

Abstract
Background
The network pathophysiology underlying the motor symptoms of Parkinson's disease (PD) is poorly understood. In models of late‐stage PD, there is significant cell‐specific remodeling of corticostriatal, axospinous glutamatergic synapses on principal spiny projection neurons (SPNs). Neurons in the centrolateral nucleus (CLN) of the thalamus that relay cerebellar activity to the striatum also make axospinous synapses on SPNs, but the extent to which they are affected in PD has not been definitively characterized.
Objective
To fill this gap, transgenic mice in which CLN neurons express Cre recombinase were used in conjunction with optogenetic and circuit mapping approaches to determine changes in the CLN projection to SPNs in a unilateral 6‐hydroxydopamine (6‐OHDA) model of late‐stage PD.
Methods
Adeno‐associated virus vectors carrying Cre‐dependent opsin expression constructs were stereotaxically injected into the CLN of Grp‐KH288 mice in which CLN, but not parafascicular nucleus neurons, expressed Cre recombinase. The properties of this projection to identify direct pathway spiny projection neurons (dSPNs) and indirect pathway spiny projection neurons (iSPNs) were then studied in ex vivo brain slices of the dorsolateral striatum from control and 6‐OHDA lesioned mice using anatomic, optogenetic, and electrophysiological approaches.
Results
Optogenetically evoked excitatory synaptic currents in both iSPNs and dSPNs were reduced in lesioned mice; however, the reduction was significantly greater in dSPNs. In iSPNs, the reduction in evoked responses was attributable to synaptic pruning, because synaptic channelrhodopsin assisted circuit mapping (sCRACm) revealed fewer synapses per cell after lesioning. In contrast, sCRACm mapping of CLN inputs to dSPNs failed to detect any change in synapse abundance in lesioned mice. However, the ratio of currents through α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid receptors to those through N‐methyl‐D‐aspartate receptors was significantly reduced in dSPNs. Moreover, the distribution of currents evoked by optical stimulation of individual synapses shifted toward smaller amplitudes by lesioning, suggesting that they had undergone long‐term depression.
Conclusions
Taken together, our results demonstrate that the CLN projection to the striatum undergoes a pathway‐specific remodeling that could contribute to the circuit imbalance thought to drive the hypokinetic features of PD. © 2022 The Authors. Movement Disorders published by Wiley Periodicals LLC on behalf of International Parkinson and Movement Disorder Society
Keywords: thalamus, centrolateral nucleus, parafascicular nucleus, axospinous synapses, Parkinson's disease, optogenetics, synaptic channelrhodopsin assisted circuit mapping
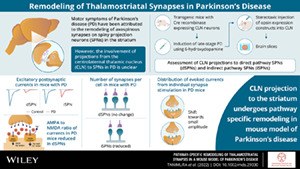
Parkinson's disease (PD) is a neurodegenerative disorder caused by progressive loss of dopaminergic neurons in the substantia nigra pars compacta (SNc) that innervate the basal ganglia. 1 The network pathophysiology underlying the cardinal motor symptoms of PD has long been attributed to an imbalance in the excitability of two striatal efferent projection systems. 2 In this framework, loss of striatal dopaminergic signaling leads to hypo‐excitability of movement promoting direct pathway spiny projection neurons (dSPNs) and hyper‐excitability of movement suppressing indirect pathway spiny projection neurons (iSPNs). Although there is unequivocal evidence that D1 dopamine receptor (D1R) signaling in dSPNs enhances excitability and D2 dopamine receptor (D2R) signaling in iSPNs suppresses excitability, both G‐protein coupled receptors (GPCRs) also regulate long‐term plasticity of corticostriatal, axospinous glutamatergic synapses. 3 To make the situation more complex, homeostatic mechanisms that serve to normalize deviations in neuronal activity undoubtedly are triggered by dopamine depletion. Indeed, following unilateral lesions of the dopaminergic innervation of the striatum, iSPNs decrease their intrinsic excitability and prune axospinous glutamatergic synapses formed by cortical pyramidal neurons, whereas dSPNs increase their intrinsic excitability and maintain glutamatergic synaptic density, at least initially. 4 , 5 , 6 , 7
Although there have been many studies focused on alterations in corticostriatal connectivity in PD models, less attention has been paid to thalamostriatal connectivity, particularly that arising from the intralaminar nuclei. The parafascicular nucleus (PFN) and the centrolateral nucleus (CLN) are the two most prominent intralaminar nuclei to innervate the striatum. PFN glutamatergic neurons are part of the reticular activating system that participates in arousal and the response to salient environmental events. 8 , 9 The PFN projection to the striatum plays a role in controlling reaction times and set shifting with changing environmental contingencies. 10 , 11 Following dopamine depleting lesions, the functional connectivity of PFN neurons with iSPNs is enhanced, contributing to the imbalance in direct and indirect pathways and motor deficits. 12
How the CLN projection to the striatum is altered in PD models is less clear. The CLN receives movement‐related information arising from the parietal cortex and the cerebellum. 13 , 14 , 15 In rodents, CLN lesions and selective disruption of the cerebellar projection to CLN impairs motor performance. 16 , 17 Unlike PFN neurons, glutamatergic CLN neurons make axospinous synapses preferentially on spiny projection neurons (SPNs). 18 Stimulation of CLN axons evoke excitatory postsynaptic currents (EPSCs) that are dominated by α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid receptors (AMPARs). 19 Recently, using mice expressing Cre recombinase under control of the vesicular glutamate transporter type 2 (vGluT2) promoter, Parker et al 20 reported that following dopamine depleting lesions, the functional connectivity of intralaminar thalamic neurons with dSPNs was preferentially reduced, potentially contributing to the imbalance between the direct and indirect pathways. Given our previous work using intersectional genetic strategies to specifically identify PFN neurons, our working hypothesis was that the change in synaptic strength reported by Parker et al 20 reflected a CLN‐specific remodeling. To test this hypothesis, adeno‐associated virus (AAV) vectors carrying Cre‐dependent opsin expression constructs were stereotaxically injected into the CLN of Grp‐KH288 mice in which CLN, but not PFN neurons expressed Cre recombinase. 21 The properties of this projection to identified dSPNs and iSPNs were then studied in ex vivo brain slices from control and 6‐hydroxydopamine (6‐OHDA) lesioned mice using anatomic, optogenetic, and electrophysiological approaches. These studies revealed that CLN connectivity with both dSPNs and iSPNs was remodeled in the parkinsonian state, but that there was a preferential loss of synaptic strength with dSPNs.
Methods
Animals
All experiments were performed in accordance with Northwestern University Institutional Animal Care and Use Committee guidelines. To identify intralaminar CLN neurons, the Grp‐KH288 Cre mouse line (Tg(Grp‐cre)KH288Gsat/Mmucd, MMRRC) was used. 12 , 21 To identify dSPNs and iSPNs, Grp‐KH288 mice were crossed with either the bacterial artificial chromosome (BAC) Drd1‐tdTomato or Drd2‐eGFP lines. 22 Two‐ to 4‐month‐old heterozygous mice were used for all experiments. For cell density analysis, Grp‐KH288 mice were crossed with Ai14 mice, resulting in tdTomato expression by CLN neurons. 23
Stereotaxic Injections
Stereotaxic injections into the striatum and caudal intralaminar thalamic nuclei were performed with the guidance of Angle Two software (Leica Biosystems, Buffalo Grove, IL). Mice were anesthetized with isoflurane. The position of bregma and lambda were provided to the software, and then a glass pipette was slowly inserted through a burr hole to the coordinates calculated by the software. The AAV virus was slowly injected by using a microinjector (NARISHIGE IM300, Japan) or syringe into the CLN; coordinates relative to bregma (in mm) were: lateral, 0.78; posterior, 1.34; depth, 3.00/2.80. Mice were analyzed 4 weeks after virus injection. All mice included in the study had postmortem verification of the viral injection into the CLN.
Unilateral Model of PD
Medial forebrain bundle (MFB) injection of 6‐OHDA was performed as previously described. 5 , 12 Briefly, 3.5 μg of 6‐OHDA HCl was dissolved in 1 μL of sterile saline with 0.02% ascorbic acid; the solution was injected into the MFB at the following coordinates (relative to bregma): lateral, 1.07 mm; posterior, 0.70 mm; and depth, 4.87 mm. Sham‐operated mice underwent identical surgical procedures and were injected 1 μL of a vehicle (saline with 0.02% ascorbic acid). Mice were supplemented with saline injections and high‐fat/high‐sucrose foods as needed based on prior studies. 24 Three to 4 weeks post‐injection, mice were subjected to a forelimb asymmetry test to verify the extent of the lesion. 5 , 25 In brief, mice were placed in a 500 mL glass beaker for 3 minutes (mice were not acclimated to the testing room or beaker) and the number of ipsilateral and contralateral (to lesioned hemisphere) forelimb weight‐bearing paw contacts were counted. Subjects were eliminated from the experimental group if they had more than 40% contralateral paw contacts. All experiments were conducted 3 to 4 weeks post‐injection. At this point, over 90% of striatal tyrosine hydroxylase (TH) immunoreactivity has typically been lost. 26
Slice Preparation
Mice were anaesthetized with a mixture of ketamine (50.0 mg/kg) and xylazine (4.5 mg/kg), and transcardially perfused with a modified artificial CSF (ACSF) containing (in mM): 124.0 NaCl, 3.0 KCl, 26.0 NaHCO3, 1.0 NaH2PO4, 1.0 CaCl2, 2.0 MgCl2, 16.6 glucose, and equilibrated with 95% oxygen and 5% CO2. Parasagittal slices (250 μm thick) were prepared using a vibratome (Leica VT 1200, Leica Biosystems, Buffalo Grove, IL) in ice‐cold low‐calcium ACSF. Slices were incubated at 34°C for 30 minutes in modified ACSF containing 2 mM CaCl2 and 1 mM MgCl2 and then transferred to a holding chamber at room temperature.
Electrophysiology
A recording chamber mounted on an Olympus BX 50 WI (Olympus, Japan) was filled with oxygenated modified ACSF containing 2 mM CaCl2 and 1 mM MgCl2 with 10 μM SR 95531 hydrobromide (GABAzine) and 2 μM CGP55845 hydrochloride (CGP) to block GABAA and GABAB receptors, respectively. Patch pipettes were pulled from thick‐walled borosilicate glass pipettes on a Sutter P‐1000 (Sutter instrument, Novato, CA). Pipette resistance was typically 4 to 5 MΩ when filled with an internal solution. The internal solution for voltage‐clamp recording contained (in mM): 120 CsMeSO3, 15 CsCl, 8 NaCl, 10 TEA‐Cl, 10 HEPES, 3 QX134‐Cl, 0.2 EGTA, 2 Mg‐ATP, and 0.3 Na‐GTP (pH 7.35 with CsOH). SPNs in the dorsolateral striatum were identified using infrared differential interference contrast on an upright Olympus microscope with a 60×/0.90 NA water‐dipping objective and a cooled CCD camera (CoolSnap HQ, Photometrics, Tucson, ARI) controlled with Metamorph software (Molecular Device, San Jose, CA). tdTomato and eGFP expressing dSPNs and iSPNs (respectively) were identified using epifluorescence microscopy. For dual patch recording, dSPN and iSPN pairs were within 70 μm of each other and at a similar depth from the slice surface. Recordings were obtained using a MultiClamp 700A amplifier and pClamp 10 software (Molecular Devices, San Jose, CA). Data were acquired at 100 kHz, filtered at 10 kHz using an 8‐pole Bessel filter, and digitized using a DigiData 1440 16‐bit A/D converter. Somatic access resistance <20 MΩ was monitored and cells with unstable access resistance (>20% change) were discarded. Cells were held at −70 mV. Membrane potentials were not adjusted for the liquid junction potential. All compounds were purchased from TOCRIS and SIGMA.
Optogenetics (Single Spot)
To express opsins selectively in CLN neurons, AAV9‐hSyn‐DIO‐ChR2(H134R)‐EYFP‐WRE‐pA, 2.47E+13 vg/mL (Virovek, Hayward, CA) or AAV9‐EF1a‐DIO‐Chronos‐eGFP vectors, 1.6E+13 vg/mL (Penn vector core, Philadelphia, PA) were stereotaxically injected into the thalamus (see above). To activate opsin‐containing containing axons in the dorsolateral striatum, a 473‐nm laser‐based photo‐stimulation system with mechanical shutter and electro‐optical modulator was used. 27 The 5×/0.15 NA objective was used with ~700 μm diameter illumination spot size. The sample power values used were: 0.98, 2.04, 4.41, 5, or 8.35 mW. Three to five optically evoked EPSCs were averaged, with a 5‐ms pulse duration and 30 to 50 seconds inter‐stimulus interval.
CLN Input Mapping
A modified version of the synaptic channelrhodopsin assisted circuit mapping (sCRACm) method 28 was created using custom integration software (WinFluor, John Dempster) added onto a Prairie Ultima‐Photolysis (Prairie Technologies, Middleton, WI) two‐photon laser scanning microscope to map the functional connectivity of CLN neurons with SPNs. 12 dSPNs and iSPNs were identified by somatic tdTomato or eGFP fluorescence using the 2PLSM system 4 with 950 nm from a Chameleon Ultra‐1 laser (Coherent, Mountain View, CA). Cells were patched using video microscopy with a Hitachi CCD camera and an Olympus 40×/0.8 NA water‐dipping lens. Whole‐cell patch‐clamp recordings were performed with Cs‐based internal containing Alexa Fluor 568: (in mM) 120 CsMeSO3, 15 CsCl, 8 NaCl, 10 TEA‐Cl, 10 HEPES, 3 QX134‐Cl, 0.2 EGTA, 2 Mg‐ATP, 0.3 Na‐GTP, and 0.05 Alexa Fluor 568 hydrazide sodium salt (Thermo‐Fisher Scientific, Waltham, MA) (pH was adjusted to 7.35 with CsOH) for dendritic visualization. A total of 1 μM tetrodotoxin (TTX), 100 μM 4‐aminopyridine (4 AP), 10 μM GABAzine, 2 μM CGP, and 50 μM D‐AP5 were added to the normal ACSF. Cells were held at −70 mV. Next, under computer control, a 473 nm laser beam (Aurora launch, Prairie Technologies) was flashed (1 ms duration, 1 second inter‐spot interval) through a 20×/0.40 NA water‐dipping lens (600 × 600 μm field of view with zoom = 1) at a predefined series of spots along a set of six concentric circles (35 μm radial spacing; 420 μm max diameter) spanning the dendritic tree while continuously monitoring somatic EPSCs. The blue laser was directed to each spot in a predetermined, spatially separated order (83 total spots; 4, 11, 11, 15, 19, and 23 per ring). Each full cycle, and therefore, each spot, was stimulated 25 to 30 times per cell. The sample power was held to ~0.1 mW, where the half‐amplitude spot at the plane of the cell in the slice (~50–70 μm below the surface) was estimated to be 23 μm diameter. Events evoked within 50 ms of the stimulus and with an amplitude >5 SDs above the distribution of points in the preceding 100 ms were counted. At the end of the experiments, Alexa Fluor 568 signals were acquired using 810 nm excitation to create a Z‐stack of the cell (with 0.36 μm2 pixels, 10 μs dwell time, and 1 μm z‐steps) and used to construct a maximum projection image. Locations where stimuli evoked a response at least 10% of the time were mapped onto this image.
Imaging
Coronal or sagittal slices (100–250 μm) were mounted onto glass slides. Images were acquired on an automated confocal microscope (FV 10i, Olympus, Japan) with 10×/0.4NA lens. Images were adjusted in ImageJ (NIH, Bethesda, MD) for brightness, contrast, and pseudo‐coloring.
Estimation of CLN Neuronal Density
Coronal CL containing brain slices (50‐μm thickness) were obtained from fixed brains of Grp‐KH288/Ai14 mice using a vibratome (Leica VT 1200, Leica Biosystems, Buffalo Grove, IL). Brain slices were washed with phosphate‐buffered saline (PBS), then incubated with PBS containing DAPI (NucBlue Fixed cell stain, Thermo‐Fisher Scientific, Waltham, MA) for 20 minutes. Single plane images were acquired on a confocal microscope (FV 10i‐DUC, Olympus, Japan) with 60×/1.35NA lens. Two randomly placed regions of interest (ROIs) were positioned in the CLN in each section that was quantified. The optical fractionator method and unbiased counting rules are used to estimate the number of Ai14 positive cells in each ROI and in the CLN. 26 , 29 tdTomato (Ai14 reporter) and DAPI‐positive neurons were counted in ImageJ (NIH) using the cell‐counting tool.
Data Analysis
Data analysis was performed with Clampfit 10.3 (Molecular Devices, San Jose, CA), Igor Pro 5.0 (WaveMetrics, Lake Oswego, OR), or MATLAB R2017b (The MathWorks, Natick, MA). Statistical analyses were performed using MATLAB. Experimenters were not blinded to the experimental condition (6‐OHDA lesioning). Sample sizes were based on previous studies using similar approaches. 12 , 27 Data were represented as median and first and third quartiles. Box and whisker plots show median, first/third quartiles, and maximum/minimum values. Statistical evaluations were performed using the nonparametric rank‐sum test for independent samples. For analysis of cumulative probability, the non‐parametric Kolmogorov–Smirnov (KS) test was used.
Results
CLN Neurons Were Not Lost in 6‐OHDA Lesioned Mice
Postmortem analysis of the brains of PD patients has revealed that intralaminar neurons, including those in the CLN, are lost. 30 , 31 , 32 However, this has not been found in rodent 6‐OHDA models of PD. 12 , 20 , 33 , 34 To specifically determine whether CLN neurons were lost in our mouse 6‐OHDA model, Grp‐KH288 mice were crossed into the Ai14 reporter line, in which tdTomato expression is controlled by Cre recombinase, 21 and then, unilaterally lesioned. Three to 4 weeks later, mice were sacrificed and brain sections through the CLN were studied using confocal microscopy and stereological methods (see Methods). The number of tdTomato‐positive CLN neurons was indistinguishable in control and lesioned mice (Fig. 1D). In addition to confirming previous work, these experiments suggest that any alteration in CLN‐evoked responses was not attributable to neurodegeneration.
FIG 1.
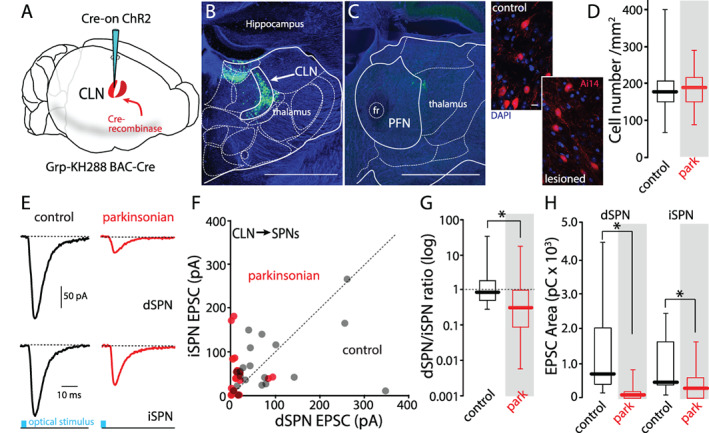
CLN‐evoked responses were smaller in both iSPNs and dSPNs from parkinsonian mice. (A) Schematic diagram of injection scheme. Cre‐on ChR2 was injected into the CLN of the Grp‐KH288 Cre mouse. (B) Confocal image of the coronal slice containing CLN from a D1‐tdTomato/Grp‐KH288 Cre mouse injected with Cre‐on ChR2. ChR2‐eYFP was expressed by CLN neurons. (C) A confocal image of the coronal slice containing PFN from the same brain is shown in (B). Scale bars: 1 mm. (D) Left: confocal images showing tdTomato‐positive CLN neurons in control and 6‐OHDA lesioned mice. Right: box plots of the density of tdTomato‐positive neurons in CLN. Control (N = 4), 6‐OHDA (N = 5). Control median value: 117, parkinsonian median value: 189. Differences between groups were not significant (P = 0.531, rank‐sum test). Scale bar: 10 μm. (E–H) Dual whole‐cell patch patch‐clamp recording from a dSPN and an iSPN. CLN axons were activated by large area optical stimulation. (E) Representative traces showing CLN‐EPSCs evoked by laser stimulation (8.35 mW, 5 ms). (F) Scatter plots of peak amplitudes of paired dSPN and iSPN CLN‐ EPSCs from control (black) and parkinsonian (red) mice. Filled black circles are control mice (n = 17 pairs, N = 10) and filled red circles are parkinsonian mice (n = 21 pairs, N = 8). (G) Box plots of log [dSPN/iSPN] ratios from scatter plot in (F). Median dSPN/iSPN ratios; control: 0.88, parkinsonian: 0.33. Control vs. parkinsonian; P = 0.022, rank‐sum test. (H) Box plots of EPSC areas from scatter plots in (F). Median charge in dSPN (pC); control: 714.41, parkinsonian: 97.92, control vs. parkinsonian; P < 0.001, rank‐sum test. Median charge in iSPN (pC); control: 467.46, parkinsonian: 297.54. Control vs. parkinsonian; P = 0.034, rank‐sum test. [Color figure can be viewed at wileyonlinelibrary.com]
CLN‐evoked Synaptic Responses in Both iSPNs and dSPNs Were Reduced in Lesioned Mice
To study CLN connectivity with identified dSPNs and iSPNs, Grp‐KH288 transgenic mice were crossed with mice expressing tdTomato under control of the Drd1a promoter or enhanced green fluorescent protein (eGFP) under control of the Drd2 promoter. 22 , 35 Stereotaxic thalamic injection of an AAV vector carrying a Cre‐dependent channelrhodopsin 2 (ChR2) expression construct into this crossed line led to the selective expression of ChR2 in CLN neurons (Fig. 1A–C). Although ChR2 expression often extended into the lateral habenula (LH) from the CLN, LH neurons do not project to the dorsal striatum (Allen Brain Atlas; https://portal.brain-map.org). In ex vivo brain slices from these mice, full‐field light pulses (8.35 mW, 5 ms duration) evoked EPSCs in both iSPNs and dSPNs (Fig. 1E,F). In slices from control mice, optically evoked EPSCs in neighboring iSPNs and dSPNs were similar in amplitude (Fig. 1E–G). The ratios of EPSC amplitudes in dSPNs and nearby iSPNs were near 1 (Fig. 1F,G). However, in slices from mice, 3 to 4 weeks after near‐complete unilateral 6‐OHDA lesions of nigrostriatal dopaminergic fibers, the amplitude and area of CLN‐evoked EPSCs were smaller in both iSPNs and dSPNs (Fig. 1E,F,H). However, the reduction in EPSC amplitude was more pronounced in dSPNs and the amplitude ratio for neighboring neurons (dSPN/iSPN) fell significantly below 1 (Fig. 1G).
The Reduction of CLN‐Evoked Responses in iSPNs Was Attributable to Spine Pruning
To determine the mechanisms mediating the change in EPSC amplitude in lesioned mice, physiological approaches were used. At the outset, iSPNs were studied. First, the relationship between optical stimulus intensity and evoked EPSCs was determined. In iSPNs from lesioned mice, CLN‐evoked EPSCs were smaller across the full spectrum of light intensities (Fig. 2A). Moreover, the ratio of AMPAR currents to NMDAR currents was not significantly different in control and lesioned iSPNs, suggesting that conventional forms of postsynaptic depression were not responsible for the shift (Fig. 2B). That said, the variance in these estimates was large and a small difference between groups could have been missed because of our limited sample size.
FIG 2.
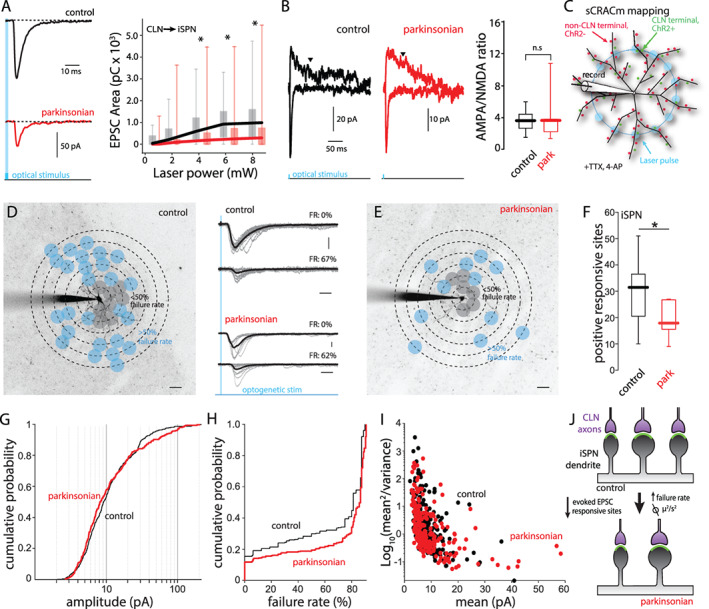
The reduction of CLN‐evoked responses on iSPN was attributable to synaptic pruning. (A) Input–output curves of CLN‐evoked EPSCs in iSPNs from control (black) and parkinsonian (red) mice. Left: Representative traces of CLN‐evoked EPSCs. Right: CLN‐EPSC areas (pC) are plotted against laser intensity. Control vs. parkinsonian (laser power [mW]): P = 0.033 (8.35), P = 0.023 (6.25), P = 0.026 (4.41), P = 0.089 (2.04), P = 0.077 (0.98), rank‐ sum test. Control: n = 20, N = 9. Parkinsonian: n = 26, N = 10. (B) AMPA/NMDA ratios of CLN synapses on iSPNs from control and parkinsonian mice. Left: representative traces showing CLN‐EPSCs evoked by large area stimulation. AMPA EPSCs were measured by peak amplitude at −70 mV. NMDA EPSCs were measured at +40 mV. Downward arrowheads indicate the time point (200 ms from stimulation) when NMDA EPSCs were measured. Right: box plots of AMPA/NMDA ratios of CLN synapses on iSPN from control and parkinsonian mice. Control vs. parkinsonian; P = 0.6943, rank‐sum test. Control: n = 8, N = 4. Parkinsonian: n = 7, N = 4. (C) Schematic diagram of sCRACm CLN mapping. The diagram shows the example of blue laser stimulation on a middle concentric circle. (D and E) Example CLN input mappings from iSPNs of control (D) and parkinsonian (E). Projection image superimposed by mapping grid. Gray dots indicate responsive sites with <50% failure rates. Blue dots indicate responsive sites with 51% < failure rate < 90%. Scale bar; 20 μm. Middle: example traces with failure rate (FR). Gray thin lines are evoked responses at a typical stimulation spot. Thick black lines are averages of the individual traces. Scale bar: 20 pA and 20 ms. (F) Box plots of responsive sites in iSPN from control and parkinsonian mice. Control median value; 31.5, parkinsonian median value; 19.5. Control vs. parkinsonian; P = 0.0258, rank‐sum test. Control: n = 14, N = 12, parkinsonian: n = 14, N = 8. (G) Cumulative probability of CLN‐iSPN EPSC peak amplitudes obtained from input mapping experiments. Control vs. parkinsonian; P = 0.2634, KS test. Control: event number: n = 469, parkinsonian: event number: n = 298. (H) Cumulative probability of failure rates of CLN‐iSPN EPSC. P < 0.001, KS test. Control: event number: n = 913, parkinsonian: event number: n = 1162. (I) Variance plots of CLN‐iSPN EPSC. The events with over 50% failure rates were analyzed. Control: event number: n = 234, parkinsonian: event number: n = 232. (J) Summary diagram of the change of CLN synapses on iSPN. [Color figure can be viewed at wileyonlinelibrary.com]
To better understand why CLN‐evoked currents were reduced in parkinsonian mice, a variant of the sCRACm approach was used (Fig. 2C). 28 This approach provides information about not only the spatial distribution of synapses, but also their relative strength. 5 , 12 Because previous work had shown 6‐OHDA lesioning led to pruning of iSPN corticostriatal synapses, 4 , 5 , 36 it was reasonable to hypothesize that the same was true for CLN axospinous synapses. Indeed, sCRACm assessment of CLN connectivity with iSPNs revealed a significant loss of functional connections following lesioning (Fig. 2D–F). The distribution of individual CLN‐evoked EPSC amplitudes was not significantly different in cells from control and lesioned mice (Fig. 2G), although it did appear that a subset of synapses increased in strength in lesioned mice, as reported for corticostriatal synapses. 5 An examination of failure rates at sites with at least a 10% response rate revealed that in lesioned mice, failure rates rose in iSPNs (Fig. 2H). The simplest interpretation of this result is that pruning of synapses led to reduction in the number of stimulated synapses at many sites, leading to increased failure rates at those sites. This effect should have been prominent at proximal synapses where the failure rate was <50% and evoked responses were commonly not unitary in appearance (eg, Fig. 2D bottom right). To provide an independent assessment of whether lesioning reduced CLN‐evoked responses in iSPNs through a presynaptic mechanism, variance analysis was used. 37 A reduction in presynaptic release probability or the number of vesicular release sites results in a center of mass shift toward the origin in plots of mean2/variance against mean EPSC amplitude. To focus this analysis on sites where it was likely that there was a single synapse, it was restricted to sites with failure rates between 50% and 90%, which is in the range found at single synapses in the brain. 38 , 39 There was no discernible shift in the distribution of CLN synaptic responses in iSPNs following 6‐OHDA lesioning (Fig. 2I). The lack of change in AMPAR/NMDAR current ratio, or the distribution of EPSC amplitudes, or in the relationship between mean EPSC amplitude and EPSC variance together with the reduction in responsive sites in sCRACm experiments clearly points to synaptic pruning as being responsible for the drop in CLN‐evoked responses in iSPNs (Fig. 2J).
The Reduction of CLN‐Evoked Responses in dSPNs Was Attributable to a Postsynaptic Depression
As with iSPNs, examination of the relationship between stimulus power and evoked EPSCs revealed a reduction in synaptic strength that was independent of light intensity (Fig. 3A). However, in contrast to the situation in iSPNs, the ratio of CLN‐evoked AMPAR currents to NMDAR currents was significantly smaller in dSPNs from lesioned mice (Fig. 3B), pointing to a postsynaptic depression. 40 Consistent with this inference, CLN‐evoked paired‐pulse ratios were unchanged in dSPNs from lesioned mice (Fig. 3C,D). To determine if the number of synapses changed following lesioning, the sCRACm approach was used. Again, unlike the situation with iSPNs, there was no discernible change in the number or distribution of CLN synapses on dSPNs from lesioned mice (Fig. 3E–G).
FIG 3.
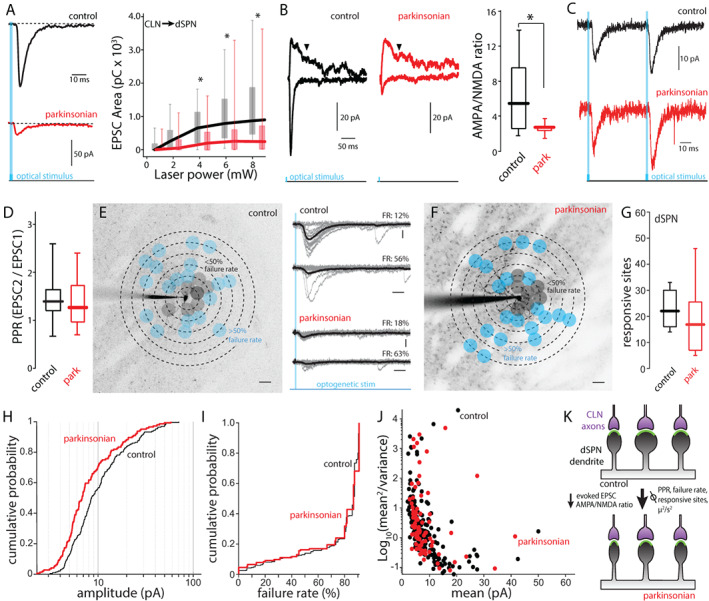
The reduction of CLN‐evoked responses on dSPN was attributable to postsynaptic mechanisms. (A) Input–output curves of CLN‐evoked EPSCs in dSPNs from control (black) and parkinsonian (red) mice. Left: Representative traces of CLN‐evoked EPSCs. Right: CLN‐EPSC areas (pC) are plotted against laser intensity. Control vs. parkinsonian (laser power [mW]): P < 0.001 (8.35), P < 0.001 (6.25), P = 0.006 (4.41), P = 0.293 (2.04), P = 0.438 (0.98), rank‐sum test. Control: n = 30, N = 14. Parkinsonian: n = 25, N = 9. (B) AMPA/NMDA ratios of CLN synapses on dSPNs from control and parkinsonian mice. Left: representative traces showing CLN‐EPSCs evoked by whole field stimulation. AMPA EPSCs were measured by peak amplitude at −70 mV. NMDA EPSCs were measured at +40 mV. Downward arrowheads indicate the time point (200 ms from stimulation) when NMDA EPSCs were measured. Right: box plots of AMPA/NMDA ratios of CL synapses on dSPN from control and parkinsonian mice. Control vs. parkinsonian; P = 0.0275, rank‐sum test. Control: n = 11, N = 8. Parkinsonian: n = 5, N = 5. (C and D) Paired‐pulse ratios of CLN‐evoked EPSCs in dSPNs. (C) Representative traces of CLN‐dSPN EPSC when CL axons are optically stimulated with 50 ms interval. (D) Box plots of paired‐pulse ratios at 20 Hz stimulation. P = 0.633, rank‐sum test. Control: n = 18, N = 11, parkinsonian: n = 11, N = 8. (E and F) Examples of CLN input mappings from dSPNs of control (E) and parkinsonian (F). Projection image superimposed by mapping grid. Gray dots indicate responsive sites with <50% failure rates. Blue dots indicate responsive sites with 51% < failure rate < 90%. Scale bar; 20 μm. Middle: example traces with failure rate (FR). Gray thin lines are evoked responses at a typical stimulation spot. Thick black lines are averages of the individual traces. Scale bar: 10 pA and 20 ms. (G) Box plots of the number of responsive sites in dSPN from control and parkinsonian mice. Control median value; 22, parkinsonian median value; 17. Control vs. parkinsonian; P = 0.2162, rank‐sum test. Control: n = 12, N = 7, parkinsonian: n = 8, N = 6. (H) Cumulative probability of CLN‐dSPN EPSC peak amplitudes obtained from input mapping experiments. Control vs. parkinsonian; P < 0.001, KS test. Control: event number: n = 192, parkinsonian: event number: n = 144. (I) Cumulative probability of failure rates of CLN‐dSPN EPSC. P = 0.066, KS test. Control: event number: n = 286, parkinsonian: event number: n = 154. (J) Variance plots of CLN‐dSPN EPSC. The events with over 50% failure rates were analyzed. Control: event number: n = 193, parkinsonian: event number: n = 89. (K) Summary diagram of the change of synaptic strength after 6‐OHDA lesion. [Color figure can be viewed at wileyonlinelibrary.com]
Two other observations were consistent with a postsynaptic mechanism being responsible for the reduction in CLN‐evoked responses in dSPNs from lesioned mice. First, the distribution of CLN‐evoked EPSC amplitudes in the sCRACm experiments shifted toward smaller amplitudes after lesioning (Fig. 3H). Second, there was no difference in failure rates of CLN synapses between groups (Fig. 3I) and the mean2/variance versus mean EPSC amplitude plot did not shift in dSPNs from parkinsonian mice (Fig. 3J). Taken together, these results are consistent with the hypothesis that CLN synapses are retained in the month after 6‐OHDA lesioning, but that they undergo a form of postsynaptic depression (Fig. 3K).
Discussion
There are two main conclusions to be drawn from the described studies. First, following a near‐complete, unilateral 6‐OHDA lesion of the dopaminergic innervation of the dorsolateral striatum, the responsiveness of SPNs to activation of the CLN projection falls; however, this attenuation is more profound in dSPNs than neighboring iSPNs. Second, the mechanisms responsible for the attenuation of CLN responses differed in iSPNs and dSPNs. In iSPNs, a significant fraction of CLN axospinous synapses were eliminated, leading to a drop in functional connectivity with lesioning. In dSPNs, there was no discernible change in the number of CLN synapses, but the average strength of individual synapses fell, leading to a profound reduction in overall responsiveness to CLN activity. Taken together, our results are consistent with the conclusion that alterations in the thalamostriatal circuitry following dopamine depleting lesions contribute to the imbalance in the activity of dSPNs and iSPNs, which is thought to underlie the hypokinetic features of PD. 2
Unilateral 6‐OHDA Lesioning Did Not Induce CLN Degeneration
In contrast to what has been found in postmortem analysis of PD brains and brains of MPTP‐treated primates, 30 , 31 , 32 , 41 there was no discernible loss of CLN neurons following unilateral, 6‐OHDA lesioning of dopaminergic neurons in mice, in agreement with previous studies in rodents. 20 , 42 Although neuronal loss in PD is widespread and attributable to factors other than dopamine depletion (eg, α‐synuclein toxicity and bioenergetic stress), 43 the lack of alignment with the primate MPTP literature is somewhat puzzling, because both this model and the 6‐OHDA model rely on a toxin that targets dopaminergic neurons. The difference may stem from the fact that MPTP is systemically administered (leading to damage that is more widespread) or that the assessment of cell loss in the primate models is made many months after toxin administration. Dopamine depletion induces a cascade of events, which may take months to stabilize, even in rodents. 44 For example, in rodents, a reduction in dSPN spine density is not evident 1 month after MFB 6‐OHDA lesioning, but is discernible 2 to 3 months after the lesion, 45 as in primates following MPTP treatment. 46 Therefore, it is possible that with longer survival times, CLN neurons might be lost in the rodent PD model. Regardless, at the time point examined, the change in functional connectivity of SPNs cannot be attributed to CLN degeneration.
Dopamine Depletion Induced Cell Type‐Specific Remodeling of CLN Connectivity with SPNs
Previous work has shown that lesioning dopaminergic neurons triggers a complex set of synaptic adaptations in SPNs. For example, following near complete striatal dopamine depletion following 6‐OHDA lesions, the dendritic arbors of both iSPNs and dSPNs are modestly reduced in size. 5 Although this adaptation may account in part for the reduction in SPN responsiveness to optogenetic stimulation of CLN axons following lesioning, it does not account for other changes in CLN‐evoked synaptic events, including differences those evoked in iSPNs and dSPNs.
Perhaps the most prominent cell‐type‐specific striatal adaptation seen following dopamine depletion is the pruning of iSPN corticostriatal, axospinous synapses. 4 , 5 , 6 , 7 , 36 This can be viewed as a form of adaptive, homeostatic plasticity engaged by the disinhibition of iSPNs following the loss of tonic D2R signaling. 5 , 47 Our sCRACm experiments suggest that a similar percentage (30%–50%) of CLN axospinous synapses on iSPNs were eliminated after depletion. The inference that elimination of synapses was responsible for the reduction in CLN‐evoked EPSCs in iSPNs, as opposed to presynaptic or postsynaptic depression, is also consistent with the lack of change AMPAR/NMDAR current ratio, or in the CLN EPSC amplitude distribution or in the relationship between mean EPSC amplitude and current variance. The one caveat to this conclusion was the modest increase in failure rates seen with spot illumination in the sCRACm experiments. Although an increase in failure rates is consistent with presynaptic depression, it also could be a consequence of synaptic pruning, which resulted in fewer sites of stimulation where more than one synapse was activated.
In contrast to iSPNs, there was no discernible loss of CLN synapses on dSPNs following dopamine depletion. However, the functional strength of the pathway declined more dramatically than did the CLN projection to iSPNs. This decline was attributable to a reduction in the postsynaptic strength of individual CLN synaptic connections with dSPNs. In agreement with previous work, 20 this reduction appeared to be the consequence of depletion‐triggered postsynaptic depression, because the ratio of AMPAR to NMDAR currents at CLN synapses declined and the amplitude distribution of sCRACm EPSCs shifted toward smaller amplitudes, but paired‐pulse ratio, failure rates, and the relationship between mean EPSC amplitude and variance did not change. These changes mirror those found at dSPN corticostriatal synapses in PD models. 5 At dSPN corticostriatal synapses, the depletion‐induced long‐term depression (LTD) of synaptic transmission is attributable to the loss of D1R signaling and the elevation of depression‐promoting M4 muscarinic receptor (M4R) signaling. 48 However, whether this mechanism is responsible for the change at CLN synapses is uncertain, particularly given the absence of M4R modulated endocannabinoid‐dependent LTD at this synapse. 49 Another form of LTD, which is expressed at thalamostriatal synapses, is dependent on nitric oxide (NO) stimulation of cyclic guanosine monophosphate (cGMP) signaling in SPNs. 50 Interestingly, the NO in this pathway is generated by a class of striatal GABAergic interneurons that are not responsive to thalamic stimulation, 51 making CLN stimulation alone an ineffective strategy for inducing it. 19 Clearly, additional studies are needed to sort out the mechanisms controlling the plasticity at this synapse and how it is affected by dopamine depletion.
The Contribution of Plasticity at CLN Synapses to Striatal Pathophysiology in PD
Our studies are consistent with the proposition that remodeling of the thalamic intralaminar projection to SPNs contributes to the imbalance in striatal efferent pathways thought to underlie movement deficits in the classic model of late‐stage PD. 2 That is, loss of striatal dopaminergic signaling leads to hypo‐excitability of movement promoting dSPNs and hyper‐excitability of movement suppressing iSPNs. Previous work targeting both the PFN and CLN using vGlut2‐Cre mice suggested depression of excitatory thalamostriatal synapses on dSPNs contributed to this imbalance and found that that chemogenetic or optogenetic suppression of thalamic activity alleviated open‐field deficits produced by unilateral 6‐OHDA lesions. 20 Subsequent work using mouse intersectional genetics to isolate the PFN projection to the striatum failed to see any evidence that axodendritic PFN synapses on iSPNs or dSPNs were altered following 6‐OHDA lesioning, but did find enhanced PFN coupling with striatal cholinergic interneurons (ChIs) 12 —interneurons long implicated in the imbalance in striatal efferent pathway imbalance and motor symptoms of PD. 52 Complementing the well‐described muscarinic acetylcholine receptor mechanisms, ChIs were also found selectively boosted glutamate release by PFN synapses on iSPNs through presynaptic nicotinic acetylcholine receptors. In both the Parker et al20 and Tanimura et al12 studies, suppressing the activity of thalamic neurons alleviated the motor deficits accompanying unilateral 6‐OHDA lesioning, but it is likely that this effect stemmed from the ability of PFN neurons to drive ChI spiking, rather than by directly altering the activity of iSPNs or dSPNs.
The discovery that CLN connectivity with SPNs changes in a way that mirrors that seen at corticostriatal axospinous synapses, but not that at PFN synapses, is potentially an important piece in the puzzle of the network mechanisms underlying PD motor symptoms. The CLN is a bridge between the striatum and cerebellum. Our results suggest that this bridge is disrupted in PD, particularly the component linked to dSPNs (and movement initiation). This disruption could mimic lesioning the CLN or its input, both of which impair motor performance. 16 , 17 It also is tempting to speculate that the cerebellar hyperactivity reported in PD patients might be caused, at least in part, by this functional disconnection. 53 Last, given the parallels between the effects of dopamine depleting lesions on CLN and cortical synapses on SPNs, it will be of considerable interest to determine how levodopa therapy remodels the functional connectivity of CLN with the striatum given the apparent importance of re‐established axospinous synapses to the emergence of levodopa‐induced dyskinesia. 54
Author Roles
(1) Research project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript: A. Writing of the First Draft, B. Review and Critique.
A.T.: 1A‐C, 2A‐C, 3A‐B
W.S.: 1A, 3B
D.W.: 1B, 3B
D.J.S.: 1A, 2C, 3A‐B
Financial Disclosures of All Authors (for the Preceding 12 Months)
Financial support provided by grants from JPB Foundation, Flanagan Foundation, MJFF, Bumpus Foundation, CHDI Foundation, US Department of Defense, National Institutes of Health.
Supporting information
Appendix S1 Supporting Information.
Acknowledgments
Dr. John Dempster provided and supported the WinFluor ephys integration software with Nicholas Schwarz adding the PhotoStimulusEditor module to enable the sCRACM experiments. This work was supported by grants from the JPB Foundation and National Institutes of Health (NS034696).
Relevant conflicts of interest/financial disclosures: The authors report no financial or commercial relationships that could create a conflict of interest with respect to this work.
Funding agencies: National Institutes of Health (NS034696), JPB Foundation, Flanagan Foundation.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J, et al. Parkinson disease. Nat Rev Dis Primers 2017;3:17013 [DOI] [PubMed] [Google Scholar]
- 2. Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci 1989;12(10):366–375. [DOI] [PubMed] [Google Scholar]
- 3. Gerfen CR, Surmeier DJ. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci 2011;34(1):441–466. Available from:. http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=21469956&retmode=ref&cmd=prlinks [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Day M, Wang Z, Ding J, An X, Ingham CA, Shering AF, et al. Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models. Nat Neurosci 2006;9(2):251–259. Available from:. http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=16415865&retmode=ref&cmd=prlinks [DOI] [PubMed] [Google Scholar]
- 5. Fieblinger T, Graves SM, Sebel LE, Alcacer C, Plotkin JL, Gertler TS, et al. Cell type‐specific plasticity of striatal projection neurons in parkinsonism and L‐DOPA‐induced dyskinesia. Nat Commun 2014;5(1):5316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schuster S, Doudnikoff E, Rylander D, Berthet A, Aubert I, Ittrich C, et al. Antagonizing L‐type Ca2+ channel reduces development of abnormal involuntary movement in the rat model of L‐3,4‐dihydroxyphenylalanine‐induced dyskinesia. Biol Psychiatry 2009;65(6):518–526. [DOI] [PubMed] [Google Scholar]
- 7. Nishijima H, Suzuki S, Kon T, Funamizu Y, Ueno T, Haga R, et al. Morphologic changes of dendritic spines of striatal neurons in the levodopa‐induced dyskinesia model. Mov Disord Off J Mov Disord Soc 2014;29(3):336–343. [DOI] [PubMed] [Google Scholar]
- 8. Lacey CJ, Bolam JP, Novel MPJ. Distinct operational principles of intralaminar thalamic neurons and their striatal projections. J Neurosci 2007;27(16):4374–4384. Available from:. http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=17442822&retmode=ref&cmd=prlinks [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Smith Y, Galvan A, Ellender TJ, Doig N, Villalba RM, Huerta‐Ocampo I, et al. The thalamostriatal system in normal and diseased states. Front Syst Neurosci 2014;8:5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Aoki S, Liu AW, Zucca A, Zucca S, Wickens JR. Role of striatal cholinergic interneurons in set‐shifting in the rat. J Neurosci Off J Soc Neurosci 2015;35(25):9424–9431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bradfield LA, Bertran‐Gonzalez J, Chieng B, Balleine BW. The thalamostriatal pathway and cholinergic control of goal‐directed action: interlacing new with existing learning in the striatum. Neuron 2013;79(1):153–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tanimura A, Du Y, Kondapalli J, Wokosin DL, Surmeier DJ. Cholinergic interneurons amplify Thalamostriatal excitation of striatal indirect pathway neurons in Parkinson's disease models. Neuron 2019;101(3):444–458. [DOI] [PubMed] [Google Scholar]
- 13. Akert K, Monakow KH. Relationships of precentral premotor and prefrontal cortex to the mediodorsal and intralaminar nuclei of the monkey thalamus. Acta Neurobiol Exp 1980;40(1):7–25. [PubMed] [Google Scholar]
- 14. Angaut P, Cicirata F, Serapide F. Topographic organization of the cerebellothalamic projections in the rat. An autoradiographic study. Neuroscience 1985;15(2):389–401. [DOI] [PubMed] [Google Scholar]
- 15. Aumann TD, Rawson JA, Finkelstein DI, Horne MK. Projections from the lateral and interposed cerebellar nuclei to the thalamus of the rat: a light and electron microscopic study using single and double anterograde labelling. J Comp Neurol 1994;349(2):165–181. [DOI] [PubMed] [Google Scholar]
- 16. Jeljeli M, Strazielle C, Caston J, Lalonde R. Effects of centrolateral or medial thalamic lesions on motor coordination and spatial orientation in rats. Neurosci Res 2000;38(2):155–164. [DOI] [PubMed] [Google Scholar]
- 17. Sakayori N, Kato S, Sugawara M, Setogawa S, Fukushima H, Ishikawa R, et al. Motor skills mediated through cerebellothalamic tracts projecting to the central lateral nucleus. Mol Brain 2019;12(1):13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Smith Y, Raju DV, Pare J‐F, Sidibe M. The thalamostriatal system: a highly specific network of the basal ganglia circuitry. Trends Neurosci 2004;27(9):520–527. Available from:. http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=15331233&retmode=ref&cmd=prlinks [DOI] [PubMed] [Google Scholar]
- 19. Ellender TJ, Harwood J, Kosillo P, Capogna M, Bolam JP. Heterogeneous properties of central lateral and parafascicular thalamic synapses in the striatum. J Physiol 2013;591(Pt 1):257–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Parker PRL, Lalive AL, Kreitzer AC. Pathway‐specific remodeling of thalamostriatal synapses in parkinsonian mice. Neuron 2016;89(4):734–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gerfen CR, Paletzki R, Heintz N. GENSAT BAC Cre‐recombinase driver lines to study the functional organization of cerebral cortical and basal ganglia circuits. Neuron 2013;80(6):1368–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, et al. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nat Cell Biol 2003;425(6961):917–925. Available from:. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=14586460 [DOI] [PubMed] [Google Scholar]
- 23. Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, et al. A robust and high‐throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 2009;13(1):133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Francardo V, Recchia A, Popovic N, Andersson D, Nissbrandt H, Cenci MA. Impact of the lesion procedure on the profiles of motor impairment and molecular responsiveness to L‐DOPA in the 6‐hydroxydopamine mouse model of Parkinson's disease. Neurobiol Dis 2011;42(3):327–340. [DOI] [PubMed] [Google Scholar]
- 25. Schallert T, Fleming SM, Leasure JL, Tillerson JL, Bland ST. CNS plasticity and assessment of forelimb sensorimotor outcome in unilateral rat models of stroke, cortical ablation, parkinsonism and spinal cord injury. Neuropharmacology 2000;39(5):777–787. [DOI] [PubMed] [Google Scholar]
- 26. Ilijic E, Guzman JN, Surmeier DJ. The L‐type channel antagonist isradipine is neuroprotective in a mouse model of Parkinson's disease. Neurobiol Dis 2011;43(2):364–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kress GJ, Yamawaki N, Wokosin DL, Wickersham IR, Shepherd GMG, Surmeier DJ. Convergent cortical innervation of striatal projection neurons. Nat Neurosci 2013;16(6):665–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Petreanu L, Mao T, Sternson SM, Svoboda K. The subcellular organization of neocortical excitatory connections. Nature 2009;457(7233):1142–1145. Available from:. http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=19151697&retmode=ref&cmd=prlinks [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. West MJ, Slomianka L, Gundersen HJG. Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anatomical Rec 1991;231(4):482–497. [DOI] [PubMed] [Google Scholar]
- 30. Halliday GM. Thalamic changes in Parkinson's disease. Parkinsonism Relat D 2009. Dec;15(Suppl 3):S152–S155. [DOI] [PubMed] [Google Scholar]
- 31. Heinsen H, Rüb U, Gangnus D, Jungkunz G, Bauer M, Ulmar G, et al. Nerve cell loss in the thalamic centromedian‐parafascicular complex in patients with Huntington's disease. Acta Neuropathol 1996;91(2):161–168. [DOI] [PubMed] [Google Scholar]
- 32. Xuereb JH, Perry RH, Candy JM, Perry EK, Marshall E, Bonham JR. Nerve cell loss in the thalamus in Alzheimer's disease and Parkinson's disease. Brain J Neurol 1991;114(Pt 3):1363–1379. [PubMed] [Google Scholar]
- 33. Orieux G, Francois C, Féger J, Yelnik J, Vila M, Ruberg M, et al. Metabolic activity of excitatory parafascicular and pedunculopontine inputs to the subthalamic nucleus in a rat model of Parkinson's disease. Neuroscience 2000;97(1):79–88. [DOI] [PubMed] [Google Scholar]
- 34. Watson GDR, Hughes RN, Petter EA, Fallon IP, Kim N, Severino FPU, et al. Thalamic projections to the subthalamic nucleus contribute to movement initiation and rescue of parkinsonian symptoms. Sci Adv 2021;7(6):eabe9192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Heintz N. Bac to the future: the use of Bac transgenic mice for neuroscience research. Nat Rev Neurosci 2001;2(12):861–870. Available from:. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=11733793 [DOI] [PubMed] [Google Scholar]
- 36. Suarez LM, Solis O, Aguado C, Lujan R, Moratalla R. L‐DOPA oppositely regulates synaptic strength and spine morphology in D1 and D2 striatal projection neurons in dyskinesia. Cereb Cortex New York NY 2016;26(11):4253–4264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Malinow R, Tsien RW. Presynaptic enhancement shown by whole‐cell recordings of long‐term potentiation in hippocampal slices. Nature 1990;346(6280):177–180. [DOI] [PubMed] [Google Scholar]
- 38. Allen C, Stevens CF. An evaluation of causes for unreliability of synaptic transmission. Proc Natl Acad Sci 1994;91(22):10380–10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Franks KM, Stevens CF, Sejnowski TJ. Independent sources of quantal variability at single glutamatergic synapses. J Neurosci 2003;23(8):3186–3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Luscher C, Malenka RC. NMDA receptor‐dependent long‐term potentiation and long‐term depression (LTP/LTD). Cold Spring Harbor Persp Biol 2012;4(6):a005710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Villalba RM, Wichmann T, Smith Y. Neuronal loss in the caudal intralaminar thalamic nuclei in a primate model of Parkinson's disease. Brain Struct Func 2014. Jan;219(1):381–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Aymerich MS, Barroso‐Chinea P, Pérez‐Manso M, Muñoz‐Patiño AM, Moreno‐Igoa M, González‐Hernández T, et al. Consequences of unilateral nigrostriatal denervation on the thalamostriatal pathway in rats. Eur J Neurosci 2006;23(8):2099–2108. [DOI] [PubMed] [Google Scholar]
- 43. Surmeier DJ, Obeso JA, Halliday GM. Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci 2017;18(2):101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schwarting RK. The unilateral 6‐hydroxydopamine model in behavioral brain research. Analysis of functional deficits, recovery and treatments. Prog Neurobiol 1996;50(275):331 [DOI] [PubMed] [Google Scholar]
- 45. Graves SM, Surmeier DJ. Delayed spine pruning of direct pathway spiny projection neurons in a mouse model of Parkinson's disease. Front Cell Neurosci 2019;13:32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Villalba RM, Lee H, Smith Y. Dopaminergic denervation and spine loss in the striatum of MPTP‐treated monkeys. Exp Neurol 2009;215(2):220–227. Available from:. http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=18977221&retmode=ref&cmd=prlinks [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Turrigiano GG. Homeostatic plasticity in neuronal networks: the more things change, the more they stay the same. Trends Neurosci 1999;22(5):221–227. [DOI] [PubMed] [Google Scholar]
- 48. Shen W, Plotkin JL, Francardo V, Ko WKD, Xie Z, Li Q, et al. M4 muscarinic receptor signaling ameliorates striatal plasticity deficits in models of L‐DOPA‐induced dyskinesia. Neuron 2015;88(4):762–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wu Y‐W, Kim J‐I, Tawfik VL, Lalchandani RR, Scherrer G, Ding JB. Input‐ and cell‐type‐specific endocannabinoid‐dependent LTD in the striatum. Cell Reports 2015;10(1):75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rafalovich IV, Melendez AE, Plotkin JL, Tanimura A, Zhai S, Surmeier DJ. Interneuronal nitric oxide signaling mediates post‐synaptic long‐term depression of striatal glutamatergic synapses. Cell Reports 2015;13(7):1336–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Melendez‐Zaidi AE, Lakshminarasimhah H, Surmeier DJ. Cholinergic modulation of striatal nitric oxide‐producing interneurons. Eur J Neurosci 2019;50(11):3713–3731. [DOI] [PubMed] [Google Scholar]
- 52. Bohnen NI, Albin RL. The cholinergic system and Parkinson disease. Behav Brain Res 2011;221(2):564–573. Available from:. http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=20060022&retmode=ref&cmd=prlinks [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wu T, Hallett M. The cerebellum in Parkinson's disease. Brain 2013;136(3):696–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Steece‐Collier K, Stancati JA, Collier NJ, Sandoval IM, Mercado NM, Sortwell CE, et al. Genetic silencing of striatal CaV1.3 prevents and ameliorates levodopa dyskinesia. Mov Disord 2019;34(5):697–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Supporting Information.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.