

Summary
Although transcription is an essential cellular process, it is paradoxically also a well-recognized cause of genomic instability. R-loops, non-B DNA structures formed when nascent RNA hybridizes to DNA to displace the non-template strand as ssDNA, are partially responsible for this instability. Yet recent work has begun to elucidate regulatory roles for R-loops in maintaining the genome. In this review, we discuss the cellular contexts in which R-loops contribute to genomic instability, particularly during DNA replication and double-strand break (DSB) repair. We also summarize the evidence that R-loops participate as an intermediate during repair and may influence pathway choice to preserve genomic integrity. Finally, we discuss the immunogenic potential of R-loops and highlight their links to disease should they become pathogenic.
While R-loops are a well-recognized source of genomic instability, replication stress, and DNA damage, there is a growing appreciation for how they contribute to genome maintenance. Here, Brickner et. al. navigate this dichotomy and discuss the consequences of R-loop dysregulation, highlighting links to the immune response and disease progression.
Introduction
How cells respond to the host of endogenous and exogenous insults that threaten to damage their genomes is particularly important to preserve genome stability. Loss of genome integrity can lead to a host of complications, from cancers to neurological disorders (Aguilera and Gómez-González, 2008). Paradoxically, transcription, a fundamental cellular process, can contribute to genomic instability. Although transcription-associated instability can result from many mechanisms, including increased DNA accessibility and interference between replication and transcription, the deregulated formation of non-B DNA structures known as R-loops has emerged as a potent source of instability (Crossley et al., 2019; García-Muse and Aguilera, 2019). R-loops are thought to occur when a nascently transcribed RNA strand invades duplex DNA behind RNA polymerases and hybridizes to the template DNA, forming an RNA-DNA hybrid (hybrid) (Figure 1). Consequently, the non-template DNA is displaced as a strand of single-stranded DNA (ssDNA). While R-loops are generally thought to form behind the polymerase, some evidence suggests that they can form anterior to RNA Polymerase II (RNAPII) during polymerase backtracking (Zatreanu et al., 2019). Hybrids can also form without displacing ssDNA, for example during Okazaki fragment synthesis. Importantly, most techniques to detect so-called R-loops, outside of bisulfite-based sequencing techniques, probe for the hybrid itself and thus cannot readily distinguish between a bona fide R-loop or a hybrid (Chédin et al., 2021). Hence in some scenarios, it is not clear what structure is actually formed. In this review, we will discuss the roles of both hybrids alone as well as R-loops, referring to the specific structure when it is clear which is known to form.
Figure 1.
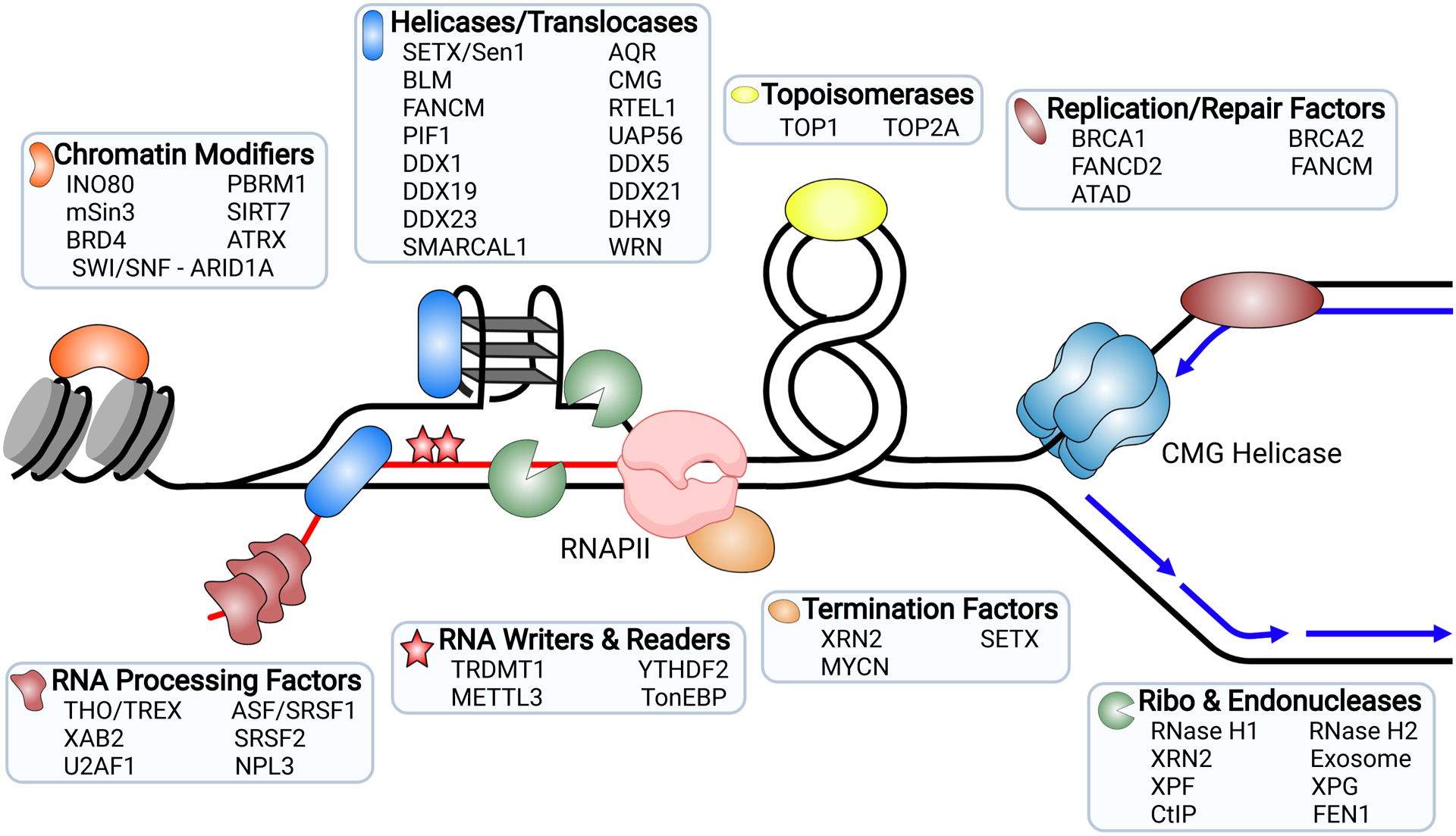
Factors that Suppress and Resolve R-loops. Factors that prevent the formation of R-loops or play a role in their resolution as referenced throughout the text. The black line is parental DNA, blue lines are daughter DNA, and RNA is depicted by red lines. The stacked structure on the ssDNA strand of the R-loop represents a G-quadruplex.
The use of imaging and sequencing-based techniques has allowed researchers to quantify R-loop levels and characterize their genomic occupancy in cells (Chédin et al., 2021; Crossley et al., 2020, 2021; Paull, 2019). These studies have shown that hybrid formation is widespread, with hybrids occupying 5–10% of the genome, and single-molecule experiments suggest they vary in length from under 60 base pairs to several kilobases. Initial attempts to quantify hybrids in unperturbed HeLa cells have put a lower estimate on their abundance at ~300 R-loops/cell, with an average half-life of 11 minutes. This suggests that cells resolve on the order of 27,000 R-loops per day (Crossley et al., 2020; Sanz et al., 2016). As products of transcription, R-loops are frequently genic in nature, although they have been found at centromeres, telomeres and other repetitive genomic regions, as well as enhancers (Liu et al., 2021a; Niehrs and Luke, 2020; Tan and Lan, 2020). They also occupy different genomic regions depending upon growth conditions or cell type. Furthermore, they tend to form in regions of high GC content and GC-skew, and the displaced ssDNA is prone to form secondary structures such as G-quadruplexes (G4s) (Castillo-Guzman and Chédin, 2021).
As dynamic structures, R-loops can be controlled at many levels, including in their formation and resolution (Figure 1). Multiple factors and pathways are involved in this regulation (reviewed in Bayona-Feliu and Aguilera, 2021; Crossley et al., 2019; García-Muse and Aguilera, 2019). These include RNA processing pathways and export machinery, which suppress hybrid formation by sequestering nascent RNA from the template, promoting chromatin compaction, and packaging and removing RNA from the nucleus. Topoisomerases also suppress hybrid formation by minimizing supercoiling. Modification of the RNA strand of the hybrid by the RNA methyltransferases METTL3 or TRDMT1 helps regulate R-loop formation (Abakir et al., 2020; Chen et al., 2020; Zhang et al., 2020). Once formed, a host of additional proteins resolve or degrade R-loops. Two nucleases dedicated to resolving R-loops in mammalian cells, RNase H1 and RNase H2, specifically degrade the RNA moiety of an RNA-DNA hybrid. RNase H2 also acts in ribonucleotide excision repair, removing single mis-incorporated ribonucleotides from DNA (Hyjek et al., 2019). Numerous helicases can also unwind R-loops and hybrids once formed, and several proteins that are involved in separate processes, such as chromatin remodelers and DNA repair nucleases, have been implicated in their resolution (García-Muse and Aguilera, 2019; Paull, 2019; Uruci et al., 2021). The many factors implicated in R-loop formation and resolution suggest the cell needs to dynamically balance these processes.
R-loops have both beneficial and harmful impacts on the cell. They are important intermediates in immunoglobulin class-switch recombination in B-cells and in mitochondrial replication (Yu et al., 2003). They have also been implicated in regulating gene expression, where they can prevent DNA methylation of promoter CpG islands and subsequent gene silencing, regulate transcriptional termination, and regulate the recruitment of chromatin remodelers (Bayona-Feliu and Aguilera, 2021; Crossley et al., 2019). Recent work has highlighted the importance of R-loops in maintaining telomeric and centromeric integrity (Liu et al., 2021a; Niehrs and Luke, 2020), and also suggests that R-loops or hybrids formed at DNA double-strand breaks (DSB) help promote DNA repair (Marnef and Legube, 2021). By contrast, R-loops can act as a source of DNA damage and genomic instability. As first shown upon perturbation of genes involved in RNA biogenesis and export, R-loop accumulation can dramatically increase DNA damage, specifically DSBs (Huertas and Aguilera, 2003; Li and Manley, 2005; Paulsen et al., 2009). Furthermore, overexpression of RNase H1 in cells can rescue R-loop induced damage in many scenarios. Precisely how R-loops increase DNA damage and genome instability is still a matter of intense investigation, and different mechanisms may be at play depending on the context in which the R-loop or hybrid forms.
In this review, we will discuss the mechanisms by which R-loops may either promote or prevent genome maintenance and stability, focusing primarily on eukaryotic processes. Excellent reviews on their physiological roles have been published, and we refer the reader to these and others on the factors that regulate R-loop formation (Bayona-Feliu and Aguilera, 2021; Crossley et al., 2019; García-Muse and Aguilera, 2019; Niehrs and Luke, 2020; Zong et al., 2020). Here, we will focus on how R-loops cause genomic instability, emphasizing their impact on DNA replication. Additionally, we will highlight advances in how R-loops are thought to promote DSB repair. Finally, we will discuss the pathological consequences of R-loop dysregulation.
R-Loops and the Replication Stress Response
During S-phase, replication forks may encounter R-loops, leading to replication stress and DNA damage. Although mechanisms exist to spatially and temporally separate transcription and replication, conflicts between these processes may occur at some frequency since they take place on the same DNA template (Hamperl and Cimprich, 2016; Lalonde et al., 2021). Indeed, numerous studies demonstrating the impact of R-loops on replication stress responses support the idea that R-loops are a source of replication stress and genome instability (reviewed in Crossley et al., 2019; García-Muse and Aguilera, 2019; Lalonde et al., 2021; Rinaldi et al., 2020). Perturbations that alter R-loop processing genes generally cause the most DNA damage in S-phase cells, suggesting that the R-loop itself affects fork progression. Early studies also demonstrated that replication forks slow at sites corresponding to an induced R-loop and that RNase H1 overexpression can restore normal fork progression in cells with deregulated R-loop formation (Gan et al., 2011; Gómez-González et al., 2011; Wellinger et al., 2006). Extending this further are observations showing that RNase H1 expression reverses fork slowing and/or fork asymmetry caused by hormone stimulation, bacterial infection, oncogene-induced replication stress, and Top1 mutations, all of which can induce R-loop accumulation (Bauer et al., 2020; Kotsantis et al., 2016; Stork et al., 2016; Tuduri et al., 2009). In recent years, much emphasis has focused on understanding the molecular mechanism by which these structures induce a block and how these barriers can be overcome.
R-loop-induced fork stalling: the nature of the block
One important question concerns the impact of replication fork orientation relative to an R-loop. Forks can encounter the transcription machinery in a head-on (HO) or co-directional (CD) orientation, leading to distinct outcomes (Figure 2A). In mammalian cells, transcription induction on an episome containing an R-loop forming region exacerbated the damage associated with both HO and CD transcription-replication conflicts (TRCs) as compared to an episome with transcription but no R-loop induction (Hamperl et al., 2017). This suggests the R-loop or R-loop-stalled transcription machinery is more of a problem than transcription itself. Moreover, although both HO and CD encounters led to DNA damage, particularly when transcription was strongly induced, HO conflicts were more detrimental. Different damage signaling pathways were also induced: HO encounters activated ATR rather than ATM, which was instead induced by CD conflicts (Figure 2A). ATM activation is likely a result of DSB formation at R-loops, but how ATR is activated is unclear. It could involve recognition of ssDNA arising at the stalled fork, on the R-loop itself, or from DSB formation and resection. As RPA binds to the ssDNA in an R-loop and recruits RNase H1 (Nguyen et al., 2017b), one possibility is that it could also recruit the ATR-ATRIP complex, contributing to ATR activation. In cells in which splicing was inhibited or splicing factors were downregulated, ATR was activated as opposed to ATM (Chen et al., 2018; Matos et al., 2020; Nguyen et al., 2018). This could indicate that fork stalling induced by HO encounters is the more problematic issue in human cells. Importantly, the activation of these kinases, in particular ATR, has been shown to prevent genome instability through multiple mechanisms described further herein.
Figure 2.
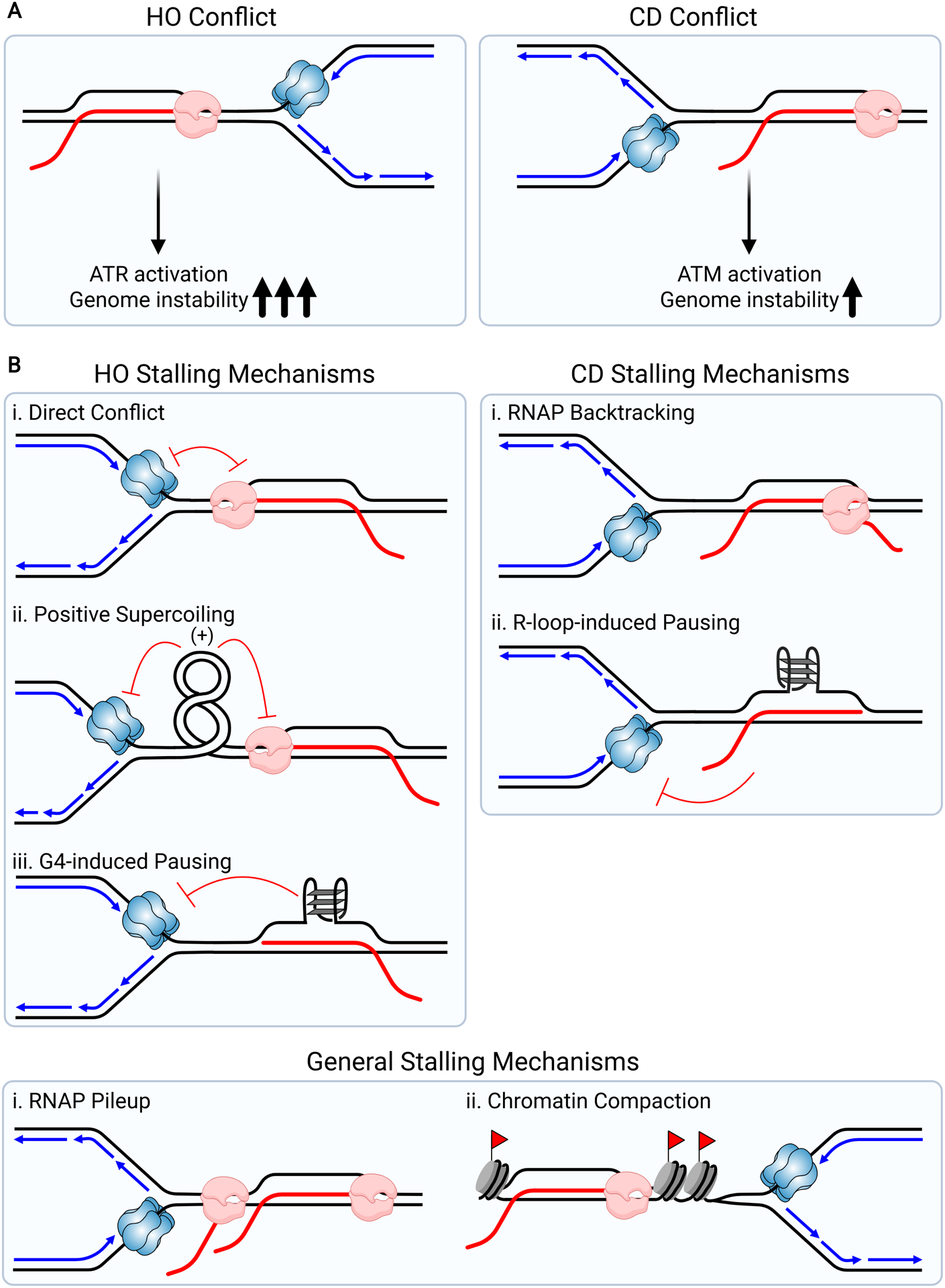
Factors Contributing to Replication Fork Stalling at R-loops. (A) Illustrates the two orientations in which a replication fork can encounter a R-loop, leading to a TRC. Head-on conflicts (HO, left) activate ATR and codirectional conflicts (CD, right) activate ATM. HO conflicts with R-loops are associated with greater genome instability. (B) Illustrates potential mechanisms that cause fork slowing in the HO (left) and CD (right) orientation. Flags represent epigenetic markers that promote chromatin compaction.
The molecular events that occur when a fork meets the transcription machinery in the HO and CD scenarios are distinct, providing some insight into the different cellular responses. In the HO orientation, the CMG helicase, which translocates on the leading strand in eukaryotes, approaches an R-loop from the opposite strand that is engaged by the transcription machinery (Figure 2B). In fact, the orientation of this approach has led some to envision that R-loops may not pose a problem for the fork. However, RNA polymerase normally engages both DNA strands, making this protein complex a likely block to CMG and/or the replicative polymerase (Barnes et al., 2015). Converging transcription and replication machineries could also lead to topological stress in the HO orientation, preventing further progression of each process. Both replication and transcription cause positive supercoiling to accumulate ahead of the respective machineries and negative supercoiling to accumulate behind them. When converging, the cumulative topological stress between the machineries may become too large to be compensated by topoisomerase activity, causing the fork to stall (Figure 2B). Furthermore, the negative supercoiling that builds up behind the replisome could promote hybrid formation in this orientation. Indeed, HO conflicts seem to enhance hybrid formation (Hamperl et al., 2017). Work in B. subtilis supports this idea and also provides insight into the mechanism by which stalling occurs. Here surprisingly, the effects of a HO vs CD encounter are similar to eukaryotic organisms, despite movement of the replicative helicase on the lagging strand (Lang et al., 2017). HO encounters induced by RNaseH loss and R-loop stabilization led to cell death and a complete failure of DNA replication in this system. Furthermore, it is topoisomerase II-dependent resolution of the positive supercoiling that promotes R-loop formation by allowing diffusion of negative supercoils behind RNAPII (Lang and Merrikh, 2021). By contrast, upon CD conflicts topological stress would be relieved. Nevertheless, in certain contexts CD encounters can be problematic. RNA polymerase may be stabilized by backtracking and pose a stronger block to fork progression (Figure 2B). Consistent with this idea, RECQL5, a helicase that associates with RNAPI and RNAPII, suppresses backtracking to reduce the incidence of TRCs and genome instability (Saponaro et al., 2014; Urban et al., 2016). Similarly, increased backtracking leads to R-loop dependent DNA damage in bacteria (Dutta et al., 2011).
There are scenarios in which an R-loop can contribute to damage regardless of orientation. For instance, RNAPII accumulation at a stalled or backtracked polymerase could exacerbate both CD and HO conflicts (Figure 2B). Additional factors may also contribute to orientation-independent damage. Studies in yeast and mammalian cells suggest that chromatin condensation contributes to the ability of R-loops to cause DNA damage. R-loop accumulation is associated with histone modifications that promote chromatin compaction, including H3S10 phosphorylation (H3S10P) and H3K9 dimethylation. Moreover, histone H3S10 phospho-mutants do not accumulate DNA damage, despite increased R-loop formation (Castellano-Pozo et al., 2013; García-Pichardo et al., 2017). One possibility is that chromatin condensation driven by these modifications contributes to R-loop-associated fork stalling (Figure 2B). Of note, R-loops themselves are not thought to be bound by nucleosomes, so these modifications are likely to be either adjacent to the R-loop, as part of a larger chromatin domain or be temporally distinct (Dunn and Griffith, 1980). By contrast, work in mammalian cells showed that H3S10P can also be protective. In this scenario, the oncogene MYCN activates Aurora A to phosphorylate H3S10, ultimately suppressing R-loop formation and TRCs (Roeschert et al., 2021). While these results seem discrepant, how R-loops promote or are regulated by chromatin condensation may be context specific, and Aurora A-mediated H3S10P may be limited to MYCN-driven genes, where avoiding damage is important to support tumorigenesis.
Another question is whether it is the R-loop alone or the stalled RNA polymerase together with the R-loop that challenges replication fork progression. As there are mechanisms for removal of stalled RNAP in the cell, both scenarios should be considered. Recent observations bring new insight to this question. Using a reconstituted Saccharomyces cerevisiae (S. cerevisiae, budding yeast)-based replication system, replication forks have been challenged by deproteinated or “naked” R-loops lacking RNA polymerase and containing a G4 on the non-template strand (Kumar et al., 2021). In this context, leading strand replication forks pause at both HO and CD R-loops but can bypass these structures and restart DNA replication on the leading strand downstream of the R-loop. Using RNase H1 to probe the hybrid’s role in these pauses, it was shown that leading strand hybrids (CD conflict) directly impede fork progression (Figure 2B). Surprisingly, however, fork stalling was still observed following RNase H1 expression when the hybrid was on the lagging strand (HO conflict). This observation, taken together with subsequent studies, suggests that in the HO orientation, the G4 formed on the leading strand as a result of R-loop formation on the lagging strand is the true cause of the fork block (Figure 2B). Hence, G4 formation in the displaced ssDNA of an R-loop may be an important determinant of an R-loop’s impact on the replication fork. Studies using a bacterial replication system are generally consistent with these findings, supporting the idea that naked R-loops can be bypassed in both orientations. They also demonstrate that a RNAP-bound R-loop poses a greater threat to fork progression than a naked R-loop, particularly in the HO orientation (Brüning and Marians, 2020). Although biochemical studies with a RNAP-bound R-loop have not yet been done in higher eukaryotes, a dCas9-associated R-loop also blocks the fork in both orientations using the reconstituted S. cerevisiae-based replication system (Schauer et al., 2020).
Mechanisms for restart
In principle, there are several ways for replication to continue when a fork encounters an R-loop. These include bypass of the R-loop and replication restart, removal of the block, either through R-loop resolution or removal of RNAPII, completion of DNA replication through use of dormant origins, or alternatively various TRC avoidance mechanisms, all of which are discussed below.
i. Fork bypass of R-loops and replication restart
It seems likely that cells require some sort of R-loop bypass mechanism to complete the replication of long genes, which can take more than one cell cycle to transcribe. Biochemical studies using the budding yeast system described above provide some basic insight into how this bypass or fork restart may occur (Kumar et al., 2021). In the CD orientation, the CMG/replisome can translocate over a naked RNA-DNA hybrid if the 5’-RNA end is flush, and if a free 3’-OH is present on the RNA strand, DNA polymerase α can extend it, allowing fork progression (Figure 3A). By contrast, if the 5’-end of the RNA is not annealed to the DNA template, CMG can unwind the flap-containing structure (Figure 3B). How the presence of RNAPII and other transcriptional regulators would affect the outcome is unclear. When RNA polymerase is present, CMG translocation along the hybrid duplex would bring it in closer proximity to the stalled RNAPII-hybrid complex without resolving the hybrid. In this case, restart would require either bypass and later dissolution of this complex, or alternatively, RNAPII removal and RNA extension using Pol α or another polymerase (Figure 3C). Alternatively, if CMG unwinds the hybrid containing the free 5’-end, it would effectively resolve that hybrid, freeing RNA polymerase to continue transcription (Figure 3D). As these machines move at similar speeds in eukaryotic cells, RNA polymerase could continue with the helicase behind it (Conti et al., 2007; Muniz et al., 2021). Whether this can occur in the genome is unclear but it is consistent with observations that R-loop levels are decreased in S-phase when the fork meets a CD R-loop in an episomal system (Hamperl et al., 2017).
Figure 3.
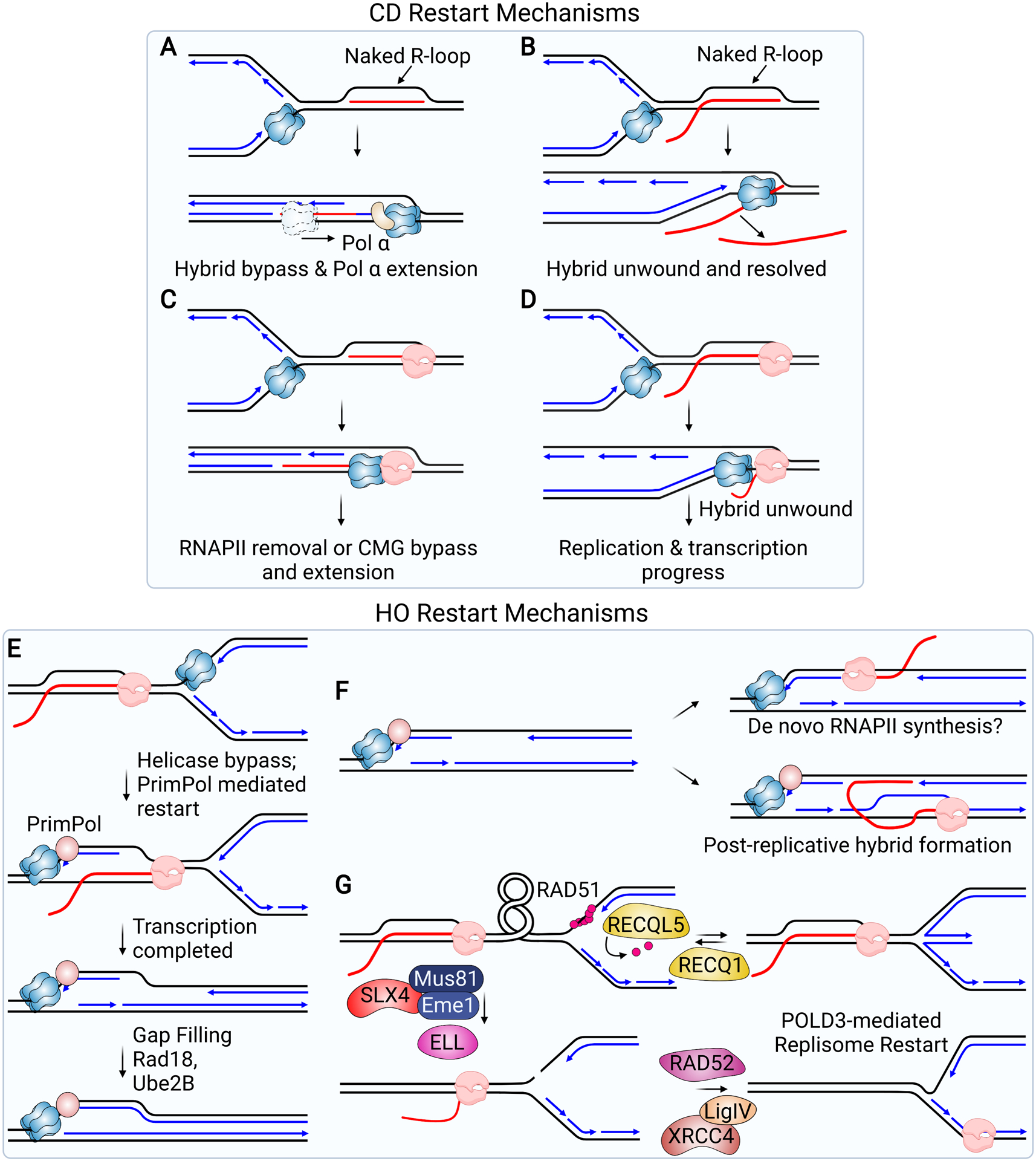
Models for the Restart of Replication Forks Stalled at R-loops. (A-D) illustrate potential bypass mechanisms in the CD orientation. (A) The CMG helicase translocates over and bypasses a “naked” hybrid when the 5’-end of the RNA strand is annealed to the DNA. Once the hybrid is bypassed, the RNA strand can be extended by Pol α. (B) The CMG helicase unwinds a hybrid when the 5’-end of the RNA strand is exposed as a flap, allowing fork progression to continue. (C) CMG translocation over the hybrid would lead to its arrest at RNAP. Fork progression at this point would require CMG to bypass the stalled RNAP or for RNAP to be removed. (D) CMG unwinding of a hybrid bound to RNAP would allow RNAP to continue and the replication fork to progress behind transcription. (E-G) illustrate potential restart mechanisms in the HO orientation. (E) CMG bypass of RNAP would leave ssDNA exposed, allowing PrimPol recruitment. Primer synthesis by PrimPol allows the fork to continue, leaving behind ssDNA that can be replicated by a gap-filling mechanism once transcription is complete. (F) ssDNA resulting from bypass and repriming or a failure to reprime, and which persists behind the replication fork, could serve as a template for the formation of hybrids. Hybrids could form through de novo synthesis by RNAP (top) or through the association of nascent RNA resulting from transcription after the fork has passed (bottom). (G) Restart of a stalled fork initiated by SLX4 and MUS81-mediated fork cleavage is thought to relieve the torsional stress, resulting in resolution of the R-loop formed during a HO conflict. Religation of the fork by XRCC4/LIG4 and ELL-mediated restart of transcription allows both replication and transcription to continue. Prior to fork cleavage, RAD51 may promote fork reversal. RECQ1 is needed to reset the fork and RECQ5 promotes removal of RAD51.
In the HO orientation, a fork can progress past a naked R-loop on the lagging strand as long as no secondary structure is present on the leading strand to impede unwinding or polymerase progression. If such a structure is present, the fork can restart downstream of the impediment, generating a gap. However, when RNAPII is present, restart is more complex. In bacteria, a full halt to DNA replication is observed in the HO orientation, and in mammalian cells HO conflicts lead to ATR activation, which is indicative of fork stalling. Since RNAPII interacts with both strands of duplex DNA, some alternative restart mechanism would need to be employed. If CMG could bypass the RNAPII block as it can bypass protein-DNA crosslinks (Huang et al., 2013; Sparks et al., 2019), restart might happen using PrimPol, an enzyme with both primase and polymerase activities (Figure 3E). Indeed, PrimPol suppresses extensive R-loop formation at GAA repeats, where R-loops can promote formation of a triplex structure (Šviković et al., 2019). PrimPol may mediate repriming in the wake of the uncoupled replicative helicase, suppressing ssDNA formation and hybrid formation on that ssDNA. Consistent with this idea, two factors which minimize ssDNA formation by promoting gap filling during post-replicative repair, RAD18 and UBE2B, also suppress hybrid formation in a post-replicative fashion (Figure 3E) (Barroso et al., 2019). These observations suggest that ssDNA forming behind the fork as a result of gap formation and/or gap resection may be a template for hybrid formation. In this context, RNAs transcribed on the other strand could bind to form a hybrid, or RNAPII could be recruited to the DNA for de novo RNA synthesis and hybrid formation (Figure 3F) (Barroso et al., 2019; Delamarre et al., 2020; Kemiha et al., 2021). As RNAPII can initiate transcription on ssDNA in the absence of a promoter sequence in vitro and has been shown to act on the ssDNA of an R-loop to initiate antisense transcription (Kadesch and Chamberlin, 1982; Tan-Wong et al., 2019), it could bind DNA gaps to synthesize RNA behind the fork.
The nuclease MUS81 also plays a role in fork restart at R-loops, initiating a fork cleavage and religation cycle that restores both transcription and replication (Figure 3G) (Chappidi et al., 2020). SLX4, MUS81 and its partner protein EME1 are proposed to cleave the fork, relieving torsional stress and resolving the R-loop generated by a HO conflict. Following ELL-mediated transcriptional restart and LIG4/XRCC4-mediated fork religation, replication restarts, allowing replication and transcription machineries to traverse each other. MUS81-EME1 acts with RAD52 and POLD3 in this process to promote fork restart at R-loops. These factors are proposed to mediate parental DNA strand annealing and DNA synthesis respectively, promoting a form of semi-conservative DNA replication. Prior to initiating this restart pathway, RAD51-mediated replication fork reversal may also occur (Figure 3G) to stabilize the stalled fork (Chappidi et al., 2020). When this occurs, RECQ1 and RECQ5 are required to reset the reversed fork and remove RAD51 at the R-loop stalled fork, respectively, such that cleavage and restart can occur.
ii. Resolution of the R-loop or transcription block
Fork bypass of the R-loop is one mechanism for allowing both continued replication and transcription, but RNA polymerase removal, transcription termination and auxiliary helicases can also facilitate fork progression. While these events could occur in all cell cycle phases, there are instances in which they are coupled to replication fork progression. In budding yeast, RNAP mutants with increased chromatin binding capacity exhibit increased TRC-associated DNA damage and genomic instability, suggesting the persistence of RNAP on chromatin is problematic for the fork (Felipe-Abrio et al., 2015). Mec1, the budding yeast ortholog of ATR, regulates the chromatin remodeler Ino80 and the transcription complex PAF1 to remove chromatin-bound RNAPII at certain transcribed genes upon HU-induced replication stress (Figure 4A) (Poli et al., 2016). This promotes fork progression and restart, consistent with the idea that RNAPII removal helps to resolve TRCs. Yeast RNAPIII may also be regulated by Mec1 to limit hybrid accumulation and control its chromatin binding (Hurst et al., 2021).
Figure 4.
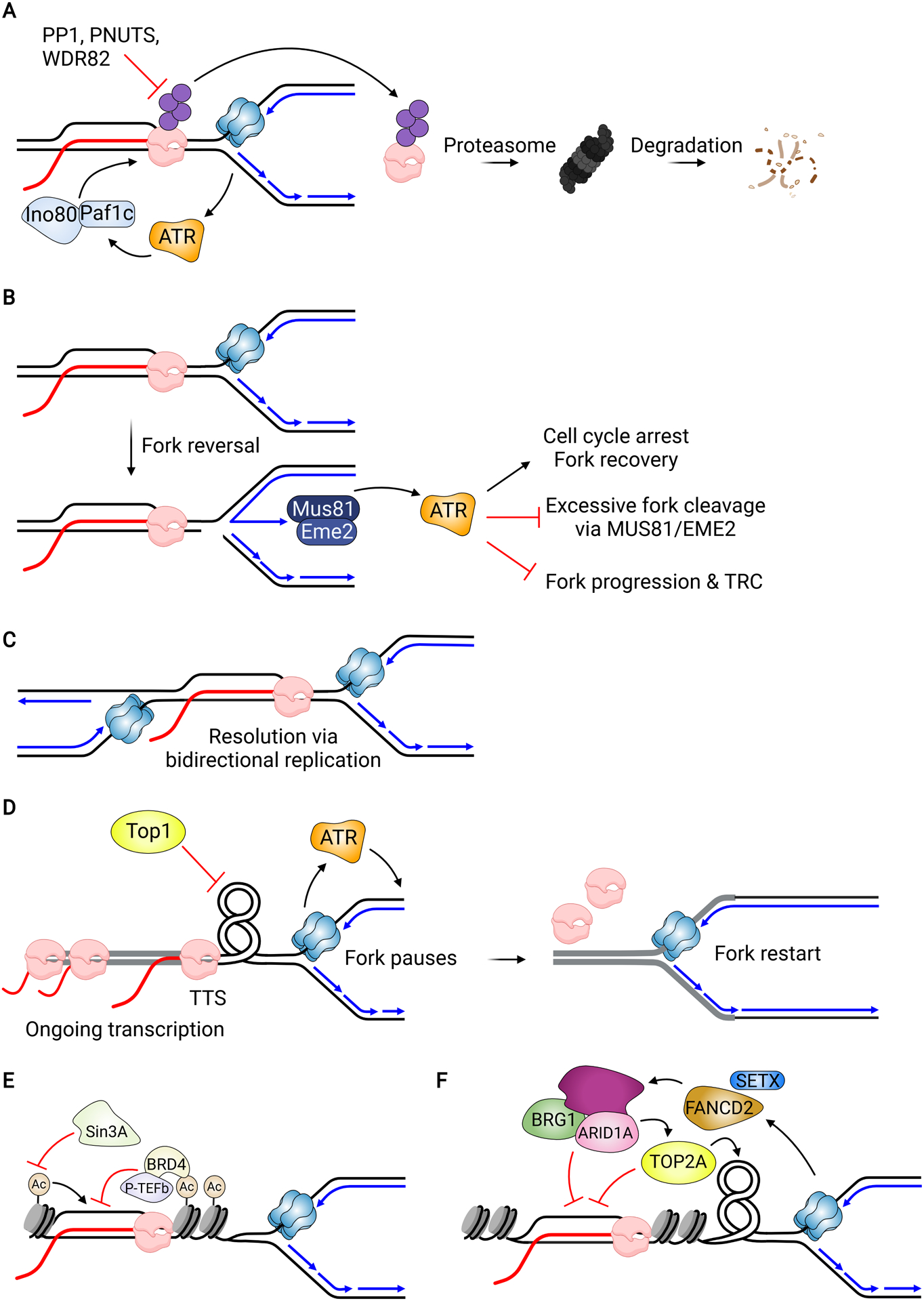
Potential Mechanisms for Resolution of R-loop Induced Transcription-Replication Conflicts. (A) In budding yeast, RNAP removal and proteasomal degradation can be initiated by activation of ATR and mediated by Paf1c and Ino80. RNAP phosphorylation also promotes its degradation, which is counteracted by the protein phosphatase PP1 and its regulatory subunit PNUTS and WDR82. (B) ATR activation at stalled forks is dependent upon MUS81-EME2-mediated fork cleavage, which is needed to generate ssDNA. Activation prevents additional and excessive fork cleavage by MUS81 and suppresses TRC formation. Other characterized functions of ATR may also promote fork recovery. (C) A fork stalled at an HO conflict can be rescued by a fork approaching the R-loop from the CD orientation. (D) Fork pausing at the 3’-end of a gene (highlighted in gray) located near an origin allows time for transcription to complete before the fork progresses into the gene. The converging fork and transcription lead to torsional stress that is relieved by TOP1. Pausing also activates ATR, which may reinforce the slowdown by inhibiting elongation. (E) Histone deacetylation suppresses R-loop formation in diverse ways, preventing genome instability. (F) SWI/SNF complexes including the subcomplex containing ARID1A act in a pathway with FANCD2 and SETX to suppress R-loop formation and TRCs. ARID1A recruits TOP2A to forks to relieve torsional stress.
Mechanisms for RNAPII removal after transcription stress are also known in higher eukaryotes. For example, one mechanism for RNAPII removal involves its ubiquitination and degradation, which can occur following UV damage or other types of transcription blocking lesions (reviewed in Noe Gonzalez et al., 2021). RNAPII phosphorylation at S5 was shown to prevent its degradation and turnover. Depletion of factors that promote S5 dephosphorylation, including the protein phosphatase PP1 and its regulatory subunits PNUTS and WDR82, increase RNAPII stability on chromatin and promote TRCs (Figure 4A) (Landsverk et al., 2020). Furthermore, INO80 can promote R-loop turnover in mammalian cells when it is tethered to an R-loop site, and its knockdown increases R-loop formation (Prendergast et al., 2020). Whether these pathways help restart forks stalled at R-loop associated TRCs is still unclear. It is also unclear if ATR regulates any of these pathways in a manner analogous to what occurs in yeast, although ATR-dependent repression of transcription in the vicinity of stalled forks, which could suppress TRCs and R-loop formation, has been reported (Im et al., 2014).
Although ATR’s role in controlling transcription is poorly understood, it has better known roles in fork stabilization and restart that may be important at R-loop stalled forks. Interestingly, MUS81 may cooperate with ATR in a negative feedback loop that protects cells from R-loop induced damage (Matos et al., 2020). MUS81, acting with its partner protein EME2, generates ssDNA at stalled forks through fork cleavage, activating ATR (Figure 4B). This activation suppresses additional R-loop formation and prevents fork collapse mediated by the MUS81-EME1 complex. As ATR inhibitor treatment led to more DSB formation when R-loops accumulate, as well as a defect in fork restart and increased TRCs, it seems likely that ATR activation serves multiple roles in this context. Though some may be related to ATR’s suppression of origin firing and arrest of cell cycle progression, ATR may also suppress the activity of other enzymes, including the MUS81-EME1 complex. Alternatively, or in parallel, it could suppress the restart of forks cleaved by MUS81-EME1. Clearly further work is required to sort out this complex pathway and elucidate how MUS81-EME1 can have both beneficial (as in the fork cleavage and religation model) and toxic roles.
Transcription termination is another mechanism for RNAPII removal, and in fact, termination defects are associated with R-loop formation and genome instability (Hatchi et al., 2015; Morales et al., 2016; Skourti-Stathaki et al., 2011). Numerous mechanisms for premature termination following transcription stress have been described that involve RNA cleavage and RNAPII disengagement (reviewed in Noe Gonzalez et al., 2021). In these cases, specialized helicases and nucleases, such as SETX (Hatchi et al., 2015; Skourti-Stathaki et al., 2011), DHX9 (Cristini et al., 2018), DDX5 (Mersaoui et al., 2019), and XRN2 may play important roles (Skourti-Stathaki et al., 2011).
MYCN, an oncogenic transcription factor often amplified in neuroblastomas, may have an analogous function, acting as a transcription terminator to resolve replication blocks. In MYCN-driven cells, MYCN acts via a two-tiered mechanism to facilitate RNAPII removal. First, MYCN recruits the exosome to stalled and backtracked RNAPII to promote transcription termination and to prevent fork stalling and DSB formation (Papadopoulos et al., 2022). Should breaks occur, MYCN indirectly recruits BRCA1 to promoter-proximal pause sites, which further recruits mRNA decapping complexes to suppress R-loop formation at these regions. Although this occurs in a cell cycle independent manner, it could also limit TRCs by suppressing the accumulation of stalled RNAPII (Herold et al., 2019; Papadopoulos et al., 2022).
Finally, SETX and other auxiliary helicases and translocases may help remove RNAP and/or the R-loop outside the context of termination and play a more direct role in the resolution of TRCs (Figure 1). SETX is associated with stalled replication forks in mammalian cells and in budding yeast, and its loss leads to increased R-loop formation and genome instability (Alzu et al., 2012; Costantino and Koshland, 2018; Skourti-Stathaki et al., 2011; Yüce and West, 2013). The FANCM helicase/translocase, or its yeast homolog Mph1, also plays a role at TRCs to suppress R-loop formation. FANCM’s ability to resolve R-loops in vitro helps to release RNAP from transcriptional arrest or promote its removal from chromatin (Lafuente-Barquero et al., 2017; Schwab et al., 2015). Furthermore, FANCM may be important to resolve TRCs at telomeres in ALT cells by disrupting TERRA R-loops (Pan et al., 2019). RTEL1 can also act in a pathway with the polymerase δ-associated factor Poldip3 to resolve R-loops formed at TRCs. This may be particularly important at common fragile sites and other loci that are difficult to replicate or which may form G4s (Björkman et al., 2020; Kotsantis et al., 2020; Wu et al., 2020). Similarly, the Pif1 helicase, which translocates on the strand opposite the CMG helicase, can assist in removing R-loops bound by Cas9 in both orientations, raising the possibility it could help remove RNAPII at forks (Schauer et al., 2020). A host of other helicases and translocases that can resolve R-loops have been identified that may also play a role in this process, including AQR, BLM, WRN, SMARCAL1, DDX1, DDX5, DDX19, DDX21, DDX23, DHX9, and UAP56 (Figure 1) (Chakraborty and Grosse, 2011; Chang et al., 2017a; Cristini et al., 2018; Hodroj et al., 2017; Kim et al., 2020; Marabitti et al., 2019, 2020; Matson and Zou, 2020; Mersaoui et al., 2019; Pérez-Calero et al., 2020; Pugliese et al., 2019; Sollier et al., 2014; Song et al., 2017; Sridhara et al., 2017; Tan et al., 2020a). Interestingly, the PCNA unloader ATAD5 may regulate the action of several of these helicases, helping recruit them to the replication fork to suppress R-loops and TRCs (Kim et al., 2020). Why the activity of so many helicases is required and how their activity is coordinated remain open questions.
iii. Origin firing and genome organization
Another mechanism for the rescue of forks stalled at R-loops is related to eukaryotic genome organization and the bidirectional nature of origin firing. In prokaryotes, most genes are replicated in a codirectional fashion. Those oriented for replication in a head-on fashion are generally stress responsive genes for which there is pressure to select for beneficial adaptive mutations that promote stress tolerance or increased virulence (Lang et al., 2017). Similarly, origins are often located upstream of the promoter regions of highly expressed genes in higher eukaryotes. Hence, replication is often codirectional with transcription, an arrangement that minimizes the problems associated with TRCs (Chen et al., 2019; Petryk et al., 2016). As forks approach a gene from both directions, a fork approaching from the opposite direction may complete replication if R-loop-induced fork stalling occurs (Figure 4C) (Brambati et al., 2018). Many TRCs in the eukaryotic genome could be resolved by the very nature of this genomic organization.
iv. TRC avoidance by fork pausing
Active fork pausing may be a(nother) mechanism to avoid a TRC. Recent work suggests that forks may pause in the HO orientation at the 3’-end of genes containing R-loops when located in close proximity to an origin (Figure 4D) (Promonet et al., 2020). This pausing may be an active process that can prevent TRCs by allowing for the completion of transcription, which has been shown to occur in bursts (Tantale et al., 2016). ATR-dependent RPA phosphorylation is seen specifically at these R-loop-containing transcription termination sites within the genome, and may result from the fork pausing that arises in response to topological stress building up upon convergence of RNA and DNA polymerases (Promonet et al., 2020). While ATR activation could help to slow fork progression, it may also promote RNAPII displacement ahead of the fork in a manner analogous to what has been reported in S. cerevisiae (Poli et al., 2016). Interestingly, H3K4 methylation in yeast, which is abundant in highly transcribed genes, acts in a similar manner to suppress TRCs in checkpoint mutants by slowing the rate of DNA replication (Chong et al., 2020). Like fork pausing, altering fork speed could allow replisomes to avoid the transcription machinery.
v. TRC avoidance by chromatin remodeling
Cells also alter the local chromatin state to avoid R-loop associated TRCs (Bayona-Feliu and Aguilera, 2021). Among the factors involved, several regulate histone post-translational modifications, such as acetylation. Initial observations revealed that mutation of THO, mSin3 and other proteins involved in regulating histone lysine deacetylation leads to R-loop accumulation and R-loop dependent DNA damage (Salas-Armenteros et al., 2017; Wahba et al., 2011). Several other chromatin regulators have also been implicated in suppressing R-loop formation and TRCs (Figure 4E). Whether they resolve the R-loop at a TRC or simply prevent R-loop formation to avoid TRCs is not clear. Among those factors involved are the yeast SIRT6 homologs Hst3 and 4 (Feldman and Peterson, 2019), the mammalian SIRT7 (Song et al., 2017), and BRD4, a bromodomain containing protein that recognizes acetylated lysines (Edwards et al., 2020; Lam et al., 2020). BRD4 may prevent R-loop formation through its interaction with the elongation factor, pTEFb. Changes to histone acetylation status may therefore control R-loop levels by altering chromatin compaction and suppressing transcription, thereby minimizing the ability of the nascent RNA to interact with the DNA template upon TRCs.
Several proteins directly involved in chromatin remodeling, which is important for nucleosome positioning and chromatin relaxation, may also suppress R-loop associated TRCs (Figure 4F). Among these are components of the SWI/SNF complex and its catalytic subunit BRG1, which appears to act with FANCD2 and SETX to suppress R-loop formation and TRCs. BRG1 is found at stalled forks and limits chromatin accessibility at R-loops sites, suggesting it may reduce the ability of nascent RNA to bind to the template DNA (Bayona-Feliu et al., 2021). Members of the cBAF and PBAF SWI/SNF complex subtypes, ARID1A and PBRM1 respectively, may also be involved (Chabanon et al., 2021; Tsai et al., 2021). ARID1A helps to recruit topoisomerase IIa (TOP2A) to chromatin and may suppress R-loop associated TRCs by promoting the resolution of topological stress at the conflicts. Finally, the remodelers INO80 (Poli et al., 2016; Prendergast et al., 2020), and ATRX have been implicated in TRC resolution and/or R-loop suppression (Nguyen et al., 2017a; Yan et al., 2022).
R-loops and fork progression
An emerging question in the field is whether fork slowing at an R-loop is actually problematic for the cell or whether post-replicative hybrid formation is an additional culprit (Kemiha et al., 2021). Although the idea that forks slow upon approaching an R-loop is certainly the dominant model, it is important to better establish whether slowing and fork reversal actually occurs in front of the R-loop. Genetic studies are consistent with the involvement of fork reversal factors, but forks reversed at an R-loop have not yet been observed by electron microscopy. Independent verification by this or another single-molecule method is needed. One intriguing possibility is that fork slowing is alternatively, or additionally, due to post-replicative hybrid formation on the ssDNA that forms upon bypass of the R-loop (Figure 3F).
Recent data raise questions about the idea that R-loops pose a direct block to fork progression. Loss of many R-loop resolution factors leads to fork slowing and asymmetric fork progression as measured by DNA fiber assays, indicating that not all forks stall. Consistent with the idea that R-loops are involved in this slowing, the block is relieved by RNaseH expression (Salas-Armenteros et al., 2017; Tuduri et al., 2009). Surprisingly, however, most forks slow under these conditions, despite the fact that not all will be in the vicinity of an R-loop. This suggests there may be some type of global mechanism for fork slowing. Local slowing at R-loops may activate a checkpoint, which in turn slows global elongation. Evidence for such a global fork response has been reported under other conditions: ATR globally slows fork progression by regulating fork reversal in response to interstrand crosslinks (Mutreja et al., 2018), and checkpoint dependent control of both initiation and elongation have been observed (Bacal et al., 2018; Can et al., 2019; Seiler et al., 2007).
How hybrids formed behind the replication fork would slow DNA replication is not clear. One possibility is that they interfere with homologous recombination-mediated fork restart if not efficiently removed. Alternatively, they could suppress histone deposition, leading to other changes that promote fork slowing. Clearly, further work is needed to investigate this and other possibilities.
Replication-Independent Mechanisms for R-Loop Induced DNA Damage and Genome Stability
Although the impact of R-loops on genome stability is largely related to their effects on DNA replication, R-loops form outside of S-phase and may also threaten genome stability in a replication-independent manner (Hamperl et al., 2017; San Martin-Alonso et al., 2021). One mechanism by which R-loops could cause damage outside of S-phase involves nucleolytic processing. The endonuclease activity of two factors involved in nucleotide excision repair, XPF and XPG, is responsible for the DNA DSBs formed when R-loops are induced, and the absence of XPG increases cellular R-loop levels (Makharashvili et al., 2018; Sollier et al., 2014; Stork et al., 2016). This suggests that these flap-endonucleases process these structures, although the mechanism by which they act is not well understood (Figure 5A). Should XPF and XPG cleave opposite strands of the R-loop, this processing would directly generate a DSB. Conversely, XPF and XPG may cleave the same strand upstream and downstream of the hybrid, respectively, generating a single strand break (SSB). If two R-loops in close proximity are processed in this way, it could produce a DSB outside of S-phase. We note however that some DSBs arising from XPF and XPG-mediated processing may not be S-phase independent, as the SSB could remain until the cell progresses into S-phase, where it leads to DSB formation upon encountering the replication fork. It was shown that the splicing factor XAB2 helps recruit XPG and XPF to R-loops, suggesting that these factors might act outside the context of classical nucleotide excision repair (Goulielmaki et al., 2021).
Figure 5.
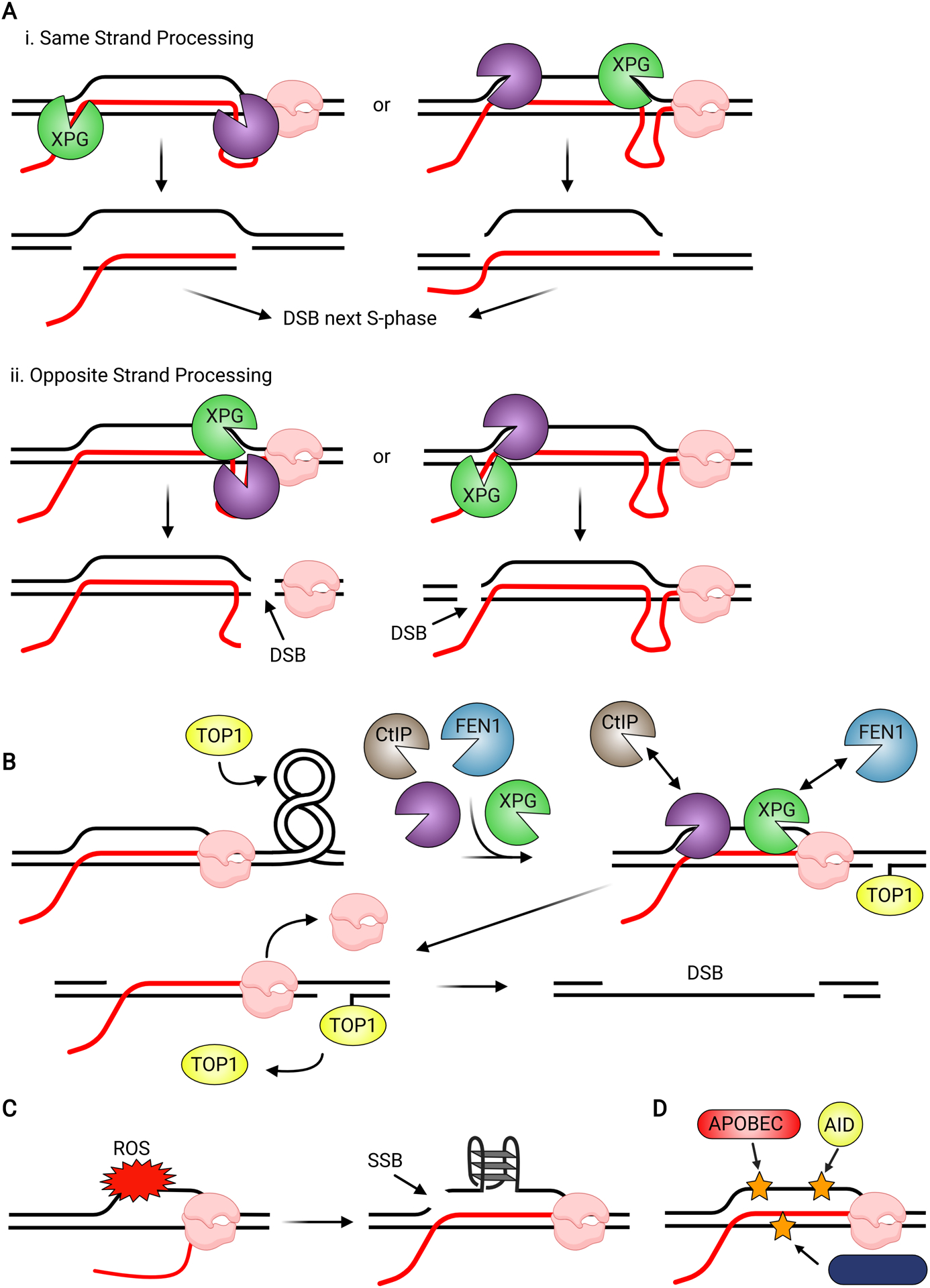
Potential Mechanisms of Replication-Independent R-loop-mediated DNA Damage. (A) The TC-NER nucleases XPG and XPF initiate R-loop processing by cleaving the 5’ or 3’-flap of the R-loop-template DNA junction, respectively. Cleavage may occur on either the template strand, excising the hybrid, or on the non-template strand, excising the displaced ssDNA to generate a single-strand break. Conversely, XPG and XPF may cleave opposing DNA strands of the R-loop, generating a DSB either 5’ or 3’ to the R-loop. (B) R-loop processing by several nucleases and TOP1-mediated DNA nicking in response to camptothecin generate two SSBs on opposing DNA strands, which mimic a DSB. (C) The displaced ssDNA of a transcription bubble is more accessible to damage from reactive oxygen species (ROS). ROS-generated SSBs in the displaced ssDNA promote unscheduled R-loops and G4s. (D) AID and APOBEC proteins can convert cytosine to uracil in the displaced ssDNA. Downstream processing of uracil results in an abasic site, which can cause SSBs. Additionally, the RNA editor ADAR has some activity towards the DNA strand of the hybrid. Base modifications are represented by stars.
R-loops also contribute to DSB formation in quiescent cells treated with camptothecin (CPT), a topoisomerase I (TOP1) inhibitor. R-loops induced by CPT contain a trapped TOP1 cleavage complex. Proteolytic removal of this complex leads to a SSB on one strand, while R-loop processing on the other strand generates a second SSB (Figure 5B). In this case, the endonucleases XPF, XPG, FEN1 and MRE11 all contribute to break formation (Cristini et al., 2019). Similarly, the flap endonuclease activity of the Sae2/CtIP protein, normally involved in DSB resection, can suppress R-loop and DNA break formation in both untreated and CPT-treated human cells (Makharashvili et al., 2018). In all cases, whether nucleolytic processing is required for repair/removal of the R-loop or whether the nucleases are acting in a gratuitous and pathological fashion simply because the structure is accessible for processing is not clear. The accumulation of R-loops in the absence of CtIP and XPG suggests that this may be a step in the repair of R-loops (Makharashvili et al., 2018; Sollier et al., 2014), although with CPT, processing may be more pathological. In either case, the nature of repair could depend on the cell cycle stage, with recombination initiated after DSB formation in S-phase, and some sort of R-loop excision or unwinding initiated by break formation outside of S-phase. Clearly further work is needed to understand how these nucleases contribute to both repair and genome instability. As the ssDNA of an R-loop structure is no longer protected by the other DNA strand or bound proteins, it may also be more accessible and prone to damage via endogenous metabolites such as reactive oxygen species (ROS) or certain enzymatic activities. For instance, DNA nicks, such as those generated by ROS, can stabilize and promote R-loop formation (Roy et al., 2010). Targeting ROS to actively transcribed genes or telomeres promotes G4 and R-loop formation, resulting in replication-independent damage at these sites (Figure 5C) (Tan et al., 2020a, 2020b; Teng et al., 2018). Furthermore, damage in the extruded ssDNA could arise due to the action of cytidine deaminases such as the APOBEC proteins or AID (Saini et al., 2017; Yu et al., 2003; Zheng et al., 2017). The resulting uracil that forms would be subject to glycosylase processing and base excision repair, ultimately causing SSB formation (Figure 5D). Indeed, AID acts on R-loops in the switch regions during immunoglobulin class-switch recombination (Yu et al., 2003). RNA editing enzymes, such as ADAR1, may also modify the DNA moiety of a hybrid, providing an alternative mechanism of SSB formation (Zheng et al., 2017).
R-loops and Mitochondrial Genomic Stability
The impact of R-loops on genome stability is not restricted to the nuclear genome but extends to the mitochondrial DNA (mtDNA), where R-loops are important intermediates in mtDNA replication (Holt, 2019). Mitochondrial DNA is circular and consists of a heavy (H) and a light (L) strand, each of which contain an origin, OriH and OriL, respectively. Intriguingly, mtDNA replication is predominantly asynchronous, beginning on the H-strand. Before replication, R-loops form across OriH (Holt, 2019) then are processed by RNase H1 to generate the RNA primer from which the replicative polymerase initiates nascent DNA synthesis (Lima et al., 2016; Posse et al., 2019). Synthesis of the L-strand begins once replication of the H-strand reaches OriL (Falkenberg, 2018). Thus, mitochondrial R-loop regulation is vital to ensure that the mtDNA is properly replicated and that mitochondrial levels are maintained.
As a regulator of mtDNA replication, RNase H1 is particularly important in maintaining mitochondrial genome stability, and its levels need to be properly regulated. In Arabidopsis thaliana, loss of the RNase H1 homologs AtRNH1B and AtRNH1C caused mitochondrial R-loop accumulation and elevated levels of mitochondrial recombination. R-loop accumulation reduced mitochondrial copy number and caused embryonic lethality, suggesting that the mitochondrial genome is unstable (Cheng et al., 2021). Loss of RNase H1 in the liver of mice also led to R-loop-induced mitochondrial genomic instability. This instability was associated with mitochondrial dysfunction, including altered fusion and fission dynamics, and liver degeneration (Lima et al., 2016). In contrast, expression of catalytically inactive RNase H1 mutants in human cells reduced R-loop levels (Akman et al., 2016). In these cells, mitochondria aggregation was observed, indicating that mtDNA segregation is impaired, another feature of genomic instability. Perturbations of other genes may also cause RNase H1-mediated mitochondrial dysfunction. In BRCA2-deficient cells, ROS levels are elevated, generating 8-oxoguanine. This inhibits RNase H1 recruitment to OriH, thus mimicking RNase H1-deficiency (Renaudin et al., 2021). These cells accumulate R-loops in the mtDNA, leading to its instability. While the molecular mechanism by which RNase H1 loss destabilizes the mitochondrial genome is not clear, recent work suggests that mtDNA replication initiation is promiscuous in the absence of RNase H1, which likely accounts for this instability (Posse et al., 2019). Additionally, persistent hybrids can interfere with initiation and progression of L-strand DNA synthesis (Chang et al., 2020). Further investigation into this mechanism is required.
In addition to RNase H1, several other factors are implicated in maintaining the mtDNA integrity. In mammalian cells, members of the mitochondrial degradosome, such as SUV3 and PNPase, act analogously to RNase H1 to prevent R-loop accumulation in the mtDNA. In the absence of the degradosome, mtDNA synthesis was impaired and mitochondrial DNA content was lost, suggesting that the degradosome maintains mitochondrial genome stability (Silva et al., 2018). Proper localization of the helicase RECQ4 to the mitochondria is also required to regulate R-loop formation and resolution and maintain genome stability (Chang et al., 2020). As mitochondrial dysfunction is associated with multiple diseases, delineating how different R-loop resolution factors contribute to the maintenance of the mitochondria is imperative.
R-loops and DNA Break Repair
Double-strand breaks (DSBs) are one of the most toxic forms of DNA damage and are a primary driver of genome instability. Although early observations showed that unscheduled R-loops can cause DSB formation, DSBs have also been shown to elicit elevated R-loop levels. Cells employ a variety of strategies to repair DSBs, including homologous recombination (HR) and non-homologous end joining (NHEJ) (Chang et al., 2017b; Chapman et al., 2012). During HR, DSB ends are resected by nucleases, generating ssDNA. RPA and RAD51 bind to ssDNA and facilitate strand invasion into the sister chromatid, providing a template for error-free repair of the break. HR is primarily restricted to the S and G2 phases of the cell cycle, consistent with the requirement for a sister chromatid. During NHEJ, which is potentially error-prone, DNA DSB ends are initially recognized by the KU complex and a host of other factors, including 53BP1, that initiate processing of damaged ends but protect them from extensive resection prior to religation. Importantly, there is growing recognition that R-loops may directly influence the DSB repair process. Indeed, evidence exists that, while their deregulation can impede DSB repair, R-loops and hybrids are important intermediates that facilitate this process.
R-loops hinder DNA repair and augment genomic instability
The role of R-loops and hybrids in DSB repair is debated. On one hand, hybrids have been proposed to hinder the repair of breaks once formed. Recent work using a reporter in budding yeast where DSBs were induced at transcriptionally active or silenced regions demonstrated that hybrids accumulate at transcriptionally active breaks (Ortega et al., 2021). When R-loop resolution factors were lost, transcription impaired recombination and increased genome instability at induced DSBs. This effect could be rescued by overexpressing RNase H1, clearly suggesting that hybrids can interfere with DSB repair processes.
Although mechanisms by which hybrids impede repair are not fully understood, several studies suggest they may alter end resection processes. DNA ends are recognized by MRE11-RAD50-NBS1 (MRN) and CtIP, which initiate resection by nicking the DNA on either side of the break (Reginato and Cejka, 2020). EXO1 and DNA2 subsequently promote long-range resection, in a process requiring the BLM helicase (Nimonkar et al., 2011; Sturzenegger et al., 2014). The loss of certain R-loop resolution factors, such as DDX5, DHX9, and HNRNPD/SAF-A leads to hybrid accumulation and impaired end resection at induced DSBs (Figure 6A) (Alfano et al., 2019; Matsui et al., 2020; Sessa et al., 2021; Yu et al., 2020). In each case, hybrid retention diminished RPA and RAD51 loading and was associated with increased genomic instability. Therefore, accumulated hybrids at break sites may block nucleases from binding to the DNA ends, preventing resection. They may also mask ssDNA recognition by RPA, thereby inhibiting downstream RAD51 nucleofilament formation and homology search. In support of the former, in vitro experiments demonstrated that long hybrids at DNA ends cannot be unwound by BLM and inhibit EXO1 activity (Daley et al., 2020).
Figure 6.
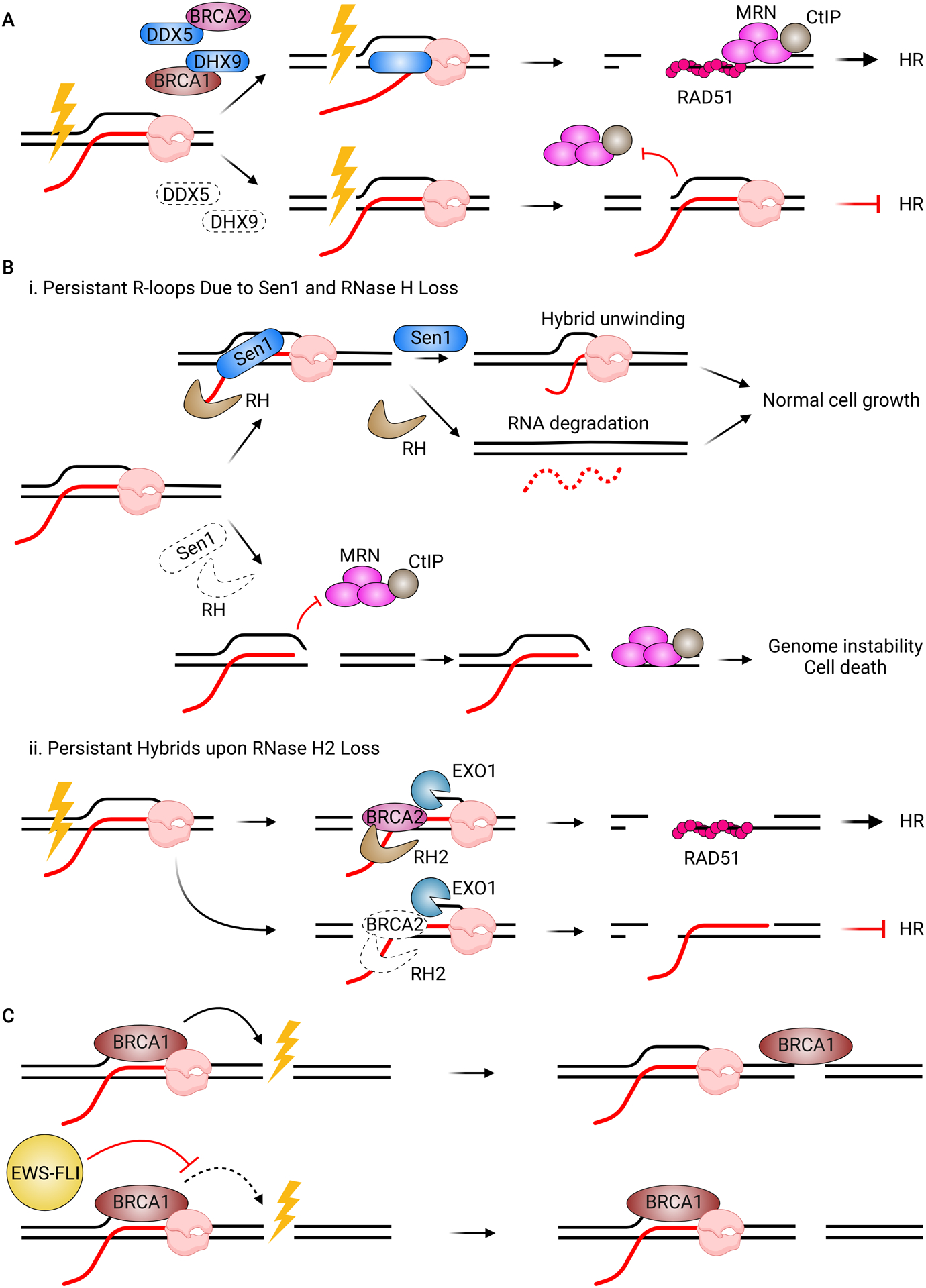
Unscheduled R-loops Interfere with DNA Repair. (A) Various helicases, including DDX5 and DHX9, act with BRCA1 to resolve hybrids that form when a DSB occurs in an actively transcribed locus. Loss of DDX5 or DHX9 results in a persistent R-loop at the break site, which inhibits MRN/CtIP-mediated end resection, leading to HR defects. (B) (i) In budding yeast, Sen1 and RNase H (RH) resolve hybrids. When both factors are lost, persistent R-loops result in a DSB forming downstream of the R-loop. While resection can occur normally on the hybrid-free end, the R-loop blocks MRN/CtIP activity, preventing resection and resulting in genomic instability and cell death. (ii) In wild-type cells, BRCA2 recognizes an R-loop near a DSB and recruits RNase H2 (RH2) and EXO1. EXO1 resects the DNA strand, while RNase H2 degrades the RNA, promoting RAD51 nucleofilament formation and proper HR. Upon loss of BRCA2 or RNase H2, the hybrid persists, blocking RAD51 loading and impairing HR. (C) In normal cells, BRCA1 associates with RNAP to promote transcription termination and resolve R-loops at termination sites. Upon DSB induction, BRCA1 dissociates from RNAP and traverses to the break site to promote repair. In Ewing’s Sarcoma patients, EWS-FLI prevents BRCA1 from dissociating with RNAP, even in the presence of DNA damage.
The impact of RNase H1 and RNase H2 on end resection further supports a model whereby unscheduled hybrids inhibit resection. In budding yeast, persistent R-loops caused by co-depletion of RNaseH and Sen1, the yeast ortholog of SETX, led to DSB formation downstream of the R-loop, leaving only one end of the break with a hybrid (Amon and Koshland, 2016; Costantino and Koshland, 2018). While resection proceeded normally on the hybrid-free end, it was inhibited at the hybrid-containing end. It seems likely that the hybrid needs to be removed for resection to occur (Figure 6B). In mammalian cells, RNase H2 has also been implicated in removing hybrids at DSB sites to allow RAD51 loading (Figure 6B) (D’Alessandro et al., 2018). Consistent with this result, RNase H1/2 overexpression in Saccharomyces pombe (S. pombe) enhanced resection (Ohle et al., 2016). Taken together, these studies suggest that hybrid clearance is a common event at DSB sites, but if hybrids become deregulated, they can interfere with resection, leading to defects in repair and genome instability.
Interestingly, a number of well-known HR proteins interact with factors implicated in hybrid turnover at DSBs. For instance, BRCA2 stimulates the helicase activity of DDX5 toward hybrids, and perturbing this interaction prevents DDX5-mediated R-loop dissolution (Sessa et al., 2021). Similarly, the deubiquitinase USP42 promotes DHX9 activity towards R-loops at DSBs and is needed to recruit BRCA1 to break sites (Matsui et al., 2020). The close links between these HR and R-loop processing factors suggests that hybrid resolution is an integral part of DSB repair.
Another, albeit indirect, mechanism by which R-loops may impede repair is by sequestering repair proteins and preventing their recruitment to DSB sites. For instance, R-loops accumulate in cells expressing EWS-FLI, the fusion protein driving Ewing’s sarcoma, in response to pro-tumorigenic transcription (Gorthi et al., 2018). In EWS-FLI cells, BRCA1 remained associated with R-loop sites and could not relocalize to DSB sites after damage induction, while BRCA1 recruitment to DSBs was not altered in normal cells (Gorthi et al., 2018). Unscheduled R-loops in patient cells may sequester BRCA1, preventing repair (Figure 6C). Of note, auxin treatment in A. thaliana induces expression of the long non-coding RNA APOLO (Ariel et al., 2014, 2020). APOLO can invade duplexed DNA distal to its site of transcription to form R-loops, which are recognized by Polycomb Repressive Complexes (PRCs). APOLO sequesters PRCs away from auxin-responsive genes, upregulating their expression. As protein sequestration by APOLO represents a normal developmental process while sequestration by EWS-FLI is pathological, there may be multiple scenarios in which R-loops sequester proteins to control their function.
R-loops mediate the DNA damage response
In contrast to the idea that R-loops impede DSB repair is their emerging role in promoting the DNA damage response (DDR) and DNA repair (Marnef and Legube, 2021; Paull, 2019). In fact, hybrids have been proposed to be required for both HR and NHEJ, and their presence may affect repair pathway choice.
i. Non-homologous end joining
In certain contexts, R-loops may be involved in NHEJ. SETX loss triggered an increase in 53BP1 foci while inhibiting RAD51 foci formation (Cohen et al., 2018). These changes were accompanied by increased translocations, suggesting that hybrid persistence may favor error-prone repair. In agreement, Sen1 loss led to increased KU occupancy at break ends, activating an alternative resection pathway that promotes classical and microhomology-mediated NHEJ (Rawal et al., 2020). Interestingly, KU exhibits robust RNA and DNA binding activity, and weakly binds hybrids in extracts (Wang et al., 2018). In conjunction with the RNA binding protein RBM14, KU facilitated RNAPII and 53BP1 recruitment to DSBs, as well as hybrid formation (Jang et al., 2020; Schellenbauer et al., 2021; Simon et al., 2017). It is therefore possible that KU promotes both the formation and stabilization of hybrids at DSBs, creating a positive feedback loop that promotes NHEJ (Figure 7A). Of note, loss of other R-loop resolution factors, such as DDX1 or EXOSC10, augmented NHEJ efficiency at the cost of HR efficiency, and loss of USP42/DHX9 increased 53BP1 foci (Domingo-Prim et al., 2019; Li et al., 2016; Matsui et al., 2020). Excessive hybrids may therefore favor NHEJ in these contexts. By contrast, XRN-depleted glioblastoma cells, which also accumulate R-loops, exhibit decreased KU binding and impaired NHEJ, suggesting not all instances of unscheduled hybrids promote NHEJ (Dang and Morales, 2020).
Figure 7.
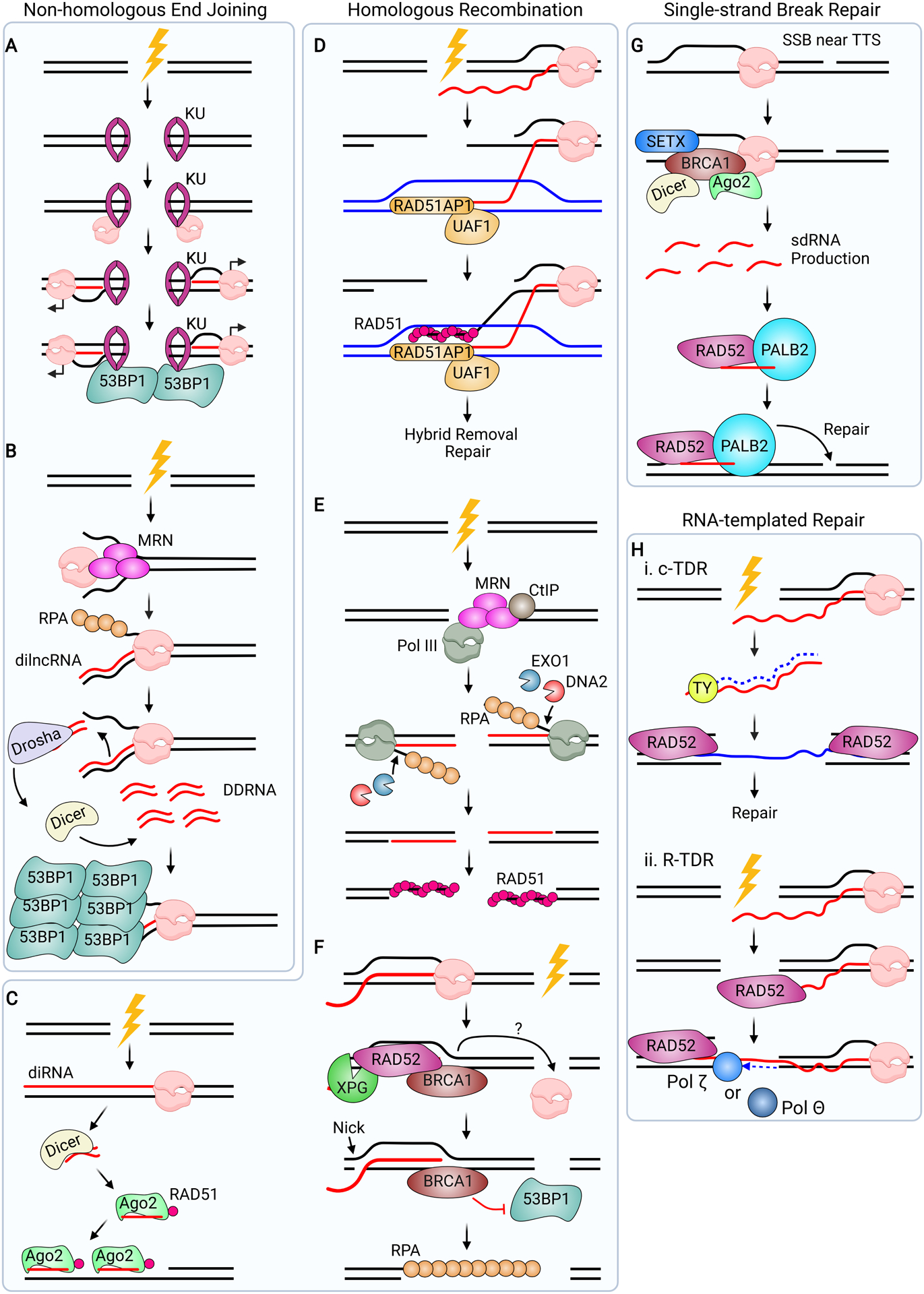
Roles for R-loops and Hybrids in DNA Repair Pathways. (A) During NHEJ, KU recognizes the DSB break ends and recruits RNAPII. RNAPII may transcribe de novo RNA from the break site and recruit 53BP1. (B) MRN binds to DNA ends and melts the duplexed DNA. MRN also recruits RNAPII, which initiates de novo RNA synthesis, forming dilncRNAs. These RNAs are consecutively processed by Drosha and Dicer, generating DDRNAs (red dsRNAs). DDRNAs promote 53BP1 recruitment and aggregation, and NHEJ repair. (C) During HR, RNAPII synthesizes diRNA at the break site. diRNAs are processed by Dicer, and Ago2 interacts with the product. Ago2 binds RAD51 and localizes it to the break site, likely forming a hybrid between the processed diRNA and the resected end. (D) When a DSB occurs in an actively transcribed region, a complex of RAD51AP1 and UAF1 facilitates ssRNA strand invasion into the donor DNA (blue). This stimulates RAD51-mediated ssDNA invasion into the donor template. The resulting structure is called a DR-loop. After hybrid removal, HR proceeds as normal. (E) MRN/CtIP recognize DSB ends and recruit RNAPIII, which initiates de novo RNA synthesis. The DNA flap is excised by EXO1 and DNA2. RNAPIII-mediated hybrid formation promotes RAD51 loading. (F) An R-loop forms at a break site when a DSB occurs in an actively transcribed locus. RAD52 recognizes the R-loop and recruits XPG and BRCA1 to the break site. BRCA1 inhibits 53BP1 at the break, while XPG cleaves the junction 5’ to the hybrid, initiating resection and RPA loading. (G) When a SSB occurs near an R-loop that forms at a transcription termination site, BRCA1 recruits SETX, Dicer, and Ago2 to the SSB, which generates sdRNAs. These RNAs are recognized by the RAD52/PALB2 complex, which localize to the SSB site and promote repair. (H) Should a DSB occur in a transcribed region, RNA can mediate repair via at least two distinct mechanisms. (i) The reverse transcriptase TY generates cDNA from the RNA transcript. RAD52 then promotes invasion of the cDNA into the break ends, and downstream processing occurs to complete repair. This pathway is called cDNA-templated repair (c-TDR). (ii) During RNA-templated repair (R-TDR), RAD52 promotes nascent RNA invasion into the upstream break end, creating an RNA bridge. The DNA Polymerases Polθ or Polζ can synthesize DNA from the template, repairing the break.
Another mechanism by which hybrids and RNAs may promote NHEJ at a DSB is through the RNAi machinery (Figure 7B). Initial investigations found that the RNAi factors Drosha and Dicer process RNAs generated following break induction into DSB-induced small non-coding double-stranded RNAs, termed DDRNAs (Francia et al., 2012; Michelini et al., 2018). DDRNAs likely derive from the break site, as they have the same sequence as the damaged locus. More recent evidence suggests that DDRNAs originate from precursor RNAs, called damage-induced long non-coding RNAs (dilncRNAs) (Michelini et al., 2017; Pessina et al., 2019). De novo RNAPII transcription at a DSB leads to synthesis of dilncRNAs, which are processed into pre-DDRNAs by Drosha and then into DDRNAs by Dicer (Michelini et al., 2017, 2018). How dilncRNAs become double stranded is not well understood, but dilncRNAs can anneal to the DNA ends from which they derive, forming bona fide RNA-DNA hybrids. Loss of either Dicer or Drosha, or RNAPII inhibition, precludes the formation of 53BP1 or RIF1 condensates after DSB induction (Francia et al., 2016; Lu et al., 2018; Michelini et al., 2017). It is likely that NHEJ is compromised in these cells, as phase separation of repair proteins is a prerequisite DNA repair (Fijen and Rothenberg, 2021). These observations suggest a model whereby hybrids shift the balance of repair pathway choice towards NHEJ, by helping to both restrict resection and recruit NHEJ factors.
Hybrids may regulate alternative repair pathways, such as microhomology-mediated end joining, by controlling the localization and clustering of DSBs at the nuclear periphery. DSBs relocate to the nuclear periphery in order to perform such alternative repair pathways (Caridi et al., 2017). mRNA processing and export proteins, which suppress hybrid formation, also localize with nucleoporins and the nuclear periphery (García-Benítez et al., 2017), suggesting that hybrids may normally localize to the nuclear periphery as part of their resolution. Additionally, RNAs containing the m6A modification, which is also observed on hybrids, promote DSB clustering and phase separation (Garcia-Jove Navarro et al., 2019; Ries et al., 2019). Thus, hybrid-driven DSB clustering may be another mechanism by which hybrids promote repair, which has indeed been observed for NHEJ (Pessina et al., 2019). Exactly how they promote clustering, however, remains unclear.
ii. Homologous recombination
R-loops and hybrids also have been reported to direct cells towards homologous recombination. Early studies in S. pombe demonstrated that hybrids were needed transiently at DSBs to restrain resection and modulate RPA loading for efficient HR (Ohle et al., 2016). While in apparent contrast to its role in inhibiting resection, these findings may indicate that hybrid formation and turnover is needed to control resection. Another class of small RNAs, called DSB-induced small RNAs (diRNAs), may also regulate repair through hybrid formation (Figure 7C) (Gao et al., 2014). diRNAs map to sequences distal to a break and are thus thought to be distinct from dilncRNAs/DDRNAs. Here, the RNAi processing protein Ago2, but not Drosha or Dicer, recognizes diRNAs and mediates RAD51 recruitment to DSBs, promoting HR. While the exact mechanism by which Ago2 regulates RAD51 is not clear, it has been hypothesized that the diRNA-Ago2-RAD51 complex hybridizes to free DNA ends, forming a hybrid that drives its recruitment. Of note, Drosha processing has also been implicated in regulating HR, and its loss diminishes BRCA1 and RAD51 loading (Lu et al., 2018). As such, how small RNAs and hybrids help regulate repair requires further investigation.
Further supporting a role for R-loops in HR is work using a novel HR reporter in mammalian cells which revealed that HR was stimulated by local active transcription (Ouyang et al., 2021). HR was dependent upon the RAD51-associated protein 1 (RAD51AP1) and UAF1 protein complex, which were originally found to stimulate RAD51-mediated strand invasion (Liang et al., 2016; Ouyang et al., 2021). RAD51AP1 was shown to promote invasion of ssRNA into the template DNA, suggesting that RNA could invade the sister chromatid to form a type trans R-loop (Figure 7D). Concurrent invasion of ssDNA at these regions also occurs, forming a DR-loop. DR-loops enhanced RAD51-mediated invasion of the template DNA by ssDNA to generate a D-loop, an important intermediate in DSB repair. While it is tempting to speculate that de novo transcription drives DR-loop formation in this context, a tethered RNA molecule was sufficient to robustly stimulate DR-loop-driven HR. This result leaves open the possibility that the RNA transcripts involved in DR-loop formation have already been transcribed at the respective locus.
Recent evidence also implicates RNAPIII-driven hybrid synthesis in the regulation of DNA repair (Liu et al., 2021b). Indeed, RNAPIII, which transcribes transfer RNAs and other small RNAs, but not RNAPII, was recruited to DSBs, where it transcribed de novo hybrids in an MRE11 and CtIP-dependent manner from the 3’-strand (Figure 7E). Loss of RNAPIII activity and hybrid formation impaired RPA and RAD51 loading, suggesting that hybrid formation is necessary for HR. It was also proposed that RNAPIII-dependent transcription protects the 3’ DNA strand from exonucleolytic attack and the loss of genetic material, thereby maintaining genome stability.
R-loops can also propagate an alternative HR pathway known as transcription-associated homologous recombination repair (TA-HRR) (Yasuhara et al., 2018). During TA-HRR, R-loops that form near break sites in actively transcribed loci are recognized by RAD52, which promotes XPG-mediated hybrid removal to generate ssDNA in what might be considered the first step of resection (Figure 7F). Downstream HR factors, such as BRCA1, are also recruited by RAD52 to prevent the binding of RIF1 and 53BP1. A related model proposes that RAD52 recruitment following localized ROS damage requires hybrid recognition by the TC-NER factor CSB (Teng et al., 2018). After binding ROS-stabilized hybrids, RAD52 recruits RAD51 to promote strand invasion and HR. R-loops have also been shown to recruit RAD52 and CSB to ROS-induced breaks that occur near telomeres to promote their repair (Tan et al., 2020b), although this is more akin to break-induced repair (BIR) rather than canonical HR. Together, these works illuminate the need for transcription and R-loops to promote faithful DNA repair, particularly in highly transcribed regions.
iii. Single-strand break repair
A third class of damage-induced RNAs, single-stranded, DNA damage-associated sRNAs (sdRNAs) has been identified and implicated in SSB repair (Figure 7G) (Hatchi et al., 2021). In this pathway, BRCA1 and its partner BARD1, in conjunction with Dicer and Ago1, are thought to promote sdRNA synthesis at SSBs found at transcription termination sites (TTSs). BRCA1/BARD1 may then transfer the sdRNA moiety to the PALB2/RAD52 complex, which facilitates repair of the SSB. While the link between R-loops and sdRNA synthesis is unresolved, it is likely that R-loops have an important role in this pathway. sdRNAs map to SETX and BRCA1 occupied TTSs, which are prone to R-loop formation. Furthermore, previous work demonstrated that BRCA1 recruits SETX to TTSs to promote the repair of R-loop induced SSBs at these sites (Hatchi et al., 2015). Additional work is required to elucidate the role of R-loops in promoting SSB repair.
iv. RNA-templated DNA repair
An RNA molecule itself may be used as a template to direct the repair of the gene from which it is derived (Figure 7H). In budding yeast, reverse transcriptase can synthesize complementary DNA (cDNA) from an existing mRNA, which is then used as the homologous template for repair in a process called cDNA-templated DSB repair (Keskin et al., 2014; Meers et al., 2020). Conversely, the RNA itself can serve as the template during a process called RNA-templated repair (R-TDR). During R-TDR, the RNA molecule hybridizes with the free ssDNA end via an inverse strand exchange mechanism stimulated by RAD52, forming a hybrid. DNA is then directly synthesized from the RNA by Polymerase ζ (Mazina et al., 2017; Meers et al., 2020). Of note, Polθ can also reverse-transcribe cDNA from an RNA template (Chandramouly et al., 2021). Importantly, only RNA transcribed at the same locus as the break, and not homologous RNA transcribed at a distal locus, can undergo inverse strand exchange (Keskin et al., 2014; Meers et al., 2020). RAD52 and CSB also recognize hybrids to drive RNA-directed recombination in G0/G1 mammalian cells, providing additional context in which RNA could be used to drive error-free repair (Wei et al., 2015).
RNA-DNA hybrid formation at DSB sites
To resolve questions regarding how R-loops facilitate repair, the mechanism by which hybrids form at break sites must be determined. Specifically, whether hybrids are products of de novo transcription that occurs after the break forms or whether they result from preexisting transcription at the break site remains an intriguing question.
In support of the first hypothesis, recent work provides evidence of de novo transcription at DSBs in mammalian cells (Liu et al., 2021b; Michelini et al., 2017). In response to damage, MRN can recruit the transcriptional preinitiation complex (PIC), including the transcriptional coactivator Mediator and the RNAPII kinases CDK7 and CDK9, to DNA ends (Pessina et al., 2019; Sharma et al., 2021). During normal transcription, the PIC, CDK7, and CDK9 act in concert to phosphorylate and activate RNAPII, and they may have a similar role at DSBs (Larochelle et al., 2012). The PIC, in conjunction with MRN, promotes RNAPII association with break sites, resulting in local dilncRNA synthesis (Pessina et al., 2019). It is thought that dilncRNAs hybridize with DNA at the break site, accounting for the hybrids observed at DSBs. Surprisingly, MRN’s nuclease activity seems to be dispensable for RNAPII recruitment, which instead requires MRN’s DNA melting activity (Sharma et al., 2021). Consistent with this finding, work in S. pombe demonstrates MRN-dependent RNAPII-mediated hybrid formation at DSBs, although in this case MRN’s end processing activity was required (Ohle et al., 2016). Similarly, the synthesis of diRNA and sdRNA appears to occur de novo at DSBs (Gao et al., 2014; Hatchi et al., 2021). Work in mammalian cells also supports a role for RNAPIII in hybrid generation at break sites (Liu et al., 2021b). RNAPIII localization to DSBs required the nuclease activities of MRN as well as CtIP (Liu et al., 2021b). In each instance, de novo hybrid formation promotes the recruitment of downstream repair factors to break sites, providing insight into the potential mechanisms by which hybrids propagate repair signaling pathways.
While these works support the concept of de novo RNAPII transcription and hybrid formation at break sites, the exact mechanism by which processed DNA ends support transcription is still unclear. There are mechanisms for RNAP-mediated transcription at ssDNA sites lacking a promoter sequence (Kadesch and Chamberlin, 1982). It is also likely that various helicases are required to promote new transcription and hybrid formation at DSBs. For example, the helicase UPF1 promotes hybrid formation at regions where a DSB has been induced (Ngo et al., 2021). Although further studies are needed, UPF1 was proposed to both unwind dsDNA to promote RNAPII elongation and to translocate along the nascent RNA to remove RNA binding proteins, both of which would facilitate hybrid formation.
Alternatively, R-loops observed at DSBs may be an unwanted consequence of altered transcriptional dynamics in response to break induction. Indeed, R-loops may arise due to changes in promoter proximal pausing or termination even in the absence of damage (Shivji et al., 2018; Skourti-Stathaki et al., 2011; Zhang et al., 2017). As such, DSB-induced transcriptional repression, which occurs both globally and locally, could drive R-loop formation (Shanbhag et al., 2010; Silva and Ideker, 2019). Consistent with this idea, hybrids were elevated only at DSBs occurring in actively transcribed regions upon SETX loss (Cohen et al., 2018). DRIP-seq analysis similarly revealed elevated hybrid occupancy specifically at DSBs in transcriptionally active loci (Bader and Bushell, 2020). This finding was corroborated by mNET-seq, which showed that elongating transcripts are only found at DSBs induced in actively transcribed genes (Burger et al., 2019). While the above techniques utilize enzyme-mediated DSB formation, DSBs induced by ROS also stimulate R-loop formation in a transcription-dependent manner (Teng et al., 2018). These findings suggest that DSBs in actively transcribed loci may induce hybrid formation by either changing where the polymerase resides within that gene or slowing elongation rates. R-loops and hybrids may arise as a consequence of these altered dynamics.
R-loops and DSB crosstalk: in trans or in cis?
While there is a general consensus that R-loops can form in cis at sites of DNA breaks, an intriguing possibility is that these structures can also form in trans. Recent work has shown that trans R-loops can form in a variety of contexts and these R-loops may have physiological roles. As described previously, in vitro experiments that demonstrate RAD51AP1-mediated RNA invasion of template DNA using a ssRNA oligo suggest that trans-forming R-loops may help promote DSB repair (Ouyang et al., 2021). Another example of trans R-loop formation is the well-known telomeric lncRNA TERRA, which can invade short telomeric repeats in trans in a RAD51-dependent manner to maintain telomere integrity (Feretzaki et al., 2020). Furthermore, the APOLO lncRNA in A. thaliana can form trans R-loops via a mechanism described previously (Ariel et al., 2020). Whether trans hybrid formation is consistently associated with a physiological role remains to be determined, but these works highlight the capability of RNA to invade DNA in trans.
Less well-known, however, is the propensity for trans-forming R-loops to drive DSB formation and genome instability. An important example of this is trans R-loop formation during CRISPR/Cas9-guided DSB induction (Jinek et al., 2012; Nishimasu et al., 2014). Here, Cas9 opens the DNA duplex, allowing the guide RNA to base pair with its complementary DNA sequence and displace ssDNA. As hybrids direct Cas9 nuclease activity, they represent a source of DNA damage in this context. Intriguingly, trans R-loops have also been directly implicated as a source of genomic instability. Initial studies using a yeast artificial chromosome (YAC) system showed that Rad51 facilitated the trans invasion of RNA transcribed from the yeast genome into YAC DNA, forming an R-loop that ultimately led to YAC loss, a readout for genomic instability (Wahba et al., 2013). Other studies, however, found that only cis R-loops generate DNA damage, and that this occurs in a RAD51-independent process (Lafuente-Barquero et al., 2020).
R-Loops and Immune Signaling
Given that R-loops cause DNA damage, it seems likely that they may also have an effect on the innate immune response, a pathway known to protect cells from pathogens and viruses by detecting foreign nucleic acids in the cytoplasm (Zhou et al., 2017). Normally, recognition of nucleic acids through pattern recognition receptors, including the Toll Like Receptors (TLRs) and the cGAS/STING pathway, activates downstream inflammatory signaling mediated by IRF3, NF-kB, and MAPK (Brubaker et al., 2015; Takeuchi and Akira, 2010; Zhou et al., 2017). Recognition of self-nucleic acids resulting from DNA damage and replication stress has also been shown to induce innate immune signaling (Hopfner and Hornung, 2020; Ragu et al., 2020). Although the mechanism by which damage activates innate immune signaling is not fully understood, several sources of endogenous nucleic acids that activate this response have been identified in the cytoplasm. For example, micronuclei arising from DNA damage, ssDNA arising from stalled fork processing, and mitochondrial RNA released after damage-induced mitochondrial herniation can activate cGAS or RIG-I (Coquel et al., 2018; Harding et al., 2017; Mackenzie et al., 2017; Tigano et al., 2021).
These studies raise the question of whether R-loops may also contribute to innate immune signaling. Consistent with such an idea, RNase H1 overexpression reduced the level of cytosolic DNA and the interferon response in tumor cells (Shen et al., 2015) and in cells in which R-loop accumulation was induced by DNA damage (Chatzidoukaki et al., 2021). Similarly, loss of the DEAD-box helicase 41 (DDX41) led to RNase H-reversible nuclear R-loop accumulation and the expression of genes involved in the inflammatory response in both zebrafish embryos and human cells (Weinreb et al., 2021). Intriguingly, inflammatory signaling driven by excessive R-loop production was also shown to increase hematopoietic stem and progenitor cell production in zebrafish models, raising the possibility that R-loops may control developmental processes through the inflammatory response. Factors that dampen the inflammatory response induced by hybrids also exist. Hybrid binding to lysyl tRNA synthetase reduces the inflammatory response through two mechanisms: by suppressing the interaction of hybrids with cGAS, thereby reducing cGAMP production in cells, and by stimulating diadenosine tetraphosphate production, which can act as an antagonist to STING and reduce STING-dependent signaling (Guerra et al., 2020).
Whether hybrids directly activate the innate immune response or whether the activation is a result of the damage produced by hybrids is still not clear. However, there are hints they might be in a position to directly activate innate immune responses. First, hybrids have been detected in the cytoplasm of virus-free cells by immunofluorescence (Koo et al., 2015). Recent work, however, has demonstrated that the majority of signal from the S9.6 antibody, which recognizes dsRNA and hybrid structures, derives from dsRNA and not hybrid recognition (Crossley et al., 2021; Smolka et al., 2021). As such, further characterization is needed to verify these species as hybrids. Second, the transfection of in vitro generated synthetic RNA-DNA hybrids has been shown to induce a cGAS- or TLR9-dependent response, depending on the cell type (Mankan et al., 2014; Rigby et al., 2014). Synthetic hybrids can also activate cGAS in vitro (Mankan et al., 2014). Although these observations suggest hybrids could directly interact with pattern recognition receptors in cells, additional work will be needed to verify this idea and to understand the mechanism of this activation.
While the accumulation of R-loops is associated with the induction of an inflammatory response, there is also evidence that increased inflammatory signaling induces R-loop formation. NF-kB activation is a downstream effector of the inflammatory response that can be activated by the HTLV-1 transactivator/oncoprotein Tax to promote cellular senescence. In adult T leukemia (ATL) cells, NF-kB activation by TAX leads to R-loop dependent DNA damage and senescence in a pathway mediated by XPF and XPG. Many ATL cells are deficient for these endonuclease activities, which may help them proliferate (He et al., 2021). Similarly, NF-kB-mediated innate immune signaling activated by Helicobacter pylori infection causes R-loop-induced replication stress, and cancers associated with this infection demonstrate higher mutational burden (Bauer et al., 2020).
These findings illustrate how R-loops could contribute to immune signaling, but whether R-loop formation is causative for or consequential to innate immune signaling is still unclear. One can envision an R-loop-dependent positive feedback loop, whereby hybrids activate cGAS/STING to elicit an immune response, which further drives R-loop formation during the change in transcription.
R-Loop Pathology
Deregulation of R-loop levels has also been associated with multiple human diseases, including cancers, neurological disorders and autoimmune diseases (Table 1). In these situations, R-loops may drive genome instability, alter gene expression or alter cellular signaling pathways. Understanding the mechanisms behind R-loop-driven disease could reveal novel therapeutic opportunities. Below, we provide an update on R-loop regulation and disease connections, though we also refer the reader to other in-depth reviews (Groh and Gromak, 2014; Richard and Manley, 2017).
Table 1.
R-loop resolution and suppression factors and their links to disease. Bold denotes a gain-of-function mutation for that factor associated with disease, while italics signifies a loss-of-function mutation.
| Gene | Description | Disease | Reference |
|---|---|---|---|
| ADAR1 | Edits mismatches to promote RNase H2 R-loop resolution | Telomerase-Reactivated Cancers | (Shiromoto et al., 2021) |
| BRCA1 | Suppresses satellite RNA production to suppress R-loops; also sequestered by satellite RNAs preventing its proper functioning | Breast and Ovarian Cancers | (Zhu et al., 2018) |
| Resolves R-loops at transcription termination sites (with SETX) | (Hatchi et al., 2015) | ||
| Limits RNAPII proximal pausing, preventing instability | (Zhang et al., 2017) | ||
| Limits the ESR1 R-loop to maintain neighboring gene transcription | (Chiang et al., 2019) | ||
| BRCA2 | Resolves R-loops and limits genomic instability | Breast and Ovarian Cancer | (Bhatia et al., 2014) |
| Depletion by aldehydes increases replication stress and instability | (Tan et al., 2017) | ||
| Promotes the transition of RNAPII from an initiating to an elongating complex | (Shivji et al., 2018) | ||
| C9orf72 | R-loops formed over this region interfere with transcription and promote disease pathologies | ALS/Frontotempor al Dementia (FTD) | (Haeusler et al., 2014; Reddy et al., 2014) |
| CDCA7/HELLS | Promotes methylation at pericentromeres to suppress R-loops | ICF Syndrome | (Unoki et al., 2020) |
| CSB | Processes R-loops to limit levels | Cockayne Syndrome (CS) | (Sollier et al., 2014) |
| Dicer | Prevents R-loop associated genome instability | Embryonal Tumors with Multilayered Rosettes (ETMR) | (Lambo et al., 2019) |
| DDX41 | Recognizes viral hybrids to initiate immune signaling | Human Immunodeficiency Virus (HIV) | (Stavrou et al., 2018) |
| Regulates alternative splicing | Cervical Cancer | (Qin et al., 2021) | |
| Suppresses R-loops and R-loop-induced immune signaling | Myelodysplastic Syndromes | (Weinreb et al., 2021) | |
| Estrogen | Induces transcription and R-loops, leading to instability | Breast/Ovarian Cancer | (Stork et al., 2016) |
| EWS-FLI | Promotes R-loop formation and prevents BRCA1 localization to damage sites | Ewing’s Sarcoma | (Gorthi et al., 2018) |
| FANCM FANCD2 |
Resolves TERRA R-loops to limit genome instability and regulate ALT activity | ALT cancers | (Pan et al., 2019; Silva et al., 2019) |
| Suppresses R-loops and TRCs to maintain genomic stability | Fanconi Anemia | (García-Rubio et al., 2015; Schwab et al., 2015) | |
| FXN FMR1 |
R-loops in these genes lead to transcriptional silencing | Fragile X Syndrome Friedreich Ataxia | (Colak et al., 2014; Groh et al., 2014; Loomis et al., 2014) |
| H. pylori | Activates Nf-kB-driven expression, leading to TRCs and damage | Gastric Cancer | (Bauer et al., 2020) |
| HRAS | Increases RNA synthesis and R-loops leading to increased damage and replication stress | Many Cancers | (Kotsantis et al., 2016) |
| INO80 | Remodels chromatin to suppress R-loops and prevent TRCs | Multiple Cancers | (Prendergast et al., 2020) |
| MDM2/RNF2 | Ubiquitinates H2A to increase replication and prevent R-loops | Sarcomas | (Klusmann et al., 2018) |
| MYCN | MYCN recruits BRCA1 to release RNAPII from proximal pause to prevent R-loops | Neuronal and Neuroendocrine Cancers (eg. Neuroblastoma) | (Herold et al., 2019) |
| RECQ4 | Resolves mitochondrial R-loops to promote mtDNA replication | Rothmund-Thomson Syndrome, RAPADILINO, Baller-Gerold Syndrome | (Chang et al., 2020) |
| RNase H2 | Suppress R-loop formation and genomic instability | Aicardi-Goutières Syndrome (AGS) | (Giordano et al., 2022; Mackenzie et al., 2016) |
| SAMHD1 | Suppress R-loop accumulation and R-loop induced TRCs | AGS | (Lim et al., 2015; Park et al., 2021) |
| SAN1 | Interacts with SETX to resolve R-loops | Cardiomyopathies | (Liu et al., 2021c) |
| SETX | Resolves R-loops to prevent instability | AOA2 | (Becherel et al., 2015) |
| Resolves R-loops to prevent aberrant TGF-β signaling | Amyotrophic Lateral Sclerosis (ALS4) | (Grunseich et al., 2018) | |
| Interacts with ZPR1 to resolve R-loops | Amyotrophic Lateral Sclerosis (ALS4) | (Kannan et al., 2022) | |
| Promotes meiosis | Infertility | (Becherel et al., 2013) | |
| SF3B1 U2AF1 SRSF2 |
Maintain splicing to prevent R-loops | Myelodysplastic Syndromes | (Cheruiyot et al., 2021; Sorrells et al., 2018) |
| Promotes RNAPII release from promoter pause sites | (Chen et al., 2018) | ||
| When lost, R-loops activate ATR to promote survival | (Nguyen et al., 2018) | ||
| SMN1 | Promotes splicing to prevent R-loops and instability | SMA | (Jangi et al., 2017) |
| Snord116 | Loss of R-loops in this region leads to altered imprinting and chromatin condensation | Prader-Willi Syndrome | (Powell et al., 2013) |
| SS18-SSX | Increases R-loop formation | Synovial Sarcoma | (Jones et al., 2017) |
| TDP-43 | Suppress hybrid formation and replication stress | ALS/FTD | (Giannini et al., 2020; Wood et al., 2020) |
| TDRD3/TOP3B | Induces negative supercoiling and increases R-loop resolution via DHX9 to prevent c-MYC/IgH translocations | Multiple Myeloma Burkhitt’s Lymphoma | (Yang et al., 2014; Yuan et al., 2021) |
| TERRA | DNMT3B mutations in ICF cells elevate TERRA, increasing R-loops and causing telomere shortening | Immunodeficiency, Centromere instability and facial anomalies syndrome (ICF) | (Sagie et al., 2017) |
| Increased production leads to increased R-loops and recombination to lengthen telomeres, mediated by RNase H1 | Alternative-Lengthening of Telomeres (ALT)-driven cancers | (Arora et al., 2014) | |
| TonEBP | Recruits METTL3 to resolve R-loops and maintain stability | Hepatocellular carcinoma | (Kang et al., 2021; Ye et al., 2021) |
| TPR | Prevents R-loop formation and maintains fork progression | Ovarian cancers Others | (Kosar et al., 2021) |
| TREX (Transcription-export) complex | Prevents R-loop formation and DNA breaks; sequestered and inhibited by KSHV ORF57 | Kaposi’s Sarcoma | (Jackson et al., 2014) |
| TREX1 | Suppress R-loop formation and maintain genome stability; maintain DNA methylation | AGS | (Giordano et al., 2022; Lim et al., 2015) |
| XPF | Processes R-loops to limit levels | XP | (Cristini et al., 2019; Goulielmaki et al., 2021; Makharashvili et al., 2018; Sollier et al., 2014) |
| Processes R-loops to limit cytosolic ssDNA accumulation and inflammation | Chronic Pancreatitis | (Chatzidoukaki et al., 2021) | |
| XPG | Processes R-loops to limit their levels | Xeroderma Pigmentosum (XP) | (Cristini et al., 2019; Goulielmaki et al., 2021; Makharashvili et al., 2018; Sollier et al., 2014). |
| ZPR1 | Interacts with SETX to resolve R-loops | ALS4 | (Kannan et al., 2022) |
| Regulates SETX and SMN2 levels to promote R-loop resolution | Spinal Muscular Atrophy (SMA) | (Kannan et al., 2020, 2022) |
Cancers
The ability of R-loops to cause replication stress and DNA damage, two hallmarks of cancer, suggest these structures could drive tumorigenesis. Indeed, several oncogenes and proliferation factors promote R-loop formation and R-loop associated DNA damage, as do loss-of-function mutations in tumor suppressors and other genes that suppress and resolve R-loops or TRCs (Table 1). For example, estrogen and HRAS both drive R-loop-dependent DNA damage, and mutations are found in genes where estrogen-responsive R-loops form (Stork et al. 2016; Kotsantis et al. 2016). Dicer mutations are also associated with R-loop induced chromosomal instability in pediatric brain tumors (Lambo et al., 2019). Similarly, the Cyclin E and MYC oncogenes promote early S phase entry, leading to intra-genic activation of origins that are normally deactivated by G1-phase transcription. This leads to an increase in HO TRCs and chromosomal rearrangements at sites altered in human cancers (Macheret and Halazonetis, 2018). As HO TRCs and some oncogenes cause R-loop accumulation (Table 1), R-loops may drive the genome instability occurring at these sites. R-loop-driven instability may promote both specific mutational signatures that are associated with specific cancers and the evolution of tumorigenic traits.
R-loops may also promote tumorigenesis by supporting telomere maintenance. In ALT-driven cancers, upregulation of the TERRA RNA and subsequent R-loop formation promotes telomeric recombination to lengthen telomeres (Table 1). In ALT-driven cancers, R-loop and G4 formation is also interdependent, and these structures appear to act synergistically in driving ALT (Yang et al., 2021). Chromatin remodeling at TERRA may also link R-loops to telomere maintenance. For example, mutation of the histone chaperone ATRX, which normally suppresses TERRA expression, is common in ALT cancers (Clynes et al., 2015; Flynn et al., 2015; Goldberg et al., 2010).
How R-loop deregulation contributes to tumorigenesis is complex. At one extreme, low R-loops levels could drive mutation, but high R-loop levels may cause excessive damage and cell death. As discussed, increased DNA damage activates immune signaling and elicits an innate immune response (Coquel et al., 2018; Harding et al., 2017), leading to chronic inflammation, which can drive tumorigenesis. If left unchecked, inflammation may also lead to apoptosis or elimination of the tumor cell by the immune system. Cancer cells may adapt to this state, downregulating the immune system or promoting a tolerance to R-loops. In patients with BRCA1/2-deficient breast cancer, high levels of RNF168, a factor that recruits the R-loop resolving helicase DHX9 to R-loop prone loci, are associated with poor prognosis. Moreover, RNF168 loss in a BRCA1/2 mouse prevents tumorigenesis (Patel et al., 2021). R-loop levels may need to be regulated even when pathologic in nature, and understanding how R-loops activate and avoid immune responses could provide insight into ways to improve immunotherapy.
Importantly, elevated R-loop levels and R-loop associated DNA damage proffer additional avenues by which therapies can capitalize upon heightened instability in cancer cells. For instance, mutations in splicing factors causative for myelodysplastic syndromes and acute myeloid leukemia initiate R-loop dependent ATR signaling (Nguyen et al., 2018). ATR inhibitor treatment exacerbated DNA damage and led to cell death, making ATR inhibition an attractive therapy for diseases harboring such mutations. As ATR is activated by R-loops, its inhibition may represent a broadly generalizable therapeutic strategy for many diseases that augment R-loop levels. Splicing factor mutations also increase cellular sensitivity to Chk1 and PARP inhibition, providing alternative therapies (Lappin et al., 2022; Singh et al., 2020). Interestingly, some of these mutations result in a BRCA1-like phenotype, suggesting there may be overlap in therapeutic strategies between BRCA1-driven diseases and R-loop diseases (Lappin et al., 2022). In addition, inhibiting the RNA degradation machinery, which also results in excessive R-loop formation and damage, may be effective in cells already bearing a high R-loop burden (Cheruiyot et al., 2021).
Neurological Disorders
Alterations in R-loop levels are also strongly associated with several neurological disorders (reviewed in Perego et al., 2019). One such factor mutated in disease on which a number of studies have focused is SETX. Activating and loss-of-function mutations in SETX, found in amyotrophic lateral sclerosis type 4 (ALS4) and ataxia with oculomotor apraxia2 (AOA2), respectively, are thought to drive disease progression (Table 1). Hyperactive SETX reduces R-loop levels, leading to increased gene silencing in ALS4 patients, including at one gene that suppresses TGF-β expression. Increased TGF-β signaling is thought to ultimately cause neuronal death (Grunseich et al., 2018). Conversely, SETX loss in AOA2 results in elevated R-loops at promoter-proximal regions, changes in transcription and chromosomal instability in gene bodies and at common fragile sites (Becherel et al., 2015; Kanagaraj et al., 2022). R-loops in AOA2 cells are processed via an auxiliary pathway involving CSB, XPG and RAD52, which may contribute to the observed genomic instability (Kanagaraj et al., 2022). Other studies have focused on regulators of SETX demonstrating that SETX protein levels are regulated by ubiquitin-mediated degradation, controlled by the E3 ligase KEAP1 and the deubiquitinase USP11 (Jurga et al., 2021). Targeting USP11 in ALS4 patients to limit the activity of mutated SETX may present a viable therapy, and applications in cancer cells can also be envisioned to overload cells with R-loops.
Emerging evidence also supports a role for R-loops in neurodegenerative diseases associated with repeat instability, including trinucleotide repeat (TNR) expansion or deletion. This includes Huntington’s Disease, Fragile X syndrome, Friedreich’s ataxia, myotonic dystrophy and several motor neuron disorders and ataxias (reviewed in Groh and Gromak, 2014; Malik et al., 2021). TNRs are hotspots for R-loop formation, as they are prone to form secondary structures in the ssDNA which stabilize hybrids on the template strand (Loomis et al., 2014). Interestingly, R-loops contribute to repeat instability by promoting both repeat expansion and deletion. Transcription through GAA repeat regions results in repeat expansion upon RNase H1/2 loss in budding yeast. Although the mechanism is not fully understood, it is likely that R-loops formed at these repeats impede the replisome, causing fork stalling, collapse and then BIR, which leads to TNR expansion (Neil et al., 2018).
Conversely, R-loops drive TNR contraction at CAG repeats. In this case, it is thought that hairpin structures which form in the displaced ssDNA of the R-loop are targeted by cytosine deaminases and downstream repair factors generating abasic sites or DNA nicks. Subsequent removal of the RNA strand of the R-loop allows for additional hairpin formation in the template strand, which allows for repeat deletions when bypassed by Polβ during break repair (Laverde et al., 2020). Hybrid removal can also lead to the formation of mismatches, which are subject to processing by the MutLγ complex, driving break formation and repeat loss (Su and Freudenreich, 2017). It will be interesting to see how R-loops differentially regulate changes in TNR tracts depending upon the type of repeat.
R-loops in repeat regions can also alter the chromatin landscape leading to disease-associated transcriptional silencing. For example, in Friedreich’s ataxia and Fragile X syndrome, stable R-loops in the repeat region of the frataxin (FXN) and FMR genes promote H3K9 dimethylation, a marker of transcriptional repression (Groh et al., 2014). Loss of expression of these genes may be an important driver of neurodegenerative disorders like Fragile X Syndrome. Hence, induction of chromatin repression represents an additional mechanism by which R-loops can drive neurodegeneration.
The extent to which R-loops are responsible for the progression of these different neurodegenerative disorders remains unresolved. Many animal models that recapitulate these disease mutations do not demonstrate elevated R-loop levels and fail to result in similar phenotypic abnormalities (Becherel et al., 2013). Deciphering these discrepancies will be important to further our understanding of these diseases and illuminate the propensity for R-loops to be targeted as a therapy. Furthermore, as these are diseases of post-mitotic cells, the pathological effects of R-loops in these situations should be independent of replication. However, it remains possible that events occurring in dividing cells during development could affect the progeny, by altering their epigenetic state or some other feature. Clearly, further work will be required to address this issue.
Autoimmune Disease
Aicardi Goutières Syndrome (AGS) is an inflammatory/autoimmune disorder associated with the accumulation of nucleic acid fragments in the brain that can be caused by a number of genes involved in processing RNA and DNA, including SAMHD1, RNaseH2, TREX1, and ADAR1 (Table 1). Interestingly, mutations in these genes also lead to increased innate immune signaling, through a mechanism that is not well understood. For instance, loss of SAMHD1 leads to aberrant replication fork processing and the release of ssDNA into the cytosol, activating cGAS-STING signaling (Coquel et al., 2018). Similarly, loss of RNase H2 activates cGAS and is associated with increased micronuclei formation (Giordano et al., 2022; Mackenzie et al., 2016). Perturbation of ADAR1 also activates immune signaling, though this is thought to be through increased dsRNA (Guo et al., 2021; Inoue et al., 2021). However, perturbation of SAMHD1, RNase H2, TREX1, or ADAR1 also elevates R-loop levels (Giordano et al., 2022; Park et al., 2021). As emerging evidence demonstrates that R-loops can activate immune signaling, it will be interesting to assess the extent to which R-loop deregulation is associated with the inflammation observed in AGS patients.
Concluding Remarks
The works described herein have begun to decipher the dichotomous role for R-loops in maintaining genomic integrity. In the context of replication, whether R-loops directly impede fork progression and/or whether they also affect forks post-replicatively requires further investigation. How forks can bypass R-loops is also of great interest. Although our knowledge of this process is increasing, many questions remain about whether RNAPII is bypassed or removed and whether this is context-dependent. Which R-loops threaten the fork is also an outstanding question. The pervasive nature of R-loops and their physiological roles suggest the cell can tolerate them under most circumstances, and recent studies indicate only a small subset may cause replication stress under normal circumstances. Further work will be needed to understand R-loop tolerance, and could be important for understanding how cancer cells may tolerate high levels of R-loops. It seems likely that potential positive roles for hybrids at the fork may also emerge in the years to come, given the similarities in processing stalled forks and DSBs. Consistent with this idea, it was proposed that hybrids may form de novo at reversed replication forks (Adeyemi et al., 2021). Protexin, a complex of SCA1 and the Rev3 polymerase, recruits RNAPII to the reversed fork to protect the fork from degradation, similar to what has been observed at DSBs.
Whether R-loops interfere with or actually mediate DNA repair also necessitates further investigation. Indeed, it is likely that both are true. They may be crucial intermediates in the DDR and in controlling DNA repair, but their removal may also be necessary to complete repair. The extent in which R-loops play a role in pathway choice is still unclear. Does an R-loop pre-dispose a cell to repair damage via one pathway over the other, or do they inhibit certain forms of repair as a consequence of being in the wrong place at the wrong time? As most studies focus on how R-loops affect repair when their formation or turnover is deregulated, the problems observed are likely to result from being unable to remove unscheduled R-loops. As studies are beginning to implicate R-loops as intermediates in both NHEJ and HR, it will be interesting to see in what contexts R-loops favor one pathway over another, and what determines which damage-induced RNA species is formed.
Another important question is why so many factors contribute to R-loop prevention and resolution, and to what extent these factors cooperate to regulate R-loop levels? The field also needs to move past simple analysis of hybrid levels at a few common genes or by imaging. Though a herculean effort, using quantitative approaches to analyze the genomic distribution of unscheduled R-loops could provide insight into this question (Crossley et al., 2020; Šviković et al., 2019). Such large-scale studies could reveal the differential contexts in which these factors might act, such as specifically at long genes, only when a certain chromatin mark is present, or during a specific stage of the cell cycle. This may be more challenging than it appears, however, as many R-loop processing proteins have roles in transcription, which also alters R-loop levels. It is also important to discern whether the ability of so many different cellular perturbations, ranging from distinct forms of DNA damage to disruption of RNA processing and replication factors, may cause R-loops to accumulate for indirect reasons. Many of these stressors cause replication and transcription stress, both of which may exacerbate the other. Thus, R-loop accumulation could reflect replication stress itself and even be a marker of DNA damage or genome instability. For instance, R-loops could arise as a result of replication stress due to the firing of dormant origins, which could increase HO conflicts by disrupting the normal replication program, or they could arise due to the increased ssDNA formation associated with replication stress. The high tolerance of cells to physiological R-loops in general indicates they are not normally a problem.
In this context, it will also be important to determine which unscheduled R-loops present a problem and contribute to genomic instability. One interesting possibility is that the analysis of sites of DNA damage will reveal the different contexts in which R-loop resolution factors are most important. Will the same subset pose a threat following different perturbations or will different R-loops contribute to damage and genome instability under different conditions? As methods for capturing and sequencing R-loops and hybrids have improved, it is more apparent that hybrids are not uniform structures but are rather heterogeneous. Recent insights suggest that R-loops exist in at least two distinct subclasses: pausing-associated and elongating-associated R-loops (Castillo-Guzman and Chédin, 2021). A subclass of R-loops that cause genomic instability may also exist, and if so, what intrinsic properties of those R-loops lead to their pathological nature? A class of RNase H-resistant R-loops was recently identified, which may provide hints into what makes some R-loops harmful (Crossley et al., 2020). R-loops that drive damage may be more intrinsically stable or more likely to contain secondary structures, such as G4s, in the displaced ssDNA, RNA, or DNA near the R-loop that confers resistance to certain resolution pathways. Alternatively, they could be associated with specific chromatin states that make them less accessible to non-damaging forms of R-loop resolution. Along these lines, are the R-loops that cause damage more likely to be processed by nucleases, and does this processing represent an auxiliary, last resort pathway to reduce R-loop levels? Investigation into each of these questions could significantly advance our understanding of R-loop biology.
Finally, the strong link between different diseases and R-loop formation suggests that manipulating and targeting R-loop pathways could lead to novel therapeutic approaches. Additionally, as products of transcription, the distinct perturbations that elevate R-loops and drive cancer could have different profiles, rendering the characterization of R-loops from a tumor a useful diagnostic tool.
Acknowledgements
We thank Stephan Hamperl, Madzia Crossley and Theresa Endres for critically reading this manuscript. We apologize to all authors whose work could not be discussed due to space constraints. This work was supported by the Jane Coffin Childs Memorial Fund for Medical Research [61-1755 to JRB] and the National Institutes of Health [GM119334 to KAC]. KAC is an American Cancer Society Research Professor. All figures were created with BioRender.com.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
Karlene Cimprich is a member of the Advisory Board for Molecular Cell.
References
- Abakir A, Giles TC, Cristini A, Foster JM, Dai N, Starczak M, Rubio-Roldan A, Li M, Eleftheriou M, Crutchley J, et al. (2020). N6-methyladenosine regulates the stability of RNA:DNA hybrids in human cells. Nat. Genet 52, 48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adeyemi RO, Willis NA, Elia AEH, Clairmont C, Li S, Wu X, D’Andrea AD, Scully R, and Elledge SJ (2021). The Protexin complex counters resection on stalled forks to promote homologous recombination and crosslink repair. Mol. Cell 81, 4440–4456.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilera A, and Gómez-González B (2008). Genome instability: a mechanistic view of its causes and consequences. Nat. Rev. Genet 9, 204–217. [DOI] [PubMed] [Google Scholar]
- Akman G, Desai R, Bailey LJ, Yasukawa T, Dalla Rosa I, Durigon R, Holmes JB, Moss CF, Mennuni M, Houlden H, et al. (2016). Pathological ribonuclease H1 causes R-loop depletion and aberrant DNA segregation in mitochondria. Proc. Natl. Acad. Sci. U. S. A 113, E4276–E4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfano L, Caporaso A, Altieri A, Dell’Aquila M, Landi C, Bini L, Pentimalli F, and Giordano A (2019). Depletion of the RNA binding protein HNRNPD impairs homologous recombination by inhibiting DNA-end resection and inducing R-loop accumulation. Nucleic Acids Res. 47, 4068–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alzu A, Bermejo R, Begnis M, Lucca C, Piccini D, Carotenuto W, Saponaro M, Brambati A, Cocito A, Foiani M, et al. (2012). Senataxin associates with replication forks to protect fork integrity across RNA-polymerase-II-transcribed genes. Cell 151, 835–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amon JD, and Koshland D (2016). RNase H enables efficient repair of R-loop induced DNA damage. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariel F, Jegu T, Latrasse D, Romero-Barrios N, Christ A, Benhamed M, and Crespi M (2014). Noncoding transcription by alternative RNA polymerases dynamically regulates an auxin-driven chromatin loop. Mol. Cell 55, 383–396. [DOI] [PubMed] [Google Scholar]
- Ariel F, Lucero L, Christ A, Mammarella MF, Jegu T, Veluchamy A, Mariappan K, Latrasse D, Blein T, Liu C, et al. (2020). R-Loop Mediated trans Action of the APOLO Long Noncoding RNA. Mol. Cell 77, 1055–1065.e4. [DOI] [PubMed] [Google Scholar]
- Arora R, Lee Y, Wischnewski H, Brun CM, Schwarz T, and Azzalin CM (2014). RNaseH1 regulates TERRA-telomeric DNA hybrids and telomere maintenance in ALT tumour cells. Nat. Commun 5, 5220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacal J, Moriel-Carretero M, Pardo B, Barthe A, Sharma S, Chabes A, Lengronne A, and Pasero P (2018). Mrc1 and Rad9 cooperate to regulate initiation and elongation of DNA replication in response to DNA damage. EMBO J. 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader AS, and Bushell M (2020). DNA:RNA hybrids form at DNA double-strand breaks in transcriptionally active loci. Cell Death Dis. 11, 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes CO, Calero M, Malik I, Graham BW, Spahr H, Lin G, Cohen AE, Brown IS, Zhang Q, Pullara F, et al. (2015). Crystal Structure of a Transcribing RNA Polymerase II Complex Reveals a Complete Transcription Bubble. Mol. Cell 59, 258–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barroso S, Herrera-Moyano E, Muñoz S, García-Rubio M, Gómez-González B, and Aguilera A (2019). The DNA damage response acts as a safeguard against harmful DNA-RNA hybrids of different origins. EMBO Rep. 20, e47250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer M, Nascakova Z, Mihai A-I, Cheng PF, Levesque MP, Lampart S, Hurwitz R, Pfannkuch L, Dobrovolna J, Jacobs M, et al. (2020). The ALPK1/TIFA/NF-κB axis links a bacterial carcinogen to R-loop-induced replication stress. Nat. Commun 11, 5117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayona-Feliu A, and Aguilera A (2021). The role of chromatin at transcription-replication conflicts as a genome safeguard. Biochem. Soc. Trans 49, 2727–2736. [DOI] [PubMed] [Google Scholar]
- Bayona-Feliu A, Barroso S, Muñoz S, and Aguilera A (2021). The SWI/SNF chromatin remodeling complex helps resolve R-loop-mediated transcription-replication conflicts. Nat. Genet 53, 1050–1063. [DOI] [PubMed] [Google Scholar]
- Becherel OJ, Yeo AJ, Stellati A, Heng EYH, Luff J, Suraweera AM, Woods R, Fleming J, Carrie D, McKinney K, et al. (2013). Senataxin plays an essential role with DNA damage response proteins in meiotic recombination and gene silencing. PLoS Genet. 9, e1003435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becherel OJ, Sun J, Yeo AJ, Nayler S, Fogel BL, Gao F, Coppola G, Criscuolo C, De Michele G, Wolvetang E, et al. (2015). A new model to study neurodegeneration in ataxia oculomotor apraxia type 2. Hum. Mol. Genet 24, 5759–5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia V, Barroso SI, García-Rubio ML, Tumini E, Herrera-Moyano E, and Aguilera A (2014). BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 511, 362–365. [DOI] [PubMed] [Google Scholar]
- Björkman A, Johansen SL, Lin L, Schertzer M, Kanellis DC, Katsori A-M, Christensen ST, Luo Y, Andersen JS, Elsässer SJ, et al. (2020). Human RTEL1 associates with Poldip3 to facilitate responses to replication stress and R-loop resolution. Genes Dev. 34, 1065–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambati A, Zardoni L, Achar YJ, Piccini D, Galanti L, Colosio A, Foiani M, and Liberi G (2018). Dormant origins and fork protection mechanisms rescue sister forks arrested by transcription. Nucleic Acids Res. 46, 1227–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brubaker SW, Bonham KS, Zanoni I, and Kagan JC (2015). Innate immune pattern recognition: a cell biological perspective. Annu. Rev. Immunol 33, 257–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brüning J-G, and Marians KJ (2020). Replisome bypass of transcription complexes and R-loops. Nucleic Acids Res. 48, 10353–10367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger K, Schlackow M, and Gullerova M (2019). Tyrosine kinase c-Abl couples RNA polymerase II transcription to DNA double-strand breaks. Nucleic Acids Res. 47, 3467–3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Can G, Kauerhof AC, Macak D, and Zegerman P (2019). Helicase Subunit Cdc45 Targets the Checkpoint Kinase Rad53 to Both Replication Initiation and Elongation Complexes after Fork Stalling. Mol. Cell 73, 562–573.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caridi PC, Delabaere L, Zapotoczny G, and Chiolo I (2017). And yet, it moves: nuclear and chromatin dynamics of a heterochromatic double-strand break. Philos. Trans. R. Soc. Lond. B Biol. Sci 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano-Pozo M, Santos-Pereira JM, Rondón AG, Barroso S, Andújar E, Pérez-Alegre M, García-Muse T, and Aguilera A (2013). R loops are linked to histone H3 S10 phosphorylation and chromatin condensation. Mol. Cell 52, 583–590. [DOI] [PubMed] [Google Scholar]
- Castillo-Guzman D, and Chédin F (2021). Defining R-loop classes and their contributions to genome instability. DNA Repair 106, 103182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabanon RM, Morel D, Eychenne T, Colmet-Daage L, Bajrami I, Dorvault N, Garrido M, Meisenberg C, Lamb A, Ngo C, et al. (2021). PBRM1 Deficiency Confers Synthetic Lethality to DNA Repair Inhibitors in Cancer. Cancer Res. 81, 2888–2902. [DOI] [PubMed] [Google Scholar]
- Chakraborty P, and Grosse F (2011). Human DHX9 helicase preferentially unwinds RNA-containing displacement loops (R-loops) and G-quadruplexes. DNA Repair 10, 654–665. [DOI] [PubMed] [Google Scholar]
- Chandramouly G, Zhao J, McDevitt S, Rusanov T, Hoang T, Borisonnik N, Treddinick T, Lopezcolorado FW, Kent T, Siddique LA, et al. (2021). Polθ reverse transcribes RNA and promotes RNA-templated DNA repair. Sci Adv 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C-W, Xu X, Li M, Xin D, Ding L, Wang Y-T, and Liu Y (2020). Pathogenic mutations reveal a role of RECQ4 in mitochondrial RNA:DNA hybrid formation and resolution. Sci. Rep 10, 17033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang EY-C, Novoa CA, Aristizabal MJ, Coulombe Y, Segovia R, Chaturvedi R, Shen Y, Keong C, Tam AS, Jones SJM, et al. (2017a). RECQ-like helicases Sgs1 and BLM regulate R-loop-associated genome instability. J. Cell Biol 216, 3991–4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HHY, Pannunzio NR, Adachi N, and Lieber MR (2017b). Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol 18, 495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman JR, Taylor MRG, and Boulton SJ (2012). Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 47, 497–510. [DOI] [PubMed] [Google Scholar]
- Chappidi N, Nascakova Z, Boleslavska B, Zellweger R, Isik E, Andrs M, Menon S, Dobrovolna J, Balbo Pogliano C, Matos J, et al. (2020). Fork Cleavage-Religation Cycle and Active Transcription Mediate Replication Restart after Fork Stalling at Co-transcriptional R-Loops. Mol. Cell 77, 528–541.e8. [DOI] [PubMed] [Google Scholar]
- Chatzidoukaki O, Stratigi K, Goulielmaki E, Niotis G, Akalestou-Clocher A, Gkirtzimanaki K, Zafeiropoulos A, Altmüller J, Topalis P, and Garinis GA (2021). R-loops trigger the release of cytoplasmic ssDNAs leading to chronic inflammation upon DNA damage. Sci Adv 7, eabj5769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chédin F, Hartono SR, Sanz LA, and Vanoosthuyse V (2021). Best practices for the visualization, mapping, and manipulation of R-loops. EMBO J. e106394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Yang H, Zhu X, Yadav T, Ouyang J, Truesdell SS, Tan J, Wang Y, Duan M, Wei L, et al. (2020). m5C modification of mRNA serves a DNA damage code to promote homologous recombination. Nat. Commun 11, 2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Chen J-Y, Huang Y-J, Gu Y, Qiu J, Qian H, Shao C, Zhang X, Hu J, Li H, et al. (2018). The Augmented R-Loop Is a Unifying Mechanism for Myelodysplastic Syndromes Induced by High-Risk Splicing Factor Mutations. Mol. Cell 69, 412–425.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y-H, Keegan S, Kahli M, Tonzi P, Fenyö D, Huang TT, and Smith DJ (2019). Transcription shapes DNA replication initiation and termination in human cells. Nat. Struct. Mol. Biol 26, 67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L, Wang W, Yao Y, and Sun Q (2021). Mitochondrial RNase H1 activity regulates R-loop homeostasis to maintain genome integrity and enable early embryogenesis in Arabidopsis. PLoS Biol. 19, e3001357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheruiyot A, Li S, Nonavinkere Srivatsan S, Ahmed T, Chen Y, Lemacon DS, Li Y, Yang Z, Wadugu BA, Warner WA, et al. (2021). Nonsense-Mediated RNA Decay Is a Unique Vulnerability of Cancer Cells Harboring SF3B1 or U2AF1 Mutations. Cancer Res. 81, 4499–4513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang H-C, Zhang X, Li J, Zhao X, Chen J, Wang HT-H, Jatoi I, Brenner A, Hu Y, and Li R (2019). BRCA1-associated R-loop affects transcription and differentiation in breast luminal epithelial cells. Nucleic Acids Res. 47, 5086–5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong SY, Cutler S, Lin J-J, Tsai C-H, Tsai H-K, Biggins S, Tsukiyama T, Lo Y-C, and Kao C-F (2020). H3K4 methylation at active genes mitigates transcription-replication conflicts during replication stress. Nat. Commun 11, 809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clynes D, Jelinska C, Xella B, Ayyub H, Scott C, Mitson M, Taylor S, Higgs DR, and Gibbons RJ (2015). Suppression of the alternative lengthening of telomere pathway by the chromatin remodelling factor ATRX. Nat. Commun 6, 7538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, Puget N, Lin Y-L, Clouaire T, Aguirrebengoa M, Rocher V, Pasero P, Canitrot Y, and Legube G (2018). Senataxin resolves RNA:DNA hybrids forming at DNA double-strand breaks to prevent translocations. Nat. Commun 9, 533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colak D, Zaninovic N, Cohen MS, Rosenwaks Z, Yang W-Y, Gerhardt J, Disney MD, and Jaffrey SR (2014). Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science 343, 1002–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti C, Saccà B, Herrick J, Lalou C, Pommier Y, and Bensimon A (2007). Replication fork velocities at adjacent replication origins are coordinately modified during DNA replication in human cells. Mol. Biol. Cell 18, 3059–3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coquel F, Silva M-J, Técher H, Zadorozhny K, Sharma S, Nieminuszczy J, Mettling C, Dardillac E, Barthe A, Schmitz A-L, et al. (2018). SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature 557, 57–61. [DOI] [PubMed] [Google Scholar]
- Costantino L, and Koshland D (2018). Genome-wide Map of R-Loop-Induced Damage Reveals How a Subset of R-Loops Contributes to Genomic Instability. Mol. Cell 71, 487–497.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristini A, Groh M, Kristiansen MS, and Gromak N (2018). RNA/DNA Hybrid Interactome Identifies DXH9 as a Molecular Player in Transcriptional Termination and R-Loop-Associated DNA Damage. Cell Rep. 23, 1891–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristini A, Ricci G, Britton S, Salimbeni S, Huang S-YN, Marinello J, Calsou P, Pommier Y, Favre G, Capranico G, et al. (2019). Dual Processing of R-Loops and Topoisomerase I Induces Transcription-Dependent DNA Double-Strand Breaks. Cell Rep. 28, 3167–3181.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossley MP, Bocek M, and Cimprich KA (2019). R-Loops as Cellular Regulators and Genomic Threats. Mol. Cell 73, 398–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossley MP, Bocek MJ, Hamperl S, Swigut T, and Cimprich KA (2020). qDRIP: a method to quantitatively assess RNA-DNA hybrid formation genome-wide. Nucleic Acids Res. 48, e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossley MP, Brickner JR, Song C, Zar SMT, Maw SS, Chédin F, Tsai M-S, and Cimprich KA (2021). Catalytically inactive, purified RNase H1: A specific and sensitive probe for RNA-DNA hybrid imaging. J. Cell Biol 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Alessandro G, Whelan DR, Howard SM, Vitelli V, Renaudin X, Adamowicz M, Iannelli F, Jones-Weinert CW, Lee M, Matti V, et al. (2018). BRCA2 controls DNA:RNA hybrid level at DSBs by mediating RNase H2 recruitment. Nat. Commun 9, 5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daley JM, Tomimatsu N, Hooks G, Wang W, Miller AS, Xue X, Nguyen KA, Kaur H, Williamson E, Mukherjee B, et al. (2020). Specificity of end resection pathways for double-strand break regions containing ribonucleotides and base lesions. Nat. Commun 11, 3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang TT, and Morales JC (2020). XRN2 Links RNA:DNA Hybrid Resolution to Double Strand Break Repair Pathway Choice. Cancers 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delamarre A, Barthe A, de la Roche Saint-André C, Luciano P, Forey R, Padioleau I, Skrzypczak M, Ginalski K, Géli V, Pasero P, et al. (2020). MRX Increases Chromatin Accessibility at Stalled Replication Forks to Promote Nascent DNA Resection and Cohesin Loading. Mol. Cell 77, 395–410.e3. [DOI] [PubMed] [Google Scholar]
- Domingo-Prim J, Endara-Coll M, Bonath F, Jimeno S, Prados-Carvajal R, Friedländer MR, Huertas P, and Visa N (2019). EXOSC10 is required for RPA assembly and controlled DNA end resection at DNA double-strand breaks. Nat. Commun 10, 2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn K, and Griffith JD (1980). The presence of RNA in a double helix inhibits its interaction with histone protein. Nucleic Acids Res. 8, 555–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta D, Shatalin K, Epshtein V, Gottesman ME, and Nudler E (2011). Linking RNA polymerase backtracking to genome instability in E. coli. Cell 146, 533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards DS, Maganti R, Tanksley JP, Luo J, Park JJH, Balkanska-Sinclair E, Ling J, and Floyd SR (2020). BRD4 Prevents R-Loop Formation and Transcription-Replication Conflicts by Ensuring Efficient Transcription Elongation. Cell Rep. 32, 108166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkenberg M (2018). Mitochondrial DNA replication in mammalian cells: overview of the pathway. Essays Biochem. 62, 287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman JL, and Peterson CL (2019). Yeast Sirtuin Family Members Maintain Transcription Homeostasis to Ensure Genome Stability. Cell Rep. 27, 2978–2989.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felipe-Abrio I, Lafuente-Barquero J, García-Rubio ML, and Aguilera A (2015). RNA polymerase II contributes to preventing transcription-mediated replication fork stalls. EMBO J. 34, 236–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feretzaki M, Pospisilova M, Valador Fernandes R, Lunardi T, Krejci L, and Lingner J (2020). RAD51-dependent recruitment of TERRA lncRNA to telomeres through R-loops. Nature 587, 303–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fijen C, and Rothenberg E (2021). The evolving complexity of DNA damage foci: RNA, condensates and chromatin in DNA double-strand break repair. DNA Repair 105, 103170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn RL, Cox KE, Jeitany M, Wakimoto H, Bryll AR, Ganem NJ, Bersani F, Pineda JR, Suvà ML, Benes CH, et al. (2015). Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science 347, 273–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francia S, Michelini F, Saxena A, Tang D, de Hoon M, Anelli V, Mione M, Carninci P, and d’Adda di Fagagna F (2012). Site-specific DICER and DROSHA RNA products control the DNA-damage response. Nature 488, 231–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francia S, Cabrini M, Matti V, Oldani A, and d’Adda di Fagagna F (2016). DICER, DROSHA and DNA damage response RNAs are necessary for the secondary recruitment of DNA damage response factors. J. Cell Sci 129, 1468–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan W, Guan Z, Liu J, Gui T, Shen K, Manley JL, and Li X (2011). R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes Dev. 25, 2041–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Wei W, Li M-M, Wu Y-S, Ba Z, Jin K-X, Li M-M, Liao Y-Q, Adhikari S, Chong Z, et al. (2014). Ago2 facilitates Rad51 recruitment and DNA double-strand break repair by homologous recombination. Cell Res. 24, 532–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Benítez F, Gaillard H, and Aguilera A (2017). Physical proximity of chromatin to nuclear pores prevents harmful R loop accumulation contributing to maintain genome stability. Proc. Natl. Acad. Sci. U. S. A 114, 10942–10947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Jove Navarro M, Kashida S, Chouaib R, Souquere S, Pierron G, Weil D, and Gueroui Z (2019). RNA is a critical element for the sizing and the composition of phase-separated RNA-protein condensates. Nat. Commun 10, 3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Muse T, and Aguilera A (2019). R Loops: From Physiological to Pathological Roles. Cell 179, 604–618. [DOI] [PubMed] [Google Scholar]
- García-Pichardo D, Cañas JC, García-Rubio ML, Gómez-González B, Rondón AG, and Aguilera A (2017). Histone Mutants Separate R Loop Formation from Genome Instability Induction. Mol. Cell 66, 597–609.e5. [DOI] [PubMed] [Google Scholar]
- García-Rubio ML, Pérez-Calero C, Barroso SI, Tumini E, Herrera-Moyano E, Rosado IV, and Aguilera A (2015). The Fanconi Anemia Pathway Protects Genome Integrity from R-loops. PLoS Genet. 11, e1005674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannini M, Bayona-Feliu A, Sproviero D, Barroso SI, Cereda C, and Aguilera A (2020). TDP-43 mutations link Amyotrophic Lateral Sclerosis with R-loop homeostasis and R loop-mediated DNA damage. PLoS Genet. 16, e1009260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano AMS, Luciani M, Gatto F, Abou Alezz M, Beghè C, Della Volpe L, Migliara A, Valsoni S, Genua M, Dzieciatkowska M, et al. (2022). DNA damage contributes to neurotoxic inflammation in Aicardi-Goutières syndrome astrocytes. J. Exp. Med 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg AD, Banaszynski LA, Noh K-M, Lewis PW, Elsaesser SJ, Stadler S, Dewell S, Law M, Guo X, Li X, et al. (2010). Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 140, 678–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-González B, García-Rubio M, Bermejo R, Gaillard H, Shirahige K, Marín A, Foiani M, and Aguilera A (2011). Genome-wide function of THO/TREX in active genes prevents R-loop-dependent replication obstacles. EMBO J. 30, 3106–3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorthi A, Romero JC, Loranc E, Cao L, Lawrence LA, Goodale E, Iniguez AB, Bernard X, Masamsetti VP, Roston S, et al. (2018). EWS-FLI1 increases transcription to cause R-loops and block BRCA1 repair in Ewing sarcoma. Nature 555, 387–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulielmaki E, Tsekrekou M, Batsiotos N, Ascensão-Ferreira M, Ledaki E, Stratigi K, Chatzinikolaou G, Topalis P, Kosteas T, Altmüller J, et al. (2021). The splicing factor XAB2 interacts with ERCC1-XPF and XPG for R-loop processing. Nat. Commun 12, 3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groh M, and Gromak N (2014). Out of balance: R-loops in human disease. PLoS Genet. 10, e1004630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groh M, Lufino MMP, Wade-Martins R, and Gromak N (2014). R-loops associated with triplet repeat expansions promote gene silencing in Friedreich ataxia and fragile X syndrome. PLoS Genet. 10, e1004318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunseich C, Wang IX, Watts JA, Burdick JT, Guber RD, Zhu Z, Bruzel A, Lanman T, Chen K, Schindler AB, et al. (2018). Senataxin Mutation Reveals How R-Loops Promote Transcription by Blocking DNA Methylation at Gene Promoters. Mol. Cell 69, 426–437.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra J, Valadao A-L, Vlachakis D, Polak K, Vila IK, Taffoni C, Prabakaran T, Marriott AS, Kaczmarek R, Houel A, et al. (2020). Lysyl-tRNA synthetase produces diadenosine tetraphosphate to curb STING-dependent inflammation. Sci Adv 6, eaax3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Wiley CA, Steinman RA, Sheng Y, Ji B, Wang J, Zhang L, Wang T, Zenatai M, Billiar TR, et al. (2021). Aicardi-Goutières syndrome-associated mutation at ADAR1 gene locus activates innate immune response in mouse brain. J. Neuroinflammation 18, 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeusler AR, Donnelly CJ, Periz G, Simko EAJ, Shaw PG, Kim M-S, Maragakis NJ, Troncoso JC, Pandey A, Sattler R, et al. (2014). C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 507, 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamperl S, and Cimprich KA (2016). Conflict Resolution in the Genome: How Transcription and Replication Make It Work. Cell 167, 1455–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamperl S, Bocek MJ, Saldivar JC, Swigut T, and Cimprich KA (2017). Transcription-Replication Conflict Orientation Modulates R-Loop Levels and Activates Distinct DNA Damage Responses. Cell 170, 774–786.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, and Greenberg RA (2017). Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548, 466–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatchi E, Skourti-Stathaki K, Ventz S, Pinello L, Yen A, Kamieniarz-Gdula K, Dimitrov S, Pathania S, McKinney KM, Eaton ML, et al. (2015). BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol. Cell 57, 636–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatchi E, Goehring L, Landini S, Skourti-Stathaki K, DeConti DK, Abderazzaq FO, Banerjee P, Demers TM, Wang YE, Quackenbush J, et al. (2021). BRCA1 and RNAi factors promote repair mediated by small RNAs and PALB2-RAD52. Nature 591, 665–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Pasupala N, Zhi H, Dorjbal B, Hussain I, Shih H-M, Bhattacharyya S, Biswas R, Miljkovic M, Semmes OJ, et al. (2021). NF-κB-induced R-loop accumulation and DNA damage select for nucleotide excision repair deficiencies in adult T cell leukemia. Proc. Natl. Acad. Sci. U. S. A 118, e2005568118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold S, Kalb J, Büchel G, Ade CP, Baluapuri A, Xu J, Koster J, Solvie D, Carstensen A, Klotz C, et al. (2019). Recruitment of BRCA1 limits MYCN-driven accumulation of stalled RNA polymerase. Nature 567, 545–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodroj D, Recolin B, Serhal K, Martinez S, Tsanov N, Abou Merhi R, and Maiorano D (2017). An ATR-dependent function for the Ddx19 RNA helicase in nuclear R-loop metabolism. EMBO J. 36, 1182–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt IJ (2019). The mitochondrial R-loop. Nucleic Acids Res. 47, 5480–5489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopfner K-P, and Hornung V (2020). Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat. Rev. Mol. Cell Biol 21, 501–521. [DOI] [PubMed] [Google Scholar]
- Huang J, Liu S, Bellani MA, Thazhathveetil AK, Ling C, de Winter JP, Wang Y, Wang W, and Seidman MM (2013). The DNA translocase FANCM/MHF promotes replication traverse of DNA interstrand crosslinks. Mol. Cell 52, 434–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huertas P, and Aguilera A (2003). Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol. Cell 12, 711–721. [DOI] [PubMed] [Google Scholar]
- Hurst V, Challa K, Jonas F, Forey R, Sack R, Seebacher J, Schmid CD, Barkai N, Shimada K, Gasser SM, et al. (2021). A regulatory phosphorylation site on Mec1 controls chromatin occupancy of RNA polymerases during replication stress. EMBO J. 40, e108439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyjek M, Figiel M, and Nowotny M (2019). RNases H: Structure and mechanism. DNA Repair 84, 102672. [DOI] [PubMed] [Google Scholar]
- Im J-S, Keaton M, Lee KY, Kumar P, Park J, and Dutta A (2014). ATR checkpoint kinase and CRL1βTRCP collaborate to degrade ASF1a and thus repress genes overlapping with clusters of stalled replication forks. Genes Dev. 28, 875–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M, Nakahama T, Yamasaki R, Shibuya T, Kim JI, Todo H, Xing Y, Kato Y, Morii E, and Kawahara Y (2021). An Aicardi-Goutières Syndrome-Causative Point Mutation in Adar1 Gene Invokes Multiorgan Inflammation and Late-Onset Encephalopathy in Mice. J. Immunol 207, 3016–3027. [DOI] [PubMed] [Google Scholar]
- Jackson BR, Noerenberg M, and Whitehouse A (2014). A novel mechanism inducing genome instability in Kaposi’s sarcoma-associated herpesvirus infected cells. PLoS Pathog. 10, e1004098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang Y, Elsayed Z, Eki R, He S, Du K-P, Abbas T, and Kai M (2020). Intrinsically disordered protein RBM14 plays a role in generation of RNA:DNA hybrids at double-strand break sites. Proc. Natl. Acad. Sci. U. S. A 117, 5329–5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jangi M, Fleet C, Cullen P, Gupta SV, Mekhoubad S, Chiao E, Allaire N, Bennett CF, Rigo F, Krainer AR, et al. (2017). SMN deficiency in severe models of spinal muscular atrophy causes widespread intron retention and DNA damage. Proc. Natl. Acad. Sci. U. S. A 114, E2347–E2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, and Charpentier E (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SE, Fleuren EDG, Frankum J, Konde A, Williamson CT, Krastev DB, Pemberton HN, Campbell J, Gulati A, Elliott R, et al. (2017). ATR Is a Therapeutic Target in Synovial Sarcoma. Cancer Res. 77, 7014–7026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurga M, Abugable AA, Goldman ASH, and El-Khamisy SF (2021). USP11 controls R-loops by regulating senataxin proteostasis. Nat. Commun 12, 5156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadesch TR, and Chamberlin MJ (1982). Studies of in vitro transcription by calf thymus RNA polymerase II using a novel duplex DNA template. J. Biol. Chem 257, 5286–5295. [PubMed] [Google Scholar]
- Kanagaraj R, Mitter R, Kantidakis T, Edwards MM, Benitez A, Chakravarty P, Fu B, Becherel O, Yang F, Lavin MF, et al. (2022). Integrated genome and transcriptome analyses reveal the mechanism of genome instability in ataxia with oculomotor apraxia 2. Proc. Natl. Acad. Sci. U. S. A 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HJ, Cheon NY, Park H, Jeong GW, Ye BJ, Yoo EJ, Lee JH, Hur J-H, Lee E-A, Kim H, et al. (2021). TonEBP recognizes R-loops and initiates m6A RNA methylation for R-loop resolution. Nucleic Acids Res. 49, 269–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan A, Jiang X, He L, Ahmad S, and Gangwani L (2020). ZPR1 prevents R-loop accumulation, upregulates SMN2 expression and rescues spinal muscular atrophy. Brain 143, 69–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan A, Cuartas J, Gangwani P, Branzei D, and Gangwani L (2022). Mutation in senataxin alters the mechanism of R-loop resolution in amyotrophic lateral sclerosis 4. Brain. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemiha S, Poli J, Lin Y-L, Lengronne A, and Pasero P (2021). Toxic R-loops: Cause or consequence of replication stress? DNA Repair 107, 103199. [DOI] [PubMed] [Google Scholar]
- Keskin H, Shen Y, Huang F, Patel M, Yang T, Ashley K, Mazin AV, and Storici F (2014). Transcript-RNA-templated DNA recombination and repair. Nature 515, 436–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Kang N, Park SH, Wells J, Hwang T, Ryu E, Kim B-G, Hwang S, Kim S-J, Kang S, et al. (2020). ATAD5 restricts R-loop formation through PCNA unloading and RNA helicase maintenance at the replication fork. Nucleic Acids Res. 48, 7218–7238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klusmann I, Wohlberedt K, Magerhans A, Teloni F, Korbel JO, Altmeyer M, and Dobbelstein M (2018). Chromatin modifiers Mdm2 and RNF2 prevent RNA:DNA hybrids that impair DNA replication. Proc. Natl. Acad. Sci. U. S. A 115, E11311–E11320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo CX, Kobiyama K, Shen YJ, LeBert N, Ahmad S, Khatoo M, Aoshi T, Gasser S, and Ishii KJ (2015). RNA polymerase III regulates cytosolic RNA:DNA hybrids and intracellular microRNA expression. J. Biol. Chem 290, 7463–7473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosar M, Giannattasio M, Piccini D, Maya-Mendoza A, García-Benítez F, Bartkova J, Barroso SI, Gaillard H, Martini E, Restuccia U, et al. (2021). The human nucleoporin Tpr protects cells from RNA-mediated replication stress. Nat. Commun 12, 3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotsantis P, Silva LM, Irmscher S, Jones RM, Folkes L, Gromak N, and Petermann E (2016). Increased global transcription activity as a mechanism of replication stress in cancer. Nat. Commun 7, 13087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotsantis P, Segura-Bayona S, Margalef P, Marzec P, Ruis P, Hewitt G, Bellelli R, Patel H, Goldstone R, Poetsch AR, et al. (2020). RTEL1 Regulates G4/R-Loops to Avert Replication-Transcription Collisions. Cell Rep. 33, 108546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar C, Batra S, Griffith JD, and Remus D (2021). The interplay of RNA:DNA hybrid structure and G-quadruplexes determines the outcome of R-loop-replisome collisions. Elife 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafuente-Barquero J, Luke-Glaser S, Graf M, Silva S, Gómez-González B, Lockhart A, Lisby M, Aguilera A, and Luke B (2017). The Smc5/6 complex regulates the yeast Mph1 helicase at RNA-DNA hybrid-mediated DNA damage. PLoS Genet. 13, e1007136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafuente-Barquero J, García-Rubio ML, Martin-Alonso MS, Gómez-González B, and Aguilera A (2020). Harmful DNA:RNA hybrids are formed in cis and in a Rad51-independent manner. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalonde M, Trauner M, Werner M, and Hamperl S (2021). Consequences and Resolution of Transcription-Replication Conflicts. Life 11, 637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam FC, Kong YW, Huang Q, Vu Han T-L, Maffa AD, Kasper EM, and Yaffe MB (2020). BRD4 prevents the accumulation of R-loops and protects against transcription-replication collision events and DNA damage. Nat. Commun 11, 4083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambo S, Gröbner SN, Rausch T, Waszak SM, Schmidt C, Gorthi A, Romero JC, Mauermann M, Brabetz S, Krausert S, et al. (2019). The molecular landscape of ETMR at diagnosis and relapse. Nature 576, 274–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landsverk HB, Sandquist LE, Bay LTE, Steurer B, Campsteijn C, Landsverk OJB, Marteijn JA, Petermann E, Trinkle-Mulcahy L, and Syljuåsen RG (2020). WDR82/PNUTS-PP1 Prevents Transcription-Replication Conflicts by Promoting RNA Polymerase II Degradation on Chromatin. Cell Rep. 33, 108469. [DOI] [PubMed] [Google Scholar]
- Lang KS, and Merrikh H (2021). Topological stress is responsible for the detrimental outcomes of head-on replication-transcription conflicts. Cell Rep. 34, 108797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang KS, Hall AN, Merrikh CN, Ragheb M, Tabakh H, Pollock AJ, Woodward JJ, Dreifus JE, and Merrikh H (2017). Replication-Transcription Conflicts Generate R-Loops that Orchestrate Bacterial Stress Survival and Pathogenesis. Cell 170, 787–799.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappin KM, Barros EM, Jhujh SS, Irwin GW, McMillan H, Liberante FG, Latimer C, La Bonte MJ, Mills KI, Harkin DP, et al. (2022). Cancer-Associated SF3B1 Mutations Confer a BRCA-Like Cellular Phenotype and Synthetic Lethality to PARP Inhibitors. Cancer Res. 82, 819–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larochelle S, Amat R, Glover-Cutter K, Sansó M, Zhang C, Allen JJ, Shokat KM, Bentley DL, and Fisher RP (2012). Cyclin-dependent kinase control of the initiation-to-elongation switch of RNA polymerase II. Nat. Struct. Mol. Biol 19, 1108–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laverde EE, Lai Y, Leng F, Balakrishnan L, Freudenreich CH, and Liu Y (2020). R-loops promote trinucleotide repeat deletion through DNA base excision repair enzymatic activities. J. Biol. Chem 295, 13902–13913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, and Manley JL (2005). Inactivation of the SR protein splicing factor ASF/SF2 results in genomic instability. Cell 122, 365–378. [DOI] [PubMed] [Google Scholar]
- Li L, Germain DR, Poon H-Y, Hildebrandt MR, Monckton EA, McDonald D, Hendzel MJ, and Godbout R (2016). DEAD Box 1 Facilitates Removal of RNA and Homologous Recombination at DNA Double-Strand Breaks. Mol. Cell. Biol 36, 2794–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang F, Longerich S, Miller AS, Tang C, Buzovetsky O, Xiong Y, Maranon DG, Wiese C, Kupfer GM, and Sung P (2016). Promotion of RAD51-Mediated Homologous DNA Pairing by the RAD51AP1-UAF1 Complex. Cell Rep. 15, 2118–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim YW, Sanz LA, Xu X, Hartono SR, and Chédin F (2015). Genome-wide DNA hypomethylation and RNA:DNA hybrid accumulation in Aicardi-Goutières syndrome. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima WF, Murray HM, Damle SS, Hart CE, Hung G, De Hoyos CL, Liang X-H, and Crooke ST (2016). Viable RNaseH1 knockout mice show RNaseH1 is essential for R loop processing, mitochondrial and liver function. Nucleic Acids Res. 44, 5299–5312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Liu Y, Shi Q, Su H, Wang C, Birchler JA, and Han F (2021a). Emerging roles of centromeric RNAs in centromere formation and function. Genes Genomics 43, 217–226. [DOI] [PubMed] [Google Scholar]
- Liu S, Hua Y, Wang J, Li L, Yuan J, Zhang B, Wang Z, Ji J, and Kong D (2021b). RNA polymerase III is required for the repair of DNA double-strand breaks by homologous recombination. Cell 184, 1314–1329.e10. [DOI] [PubMed] [Google Scholar]
- Liu Z, Gao X, Zhou Z, Kang SW, Yang Y, Liu H, Zhang C, Wen Z, Rao X, Wang D, et al. (2021c). San1 deficiency leads to cardiomyopathy due to excessive R-loop-associated DNA damage and cardiomyocyte hypoplasia. Biochim. Biophys. Acta Mol. Basis Dis 1867, 166237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomis EW, Sanz LA, Chédin F, and Hagerman PJ (2014). Transcription-associated R-loop formation across the human FMR1 CGG-repeat region. PLoS Genet. 10, e1004294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W-T, Hawley BR, Skalka GL, Baldock RA, Smith EM, Bader AS, Malewicz M, Watts FZ, Wilczynska A, and Bushell M (2018). Drosha drives the formation of DNA:RNA hybrids around DNA break sites to facilitate DNA repair. Nat. Commun 9, 532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macheret M, and Halazonetis TD (2018). Intragenic origins due to short G1 phases underlie oncogene-induced DNA replication stress. Nature 555, 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie KJ, Carroll P, Lettice L, Tarnauskaitė Ž, Reddy K, Dix F, Revuelta A, Abbondati E, Rigby RE, Rabe B, et al. (2016). Ribonuclease H2 mutations induce a cGAS/STING-dependent innate immune response. EMBO J. 35, 831–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie KJ, Carroll P, Martin C-A, Murina O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch A, et al. (2017). cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548, 461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makharashvili N, Arora S, Yin Y, Fu Q, Wen X, Lee J-H, Kao C-H, Leung JW, Miller KM, and Paull TT (2018). Sae2/CtIP prevents R-loop accumulation in eukaryotic cells. Elife 7, e42733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik I, Kelley CP, Wang ET, and Todd PK (2021). Molecular mechanisms underlying nucleotide repeat expansion disorders. Nat. Rev. Mol. Cell Biol 22, 589–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mankan AK, Schmidt T, Chauhan D, Goldeck M, Höning K, Gaidt M, Kubarenko AV, Andreeva L, Hopfner K-P, and Hornung V (2014). Cytosolic RNA:DNA hybrids activate the cGAS-STING axis. EMBO J. 33, 2937–2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marabitti V, Lillo G, Malacaria E, Palermo V, Sanchez M, Pichierri P, and Franchitto A (2019). ATM pathway activation limits R-loop-associated genomic instability in Werner syndrome cells. Nucleic Acids Res. 47, 3485–3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marabitti V, Lillo G, Malacaria E, Palermo V, Pichierri P, and Franchitto A (2020). Checkpoint Defects Elicit a WRNIP1-Mediated Response to Counteract R-Loop-Associated Genomic Instability. Cancers 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marnef A, and Legube G (2021). R-loops as Janus-faced modulators of DNA repair. Nat. Cell Biol 23, 305–313. [DOI] [PubMed] [Google Scholar]
- Matos DA, Zhang J-M, Ouyang J, Nguyen HD, Genois M-M, and Zou L (2020). ATR Protects the Genome against R Loops through a MUS81-Triggered Feedback Loop. Mol. Cell 77, 514–527.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matson JP, and Zou L (2020). A genome-wide and cotranscriptional suppressor of R loops. Genes Dev. 34, 863–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui M, Sakasai R, Abe M, Kimura Y, Kajita S, Torii W, Katsuki Y, Ishiai M, Iwabuchi K, Takata M, et al. (2020). USP42 enhances homologous recombination repair by promoting R-loop resolution with a DNA-RNA helicase DHX9. Oncogenesis 9, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazina OM, Keskin H, Hanamshet K, Storici F, and Mazin AV (2017). Rad52 Inverse Strand Exchange Drives RNA-Templated DNA Double-Strand Break Repair. Mol. Cell 67, 19–29.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meers C, Keskin H, Banyai G, Mazina O, Yang T, Gombolay AL, Mukherjee K, Kaparos EI, Newnam G, Mazin A, et al. (2020). Genetic Characterization of Three Distinct Mechanisms Supporting RNA-Driven DNA Repair and Modification Reveals Major Role of DNA Polymerase ζ. Mol. Cell 79, 1037–1050.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mersaoui SY, Yu Z, Coulombe Y, Karam M, Busatto FF, Masson J-Y, and Richard S (2019). Arginine methylation of the DDX5 helicase RGG/RG motif by PRMT5 regulates resolution of RNA:DNA hybrids. EMBO J. 38, e100986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelini F, Pitchiaya S, Vitelli V, Sharma S, Gioia U, Pessina F, Cabrini M, Wang Y, Capozzo I, Iannelli F, et al. (2017). Damage-induced lncRNAs control the DNA damage response through interaction with DDRNAs at individual double-strand breaks. Nat. Cell Biol 19, 1400–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelini F, Jalihal AP, Francia S, Meers C, Neeb ZT, Rossiello F, Gioia U, Aguado J, Jones-Weinert C, Luke B, et al. (2018). From “Cellular” RNA to “Smart” RNA: Multiple Roles of RNA in Genome Stability and Beyond. Chem. Rev 118, 4365–4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales JC, Richard P, Patidar PL, Motea EA, Dang TT, Manley JL, and Boothman DA (2016). XRN2 Links Transcription Termination to DNA Damage and Replication Stress. PLoS Genet. 12, e1006107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muniz L, Nicolas E, and Trouche D (2021). RNA polymerase II speed: a key player in controlling and adapting transcriptome composition. EMBO J. 40, e105740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutreja K, Krietsch J, Hess J, Ursich S, Berti M, Roessler FK, Zellweger R, Patra M, Gasser G, and Lopes M (2018). ATR-Mediated Global Fork Slowing and Reversal Assist Fork Traverse and Prevent Chromosomal Breakage at DNA Interstrand Cross-Links. Cell Rep. 24, 2629–2642.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neil AJ, Liang MU, Khristich AN, Shah KA, and Mirkin SM (2018). RNA-DNA hybrids promote the expansion of Friedreich’s ataxia (GAA)n repeats via break-induced replication. Nucleic Acids Res. 46, 3487–3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo GHP, Grimstead JW, and Baird DM (2021). UPF1 promotes the formation of R loops to stimulate DNA double-strand break repair. Nat. Commun 12, 3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen DT, Voon HPJ, Xella B, Scott C, Clynes D, Babbs C, Ayyub H, Kerry J, Sharpe JA, Sloane-Stanley JA, et al. (2017a). The chromatin remodelling factor ATRX suppresses R-loops in transcribed telomeric repeats. EMBO Rep. 18, 914–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HD, Yadav T, Giri S, Saez B, Graubert TA, and Zou L (2017b). Functions of Replication Protein A as a Sensor of R Loops and a Regulator of RNaseH1. Mol. Cell 65, 832–847.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HD, Leong WY, Li W, Reddy PNG, Sullivan JD, Walter MJ, Zou L, and Graubert TA (2018). Spliceosome Mutations Induce R Loop-Associated Sensitivity to ATR Inhibition in Myelodysplastic Syndromes. Cancer Res. 78, 5363–5374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehrs C, and Luke B (2020). Regulatory R-loops as facilitators of gene expression and genome stability. Nat. Rev. Mol. Cell Biol 21, 167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimonkar AV, Genschel J, Kinoshita E, Polaczek P, Campbell JL, Wyman C, Modrich P, and Kowalczykowski SC (2011). BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 25, 350–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimasu H, Ran FA, Hsu PD, Konermann S, Shehata SI, Dohmae N, Ishitani R, Zhang F, and Nureki O (2014). Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 156, 935–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noe Gonzalez M, Blears D, and Svejstrup JQ (2021). Causes and consequences of RNA polymerase II stalling during transcript elongation. Nat. Rev. Mol. Cell Biol 22, 3–21. [DOI] [PubMed] [Google Scholar]
- Ohle C, Tesorero R, Schermann G, Dobrev N, Sinning I, and Fischer T (2016). Transient RNA-DNA Hybrids Are Required for Efficient Double-Strand Break Repair. Cell 167, 1001–1013.e7. [DOI] [PubMed] [Google Scholar]
- Ortega P, Mérida-Cerro JA, Rondón AG, Gómez-González B, and Aguilera A (2021). DNA-RNA hybrids at DSBs interfere with repair by homologous recombination. Elife 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang J, Yadav T, Zhang J-M, Yang H, Rheinbay E, Guo H, Haber DA, Lan L, and Zou L (2021). RNA transcripts stimulate homologous recombination by forming DR-loops. Nature 594, 283–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Chen Y, Biju B, Ahmed N, Kong J, Goldenberg M, Huang J, Mohan N, Klosek S, Parsa K, et al. (2019). FANCM suppresses DNA replication stress at ALT telomeres by disrupting TERRA R-loops. Sci. Rep 9, 19110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos D, Solvie D, Baluapuri A, Endres T, Ha SA, Herold S, Kalb J, Giansanti C, Schülein-Völk C, Ade CP, et al. (2022). MYCN recruits the nuclear exosome complex to RNA polymerase II to prevent transcription-replication conflicts. Mol. Cell 82, 159–176.e12. [DOI] [PubMed] [Google Scholar]
- Park K, Ryoo J, Jeong H, Kim M, Lee S, Hwang S-Y, Ahn J, Kim D, Moon HC, Baek D, et al. (2021). Aicardi-Goutières syndrome-associated gene SAMHD1 preserves genome integrity by preventing R-loop formation at transcription-replication conflict regions. PLoS Genet. 17, e1009523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel PS, Abraham KJ, Guturi KKN, Halaby M-J, Khan Z, Palomero L, Ho B, Duan S, St-Germain J, Algouneh A, et al. (2021). RNF168 regulates R-loop resolution and genomic stability in BRCA1/2-deficient tumors. J. Clin. Invest 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull TT (2019). RNA-DNA hybrids and the convergence with DNA repair. Crit. Rev. Biochem. Mol. Biol 54, 371–384. [DOI] [PubMed] [Google Scholar]
- Paulsen RD, Soni DV, Wollman R, Hahn AT, Yee M-C, Guan A, Hesley JA, Miller SC, Cromwell EF, Solow-Cordero DE, et al. (2009). A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol. Cell 35, 228–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perego MGL, Taiana M, Bresolin N, Comi GP, and Corti S (2019). R-Loops in Motor Neuron Diseases. Mol. Neurobiol 56, 2579–2589. [DOI] [PubMed] [Google Scholar]
- Pérez-Calero C, Bayona-Feliu A, Xue X, Barroso SI, Muñoz S, González-Basallote VM, Sung P, and Aguilera A (2020). UAP56/DDX39B is a major cotranscriptional RNA-DNA helicase that unwinds harmful R loops genome-wide. Genes Dev. 34, 898–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessina F, Giavazzi F, Yin Y, Gioia U, Vitelli V, Galbiati A, Barozzi S, Garre M, Oldani A, Flaus A, et al. (2019). Functional transcription promoters at DNA double-strand breaks mediate RNA-driven phase separation of damage-response factors. Nat. Cell Biol 21, 1286–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petryk N, Kahli M, d’Aubenton-Carafa Y, Jaszczyszyn Y, Shen Y, Silvain M, Thermes C, Chen C-L, and Hyrien O (2016). Replication landscape of the human genome. Nat. Commun 7, 10208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poli J, Gerhold C-B, Tosi A, Hustedt N, Seeber A, Sack R, Herzog F, Pasero P, Shimada K, Hopfner K-P, et al. (2016). Mec1, INO80, and the PAF1 complex cooperate to limit transcription replication conflicts through RNAPII removal during replication stress. Genes Dev. 30, 337–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posse V, Al-Behadili A, Uhler JP, Clausen AR, Reyes A, Zeviani M, Falkenberg M, and Gustafsson CM (2019). RNase H1 directs origin-specific initiation of DNA replication in human mitochondria. PLoS Genet. 15, e1007781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell WT, Coulson RL, Gonzales ML, Crary FK, Wong SS, Adams S, Ach RA, Tsang P, Yamada NA, Yasui DH, et al. (2013). R-loop formation at Snord116 mediates topotecan inhibition of Ube3a-antisense and allele-specific chromatin decondensation. Proc. Natl. Acad. Sci. U. S. A 110, 13938–13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prendergast L, McClurg UL, Hristova R, Berlinguer-Palmini R, Greener S, Veitch K, Hernandez I, Pasero P, Rico D, Higgins JMG, et al. (2020). Resolution of R-loops by INO80 promotes DNA replication and maintains cancer cell proliferation and viability. Nat. Commun 11, 4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Promonet A, Padioleau I, Liu Y, Sanz L, Biernacka A, Schmitz A-L, Skrzypczak M, Sarrazin A, Mettling C, Rowicka M, et al. (2020). Topoisomerase 1 prevents replication stress at R-loop-enriched transcription termination sites. Nat. Commun 11, 3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugliese GM, Salaris F, Palermo V, Marabitti V, Morina N, Rosa A, Franchitto A, and Pichierri P (2019). Inducible SMARCAL1 knockdown in iPSC reveals a link between replication stress and altered expression of master differentiation genes. Dis. Model. Mech 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin K, Jian D, Xue Y, Cheng Y, Zhang P, Wei Y, Zhang J, Xiong H, Zhang Y, and Yuan X (2021). DDX41 regulates the expression and alternative splicing of genes involved in tumorigenesis and immune response. Oncol. Rep 45, 1213–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragu S, Matos-Rodrigues G, and Lopez BS (2020). Replication Stress, DNA Damage, Inflammatory Cytokines and Innate Immune Response. Genes 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawal CC, Zardoni L, Di Terlizzi M, Galati E, Brambati A, Lazzaro F, Liberi G, and Pellicioli A (2020). Senataxin Ortholog Sen1 Limits DNA:RNA Hybrid Accumulation at DNA Double-Strand Breaks to Control End Resection and Repair Fidelity. Cell Rep. 31, 107603. [DOI] [PubMed] [Google Scholar]
- Reddy K, Schmidt MHM, Geist JM, Thakkar NP, Panigrahi GB, Wang Y-H, and Pearson CE (2014). Processing of double-R-loops in (CAG)·(CTG) and C9orf72 (GGGGCC)·(GGCCCC) repeats causes instability. Nucleic Acids Res. 42, 10473–10487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reginato G, and Cejka P (2020). The MRE11 complex: A versatile toolkit for the repair of broken DNA. DNA Repair 91–92, 102869. [DOI] [PubMed] [Google Scholar]
- Renaudin X, Lee M, Shehata M, Surmann E-M, and Venkitaraman AR (2021). BRCA2 deficiency reveals that oxidative stress impairs RNaseH1 function to cripple mitochondrial DNA maintenance. Cell Rep. 36, 109478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard P, and Manley JL (2017). R Loops and Links to Human Disease. J. Mol. Biol 429, 3168–3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ries RJ, Zaccara S, Klein P, Olarerin-George A, Namkoong S, Pickering BF, Patil DP, Kwak H, Lee JH, and Jaffrey SR (2019). m6A enhances the phase separation potential of mRNA. Nature 571, 424–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigby RE, Webb LM, Mackenzie KJ, Li Y, Leitch A, Reijns MAM, Lundie RJ, Revuelta A, Davidson DJ, Diebold S, et al. (2014). RNA:DNA hybrids are a novel molecular pattern sensed by TLR9. EMBO J. 33, 542–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldi C, Pizzul P, Longhese MP, and Bonetti D (2020). Sensing R-Loop-Associated DNA Damage to Safeguard Genome Stability. Front Cell Dev Biol 8, 618157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeschert I, Poon E, Henssen AG, Garcia HD, Gatti M, Giansanti C, Jamin Y, Ade CP, Gallant P, Schülein-Völk C, et al. (2021). Combined inhibition of Aurora-A and ATR kinase results in regression of MYCN-amplified neuroblastoma. Nat Cancer 2, 312–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy D, Zhang Z, Lu Z, Hsieh C-L, and Lieber MR (2010). Competition between the RNA transcript and the nontemplate DNA strand during R-loop formation in vitro: a nick can serve as a strong R-loop initiation site. Mol. Cell. Biol 30, 146–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagie S, Toubiana S, Hartono SR, Katzir H, Tzur-Gilat A, Havazelet S, Francastel C, Velasco G, Chédin F, and Selig S (2017). Telomeres in ICF syndrome cells are vulnerable to DNA damage due to elevated DNA:RNA hybrids. Nat. Commun 8, 14015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini N, Roberts SA, Sterling JF, Malc EP, Mieczkowski PA, and Gordenin DA (2017). APOBEC3B cytidine deaminase targets the non-transcribed strand of tRNA genes in yeast. DNA Repair 53, 4–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas-Armenteros I, Pérez-Calero C, Bayona-Feliu A, Tumini E, Luna R, and Aguilera A (2017). Human THO-Sin3A interaction reveals new mechanisms to prevent R-loops that cause genome instability. EMBO J. 36, 3532–3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- San Martin-Alonso M, Soler-Oliva ME, García-Rubio M, García-Muse T, and Aguilera A (2021). Harmful R-loops are prevented via different cell cycle-specific mechanisms. Nat. Commun 12, 4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz LA, Hartono SR, Lim YW, Steyaert S, Rajpurkar A, Ginno PA, Xu X, and Chédin F (2016). Prevalent, Dynamic, and Conserved R-Loop Structures Associate with Specific Epigenomic Signatures in Mammals. Mol. Cell 63, 167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saponaro M, Kantidakis T, Mitter R, Kelly GP, Heron M, Williams H, Söding J, Stewart A, and Svejstrup JQ (2014). RECQL5 controls transcript elongation and suppresses genome instability associated with transcription stress. Cell 157, 1037–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauer GD, Spenkelink LM, Lewis JS, Yurieva O, Mueller SH, van Oijen AM, and O’Donnell ME (2020). Replisome bypass of a protein-based R-loop block by Pif1. Proc. Natl. Acad. Sci. U. S. A 117, 30354–30361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schellenbauer A, Guilly M-N, Grall R, Le Bars R, Paget V, Kortulewski T, Sutcu H, Mathé C, Hullo M, Biard D, et al. (2021). Phospho-Ku70 induced by DNA damage interacts with RNA Pol II and promotes the formation of phospho-53BP1 foci to ensure optimal cNHEJ. Nucleic Acids Res. 49, 11728–11745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab RA, Nieminuszczy J, Shah F, Langton J, Lopez Martinez D, Liang C-C, Cohn MA, Gibbons RJ, Deans AJ, and Niedzwiedz W (2015). The Fanconi Anemia Pathway Maintains Genome Stability by Coordinating Replication and Transcription. Mol. Cell 60, 351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler JA, Conti C, Syed A, Aladjem MI, and Pommier Y (2007). The intra-S-phase checkpoint affects both DNA replication initiation and elongation: single-cell and -DNA fiber analyses. Mol. Cell. Biol 27, 5806–5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessa G, Gómez-González B, Silva S, Pérez-Calero C, Beaurepere R, Barroso S, Martineau S, Martin C, Ehlén Å, Martínez JS, et al. (2021). BRCA2 promotes DNA-RNA hybrid resolution by DDX5 helicase at DNA breaks to facilitate their repair‡. EMBO J. 40, e106018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanbhag NM, Rafalska-Metcalf IU, Balane-Bolivar C, Janicki SM, and Greenberg RA (2010). ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 141, 970–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Anand R, Zhang X, Francia S, Michelini F, Galbiati A, Williams H, Ronato DA, Masson J-Y, Rothenberg E, et al. (2021). MRE11-RAD50-NBS1 Complex Is Sufficient to Promote Transcription by RNA Polymerase II at Double-Strand Breaks by Melting DNA Ends. Cell Rep. 34, 108565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen YJ, Le Bert N, Chitre AA, Koo CX, Nga XH, Ho SSW, Khatoo M, Tan NY, Ishii KJ, and Gasser S (2015). Genome-derived cytosolic DNA mediates type I interferon-dependent rejection of B cell lymphoma cells. Cell Rep. 11, 460–473. [DOI] [PubMed] [Google Scholar]
- Shiromoto Y, Sakurai M, Minakuchi M, Ariyoshi K, and Nishikura K (2021). ADAR1 RNA editing enzyme regulates R-loop formation and genome stability at telomeres in cancer cells. Nat. Commun 12, 1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivji MKK, Renaudin X, Williams ÇH, and Venkitaraman AR (2018). BRCA2 Regulates Transcription Elongation by RNA Polymerase II to Prevent R-Loop Accumulation. Cell Rep. 22, 1031–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva E, and Ideker T (2019). Transcriptional responses to DNA damage. DNA Repair 79, 40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva B, Pentz R, Figueira AM, Arora R, Lee YW, Hodson C, Wischnewski H, Deans AJ, and Azzalin CM (2019). FANCM limits ALT activity by restricting telomeric replication stress induced by deregulated BLM and R-loops. Nat. Commun 10, 2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva S, Camino LP, and Aguilera A (2018). Human mitochondrial degradosome prevents harmful mitochondrial R loops and mitochondrial genome instability. Proc. Natl. Acad. Sci. U. S. A 115, 11024–11029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon NE, Yuan M, and Kai M (2017). RNA-binding protein RBM14 regulates dissociation and association of non-homologous end joining proteins. Cell Cycle 16, 1175–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S, Ahmed D, Dolatshad H, Tatwavedi D, Schulze U, Sanchi A, Ryley S, Dhir A, Carpenter L, Watt SM, et al. (2020). SF3B1 mutations induce R-loop accumulation and DNA damage in MDS and leukemia cells with therapeutic implications. Leukemia 34, 2525–2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skourti-Stathaki K, Proudfoot NJ, and Gromak N (2011). Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol. Cell 42, 794–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolka JA, Sanz LA, Hartono SR, and Chédin F (2021). Recognition of RNA by the S9.6 antibody creates pervasive artifacts when imaging RNA:DNA hybrids. J. Cell Biol 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollier J, Stork CT, García-Rubio ML, Paulsen RD, Aguilera A, and Cimprich KA (2014). Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol. Cell 56, 777–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C, Hotz-Wagenblatt A, Voit R, and Grummt I (2017). SIRT7 and the DEAD-box helicase DDX21 cooperate to resolve genomic R loops and safeguard genome stability. Genes Dev. 31, 1370–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrells S, Nik S, Casey MJ, Cameron RC, Truong H, Toruno C, Gulfo M, Lowe A, Jette C, Stewart RA, et al. (2018). Spliceosomal components protect embryonic neurons from R-loop-mediated DNA damage and apoptosis. Dis. Model. Mech 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks JL, Chistol G, Gao AO, Räschle M, Larsen NB, Mann M, Duxin JP, and Walter JC (2019). The CMG Helicase Bypasses DNA-Protein Cross-Links to Facilitate Their Repair. Cell 176, 167–181.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridhara SC, Carvalho S, Grosso AR, Gallego-Paez LM, Carmo-Fonseca M, and de Almeida SF (2017). Transcription Dynamics Prevent RNA-Mediated Genomic Instability through SRPK2-Dependent DDX23 Phosphorylation. Cell Rep. 18, 334–343. [DOI] [PubMed] [Google Scholar]
- Stavrou S, Aguilera AN, Blouch K, and Ross SR (2018). DDX41 Recognizes RNA/DNA Retroviral Reverse Transcripts and Is Critical for In Vivo Control of Murine Leukemia Virus Infection. MBio 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stork CT, Bocek M, Crossley MP, Sollier J, Sanz LA, Chédin F, Swigut T, and Cimprich KA (2016). Co-transcriptional R-loops are the main cause of estrogen-induced DNA damage. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturzenegger A, Burdova K, Kanagaraj R, Levikova M, Pinto C, Cejka P, and Janscak P (2014). DNA2 cooperates with the WRN and BLM RecQ helicases to mediate long-range DNA end resection in human cells. J. Biol. Chem 289, 27314–27326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su XA, and Freudenreich CH (2017). Cytosine deamination and base excision repair cause R-loop-induced CAG repeat fragility and instability in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U. S. A 114, E8392–E8401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šviković S, Crisp A, Tan-Wong SM, Guilliam TA, Doherty AJ, Proudfoot NJ, Guilbaud G, and Sale JE (2019). R-loop formation during S phase is restricted by PrimPol-mediated repriming. EMBO J. 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi O, and Akira S (2010). Pattern recognition receptors and inflammation. Cell 140, 805–820. [DOI] [PubMed] [Google Scholar]
- Tan J, and Lan L (2020). The DNA secondary structures at telomeres and genome instability. Cell Biosci. 10, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J, Wang X, Phoon L, Yang H, and Lan L (2020a). Resolution of ROS-induced G-quadruplexes and R-loops at transcriptionally active sites is dependent on BLM helicase. FEBS Lett. 594, 1359–1367. [DOI] [PubMed] [Google Scholar]
- Tan J, Duan M, Yadav T, Phoon L, Wang X, Zhang J-M, Zou L, and Lan L (2020b). An R-loop-initiated CSB-RAD52-POLD3 pathway suppresses ROS-induced telomeric DNA breaks. Nucleic Acids Res. 48, 1285–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan SLW, Chadha S, Liu Y, Gabasova E, Perera D, Ahmed K, Constantinou S, Renaudin X, Lee M, Aebersold R, et al. (2017). A Class of Environmental and Endogenous Toxins Induces BRCA2 Haploinsufficiency and Genome Instability. Cell 169, 1105–1118.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tantale K, Mueller F, Kozulic-Pirher A, Lesne A, Victor J-M, Robert M-C, Capozi S, Chouaib R, Bäcker V, Mateos-Langerak J, et al. (2016). A single-molecule view of transcription reveals convoys of RNA polymerases and multi-scale bursting. Nat. Commun 7, 12248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan-Wong SM, Dhir S, and Proudfoot NJ (2019). R-Loops Promote Antisense Transcription across the Mammalian Genome. Mol. Cell 76, 600–616.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng Y, Yadav T, Duan M, Tan J, Xiang Y, Gao B, Xu J, Liang Z, Liu Y, Nakajima S, et al. (2018). ROS-induced R loops trigger a transcription-coupled but BRCA1/2-independent homologous recombination pathway through CSB. Nat. Commun 9, 4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tigano M, Vargas DC, Tremblay-Belzile S, Fu Y, and Sfeir A (2021). Nuclear sensing of breaks in mitochondrial DNA enhances immune surveillance. Nature 591, 477–481. [DOI] [PubMed] [Google Scholar]
- Tsai S, Fournier L-A, Chang EY-C, Wells JP, Minaker SW, Zhu YD, Wang AY-H, Wang Y, Huntsman DG, and Stirling PC (2021). ARID1A regulates R-loop associated DNA replication stress. PLoS Genet. 17, e1009238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuduri S, Crabbé L, Conti C, Tourrière H, Holtgreve-Grez H, Jauch A, Pantesco V, De Vos J, Thomas A, Theillet C, et al. (2009). Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat. Cell Biol 11, 1315–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unoki M, Sharif J, Saito Y, Velasco G, Francastel C, Koseki H, and Sasaki H (2020). CDCA7 and HELLS suppress DNA:RNA hybrid-associated DNA damage at pericentromeric repeats. Sci. Rep 10, 17865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban V, Dobrovolna J, Hühn D, Fryzelkova J, Bartek J, and Janscak P (2016). RECQ5 helicase promotes resolution of conflicts between replication and transcription in human cells. J. Cell Biol 214, 401–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uruci S, Lo CSY, Wheeler D, and Taneja N (2021). R-Loops and Its Chro-Mates: The Strange Case of Dr. Jekyll and Mr. Hyde. Int. J. Mol. Sci 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahba L, Amon JD, Koshland D, and Vuica-Ross M (2011). RNase H and multiple RNA biogenesis factors cooperate to prevent RNA:DNA hybrids from generating genome instability. Mol. Cell 44, 978–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahba L, Gore SK, and Koshland D (2013). The homologous recombination machinery modulates the formation of RNA-DNA hybrids and associated chromosome instability. Elife 2, e00505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang IX, Grunseich C, Fox J, Burdick J, Zhu Z, Ravazian N, Hafner M, and Cheung VG (2018). Human proteins that interact with RNA/DNA hybrids. Genome Res. 28, 1405–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L, Nakajima S, Böhm S, Bernstein KA, Shen Z, Tsang M, Levine AS, and Lan L (2015). DNA damage during the G0/G1 phase triggers RNA-templated, Cockayne syndrome B-dependent homologous recombination. Proc. Natl. Acad. Sci. U. S. A 112, E3495–E3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreb JT, Ghazale N, Pradhan K, Gupta V, Potts KS, Tricomi B, Daniels NJ, Padgett RA, De Oliveira S, Verma A, et al. (2021). Excessive R-loops trigger an inflammatory cascade leading to increased HSPC production. Dev. Cell 56, 627–640.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellinger RE, Prado F, and Aguilera A (2006). Replication fork progression is impaired by transcription in hyperrecombinant yeast cells lacking a functional THO complex. Mol. Cell. Biol 26, 3327–3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood M, Quinet A, Lin Y-L, Davis AA, Pasero P, Ayala YM, and Vindigni A (2020). TDP-43 dysfunction results in R-loop accumulation and DNA replication defects. J. Cell Sci 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Bhowmick R, Vogel I, Özer Ö, Ghisays F, Thakur RS, Sanchez de Leon E, Richter PH, Ren L, Petrini JH, et al. (2020). RTEL1 suppresses G-quadruplex-associated R-loops at difficult-to-replicate loci in the human genome. Nat. Struct. Mol. Biol 27, 424–437. [DOI] [PubMed] [Google Scholar]
- Yan Q, Wulfridge P, Doherty J, Fernandez-Luna JL, Real PJ, Tang H-Y, and Sarma K (2022). Proximity labeling identifies a repertoire of site-specific R-loop modulators. Nat. Commun 13, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SY, Chang EYC, Lim J, Kwan HH, Monchaud D, Yip S, Stirling PC, and Wong JMY (2021). G-quadruplexes mark alternative lengthening of telomeres. NAR Cancer 3, zcab031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, McBride KM, Hensley S, Lu Y, Chédin F, and Bedford MT (2014). Arginine methylation facilitates the recruitment of TOP3B to chromatin to prevent R loop accumulation. Mol. Cell 53, 484–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuhara T, Kato R, Hagiwara Y, Shiotani B, Yamauchi M, Nakada S, Shibata A, and Miyagawa K (2018). Human Rad52 Promotes XPG-Mediated R-loop Processing to Initiate Transcription-Associated Homologous Recombination Repair. Cell 175, 558–570.e11. [DOI] [PubMed] [Google Scholar]
- Ye BJ, Kang HJ, Lee-Kwon W, Kwon HM, and Choi SY (2021). PARP1-mediated PARylation of TonEBP prevents R-loop-associated DNA damage. DNA Repair 104, 103132. [DOI] [PubMed] [Google Scholar]
- Yu K, Chédin F, Hsieh C-L, Wilson TE, and Lieber MR (2003). R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nat. Immunol 4, 442–451. [DOI] [PubMed] [Google Scholar]
- Yu Z, Mersaoui SY, Guitton-Sert L, Coulombe Y, Song J, Masson J-Y, and Richard S (2020). DDX5 resolves R-loops at DNA double-strand breaks to promote DNA repair and avoid chromosomal deletions. NAR Cancer 2, zcaa028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W, Al-Hadid Q, Wang Z, Shen L, Cho H, Wu X, and Yang Y (2021). TDRD3 promotes DHX9 chromatin recruitment and R-loop resolution. Nucleic Acids Res. 49, 8573–8591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yüce Ö, and West SC (2013). Senataxin, defective in the neurodegenerative disorder ataxia with oculomotor apraxia 2, lies at the interface of transcription and the DNA damage response. Mol. Cell. Biol 33, 406–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zatreanu D, Han Z, Mitter R, Tumini E, Williams H, Gregersen L, Dirac-Svejstrup AB, Roma S, Stewart A, Aguilera A, et al. (2019). Elongation Factor TFIIS Prevents Transcription Stress and R-Loop Accumulation to Maintain Genome Stability. Mol. Cell 76, 57–69.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Chen L, Peng D, Jiang A, He Y, Zeng Y, Xie C, Zhou H, Luo X, Liu H, et al. (2020). METTL3 and N6-Methyladenosine Promote Homologous Recombination-Mediated Repair of DSBs by Modulating DNA-RNA Hybrid Accumulation. Mol. Cell 79, 425–442.e7. [DOI] [PubMed] [Google Scholar]
- Zhang X, Chiang H-C, Wang Y, Zhang C, Smith S, Zhao X, Nair SJ, Michalek J, Jatoi I, Lautner M, et al. (2017). Attenuation of RNA polymerase II pausing mitigates BRCA1-associated R-loop accumulation and tumorigenesis. Nat. Commun 8, 15908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Lorenzo C, and Beal PA (2017). DNA editing in DNA/RNA hybrids by adenosine deaminases that act on RNA. Nucleic Acids Res. 45, 3369–3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, He C, Wang L, and Ge B (2017). Post-translational regulation of antiviral innate signaling. Eur. J. Immunol 47, 1414–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q, Hoong N, Aslanian A, Hara T, Benner C, Heinz S, Miga KH, Ke E, Verma S, Soroczynski J, et al. (2018). Heterochromatin-Encoded Satellite RNAs Induce Breast Cancer. Mol. Cell 70, 842–853.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong D, Oberdoerffer P, Batista PJ, and Nussenzweig A (2020). RNA: a double-edged sword in genome maintenance. Nat. Rev. Genet 21, 651–670. [DOI] [PubMed] [Google Scholar]