

Abstract
Background:
Administration of anti-fibrinolytic medications, including tranexamic acid (TXA), may reduce head injury-related mortality. The effect of these medications on post-TBI inflammatory response is unknown. The goal of this study was to investigate the role of available anti-fibrinolytic medications on both systemic and cerebral inflammation after TBI.
Methods:
An established murine weight drop model was utilized to induce a moderate TBI. Mice were administered 1, 10, or 100mg/kg TXA, 400mg/kg aminocaproic acid (amicar), 100 KIU/kg aprotonin, or equivalent volume of normal saline (NS) 10 minutes after recovery. Mice were euthanized at 1, 6, or 24 hours. Serum and cerebral tissue were analyzed for neuron-specific enolase and inflammatory cytokines. Hippocampal histology was evaluated at 30 days for phosphorylated tau (p-tau) accumulation.
Results:
One hour after TBI, mice given TXA displayed decreased cerebral cytokine concentrations of TNF-α, and by 24 hours displayed decreased concentrations of cerebral TNF-α, IL-6, and MCP-1 compared to TBI+NS. However, serum concentrations of TNF-α and MIP1-α were significantly elevated from 1 to 24 hours in TBI+TXA groups compared to TBI+NS. The concentration of p-tau was significantly decreased in a dose dependent manner in TBI+TXA groups compared to TBI+NS. By contrast, aminocaproic acid administration increased cerebral cytokine levels of IL-6 1 hour after TBI, with serum elevations noted in TNF-α, MIP1-α, and MCP-1 at 24 hours compared to TBI+NS. Aprotonin administration increased serum TNF-α, IL-6, MIP1-α from 1 to 24 hours without differences in cerebral cytokines compared to TBI+NS.
Conclusions:
TXA administration may provide acute neuro-inflammatory protection in a dose-dependent manner. Amicar administration may be detrimental after TBI with increased cerebral and systemic inflammatory effects. Aprotonin administration may increase systemic inflammation without significant contributions to neuroinflammation. While no antifibrinolytic medication improved systemic inflammation, these data suggest that TXA may provide the most beneficial inflammatory modulation after TBI.
Level of Evidence:
Basic Science paper
Keywords: TXA, antifibrinolytics, traumatic brain injury, cytokine, inflammation
Media Summary:
The goal of this study was to investigate the role of current antifibrinolytic medications in both systemic and neuroinflammation after TBI. We found that TXA administration may provide the most beneficial cerebral inflammatory modulation after isolated TBI.
Background
Traumatic brain injury (TBI) is a widely recognized global health issue with increasing annual occurrence. Current management strategies to prevent the secondary brain injury that occurs after initial insult lack standardization. Previous investigations into the coagulopathy of trauma and more specifically acute hyperfibrinolysis after TBI have introduced an increasing role for antifibrinolytic medications (1). Recent research on the pathophysiology of TBI has revealed that the inflammatory response to primary brain injury is widespread and can disseminate to many areas of the brain that are remote from the initial injury site (2). The primary brain injury subsequently initiates a secondary cerebral insult through free radical generation and neuronal excitotoxicity that can persist for months to years (2, 3). One of the prevailing theories regarding the efficacy of antifibrinolytic medications in the improvement of mortality after TBI revolves around the attenuation of systemic inflammation (4). A proposed mechanism of inflammation modulation is through the direct inhibition or blockade of plasmin formation. Plasmin has been shown to induce the systemic inflammatory response by stimulating the release of various proinflammatory cytokine/chemokines such as interleukin-1 (IL-1), interleukin 6 (IL-6), tumor necrosis factor alpha (TNF-α), and tissue factor (5, 6). Inactivation of these cytokines has been shown to dampen post-traumatic systemic inflammation. However, the theory of systemic inflammatory attenuation secondary to antifibrinolytic therapies has yet to be validated.
Investigations into the use of antifibrinolytic therapies, such as tranexamic acid (TXA), aminocaproic acid (Amicar), and aprotonin (Trasylol), in the acute management of TBI have noted significant improvements in brain related mortality (4, 7). The Clinical Randomization of an Antifibrinolytic in Significant intracranial Hemorrhage 3 (CRASH-3) trial provided evidence to support the early use (<3hrs) of TXA in TBI patients by demonstrating improved brain injury related mortality (7). Additional studies addressing the prehospital dose and timing of TXA administration revealed that there were no significant differences in 28 day mortality between patients who received 1g versus a 2g bolus of TXA within 3 hours of initial injury (8). Similarly, aminocaproic acid has been shown to be beneficial when given prior to brain injury in a rodent model, with notable improvements in neurobehavioral function and overall brain injury by 24 hours (9). Evaluation of the effects of aprotonin after TBI demonstrated that cerebral protease activity was inhibited after aprotonin administration and survival was significantly improved in a small human cohort study (10). Although these studies provide some evidence to support that acute administration of antifibrinolytic medications is safe and can be effective for TBI patients, the mechanism behind this stated benefit has yet to be investigated.
Despite the current clinical and pre-clinical literature on the potential inflammatory impact of antifibrinolytic medications, the available pharmacologic options have not been directly compared in a standardized model of TBI. A majority of the current literature regarding antifibrinolytic therapy administration in traumatically injured patients has utilized TXA. It is important to note that one of the main reasons TXA was chosen over other antifibrinolytic therapies in major studies such as the Clinical Randomization of an Antifibrinolytic in Significant Hemorrhage 2 (CRASH-2) trial was because of the elevated cost of and the possible allergic reaction found with aprotonin administration (11). Therefore, we investigated all three commercially available antifibrinolytic therapies in an established murine model of TBI. The goal of this study was to investigate the role of current antifibrinolytic medications in both systemic and neuroinflammation after TBI. We hypothesized that administration of antifibrinolytic medications, particularly TXA, would show an improvement in both systemic and neuroinflammation after TBI.
Methods
Animal and TBI Injury Model
The University of Cincinnati Institutional Animal Care and Use Committee approved all animal experimentation. Animals were 9–10 week old male C57BL6/J mice with a mean weight of 25.6 ± 2.1g acquired from Jackson Laboratory (Bar Harbor, Maine). Only male mice were utilized for this study to establish consistency and to control for the potential neuroprotective effects of both estrogen and progesterone (12). Mice were acclimated for 1 week prior to experimentation in a sterile controlled housing environment with unlimited access to food and water. This study conforms with the ARRIVE guidelines and a complete checklist has been uploaded as Supplemental Digital Content (SDC-1). Mice that did not acutely recover neurologically from the TBI (n=8) and mice that did not survive to their allocated timepoint of euthanasia (n=4) were excluded from serum and tissue analyses.
Mice underwent a controlled weight drop injury, as previously described, to induce a TBI of moderate severity, as defined by righting reflex response times (13–16). In brief, mice were anesthetized with 2% inhaled isoflurane in 100% oxygen at 1 L/min for approximately 2 minutes. After determining that an appropriate level of anesthesia was obtained, mice were placed in a prone position onto the TBI platform. The head was centered under a 400g cylindrical rod that was dropped from 2.5 cm above the surface of the table to induce a global concussive blunt TBI (13–15). All mice were observed until appropriate respiratory and neurologic recovery had been achieved. Sham TBI mice were anesthetized and positioned similarly on the TBI platform but were not subjected to the TBI.
Pharmacologic Intervention
Mice were randomized into 12 groups; 6 with TBI (n=5 per group), 6 with sham TBI (n=5 per group) and were given 1 of 6 drug treatments (tranexamic acid [1mg/kg, 10mg/kg, 100mg/kg] (Athenex, Schaumburg, IL), aminocaproic acid [400mg/kg](Hospira, Lake Forest, IL), aprotonin [100 KIU/kg]) (Sigma Aldrich, St Louis, MO), or normal saline via intraperitoneal injection 10 minutes after recovery from TBI. The dose of TXA used was based upon our previous experimentation in a murine TBI model that had established a dose response curve administering 1,10, and 100mg/kg TXA (17–19). Similarly, the selected doses of amicar and aprotonin were based upon previous studies in murine models (19, 20). Strategies were employed to minimize confounding data, including assimilation of all animals in each of the study cohorts in the same cage prior to injury induction and drug treatment. No immediate adverse events, including seizure activity, were noted after administration of any pharmaceutical agent.
Cytokine Evaluation
Mice were sacrificed at 1, 6, or 24 hours (n=5 per group at each time point) for serum and brain tissue cytokine analysis. Mice were anesthetized with 100mg ketamine/16mg xylazine via intraperitoneal injection and then whole blood was collected via intra-cardiac puncture. Whole blood was placed into serum separator tubes (BD Bioscience, San Diego, California) and centrifuged at 1000g for 10 minutes and serum was collected. Brain tissue collection occurred subsequent to blood collection based upon a previously described protocol(21). In brief, cerebral tissue was removed via craniotomy, separated from the cerebellum and immediately frozen using liquid nitrogen. The thawed brain tissue was homogenized with PBS buffer, 10mM EDTA, 2mM PMSF, 0.1mg/mL soybean trypsin inhibitor, 1.0mg/mL BSA, 0.02% sodium azide, 10uL PIC (1mg/mL leupeptin, 1mg/mL aprotonin, 1mg/mL pepstatin). After 2 rounds of centrifugation at 10,000g for 20 minutes, supernatant protein content was normalized using a BCA Protein Assay Kit (Thermo Scientific, Rockford, IL).
Serum and cerebral tissue samples were analyzed by Multiplex enzyme linked immunosorbent assay (ELISA), including interleukin 1 alpha (IL-1a), interleukin 1 beta (IL-1b), interleukin 2 (IL-2), interleukin 3 (IL-3), interleukin 4 (IL-4), interleukin 6 (IL-6), interleukin 10 (IL-10), interleukin 12 (IL-12), interleukin 17 (IL-17), monocyte chemoattractant protein 1 (MCP-1), tumor necrosis factor alpha (TNF-α), granulocyte-macrophage colony stimulated factor (GM-CSF), macrophage inflammatory protein 1 alpha (MIP-1α), and Regulated upon Activation, Normal T Cell Expressed and Presumably Secreted (RANTES) (Quansys Biosciences, Logan, UT). These cytokine and chemokines were selected as a comprehensive post-injury inflammatory profile to demonstrate the systemic and cerebral inflammatory responses after TBI(16, 17). Serum neuron specific enolase (NSE) was additionally analyzed by ELISA as a systemic biomarker of acute cerebral injury (Abcam, Cambridge, MA).
Histologic Evaluation of hippocampal phosphorylated tau (p-tau) protein accumulation
Following cytokine analysis demonstrating potential TXA benefits, an additional cohort of mice was survived to 30 days after TBI and treatment with either 1,10, or 100mg/kg TXA. Mice were anesthetized with 100mg ketamine/16mg xylazine via intraperitoneal injection and perfused with 10% formalin via intracardiac puncture. Brains were all removed and fixed in 10% formalin for 48 hours, dehydrated and embedded in paraffin for immunohistochemistry. Coronal sections were taken at 5μm slices. Immunohistochemistry staining for phosphorylated tau protein (p-tau) was performed according to a previously described method (22). Slides were stained with rabbit polyclonal anti-tau (phosphor S262) antibody (ab131354; Abcam, Cambridge, UK). Images were captured on Nikon AIR GaAsP inverted microscope (Nikon Corporation, Tokyo, Japan) and quantified using cell counter features on Image J software. Researchers were blinded to the treatment groups during histologic imaging and cellular counting.
Statistical Analysis
For sample size calculation, the primary outcome measure utilized was cerebral IL-6 based on the study interest in the neuroinflammatory response. Using preliminary data demonstrating that cerebral IL-6 levels were 40% lower in sham compared to TBI mice with a 20% variance, a minimum sample size of 4 animals per group was established. All statistical analyses were performed with Prism 6 (GraphPad Software, La Jolla, California). T-tests were used to compare cytokine concentrations in response to each drug administration after TBI. A p value of less than 0.05 was considered significant.
Results
Serum Cytokine Analysis
Serum concentrations of TNF-α were significantly elevated in TBI+TXA 100mg/kg and TBI+aprotonin at 1 and 24 hours after TBI compared to TBI+NS (Figure 1). TNF-α was also noted to be elevated 1 hour after TBI in TBI+TXA 10mg/kg compared to TBI+NS (Figure 1). Similarly, serum concentrations of TNF-α were significantly elevated 24 hours after TBI in TBI+amicar mice compared to TBI+NS group (Figure 1). There were no significant differences in serum TNF-α 1–24 hours after TBI between TBI+NS and sham TBI+NS (Figure 1). Similarly, there were no significant differences in serum IL-6 between TBI+NS and sham TBI+NS cohorts (Figure 2). By contrast, serum concentrations of IL-6 were significantly elevated in TBI+TXA 10mg/kg 1 and 6 hours after TBI compared to TBI+NS group (Figure 2). IL-6 levels were also noted to be elevated in TBI+TXA 100mg/kg but only at 1 hour after TBI compared to TBI+NS (Figure 2). Mice receiving TBI+aprotonin also displayed significant elevations in serum IL-6 at 6 and 24 hours after TBI compared to TBI+NS group (Figure 2). Serum concentrations of MIP-1α were significantly elevated in TBI+NS at 1 and 24 hours after TBI compared to sham TBI+NS cohort (Figure 3). At 1,6, and 24 hours after TBI, serum concentrations of MIP-1α were significantly elevated in TBI+TXA 10mg/kg, TBI+TXA 100mg/kg, and TBI+aprotonin mice compared to TBI+NS (Figure 3). MIP-1α levels were also noted to be elevated in TBI+amicar cohort at 6 and 24 hours after TBI compared to TBI+NS group (Figure 3).
Figure 1: Serum TNF-α levels are significantly elevated 1 and 24 hours after TXA and aprotonin administration after isolated TBI.
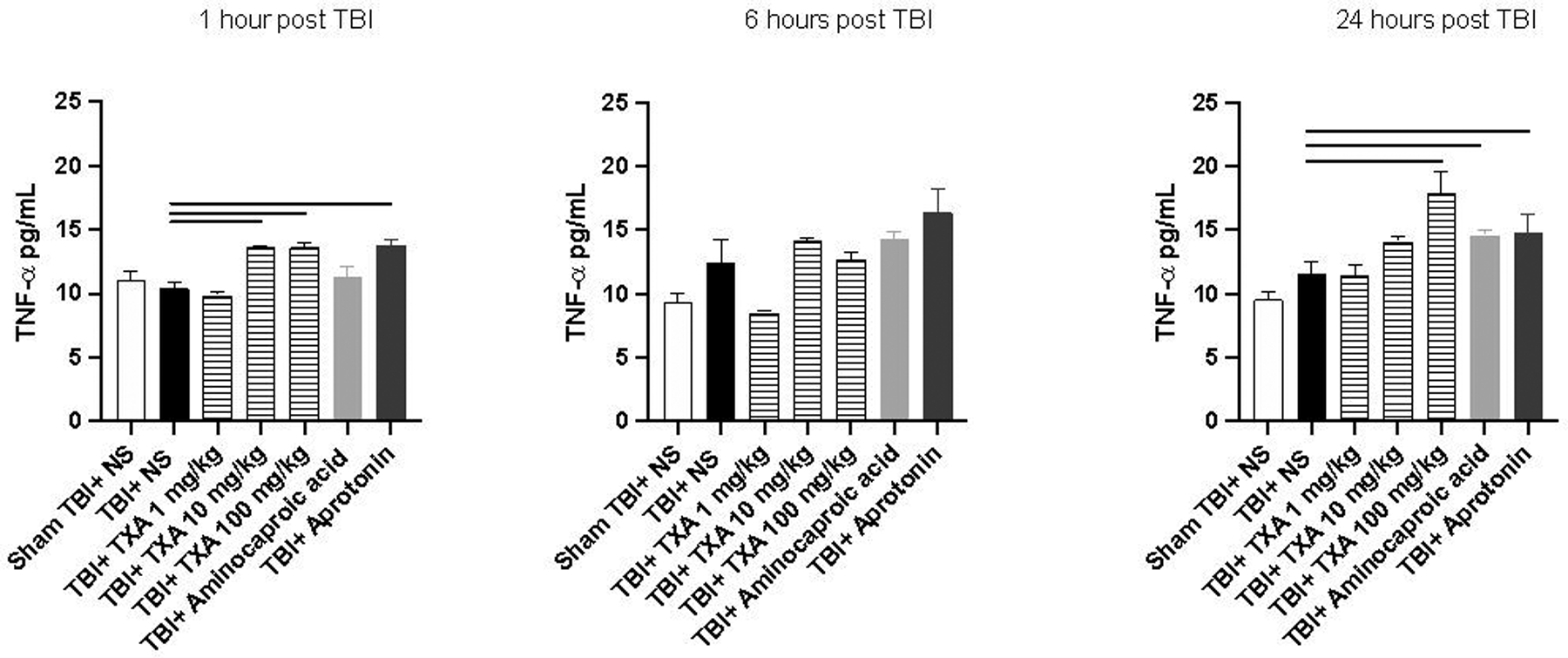
Solid line indicates significant difference between groups, p<0.05
Figure 2: TXA administration after TBI causes an acute elevation of serum IL-6, whereas aprotonin causes a delayed elevation of serum IL-6.
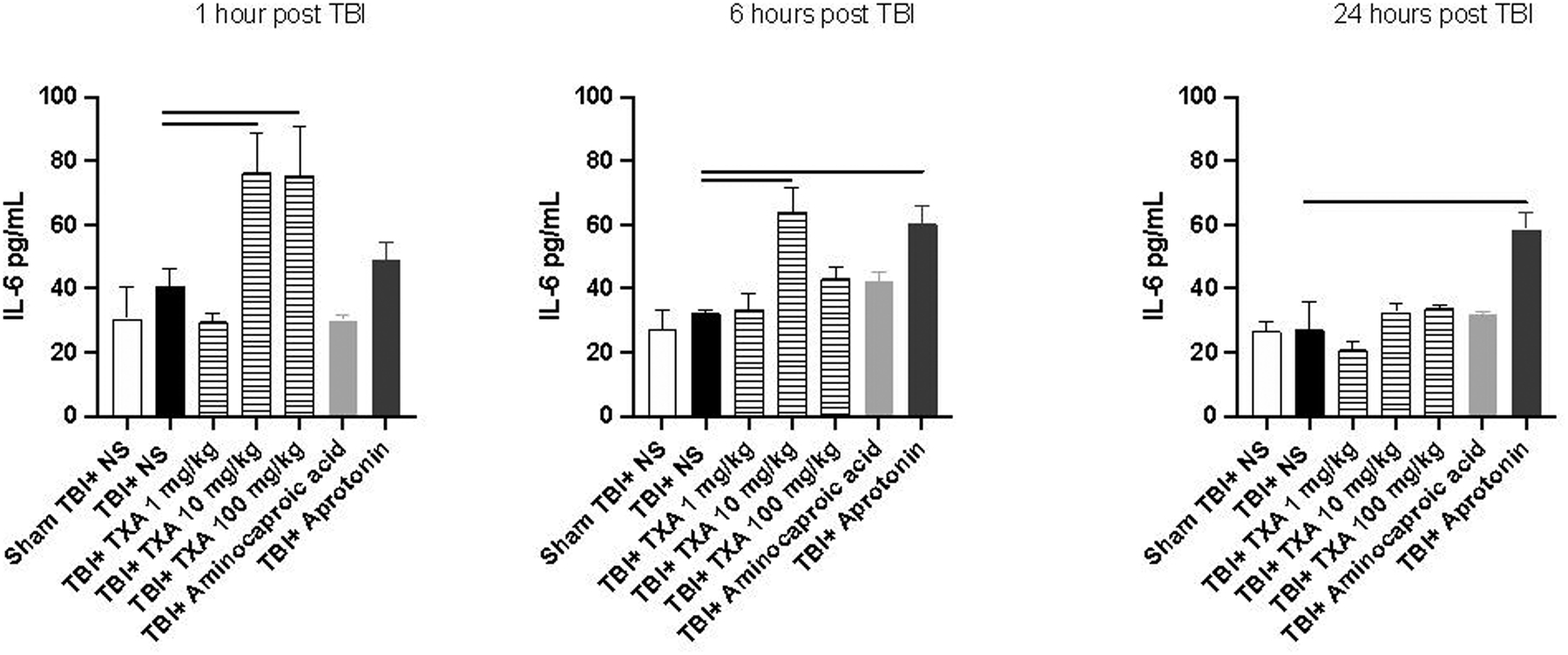
Solid line indicates significant difference between groups, p<0.05
Figure 3: Serum concentrations of MIP1-α are significantly elevated from 1 to 24 hours after administration of TXA and from 6 to 24 hours after administration of aminocaproic acid and aprotonin after TBI.
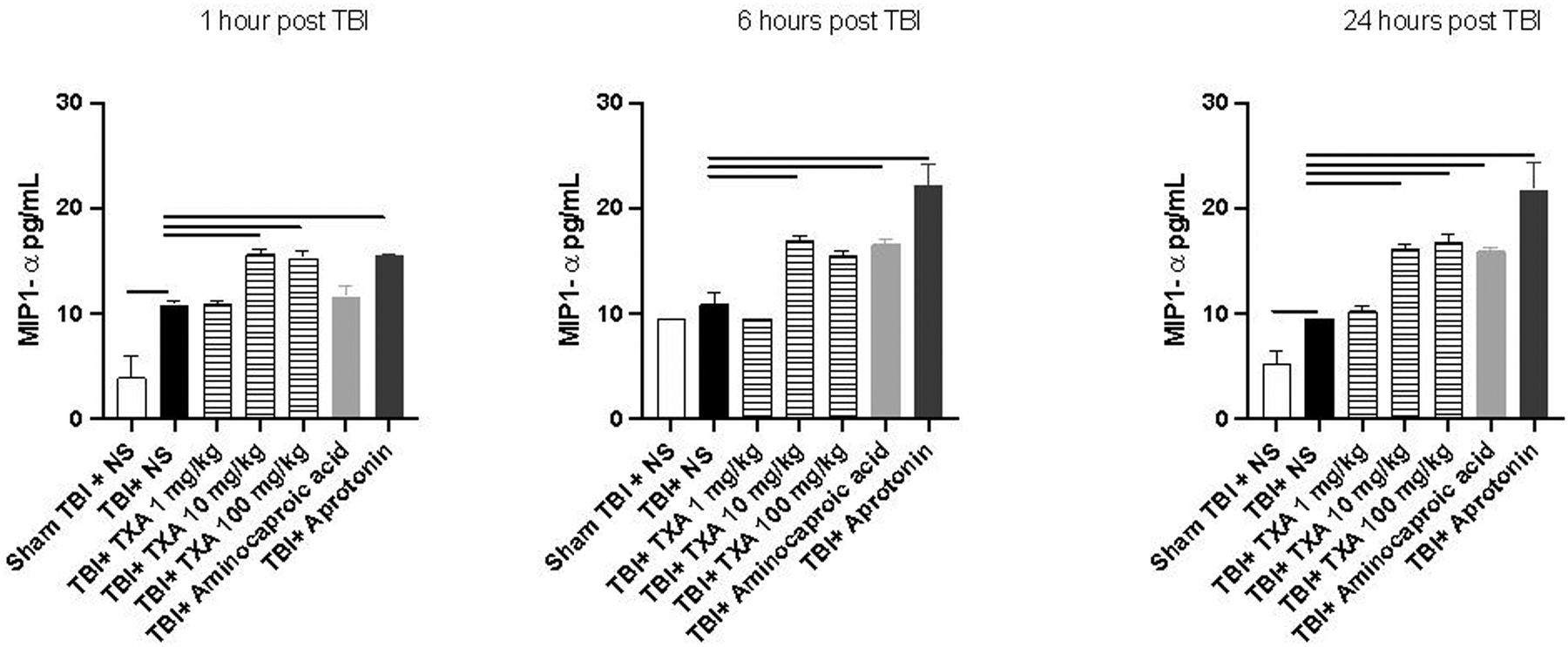
Solid line indicates significant difference between groups, p<0.05
MCP-1 serum concentrations were significantly elevated in TBI+TXA 100mg/kg, TBI+amicar, and TBI+aprotonin 24 hours after TBI compared to TBI+NS (SDC-2). Interestingly, serum MCP-1 levels decreased in TBI+TXA 1mg/kg 6 hours after TBI compared to TBI+NS. By contrast, significant elevations of MCP-1 were noted in the TBI+TXA 10mg/kg group compared to TBI+NS at 6 hours (SDC-2). Serum concentrations of IL-1b were significantly elevated in TBI+aprotonin 6 hours after TBI and TBI+TXA 100mg/kg 24 hours after TBI compared to TBI+NS (SDC-3). IL-12 levels were significantly elevated in TBI+TXA 10mg/kg 1 hour after TBI compared to TBI+NS (SDC-4). In addition, IL-12 levels were elevated in TBI+amicar 6 hours after TBI compared to TBI+NS (SDC-4).
There were no significant or consistent differences in the serum concentrations of IL-1α, IL-1b, IL-2, IL-3, IL-4, IL-6, IL-10, IL-12, IL-17, MCP-1, TNF-α, MIP-1α, GM-CSF, and RANTES between the sham TBI treatment groups at 1, 6, or 24 hours compared to sham+NS (data not shown).
Similarly, there were no significant or consistent differences in serum cytokine concentrations of IL-1α, IL-2, IL-3, IL-4, IL-10, IL-17, GM-CSF, and RANTES between TBI mice treated with any of the antifibrinolytic therapies compared to TBI+NS group at 1,6, and 24 hours (data not shown).
Brain Cytokine Analysis
One and 6 hours after TBI cerebral TNF-α levels were significantly elevated in TBI+ NS group compared to sham TBI +NS mice (Figure 4). Cerebral TNF-a levels were significantly reduced 1 hour after TBI within TBI+TXA 100mg/kg, TBI+TXA 10mg/kg, and TBI+aprotonin cohorts as compared to TBI+NS (Figure 4). Similarly, TNF-a levels were further reduced 24 hours after TBI in TBI+TXA 100mg/kg cohort compared to TBI+NS (Figure 4).
Figure 4: The levels of cerebral TNF-α after isolated TBI were significantly decreased acutely after TXA and aprotonin administration.
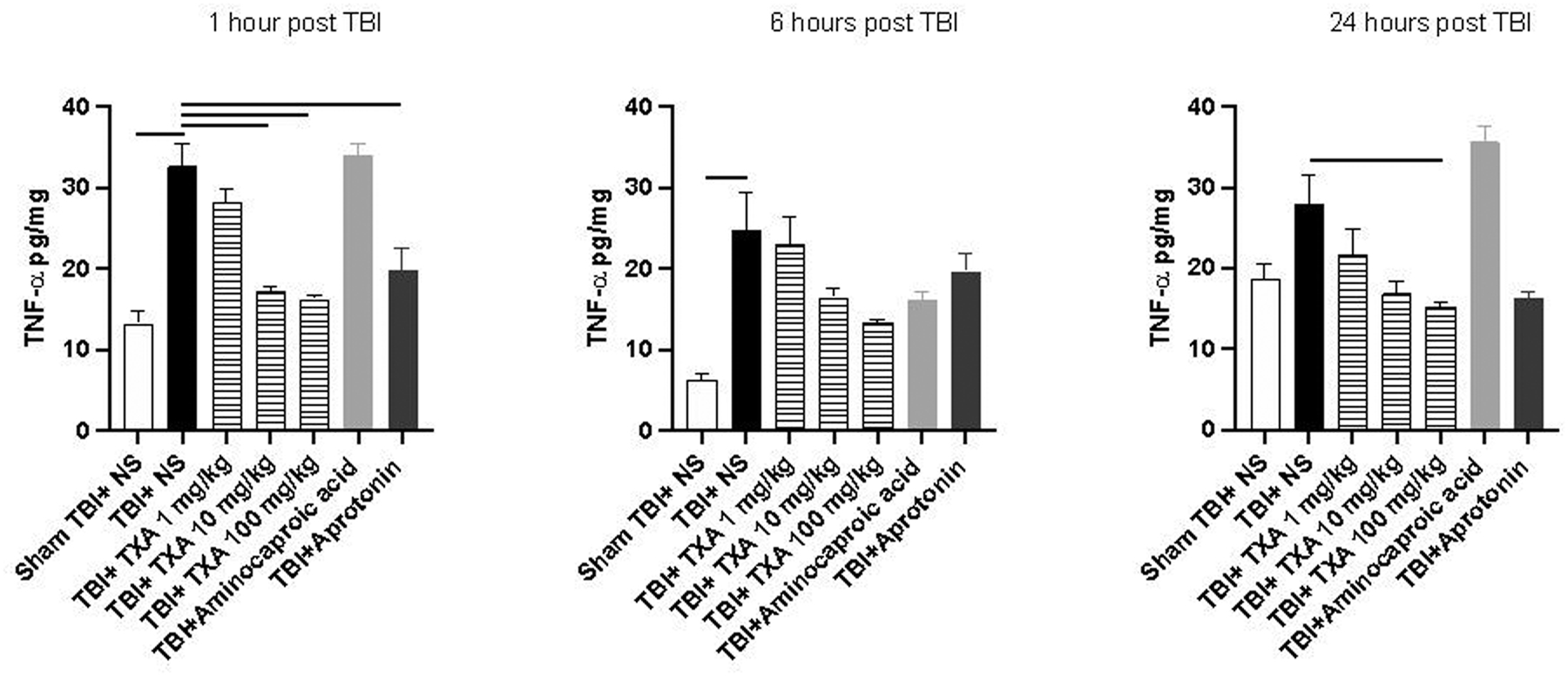
Solid line indicates significant difference between groups, p<0.05
Of note, cerebral IL-6 concentrations were significantly reduced in TBI+TXA 100mg/kg cohort at 1 and 24 hours after TBI compared to TBI+NS (Figure 5). Furthermore, cerebral IL-6 concentrations were significantly reduced in TBI+TXA 1 and 10mg/kg 24 hours after TBI compared to TBI+NS (Figure 5). Additionally, cerebral IL-6 was significantly elevated 1 hour after TBI in TBI+NS mice compared to sham TBI+NS mice (Figure 5). TBI+amicar mice conversely displayed significant elevations in cerebral IL-6 levels 1 hour after TBI with a similar trend noted at 24 hours compared to TBI+NS (Figure 5). Cerebral MCP-1 levels were significantly reduced in TBI+TXA 10mg/kg 1 and 24 hours after TBI compared to TBI+NS (Supplemental Digital Content 4). Similar reductions were noted in cerebral MCP-1 levels in TBI+TXA 100mg/kg and TBI+aprotonin 1 hour after TBI and TBI+TXA1mg/kg 24 hour after TBI compared to TBI+NS (SDC-5).
Figure 5: Cerebral IL-6 concentrations were significantly decreased after TXA administration 1 and 24 hours after TBI whereas aminocaproic acid administration increased cerebral IL-6 levels acutely after TBI.
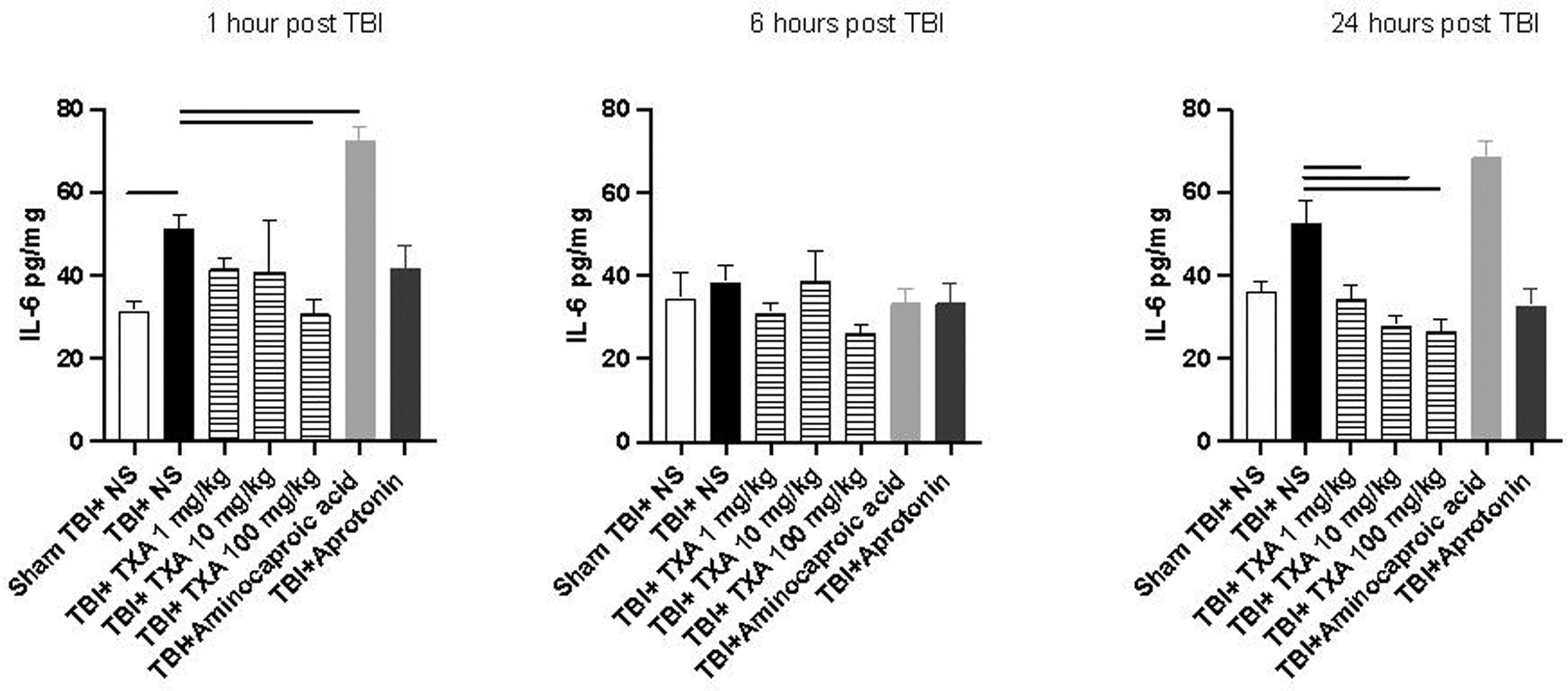
Solid line indicates significant difference between groups, p<0.05
Cerebral cytokine concentrations of IL-1α, IL-1b, IL-2, IL-3, IL-4, IL-6, IL-10, IL-12, IL-17, MCP-1, MIP-1α, GM-CSF, and RANTES did not display significant or consistent differences between sham TBI mice treated with antifibrinolytic therapy compared to sham TBI+NS (data not shown). There was a significant increase noted in the concentration of cerebral TNF-α in sham TBI+TXA 100 mg/kg at 1 and 6 hours compared to sham TBI+NS. Similarly, there was an increase in TNF-α within sham TBI+amicar 1 hour after sham injury compared to sham TBI+NS (data not shown).
Cerebral cytokine concentrations of IL-1α, IL-1b, IL-2, IL-3, IL-4, IL-10, IL-12, IL-17, MIP-1 α, GM-CSF, and RANTES did not display any consistent elevations in TBI+TXA treated cohorts or TBI+aprotonin mouse cohort compared to TBI+NS (data not shown). Conversely, TBI+amicar mice demonstrated significant elevations in cerebral concentrations of IL-1 α, IL-1b, IL-2, IL-3, IL-4, IL-12, IL-17, MCP-1, and MIP-1 α at 1 and 24 hours after TBI compared to TBI+NS (data not shown).
Serum Biomarker of Injury Severity
Serum NSE levels were significantly elevated in TBI+NS group compared to sham TBI+NS group 1 hour after TBI, with differences resolving by 6 hours (Figure 6A–B). NSE levels were noted to display a significant decrease in TBI+TXA 10mg/kg and 100mg/kg compared to TBI+NS 1 hour after TBI which did not persist to 24 hours (Figure 6A–C). Serum NSE levels increased 6 hours after TBI in TBI+TXA 10mg/kg compared to TBI+NS (Figure 6B). Mice within the TBI+TXA 1mg/kg cohort did not display differences in serum NSE from 1–24 hours compared to TBI+NS (Figure 6A–C). Serum NSE levels were significantly elevated in TBI+aprotonin mice both at 6 and 24 hours after TBI compared to TBI+NS group (Figure 6B–C). TBI+amicar groups did not display significant or consistent differences in serum NSE from 1 to 24 hours after TBI compared to TBI+NS mice (Figure 6A–C).
Figure 6A-E: Serum NSE concentrations decreased acutely after TXA administration 1 hour after TBI with notable elevations in NSE after aprotonin administration 6 and 24 hours after TBI. Hippocampal p-tau levels were significantly decreased after TXA administration 30 days after isolated TBI. Representative immunohistochemistry images of phosphorylated tau protein.
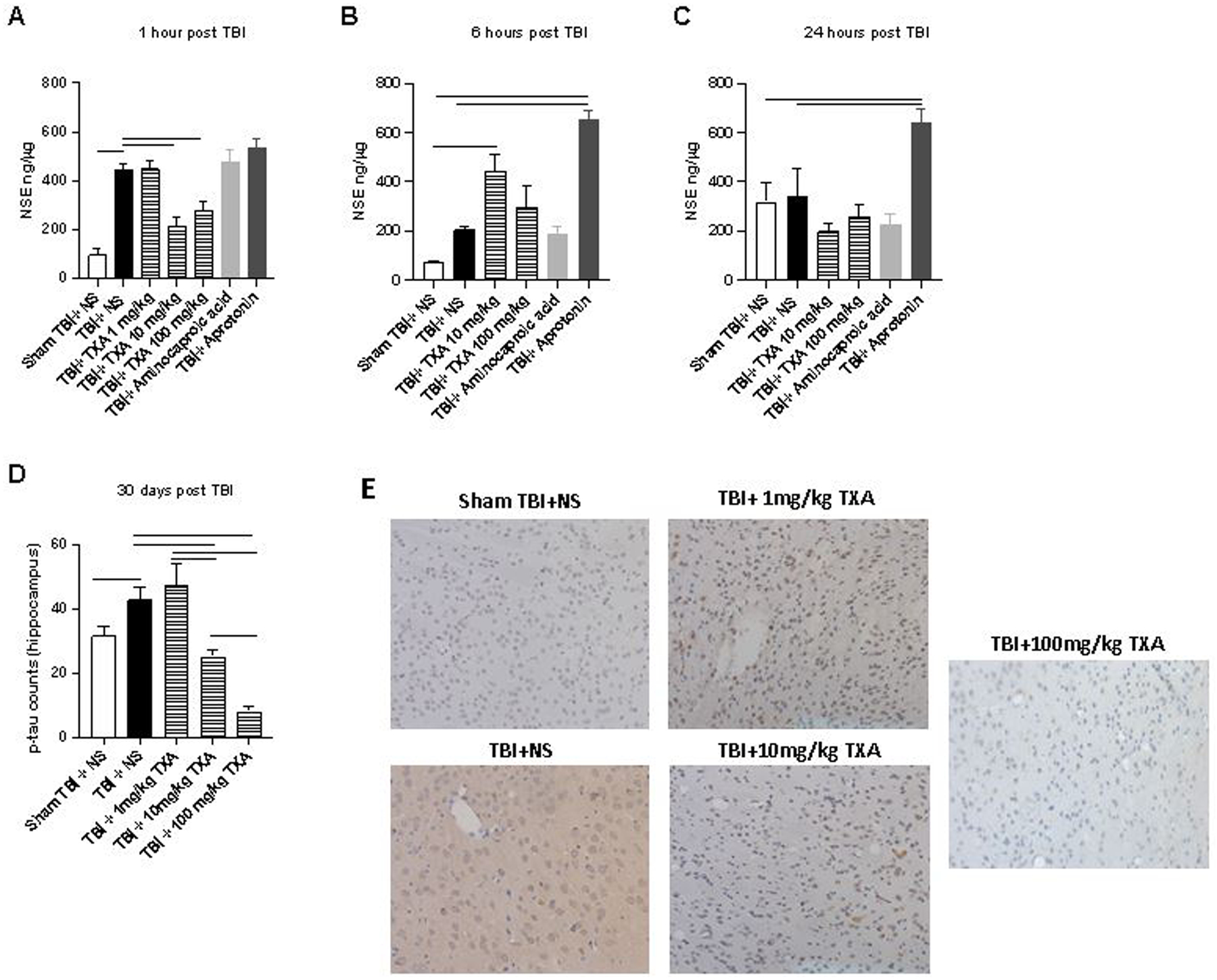
Solid line indicates significant difference between groups, p<0.05.
Histologic Evaluation of hippocampal p-tau at 30 days
P-tau accumulation was noted to be significantly elevated in TBI+NS group (42.8 ± 17.9) compared to sham TBI+NS group (31.5 ± 14.5, p<0.05; Figure 6D). TXA demonstrated a dose dependent decrease in hippocampal p-tau accumulation, with TBI+TXA 10mg/kg (25.0± 11.5) and TBI+TXA 100mg/kg (8.04± 8.6) cohorts noted to have significantly less accumulation of hippocampal p-tau compared to TBI+NS group after 30 days (Figure 6D).
Discussion
In this study we investigated the systemic and cerebral inflammatory effects of antifibrinolytic therapies, including TXA, amicar, and aprotonin, after TBI in a murine model. We identified that TXA administration at both 10 and 100mg/kg was neuroprotective with significant reductions noted in both cerebral cytokines and 30-day hippocampal p-tau accumulation, however, induced an elevated systemic inflammatory response after TBI. Amicar administration may be detrimental for isolated TBI patients with noted elevations in cerebral cytokines and inconsistent effects on systemic inflammatory biomarkers. Aprotonin administration displayed elevated systemic inflammatory markers with inconsistent neuroinflammatory changes after TBI. Although TXA administration may cause an augmented acute systemic inflammatory response, there is evidence to support neuroprotection with higher doses.
The use of antifibrinolytic therapy in traumatically injured patients has been proven effective in preventing death secondary to hemorrhage and may improve brain-related mortality in mild or moderate TBI patients, however, the mechanism of this stated benefit and the systemic effects these medications may have has yet to be investigated. The current prevailing theory regarding the mechanism of improved mortality secondary to antifibrinolytic therapy administration is via immunomodulation and the inhibition of plasmin (4, 23, 24). Interestingly, we found that TXA administration did not attenuate the systemic inflammatory response, rather TXA appeared to increase the acute release of systemic inflammatory mediators. Wang et al. showed that repeated doses of TXA prior to total knee arthroplasty decreased serum levels of erythrocyte sedimentation rate (ESR) and IL-6 compared to placebo from 10 to 24 hours post-operatively(25). Although this study provides evidence to support a possible immunomodulatory effect of TXA, the patients received multiple oral doses of TXA along with a single intra-articular injection of TXA which may have attenuated the systemic response at a higher cumulative dose received (25). Further, Draxler et al. found that in patients undergoing cardiac surgery, pre-operative administration of TXA led to decreased levels of serum IL-1b post-operative day 1, without significant alteration in other systemic cytokines such as TNF-a, IL-6, MCP-1, and interferon gamma (IFN-γ)(26). This study was performed on a small sample of 41 patients and was performed in a surgical setting where other immunomodulating medications could have been given both pre- and post-operatively that could have led to the investigators detecting changes in systemic inflammatory biomarkers not isolated to administration of antifibrinolytic medications alone(26). By contrast, Grant et al. found that administration of a single intravenous dose of TXA and single intra-articular dose of TXA immediately following knee arthroplasty increased plasma levels on post-operative day 2 of MCP-1, TNF-a, and IL-6, with IL-6 levels being 1.8 times higher than those who did not receive TXA, further corroborating our results (27). Although the proposed mechanism of TXA after TBI and traumatic injury was through systemic immunomodulation, our data would suggest that the systemic inflammatory response may actually be augmented rather than attenuated. Therefore, more injury, organism, and cell specific investigations are warranted to understand the mechanisms of action for the role of TXA in post-injury inflammation.
Currently there are limited strategies to employ in the prevention of secondary brain injury after the initial TBI. Previous studies into the mechanism of secondary brain injury have shown that moderate acute post-traumatic inflammation may be beneficial due to the promotion of debris clearance and cell regeneration (28, 29). However, when inflammation persists, neuronal cell death and progressive neurodegeneration may occur (28). Some of the cerebral inflammatory mediators shown to be important in modulating neuroinflammation after TBI include TNF-a, INF-γ, IL-1β, IL-6, IL-10 (28). Our study provides evidence that TXA administration may blunt the post-TBI cerebral cytokine release of IL-6, TNF-a and MCP-1 with a notable reduction in 30-day p-tau accumulation which is a known marker of chronic traumatic encephalopathy (CTE) (30). Hiramoto et al. provided evidence that TXA and aprotonin administration ameliorated the aging induced decline in memory and learning ability with notable reductions in cerebral IL-1β and TNF-a in TXA administered mice (31). Although this study was not performed on TBI subjects it did demonstrate that administration of antifibrinolytic medications may alter cerebral cytokine profiles. Yoshizaki et al. found that administration of TXA after acute spinal cord injury decreased expression of TNF-a and toll like receptor 4 (TLR-4) within spinal cord sections while also significantly reducing the number of apoptotic cells in spinal cord injured subjects suggesting that antifibrinolytic medications may also be protective in the central nervous system (32). Conversely, our group previously found that there were no significant neuroinflammatory changes with TXA administration by 24 hours after TBI. This study, although similar, administered only 10mg/kg TXA which may have limited the ability to induce neuroinflammatory changes. An important difference between the current study and the previous study is that the previous study utilized a combined model of hemorrhagic shock with TBI, which also may have affected the systemic and cerebral inflammatory responses. In the future, we plan to utilize a polytrauma model combining TBI and hemorrhagic shock to investigate if the additional endothelial damage associated with hemorrhagic shock affects the systemic and cerebral inflammatory responses to antifibrinolytic therapy.
The antifibrinolytic medications utilized in this study have varying mechanisms of action leading to their opposing effects on both systemic and cerebral inflammation. The two major subtypes of antifibrinolytic agents are the synthetic lysine analogs (TXA and aminocaproic acid) and a protease inhibitor (aprotonin) (33). Tranexamic acid is a potent lysine analog that binds to the lysine binding site of plasminogen thereby inhibiting its ability to bind fibrin and preventing fibrinolysis (33). Molecular examination of TXA has displayed that TXA is 7–10 times more potent than aminocaproic acid and is able to provide more sustained systemic antifibrinolytic activity (34). Further, studies have revealed that TXA and amicar may act on glycine receptors within the central and peripheral nervous system however the pharmacokinetics of these drugs within the brain and the influence they have on cerebral function warrant further investigation in the setting of both the intact and traumatically disrupted blood brain barrier (BBB) (35). By contrast, aprotonin is a diverse protease inhibitor that is extracted from bovine lung. Aprotonin has been shown to reversibly interact with various proteins throughout the body leading to profound antifibrinolytic effects most notably through inhibition of the factor XIIa mediated kallikrein activation and inhibition of plasmin generation via direct inhibition (33, 36). Although these medications have been extensively examined for their effects on coagulation there is very limited data regarding the effects on systemic inflammation. Previous work on the integrity and composition of the BBB identified that plasmin may disrupt the BBB though enhancement of matrix metalloproteinase activity and other cellular factors leading to brain edema and worsening hemorrhage in the setting of traumatic injury (37, 38). Medications such as TXA, aminocaproic acid, and aprotonin may help aid in BBB integrity though inhibition of plasmin, however, the mechanism and effects of cerebral inflammation after injury remains unclear. Further investigation into the mechanism of current antifibrinolytic agents within the trauma population is needed to better understand the systemic effects these medications have on patient physiology and overall recovery after injury.
Our study does have several limitations. First, we evaluated systemic and neuroinflammatory markers up to 24 hours after TBI leaving the possibility for further changes that could occur later after the induced injury. Second, the mice were injected with a single dose of antifibrinolytic medication. Clinically, patients may receive repeated doses of such medications after traumatic injury, so we may have not observed changes that could occur with a multi-dose regimen. Third, we performed a cytokine analysis after an isolated TBI model as an initial study of the inflammatory response without addressing polytraumatic injury. In the future we plan to investigate systemic inflammation in a polytrauma model as we recognize that a majority of traumatically injured patients sustain multiple injuries. Fourth, for the evaluation of cerebral changes consistent with chronic traumatic encephalopathy at 30 days we investigated the accumulation of hippocampal p-tau, as has been shown previously to be elevated after isolated TBI (22). However, we have not yet evaluated cortical thinning or the microglial and astrocyte response as additional markers of the cerebral response to injury. Lastly, we created a dose response curve of 1,10, and 100mg/kg for TXA administration but only administered a single concentration of both amicar and aprotonin. Although the concentrations used in this study were based on previous literature, multiple doses of each medication could help further investigation into the optimal dose of antifibrinolytic medications after TBI (19, 20).
In conclusion, in this study we identified that TXA administration may provide acute neuro-inflammatory protection in a dose dependent manner. These data also provide evidence to support that amicar administration may be detrimental after isolated TBI with increased systemic and neuroinflammatory effects. Aprotonin administration may be beneficial after isolated TBI, however further investigation is needed. Taken together, of the current antifibrinolytic medications, we found that TXA administration may provide the most beneficial cerebral inflammatory modulation after isolated TBI.
Supplementary Material
Supplemental Digital Content 1: ARRIVE guidelines
Supplemental Digital Content 2: Serum MCP-1 concentrations were significantly elevated 6 to 24 hours after TBI with TXA administration. Solid line indicates significant difference between groups, p<0.05
Supplemental Digital Content 3: Serum IL-1b levels were significantly elevated 6 hours after TBI with aprotonin administration and 24 hours after TBI with TXA administration. Solid line indicates significant difference between groups, p<0.05
Supplemental Digital Content 4: Serum IL-12 concentrations were significantly elevated acutely after TBI and TXA administration with an elevation at 6 hours noted after TBI and aminocaproic acid administration. Solid line indicates significant difference between groups, p<0.05
Supplemental Digital Content 5: TXA administration significantly decreased the concentration of cerebral MCP-1 1 and 24 hours after TBI. Solid line indicates significant difference between groups, p<0.05
Funding:
NIH T32GM008478-30; NIH R01 GM124156-01A1
NIH- Ruth Kirschstein T32GM008478-30
Footnotes
This data will be presented as an oral presentation at the 35th EAST Annual Scientific Assembly January 11–15, 2022 in Austin, Texas.
Authors have no conflicts of interest to disclose.
Disclosures: The stated authors have no conflicts of interest to disclose
References
- 1.Nakae R, Takayama Y, Kuwamoto K, Naoe Y, Sato H, Yokota H. Time Course of Coagulation and Fibrinolytic Parameters in Patients with Traumatic Brain Injury. J Neurotrauma. 2016;33(7):688–95. [DOI] [PubMed] [Google Scholar]
- 2.Shi K, Zhang J, Dong J-f, Shi F-D. Dissemination of brain inflammation in traumatic brain injury. Cell Mol Immunol. 2019;16(6):523–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jassam YN, Izzy S, Whalen M, McGavern DB, El Khoury J. Neuroimmunology of Traumatic Brain Injury: Time for a Paradigm Shift. Neuron. 2017;95(6):1246–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Napolitano LM, Cohen MJ, Cotton BA, Schreiber MA, Moore EE. Tranexamic acid in trauma: how should we use it? J Trauma Acute Care Surg. 2013;74(6):1575–86. [DOI] [PubMed] [Google Scholar]
- 5.Syrovets T, Simmet T. Novel aspects and new roles for the serine protease plasmin. Cell Mol Life Sci. 2004;61(7–8):873–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Medcalf RL. Fibrinolysis, inflammation, and regulation of the plasminogen activating system. J Thromb Haemost. 2007;5 Suppl 1:132–42. [DOI] [PubMed] [Google Scholar]
- 7.collaborators C-t. Effects of tranexamic acid on death, disability, vascular occlusive events and other morbidities in patients with acute traumatic brain injury (CRASH-3): a randomised, placebo-controlled trial. Lancet. 2019;394(10210):1713–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rowell SE, Meier EN, McKnight B, Kannas D, May S, Sheehan K, et al. Effect of Out-of-Hospital Tranexamic Acid vs Placebo on 6-Month Functional Neurologic Outcomes in Patients With Moderate or Severe Traumatic Brain Injury. JAMA. 2020;324(10):961–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Komanapalli ES, Sherchan P, Rolland W 2nd, Khatibi N, Martin RD, Applegate RL 2nd,et al. Epsilon Aminocaproic Acid Pretreatment Provides Neuroprotection Following Surgically Induced Brain Injury in a Rat Model. Acta Neurochir Suppl. 2016;121:311–5. [DOI] [PubMed] [Google Scholar]
- 10.Auer LM, Marth E, Heppner F, Holasek A. Proteolytic enzyme activity in patients with severe head injury and the effect of a proteinase inhibitor. Acta Neurochir (Wien). 1979;49(3–4):207–17. [DOI] [PubMed] [Google Scholar]
- 11.Roberts I, Shakur H, Coats T, Hunt B, Balogun E, Barnetson L, et al. The CRASH-2 trial: a randomised controlled trial and economic evaluation of the effects of tranexamic acid on death, vascular occlusive events and transfusion requirement in bleeding trauma patients. Health Technol Assess. 2013;17(10):1–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brotfain E, Gruenbaum SE, Boyko M, Kutz R, Zlotnik A, Klein M. Neuroprotection by Estrogen and Progesterone in Traumatic Brain Injury and Spinal Cord Injury. Curr Neuropharmacol. 2016;14(6):641–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morris MC, Bercz A, Niziolek GM, Kassam F, Veile R, Friend LA, et al. UCH-L1 is a Poor Serum Biomarker of Murine Traumatic Brain Injury After Polytrauma. J Surg Res. 2019;244:63–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morris MC, Kassam F, Bercz A, Beckmann N, Schumacher F, Gulbins E, et al. The Role of Chemoprophylactic Agents in Modulating Platelet Aggregability After Traumatic Brain Injury. J Surg Res. 2019;244:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morris MC, Singer KE, Niziolek GM, McGlone E, Veile R, Friend LA, et al. Oxygenation extremes after traumatic brain injury transiently affect coagulation. Thromb Res. 2020;186:58–63. [DOI] [PubMed] [Google Scholar]
- 16.Singer KE, Wallen TE, Morris MC, McGlone E, Stevens-Topie S, Earnest R, et al. Postinjury treatments to make early tactical aeromedical evacuation practical for the brain after TBI. J Trauma Acute Care Surg. 2021;91(2S Suppl 2):S89–S98. [DOI] [PubMed] [Google Scholar]
- 17.Boudreau RM, Johnson M, Veile R, Friend LA, Goetzman H, Pritts TA, et al. Impact of tranexamic acid on coagulation and inflammation in murine models of traumatic brain injury and hemorrhage. J Surg Res. 2017;215:47–54. [DOI] [PubMed] [Google Scholar]
- 18.Shiraishi Y, Kimura A, Matsuo O, Sakata Y, Takeshita K, Ohmori T. Short-term inhibition of fibrinolytic system restores locomotor function after spinal cord injury in mice. Sci Rep-Uk. 2019;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cuellar JM, Yoo A, Tovar N, Coelho PG, Jimbo R, Vandeweghe S, et al. The effects of Amicar and TXA on lumbar spine fusion in an animal model. Spine (Phila Pa 1976). 2014;39(19):E1132–7. [DOI] [PubMed] [Google Scholar]
- 20.Hruby Z, Wendycz D, Kopec W, Czerchawski L, Jozefowiak M, Rabczynski J. Effect of antiproteolytic drugs: epsilon-aminocaproic acid (EACA) and aprotinin on experimental anti-GBM nephritis. Nephrol Dial Transplant. 1996;11(1):32–9. [PubMed] [Google Scholar]
- 21.Goodman MD, Makley AT, Huber NL, Clarke CN, Friend LAW, Schuster RM, et al. Hypobaric Hypoxia Exacerbates the Neuroinflammatory Response to Traumatic Brain Injury. J Surg Res. 2011;165(1):30–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Niziolek GM, Boudreau RM, Baker J, Friend LA, Makley AT, Edwards MJ, et al. Acid Sphingomyelinase Inhibition Mitigates Histopathological and Behavioral Changes in a Murine Model of Traumatic Brain Injury. J Neurotrauma. 2020;37(17):1902–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levy JH. Antifibrinolytic therapy: new data and new concepts. Lancet. 2010;376(9734):3–4. [DOI] [PubMed] [Google Scholar]
- 24.Roberts I, Prieto-Merino D, Manno D. Mechanism of action of tranexamic acid in bleeding trauma patients: an exploratory analysis of data from the CRASH-2 trial. Crit Care. 2014;18(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang D, Luo ZY, Yu ZP, Liu LX, Chen C, Meng WK, et al. The antifibrinolytic and anti-inflammatory effects of multiple doses of oral tranexamic acid in total knee arthroplasty patients: a randomized controlled trial. J Thromb Haemost. 2018;16(12):2442–53. [DOI] [PubMed] [Google Scholar]
- 26.Draxler DF, Yep K, Hanafi G, Winton A, Daglas M, Ho H, et al. Tranexamic acid modulates the immune response and reduces postsurgical infection rates. Blood Adv. 2019;3(10):1598–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grant AL, Letson HL, Morris JL, McEwen P, Hazratwala K, Wilkinson M, et al. Tranexamic acid is associated with selective increase in inflammatory markers following total knee arthroplasty (TKA): a pilot study. J Orthop Surg Res. 2018;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Simon DW, McGeachy MJ, Bayir H, Clark RS, Loane DJ, Kochanek PM. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat Rev Neurol. 2017;13(3):171–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frugier T, Morganti-Kossmann MC, O’Reilly D, McLean CA. In situ detection of inflammatory mediators in post mortem human brain tissue after traumatic injury. J Neurotrauma. 2010;27(3):497–507. [DOI] [PubMed] [Google Scholar]
- 30.Sacramento CB, Sondhi D, Rosenberg JB, Chen A, Giordano S, Pey E, et al. Anti-Phospho-Tau Gene Therapy for Chronic Traumatic Encephalopathy. Hum Gene Ther. 2020;31(1–2):57–69. [DOI] [PubMed] [Google Scholar]
- 31.Hiramoto K, Yamate Y, Matsuda K, Sugiyama D, Iizuka Y. Tranexamic Acid Improves Memory and Learning Abilities in Aging Mice. J Exp Pharmacol. 2020;12:653–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoshizaki S, Kijima K, Hara M, Saito T, Tamaru T, Tanaka M, et al. Tranexamic acid reduces heme cytotoxicity via the TLR4/TNF axis and ameliorates functional recovery after spinal cord injury. J Neuroinflammation. 2019;16(1):160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Levy JH, Koster A, Quinones QJ, Milling TJ, Key NS. Antifibrinolytic Therapy and Perioperative Considerations. Anesthesiology. 2018;128(3):657–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nilsson IM. Clinical pharmacology of aminocaproic and tranexamic acids. J Clin Pathol Suppl (R Coll Pathol). 1980;14:41–7. [PMC free article] [PubMed] [Google Scholar]
- 35.Lecker I, Wang DS, Romaschin AD, Peterson M, Mazer CD, Orser BA. Tranexamic acid concentrations associated with human seizures inhibit glycine receptors. J Clin Invest. 2012;122(12):4654–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Greilich PE, Brouse CF, Whitten CW, Chi L, Dimaio JM, Jessen ME. Antifibrinolytic therapy during cardiopulmonary bypass reduces proinflammatory cytokine levels: a randomized, double-blind, placebo-controlled study of epsilon-aminocaproic acid and aprotinin. J Thorac Cardiovasc Surg. 2003;126(5):1498–503. [DOI] [PubMed] [Google Scholar]
- 37.Aoki T, Sumii T, Mori T, Wang X, Lo EH. Blood-brain barrier disruption and matrix metalloproteinase-9 expression during reperfusion injury: mechanical versus embolic focal ischemia in spontaneously hypertensive rats. Stroke. 2002;33(11):2711–7. [DOI] [PubMed] [Google Scholar]
- 38.Yamaguchi M, Jadhav V, Obenaus A, Colohan A, Zhang JH. Matrix metalloproteinase inhibition attenuates brain edema in an in vivo model of surgically-induced brain injury. Neurosurgery. 2007;61(5):1067–75; discussion 75–6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Digital Content 1: ARRIVE guidelines
Supplemental Digital Content 2: Serum MCP-1 concentrations were significantly elevated 6 to 24 hours after TBI with TXA administration. Solid line indicates significant difference between groups, p<0.05
Supplemental Digital Content 3: Serum IL-1b levels were significantly elevated 6 hours after TBI with aprotonin administration and 24 hours after TBI with TXA administration. Solid line indicates significant difference between groups, p<0.05
Supplemental Digital Content 4: Serum IL-12 concentrations were significantly elevated acutely after TBI and TXA administration with an elevation at 6 hours noted after TBI and aminocaproic acid administration. Solid line indicates significant difference between groups, p<0.05
Supplemental Digital Content 5: TXA administration significantly decreased the concentration of cerebral MCP-1 1 and 24 hours after TBI. Solid line indicates significant difference between groups, p<0.05