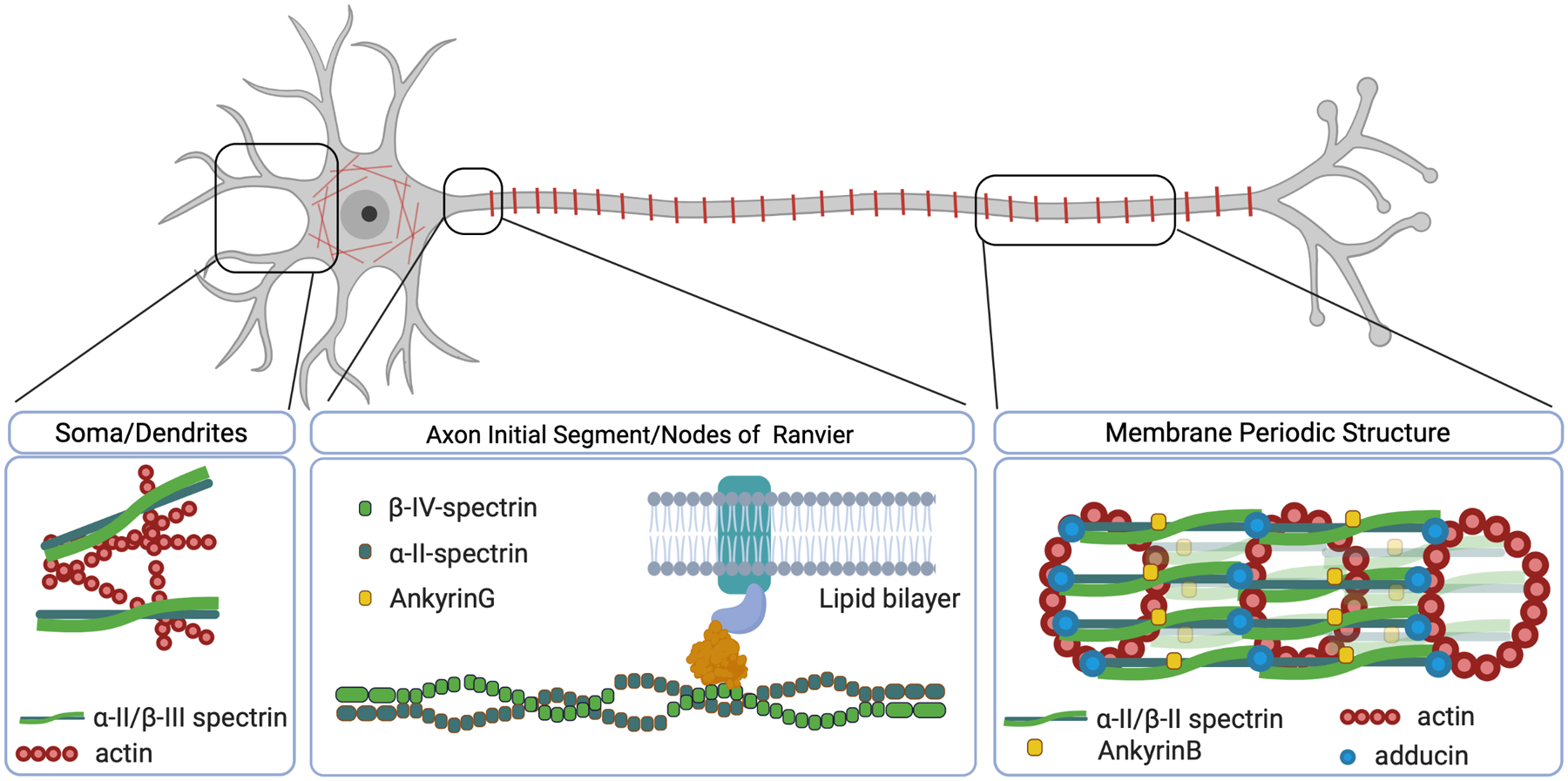
Left, the mesh-like structure formed in the somatodendritic area. Middle, the scaffolding role of the α-II/beta-IV spectrin complex. Right, Membrane Periodic Structure (MPS) with actin rings along the distal axon.
The exact genotype-phenotype correlations remain enigmatic in SPTAN1-related diseases. At least 35 different mutations in SPTAN1 scattered across the entire gene have been identified, but why these mutations affect diverse neuronal subtypes at different developmental timepoints remains unexplained (Figure 2). Several β-spectrins have also been implicated in neuronal disease. SPTBN1 (β-II-spectrin) mutations have been associated with intellectual disability and autism, while SPTBN2 (β-III-spectrin) mutations cause both dominant and recessive cerebellar ataxia.15–17 Furthermore, SPTBN4 (β-IV-spectrin) is associated with a neurodevelopmental disorder with hypotonia, neuropathy and deafness.18,19 Apart from Mendelian gene-disease links, defects in spectrins are also implicated in degenerative and psychiatric conditions.20 These ‘spectrinopathies’ illustrate the vital nature of the spectrin complex in neuronal development and homeostasis.
Figure 2: Graphical representation of SPTAN1 mutations reported with conclusive Mendelian inheritance.
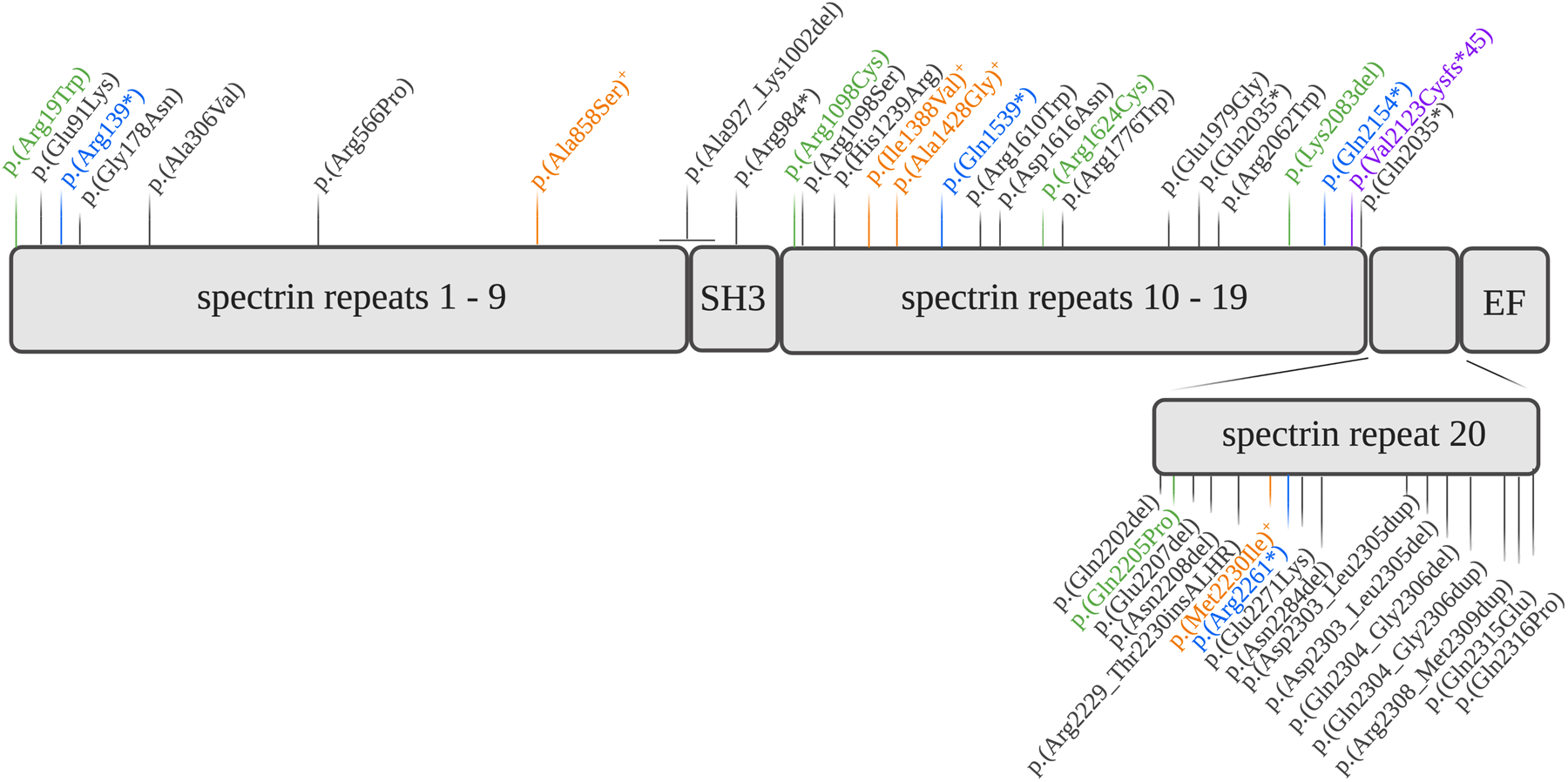
Green, mutations identified in this study. Orange, mutations identified in HSP patients. Blue, mutations identified in HMN patients. In purple, mutation causative of sensorimotor neuropathy and developmental disorder. In black, mutations associated with epilepsy, autism or intellectual disability.
Given the association of SPTBN2 with cerebellar ataxia and preliminary reports of SPTAN1 mutations in two recessive HSP families, as well as the cerebellar atrophy frequently noted in epilepsy patients carrying SPTAN1 mutations, we set out to delineate the role of SPTAN1 variants in these rare neurological disorders. Through a screen of more than 10,000 NGS datasets, we describe 22 patients from 14 families with five novel SPTAN1 variants.
Materials and methods
Patient Cohort
We used 10,000 NGS datasets of RD patients from two large international consortia and one local database. First, 3,500 HSP and ataxia NGS datasets, including 2,500 datasets from the PREPARE ataxia network and a further 1,000 HSP datasets were provided by the Genesis database.21 Genesis is a NGS data-sharing and analysis platform (https://www.tgp-foundation.org), housing a number of rare disease consortia. Additionally, the Solve-RD EU-Horizon 2020 (http://solve-rd.eu/) network includes more than 12,000 NGS datasets from which we used 3,500 datasets from patients in the European Reference Network for Rare Neurological Disorders (ERN-RND). Diseases covered in the ERN-RND include Cerebellar Ataxias and Hereditary Spastic Paraplegias, Choreatic syndromes, Dystonias, Paroxysmal Movement Disorders, Frontotemporal Dementia and Leukodystrophies. They were analyzed through the RD-Connect Genome-Phenome Analysis Platform (GPAP). Finally, 3,600 datasets were included from patients with different neurological diseases, belonging to a series of 14,500 NGS datasets generated in a diagnostic context at the Institute of Medical Genetics and Applied Genomics (IMGAG, Tübingen). Part of these datasets (>1,200) have been contributed to the Solve-RD consortium for secondary use in research setting and overlap with the NGS datasets stated above in ERN-RND. Finally, 30,000 more NGS datasets belonging to patients with other RD phenotypes are available for analysis through the abovementioned consortia. All genetic and patient data is handled and processed according to the General Data Protection Regulation (GDPR) rules set by the EU, as approved in a Data Management Plan submitted to the University of Antwerp. All patients and/or their legal representatives signed an informed consent locally, the study was approved by the Ethics Committee of the participating centers.
Genetic Studies
Conventional NGS methodology was used (see Supplementary Material). There were no relevant variants in genes known for ataxia, HSP or other Rare Neurological Disorders (RND) found for any of the patients described in this study. Filtering was performed first by searching for variants affecting conserved amino acids in the SPTAN1 gene shared across multiple families but absent from gnomAD.22 After the identification of a de novo deletion, an additional search was performed for conserved variants absent from gnomAD with a possible de novo inheritance. Where possible Sanger segregation analysis was performed (see carrier status in Figure 3 and Supplementary Figure 2). Primer sequences are available upon request. Paternity testing by DNA fingerprinting and STR analysis was performed where available (families A, B, M and N) to confirm the de novo status of variants. All SPTAN1 variants are mapped to transcript NM_001130438.2.
Figure 3: Pedigrees of families with SPTAN1 mutations.
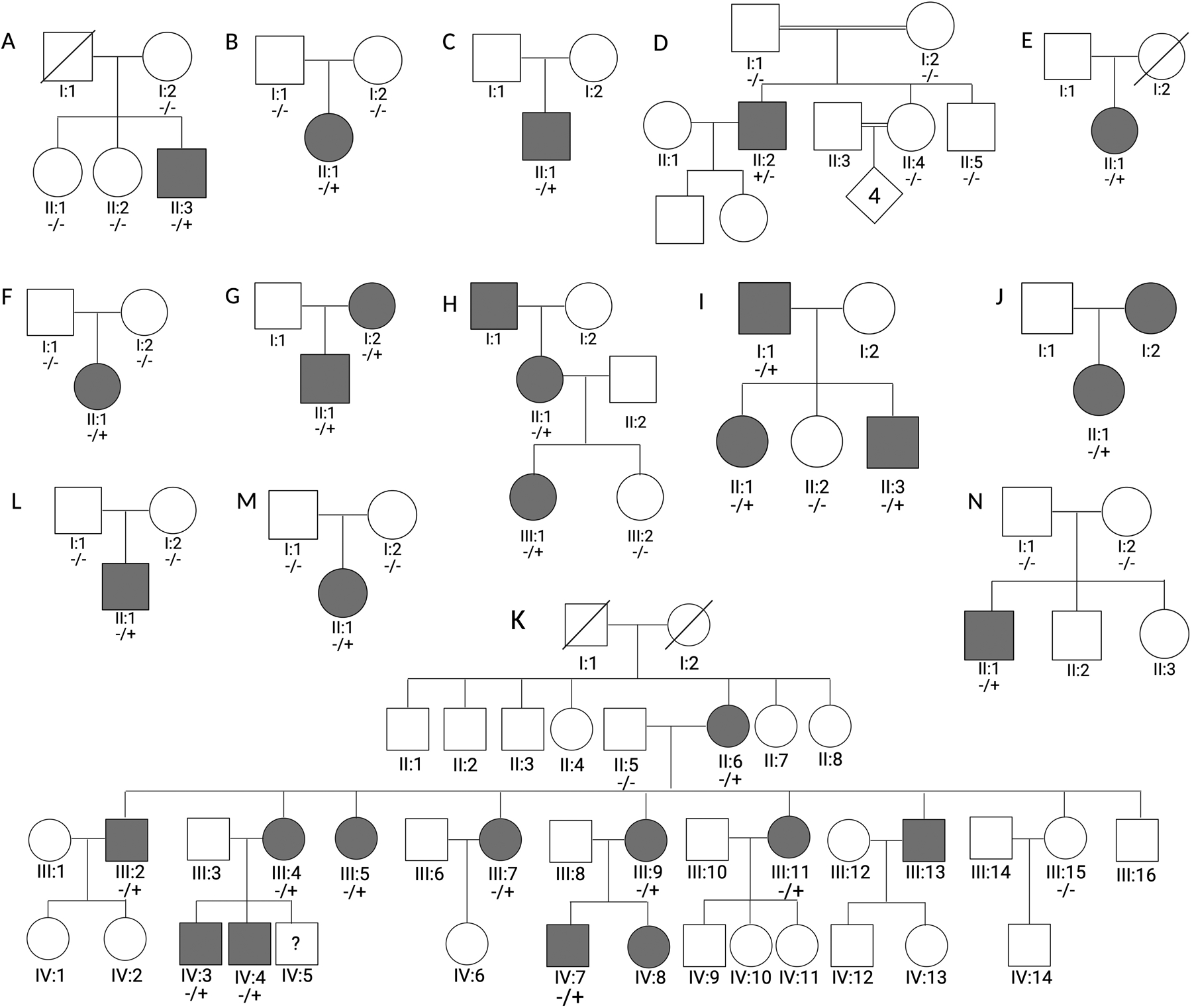
Pedigrees of families showing the segregation of SPTAN1 variants (NM_001130438.2:c.6247_6249delAAG, p.Lys2083del in families A-D), (NM_001130438.2:c.55C>T, p.Arg19Trp in families E-K), (NM_001130438.2:c.3292C>T, p.Arg1098Cys in family L), (NM_001130438.2:c.4870C>T, p.Arg1624Cys in family M), (NM_001130438.2:c.6614A>C, p.Gln2205Pro in family N); Affected (black symbols) and unaffected individuals (white symbols). Mutation results are indicated as carrier (−/+) or non-carrier (−/−).
In-depth phenotyping
Standardized in-depth phenotyping was used for all patients with a SPTAN1 variant. Clinical information was collected per patient in a cross-center harmonized fashion using a questionnaire. Phenotypes evaluated were not only limited to the specific phenotypic traits associated with HSP and ataxia but also evaluated the presence of epilepsy, intellectual disability and peripheral neuropathy, all symptoms known to be associated with other SPTAN1 variants. Representative MRI images are shown in Supplementary Figure 1. Data of performed Nerve Conduction Studies (NCS) is available in Supplementary Table 1.
Structural modelling of SPTAN1 variants
We evaluated the consequence of each SPTAN1 missense variant using three-dimensional (3D) models of the α-II-spectrin protein structure. For the p.Arg19Trp mutation, the crystal structure of the α-II-spectrin tetramerization domain was used (PDB entry 3F31).23 No experimental structure is known for other parts of the α-II-spectrin protein. Given the extensive size (2472 amino acids) of α-II-spectrin, the Phyre2 webserver was used to model at least two neighboring spectrin repeats at the location of each missense variant,24 resulting in five different 3D models (corresponding to amino acid positions 890–1170, 1339–1549, 1446–1762 and 1873–2472). Across all generated models, the top template hit used for homology modelling by Phyre2 was either PDB entry 3edv, 1u4q or 1u5p, all relevant structures of spectrin repeats.25,26 For missense variants, the impact of the variant was predicted and modelled using the Missense3D server.27 Protein structures were visualized using the UCSF ChimeraX molecular visualization program.28
Data availability
The raw NGS data cannot be made publicly available for confidentiality policies in the consortia involved. The NGS data in Solve-RD is available through the RD-Connect GPAP after an embargo period of 6 months. All other data generated during this study is available upon reasonable request.
Results
Genetic findings
Shared across Solve-RD, PREPARE and Genesis consortia as well as the IMGAG database, we analyzed in total more than 10,000 whole-exome or whole-genome sequencing datasets. We identified the c.6247_6249delAAG, p.Lys2083del variant in four sporadic probands with ataxia (patients A:II:3, B:II:1, C:II:1 and D:II:2). We also identified the c.55C>T, p.Arg19Trp SPTAN1 variant in seven probands with HSP (patients E:II:1, F:II:1, G:II:1, H:III:1, I:II:1, J:II:1 and K:III:5). Through segregation analysis, we found that the p.Arg19Trp variant follows a dominant inheritance pattern in four different families (families G, H, I and K) and arose de novo in one additional index patient (patient F:II:1). In the last two families, families E and J, a segregation pattern could not be determined, although patient J:I:2 is reported as affected and thus a dominant inheritance pattern is probable. The p.Lys2083del variant was shown to be de novo in two index patients (patients B:II:1 and D:II:2). A de novo variant was suspected but not conclusively proven in two more index patients (patients A:II:3 and C:II:1) (Figure 3). We therefore re-evaluated the patient cohort for conserved de novo variants in the SPTAN1 gene, leading to the identification of three additional SPTAN1 variants (c.3292C>T, p.Arg1098Cys; c.4870C>T, p.Arg1624Cys; c.6614A>C, p.Gln2205Pro) linked to ataxia with or without epilepsy and intellectual disability; all were demonstrated to be de novo (Figure 3, L–M). Identified variants are furthermore absent from more than 30,000 NGS datasets from patients with other RD phenotypes that do not include HSP or ataxia symptoms (see Patient Cohort).
Clinical findings
We identify 22 patients belonging to 14 families. Two recurrent mutations were found; p.Lys2083del and p.Arg19Trp. The first associated with predominant cerebellar ataxia (4/22), the latter with spastic paraplegia (15/22). Furthermore, we describe three de novo missense mutations: p.Arg1098Cys, p.Arg1624Cys and p.Gln2205Pro.
Cerebellar ataxia sub-group (p.Lys2083del mutation)
Four unrelated patients (A:II:3, B:II:1, C:II:1 and D:II:2) with a p.Lys2083del mutation were diagnosed with cerebellar ataxia. Gait instability was the initial symptom in all four patients. The overall phenotype is described as slowly progressive cerebellar ataxia and mild intellectual disability with early to juvenile disease onset (range 3–25 years). Patient B:II:1 showed a more complex phenotype with co-occurrence of hearing impairment, paroxysmal dyskinesia and dystonic movement in the upper limbs and toes as well as cervical dystonia. Also (sub-)cortical myoclonus was present responding to carbamazepine, lamotrigine and regressing fully under zonisamide. Patient D:II:2 showed signs of pyramidal tract involvement. MRI showed marked cerebellar atrophy in all patients and a thin corpus callosum in one (D:II:2).
Hereditary Spastic Paraplegia sub-group (p.Arg19Trp mutation)
This group consists of 15 patients. The onset age ranges from congenital to adolescence (0 – 13y). One multigenerational family was included (Figure 3, family K) with in total 12 affected members across three generations; detailed clinical data was available for 7 of them. The index patient (K:III:5) showed a congenital onset pure spastic paraplegia with marked leg spasticity (Ashworth 3–4). Most of the other family members showed a similar phenotype albeit with a variable grade of severity. Three patients, a mother and two of her three sons (K:III:4; K:IV:3 and K:IV:4), developed a more complex phenotype, with evidence of cerebellar and peripheral neuropathic involvement. NCS/EMG confirmed the presence of a sensory-motor mainly axonal neuropathy with possibly superimposed mild focal demyelinating features. No MRI abnormalities were present in all (7) examined patients.
In addition, four other families (G, H, I and J) were included. All showed early onset (range 6–8 years) pure spastic paraplegia and slowly progressed to moderate reduction in ambulation distance, except patient H:III:1 who is wheelchair dependent at age 40. Only the father of family J (I:I:1) had evidence of a mixed sensorimotor axonal neuropathy. Of note, patient I:II:1 had Rolandic epilepsy of maternal inheritance based on medical history. In patient G:II:1, walking difficulties similarly started at age 6, with the patient recently being diagnosed with bilateral optic atrophy.
Finally, two sporadic patients were included. Patient E:II:1 showed an early (3 years) onset spastic paraplegia with high arched feet. Patient F:II:1 showed an early onset form of spastic paraplegia complicated with distal weakness, optic neuropathy, unilateral vestibular areflexia and hypermobility.
Additional complex spastic and ataxia phenotypes
Three more patients were found to harbour a de novo SPTAN1 mutation, while displaying the different extremes of the spastic ataxia spectrum. The phenotype of patient M:II:1, bearing a de novo p.Arg1624Cys mutation, is dominated by early onset cerebellar ataxia, dystonic head movement disorder and distal muscular atrophy. Patient L:II:1, bearing a p.Arg1098Cys mutation, displays a cerebellar ataxia with severe intellectual disability and seizures. Interestingly, serial brain MRI at age 4 and 9 showed a hypoplastic cerebellum, progressive cerebellar cortical atrophy and a FLAIR-hyperintense superior cerebellar cortex. Lastly, clinical examination of patient N:II:1, bearing a de novo p.Gln2205Pro mutation, is in line with spastic cerebellar ataxic gait, epilepsy, mild intellectual disability and scoliosis. Brain MRI revealed hypoplasia of the vermis and cerebellar hemispheres together with parietal polymicrogyria.
Structural modelling of SPTAN1 variants
The SPTAN1 gene encodes the sizeable α-II-spectrin protein, which forms the functional spectrin complex through heterotetramerisation with one of four different β-spectrins.
Although initially no structural damage was predicted by the Missense3D server, we pinpointed the p.Arg19Trp mutation to a crucial location in the tetramerization interface of the spectrin complex (Figure 4). The Arginine to Tryptophan amino acid substitution results in a loss of positive charge, which could in turn result in a partial loss of this electrostatic interaction at the tetramerization interface.
Figure 4: Protein modelling of respective SPTAN1 variants.
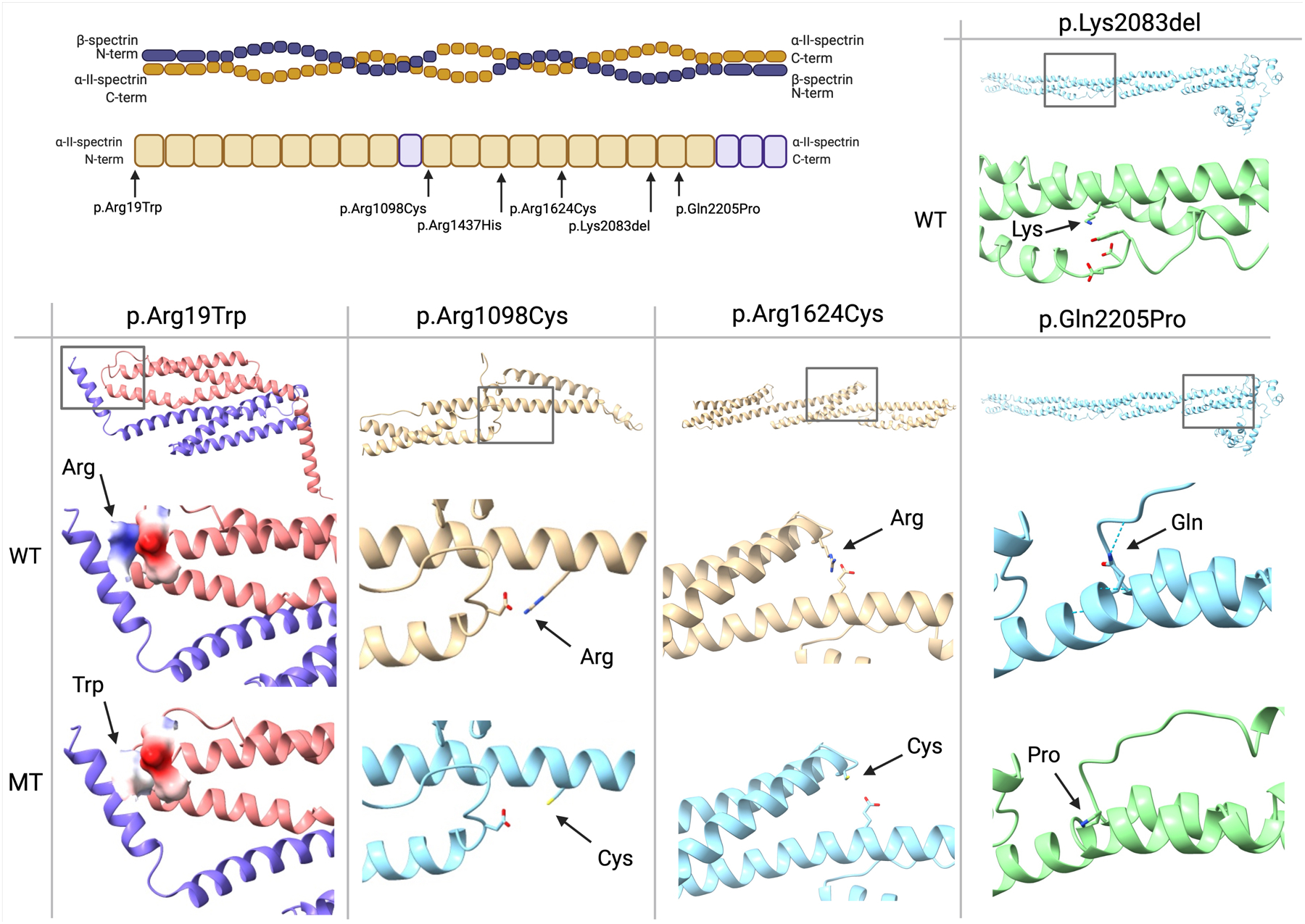
Top left panel: overview of the tetramer structure of the spectrin complex. Below, a graphical depiction of the α-II-spectrin protein structure with indication of the mutation locations. Top right panel: indication of the wild type Lys2083 residue, located across from three negatively charged residues. Bottom panel: comparison of the predicted wild type (WT) and mutant (MT) structures for every respective variant, with an overview of the location of the variant above. Positively charged residues are colored dark blue, negatively charged residues red. A hydrogen bond is indicated with a dotted line.
Recently and highly relevant to the current study, a mouse model with a spontaneous p.Arg1098Gln mutation in the Sptan1 gene was shown to develop progressive ataxia with tremors and seizures, with weakened binding of calmodulin as the proposed pathomechanism.29 Through protein homology modelling we similarly identified the Arg1098 residue to be located at a position where electrostatic interactions across the spectrin repeat are highly likely. We hypothesize that the loss of a positive charge due to the p.Arg1098Cys mutation results in the loss of stabilizing interhelical electrostatic interactions in the 10th spectrin repeat.
We identified the p.Arg1624Cys and p.Gln2205Pro substitutions to be located at similar locations in different spectrin repeats. We identify the p.Arg1624Cys mutation to be located in the 14th spectrin repeat with a possible electrostatic interaction with the Glu1549 residue. We analogously hypothesize a pathomechanism where the loss of positive charge results in destabilization of the interlinked α-helices. Although for the p.Gln2205Pro mutation there is no loss of charge resulting from the missense mutation, there is a predicted loss of hydrogen bond interlinking the Gln2205 and Cys2120 residues at the start of the 20th spectrin repeat. This bond is predicted to be lost due to the mutation (Figure 4). Furthermore, the introduction of a buried Proline is likely to destabilize the protein domain. To add to these observations, the recurrent p.Lys2083del mutation is predicted to be located across from two Aspartic acid residues (protein positions 2002 and 2003). Taken together, these observations form a common pattern, suggesting destabilization of spectrin repeats through disturbances in either electrostatic interactions or hydrogen bonds.
Discussion
The current study investigated the impact of SPTAN1 mutations in cerebellar ataxia and HSP as well as other rare neurological disorders (see Methods). Given the association of ataxia with SPTBN2, earlier reports of SPTAN1 related epilepsy syndromes showing cerebellar structural abnormalities on brain MRI and the known widespread and diverse functions of α-II-spectrin in neuronal cells, we postulated a role for SPTAN1 mutations in rare neurological disorders, most notably HSP and cerebellar ataxia.
We report five novel SPTAN1 mutations through a screen of 10,000 NGS datasets. In four HSP families, a recurrent p.Arg19Trp mutation segregates as a dominantly inherited trait, while in one additional patient this mutation occurs de novo. This finding is in contrast with earlier reported putatively recessive SPTAN1 variants causing HSP. In the first paper, segregation data presented to support his recessive inheritance pattern was limited to one trio only; recently, a second report was made of a homozygous missense variant in one family. None of these variants are reported to be recurrent.8,9 In our cohort of 10,000 NGS datasets, we were not able to identify any segregating SPTAN1 variants that fit a recessive inheritance pattern. We secondly identified a recurrent p.Lys2083del mutation in four cerebellar ataxia patients; two are de novo mutations, two occur in sporadic patients. Three other de novo missense mutations in SPTAN1 are associated with cerebellar ataxia with possible co-occurrence of epilepsy, intellectual disability and/or pyramidal signs (Table 1). Although both the p.Arg19Trp and p.Lys2083del mutations are recurrent in the very large datasets used for this study, the identification of these patients also shows the relative rarity of SPTAN1 mutations and the need to share NGS data for RD patients in large consortia.
Table 1:
Clinical characteristics of patients with SPTAN1 mutations
| SPTAN1 mutation | Patient | Inheritance | Etnicity | AAO (y) | Initial symptoms | ALE (y) | Cerebellar ataxia | Pyramidal signs | Muscle weakness | Sensory abnormalities | Intellectual disability | Epilepsy | Other features |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A:II:3 | Sporadic | Caucasian | 17 | Atactic gait | 42 | Yes: LL * | - | - | - | + | - | - | |
| B:II:1 | De novo | Caucasian | 3 | Atactic gait | 28 | Yes: LL * | +++ | - | + | + | - | Cervical dystonia, paroxysmal dyskinesia, myoclonus, hearing impairment | |
| C:II:1 | Sporadic | Caucasian | 6 | Atactic gait | 20 | Yes: LL * | - | - | - | + | - | - | |
| D:II:2 | De novo | Zaza | 25 | Atactic gait | 36 | Yes: LL * Dysarthria |
+ | - | - | - | - | - | |
| E:II:1 | Unknown | Caucasian | 3 | Gait difficulty | 15 | - | +++ * | - | - | - | - | Pes cavus | |
| F:II:1 | De novo | Caucasian | 7 | Gait difficulty | 23 | - | +++ * | LL: Distal | - | - | - | Optic neuropathy, myopia Left vestibular areflexia; Hypermobility |
|
| G:II:1 | AD | Caucasian | 6 | Gait difficulty | 28 | - | +++* | LL: Proximal | - | - | - | Pes cavus; optic neuropathy | |
| H:III:1 | AD | Caucasian | 7 | Gait difficulty | 40 | - | +++* | LL: Proximal | - | - | - | - | |
| 7 | Gait difficulty | 57 | - | +++ * | LL: Distal | + | - | - | Sensory-motor axonal neuropathy | ||||
| 8 | Gait difficulty | 15 | - | ++ * | LL: Distal | - | - | + | - | ||||
| I:II:3 | 6 | Gait difficulty | 10 | - | +++ * | - | - | - | - | - | |||
| J:II:1 | Probable AD | Caucasian | 8 | Gait difficulty | 22 | - | ++ * | LL: Distal | - | - | - | - | |
| 13 | Gait difficulty | 18 | Yes: LL | +++ * | LL: Distal | + | - | - | Sensory-motor demyelinating neuropathy; Pes cavus | ||||
| 0 | Gait difficulty | 40 | - | ++ * | LL: Distal | - | - | - | Pes cavus | ||||
| 0 | Gait difficulty | ? | - | ++ * | - | - | - | - | - | ||||
| 13 | Gait difficulty | 18 | Yes: LL | +++ * | LL: distal | + | - | - | Sensory-motor demyelinating neuropathy; Pes cavus | ||||
| 4 | Gait difficulty | 15 | - | +++ * | LL: proximal / distal | - | - | - | Sensory-motor axonal neuropathy; Pes cavus; febrile seizures; cardiac arrhythmia; patent foramen ovale | ||||
| 6 | Gait difficulty | ? | - | ++ * | - | - | - | - | - | ||||
| K:IV:8 | 6 | Gait difficulty | ? | - | ++ * | - | - | - | - | - | |||
| L:II:1 | De novo | Caucasian | 0 | Developmental delay | 15 | Yes: LL * | + | - | - | +++ | + | - | |
| M:II:1 | De novo | Caucasian | 0 | Clumsiness | 34 | Yes: LL * | - | LL: Distal | - | - | - | Dystonic head tremor | |
| N:II:1 | De novo | Serbian | 1 | Developmental delay | 26 | Yes: LL* | ++ | - | - | ++ | +++ | Scoliosis |
Abbrevations: AD = Autosomal Dominant; AAO = age at onset; ALE = age at last examination; LL = lower limbs; UL = upper limbs; −/+/++/+++ = absent / mild / moderate / severe. Predominance of clinical phenotype indicated by *
The large number of patients and families presented in our study allows us to define the contours of a very broad phenotypical spectrum linked to SPTAN1 mutations. There are two clearly delineated groups of patients with a respectively predominantly HSP phenotype linked with the p.Arg19Trp mutation and a cerebellar ataxia phenotype associated with the p.Lys2083del mutation. A third group consists of patients with more complex phenotypes, typically with cerebellar ataxia as the defining feature in the developmental delay. Two of these patients showed a more syndromic clinical phenotype with varying grades of intellectual disability and epilepsy. One of these patients (M:II:1) showed a superior cerebellar cortex hyperintense FLAIR signal, a relatively rare feature which was also reported in a patient with congenital non-progressive cerebellar ataxia due to a SPTBN2 mutation,17 hinting towards further phenotypical overlap between mutations in the different spectrin genes.
Strikingly, in a multigenerational family three out of 12 patients present with an HSP phenotype complicated by cerebellar ataxia and a sensory-motor axonal neuropathy with demyelinating features, while the other family members have a consistently pure HSP phenotype. This illustrates that the same SPTAN1 mutation, segregating within the same kinship, can result in a marked variability of the clinical expression. Generally, however, most p.Arg19Trp patients do still display a pure HSP phenotype.
The availability of a crystal structure of the tetramerization domain of SPTAN1 in addition to the well-established composition of the spectrin repeat allowed us to approach the range of mutations using in silico protein modelling. A common mechanistic theme arises when realizing that the amino acids identified in this study are located at crucial linking positions within the spectrin repeats. The p.Arg19Trp substitution creates a loss of positive charge and results in a destabilization of electrostatic interaction at the heterotetramerization site.23 Previous studies have postulated that the first helix of the α-II-spectrin protein binds with the two C-terminal helices of the β-spectrins, forming a functional spectrin repeat. This three-helix bundle was also dependent on a salt bridge formed by the positively charged Arg19 residue with an Aspartic acid residue of the β-spectrin helix.
Recently, a mouse model with a spontaneous p.Arg1098Gln mutation in the Sptan1 gene was shown to develop progressive ataxia with tremors and seizures, mimicking the phenotype seen in the patient with the p.Arg1098Cys variant in SPTAN1 and reminiscent of mice with βIII spectrin deficiency.29,30 In general, the SPTAN1 gene is moderately conserved for missense variants, with a 90% confidence interval for the observed/expected ratio of missense variants being 0.55–0.62. The Arg1098 residue has a PhyloP score of 7.1794; indicative of a conserved amino acid so both the glutamine and the cysteine substitutions of this same residue might result in similar effects. It was shown that the p.Arg1098Gln variant in the mouse Sptan1 caused a weakened binding of calmodulin, therefore enhancing the in vivo proteolysis by calpain. Through protein modelling we are analogously able to hypothesize that the loss of a positive charge induced by the p.Arg1098Cys substitution results in a loss of stabilizing electrostatic interactions.
The same pattern also applies to the p.Arg1624Cys, p.Lys2083del and p.Gln2205Pro substitutions, all targeting strategically positioned (charged) amino acids interlinking different helices in the spectrin repeats. Interestingly, interhelical interactions have been shown to be of importance in the stabilization of spectrin repeats before.26 Even more so, a mutation in dystrophin has been shown to destabilize its spectrin-like domain due to a change in electrostatic interactions, leading to Duchenne Muscular Dystrophy.31
Numerous SPTAN1 mutations have been reported previously, obscuring a thorough literature search.4,6,39–48 In order to provide at least a partial overview, we have recapitulated those with conclusive Mendelian inheritance patterns based on segregation and recurrency of mutations in Figure 2. The clinical spectrum ranges from early-onset severe epileptic syndromes, intellectual disability, HSP, spinocerebellar ataxia to late-onset mild HMN, with some patients displaying a combination of phenotypical traits. In addition, the first patient presenting with a sensory involvement was recently described.7 Although age of onset, disease severity and some phenotypical traits can differ, correlations can be made between SPTAN1 variants and the predominantly associated phenotype, such as the p.Lys2083del mutation in the cerebellar atactic phenotype and the p.Arg19Trp mutation in the HSP phenotype.
Although it remains challenging to draw clear-cut genotype-phenotype correlations for all SPTAN1 variants, several interesting trends can be delineated: C-terminal in-frame insertions, deletions and duplications are highly linked to severe, early-onset epileptic encephalopathies and West syndrome. These variants are located within the spectrin dimerization domain, and one has been shown to cause spectrin aggregates in patient-derived iPSCs.10 In contrast, nonsense mutations are seen more frequently in patients with mild HMN, where a haploinsuffiency mechanism is proposed due to nonsense-mediated decay.5 The mutations presented in the current study typically affect positively charged Arginine or Lysine residues, producing the hypothesis that the disease mechanism is driven by the disruption of stabilizing electrostatic interactions.
How SPTAN1 mutations affect different neuronal subtypes at different stages of life remains unanswered. In comparison to SPTAN1, mutations in the β-spectrins display a more limited phenotypical spectrum per gene to date. Since it is known that the composition of the spectrin complex in terms of the β-spectrin differs depending on the subcellular localization and specific neuron type, future work could uncover additional molecular evidence that results into divergent pathomechanisms between SPTAN1 variants.
In conclusion, we report recurrent de novo and dominantly inherited SPTAN1 mutations as a cause of both pure and complex HSP and cerebellar ataxia phenotypes in 22 patients from 14 families. The nature and position of the variants within the α-II-spectrin protein strongly suggests a common mechanistic theme, of spectrin repeats destabilized due to loss of electrostatic interaction or loss of hydrogen bond.
Supplementary Material
Acknowledgment
Several authors are member of the European Reference Network for Rare Neurological Diseases (ERN-RND, project N°739510) and of the European Reference Network for Rare Neuromuscular Diseases (ERN EURO-NMD, project N°870177). We thank all participating patients and their families for the cooperation in this study. We acknowledge the work of Marc Sturm who handled the bio-informatic processing of the NGS data generated at the IMGAG in Tübingen. JB and VT are part of the μNEURO Research Centre of Excellence of the University of Antwerp.This work was supported by the EJPRD PREPARE consortium, as well as the EJPRD PROSPAX consortium (EJP RD COFUND-EJP, project N°825575 to MS, RS, NB, FS, AD, and to SK and SZ as associated partners), the SPATAX network (AB, AD, RS, GS and NB), and the TreatHSP network (RS, MW, SK, ATB and TK).
Financial disclosure:
This work was supported by the EU Horizon 2020 program (Solve-RD under grant agreement N°779257), the Association Belge contre les Maladies Neuromusculaire (ABMM), and the University of Antwerp under Grant Agreement KP-BOF-2021, N°FFB210049. JB is supported by a Senior Clinical Researcher mandate of the Research Fund - Flanders (FWO) under grant agreement N°1805021N. LVdV is supported by a predoctoral fellowship of the FWO under grant agreement N°11F0921N, JDW is supported by the Goldwasser-Emsens fellowship. KL is funded by a research grant from the Damp foundation. MGP is funded by the Clinician Scientist School Luebeck (DFG-GEPRIS, project N°413535489). TBH was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, projects N°418081722 and N°433158657. This work was further supported by the Bundesministerium für Forschung und Bildung (BMBF) through funding for the TreatHSP network (N°01GM1905 to RS, SK), the National Institute of Health (NIH/NINDS) (grant N°5R01NS072248 to RS and SZ), the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, N°441409627), as part of the PROSPAX consortium under the frame of EJP RD, the European Joint Programme on Rare Diseases, under the EJP RD COFUND-EJP N°825575 (to MS, RS, NB, FS, AD, and to SK and SZ as associated partner), and the HSP Selbsthilfegruppe e.V. (grant to RS). TWR was supported by the Clinician Scientist program (Grant N°386-0-0), University of Tübingen, Medical Faculty.
Financial Disclosures of all authors
MS received consultancy honoraria from Janssen Pharmaceuticals, Ionis Pharmaceuticals and Orphazyme Pharmaceuticals, all unrelated to this work. MGP is employed by the University Medical Center Schleswig-Holstein, Campus Lübeck. KL received honoraria from Springer Medical Publishers and grants from the German Research Foundation, Movement Disorders Society, Damp Foundation, Michael J Fox Foundation and is employed by the University of Lübeck, all unrelated to the study. VT received a grant from the University of Antwerp (grant n° 38694 VT is principal investigator), a concerted action project by the University of Antwerp (grant n° 41667) and a fund from the FWO (Fonds voor Wetenschappelijk Onderzoek) (grant n°G040821N), all unrelated to this study. GS received grants from ATAXIA-UK and from Biogen, all unrelated to the current study. SZ received grants from NINDS (grant n° 5R01NS105755–03 and 5R01NS072248–10), both unrelated to this study.
Footnotes
Conflict of interest: The authors report no conflict of interest.
References
- 1.Beijer D, Baets J. The expanding genetic landscape of hereditary motor neuropathies. Brain. 2020;143(12):3540–3563. doi: 10.1093/brain/awaa311 [DOI] [PubMed] [Google Scholar]
- 2.Timmerman V, Clowes VE, Reid E. Overlapping molecular pathological themes link Charcot-Marie-Tooth neuropathies and hereditary spastic paraplegias. Exp Neurol. 2013;246:14–25. doi: 10.1016/j.expneurol.2012.01.010 [DOI] [PubMed] [Google Scholar]
- 3.M S, R S. Overcoming the divide between ataxias and spastic paraplegias: Shared phenotypes, genes, and pathways. Mov Disord. 2017;32(3):332–345. doi: 10.1002/MDS.26944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Syrbe S, Harms FL, Parrini E, et al. Delineating SPTAN1 associated phenotypes: From isolated epilepsy to encephalopathy with progressive brain atrophy. Brain. 2017;140(9):2322–2336. doi: 10.1093/brain/awx195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beijer D, Deconinck T, de Bleecker JL, et al. Nonsense mutations in alpha-II spectrin in three families with juvenile onset hereditary motor neuropathy. Brain. 2019;142(9):2605–2616. doi: 10.1093/brain/awz216 [DOI] [PubMed] [Google Scholar]
- 6.Dong HL, Chen L, Wu ZY. A novel de novo SPTAN1 nonsense variant causes hereditary motor neuropathy in a Chinese family. Brain. 2021;144(1):e11–e11. doi: 10.1093/BRAIN/AWAA357 [DOI] [PubMed] [Google Scholar]
- 7.Ylikallio E, Ritari N, Sainio M, et al. De novo SPTAN1 mutation in axonal sensorimotor neuropathy and developmental disorder. Brain. 2020;143(12):6–8. doi: 10.1093/brain/awaa344 [DOI] [PubMed] [Google Scholar]
- 8.Leveille E, Estiar MA, Krohn L, et al. SPTAN1 variants as a potential cause for autosomal recessive hereditary spastic paraplegia. J Hum Genet. 2019;64(11):1145–1151. doi: 10.1038/s10038-019-0669-2 [DOI] [PubMed] [Google Scholar]
- 9.Xie F, Chen S, Liu P, Chen X, Luo W. SPTAN1 variants likely cause autosomal recessive complicated hereditary spastic paraplegia. J Hum Genet 2021. Published online September 16, 2021:1–4. doi: 10.1038/s10038-021-00975-1 [DOI] [PubMed] [Google Scholar]
- 10.Wang Y, Ji T, Nelson AD, et al. Critical roles of αII spectrin in brain development and epileptic encephalopathy. J Clin Invest. 2018;128(2):760–773. doi: 10.1172/JCI95743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang CYM, Zhang C, Ho TSY, et al. αII spectrin forms a periodic cytoskeleton at the axon initial segment and is required for nervous system function. J Neurosci. 2017;37(47):11311–11322. doi: 10.1523/JNEUROSCI.2112-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoshimura T, Stevens SR, Leterrier C, Stankewich MC, Rasband MN. Developmental changes in expression of βIV spectrin splice variants at axon initial segments and nodes of ranvier. Front Cell Neurosci. 2017;10:304. doi: 10.3389/fncel.2016.00304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leterrier C A dual role for βiI-spectrin in axons. Proc Natl Acad Sci U S A. 2019;116(31):15324–15326. doi: 10.1073/pnas.1909789116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stevens SR, Rasband MN. Ankyrins and neurological disease. Curr Opin Neurobiol. 2021;69:51–57. doi: 10.1016/j.conb.2021.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ikeda Y, Dick KA, Weatherspoon MR, et al. Spectrin mutations cause spinocerebellar ataxia type 5. Nat Genet. 2006;38(2):184–190. doi: 10.1038/ng1728 [DOI] [PubMed] [Google Scholar]
- 16.Nicita F, Nardella M, Bellacchio E, et al. Heterozygous missense variants of SPTBN2 are a frequent cause of congenital cerebellar ataxia. Clin Genet. 2019;96(2):169–175. doi: 10.1111/cge.13562 [DOI] [PubMed] [Google Scholar]
- 17.Sancho P, Andrés‐bordería A, Gorría‐redondo N, et al. Expanding the β‐iii spectrin‐associated phenotypes toward non‐progressive congenital ataxias with neurodegeneration. Int J Mol Sci. 2021;22(5):1–11. doi: 10.3390/ijms22052505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buelow M, Süßmuth D, Smith LD, et al. Novel bi-allelic variants expand the SPTBN4-related genetic and phenotypic spectrum. Eur J Hum Genet. Published online 2021. doi: 10.1038/s41431-021-00846-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Knierim E, Gill E, Seifert F, et al. A recessive mutation in beta-IV-spectrin (SPTBN4) associates with congenital myopathy, neuropathy, and central deafness. Hum Genet. 2017;136(7):903–910. doi: 10.1007/s00439-017-1814-7 [DOI] [PubMed] [Google Scholar]
- 20.Morrow JS, Stankewich MC. The Spread of Spectrin in Ataxia and Neurodegenerative Disease HHS Public Access. J Exp Neurol. 2021;2(3):131–139. [PMC free article] [PubMed] [Google Scholar]
- 21.Gonzalez M, Falk MJ, Gai X, Postrel R, Schüle R, Zuchner S. Innovative Genomic Collaboration Using the GENESIS (GEM.app) Platform. Hum Mutat. 2015;36(10):950–956. doi: 10.1002/humu.22836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–443. doi: 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mehboob S, Song Y, Witek M, et al. Crystal structure of the nonerythroid α-spectrin tetramerization site reveals differences between erythroid and nonerythroid spectrin tetramer formation. J Biol Chem. 2010;285(19):14572–14584. doi: 10.1074/jbc.M109.080028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10(6):845–858. doi: 10.1038/nprot.2015.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hill SA, Kwa LG, Shammas SL, Lee JC, Clarke J. Mechanism of assembly of the non-covalent spectrin tetramerization domain from intrinsically disordered partners. J Mol Biol. 2014;426(1):21–35. doi: 10.1016/j.jmb.2013.08.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brenner AK, Kieffer B, Travé G, Frøystein NA, Raae AJ. Thermal stability of chicken brain a-spectrin repeat 17: A spectroscopic study. J Biomol NMR. 2012;53(2):71–83. doi: 10.1007/s10858-012-9620-y [DOI] [PubMed] [Google Scholar]
- 27.Ittisoponpisan S, Islam SA, Khanna T, Alhuzimi E, David A, Sternberg MJE. Can Predicted Protein 3D Structures Provide Reliable Insights into whether Missense Variants Are Disease Associated? J Mol Biol. 2019;431(11):2197–2212. doi: 10.1016/j.jmb.2019.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pettersen EF, Goddard TD, Huang CC, et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021;30(1):70–82. doi: 10.1002/pro.3943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miazek A, Zalas M, Skrzymowska J, et al. Age-dependent ataxia and neurodegeneration caused by an αII spectrin mutation with impaired regulation of its calpain sensitivity. Sci Rep. 2021;11(1):1–18. doi: 10.1038/s41598-021-86470-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stankewich MC, Gwynn B, Ardito T, et al. Targeted deletion of betaIII spectrin impairs synaptogenesis and generates ataxic and seizure phenotypes. Proc Natl Acad Sci U S A. 2010;107(13):6022–6027. doi: 10.1073/PNAS.1001522107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Legardinier S, Legrand B, Raguénès-Nicol C, et al. A two-amino acid mutation encountered in Duchenne muscular dystrophy decreases stability of the rod domain 23 (R23) spectrin-like repeat of dystrophin. J Biol Chem. 2009;284(13):8822–8832. doi: 10.1074/jbc.M805846200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gilissen C, Hehir-Kwa JY, Thung DT, et al. Genome sequencing identifies major causes of severe intellectual disability. Nature. 2014;511(7509):344–347. doi: 10.1038/nature13394 [DOI] [PubMed] [Google Scholar]
- 33.Rapaccini V, Esposito S, Strinati F, et al. A child with a c.6923_6928dup (p.Arg2308_Met2309dup) SPTAN1 mutation associated with a severe early infantile epileptic encephalopathy. Int J Mol Sci. 2018;19(7). doi: 10.3390/ijms19071976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stavropoulos DJ, Merico D, Jobling R, et al. Whole-genome sequencing expands diagnostic utility and improves clinical management in paediatric medicine. npj Genomic Med. 2016;1. doi: 10.1038/npjgenmed.2015.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gartner V, Markello TC, Macnamara E, et al. Novel variants in SPTAN1 without epilepsy: An expansion of the phenotype. Am J Med Genet Part A. 2018;176(12):2768–2776. doi: 10.1002/ajmg.a.40628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hamdan FF, Saitsu H, Nishiyama K, et al. Identification of a novel in-frame de novo mutation in SPTAN1 in intellectual disability and pontocerebellar atrophy. Eur J Hum Genet. 2012;20(7):796–800. doi: 10.1038/ejhg.2011.271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yavarna T, Al-Dewik N, Al-Mureikhi M, et al. High diagnostic yield of clinical exome sequencing in Middle Eastern patients with Mendelian disorders. Hum Genet. 2015;134(9):967–980. doi: 10.1007/s00439-015-1575-0 [DOI] [PubMed] [Google Scholar]
- 38.Retterer K, Juusola J, Cho MT, et al. Clinical application of whole-exome sequencing across clinical indications. Genet Med. 2016;18(7):696–704. doi: 10.1038/gim.2015.148 [DOI] [PubMed] [Google Scholar]
- 39.Saitsu H, Tohyama J, Kumada T, et al. Dominant-Negative Mutations in α-II Spectrin Cause West Syndrome with Severe Cerebral Hypomyelination, Spastic Quadriplegia, and Developmental Delay. Am J Hum Genet. 2010;86(6):881–891. doi: 10.1016/j.ajhg.2010.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Writzl K, Primec ZR, Stražišar BG, et al. Early onset West syndrome with severe hypomyelination and coloboma-like optic discs in a girl with SPTAN1 mutation. Epilepsia. 2012;53(6). doi: 10.1111/j.1528-1167.2012.03437.x [DOI] [PubMed] [Google Scholar]
- 41.Nonoda Y, Saito Y, Nagai S, et al. Progressive diffuse brain atrophy in West syndrome with marked hypomyelination due to SPTAN1 gene mutation. Brain Dev. 2013;35(3):280–283. doi: 10.1016/j.braindev.2012.05.002 [DOI] [PubMed] [Google Scholar]
- 42.Tohyama J, Nakashima M, Nabatame S, et al. SPTAN1 encephalopathy: Distinct phenotypes and genotypes. J Hum Genet. 2015;60(4):167–173. doi: 10.1038/jhg.2015.5 [DOI] [PubMed] [Google Scholar]
- 43.Ream MA, Mikati MA. Clinical utility of genetic testing in pediatric drug-resistant epilepsy: A pilot study. Epilepsy Behav. 2014;37:241–248. doi: 10.1016/j.yebeh.2014.06.018 [DOI] [PubMed] [Google Scholar]
- 44.Marco Hernández AV, Caro A, Montoya Filardi A, et al. Extending the clinical phenotype of SPTAN1: From DEE5 to migraine, epilepsy, and subependymal heterotopias without intellectual disability. Am J Med Genet Part A. Published online 2021. doi: 10.1002/AJMG.A.62507 [DOI] [PubMed] [Google Scholar]
- 45.Terrone G, Pinelli M, Bernardo P, et al. Intrafamilial variability in SPTAN1-related disorder: From benign convulsions with mild gastroenteritis to developmental encephalopathy. Eur J Paediatr Neurol. 2020;28:237–239. doi: 10.1016/J.EJPN.2020.07.008 [DOI] [PubMed] [Google Scholar]
- 46.Iossifov I, O’Roak BJ, Sanders SJ, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515(7526):216. doi: 10.1038/NATURE13908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun M, Johnson AK, Nelakuditi V, et al. Exome sequencing and targeted analysis identifies the genetic basis of disease in over 50% of patients with a wide range of ataxia-related phenotypes. Genet Med. 2019;21(1):195. doi: 10.1038/S41436-018-0007-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cappi C, Oliphant ME, Péter Z, et al. De novo damaging DNA coding mutations are associated with obsessive-compulsive disorder and overlap with Tourette’s disorder and autism. doi: 10.1016/j.biopsych.2019.09.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw NGS data cannot be made publicly available for confidentiality policies in the consortia involved. The NGS data in Solve-RD is available through the RD-Connect GPAP after an embargo period of 6 months. All other data generated during this study is available upon reasonable request.