

Abstract
BACKGROUND & AIMS
Mitochondrial dysfunction disrupts the synthesis and secretion of digestive enzymes in pancreatic acinar cells and plays a primary role in the etiology of exocrine pancreas disorders. However, the transcriptional mechanisms that regulate mitochondrial function to support acinar cell physiology are poorly understood. Here, we aim to elucidate the function of estrogen-related receptor γ (ERRγ) in pancreatic acinar cell mitochondrial homeostasis and energy production.
METHODS
Two models of ERRγ inhibition, GSK5182-treated wild-type mice and ERRγ conditional knock-out (cKO) mice, were established to investigate ERRγ function in the exocrine pancreas. To identify the functional role of ERRγ in pancreatic acinar cells, we performed histological and transcriptome analysis with the pancreas isolated from ERRγ cKO mice. To determine the relevance of these findings for human disease, we analyzed transcriptome data from multiple independent human cohorts and conducted genetic association studies for ESRRG variants in two distinct human pancreatitis cohorts.
RESULTS
Blocking ERRγ function in mice by genetic deletion or inverse agonist treatment results in striking pancreatitis-like phenotypes accompanied by inflammation, fibrosis, and cell death. Mechanistically, loss-of-ERRγ in primary acini abrogates mRNA expression and protein levels of mitochondrial oxidative phosphorylation (OXPHOS) complex genes, resulting in defective acinar cell energetics. Mitochondrial dysfunction due to ERRγ deletion further triggers autophagy dysfunction, ER stress, and production of reactive oxygen species, ultimately leading to cell death. Interestingly, ERRγ-deficient acinar cells that escape cell death acquire ductal cell characteristics indicating a role for ERRγ in acinar-to-ductal metaplasia. Consistent with our findings in ERRγ cKO mice, ERRγ expression was significantly reduced in patients with chronic pancreatitis compared to normal subjects. Furthermore, candidate locus region genetic association studies revealed multiple single nucleotide variants (SNVs) for ERRγ that associated with chronic pancreatitis.
CONCLUSIONS
Collectively, our findings highlight an essential role for ERRγ in maintaining the transcriptional program that supports acinar cell mitochondrial function and organellar homeostasis and provide a novel molecular link between ERRγ and exocrine pancreas disorders.
Keywords: ERRγ, Pancreatic acinar cells, Mitochondrial oxidative phosphorylation, Reactive oxygen species, Acinar-to-ductal metaplasia
Introduction
Acinar cells of the exocrine pancreas perform exceptionally high rates of protein translation to support the synthesis, storage, and secretion of digestive enzymes. The intense energetic demands to support these processes necessitate a robust cellular program for mitochondrial oxidative phosphorylation (OXPHOS) and energy production.1 Of note, pathological conditions, such as acute pancreatitis, associate with abnormal mitochondria, reduced cellular respiration, and diminished ATP reserves. Impaired acinar cell OXPHOS can lead to the accumulation of reactive oxygen species (ROS), ER stress, and autophagy dysfunction, and, when sustained, leads to acinar cell death in mouse models of pancreatitis.2, 3
While it is clear that high levels of energy production are crucial for normal acinar cell function, the molecular program that governs acinar cell energetics and mitochondrial function remains unknown. To address this gap, we turned our attention to a common link between several observations regarding mitochondrial function and exocrine pancreas dysfunction. First, deficiency of PPARγ coactivator 1 α (PGC-1α), a master regulator of mitochondrial biogenesis and energy metabolism, has been shown to exacerbate experimentally induced acute pancreatitis in mouse studies.4 Second, the clinical use of tamoxifen has been linked to acute pancreatitis of unknown etiology and impaired respiration of cancer cells through estrogen receptor-independent mechanisms.5, 6 Notably, both PGC-1α and tamoxifen can directly bind and regulate estrogen-related receptor γ (ERRγ), suggesting that ERRγ may play a role in acinar cell biology and energy metabolism.
ERRγ is an orphan nuclear receptor and a member of the estrogen-related receptors (ERRs) subfamily including ERRα and ERRβ. Transcriptional activity of ERRs is enhanced by PGC-1 family coactivators and together orchestrate transcriptional programs involved in the regulation of mitochondrial biogenesis, the tricarboxylic acid (TCA) cycle, and OXPHOS components.7 Notably, one or more ERRs are recruited near the transcriptional start sites of about 64% of nuclear-encoded mitochondrial genes.8 However, despite the fact that there are many overlapping target genes between ERRα and ERRγ, the two transcription factors have differential effects on regulation of target gene expression. For example, ERRα upregulates glycolytic enzymes in breast cancer to sustain aerobic glycolysis and proliferation, while ERRγ inhibits proliferation in breast cancer cells via a shift to oxidative metabolism.9–11 Furthermore, whole-body ERRα KO mice are developmentally normal, whereas whole-body ERRγ KO mice exhibit perinatal lethality due to heart failure.12, 13 Although the natural ligand for ERRγ is not known, several synthetic ligands can regulate ERRγ activity by disrupting ERRγ-cofactor interactions. For example, diethylstilbestrol (DES) and 4-hydroxytamoxifen (4-OHT), first identified as ER ligands, can also function as ERRγ inverse agonists.14
Here, we investigate the role of ERRγ in acinar cell biology and find both pharmacological inhibition of ERRγ as well as genetic ablation of ERRγ cause degenerative pancreatitis-like phenotypes accompanied by immune cell infiltration, inflammation, and fibrosis. To understand the cellular and molecular basis of the obligate requirement for ERRγ in the exocrine pancreas, we performed transcriptome analysis of gain- and loss-of-ERRγ pancreatic acini and identify that ERRγ is required for the transcriptional control of mitochondrial gene networks. Consistent with transcriptome analysis results, disrupting ERRγ function by pharmacological inhibition or genetic ablation triggered mitochondrial dysfunction. ERRγ inhibition, and the resultant mitochondrial dysfunction, also led to increased oxidative stress, impaired autophagy, and ER stress indicating that ERRγ is required for pancreatic acinar cell organellar homeostasis. In addition to the requirement for ERRγ as an essential coordinator of acinar cell metabolism, loss-of-ERRγ induces reprogramming of acinar cells to transdifferentiate into duct-like cells. Taken together, our study uncovers a previously unappreciated and crucial role for ERRγ in maintaining acinar cell metabolism, function, and identity.
Results
ERRγ is required for maintaining pancreas tissue homeostasis and function
Clinical use of tamoxifen (TAM), a selective ER modulator, has been reported to induce acute pancreatitis.5 Confirming previous reports, pancreas tissue from mice treated with TAM at higher doses, 150 and 300 mg/kg, displayed degenerative acinar cell morphology and decreased amylase content (Supplementary Figure 1A). However, a lower dose of TAM at 75 mg/kg did not show any significant effects on pancreas tissue (Supplementary Figure 1A). The molecular target(s) mediating TAM-induced pancreatitis are unclear but may involve non-ER targets such as the ERRs.5, 6 Coactivator recruitment assays for ERRs revealed that 4-hydroxytamoxifen (4-OHT) inhibits transcriptional activity of ERRβ (IC50 = 0.915 μM) and ERRγ (IC50 = 0.078 μM), but had no effect on ERRα transcriptional activity (Supplementary Figure 1B). To further test whether ERRβ and/or ERRγ may be involved in the TAM-induced pancreas phenotype, we treated mice with GSK5182, an inverse agonist of ERRβ/γ, via osmotic pump delivery (0.5 mg/mouse/day) for 1 wk (Figure 1A). Examination of pancreatic tissue from GSK5182-treated mice revealed marked atrophy of acinar structures and reduction of amylase content that closely resembles the pancreas phenotype in TAM-treated mice (Figure 1B). Of note, GSK5182 (10 μM) treatment reduced acinar amylase content in primary mouse acini, indicating that GSK5182 has a cell-autonomous effect on pancreatic acini (Figure 1C and D).
Figure 1. ERRγ is required for maintaining the pancreatic exocrine gland.
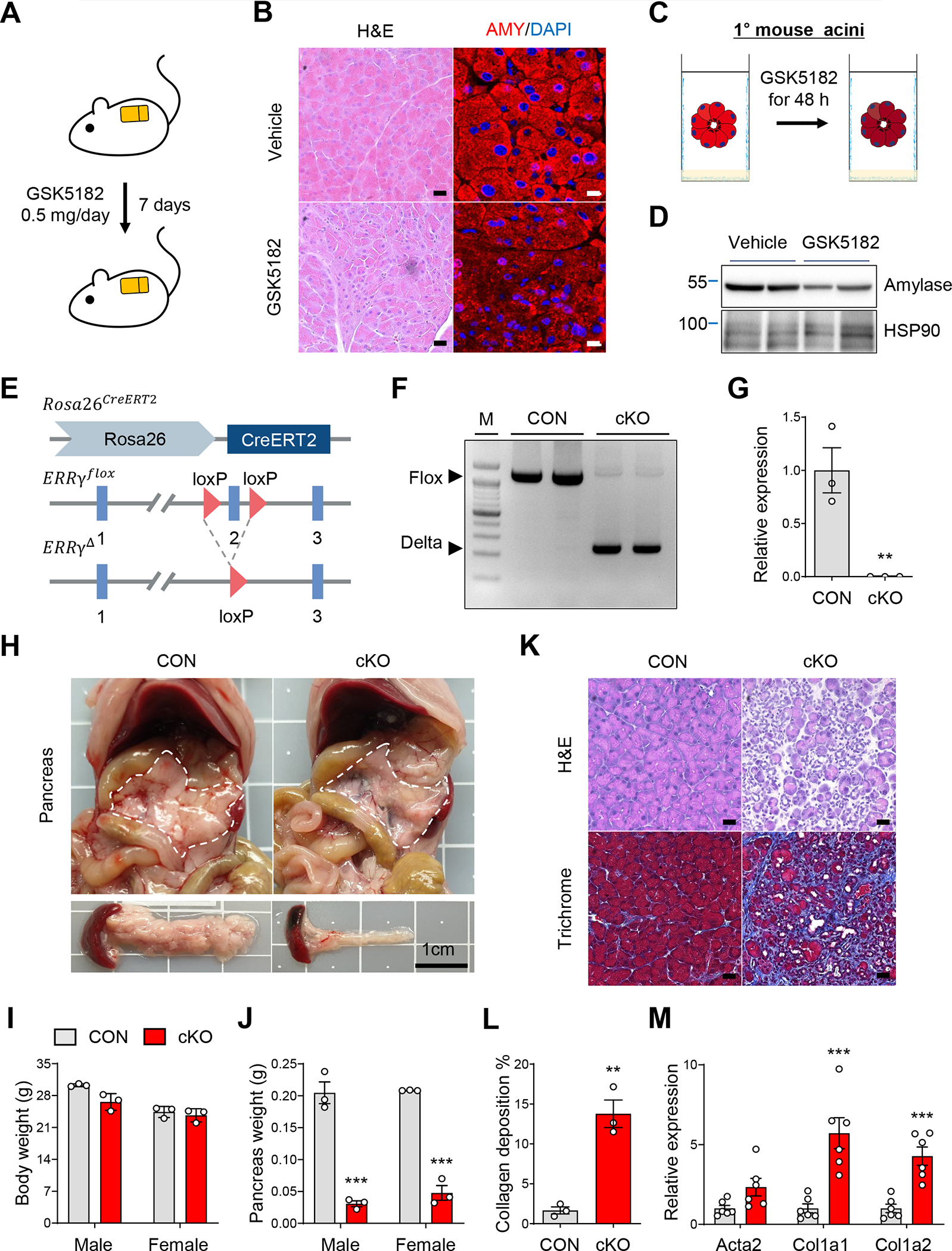
(A) Scheme of experiments. Vehicle or GSK5182 was administered to C57BL/6J mice through an osmotic pump for 1 wk (0.5 mg/day). (B) H&E and immunofluorescence staining of amylase (AMY) in pancreas from vehicle or GSK5182 treated mice. (C) Scheme of experiments. Primary acini isolated from C57BL/6J mice were treated with vehicle or GSK5182 for 48 h. (D) Western blot for amylase and HSP90 in primary acini isolated from C57BL/6J mice. (E) Genomic structure of TAM-induced ERRγ conditional KO (cKO) allele. R26, ROSA26 locus; red triangle, loxP sequence; blue box, exonic regions. (F) PCR analysis of genomic DNA showing the deletion of ERRγ in primary acini from control and ERR cKO mice. (G) Relative gene expression of ERRγ in pancreas from control and ERRγ cKO mice. (H) Gross images of pancreas from control and ERRγ cKO mice 7 d after the final TAM injection. TAM (75 mg/kg) was administered once daily by intraperitoneal injection for 5 consecutive days. Scale bar, 1 cm. (I-J) Body weights (I) and pancreas (J) from control and ERRγ cKO mice. (K) H&E staining (upper panel) and Masson’s trichrome staining (lower panel) of pancreas from control and ERRγ cKO mice. (L) Quantitation of collagen deposited area over total area in trichrome stained pancreas sections. (M) Relative gene expression of fibrosis marker genes in pancreas from control and ERRγ cKO mice. Results were normalized to 36b4. CON, control; cKO, ERRγ cKO. All data are presented as mean ± SEM. * p < 0.05, ** p < 0.01, *** p < 0.005 by student’s t-test.
Next, we sought to determine which target of GSK5182, ERRβ or ERRγ, mediates the pancreatic atrophy phenotype using genetic loss-of-ERRβ and -ERRγ models. Germline deletion of ERRβ and ERRγ in mice results in embryonic and neonatal lethality, respectively, thereby precluding analysis of adult pancreas tissue.13, 15, 16 To circumvent this early lethality, we generated ERRβ and ERRγ conditional knockout mice by crossing ERRβfl/fl and ERRγfl/fl mice with CAG-CreERT2 transgenic mice (hereafter ERRβ cKO and ERRγ cKO) (Figure 1E and Supplementary Figure 1C). We confirmed efficient deletion of ERRβ and ERRγ in mice treated with 75 mg/kg TAM by genomic DNA PCR and gene expression analysis (Figure 1F and G, data not shown). While ERRβ cKO mice did not show any appreciable pancreas phenotype, ERRγ cKO mice of both sexes exhibited severe pancreatic atrophy (Figure 1H and Supplementary Figure 1C) and decreased pancreas weight without changes in body weight (Figure 1I and J). Histological analysis of ERRγ cKO pancreas showed acinar cell dystrophy and marked fibrosis (Figure 1K and L) accompanied by upregulation of α-SMA (Acta2), collagen 1A1 (Col1a1), and collagen 1A2 (Col1a2) (Figure 1M). We next analyzed tissues other than the pancreas in ERRγ cKO mice but did not observe any histological abnormalities in metabolic organs or other exocrine glands (Supplementary Figure 2A and B). To further analyze whole body metabolic phenotypes in ERRγ cKO mice, we performed indirect calorimetry and found no significant differences in oxygen consumption, carbon dioxide production, respiratory exchange ratio (RER), and heat production in ERRγ cKO mice compared to control (Supplementary Figure 3A–E).
Next, we investigated whether ERRγ function is required in a pancreas-autonomous manner in vivo. To this end, Cre-expressing adenovirus was injected into the parenchyma of pancreas tissue of ERRγfl/fl; mTmG reporter mice (Supplementary Figure 4A). Local deletion of ERRγ in the pancreas by Cre-expressing adenovirus resulted in acinar cell dystrophy and fibrosis similar to ERRγ cKO mice (Supplementary Figure 4B). Collectively, these results uncover a striking tissue-autonomous requirement for ERRγ in the adult exocrine pancreas and suggest that ERRγ may be a target mediating TAM-induced pancreatitis.
Genetic deletion of ERRγ causes progressive degeneration of the adult exocrine pancreas
We next examined cellular and molecular changes along a time course following ERRγ ablation. Pancreas from ERRγ cKO mice harvested for histological analysis 0, 5, 8 and 11 d after the final TAM injection exhibited rapid and progressive pancreatic atrophy (Figure 2A). Notably, ERRγ cKO pancreas acini, marked by amylase expression, underwent both cell death and compensatory proliferation as shown by increased cleaved caspase-3 (CC3) and phosphor-histone H3 (P-H3), respectively, followed by the activation of fibrosis as shown by increased α-SMA (Figure 2B). Local infiltration of macrophages, T-cells and B-cells marked by F4/80, CD3, and B220, respectively, was also dramatically induced in ERRγ cKO pancreas (Supplementary Figure 5A–C). Absence of enlarged lymph nodes or spleen indicate that the inflammatory changes were confined to the pancreas (Supplementary Figure 5D). Furthermore, dexamethasone treatment had no observable effect on the ERRγ cKO pancreas phenotype, indicating that local or systemic inflammation cannot account for the degenerative changes observed in ERRγ cKO pancreas (Supplementary Figure 5E–H). Ultrastructure analysis of ERRγ cKO pancreatic tissue revealed markedly reduced zymogen granules in the acinar cells (Figure 2C) which was associated with reduced expression of acinar cell markers, amylase and elastase (Figure 2D and E). In contrast to exocrine pancreas atrophy, ERRγ cKO mice displayed a significant increase in islet size (Supplementary Figure 6A and B). Consistent with this observation, ERRγ cKO mice showed reduced blood glucose levels, improved glucose tolerance, and a trend towards increased serum insulin levels in both male and female mice (Supplementary Figure 6C–E).
Figure 2. ERRγ deletion causes rapid and progressive pancreatic atrophy.
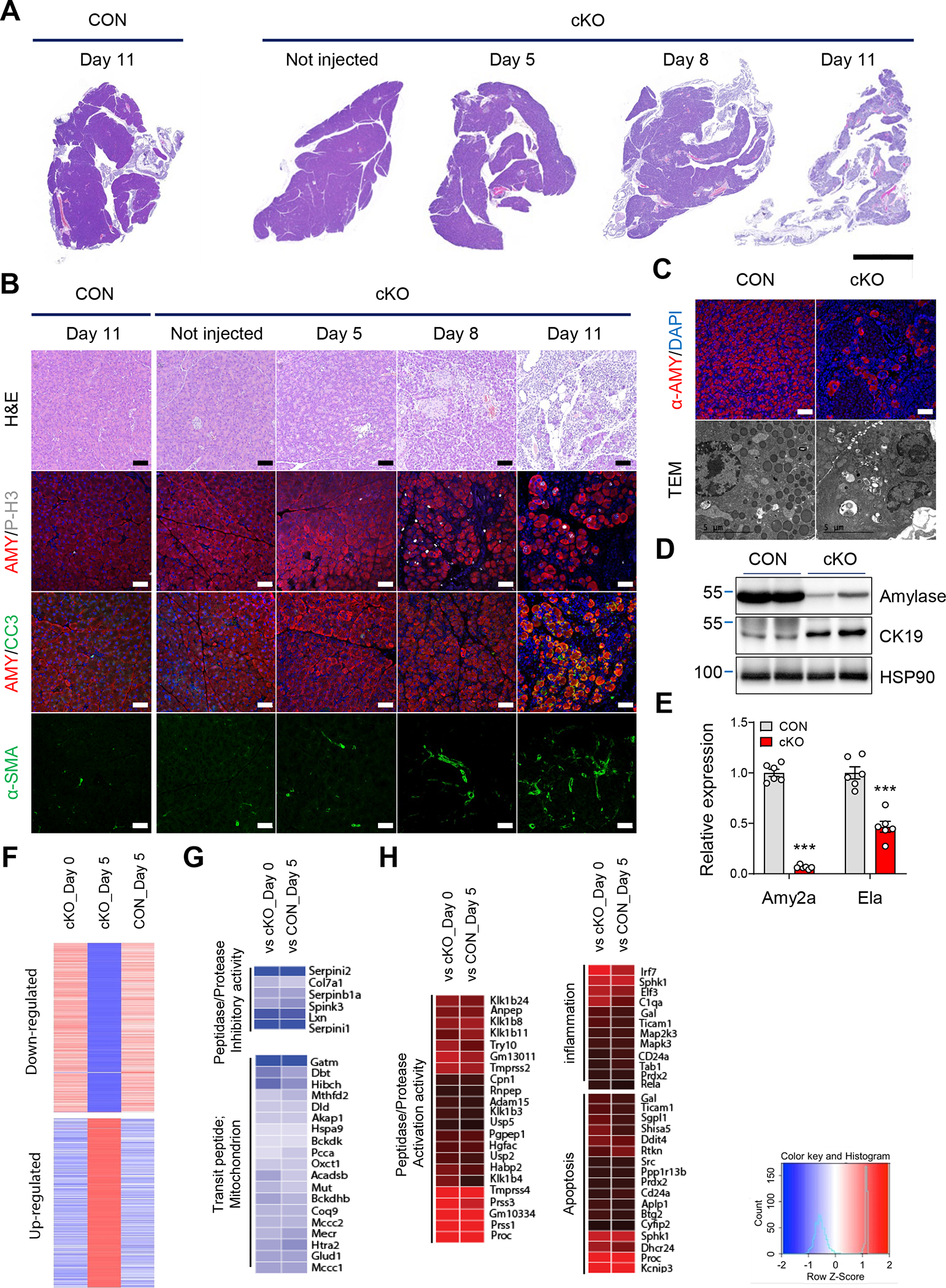
(A-B) Images of control and ERRγ cKO pancreas sections with H&E staining or fluorescence immunostaining for amylase (AMY, red), phospho-H3 (P-H3, white), cleaved caspase 3 (CC3, green), α-smooth muscle actin (α-SMA, green), and DAPI (blue). Pancreas samples obtained on indicated day after final TAM injection. (C) Immunofluorescence images for α-amylase (red) and DAPI (blue) in control and ERRγ cKO pancreas. Scale bar, 50 μm (upper panel). Transmission electronic microscopy images of pancreas from control and ERRγ cKO mice. Scale bar, 5 μm (lower panel). (D) Western blot for amylase, CK19 and HSP90 in pancreas from control and ERRγ cKO mice. (E) Relative gene expression of amylase2a (Amy2a) and elastase (Ela). (F) RNA-seq analysis of whole pancreas transcriptome. Cluster analysis of differentially expressed genes in pancreas from ERRγ cKO mice before TAM injection (cKO day 0), after the final day of TAM injection (cKO day 5), and control mice after the final day of TAM injection (CON day 5). (G-H) Gene ontology (GO) analysis of upregulated genes (G) and downregulated genes in ERRγ cKO pancreas compared to indicated sample. CON, control; cKO, ERRγ cKO. All data are presented as mean ± SEM. *** p < 0.005 by student’s t-test.
We next performed whole pancreas RNA-seq analysis to identify molecular pathways associated with the ERRγ cKO pancreas phenotype. Gene Ontology (GO) analysis revealed that ERRγ deletion causes unique transcriptional changes, which was not due to the effect of Cre expression nor tamoxifen (Figure 2F). Transcripts involved in peptidase/protease inhibitory activity and mitochondrial genes were downregulated whereas transcripts involved in peptidase/protease activation activity, inflammation, and apoptosis were upregulated by ERRγ ablation (Figure 2G and H). We performed Enrichr analysis with differentially expressed genes in ERRγ cKO pancreas to identify disease associations using the Jensen database and the Disease perturbations from GEO UP. Down regulated gene sets from RNA-seq in ERRγ cKO pancreas showed significant association with pancreatitis (Supplementary Figure 7A and B). Taken together, ERRγ cKO pancreas display progressive cellular and molecular alterations that coincide with exocrine pancreas dysfunction and pancreatitis.
ERRγ is required for maintenance of mitochondrial OXPHOS in pancreatic acinar cells
We next sought to identify the cellular and molecular processes regulated by ERRγ within pancreatic acinar cells. To investigate acinar cell-autonomous transcriptional regulation by ERRγ, we performed RNA-seq using primary acini isolated from ERRγ cKO pancreas and primary acini transduced with recombinant adenovirus expressing ERRγ. GO analysis of differentially expressed genes from loss- and gain-of-ERRγ function in primary acini revealed enrichment of genes associated with mitochondrial function (Figure 3A). For example, mitochondrial OXPHOS genes were downregulated in ERRγ cKO acini, whereas ERRγ-overexpressing acini showed opposite trends (Figure 3B and C). Downregulation of mitochondrial gene expression was accompanied by decreased mtDNA copy number (Figure 3D) and OXPHOS complex protein and transcript levels (Figure 3E, Supplementary Figure 8A) in ERRγ cKO pancreas. Interestingly, the expression of PGC-1α, a master regulator of mitochondrial biogenesis, was also decreased at both RNA (Figure 3G) and protein levels (Supplementary Figure 8B). ERRγ has been demonstrated to directly bind and regulate the expression of mitochondrial genes in various cell types.17–21 To extend these findings to pancreatic acinar cells, we conducted ChIP-qPCR with anti-ERRγ antibodies using primary acini nuclei and confirmed direct binding of ERRγ at mitochondrial gene loci, Aco2 and Sdhb (Supplementary Figure 8C).
Figure 3.
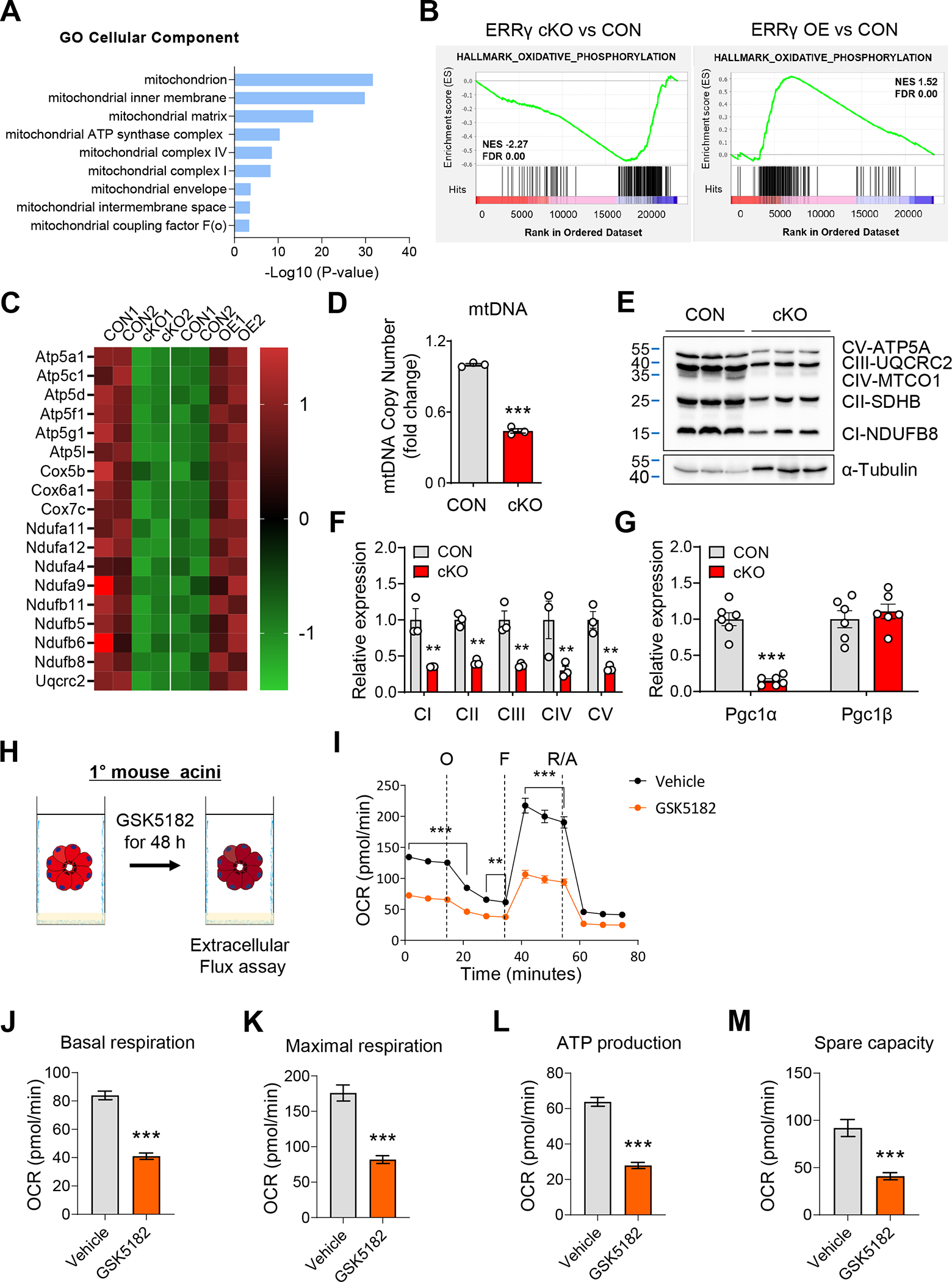
ERRγ is an essential regulator of the mitochondrial OXPHOS program in pancreatic acinar cells. (A-B) Gene ontology (GO) (A) and gene set enrichment analysis (GSEA) (B) of top rank enriched cellular components between ERRγ cKO primary acini and ERRγ adenovirus transduced (ERRγ OE) primary acini, compared to respective controls (CON). (C) Heatmap for differential gene expression in ERRγ cKO primary acini and ERRγ OE primary acini, compared to respective controls. (D-G) Pancreatic RNA, DNA, and protein were extracted from control and ERRγ cKO mice 7 d after the final TAM injection. (D) Mitochondrial DNA content was quantified by ND1 (mtDNA) / HK2 (nDNA) ratio. (E) Western blot for OXPHOS complex proteins in pancreas from control and ERRγ cKO mice. α-tubulin, loading control. (F) Quantitation of protein levels from (E) normalized to α-Tubulin expression. (G) Relative gene expression of PGC1α and PGC1β. (H) Scheme of experiments. Primary acini from C57BL/6J mice were treated with vehicle or GSK5182 (10 μM) for 48 h. (I-M) Extracellular flux analysis measurements for (I) oxygen consumption rate, (J) basal respiration, (K) maximal respiration, (L) ATP production and (M) spare capacity. CON, control; cKO, ERRγ cKO. All data are presented as mean ± SEM. ** p < 0.01, *** p < 0.005 by student’s t-test.
Next, we performed extracellular flux analysis to measure mitochondrial function in wild-type primary acini treated with vehicle or GSK5182 (Figure 3H). Pharmacological inhibition of ERRγ by GSK5182 in primary acini reduced basal respiration, maximal respiration, ATP production and spare capacity (Figure 3I–M), but did not alter extracellular acidification (Supplementary Figure 9A–F). These results indicate that ERRγ is required for maintaining mitochondrial OXPHOS function but not glycolysis in primary acini. We next measured metabolites associated with mitochondrial function and found significant increases of lactate, acetoacetate and 2-hydroxybutyrate, and 3-hydroxypropionate levels in ERRγ cKO pancreatic tissue, which is consistent with impaired mitochondrial function (Supplementary Table 1). Interestingly, metabolites involved in the initial part of the TCA cycle, e.g. citrate, cis-aconitate, and isocitrate, were increased, whereas metabolites involved in the later part of the TCA cycle, e.g., α-ketoglutarate, succinate, and fumarate were significantly decreased (Supplementary Table 1). These patterns of changes in TCA cycle intermediates and elevation of ketone body production closely mirrors what has been observed from metabolomics studies in acute pancreatitis mice models.22 Collectively, these results indicate that ERRγ plays a pivotal role in mitochondrial energy metabolism via regulation of OXPHOS gene expression in pancreatic acinar cells.
Mitochondrial dysfunction by ERRγ ablation promotes ROS accumulation in pancreatic acinar cells
Abnormal OXPHOS gene expression and cellular respiration frequently accompany structural changes in mitochondria.23, 24 Indeed, analysis of ERRγ cKO pancreas acinar cell ultrastructure revealed mitochondria with markedly reduced cristae and aberrant outer membrane contour compared to control (Figure 4A). We next evaluated ROS production in ERRγ cKO pancreas as mitochondrial dysfunction of pancreatic acinar cells is associated with the production of ROS.16 ERRγ cKO pancreas showed increased oxidative stress, as measured by increased 4-hydroxynonenal (4-HNE), a marker of oxidative stress-induced lipid peroxidation (Figure 4B). Accordingly, nuclear factor erythroid 2-related factor (Nrf2), a master regulator of antioxidant responses, and its target genes were also upregulated in ERRγ cKO pancreas (Figure 4C). Interestingly, fluorescence imaging of ERRγ cKO acini stained with mitoSOX dye revealed a distinct perinuclear clustering pattern of mitochondria (Figure 4D and Supplementary Figure 10) as well as increased mitochondrial ROS (Figure 4E). Perinuclear clustering of mitochondria has been associated with the disposal of damaged mitochondria by mitophagy.25 To analyze in vivo mitophagy, we generated ERRγ cKO mice expressing the mt-Keima transgene, which encodes a mitochondrial pH-dependent fluorescence reporter26, and performed intravital imaging of the pancreas. Red fluorescence signals, which represent mitophagy of damaged mitochondria, were increased in ERRγ cKO pancreas compared to controls (Figure 4F–H). These results demonstrate that ERRγ ablation leads to mitochondria dysfunction, increased ROS, and activated mitophagy in pancreatic acinar cells.
Figure 4. Mitochondrial dysfunction by ERRγ deletion induces ROS in pancreatic acinar cells.
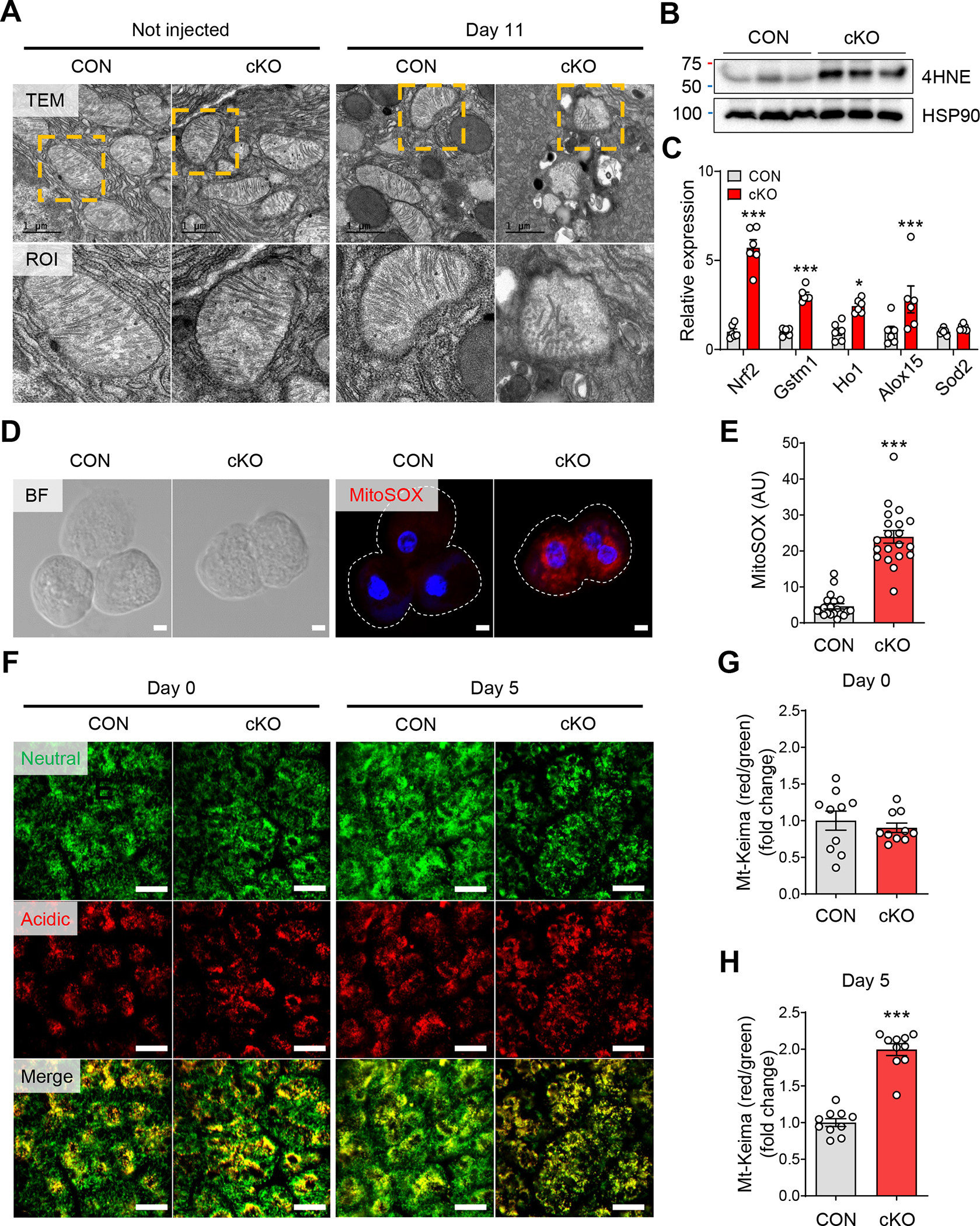
(A) Transmission electronic microscopy images of pancreas tissue showing acinar cell mitochondria from control and ERRγ cKO 0 d (left panel) and 7 d (right panel) after the final TAM injection. Scale bar, 1 μm. (B) Western blot for 4-HNE and HSP90 in pancreas from control and ERRγ cKO mice 3 d after the final TAM injection. (C) Bright field (left panel) and fluorescence (right panel) images for MitoSOX (red) and DAPI (blue) in primary acini isolated from control and ERRγ cKO mice pancreas. MitoSOX fluorescence indicates production of mitochondrial superoxide. Scale bar, 5 μm. (D) Quantitation of MitoSOX fluorescence intensity (arbitrary units). (E) Relative gene expression of antioxidant markers in pancreas from control and ERRγ cKO mice. Results were normalized to 36b4. (F) In vivo time-course imaging of pancreas in control and ERRγ cKO; mt-Keima mice. mt-Keima protein is localized in the mitochondrial matrix and displays a bimodal excitation peak that is pH-dependent. The 488 nm excitation peak of mt-Keima (green) indicates mitochondria exposed to a neutral environment. The 561 nm excitation peak of mt-Keima (red) indicates mitochondria exposed to an acidic environment. Scale bar, 50 μm. (G-H) Quantitation of the mitophagy index as calculated by red/green area of mt-Keima signals at d 0 (G) and d 5. CON, control; cKO, ERRγ cKO. All data are presented as mean ± SEM. * p < 0.05, *** p < 0.005 by student’s t-test.
Antioxidant treatment ameliorates exocrine pancreas atrophy in ERRγ cKO mice
The pronounced increase of oxidative stress observed in ERRγ cKO pancreas, prompted us to explore whether antioxidants might ameliorate the exocrine pancreas pathology due to ERRγ ablation. To test this, control and ERRγ cKO mice were fed either normal chow diet (NCD) or chow diet containing butylated hydroxyanisole (BHA), an antioxidant (Figure 5A). The decrease in pancreas weights was mitigated in ERRγ cKO mice fed BHA-diet compared to ERRγ cKO mice fed NCD (Figure 5B). Histological analysis of pancreas from ERRγ cKO mice fed with BHA-diet showed dramatic reversal of multiple pancreas phenotypes observed in ERRγ cKO pancreas, including acinar cell atrophy, amylase deficiency, cell proliferation, and cell death (Figure 5C). These data strongly suggest that oxidative stress, likely induced by mitochondrial dysfunction, is a major driver of the pancreatic pathologies observed in ERRγ cKO mice.
Figure 5. Antioxidant treatment ameliorates degenerative pancreatic phenotypes in ERRγ cKO mice.
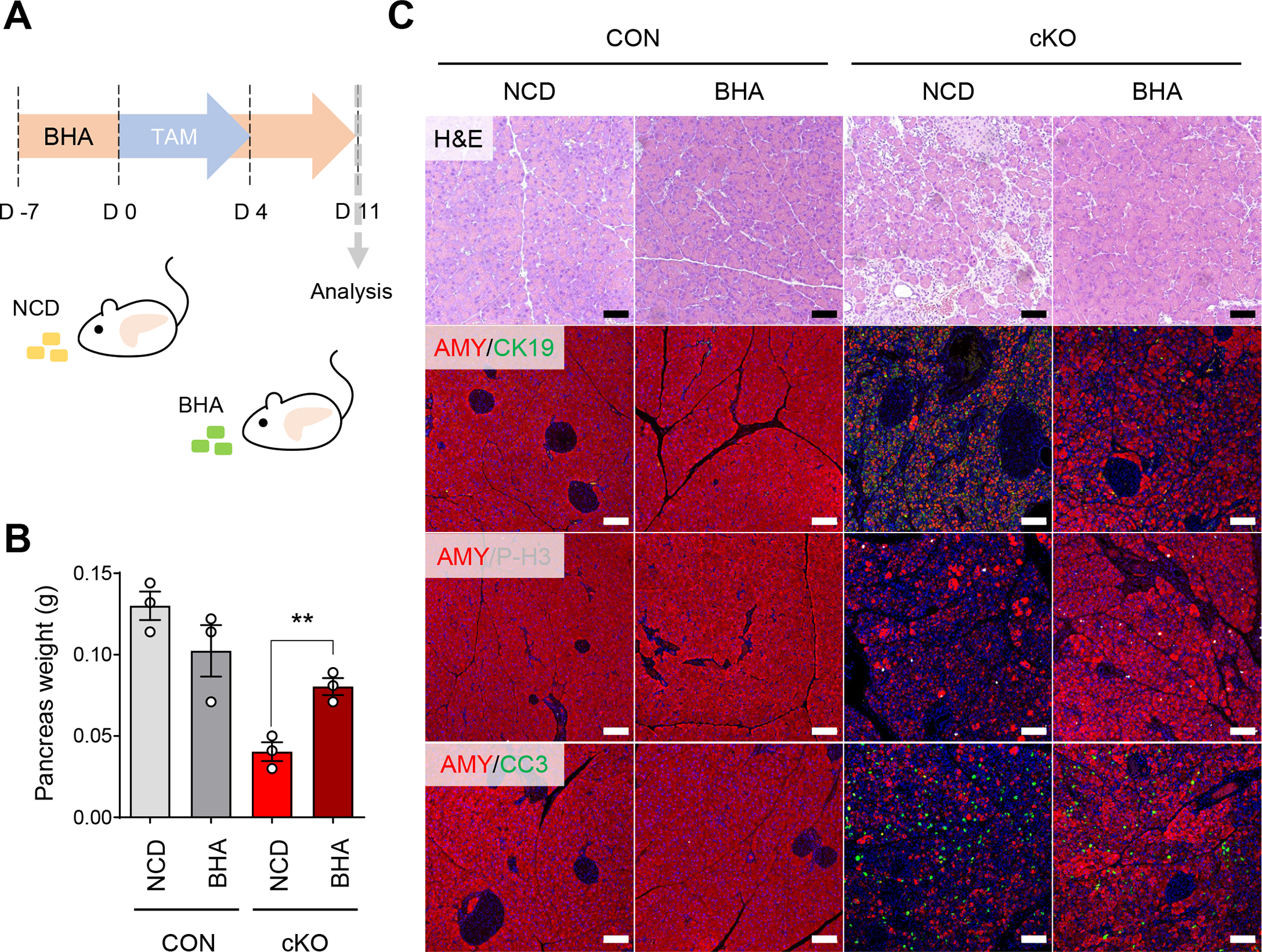
(A) Scheme of experiments. Control and ERRγ cKO mice were fed with normal chow diet (NCD) or NCD supplemented with butylated hydroxyanisole (BHA) for 1 wk prior to first TAM injection and analyzed 7 d after the final TAM injection. (B) Pancreas weights from NCD or BHA fed control and ERRγ cKO mice. (C) H&E staining and fluorescence immunostaining of pancreas in NCD or BHA fed control and ERRγ cKO mice for amylase (AMY, red), phospho-H3 (P-H3, white), cleaved caspase 3 (CC3, green) and DAPI (blue). CON, control; cKO, ERRγ cKO. Scale bar, 50 μm. All data are presented as mean ± SEM. ** p < 0.01 by student’s t-test.
Loss-of-ERRγ increases autophagy and ER stress in pancreatic acinar cells
Energy deficiency and oxidative stress caused by mitochondrial dysfunction can lead to impaired autophagy and ER stress in pancreatic acinar cells.2 Ultrastructural analysis of ERRγ cKO pancreas showed dilated rough ER and the accumulation of autophagosomes, indicating defects in protein folding and autophagy (Figure 6A). Consistent with these findings, BIP and CHOP, regulators of the unfolded protein response, and LONP1, a conserved mitochondrial protease involved in the mitochondrial unfolded protein response, were upregulated in ERRγ cKO pancreas (Figure 6B and C). We next analyzed mRNA and protein expression of autophagy pathway genes in acinar cells and pancreas from control and ERRγ cKO mice. We observed increased mRNA expression of autophagy-related genes in ERRγ cKO pancreas relative to controls (Figure 6D). LC3B-II/LC3B-I protein ratio was increased in the absence of changes in P62 protein levels in the pancreas and primary acini from ERRγ cKO mice, suggesting activated autophagy (Figure 6E). To directly visualize and quantify autophagy in ERRγ cKO pancreas, we generated ERRγ cKO mice harboring the GFP-LC3 transgene. Consistent with autophagy gene expression changes, autophagosome accumulation was increased in pancreas tissue of ERRγ cKO; GFP-LC3 transgenic mice (Figure 6F–H). Intravital imaging of pancreas tissue in ERRγ cKO; GFP-LC3 transgenic mice also showed marked increase of autophagosomes (green puncta) at day 5 after the final tamoxifen injection (Figure 6I).
Figure 6. Loss of ERRγ results in ER stress and autophagy in pancreatic acinar cells.
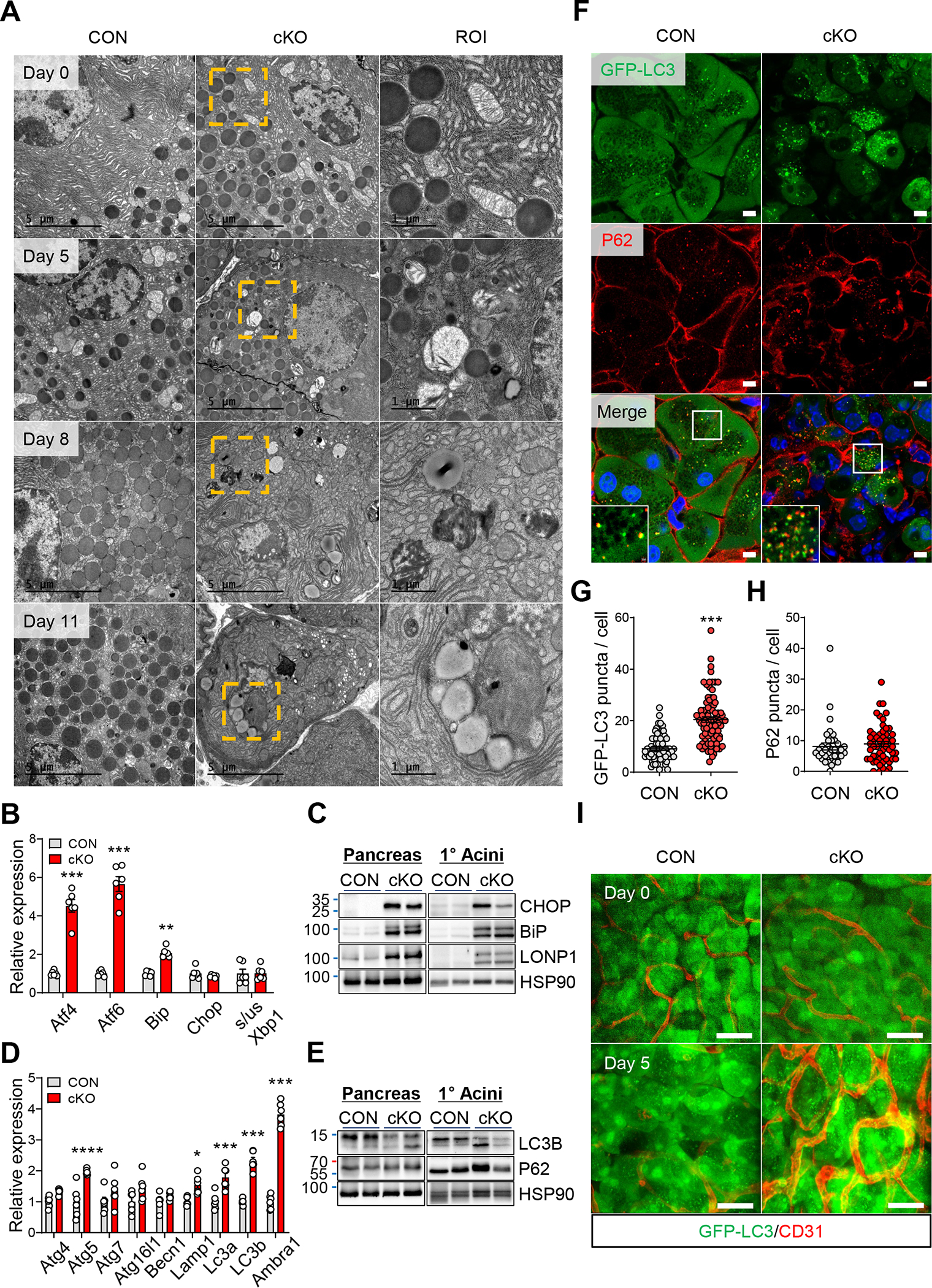
(A) Transmission electronic microscopy images of pancreas tissue showing acinar cell from control and ERRγ cKO pancreas at indicated day after the final TAM injection. Scale bar, 5 μm and 1 μm for region of interest (ROI) (B) Western blot for ER stress markers in pancreas and primary acini from ERRγ cKO mice, compared to control 4 d after the final TAM injection. (C) Relative gene expression of ER stress markers in pancreas from control and ERRγ cKO mice. Results were normalized to 36b4. (D) Western blot for autophagy markers in pancreas and primary acinar cells of ERRγ cKO mice. (E) Relative gene expression of autophagy markers in pancreas from control and ERRγ cKO mice. Results were normalized to 36b4. (F) P62 immunofluorescence staining of frozen tissue sections from control and ERRγ cKO; GFP-LC3 transgenic mice pancreas. Scale bar, 5 μm (G-H) Quantitation of GFP-LC3 puncta per cell (G) and P62 puncta per cell (H). (I) Intravital imaging control and ERRγ cKO; GFP-LC3 transgenic pancreas. GFP-LC3 puncta (green) indicate autophagosomes and CD31 (red) indicates blood vessels. CON, control; cKO, ERRγ cKO. Scale bar, 50 μm. All data are presented as mean ± SEM. * p < 0.05, ** p<0.01, *** p < 0.005 by student’s t-test.
Similar to ERRγ cKO pancreas, pharmacological inhibition of ERRγ with GSK5182 in pancreatic acini also activated autophagic influx (Supplementary Figure 11A–C) and ER stress response (Supplementary Figure 11D and E). Moreover, blocking mitochondrial OXPHOS with rotenone (Supplementary Figure 11F), a complex I inhibitor, was sufficient to activate autophagy (Supplementary Figure 11G and H) and ER stress (Supplementary Figure 11I and J), indicating an upstream role for OXPHOS in acinar cell dysfunction caused by ERRγ deficiency. These results demonstrate that mitochondrial dysfunction resulting from ERRγ ablation can subsequently disrupt organellar homeostasis, activating autophagy and ER stress in pancreatic acinar cells.
ERRγ deletion induces acinar cell reprogramming.
Pancreatic acinar cells can transdifferentiate to progenitor-like cells with ductal characteristics under stress conditions such as pancreatitis. This conversion from acinar to duct-like cells, or acinar-to-ductal metaplasia (ADM), is a reversible process to restore acinar cell function and mass, but can also contribute to the early stages of pancreatic cancer.27 In parallel with the aforementioned decrease in acinar cells, we noticed an increase of cells with ductal cell characteristics in ERRγ cKO pancreas (Figure 7A). The identity of these cells was confirmed by immunostaining of CK19, a marker for ductal cells, and SOX9, a marker for pancreatic progenitor cells (Supplementary Figure 12A). Consistent with these observations, expression of genes for maintaining acinar cell identity dramatically decreased (Supplementary Figure 12B) while ductal and progenitor markers increased in both pancreas tissue (Figure 7B) and primary acini (Figure 7C) isolated from ERRγ cKO mice compared to controls.
Figure 7. ERRγ deletion induces acinar cell reprogramming.
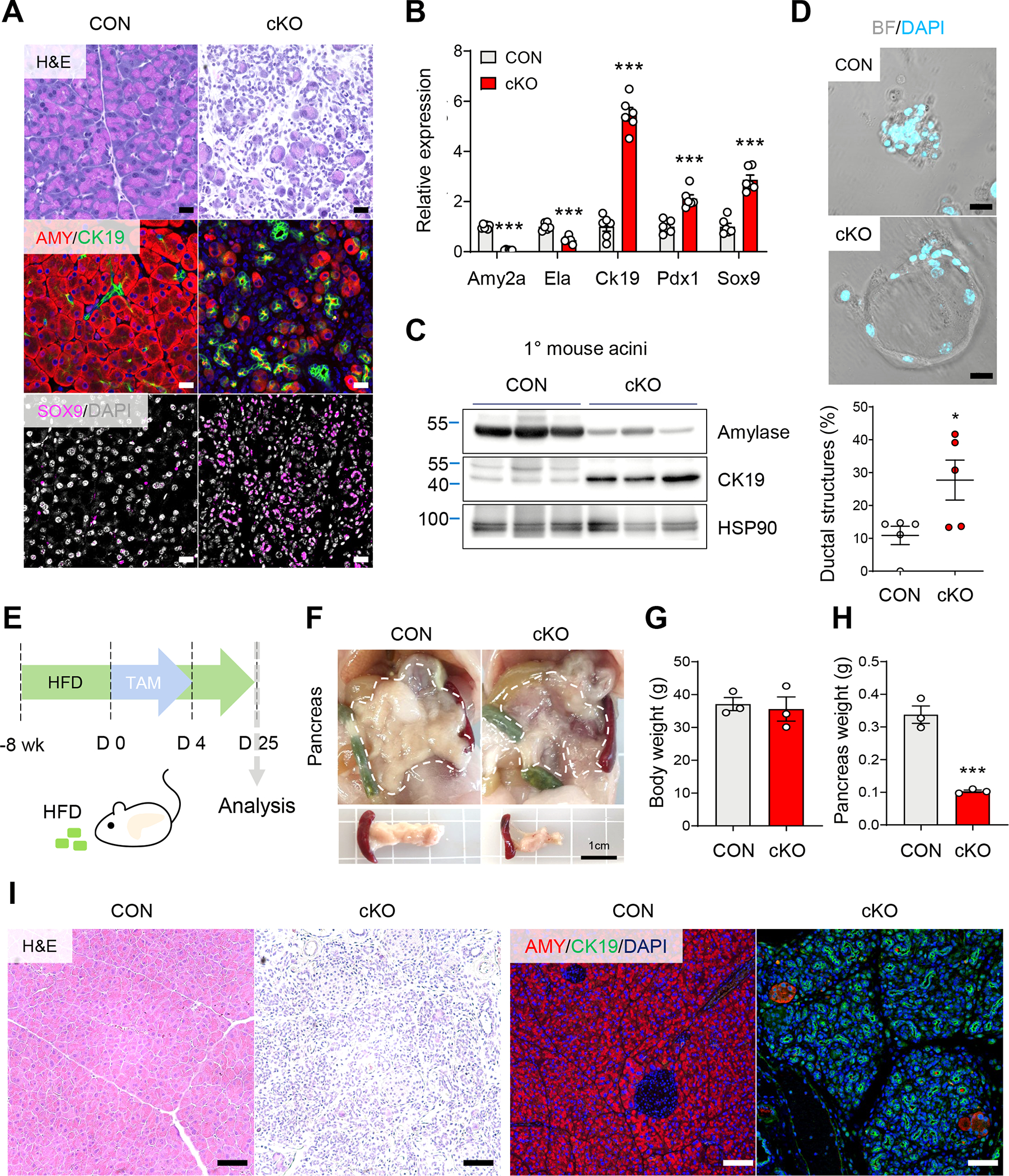
(A) H&E and immunofluorescence images for amylase (AMY, red), CK19 (green), and SOX9 (magenta) control and ERRγ cKO pancreas 7 d after the final TAM injection. Scale bar, 20 μm. (B) Relative gene expression of pancreatic lineage markers from control and ERRγ cKO mice pancreas. Results were normalized to 36b4. (C) Western blot for amylase, CK19 and HSP90 in primary acini isolated from control and ERRγ cKO mice. (D) Micrographs of ex vivo cultured primary acini on gelatin-coated wells (upper panel) and quantitation of percent duct-like cells over total cells per well (lower panel). Scale bar, 20 μm. (E) Scheme of experiments. Control and ERRγ cKO mice were fed high-fat diet (HFD) for 8 wk and then intraperitoneally injected with TAM (75mg/kg) daily for 5 consecutive days. Analysis was performed 2 wk after the final TAM injection. (F-H) gross images (F), body weights (G), and pancreas weights (H) from control and ERRγ cKO mice. Scale bar, 1 cm. (I) H&E and immunofluorescence images for amylase (red) and CK19 (green) in control and ERRγ KO pancreas. Scale bar, 50 μm. CON, control; cKO, ERRγ cKO. All data are presented as mean ± SEM. * p < 0.05, *** p < 0.005, by student’s t-test.
Ex vivo culture of primary acini on gelatin or matrigel induce transdifferentiation of acinar cells to ductal cells and serve as a model for ADM. We used this ex vivo culture system to analyze acinar-to-ductal transdifferentiation of acinar cells isolated from control and ERRγ cKO mice. At day 3 of ex vivo culture, ERRγ cKO acini showed accelerated ductal transdifferentiation compared to controls (Figure 7D). Interestingly, primary acini derived from wild-type mice downregulate ERRγ expression during ADM in ex vivo cultured acini (Supplementary Figure 12C). We next examined whether the increased number of duct-like cells may be due to ductal cell proliferation. To identify the proliferating cell type(s), we performed bromodeoxyuridine (BrdU) pulse-labeling at 7 d after the final TAM injection (Supplementary Figure 13A), which is before increased cell proliferation is observed in ERRγ cKO pancreas (Figure 2B). 4 days after the BrdU pulse, i.e. 11 d after the final TAM injection, most BrdU-positive cells co-stained with amylase but not CK19 in ERRγ cKO pancreas, suggesting that increased CK19-positive cells result from acinar cell transdifferentiation and not ductal cell proliferation (Supplementary Figure 13B). These results reveal a novel role for ERRγ in the maintenance of acinar cell identity and ADM.
As inflammation is known to affect the ADM process, we examined the effect of metabolically-induced chronic inflammation on the ERRγ cKO pancreas phenotype. To this end, mice were placed on 8 weeks of high-fat diet prior to TAM-induced ERRγ deletion and analyzed 2 weeks after the final tamoxifen injection (Figure 7E). HFD-fed ERRγ cKO mice also showed severe pancreatic atrophy (Figure 7F). Body weights of HFD-fed ERRγ cKO mice did not change (Figure 7G) but pancreas weights were decreased by ~70% (Figure 7H) compared to controls. Strikingly, ERRγ cKO pancreas exhibited a complete loss of amylase-positive acinar cells and an exaggerated ADM phenotype as shown by a massive increase of CK19-positive duct-like cells (Figure 7I). These results demonstrate that the ADM phenotype observed in ERRγ cKO pancreas is further exacerbated by metabolic inflammation, a clinically relevant modifier of pancreatic disease.
ERRγ expression is downregulated in pancreatic tissue from patients with pancreatitis.
Our findings from mouse models demonstrated ERRγ is required for exocrine pancreatic function. In order to investigate the relevance of these findings for human disease, we examined ERRγ expression in pancreas transcriptome data from two independent cohorts of normal subjects and chronic pancreatitis patients ([GSE143754] and [E-EMBL-6]).28, 29 Consistent with the pancreatitis-like phenotypes from genetic or pharmacological inhibition of ERRγ in mice, we found that ERRγ expression was significantly reduced in chronic pancreatitis compared to normal pancreas (Figure 8A and B). In both mice and humans, we did not observe significant sexual dimorphism in terms of ERRγ expression levels (Supplementary Figure 14 A and B). Next, we compared significantly downregulated genes in the pancreas of ERRγ cKO mice to genes downregulated in human pancreatitis (Figure 8C). In doing so, we identified a common signature set of 83 genes, many of which are direct targets of ERRγ based on previous ERRγ ChIP-seq datasets (GSE104905 and GSE144224), that are reduced in both ERRγ knockout mice and human pancreatitis (Figure 8C and 8D). Strikingly, Enrichr analysis with the signature set of 83 genes revealed a highly significant association with hereditary pancreatitis (Figure 8E). In addition, the common signature gene-set was predicted to be of acinar cell type, suggesting that dysregulated ERRγ expression within the acinar cells is associated with pancreatitis (Figure 8E). To further evaluate the acinar cell-specific role of ERRγ, we utilized single nucleus-RNA-seq (snRNA-seq) data from EGAD00001006396 that includes normal pancreas and pancreatitis patient samples.30 After establishing an snRNA-seq dataset with expression restoration, we extracted acinar cells and found that ERRγ expression was reduced in the pancreatitis group relative to the normal pancreas group (Figure 8F). Interestingly, we found that ERRγ target genes were significantly reduced in acinar-REG+ and acinar-s, but generally not in the acinar-i subtype of pancreatic acinar cells from pancreatitis patients (Figure 8G). Overall, these findings indicate that decreased ERRγ expression is associated with impaired pancreatic function and contribute to the pathogenesis of pancreatitis.
Figure 8. ERRγ and ERRγ-dependent genes are downregulated in human pancreatitis cohorts.
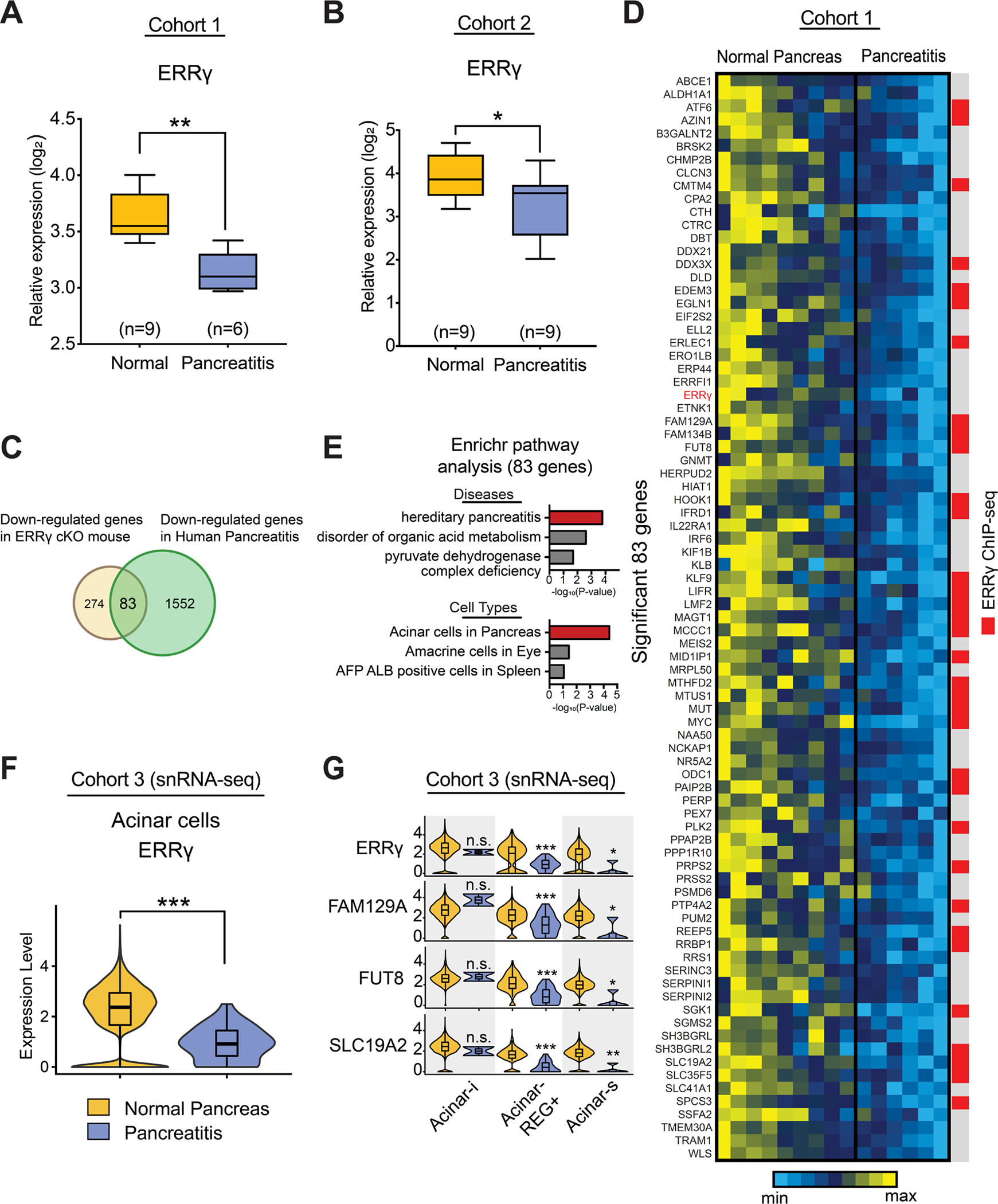
ERRγ expression in normal pancreas (n=9) and pancreatitis (n=6) from the human cohort 1 (GSE143754). The microarray data was extracted and normalized. Expression level was shown as log2 scale. (B) ERRγ expression in normal pancreas (n=9) and pancreatitis (n=9) from the human cohort 2 (E-EMBL-6). The microarray data was extracted and normalized. Expression level was shown as log2 scale. (C) Overlapping genes between down-regulated genes in ERRγ cKO mouse and down-regulated genes in human pancreatitis from human cohort 1. (D) The heatmap shows expression patterns of overlapping genes. Individual gene expression was normalized and shown as a range of min and max. Comparison of overlapping gene set with ERRγ ChIP-seq peaks was presented using red color. (E) Enrichr analysis predicts diseases and cell types. The overlapping gene set is associated with hereditary pancreatitis as a top predicted disease. The top predicted cell type was acinar cells in pancreas. The scale was presented in -log10(P-value). (F) Single nucleus RNA-seq analysis showing decreased ERRγ expression in acinar cells. The snRNA-seq object was established from the EGA dataset (EGAD00001006396). (G) In acinar-REG+ and acinar-s cells, gene expression of the expected ERRγ targets was significantly decreased, but not in acinar-i cells.
Functional genetic variants in the ESRRG locus are associated with human pancreatitis.
To determine whether functional genetic variants in the ESRRG locus, which encodes the human ERRγ gene, affect susceptibility to acute pancreatitis (AP) or chronic pancreatitis (CP), we conducted candidate locus region genetic association studies in the UK Biobank (UKBB) and the North American Pancreatitis Study II (NAPS2) cohorts. We compared pancreatitis cases and controls for differences in frequency of common single nucleotide variants (SNV) (MAF >1%) within the ESRRG exons and introns (GRCh38. Chromosome 1: 216676587…217311097 [634.511kb] plus 50 kb borders).
UKBB Cohort.
The UKBB is a population-based cohort of over 502,000 well characterized adult individuals living in England that contains genetic and phenotypic data collected on individuals aged 40 to 69, recruited from 2006–2010.31, 32 We identified patients with pancreatitis phenotypes using a two-step process, plus 24,000 random controls (see Methods). Association studies were completed on final populations of AP only (n=3229) and CP only (n=1027) and the SNVs were then filtered for association with p<0.001 (Supplementary Table 2A). Multiple individual functional SNVs were identified for association with CP that manifested potential effects on gene expression based on HaploReg v4.1.33 No SNVs in the AP cohort reached p<0.001, although 9 SNVs, representing independent functional SNVs were associated with AP at p<0.01 (Supplementary Table 3).
Evidence for effects of ESRRG genetic variants on CP were stronger than for AP, with effects based on a number of CP cases only a third the size of the number of AP cases. The lead SNV, rs201118685 (OR=0.82; p=9.80e−05) is a single nucleotide deletion at 1.5 kb 5’ of ESRRG that is rare (MAF <0.0001) but it is predicted to disrupt multiple gene regulatory elements33 and to be protective. Two of the five remaining CP-associated SNVs are intronic to the ESRRG gene (rs17669622 and rs12023399). SNV rs12023399 is in high linkage disequilibrium (LD r2 0.95–1.0) with multiple SNVs that lie within gene enhancer histone marks (including pancreatic tissue linked to rs1857413, LD r2=0.95) and multiple nucleotide binding site motifs.33 Three additional SNVs associated with CP are located in the region 5’ to the ESRRG gene (rs1502358, rs12119765, rs72743335). Both rs1502358 and rs12119765 are highly linked to multiple potentially functional SNPs. SNV rs72743335 is predicted to alter pancreatic tissue enhancer histone marks and alters two nucleotide binding motifs (Nr2f2, Pax-5).33 This SNV is also in high LD with at least 11 other SNVs that alter promoter histone marks, enhancer histone marks, DNAse protection and multiple additional nucleotide binding motifs.
NAPS2 Cohort.
The NAPS2 cohort represents three sequential, cross-sectional, case-control studies of recurrent acute pancreatitis (RAP) and CP as previously described 34–36. The subset of patients used for this additional analysis from the NAPS2 cohort was CP (n=818) and RAP+CP (n=1,277) subjects of European ancestry (EA).
The primary findings from the NAPS2 cohort for SNV associations with CP are given in Supplementary Table 2B. As seen with the UKBB cohort, there are multiple CP-associated SNVs located in gene regulatory elements or in LD with variants in gene regulatory elements.33 The strength of the effects was similar (see ORs in Supplementary Table 2A and B) although the p-values in the NAPS2 cohort were less significant. Taken together, these data demonstrated that, in two independent cohorts, multiple gene expression-altering SNVs in the ESRRG locus are associated with CP.
Discussion
ERRγ has emerged as a key regulator of cellular metabolism in a variety of tissues including muscle, brain, and insulin-secreting beta-cells.17–21 However, its role in the exocrine pancreas is unknown. Initial evidence linking ERRγ to exocrine pancreas function came from the observation that GSK5182 treatment, an inverse agonist of ERRγ, in mice induced exocrine pancreatic pathology similar to tamoxifen-induced pancreatic injury. Extending this observation, we find ERRγ cKO mice present with rapid and irreversible atrophy of the exocrine pancreas, confirming an obligate in vivo role for ERRγ in this tissue. Of note, immunosuppression by dexamethasone treatment did not alter the ERRγ cKO pancreas phenotype, whereas local deletion of ERRγ by viral Cre delivery phenocopied the ERRγ cKO pancreas. These results establish ERRγ as a tissue-autonomous regulator of adult exocrine pancreas function that cannot be compensated by related transcription factors such as ERRα or ERRβ. Furthermore, the pancreas phenotypes resulting from pharmacological inhibition or genetic deletion of ERRγ in mice suggest a potential link to human pancreatitis. Indeed, transcriptome data analysis from multiple independent cohorts revealed a significant reduction in ERRγ expression in pancreas tissues from patients with chronic pancreatitis compared to normal subjects.
The unique energetic and biosynthetic demands of acinar cell function require the concerted actions of an interdependent multi-organellar network including mitochondria, endoplasmic reticulum (ER), and the endo-lysosomal system.2, 3 We show that ERRγ is necessary and sufficient for the transcriptional control of mitochondrial gene networks, particularly OXPHOS-related genes, in pancreatic acini. Furthermore, disrupting ERRγ function not only triggered mitochondrial dysfunction but also led to increased oxidative stress, impaired autophagy, and ER stress in pancreatic acini. These results demonstrate a primary molecular function for ERRγ as a transcriptional regulator of the mitochondrial gene expression program required for pancreatic acinar cell organellar homeostasis and function.
Acinar-to-ductal metaplasia (ADM) is a regenerative process following pancreatic injury but also contributes to the development of pancreatic intraepithelial neoplasia (PanIN), a precursor lesion for pancreatic ductal adenocarcinomas.40–44 Oncogenic mutations of Kras have been shown to promote ADM and PanIN development driven by mitochondrial dysfunction and ROS production.45, 46 ERRγ cKO mice also present with an ADM phenotype that is modified by antioxidant treatment and diet-induced obesity, indicating that ROS and metabolic inflammation, respectively, are key mediators for ADM induced by ERRγ ablation. Furthermore, ERRγ cKO pancreas show decreased expression of genes involved in acinar cell development such as Ptf1a, Nr5a2, and Rbpjl (Supplementary Figure 12B), indicating a role for ERRγ in maintaining pancreatic acinar cell identity.
Reliable gene expression data sets on pancreatic tissue are very limited due to the difficulty of obtaining pancreatic tissue and also extracting high-quality RNA. Nevertheless, we analyzed ERRγ expression in two independent patient cohorts comprised of normal pancreas and chronic pancreatitis through published data sets. Consistent with our studies in mice, ERRγ expression was significantly reduced in chronic pancreatitis compared to the normal pancreas. Moreover, ERRγ gene signature module scores were dramatically decreased in the acinar cells in human pancreatitis compared to other cells (data not shown). Additionally, the human genetic studies provide new evidence that altered regulation of the ESRRG gene in humans may affect the development of chronic pancreatitis. Although no SNVs in the coding region of ESRRG were associated with AP or CP, we found multiple independent regulatory SNVs that were strongly associated with CP but not AP, with p<0.0001 being significant for a candidate gene approach rather than genome-wide significance. The identification of CP-associated SNVs linked to multiple functional regulatory elements highlights the importance of ERRγ in responding to ongoing injury or stress, rather than being a primary susceptibility gene such as CFTR47 or PRSS148. A limitation of the genetic approach is that the two cohorts (UKBB and NAPS2) used different SNV arrays that did not generally overlap within this locus, even after SNV imputation, which may explain the lack of replication. Future studies using overlapping SNV content, or whole genome sequencing approaches (to provide better coverage of the gene coding region) will give deeper insights into the spectrum of gene variant affects associated with chronic pancreatitis in the ESRRG locus.
Decades of research highlight the importance of acinar cell bioenergetics in normal physiology and pathophysiology of the exocrine pancreas. In this study, we identify an essential in vivo role for ERRγ in the control of mitochondrial gene networks supporting acinar cell energetics. In addition to this role, we show that ERRγ is required for maintaining acinar cell identity. These findings advance our understanding of the molecular programs regulating acinar cell metabolism and have therapeutic implications for relevant disorders such as pancreatitis and pancreatic cancers as ERRγ lies within a pharmacologically accessible niche.
Materials and Methods
Animal studies
ERRγ conditional knockout mice (kindly provided by Yann Herault, PHENOMIN-Mouse Clinical Institute, France, and Chul-Ho Lee, Korea Research Institute of Bioscience and Biotechnology, Korea) were crossed with R26-CreERT2 (B6.129-Gt(ROSA)26Sor tm1(Cre/ERT2)Tyj) mice (The Jackson Laboratory, Bar Harbor, Maine, USA). To induce deletion of ERRγ, tamoxifen (75 mg/kg) was intraperitoneally injected to mice daily for 5 consecutive days. Other mouse strains used in this study are listed in Supplementary Table 4. All mice were housed in a specific pathogen-free facility within the KAIST Laboratory Animal Resource Center. Mice were maintained under a 12 h light-dark cycle and given free access to food and water. All protocols for mouse experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of the Korea Advanced Institute of Science and Technology.
Dual-fluorescent Cre reporter mice and adenoviral Cre transduction of pancreas tissue
ERRγfl/fl mice were crossed with R26-mTmG reporter mice. The mTmG reporter mice express a membrane-targeted tdTomato (tdT) prior to Cre expression and express membrane-targeted GFP after Cre-mediated recombination. Mice were anesthetized with isoflurane and the pancreas was exposed via laparotomy. Control or Cre recombinant adenovirus (1.6 × 10^8 pfu) was injected at multiple sites within parenchymal regions of the pancreas with a 31-gauge needle.
Measurement of mitochondrial ROS production
4 d after the final tamoxifen injection, primary acinar cells were stained with 5 μM MitoSOX™ Red (Invitrogen, Carlsbad, CA, USA) for 10 min at 37°C. Mitochondrial ROS production was detected using confocal microscopy at 510/580 (excitation/emission) nm and the fluorescence intensity was quantified with Image J.
mtDNA Quantification
The mitochondrial DNA (mtDNA, ND1) to nuclear DNA (nDNA, HK2) ratio was measured by qRT-PCR analysis. Primers are listed in Supplementary Table 7.
Oxygen consumption rate measurement
Oxygen consumption rates of primary pancreatic acinar cells was recorded in 96-well plates using a XF96 Extracellular Flux Analyzer and the XF Cell Mito Stress Test Kit (Seahorse Bioscience, North Billerica, MA) following the manufacturer’s protocol. Briefly, primary acinar cells from C57BL/6J mice were seeded in gelatin-coated 96-well cell culture plate and subsequent treated with GSK5182 for 48 h. Cells were washed and pre-incubated in XF DMEM media (pH7.4) for 1 h prior to this assay. The Mito Stress Tests were performed according to the manufacturer’s protocol. Oligomycin (2 μM), FCCP (2 μM), and rotenone & antimycin A (0.5 μM) were added as indicated.
Intravital imaging of pancreas
Mice were anesthetized by intraperitoneal injection of a mixture of Zoletil (20 mg/kg) and xylazine (10 mg/kg) and mounted on heating pad for maintaining the body temperature at 37°C during intravital imaging. The pancreas was exposed via laparotomy and kept hydrated through repeated saline treatment during intravital imaging with an all-in-one intravital confocal microscopy (IVM-C, IVIM Technology, Daejeon, Republic of Korea). the biodistribution of autophagosome and mitophagy were detected through z-stack imaging at multiple locations. To visualize vasculature in pancreas, 25 μg anti-CD31 conjugated with Alexa Fluor 647 was injected into the tail vein of mice.
Data Availability Statement
Transcriptome datasets generated during the current study are available in GEO [GSE161757], GEO [GSE181529] and SRA [PRJNA750674].
Supplementary Material
<WHAT YOU NEED TO KNOW>
BACKGROUND AND CONTEXT
Mitochondria play crucial roles in pancreatic acinar cell energetics and physiology. However, the mechanisms regulating acinar cell mitochondrial gene expression programs are poorly understood.
NEW FINDINGS
Estrogen-related receptor γ (ERRγ) plays an essential in vivo role in the transcriptional control of mitochondrial gene networks that support acinar cell energetics, organellar homeostasis, and identity.
LIMITATIONS
Further mechanistic studies of ERRγ in relationship to exocrine pancreas disorders such as pancreatitis are required for translation to human disease.
IMPACT
ERRγ is identified to regulate the transcriptional program governing acinar cell mitochondrial function with clinical and therapeutic implications for exocrine pancreas disorders such as pancreatitis.
<LAY SUMMARY>
We identify ERRγ as a master regulator of pancreatic acinar cell mitochondrial gene networks, with novel molecular connections to human pancreatitis.
Acknowledgements
We thank H. Um, E. Ong and C. Brondos for administrative support. H. Jung, H. Juguilon and B. Collins for technical assistance. Jeanho Yun, Chounghun Kang, Yann Herault, Chul-Ho Lee, and Heung-Kyu Lee for providing mouse strains. Mara H. Sherman for helpful discussion. The authors acknowledge the contributions of the following individuals to the NAPS2 studies: C. Mel Wilcox, MD (University of Alabama, Birmingham, AL), Nalini Guda, MD (Aurora St. Luke’s Medical Center, Milwaukee, WI), Peter Banks, MD, Darwin Conwell, MD (Brigham & Women’s Hospital, Boston, MA; current affiliation, Univ. Kentucky, Lexington, KY); Simon K. Lo, MD (Cedars-Sinai Medical Center, Los Angeles, CA); Andres Gelrud MD (University of Cincinnati, Cincinnati, OH; current affiliation, GastroHealth and Miami Cancer Institute, Baptist Hospital, Miami, FL), Timothy Gardner, MD (Dartmouth-Hitchcock Medical Center, Hanover, NH); Late. John Baillie, MD (Duke University Medical Center, Durham, NC); Christopher E. Forsmark, MD (University of Florida, Gainesville, FL); Thiruvengadam Muniraj, MD, PhD (UPMC Mercy Hospital, Pittsburgh, PA; current affiliation Yale University, New Haven, CT); Stuart Sherman, MD (Indiana University, Indianapolis, IN), Vikesh Singh, MD (Johns Hopkins University, Baltimore, MD), Michele Lewis, MD (Mayo Clinic, Jacksonville, FL); Joseph Romagnuolo, MD, Robert Hawes, MD, Gregory A. Cote, MD, Christopher Lawrence, MD (Medical University of South Carolina, Charleston, SC, Current affiliation JR, Ralph H. Johnson VAMC, Charleston, SC; RH, Orlando Health Digestive Institute, Orlando, FL: GAC, Oregon Health & Science University; CL, Charleston GI, Charleston, SC); Michelle A. Anderson, MD (University of Michigan, Ann Arbor, MI; current affiliation, Mayo Clinic Arizona, Scottsdale, AZ); Stephen T. Amann, MD (North Mississippi Medical Center, Tupelo, MS); Babak Etemad, MD (Ochsner Medical Center, New Orleans, LA, current affiliation, Main Line HealthCare Interventional Gastroenterology, Wynnewood, PA); Mark DeMeo, MD (Rush University Medical Center, Chicago, IL); Michael Kochman, MD (University of Pennsylvania, Philadelphia, PA); Randall E. Brand, MD, Adam Slivka, MD PhD, David C. Whitcomb, MD PhD, Dhiraj Yadsav, MD MPH, Late. M. Michael Barmada, PhD, Jessica LaRusch, PhD, Judah N. Abberbock, PhD, Gong Tang, PhD, Phil D. Greer MS, Michael O’Connell, PhD, Kimberly Stello, Emil Bauer, Elizabeth Kennard, PhD, Stephen R. Wisniewski, PhD (University of Pittsburgh, Pittsburgh, PA); Late. Frank Burton, MD, Samer Alkaade, MD (St. Louis University, St. Louis, MO; SA current affiliation, Mercy Clinic Gastroenterology St. Louis, MO); James DiSario, MD, (University of Utah Health Science Center, Salt Lake City, UT; current affiliation Monterey Bay Gi Consultants Medical Group Inc. Monterey, CA ); Bimaljit S. Sandhu, MD (Virginia Commonwealth University, Richmond, VA; current affiliation, St Mary’s Hospital, Richmond, VA); Mary Money, MD (Washington County Hospital, Hagerstown, MD); William Steinberg, MD (Washington Medical Center, Washington, DC, retired). This research was partly supported by the NIDDK T32 DK063922-17 (DCW, BMB) and NIH DK061451 (DCW). This publication was also made possible in part by Grant Number UL1 RR024153 and UL1TR000005 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and NIH Roadmap for Medical Research (University of Pittsburgh. PI, Steven E Reis, MD). JIR was supported in part by the National Center for Advancing Translational Sciences, CTSI grant UL1TR001881, and the National Institute of Diabetes and Digestive and Kidney Disease Diabetes Research Center (DRC) grant DK063491 to the Southern California Diabetes Endocrinology Research Center. Infrastructure for the CHARGE Consortium is supported in part by the National Heart, Lung, and Blood Institute (NHLBI) grant R01HL105756. EY was supported by Japan Society for the Promotion of Science (JSPS), Kanae Medical Foundation, Allen Foundation, California Institute for Regenerative Medicine (CIRM) and Juvenile Diabetes Research Foundation (JDRF). RME was an Investigator of the Howard Hughes Medical Institute and is the March of Dimes Chair in Molecular and Developmental Biology at the Salk Institute, and was supported by grants from the Lustgarten Foundation, the NOMIS Foundation – the Science of Health, a SWCRF Investigator Award, the David C. Copley Foundation, and the Don and Lorraine Freeberg Foundation. Research reported in this publication was supported by the National Institute of Diabetes, Digestive Kidney Diseases (NIDDK) of the NIH under Award Number R01DK120480 (to RME). JMS was supported by the KAIST 2021 International Joint Research Support Program, National Research Foundation (2017M3A9G4052951, 2018R1A2A3075389, 2021M3A9G4016835, 2021R1A2C2007573) and Global Research Laboratory Program (2017K1A12013124) from the Ministry of Science and ICT of Korea. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NCRR, NIDDK, or NIH.
Abbreviations used in this paper:
- OXPHOS
oxidative phosphorylation
- ROS
reactive oxygen species
- ERRα
estrogen-related receptor α
- ERRβ
estrogen-related receptor β
- ERRγ
estrogen-related receptor γ
- PGC-1α
peroxisome proliferator-activated receptor γ coactivator-1α
- TCA cycle
tricarboxylic acid cycle
- DES
diethylstilbestrol
- 4-OHT
4-hydroxytamoxifen
- TAM
tamoxifen
- α-SMA
α smooth muscle actin
- Col1a1
collagen1A1
- Col1a2
collagen1A2
- AMY
amylase
- CC3
cleaved caspase-3
- P-H3
phosphor-histone H3
- GO
gene ontology
- 4-HNE
4-hydroxynonenal
- NRF2
nuclear factor erythroid 2
- NCD
normal chow diet
- BHA
butylated hydroxyanisole
- ADM
acinar-to-ductal metaplasia
- CK19
cytokeratin 19
- BrdU
bromodeoxyuridine
Footnotes
Competing Interests: DCW is cofounder of Ariel Precision Medicine, Pittsburgh, PA. He serves as a consultant, Chief Scientific Officer and may have equity.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Voronina SG, Barrow SL, Simpson AW, et al. Dynamic changes in cytosolic and mitochondrial ATP levels in pancreatic acinar cells. Gastroenterology 2010;138:1976–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Biczo G, Vegh ET, Shalbueva N, et al. Mitochondrial Dysfunction, Through Impaired Autophagy, Leads to Endoplasmic Reticulum Stress, Deregulated Lipid Metabolism, and Pancreatitis in Animal Models. Gastroenterology 2018;154:689–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gukovsky I, Pandol SJ, Gukovskaya AS. Organellar dysfunction in the pathogenesis of pancreatitis. Antioxid Redox Signal 2011;15:2699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perez S, Rius-Perez S, Finamor I, et al. Obesity causes PGC-1alpha deficiency in the pancreas leading to marked IL-6 upregulation via NF-kappaB in acute pancreatitis. J Pathol 2019;247:48–59. [DOI] [PubMed] [Google Scholar]
- 5.Badalov N, Baradarian R, Iswara K, et al. Drug-induced acute pancreatitis: an evidence-based review. Clin Gastroenterol Hepatol 2007;5:648–61; quiz 644. [DOI] [PubMed] [Google Scholar]
- 6.Daurio NA, Tuttle SW, Worth AJ, et al. AMPK Activation and Metabolic Reprogramming by Tamoxifen through Estrogen Receptor-Independent Mechanisms Suggests New Uses for This Therapeutic Modality in Cancer Treatment. Cancer Res 2016;76:3295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gukovskaya AS, Gukovsky I, Algul H, et al. Autophagy, Inflammation, and Immune Dysfunction in the Pathogenesis of Pancreatitis. Gastroenterology 2017;153:1212–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eichner LJ, Giguere V. Estrogen related receptors (ERRs): a new dawn in transcriptional control of mitochondrial gene networks. Mitochondrion 2011;11:544–52. [DOI] [PubMed] [Google Scholar]
- 9.Ariazi EA, Clark GM, Mertz JE. Estrogen-related receptor alpha and estrogen-related receptor gamma associate with unfavorable and favorable biomarkers, respectively, in human breast cancer. Cancer Res 2002;62:6510–8. [PubMed] [Google Scholar]
- 10.Stein RA, Chang CY, Kazmin DA, et al. Estrogen-related receptor alpha is critical for the growth of estrogen receptor-negative breast cancer. Cancer Res 2008;68:8805–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eichner LJ, Perry MC, Dufour CR, et al. miR-378( *) mediates metabolic shift in breast cancer cells via the PGC-1beta/ERRgamma transcriptional pathway. Cell Metab 2010;12:352–361. [DOI] [PubMed] [Google Scholar]
- 12.Luo J, Sladek R, Carrier J, et al. Reduced fat mass in mice lacking orphan nuclear receptor estrogen-related receptor alpha. Mol Cell Biol 2003;23:7947–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alaynick WA, Kondo RP, Xie W, et al. ERRgamma directs and maintains the transition to oxidative metabolism in the postnatal heart. Cell Metab 2007;6:13–24. [DOI] [PubMed] [Google Scholar]
- 14.Coward P, Lee D, Hull MV, et al. 4-Hydroxytamoxifen binds to and deactivates the estrogen-related receptor gamma. Proc Natl Acad Sci U S A 2001;98:8880–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luo J, Sladek R, Bader JA, et al. Placental abnormalities in mouse embryos lacking the orphan nuclear receptor ERR-beta. Nature 1997;388:778–82. [DOI] [PubMed] [Google Scholar]
- 16.Liou GY, Doppler H, DelGiorno KE, et al. Mutant KRas-Induced Mitochondrial Oxidative Stress in Acinar Cells Upregulates EGFR Signaling to Drive Formation of Pancreatic Precancerous Lesions. Cell Rep 2016;14:2325–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pei L, Mu Y, Leblanc M, et al. Dependence of hippocampal function on ERRgamma-regulated mitochondrial metabolism. Cell Metab 2015;21:628–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kida YS, Kawamura T, Wei Z, et al. ERRs Mediate a Metabolic Switch Required for Somatic Cell Reprogramming to Pluripotency. Cell Stem Cell 2015;16:547–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoshihara E, Wei Z, Lin CS, et al. ERRgamma Is Required for the Metabolic Maturation of Therapeutically Functional Glucose-Responsive beta Cells. Cell Metab 2016;23:622–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fan W, He N, Lin CS, et al. ERRgamma Promotes Angiogenesis, Mitochondrial Biogenesis, and Oxidative Remodeling in PGC1alpha/beta-Deficient Muscle. Cell Rep 2018;22:2521–2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahmadian M, Liu S, Reilly SM, et al. ERRgamma Preserves Brown Fat Innate Thermogenic Activity. Cell Rep 2018;22:2849–2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sakai A, Nishiumi S, Shiomi Y, et al. Metabolomic analysis to discover candidate therapeutic agents against acute pancreatitis. Arch Biochem Biophys 2012;522:107–20. [DOI] [PubMed] [Google Scholar]
- 23.Hackenbrock CR. Ultrastructural bases for metabolically linked mechanical activity in mitochondria. I. Reversible ultrastructural changes with change in metabolic steady state in isolated liver mitochondria. J Cell Biol 1966;30:269–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davies KM, Anselmi C, Wittig I, et al. Structure of the yeast F1Fo-ATP synthase dimer and its role in shaping the mitochondrial cristae. Proc Natl Acad Sci U S A 2012;109:13602–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Strappazzon F, Nazio F, Corrado M, et al. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ 2015;22:517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun N, Yun J, Liu J, et al. Measuring In Vivo Mitophagy. Mol Cell 2015;60:685–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Storz P Acinar cell plasticity and development of pancreatic ductal adenocarcinoma. Nat Rev Gastroenterol Hepatol 2017;14:296–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chhatriya B, Mukherjee M, Ray S, et al. Transcriptome analysis identifies putative multigene signature distinguishing benign and malignant pancreatic head mass. J Transl Med 2020;18:420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abdollahi A, Schwager C, Kleeff J, et al. Transcriptional network governing the angiogenic switch in human pancreatic cancer. Proc Natl Acad Sci U S A 2007;104:12890–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tosti L, Hang Y, Debnath O, et al. Single-Nucleus and In Situ RNA-Sequencing Reveal Cell Topographies in the Human Pancreas. Gastroenterology 2021;160:1330–1344 e11. [DOI] [PubMed] [Google Scholar]
- 31.Conroy M, Sellors J, Effingham M, et al. The advantages of UK Biobank’s open-access strategy for health research. J Intern Med 2019;286:389–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sudlow C, Gallacher J, Allen N, et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med 2015;12:e1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res 2012;40:D930–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Whitcomb DC, Yadav D, Adam S, et al. Multicenter approach to recurrent acute and chronic pancreatitis in the United States: the North American Pancreatitis Study 2 (NAPS2). Pancreatology 2008;8:520–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Conwell DL, Banks PA, Sandhu BS, et al. Validation of Demographics, Etiology, and Risk Factors for Chronic Pancreatitis in the USA: A Report of the North American Pancreas Study (NAPS) Group. Dig Dis Sci 2017;62:2133–2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilcox CM, Sandhu BS, Singh V, et al. Racial Differences in the Clinical Profile, Causes, and Outcome of Chronic Pancreatitis. Am J Gastroenterol 2016;111:1488–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Whitcomb DC, Larusch J, Krasinskas AM, et al. Common genetic variants in the CLDN2 and PRSS1-PRSS2 loci alter risk for alcohol-related and sporadic pancreatitis. Nature genetics 2012;44:1349–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dunbar E, Greer PJ, Melhem N, et al. Constant-severe pain in chronic pancreatitis is associated with genetic loci for major depression in the NAPS2 cohort. J Gastroenterol 2020;55:1000–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Phillips AE, LaRusch J, Greer P, et al. Known genetic susceptibility factors for chronic pancreatitis in patients of European ancestry are rare in patients of African ancestry. Pancreatology 2018;18:528–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Westphalen CB, Takemoto Y, Tanaka T, et al. Dclk1 Defines Quiescent Pancreatic Progenitors that Promote Injury-Induced Regeneration and Tumorigenesis. Cell Stem Cell 2016;18:441–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liou GY, Doppler H, Necela B, et al. Macrophage-secreted cytokines drive pancreatic acinar-to-ductal metaplasia through NF-kappaB and MMPs. J Cell Biol 2013;202:563–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Criscimanna A, Coudriet GM, Gittes GK, et al. Activated macrophages create lineage-specific microenvironments for pancreatic acinar- and beta-cell regeneration in mice. Gastroenterology 2014;147:1106–18 e11. [DOI] [PubMed] [Google Scholar]
- 43.Wei D, Wang L, Yan Y, et al. KLF4 Is Essential for Induction of Cellular Identity Change and Acinar-to-Ductal Reprogramming during Early Pancreatic Carcinogenesis. Cancer Cell 2016;29:324–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kopp JL, von Figura G, Mayes E, et al. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell 2012;22:737–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ogrunc M, Di Micco R, Liontos M, et al. Oncogene-induced reactive oxygen species fuel hyperproliferation and DNA damage response activation. Cell Death Differ 2014;21:998–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.DeNicola GM, Karreth FA, Humpton TJ, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011;475:106–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.LaRusch J, Jung J, General IJ, et al. Mechanisms of CFTR functional variants that impair regulated bicarbonate permeation and increase risk for pancreatitis but not for cystic fibrosis. PLoS Genetics 2014;10:e1004376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Whitcomb DC, Gorry MC, Preston RA, et al. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nature Genetics 1996;14:141–5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Transcriptome datasets generated during the current study are available in GEO [GSE161757], GEO [GSE181529] and SRA [PRJNA750674].