

Abstract
Analytical ultracentrifugation (AUC) is a critical analytical tool supporting the development and manufacture of protein therapeutics. AUC is routinely used as an assay orthogonal to size exclusion chromatography for aggregate quantitation. This article distills the experimental and analysis procedures used by the authors for sedimentation velocity AUC into a series of best-practices considerations. The goal of this distillation is to help harmonize aggregate quantitation approaches across the biopharmaceutical industry. We review key considerations for sample and instrument suitability, experimental design, and data analysis best practices and conversely, highlight potential pitfalls to accurate aggregate analysis. Our goal is to provide experienced users benchmarks against which they can standardize their analyses and to provide guidance for new AUC analysts that will aid them to become proficient in this fundamental technique.
Keywords: Analytical Ultracentrifugation, Antibody(s), Antibody drug(s), Biopharmaceutical characterization, HPLC (high performance/pressure liquid chromatography, Monoclonal antibody(s), Protein aggregation
Introduction
Protein therapeutics have rapidly progressed from an emerging to an established approach for the treatment of a wide range of human diseases. From development through administration, protein therapeutics can form higher molecular weight assemblies that are commonly referred to as ‘aggregates’. Aggregate is a broad term applied to a variety of non-native higher molecular weight assemblies. In this guidance, aggregate is defined as soluble, thermodynamically and/or kinetically stable species with a molecular weight at least twice that of the therapeutically active species, the latter typically being a monomeric IgG monoclonal antibody (mAb1). Insoluble species larger than ~60 nm, the lower limit of detection for most submicron particle characterization techniques, are considered ‘particulate matter’ and are out of scope of this guidance; quantitation and characterization of particulates is discussed elsewhere [1–3].
Minimizing the formation of soluble aggregate is key to the development of a protein therapeutic, as aggregate can reduce drug efficacy and is suspected to elicit an immune response [4]. Minimizing the risk of aggregate in protein therapeutics requires aggregate characterization by orthogonal techniques (United States Pharmacopeia (USP) chapter <1787>). A whitepaper, contributed by the AAPS Product Attribute and Biological Consequences (PABC) focus group on the application of analytical techniques to assess subvisible particles of biotherapeutics, outlines existing orthogonal techniques for aggregate and particulate matter characterization [5]. Given the substantial increase in the number of approved therapeutic proteins, with a significant portion represented by mAbs, the biopharmaceutical industry needs orthogonal techniques and best practices to guide aggregate and particle analyses [6, 7].
Several techniques are commonly used to characterize and quantify soluble protein aggregate. Size exclusion chromatography (SEC) is the primary technique employed for this purpose in development and quality control (QC) laboratories. In SEC, species are separated by their hydrodynamic size via differential exclusion from the pores of a stationary phase matrix [8]. The advantages of SEC include high throughput, sensitivity, precision, and relatively low material requirements. Limitations of SEC are that separation occurs on two phases (mobile and stationary) which requires that a sample be diluted into the mobile phase prior to loading on the SEC column. Differences in the composition of a mobile phase from that of a formulation buffer (e.g., pH, ionic strength, presence of organic solvents), the substantial dilution by the mobile phase during separation, and non-specific interactions with the stationary phase can dissociate existing aggregates or promote formation of new ones. In addition, the separation range of a typical SEC column may be insufficient to resolve larger aggregates. While there are a variety of column types available, there is frequently a balance between resolution and separation range that must be achieved. These limitations can yield inaccurate quantitation and characterization of aggregates [9, 10].
To ensure the reliability of an SEC method, various orthogonal sizing techniques are employed, including field flow fractionation (FFF), light scattering techniques, and analytical ultracentrifugation (AUC) [11]. The accuracy, precision and dynamic range of these techniques have been reviewed elsewhere, and AUC has been shown to have the broadest dynamic range with equivalent or superior quantitative ability [12, 13]. For these reasons, AUC is widely utilized during product development as an aggregate characterization and quantitation technique orthogonal to SEC. Additionally, AUC is an extensively used orthogonal technique in both industry and academia for the characterization and quantitation of protein higher order structure, assembly mechanism, and aggregates [14–17]. AUC studies can be conducted in two ways: In sedimentation equilibrium AUC (SE-AUC), the equilibrium concentration distribution of macromolecules is quantitated; in sedimentation velocity AUC (SV-AUC), the sedimentation of macromolecules is monitored in real time. Although not discussed in this guidance, SE-AUC can provide thermodynamic information on parameters such as solution molecular mass, association constants, stoichiometries, and solution nonideality of homogenous and heterogeneous preparations of molecules.
SV-AUC is the orthogonal technique of choice for aggregate characterization and quantitation for biotherapeutics. The advantages of SV-AUC are that it is a first-principles solution technique that can be carried out across a variety of solution compositions and conditions such as the matrix of a final drug substance or drug product and without confounding interactions with a stationary phase. The limitations of SV-AUC include relatively low throughput, a limited dynamic detection range, the potential for some excipients such as sugars to form gradients during sedimentation, and the need for experienced analysts to conduct and properly interpret SV-AUC studies. The goal of this article is to address the latter limitation by providing guidance on the design, execution, and analysis of aggregates in protein therapeutic preparations with a focus on monoclonal antibodies (mAb).
SV-AUC differentiates soluble species based on their hydrodynamic properties, which is essential for characterization of the kinetic and thermodynamic properties of many types of interacting systems. SV-AUC tracks the boundary between sample and solvent as the macromolecules in a solution sediment. The sedimentation coefficient(s) is a measure derived from the rate at which boundaries move down the sample column using transport models based upon the Lamm equation [18]. The buoyant mass and the frictional coefficient are correlated parameters that are derived from the relationship between shape of boundaries and their sedimentation rate. In a heterogeneous solution of different non-interacting particles, each species (or ensemble of species with similar hydrodynamic properties) has a unique and separate boundary. The accurate identification and quantitation of each species requires careful attention to the experimental conditions that influence the quality and extent of the boundaries which in turn, allows the hydrodynamic properties of each species to be extracted by analysis [19].
This article presents the authors’ recommendation of best practices for accurate aggregate quantitation by SV-AUC in samples of therapeutic antibodies or antibody-derived products. We focus on the instrumentation and analysis method that is widely used in industry for the analysis of aggregates in mAb formulations. We outline best practices that reflect the published literature and the authors’ experience. Among the key best practices articulated herein are proper instrument set up, SV run protocols, reproducible cell alignment, and rigorous data analysis [20, 21]. By sharing these best practices, we hope to facilitate the entry and training of new users in the field, improve the consistency of aggregate quantitation across the industry, and enhance the breadth and robustness of SV-AUC applications that characterize protein therapeutics.
Analytical ultracentrifuges
The majority of analytical ultracentrifuges used in the biopharmaceutical industry are manufactured and serviced by Beckman Coulter (Indianapolis, IN). The Rayleigh interferometer and a UV-Vis absorbance detector in ProteomeLab™ XL-I instruments can separately or simultaneously measure radial concentration as species sediment. The ProteomeLab™ XL-A instrument is only equipped with the absorbance scanner. The new Optima™ instrument from Beckman Coulter can also be equipped with both detection systems; see [17] for additional information about this instrument. Users of the Optima™ analytical ultracentrifuge are cautioned to monitor their data closely for signs of micro-convection as described in [22]. All Beckman analytical instruments support four- and eight-hole rotors. The best practices described herein are generally applicable across all detection systems, albeit with some specific considerations for each. The fluorescence detection system formerly manufactured by Aviv Biomedical is not used in aggregate analysis and so is not considered herein. Discussion of the applications enabled by the fluorescence optics can be found elsewhere [23–25].
Sample preparation
A successful AUC experiment begins with proper sample preparation. The optimal sample preparation depends on application and the mode of sample detection. This section outlines our suggested best practice for the preparation of mAb samples for SV-AUC aggregate analysis employing intensity or absorbance detection at 280 nm. We also briefly discuss sample preparation for other types of SV-AUC studies.
A prerequisite for accurate SV-AUC results is the calculation or measurement of the solvent density and viscosity as well as the protein extinction coefficient and partial specific volume. Use of exact values is especially important when the solvent contains uncommon excipients or when the protein is post-translationally modified; however, these parameters affect the accurate determination of sedimentation coefficients much more than the quantitation of total aggregates. SV-AUC aggregate analysis studies can often be conducted by simple dilution of a protein therapeutic into its formulation buffer [16]. However, there are special cases when sample preparation requires additional considerations. The three most common situations are:
The sample solution contains species whose absorption interferes with the intensity or absorbance tracking of the analyte (typically, 280 nm for mAbs and other proteins). In this case, sample dialysis in a buffer void of the absorbing formulation buffer excipient is required. Note that most pharmaceutically relevant formulation matrices have negligible 280 nm absorbance compared to mAbs. Alternatively, interference detection (discussed below) can be used in place of absorbance.
Thermodynamic non-ideality is expected when mAbs are formulated at low ionic strength or as self-buffered formulations [26] (e.g., Humira®). Non-ideality can be diminished in SV-AUC studies by preparing samples with an appropriate ionic strength modifier such as sodium chloride and/or a buffer; the latter is particularly important for self-buffered formulations upon dilution. A total ionic strength of ~ 25 mM is usually sufficient to neutralize long-range molecular interactions that can interfere with the sedimentation and diffusion processes during an SV-AUC run.
Co-sedimenting solutes such as sugars or polyols can form density gradients during sedimentation and potentially ‘mask’ high molecular weight species leading to underreporting of the amount of aggregate [27]. To evaluate the impact of the co-sedimenting solvent, best practice is to dialyze a sample into an appropriate buffer (e.g., the same formulation matrix without a co-sedimenting solute or a standard buffer such as PBS) and compare the amount of high molecular weight species resolved by SV-AUC for the sugar-containing and the sugar-free buffer. When SV-AUC experiments are carried out in the presence of a co-sedimenting solute, an inhomogeneous solvent model [28] must be utilized during data analysis to obtain accurate sedimentation coefficient values.
For absorbance-based detection modes, a common best practice is to target the sample concentration corresponding to an absorbance of ~ 1.0, which ensures high signal-to-noise ratio while staying within the linear range of the absorbance detector. For most mAbs with an extinction coefficient at 280 nm of ~ 1.5 (mg/mL)−1cm−1 and in 12 mm pathlength centerpieces, 1.0 OD corresponds to a loading concentration of ~ 0.6 mg/mL. SV-AUC measurements can be conducted at higher concentrations using 3 mm pathlength or thinner centerpieces. In this case, proportionally higher loading concentration (~ 2.4 mg/mL for mAbs) can be used. Much higher concentrations (up to 45 mg/mL), which are not possible to measure with absorbance detection, have been achieved with interference measurements and require analysis models accounting for nonideality [29]. Loading concentrations corresponding to absorbance of 0.5 OD or lower should be avoided for the purpose of mAb aggregate characterization and quantitation; analyzing samples at low total loading concentrations impairs the ability to detect minor species (see discussion below on the limit of detection (LOD) and the limit of quantitation (LOQ)).
Since most mAb samples are formulated at high concentration, they must be diluted or dialyzed for SV-AUC analysis with absorbance detection. Dialysis may not be necessary when absorbance detection is employed, and the dilution factor is high. For instance, dilution of a mAb formulated at nominal 150 mg/mL to 0.6 mg/mL is a 250-fold dilution and ensures practically equal sample and reference matrix composition. On the other hand, Rayleigh interference detection is based on the differential refractive index and therefore is sensitive to even the slightest differences in matrix constituents between the two sectors of the centerpiece. Extensive dialysis of the sample is strongly recommended when Rayleigh interference is employed so that all matrix constituents match between the two sectors of the centerpiece.
When dialysis is not possible, SV-AUC can still be performed but computational methods which account for sedimenting buffer species must be taken into consideration during data analysis (see below) [29, 30]. For example, when process intermediate samples are analyzed by SV-AUC, the exact composition of their buffer matrix may not be well-determined. In these situations, either a dialysis against a representative matrix is carried out, or water is used as the optical reference in combination with additional computational analysis steps to account for sedimentation of matrix constituents. Special attention must be paid when a dialysis membrane or a concentrator membrane is not permeable to an excipient (e.g., surfactants forming micelles).
To account for potential variability of the aggregate quantitation by SV-AUC, it is recommended to prepare sufficient sample volume to allow triplicate measurement of a sample. Typically, triplicates are collected in three different cells within the same run; alternatively, multiple runs can be performed.
Finally, it is important to understand the stability of the molecule at the experimental conditions (solution condition, temperature, and duration). While the majority of mAbs are stable in their respective formulation matrices, some solution and/or incubation conditions such as those used in stress studies [31, 32], can induce additional mAb fragmentation and/or aggregation. In this case, rational choice of relevant stress conditions is essential. To make a meaningful comparison between several orthogonal techniques and minimize sample storage and handling-related artifacts, care should be exercised to ensure that the samples to be analyzed undergo the same sample treatment, preparation, and storage.
Cell alignment
After assembled cells are loaded into the AUC rotor, the walls of the sample sectors should be parallel to the centrifugal force. This geometry is achieved by aligning the sector walls to the center of rotation through a process called cell alignment. Alignment is crucial for ensuring accurate quantitation of aggregate levels in mAb samples. Cell misalignment has been linked to convective flow which ultimately leads to artifacts materialized in a higher apparent aggregate content [21, 33–35]. An illustrative example in Figure 1 (taken from [35]) shows how purposefully and systematically misaligning cells in the rotor impacts aggregate quantitation. The graphs in Figure 1B and Figure 2 clearly show the impact of misalignment on SV-AUC aggregate quantitation.
Figure 1.
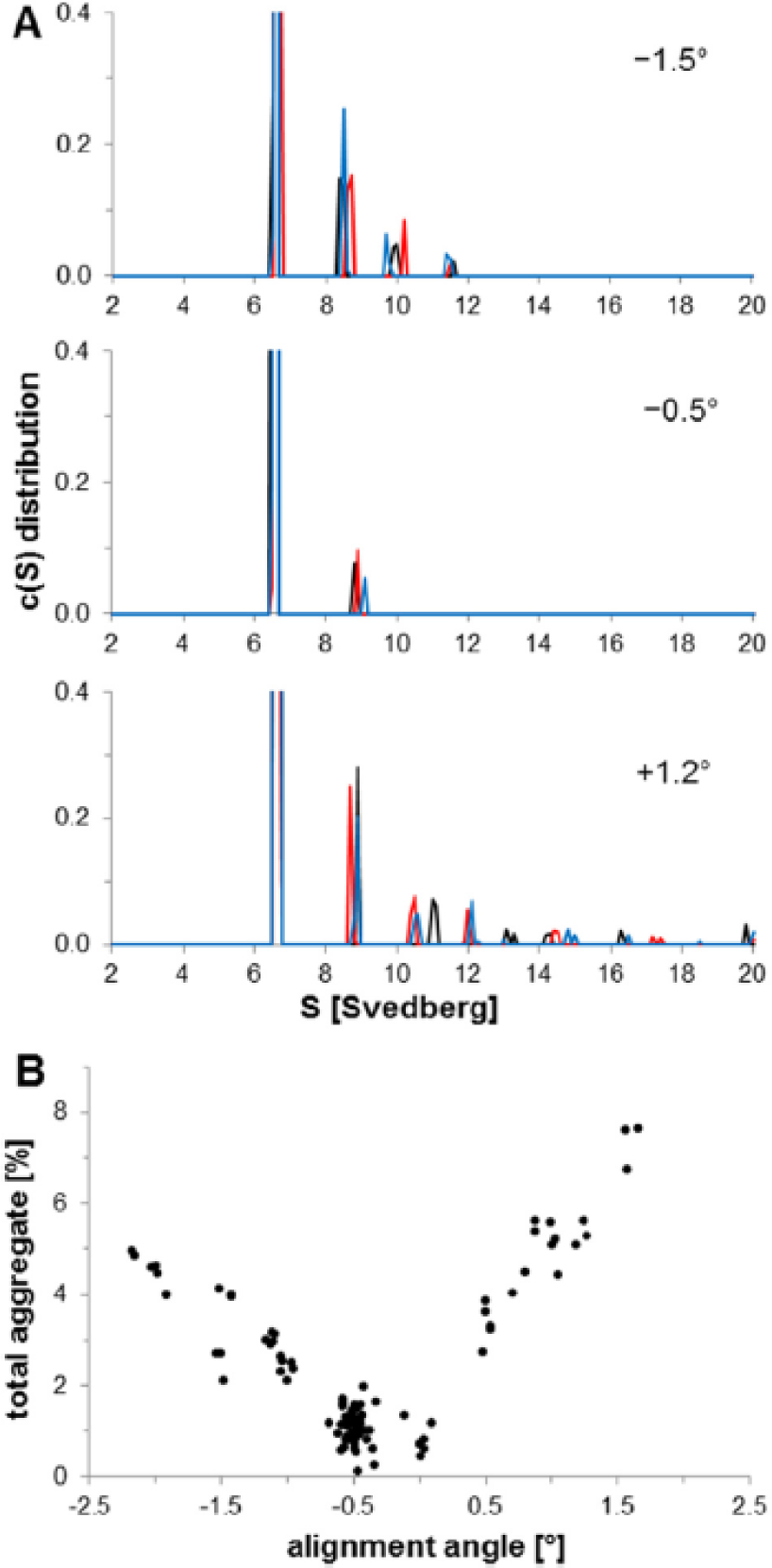
(taken from [33]) An IgG4 mAb was diluted to approximately 0.6 mg/mL and loaded into two-sector Epon-filled centerpieces equipped with sapphire windows. Cells were aligned using the OA system, and the rotor was placed into the centrifuge to equilibrate under vacuum for at least two hours at 20 °C. The mAb was sedimented using Beckman XLI analytical ultracentrifuges and An-60 Ti rotors at 60,000 rpm. A total of 50 scans per cell were collected over a period of approximately 3.5 h, sufficient to reach complete sedimentation. Sedimentation profiles were fit using the c(s) model in SEDFIT. (A) Representative IgG4 mAb c(s) distributions obtained at different alignment angles. Line colors (black, red, blue) represent c(s) distributions from three individual cells. (B) Dependence of the total measured aggregate on cell alignment. Each data point represents a result from an individual cell.
Figure 2.
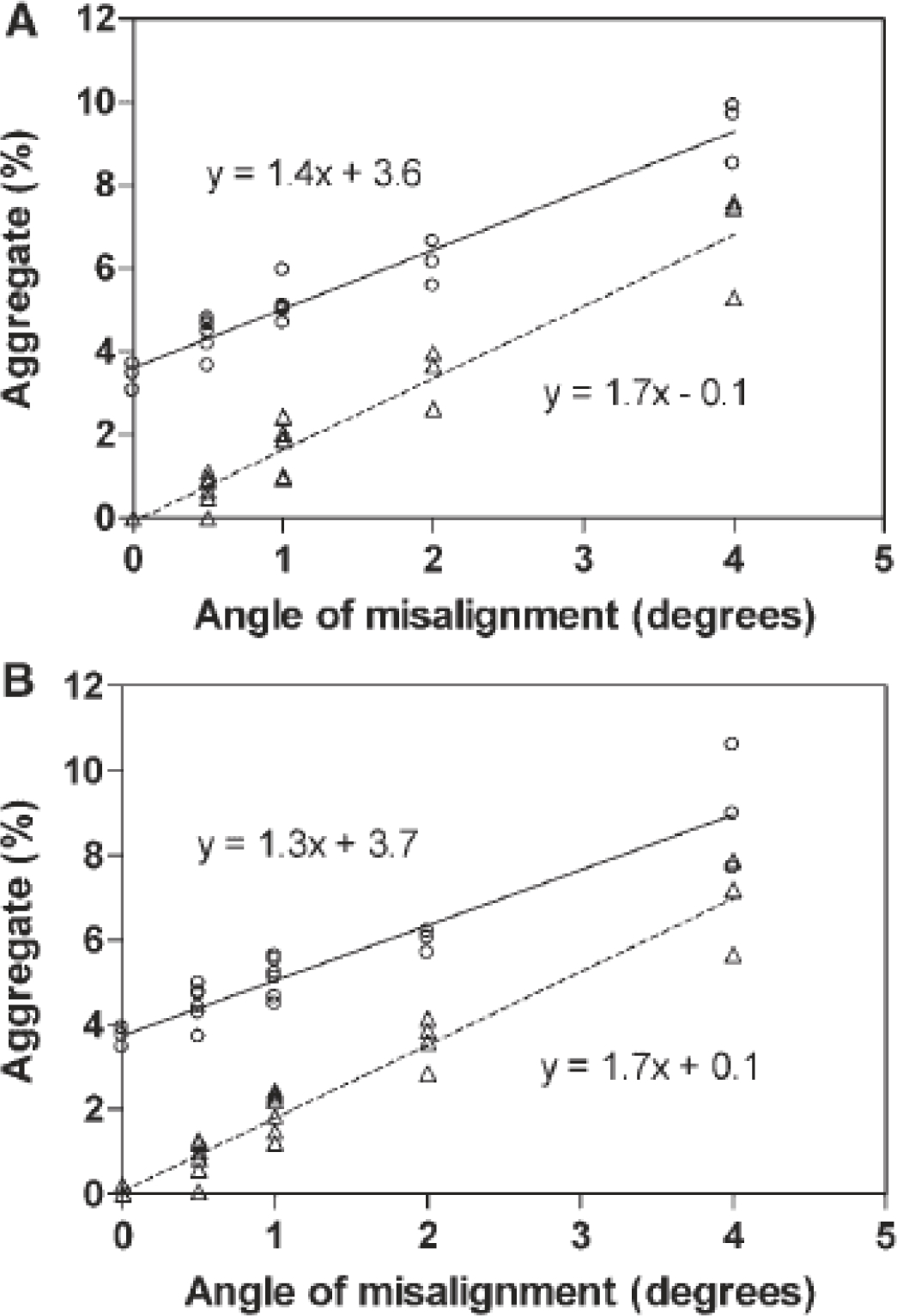
(taken from [31]) Change in detected levels of dimer and HMWS as a function of cell misalignment from the center of rotation. Analyzed (A) without TI noise decomposition and (B) with TI noise decomposition. In both panels open circles denote dimer percentage (n=3 or 6) and open triangles denote HMWS percentage (n=3 or 6). A linear fit of the relationship between angle of misalignment and dimer (solid line) and HMWS (dashed line) percentage was used to estimate the effect of misalignment on aggregate level quantitation.
Cells can be aligned visually, mechanically, or optically. Visual alignment relies on matching the scribe marks on the bottom of the rotor with those on the bottom of cell housings. Visual alignment is subjective and requires that the scribe lines be drawn with utmost accuracy during the manufacture of rotors and cell housings. Mechanical alignment is achieved with commercially available tools such as the Spin CAT manufactured by Spin Analytical, or custom-made tools described in [33]. Mechanical alignment offers higher precision in aggregate quantitation compared to visual alignment [21, 33–35]. Unlike visual alignment which depends on the scribe marks, mechanical alignment relies on the cut outs on the bottom of the cell housing to effectively align the sector walls with the center of rotation. Optical tools aim to achieve alignment by ensuring that the septum which separates both sectors of the centerpiece is parallel to the centrifugal force and that the lines drawn from the walls of the septum (or the inner walls of each sector) intersect at the center of rotation [35].
While the intermediate precision reported for optical alignment [35] and mechanical alignment [21] are similar, the former allows direct sector wall alignment without relying on the scribe marks or cut outs, thus potentially improving alignment consistency among different rotors. Note that optical alignment may by confounded by small air bubbles on sector walls which obscure light passage during measurement of alignment angles. A brief centrifugation at a low speed (e.g., 5 min at 3,000 rpm) forces small air bubbles to coalesce into a single bubble at the top of the sector and is usually sufficient to resolve this problem. As best practice, optical alignment should be used whenever possible given its higher accuracy and lowest impact on aggregate quantitation. If an optical alignment instrument is not available, use of a mechanical alignment tool is preferred over manual alignment.
Experimental temperature and temperature control
Temperature affects virtually every aspect of an AUC experiment. Sample density, viscosity, stability, and oligomerization state are all temperature dependent. Thus, rigorous analysis requires that the temperature be accurately known. In addition, temperature gradients cause convection that introduces aggregate quantitation artifacts [21, 33, 36]. Typically, the temperature must be maintained within ± 0.5 °C of the set run temperature over the course of an SV-AUC experiment. Some analysis software programs display a warning message if this tolerance is exceeded (e.g., SEDFIT, as discussed below).
AUC experiments are typically conducted at 20 °C. Sedimenting samples at 20 °C facilitates correction to the (20,w) standard condition because in this case only buffer density and viscosity need to be accounted for. Being within several degrees of ambient laboratory temperatures minimizes the time required to equilibrate the loaded rotor prior to starting a run. It is recommended to pre-equilibrate samples at 20 °C in case there is slow reversibility between species present in a sample. Insufficient temperature equilibration time in this case will cause concurrent species re-distribution during a run and introduce artifact. Best practice is to mount the loaded rotor and monochromator assembly and allow 1 – 2 hours at high vacuum to equilibrate to the 20 °C set point. Alternatively, the centrifuge with an empty rotor installed can be allowed to temperature equilibrate under vacuum overnight and run ~ 1 hour after the loaded rotor is mounted. If mAb SV-AUC analyses are conducted at temperatures approaching the high and low extremes accessible to a Beckman ProteomeLab™ XL-I or XL-A (4 and 37 °C, respectively), equilibration times will need to be longer than those noted above for 20 °C.
System suitability
According to ICH Q2(R1) ‘Validation of Analytical Procedures’, system suitability testing is an integral part of many analytical procedures and is based on the concept that the equipment, electronics, analytical operations, and samples to be analyzed constitute an integral system that should be evaluated as such [37]. Two types of system suitability checks need to be implemented for reliable and reproducible SV-AUC experiments, one for the instrument itself and another for the cell assemblies. Maintaining control charts for the performance parameters of the instrument and every cell in use is recommended for monitoring the overall performance of a given AUC system.
An instrument suitability check should include at a minimum, a radial calibration, a temperature control check, a test of detection module(s) functionality, and an evaluation of the instrument timestamp [38]. Except for radial calibration, it is recommended to have an analytical ultracentrifuge calibrated at least annually by a qualified service technician, to check for these parameters. Radial calibration is typically performed by the user whenever a rotor is changed. A recent multi-laboratory study [39] revealed systematic instrument variability, which makes instrument suitability checks a critical activity. The frequency of these checks (biweekly, monthly, or quarterly) can vary depending upon circumstances. New methods have been developed to calibrate rotor temperature by placing an integrated circuit for temperature logging either on top of a resting rotor [40], in a modified cell assembly [40] or in a modified rotor handle [41]. Absolute calibration of temperature and radial measurements in the AUC using external standards have been developed [39, 42, 43]. Periodic temperature checks can be valuable indicators of temperature control or calibration errors, but no evidence has been presented on whether or not these would influence the quantitation of aggregate species and so far have not been widely adopted in the biopharmaceutical industry.
Cell suitability refers to the ability of an assembled AUC cell to deliver reproducible results. Best practice is to keep the parts of cell assemblies together and to document their performance over time. Cells should be assembled following the manufacturer’s instructions, torqued, and checked for proper sample setup and assembly by performing a short run at low speed (e.g., 5 min at 3,000 rpm) prior to initiation of the actual run. It is convenient to do this check during the temperature equilibration; the rotor must be brought to a complete stop prior to starting the experimental run in order to reset the time and the rotation counter. Cell assemblies that are used for aggregate quantitation of mAb samples need to be routinely assessed for suitability by performing control run(s) with a protein standard. The standard may be a product-specific reference standard, representative material, or a commonly available protein (e.g., BSA or NIST mAb [44]). Typically, the mean and the standard deviation of the total level of aggregate and the s-value of the monomer can serve as control parameters for a given cell that is compared to historical and literature values, as well as to SEC results on the same protein. Since the s-value is temperature dependent, it is critical that control runs be properly equilibrated at the set temperature. Conversely, systematic deviation of the s-value from its known value can serve as an indirect suitability check for temperature.
Once a run with the actual mAb samples of interest is complete, one should critically review the data for any signs of atypical performance. While it is impossible to list all potential failure modes (Table 4 lists several common anomalies), a high standard deviation of a particular parameter may point to an outlier cell. Atypical data traces and/or large residuals may indicate instrument, cell, or sample problems.
Table 4.
Common failure modes of an SV-AUC run and their root causes.
| Error / failure | Manifestation | Potential root cause |
|---|---|---|
|
|
|
|
| Loading sample on the reference side and vice versa | Erroneous absorbance readings, potentially incorrect lamp intensity set | Analyst error |
|
|
|
|
| Cell leakage during the run | Vacuum decrease/jumps, moving meniscus, fitting artifacts, significantly unequal column heights after the run | Inappropriate cell sealing |
|
|
|
|
| Cell leakage at the start of a run at 60k/50k rpm | Moving meniscus, vacuum decrease, lack of expected data type during fitting | Inappropriate cell sealing |
|
|
|
|
| Contamination from another sample | Unexpected species in the c(s) distribution | Inappropriate cleaning of cell and centerpiece |
|
|
|
|
| Unstable temperature during the run | Best-fit meniscus is far off the optically discerned meniscus; poor fits in initial scans | Insufficient temperature equilibration time |
|
|
|
|
| High solute (reference) absorbance | Low signal-to-noise, poor fits with high RMSD | Absorbing excipients present |
Setting up an SV-AUC run
During an SV-AUC experiment, the analytes of a sample sediment in a centrifugal force field. The sedimentation behavior of a given species depends on its hydrodynamic properties that are in turn a function of the shape, size, and molecular weight of the molecule, as well as the temperature, density and viscosity of the buffer. As the SV-AUC run progresses, sedimentation boundaries broaden due to diffusion. Data scans are collected at multiple time points along the entire sample column; curve fitting globally analyzes the entire scan set, a process that increases precision and deconvolutes partially overlapping boundaries.
Information about the fastest sedimenting species is contained in the early subset of scans. Optimal information about the sedimentation of any species requires scans that follow these species to the bottom of the cell to maximally resolve differences in the velocity of migration. Thus, it is recommended to accelerate directly to the set speed and immediately initiate scanning. Analysis precision improves with increasing time-interval between the first and last scan included in the analysis (not including the scans from completely depleted solution columns) and the travel distance (i.e., a long solution column). Typically, statistical noise is reduced to a level below the ultimately unavoidable adventitious systematic errors of SV experiments at 50 – 100 scans (see below). Deconvolution of diffusion rates depend on both boundary position (mainly a function of the sedimentation coefficient and time) and change in boundary shape (mainly dependent on diffusion and the square root of time and the sedimentation resolution between species). Deconvolution of time independent (TI) noise is optimal when at least 10 – 20 baseline scans (free of any sedimenting species) are included in the scan set.
Achieving the theoretical considerations described above requires consideration of the physical properties of the sample and solute. While, a detailed discussion of the effect of excipients on the density and viscosity of solutions is beyond the scope of this article [17], solutes, particularly at high concentrations, significantly impact the sedimentation and diffusion rates of peptide, protein and aggregate. In addition, practical constraints such as instrument availability and required sample throughput may also be a factor. Rotor speed is the principal variable of an SV-AUC experiment and needs to be rationally chosen. While the information content of scans increases at higher rotor speeds, allowing more precise measurement of boundary midpoints, the total number of scans reduces thus reducing the overall data density. For Beckman XL instruments equipped with absorption optics, analyzing seven samples in the AN-50 Ti rotor at 40,000 rpm will yield 50 absorbance scans for each sample over a total experiment time of five hours, sufficient for a typical intact mAb to fully sediment. Sedimenting three samples in the AN-60 Ti rotor at the same speed will roughly double the scan density of the acquired data set. Sedimenting a single sample will further triple the scan density. Setting the rotor speed at 50,000 rpm increases the resolution between sedimenting species but reduces the number of scans per run. Having fewer scans for subsequent analysis does not necessarily create a problem, as discussed below.
Beckman Coulter charcoal-Epon centerpieces have been qualified for use at a maximum speed of 42,000 rpm; however, they are typically run in SV-AUC experiments at up to 60,000 rpm without a negative impact. On the new Optima™ centrifuge, faster scanning enables collection of a very dense raw data set or reduction of the overall time required for full sedimentation at higher rotor speed. However, as demonstrated in Figure 3 and Table 1 for BSA and the NIST mAb, a scan density above 50 scans spanning the entire column height improves the precision or resolution of a c(s) curve fit minimally if at all; at high enough scan density, systematic errors in the data acquisition start to dominate. Therefore, a delay in data collection between scans (named frequency on the Optima™ model) may be set to avoid collecting more data than needed.
Figure 3.
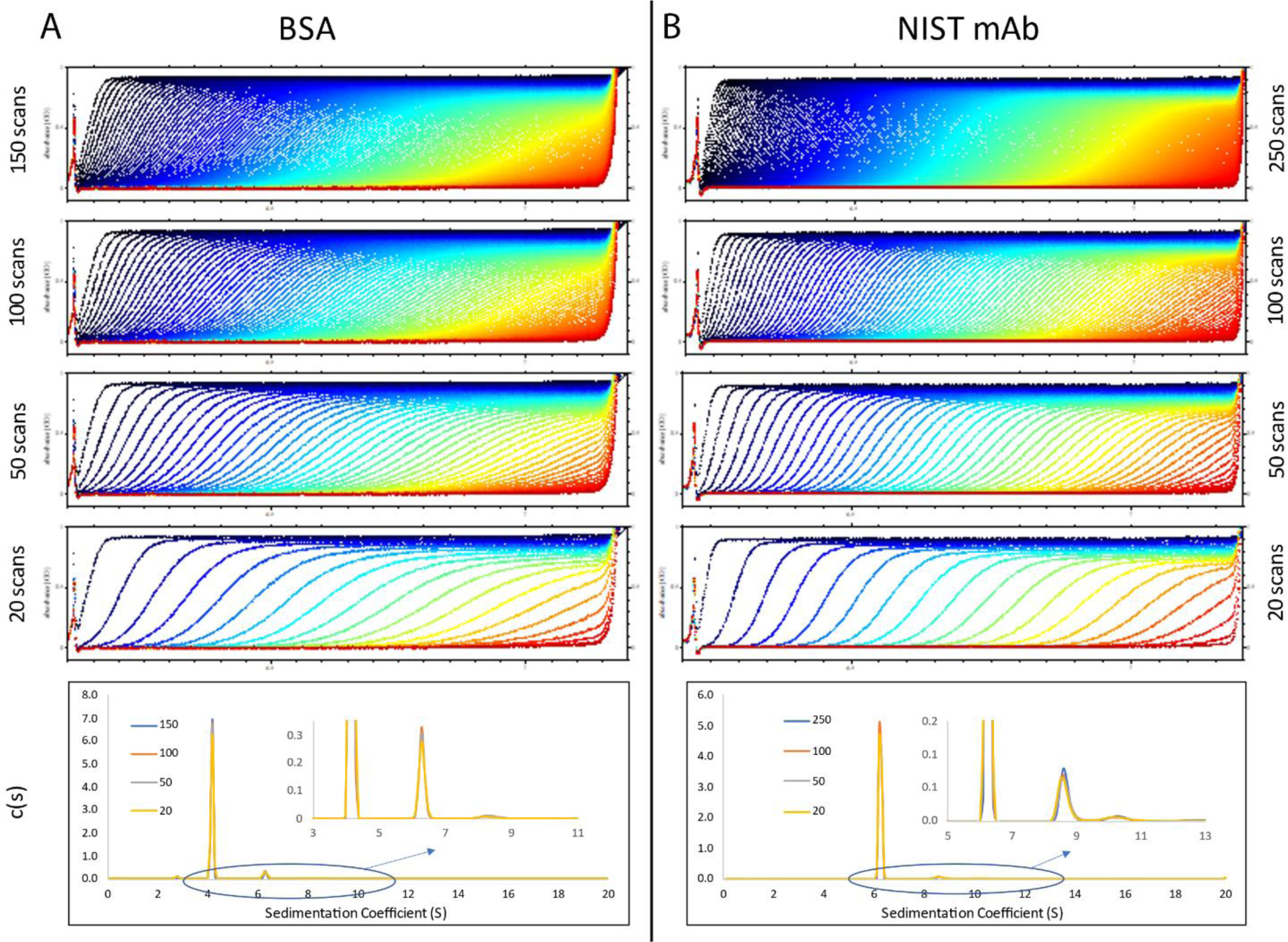
Raw SV-AUC data (top four graphs) and overlay of c(s) distributions (bottom graph) for BSA (A) and the NIST mAb (B). For BSA, 300 scans were collected and covered the full sedimentation of all species present in the sample. The four raw data displays correspond to the loading of every 2nd, 3rd, 6th, and 15th scans (from top to bottom) in SEDFIT. For the NIST mAb, 500 scans were collected and covered the full sedimentation of all species present in the sample. The four raw data displays correspond to the loading of every 2nd, 5th, 10th, and 25th scans (from top to bottom) in SEDFIT. The NIST mAb data was collected with higher scanning frequency compared to the BSA data. The c(s) distributions corresponding to the independent fitting of each of the top four raw data files are highly similar.
Table 1.
Sedimentation coefficients and relative amounts for BSA and the NIST mAb monomer and dimer species. The frictional ratios and the RMSD values corresponding to the convergence of each of the fits shown in Figure 3 are also listed.
| s (S) |
Amount (%) |
||||||
|---|---|---|---|---|---|---|---|
| Scans | Monomer | Dimer | Monomer | Dimer | f/f0 | RMSD | |
|
|
|
|
|
|
|
|
|
| BSA | 150 | 4.19 | 6.32 | 90.8 | 6.7 | 1.37 | 0.00240 |
| 100 | 4.19 | 6.31 | 90.8 | 6.8 | 1.37 | 0.00241 | |
| 50 | 4.19 | 6.31 | 90.4 | 6.8 | 1.37 | 0.00240 | |
| 20 | 4.19 | 6.31 | 90.6 | 6.8 | 1.37 | 0.00240 | |
|
| |||||||
| NIST mAb | 250 | 6.24 | 8.65 | 95.3 | 3.0 | 1.67 | 0.00232 |
| 100 | 6.24 | 8.63 | 95.5 | 3.0 | 1.67 | 0.00232 | |
| 50 | 6.24 | 8.66 | 95.3 | 3.0 | 1.67 | 0.00233 | |
| 20 | 6.24 | 8.61 | 95.0 | 3.0 | 1.67 | 0.00231 | |
Absorbance-based detection mode (intensity or absorbance at 280 nm) is typically preferred over interference for mAb aggregate quantitation because it is less prone to experimental artifacts related to baseline, noise structure, or stemming from imperfect meniscus and buffer match between the reference and the sample. Since artifacts can manifest themselves as c(s) peaks, they may negatively impact both the accuracy and precision of mAb aggregate quantitation. Atypical absorbance readings can be related to the performance of the lamp, the photomultiplier tube, or any of the other parts of the absorbance optics. In this case, troubleshooting may benefit from both types of data collected simultaneously if the centrifuge is equipped with absorbance and interference detection (e.g., Beckman ProteomeLab™ XL-I). Other data acquisition and/or analysis protocols as well as multi-speed analysis used to interrogate highly heterogeneous systems [45] are beyond the scope of this paper.
Data analysis – general considerations
Over the course of an SV-AUC run, the radial and temporal evolution of concentration profiles across the sample cell, c(r,t), is measured. A raw data set for a given cell typically consists of over 10,000 data points, which allows mathematical curve-fitting to extract quantitative information. The first step in analyzing an SV-AUC run is to examine the raw scans to evaluate of the data and diagnose aberrant behavior. Typical sedimentation profiles of an IgG mAb are shown in Figure 4. The data consist of sigmoidal-shaped boundaries that migrate and broaden over time (Fig. 4A). The presence of aggregates leads to skewing of the leading edge of the initial boundaries. Depending on the abundance and the type of species, a second, faster-sedimenting boundary (Fig. 4B) or a slightly sloping plateau region (Fig. 4C) may become evident as the run progresses.
Figure 4.
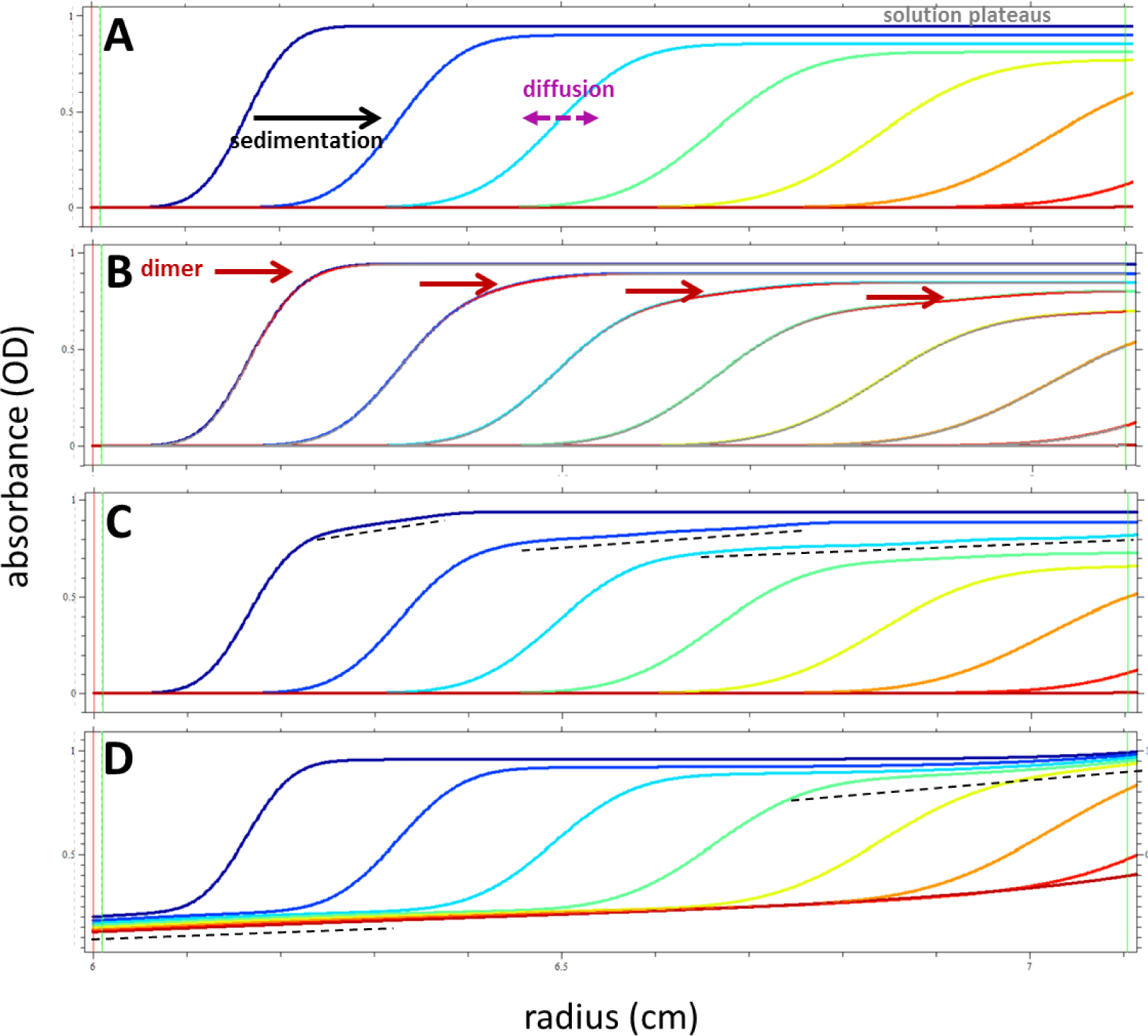
Typical features of sedimentation profiles of an IgG (150 kDa, 6.5 s) sedimenting at 45,000 rpm, in 30 min intervals (higher color temperature indicates later time). (A) The concentration boundaries migrate due to sedimentation, and increasingly broaden due to diffusion. For 100% monomer they exhibit approximately symmetric shape. Solution plateaus are flat and uniformly decreasing with time (due to radial dilution in the sector-shaped solution column). (B) For the same molecule but with 10% of signal from a dimer (here assumed to be 9 s). This causes a stretching of the leading edge of the sedimentation boundary, which later separates into a broad shoulder of a faster-sedimenting boundary. The region of the scan where dimer is present is highlighted in red – a presentation that accompanies integration in SEDFIT to enhance the understanding of the correspondence between c(s) peaks and features of the raw data. (C) The presence of dimer, trimer, and tetramer (each at 5%) causes even stronger stretching of the leading side of the sedimentation boundary, evolving into a more broadly sloping plateau (indicated by dashed lines). Due to diffusion of each species, their sedimentation boundaries cannot be separately discerned. (D) Another cause of sloping plateaus, but with different pattern, can be the presence of very small species (here 30% of 1 kDa, 1 s). This is accompanied by sloping solvent plateaus near the meniscus.
A potential limitation of any sedimentation coefficient distribution method is its basis in sedimentation modeling of non-interacting particles. For molecules that reversibly interconvert between oligomeric states with complex lifetimes much shorter than the SV-AUC experiment, what is measured is a time-average sedimentation velocity of each molecule. Because of the concentration gradients intrinsic to sedimentation boundaries, this can lead to complex sedimentation coefficient distribution patterns that are the topic of Gilbert theory, Gilbert-Jenkins theory, and effective particle theory [46]. Rapidly reversible self-association can be diagnosed from the concentration-dependence of the c(s) distributions determined as described below [46]. The interpretation of reversible self-association is outside the scope of the current work, which is focused on quantitation of oligomers and aggregates that are irreversible on the time scale of the sedimentation experiment.
Several software packages are available for quantitative trace analysis. The software DCDT+ employs a theoretical relationship between the time-derivative of the scans and the differential sedimentation coefficient distributions of non-diffusing particles, known as g*(s) [47–49]. In simple terms, a set of radial scans acquired during a small-time interval is transformed onto an axis of sedimentation coefficients and thereby converts sigmoidal boundaries into peaks. Peaks of such differential sedimentation coefficient distributions can be integrated, similar to chromatography data, thus enabling quantitation of fractional populations of species that have different sizes. Quantitation of trace oligomers is challenging with g*(s) because of limitations intrinsic to its single-species origin and characteristic distortions [49, 50], despite later extrapolations and adjustments in the graphical output implemented in the software program ULTRASCAN [51, 52]. At the same time, the g*(s) transform is a valuable tool to visualize sedimentation boundaries in the space of sedimentation coefficients as an intermediate inspection between raw data space (signal vs. radius and time) and the detailed c(s) analysis discussed below.
A sedimentation coefficients c(s) analysis implemented in the program SEDFIT [49, 53] is widely used in the biopharmaceutical industry for aggregate quantitation. In c(s), a differential sedimentation coefficient distribution is calculated by directly curve-fitting the raw sedimentation boundaries with an explicit distribution model. This analysis produces peaks similar to g*(s) except that diffusional boundary broadening is included in the sedimentation model. By fitting a set of scans representing the entire evolution of the sedimentation process, ranging from earliest to the latest scans when the sedimentation boundary has migrated past the detectable range (radii > 7.1 cm), the average degree of diffusion can be deconvoluted from the translational boundary movement. As a result, sharp peaks are obtained and baseline-resolution of features that are not visually recognizable in broad boundaries become evident.
Mathematically, the c(s) model is defined as a Fredholm integral equation, with kernels being the Lamm equation solutions (i.e., the sedimentation/diffusion master equation predicting patterns of migration and spreading of sedimentation boundaries) of single discrete species across a range of different s-values [18, 54]. The number of species that needs to be considered to describe a sedimentation coefficient distribution is on the order of 100. A direct fit would not yield meaningful results because of overparameterization and correlation. The c(s) model reduces the number of unknowns through use of a hydrodynamic scale relationship, which applies a best-fit average frictional ratio (fr,avg) of all sedimenting particles to estimate the diffusion coefficient corresponding to the species’ s-values. Regularization calculates the simplest distribution among all that provide statistically equivalent fit quality, scaled by F-statistics typically using a p-value of 0.68 – 0.95. The most used regularization is maximizing the information entropy of the resulting distribution.
These model parameters are essential for a statistically meaningful best-fit distribution. In the fit of this model, it is necessary to include appropriate baseline parameters, which are, in turn, dependent on the data structure of the optical detection. Specifically, baseline parameters consist of a constant offset, time-invariant but radial-dependent baseline, and/or time-varying but radially constant offsets in each scan [49]. The latter applies for interference data only, while the former is usually sufficient to fit absorbance data. It is essential that the c(s) fits of the entire scan data be verified by critical inspection of residuals, using radial overlays, bitmaps, runs tests, and/or tests for normal distribution [49]. Extensions for different diffusion models have been developed [49] such as two-dimensional size-and-shape distributions [55], but these extensions are not required for mAb aggregate quantitation analysis [56].
Individual species will generally yield separate peaks in a c(s) distribution. The peak areas can be integrated to determine species’ concentrations. However, the importance of keeping in mind that sedimentation coefficient distributions are calculated curves from curve-fitting the raw data cannot be overstated. For this reason, the SEDFIT display is split and shows the raw data in the upper half, and the distribution in the lower half, separated in the middle by a display of the quality of the fit. The c(s) distribution can be integrated in SEDFIT, upon which the display of the raw data is colored to highlight the boundary regions to which the integrated species contribute (Fig. 4B). Distributions can be exported to other processing software via the clipboard.
In silico simulations of raw and analyzed data are useful in experimental design. SEDFIT allows simulating sedimentation data with various models and creating artificial scan files with or without noise and baselines. These can then be subjected to data analysis to test to what extent the known input parameters are obtained. The analysis of simulated data is extremely valuable also to clarify promising experimental parameters for different systems under study.
Aggregate quantitation by c(s) model in SEDFIT
The “continuous c(s) distribution” model is typically used in SEDFIT analysis of aggregate quantitation in mAb preparations [49, 53]. Fitting the model to the data scans requires input of user-defined parameters. Before listing the parameters and the best practices for setting them, it is important to emphasize that the best fit is be obtained when a few iterations of fitting are applied in which one or another parameter is modified slightly each time. This is particularly important when a new molecule is first being analyzed during method development and/or when no other prior knowledge is available. Once a method has been developed and parameters have been set, the number of iterations required to converge the best fit is significantly reduced. Below, we list the parameters and discuss some best practices to perform the analysis.
The range of sedimentation coefficient values is the first set of values to be entered. The lower limit must be slightly greater than 0, i.e., 0.5 – 2.0 s, because very small sedimentation coefficients tend to correlate with baseline offsets. For purified mAb samples, an upper limit of 20 s is generally sufficient; samples with higher-order oligomers, stressed samples, or process intermediate samples may require a higher upper limit (often 30 – 50 s). At the convergence of the fit, SEDFIT will alert if the sedimentation coefficient range is too restrictive, especially at the upper limit.
‘Resolution’ is defined as the number of data points required to build the c(s) distribution across the sedimentation coefficient range in which the analysis is performed; in other words, resolution defines the unit step of the X-axis (s). The higher this number, the better the resolution between the different sedimenting species in the c(s) distribution. A common practice is to set the resolution at 37 (corresponding to the step of 0.5 s between 2 s and 20 s) first to enable a rapid convergence of the fit and an estimate of the frictional ratio. (Setting the resolution parameter too high at the first pass when the frictional ratio of a new molecule is unknown will result in overly long fit converge.) The resolution is increased to 181 (corresponding to the step of 0.1 s between 2 s and 20 s) in a second iteration after the frictional ratio has been estimated (see below); this fit will converge faster than if the first iteration was skipped. The first iteration at low resolution is optional if the frictional ratio of the molecule is known.
A good initial estimate of the frictional ratio for the analysis of mAbs is 1.5 – 1.6. If the frictional ratio is known through prior knowledge, inputting this value ensures a faster fit converge and the overall fitting approach becomes more consistent. No matter what value is entered for the frictional ratio, this parameter must be floated (i.e., iteratively refined) during the fit as a best practice.
The baseline is typically set at 0 and floated during the fit to account for any minor shifts in the baseline. The option to fit the TI noise reduction is always enabled to account for any time-independent features in the absorbance profile, e.g., optical window imperfections. On the other hand, fitting the RI noise adds little benefit to the quality of the fit for absorbance data, but is a must when analyzing interference data. However, in the new Optima™ ultracentrifuge, fitting both the TI and the RI noise reduction is beneficial due to slightly different radial scanning data collection.
The meniscus position is defined when the user loads the scans in SEDFIT and manually drags the red cursor on top of the optical spike in absorbance data of the sample sector. The meniscus position must always be floated; the allowable fitting range for this parameter is defined by the user by placing the dashed gray cursor ~ ±0.2 mm around the red cursor.
The bottom of the cell is typically set at 7.2 cm; this parameter is fixed unless back-diffusion must be modeled. The scan data fitting range is defined by the green cursors which are usually set by the user at 0.1 cm to the right of the meniscus (i.e., meniscus + 0.1 cm) for the lower limit and at 0.1 or 0.15 cm to the left of the bottom (i.e., bottom − 0.1 (or − 0.15 cm)) for the upper limit.
The confidence level (F-ratio) for the regularization process should be set at 0.68 (corresponding to one standard deviation). Setting it higher (i.e., 0.95 for two standard deviations) can add significant computational time. Initially calculating c(s) with a confidence level of 0.68 and then raising it to 0.95 allows in comparison of the resulting distributions an assessment of the significance of resolution between peaks. Once a fit converges, it is recommended to alternate between the Simplex and the Marquart-Levenberg regularization algorithms at least once to ensure that the fit is not caught in a local minimum. Often switching between one and the other does not significantly impact the quality of the fit in which case one cycle is enough. However, if the RMSD value significantly decreases by switching from one algorithm to another, switching needs to continue until no further change can be observed. It is important to mention that noise amplification or application of different regularization models may impact detailed peak structure and peak shape since populations of species with similar s-values are correlated. Therefore, a particular distribution value at a specific s-value is not statistically meaningful, and, similarly, fitting peak shapes to certain models is not meaningful; rather peak areas and weighted integrals are invariant and the relevant quantities to be interpreted.
As applied to aggregate quantitation, the caution noted above means that identification of individual species in a series of small aggregate peaks is not nearly as robust as quantitation of the total aggregate. There is a general hierarchy of statistical significance between the attributes of a c(s) peak: (1) The highest significance is the integral of c(s) which corresponds to the concentration of the sedimenting species represented by this peak; (2) The s-value of the peak may suffer from correlation with majority species s-value and fr,avg, and therefore is subject to more statistical variation than the integral (i.e., the concentration) of the same peak; (3) although the estimated diffusional spread based on the best-fit fr,avg allows the prediction of a molar mass, this should not be given more confidence than a general indication of the possible range of the true value, assuming the trace species of interest has similar hydrodynamic shape factors f/f0 as the major species (typically, the mAb monomer). Conversely, due to the relative lack of information on diffusional spread of each species, the use of a single frictional ratio for the entire distribution does not significantly impact integrals under other peaks.
There are several additional considerations for quantitation of mAb aggregates that can prove useful or essential in certain cases. First, when working with buffers that contain significant quantities of sedimenting co-solutes it is possible to account for the dynamic density and viscosity gradients modulating macromolecular sedimentation [28]. The application and utility of this model in the context of trace aggregate analysis has been studied in detail by Gabrielson and colleagues [27]. Second, the maximum entropy regularization can introduce a slight bias leading to underestimate low levels of aggregates and other species. This bias can be avoided by using Bayesian prior probabilities in maximizing information entropy of the distribution [57]; guidelines for its application are provided in [58]. Third, a multi-wavelength extension of c(s) [59] is sensible only when working with heterogeneous mixtures of spectrally distinct species. Fourth, an emerging opportunity is presented by a nonideal extension of c(s) [60]. This extension overcomes the concentration limit to < 1 mg/mL imposed by the current requirement in c(s) analysis for hydrodynamically ideal sedimentation. Practical experience in the application to antibody trace analysis is still limited at the present time [29]. Fifth, in a single run to study IgG-sized molecules at 40,000 rpm, it is challenging to monitor species on the order of ~100 s. The dynamic range of sizes in a single experiment can be increased up to 1,000-fold by using a gradually increasing rotor speed in a ‘gravitational sweep’ experiment [45].
The c(s) model and refinements are implemented in the data analysis software SEDFIT and SEDPHAT (provided freely by the National Institute of Biomedical Imaging and Bioengineering at the National Institutes of Health at sedfitsedphat.nibib.nih.gov/software).
Considerations for reporting SV-AUC results
Whether SV-AUC is used as a characterization tool or as a method orthogonal to SEC, SV-AUC results are best reported as individual plots of c(s) distribution functions with ranges of sedimentation coefficients integrated as the numerical results. The complete data set and analysis (e.g., the c(r,t) curves, fit lines, residual bitmaps, and residuals plots) are not always appropriate for typical reporting because they require experience for correct interpretation. In addition, experimental artifacts caused by curve fitting or triplicate measurement presentations with varying minor peaks can obfuscate the more robust overall conclusions. Instead, regulatory filings should include tabulated data of the primary reportable results from c(s) curve fits: the sedimentation coefficient values for major species, and their corresponding fraction. These values capture the percent of total signal apparently travelling between the boundary sedimentation coefficients. Typically, sedimentation coefficient data is reported to two decimal places, whereas higher molecular weight species amounts are reported to one decimal place. However, as for other analytical methods, it is the precision determined at the time of method development that ultimately dictates the number of significant figures for each of the reported results. Species present at levels below the LOD (generally, 1–2% of the total signal) should not be individually identified nor their apparent individual sedimentation coefficients reported.
When initiating a program of aggregate analysis, it is essential that studies be conducted that replicate the results documented during development of the method on the same or similar molecules. In particular, the experimental setup, the frictional ratio f/f0 that is determined, and the RMSD of the curve fitting should be comparable to the reference values. Careful tracking of expected RMSD and f/f0 values allows setting limits for acceptable ranges in subsequent studies. Experience is a powerful teacher that will allow the analyst to monitor instrument performance and determine whether the c(s) fitting model is appropriate to the protein being analyzed. RMSD values are not the sole judge of quality of the fit. The number of scans that are acquired, rotor speed, optical system, detection wavelength, and the lower and upper bounds to the sample column that is fit will affect the information content and the resulting curve fit, regularization, and expected RMSD. For example, running a sample at a lower rotor speed results in shallower boundaries and more overlap between species. However, less steep boundaries tend to contribute less noise to the curve fit because of the finite radial precision of the instrument and therefore will result in a lower RMSD value. If many scans from time points after the last major species have sedimented are included in the analysis, the average RMSD of all scans will also be reduced because scans of an empty cell always fit better than those of a moving boundary.
It is important to stress that as described in the above sections, the c(s) distribution function does not represent the raw data but rather is a fitted mathematical function. The practice of reporting c(s) distribution functions in a format like what is presented in SEC chromatograms is misleading. For instance, in a 2003 paper, aggregates present at levels ranging from 30.6% (dimer) to 0.1% (heptamer) were labeled [61]. Unfortunately, presentation of the figure in this manner caused misinterpretation of the capabilities of SV-AUC that might have led to setting unrealistic expectations. In a later publication, it was clarified that the LOD of aggregates characterization by SV-AUC was at best 1% of the total signal (depending on the aggregates size) and reproducibly detecting aggregate levels below 1–2% was hard to achieve even for experienced analysts [62]. Thus, the initial 2003 figure was misinterpreted because peaks labeled as ‘pentamer’ through ‘heptamer’ were all present at amounts lower than 2% of the total signal, below the LOD of the method followed.
Unlike in SEC, where the accuracy in the amount of total aggregate is increased by omitting peaks below the LOD, SV-AUC analysis should include the area under the peaks associated with pentamer, hexamer, and heptamer as part of total aggregate to achieve the most accurate representation of the data. In SEC, a chromatogram is the raw data and the total sum in a range is an integration of that area. In SV-AUC, however, a c(s) distribution is a deconvolution of the c(r,t) raw data where the totals are determined by a large number of data points (the plateau regions) and, thus, the totals are more accurate than the deconvolution. Both the overall amount and weight average sedimentation coefficient in a range of s-values are accurate regardless of how the curve fitting and regularization distribute that total in the final distribution [63].
SV-AUC method validation
Method validation is an essential aspect and the ultimate phase of method development as outlined in ICH Q2(R1) ‘Validation of Analytical Procedures’ [37]. The goal of method validation is to demonstrate that the analytical procedure is suitable for its intended purpose. In the context of this guidance document, SV-AUC can be considered a quantitative test for impurity content, specifically, total soluble aggregate. Once the method is developed as described in previous sections, typical validation parameters listed in Table 2 can be evaluated as outlined in ICH Q2(R1). As noted below, several of the parameters listed in Table 2 are quite difficult to achieve for SV-AUC. At a minimum the method should encompass the systems suitability testing described above and further validation described here can be undertaken as required by the needs of the specific application of the method.
Table 2.
Method validation parameters and their definitions as ipplied to the quantitation of soluble aggregates by SV-AUC.
| Method validation parameter | Definition |
|---|---|
|
|
|
| Specificity | Ability to resolve aggregates as a population of c(s) species with s-values greater than that of the monomer |
|
|
|
| Accuracy | Agreement of the measured value with the accepted true or reference value |
|
|
|
| Precision | Closeness between multiple aggregate measurements |
|
|
|
| Limit of Detection (LOD) | Lowest amount of aggregate which can be reliably detected |
|
|
|
| Limit of Quantitation (LOQ) | Lowest amount of aggregate which can be reliably quantified |
|
|
|
| Linearity | Ability of SV-AUC to detect and quantify soluble aggregates directly proportional to their concentration in the sample |
|
|
|
| Range | Interval between the highest and lowest amount of soluble aggregates where the method has the appropriate degree of precision, accuracy, and linearity |
|
|
|
| Robustness | Ability of the SV-AUC method to provide reliable quantitation with respect to small variations in method parameters |
Specificity, range, and robustness are relatively straightforward to demonstrate during the validation of a method for the aggregate quantitation in mAb samples by SV-AUC. The peaks corresponding to the higher molecular weight species are typically well-resolved from the mAb monomer within the c(s) distribution, thus demonstrating specificity. Ranges between approx. 0.5 and 13% have been used for a number of proteins including mAbs [21, 64] and are usually sufficient to cover representative levels of aggregates for a variety of nominal and stressed conditions. Method robustness for the quantitation of aggregates can be evaluated by introducing small variations in method parameters such as temperature, cell alignment etc. as described above. It should be noted that, in general, experimental setup affects the robustness of an SV-AUC method more than the data analysis [64].
The most critical and challenging aspects of an SV-AUC method validation are the determination of precision, LOD, LOQ, and the associated demonstration of linearity. ICH Q2(R1) [37] specifies three general approaches to determining the LOD and LOQ of a method:
The first approach is based on visual inspection and does not apply to aggregate quantitation by SV-AUC because the analyte is detected indirectly from the fit of the sedimentation raw scan data to the c(s) distribution model.
The second approach applies to methods in which the signal exhibits baseline noise. Here, comparing measured signals from samples with known low analyte concentrations to those of blank samples allows calculation of the signal-to-noise ratio. The lowest concentration at which the analyte can be reliably detected – typically at ~ 2:1 to 3:1 signal to noise – corresponds to the LOD. While this approach can potentially be employed for the LOD determination of an SV-AUC method, it is not straightforward. In fact, while the signal to noise can be easily estimated for absorbance scans, changes in the absorbance signal caused by the differentially sedimenting boundaries (monomer vs. aggregates) cannot be readily translated into the relative abundances of the c(s) species higher than the monomer, particularly at low concentrations of aggregates. In addition, if the sample concentration is low (< 0.5 OD total loading concentration), the signal to noise decreases and the ability to detect species present at low levels becomes problematic.
The third approach outlined in the ICH Q2(B) guideline relies on the standard deviation (σ) in the response of a given sample and the slope (a) of a standard curve generated using known concentrations of that sample. The LOD and LOQ are calculated as 3.3σ/a and as 10σ/a, respectively. While the generation of a calibration curve is time consuming and requires large sample amounts, it does not depend on the isolation of aggregate species. This approach determines the linearity and range along with the LOD and LOQ in a single set of experiments.
A framework for method validation for SV-AUC aggregate quantitation has been established. Gabrielson and Arthur describe a standard curve approach [21]. In practice, determination of specificity by testing a set of representative samples is the first required step. If the specificity is demonstrated, a dilution series of two samples with known levels (‘high’ and ‘low’) of soluble irreversible aggregates is performed to establish the LOD, LOQ, and the linearity range following the third approach listed above. At the same time, approaches other than those listed above may be acceptable. Therefore, AUC users are advised to adopt an appropriate strategy for determining LOD/LOQ based on the intended use of the SV-AUC aggregate quantitation method.
It is important to note that unlike SEC, validation parameters of a method for the aggregate quantitation by SV-AUC are not derived from a directly detected response (raw data), but rather from a c(s) distribution mathematically derived from the raw scan data. Therefore, method precision should include an evaluation of all major sources of variability (including software fitting variability) depending on the setup, sample, and analysis model used. It is strongly recommended to monitor SV-AUC method performance over time. The proposed performance attributes described in the section “System suitability” above should be routinely monitored by maintaining appropriate control charts to detect any long-term trends related to systematic offset in time stamp, centerpiece aging, gradual reduction of the instrument optics performance, etc.
Summary of recommendations
This report is a synthesis of many decades of experience conducting analytical ultracentrifugation in both the academic and biopharmaceutical arenas. Not surprisingly, much of our collective effort went into deliberating the benefits and weaknesses of independently developed aggregate analysis protocols and the relative importance of sets of parameters and practices. That our work has resulted in this best practice guide reflects a maturity in this analysis approach that we hope will result in consistent aggregate quantitation approaches across the industry. We hope that this report will serve as a reference for seasoned analysts, guidance for regulators, and an entrée to new analysts that will allow them to rapidly become proficient in quantitating the aggregate level in mAb and other protein therapeutic preparations. Below, we present a summary checklist of our best practice recommendations (Table 3). We stress that these recommendations are not meant to be prescriptive or mandatory for every molecule and/or every application. The common practice recommendations are a good starting point for the analysis of the molecule at hand. However, the specific set of conditions and parameters will always have to be considered on a case-by-case basis and will have to be guided by sound scientific judgement from the end user. While analytical ultracentrifugation can seem a daunting method to master, its repetitive application to a well characterized system is rather straightforward and can reveal a rich source of insights. However, it is essential that an effective analyst be attentive to the nuances of each system, the pitfalls of the analysis method, and the vagaries of the instrument. With experience and attention to detail, deviations from the norm become less a problem and more an opportunity to learn something new that can be beneficial to the development of novel therapeutics.
Table 3.
Summary of recommendations for the quantitation of total aggregate level in mAb samples.
| Stage | Description | First-to-try recommendation | Further options / Alternatives |
|---|---|---|---|
|
|
|
|
|
| Sample Prep | General | Load sample at approx. 1 OD (l=1.2 cm) for UV | Dialysis is required for interference |
|
|
|
|
|
| Absorbing excipient in buffer | Dialysis | ||
|
|
|
|
|
| Low ionic strength or self-buffered formulations | Add ~25 mM salt | ||
|
|
|
|
|
| Co-cute | Dialyze to remove co-solute | Run with co-solute but use inhomogeneous c(s) model | |
|
|
|
|
|
| Replicates | 3 | 2 | |
|
|
|
|
|
| Cell Alignment | Method for cell alignment | Optical (preferred) or mechanical | Manual |
|
|
|
|
|
| Temperature | Temperature | 20 °C | Other temperatures within the instrument range (4 – 37 °C |
|
|
|
|
|
| Temperature equilibration | 1–2 hours | Longer for extreme temperatures | |
|
|
|
|
|
| System suitability | Instrument check: Radial calibration | Once every time a rotor and/or counterbalance is changed | Before every run |
|
|
|
|
|
| Cell suitability | Keep cell components together, monitor performance, perform a check at 3,000 rpm | ||
|
|
|
|
|
| SV-AUC run setup | Rotor speed | 50,000 rpm | Up to 60,000 rpm for Ti-60 rotor |
|
|
|
|
|
| Detection mode | Absorbance-based at 280 nm (intensity or absorbance) | Absorbance-based at other wavelengths (e.g., 230 nm) or interference | |
|
|
|
|
|
| Fitting parameters | Resolution | Start with 37, increase to 181 (0.1 s) | Start directly with 181 (0.1 s) |
|
|
|
|
|
| s-values range | 0.1 to 20 | 2 to 20. Increase upper range for samples containing larger oligomers, in extreme cases using log-scaled division to cover very large range efficiently | |
|
|
|
|
|
| Frictional ratio | Start at 1.5 and float | Input prior knowledge value and float | |
|
|
|
|
|
| Baseline TI noise |
Set to 0 and float Floated |
||
|
|
|
|
|
| RI noise | Fixed | Floated (especially beneficial for data collected on Beckman Optima™) | |
|
|
|
|
|
| Meniscus | User defined and floated | ||
|
|
|
|
|
| Meniscus fitting range | ± 0.2 mm around meniscus | ||
|
|
|
|
|
| Bottom | 7.2 cm and fixed | Floated in presence of small MW contaminants or other species presenting back-diffusion | |
|
|
|
|
|
| Fitting range | Meniscus + 0.1 cm to Bottom − 0.1 (or − 0.15 cm) | ||
|
|
|
|
|
| F-ratio (confidence interval) | 0.68 | none | |
|
|
|
|
|
| Regularizatio n algorithm | Alternate between Simplex and Marquart Levenberg | Simplex only | |
|
|
|
|
|
| Partial specific volume | Calculated or measured | 0.73 mL/g as a reasonable approximation [62] | |
|
|
|
|
|
| Viscosity | User defined | ||
|
|
|
|
|
| Density | User defined | ||
|
|
|
|
|
| Data reporting | What should be reported | c(s) distribution, table with major species | |
|
|
|||
| Sedimentatio n coefficients | only for major species up to 2 decimal places | ||
|
|
|||
| Amounts in % | only for major species up to 1 decimal place |
Acknowledgments
This work was supported by the Intramural Research Programs of the National Institute of Biomedical Imaging and Bioengineering, NIH.
The authors of this article are volunteer members of the Biophysics Working Group assembled under the auspices of CASSS – Sharing Science Solutions, 5900 Hollis Street, Suite R3, Emeryville, CA 94608.
The authors thank Barthélemy Demeule (Genentech), Robert Kelley (Genentech), and Brandon L. Doyle (Eli Lilly and Company) for their insightful suggestions while authoring this manuscript. Michael R. De Felippis (Eli Lilly and Company) is thanked for critically reviewing the manuscript.
Footnotes
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests:
GMB is an employee of Biogen Inc.
ILB and RM are employees of Eli Lilly and Company
ESD and JCL are employees of Genentech Inc.
PR is an employee of Merck Serono S p a
MB, DH, JH and PS declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Abbreviations used: AAPS, American Association of Pharmaceutical Scientists; AUC, Analytical Ultracentrifugation; GMP, Good Manufacturing Practice; HMW, High Molecular Weight; ICH, International Council for Harmonization; LOD, Limit of Detection; LOQ, Limit of Quantitation; mAb, Monoclonal Antibody; OD, Optical Density; PABC, Product Attribute and Biological Consequences; QC, Quality Control; RMSD, Root Mean Square Deviation; SE, Sedimentation Equilibrium; SEC, Size Exclusion Chromatography; SV, Sedimentation Velocity, TI, Time Independent; USP, United States Pharmacopeia
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Narhi LO, Corvari V, Ripple DC, Afonina N, Cecchini I, Defelippis MR, Garidel P, Herre A, Koulov AV, Lubiniecki T, Mahler HC, Mangiagalli P, Nesta D, Perez-Ramirez B, Polozova A, Rossi M, Schmidt R, Simler R, Singh S, Spitznagel TM, Weiskopf A & Wuchner K (2015) Subvisible (2–100 mum) Particle Analysis During Biotherapeutic Drug Product Development: Part 1, Considerations and Strategy, J Pharm Sci. 104, 1899–1908. [DOI] [PubMed] [Google Scholar]
- 2.Corvari V, Narhi LO, Spitznagel TM, Afonina N, Cao S, Cash P, Cecchini I, DeFelippis MR, Garidel P, Herre A, Koulov AV, Lubiniecki T, Mahler HC, Mangiagalli P, Nesta D, Perez-Ramirez B, Polozova A, Rossi M, Schmidt R, Simler R, Singh S, Weiskopf A & Wuchner K (2015) Subvisible (2–100 mum) particle analysis during biotherapeutic drug product development: Part 2, experience with the application of subvisible particle analysis, Biologicals. 43, 457–73. [DOI] [PubMed] [Google Scholar]
- 3.Hubert M, Yang DT, Kwok SC, Rios A, Das TK, Patel A, Wuchner K, Antochshuk V, Junge F, Bou-Assaf GM, Cao S, Saggu M, Montrond L, Afonina N, Kolhe P, Loladze V & Narhi L (2020) A Multicompany Assessment of Submicron Particle Levels by NTA and RMM in a Wide Range of Late-Phase Clinical and Commercial Biotechnology-Derived Protein Products, J Pharm Sci. 109, 830–844. [DOI] [PubMed] [Google Scholar]
- 4.Ratanji KD, Derrick JP, Dearman RJ & Kimber I (2014) Immunogenicity of therapeutic proteins: influence of aggregation, J Immunotoxicol. 11, 99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mathaes R, Narhi L, Hawe A, Matter A, Bechtold-Peters K, Kenrick S, Kar S, Laskina O, Carpenter J, Cavicchi R, Koepf E, Lewis EN, De Silva R & Ripple D (2019) Phase-Appropriate Application of Analytical Methods to Monitor Subvisible Particles Across the Biotherapeutic Drug Product Life Cycle, AAPS J. 22, 1. [DOI] [PubMed] [Google Scholar]
- 6.Leavy O (2010) Therapeutic antibodies: past, present and future, Nat Rev Immunol. 10, 297. [DOI] [PubMed] [Google Scholar]
- 7.Walsh G (2018) Biopharmaceutical benchmarks 2018, Nat Biotechnol. 36, 1136–1145. [DOI] [PubMed] [Google Scholar]
- 8.Hong P, Koza S & Bouvier ES (2012) Size-Exclusion Chromatography for the Analysis of Protein Biotherapeutics and their Aggregates, J Liq Chromatogr Relat Technol. 35, 2923–2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carpenter JF, Randolph TW, Jiskoot W, Crommelin DJ, Middaugh CR & Winter G (2010) Potential inaccurate quantitation and sizing of protein aggregates by size exclusion chromatography: essential need to use orthogonal methods to assure the quality of therapeutic protein products, J Pharm Sci. 99, 2200–8. [DOI] [PubMed] [Google Scholar]
- 10.Gandhi AV, Pothecary MR, Bain DL & Carpenter JF (2017) Some Lessons Learned From a Comparison Between Sedimentation Velocity Analytical Ultracentrifugation and Size Exclusion Chromatography to Characterize and Quantify Protein Aggregates, J Pharm Sci. 106, 2178–2186. [DOI] [PubMed] [Google Scholar]
- 11.Mahler HC, Friess W, Grauschopf U & Kiese S (2009) Protein aggregation: pathways, induction factors and analysis, J Pharm Sci. 98, 2909–34. [DOI] [PubMed] [Google Scholar]
- 12.Liu J, Andya JD & Shire SJ (2006) A critical review of analytical ultracentrifugation and field flow fractionation methods for measuring protein aggregation, AAPS J. 8, E580–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gabrielson JP, Brader ML, Pekar AH, Mathis KB, Winter G, Carpenter JF & Randolph TW (2007) Quantitation of aggregate levels in a recombinant humanized monoclonal antibody formulation by size-exclusion chromatography, asymmetrical flow field flow fractionation, and sedimentation velocity, J Pharm Sci. 96, 268–79. [DOI] [PubMed] [Google Scholar]
- 14.Laue TM & Stafford WF 3rd (1999) Modern applications of analytical ultracentrifugation, Annu Rev Biophys Biomol Struct. 28, 75–100. [DOI] [PubMed] [Google Scholar]
- 15.Lebowitz J, Lewis MS & Schuck P (2002) Modern analytical ultracentrifugation in protein science: a tutorial review, Protein Sci. 11, 2067–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uchiyama S, Noda M & Krayukhina E (2018) Sedimentation velocity analytical ultracentrifugation for characterization of therapeutic antibodies, Biophys Rev. 10, 259–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berkowitz SA & Philo JS (2020) Chapter 9 - Characterizing biopharmaceuticals using analytical ultracentrifugation in Biophysical Characterization of Proteins in Developing Biopharmaceuticals (Second Edition) (Houde DJ & Berkowitz SA, eds) pp. 225–283, Elsevier. [Google Scholar]
- 18.Lamm O (1929) Die Differentialgleichung der Ultrazentrifugierung, Almqvist & Wiksell. [Google Scholar]
- 19.Correia JJ & Stafford WF (2015) Sedimentation Velocity: A Classical Perspective, Methods Enzymol. 562, 49–80. [DOI] [PubMed] [Google Scholar]
- 20.Pekar A & Sukumar M (2007) Quantitation of aggregates in therapeutic proteins using sedimentation velocity analytical ultracentrifugation: practical considerations that affect precision and accuracy, Anal Biochem. 367, 225–37. [DOI] [PubMed] [Google Scholar]
- 21.Gabrielson JP & Arthur KK (2011) Measuring low levels of protein aggregation by sedimentation velocity, Methods. 54, 83–91. [DOI] [PubMed] [Google Scholar]
- 22.Berkowitz SA & Laue T (2021) Boundary convection during sedimentation velocity in the Optima analytical ultracentrifuge, Anal Biochem. 631, 114306. [DOI] [PubMed] [Google Scholar]
- 23.MacGregor IK, Anderson AL & Laue TM (2004) Fluorescence detection for the XLI analytical ultracentrifuge, Biophys Chem. 108, 165–85. [DOI] [PubMed] [Google Scholar]
- 24.Kroe RR & Laue TM (2009) NUTS and BOLTS: applications of fluorescence-detected sedimentation, Anal Biochem. 390, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schuck P, Zhao H, Brautigam CA & Ghirlando R (2016) Basic Principles of Analytical Ultracentrifugation, CRC Press. [Google Scholar]
- 26.Gokarn YR, Kras E, Nodgaard C, Dharmavaram V, Fesinmeyer RM, Hultgen H, Brych S, Remmele RL Jr., Brems DN & Hershenson S (2008) Self-buffering antibody formulations, J Pharm Sci. 97, 3051–66. [DOI] [PubMed] [Google Scholar]
- 27.Gabrielson JP, Arthur KK, Kendrick BS, Randolph TW & Stoner MR (2009) Common excipients impair detection of protein aggregates during sedimentation velocity analytical ultracentrifugation, J Pharm Sci. 98, 50–62. [DOI] [PubMed] [Google Scholar]
- 28.Schuck P (2004) A model for sedimentation in inhomogeneous media. I. Dynamic density gradients from sedimenting co-solutes, Biophys Chem. 108, 187–200. [DOI] [PubMed] [Google Scholar]
- 29.Chaturvedi SK, Parupudi A, Juul-Madsen K, Nguyen A, Vorup-Jensen T, Dragulin-Otto S, Zhao H, Esfandiary R & Schuck P (2020) Measuring aggregates, self-association, and weak interactions in concentrated therapeutic antibody solutions, MAbs. 12, 1810488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao H, Brown PH, Balbo A, Fernandez-Alonso Mdel C, Polishchuck N, Chaudhry C, Mayer ML, Ghirlando R & Schuck P (2010) Accounting for solvent signal offsets in the analysis of interferometric sedimentation velocity data, Macromol Biosci. 10, 736–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hawe A, Wiggenhorn M, van de Weert M, Garbe JH, Mahler HC & Jiskoot W (2012) Forced degradation of therapeutic proteins, J Pharm Sci. 101, 895–913. [DOI] [PubMed] [Google Scholar]
- 32.Nowak C, J KC, S MD, Katiyar A, Bhat R, Sun, Ponniah G, Neill A, Mason B, Beck A & Liu H (2017) Forced degradation of recombinant monoclonal antibodies: A practical guide, MAbs. 9, 1217–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arthur KK, Gabrielson JP, Kendrick BS & Stoner MR (2009) Detection of protein aggregates by sedimentation velocity analytical ultracentrifugation (SV-AUC): sources of variability and their relative importance, J Pharm Sci. 98, 3522–39. [DOI] [PubMed] [Google Scholar]
- 34.Gabrielson JP, Arthur KK, Stoner MR, Winn BC, Kendrick BS, Razinkov V, Svitel J, Jiang Y, Voelker PJ, Fernandes CA & Ridgeway R (2010) Precision of protein aggregation measurements by sedimentation velocity analytical ultracentrifugation in biopharmaceutical applications, Anal Biochem. 396, 231–41. [DOI] [PubMed] [Google Scholar]
- 35.Doyle BL, Budyak IL, Rauk AP & Weiss WF 4th. (2017) An optical alignment system improves precision of soluble aggregate quantitation by sedimentation velocity analytical ultracentrifugation, Anal Biochem. 531, 16–19. [DOI] [PubMed] [Google Scholar]
- 36.Balbo A & Schuck P (2005). Analytical Ultracentrifugation in the Study of Protein Self-Association and Heterogeneous Protein-Protein Interactions. Paper presented at the Protein-Protein Interactions: A Molecular Cloning Manual. [Google Scholar]
- 37.Borman P & Elder D (2017) Q2(R1) Validation of Analytical Procedures in ICH Quality Guidelines pp. 127–166. [Google Scholar]
- 38.Zhao H, Ghirlando R, Piszczek G, Curth U, Brautigam CA & Schuck P (2013) Recorded scan times can limit the accuracy of sedimentation coefficients in analytical ultracentrifugation, Anal Biochem. 437, 104–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao H.Ghirlando, Alfonso R, Arisaka C, Attali F, Bain I, Bakhtina DL, Becker MM, Bedwell DF, Bekdemir GJ, Besong A, Birck TM, Brautigam C, Brennerman CA, Byron W, Bzowska O, Chaires A, Chaton JB, Colfen CT, Connaghan H, Crowley KD, Curth KA, Daviter U, Dean T, Diez WL, Ebel AI, Eckert C, Eisele DM, Eisenstein LE, England E, Escalante P, Fagan C, Fairman JA, Finn R, Fischle RM, de la Torre W, Gor JG, Gustafsson J, Hall H, Harding D, Cifre SE, Herr JG, Howell AB, Isaac EE, Jao RS, Jose SC, Kim D, Kokona SJ, Kornblatt B, Kosek JA, Krayukhina D, Krzizike E, Kusznir D, Kwon EA, Larson H, Laue A, Le Roy TM, Leech A, Lilie AP, Luger H, Luque-Ortega K, Ma JR, May J, Maynard CA, Modrak-Wojcik EL, Mok A, Mucke YF, Nagel-Steger N, Narlikar L, Noda GJ, Nourse M, Obsil A, Park T, Park CK, Pawelek JK, Perdue PD, Perkins EE, Perugini SJ, Peterson MA, Peverelli CL, Piszczek MG, Prag G, Prevelige G, Raynal PE, Rezabkova BD, Richter L, Ringel K, Rosenberg AE, Rowe R, Rufer AJ, Scott AC, Seravalli DJ, Solovyova JG, Song AS, Staunton R, Stoddard D, Stott C, Strauss K, Streicher HM, Sumida WW, J. P., et al. (2015) A multilaboratory comparison of calibration accuracy and the performance of external references in analytical ultracentrifugation, PLoS One. 10, e0126420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ghirlando R, Zhao H, Balbo A, Piszczek G, Curth U, Brautigam CA & Schuck P (2014) Measurement of the temperature of the resting rotor in analytical ultracentrifugation, Anal Biochem. 458, 37–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao H, Balbo A, Metger H, Clary R, Ghirlando R & Schuck P (2014) Improved measurement of the rotor temperature in analytical ultracentrifugation, Anal Biochem. 451, 69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.LeBrun T, Schuck P, Wei R, Yoon JS, Dong X, Morgan NY, Fagan J & Zhao H (2018) A radial calibration window for analytical ultracentrifugation, PLoS One. 13, e0201529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ghirlando R, Balbo A, Piszczek G, Brown PH, Lewis MS, Brautigam CA, Schuck P & Zhao H (2013) Improving the thermal, radial, and temporal accuracy of the analytical ultracentrifuge through external references, Anal Biochem. 440, 81–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schiel JE & Turner A (2018) The NISTmAb Reference Material 8671 lifecycle management and quality plan, Anal Bioanal Chem. 410, 2067–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ma J, Zhao H, Sandmaier J, Alexander Liddle J & Schuck P (2016) Variable Field Analytical Ultracentrifugation: II. Gravitational Sweep Sedimentation Velocity, Biophys J. 110, 103–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schuck P, Schuck PW & Zhao H (2017) Sedimentation Velocity Analytical Ultracentrifugation: Interacting Systems, CRC Press, Taylor & Francis Group. [Google Scholar]
- 47.Stafford WF 3rd (1992) Boundary analysis in sedimentation transport experiments: a procedure for obtaining sedimentation coefficient distributions using the time derivative of the concentration profile, Anal Biochem. 203, 295–301. [DOI] [PubMed] [Google Scholar]
- 48.Schuck P (2016) Sedimentation coefficient distributions of large particles, Analyst. 141, 4400–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schuck P (2016) Sedimentation Velocity Analytical Ultracentrifugation: Discrete Species and Size-Distributions of Macromolecules and Particles, CRC Press, Taylor & Francis Group. [Google Scholar]
- 50.Schuck P, Perugini MA, Gonzales NR, Howlett GJ & Schubert D (2002) Size-distribution analysis of proteins by analytical ultracentrifugation: strategies and application to model systems, Biophys J. 82, 1096–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Demeler B & van Holde KE (2004) Sedimentation velocity analysis of highly heterogeneous systems, Anal Biochem. 335, 279–88. [DOI] [PubMed] [Google Scholar]
- 52.Savelyev A, Gorbet GE, Henrickson A & Demeler B (2020) Moving analytical ultracentrifugation software to a good manufacturing practices (GMP) environment, PLoS Comput Biol. 16, e1007942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schuck P (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling, Biophys J. 78, 1606–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brown PH & Schuck P (2008) A new adaptive grid-size algorithm for the simulation of sedimentation velocity profiles in analytical ultracentrifugation, Comput Phys Commun. 178, 105–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brown PH & Schuck P (2006) Macromolecular size-and-shape distributions by sedimentation velocity analytical ultracentrifugation, Biophys J. 90, 4651–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schuck P (2010) On computational approaches for size-and-shape distributions from sedimentation velocity analytical ultracentrifugation, Eur Biophys J. 39, 1261–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brown PH, Balbo A & Schuck P (2008) A bayesian approach for quantifying trace amounts of antibody aggregates by sedimentation velocity analytical ultracentrifugation, AAPS J. 10, 481–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wafer L, Kloczewiak M & Luo Y (2016) Quantifying Trace Amounts of Aggregates in Biopharmaceuticals Using Analytical Ultracentrifugation Sedimentation Velocity: Bayesian Analyses and F Statistics, AAPS J. 18, 849–60. [DOI] [PubMed] [Google Scholar]
- 59.Balbo A, Minor KH, Velikovsky CA, Mariuzza RA, Peterson CB & Schuck P (2005) Studying multiprotein complexes by multisignal sedimentation velocity analytical ultracentrifugation, Proc Natl Acad Sci U S A. 102, 81–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chaturvedi SK, Ma J, Brown PH, Zhao H & Schuck P (2018) Measuring macromolecular size distributions and interactions at high concentrations by sedimentation velocity, Nat Commun. 9, 4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Philo J (2003) Characterizing the aggregation and conformation of protein therapeutics, American Biotechnology Laboratory. 21, 22–26. [Google Scholar]
- 62.Philo JS (2009) A critical review of methods for size characterization of non-particulate protein aggregates, Curr Pharm Biotechnol. 10, 359–72. [DOI] [PubMed] [Google Scholar]
- 63.Dam J & Schuck P (2004) Calculating sedimentation coefficient distributions by direct modeling of sedimentation velocity concentration profiles, Methods Enzymol. 384, 185–212. [DOI] [PubMed] [Google Scholar]
- 64.Doyle BL, Rauk AP, Weiss WF 4th. & Budyak IL (2020) Quantitation of soluble aggregates by sedimentation velocity analytical ultracentrifugation using an optical alignment system - Aspects of method validation, Anal Biochem. 605, 113837. [DOI] [PubMed] [Google Scholar]
- 65.Zhao H, Brown PH & Schuck P (2011) On the distribution of protein refractive index increments, Biophys J. 100, 2309–17. [DOI] [PMC free article] [PubMed] [Google Scholar]