

Abstract
Scope:
Butyrate (B) is a short-chain fatty acid produced by dietary fiber, known to inhibit histone deacetylases (HDACs) and possess cancer-preventive/anti-cancer effects. However, the role of B in metabolic rewiring, epigenomic reprogramming, transcriptomic network, NRF2 signaling and eliciting cancer-preventive effects in colorectal cancer (CRC) HCT116 cell remains unclear.
Methods and results:
Sodium butyrate (NaB) dose-dependently inhibited the growth of CRC HCT116 cells. NaB inhibited NRF2/NRF2-target genes and blocked NRF2-ARE signaling. NaB increased NRF2 negative regulator KEAP1 expression through inhibiting its promoter methylation. Associative analysis of DEGs (differentially expressed genes) from RNA-seq and DMRs (differentially methylated regions) from CpG methyl-seq identified the tumor suppressor gene ABCA1 and tumor promote gene EGR3 were correlated with their promoters’ CpG methylation indicating NaB regulates cancer markers through modulating their promoter methylation. NaB activated the mitochondrial tricarboxylic acid (TCA) cycle while inhibited the methionine metabolism which are both tightly coupled to the epigenetic machinery. NaB regulated the epigenetic enzymes/genes including DNMT1, HAT1, KDM1A, KDM1B and TET1. Altogether, B’s regulation of metabolites coupled to the epigenetic enzymes illustrates the potential underlying biological connectivity between metabolomics and epigenomics.
Conclusion:
B regulates KEAP1/NRF2 signaling, drives metabolic rewiring, CpG methylomic and transcriptomic reprogramming contributing to the overall cancer-prevention/anti-cancer effect in the CRC cell model.
Keywords: Colorectal cancer, Epigenetic, Metabolomics, Nuclear Factor Erythroid-2 Like 2 (NRF2), Sodium Butyrate
Graphical Abstract
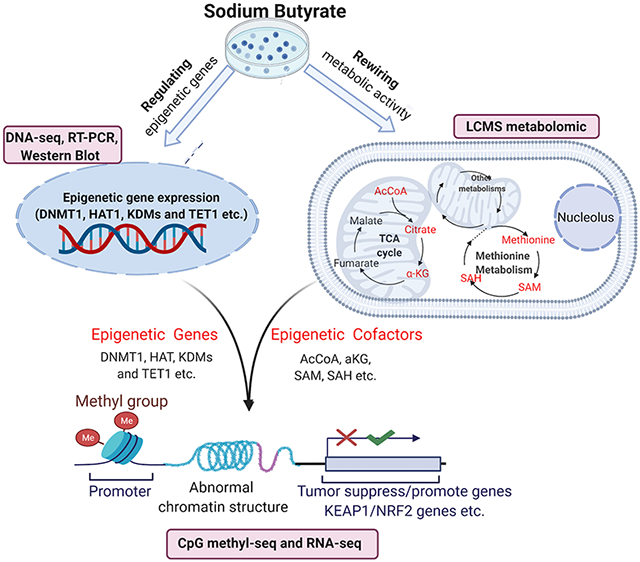
Integration of CpG Methyl-seq, RNA-seq and isotope labeling LC/MS technologies to dissect the connectivity mechanism of epigenomic CpG methylation, mRNA transcriptomic and metabolic rewiring in human colorectal cancer HCT116 cells after expose to sodium butyrate (NaB). Particularly, the role of NaB in NRF2 signaling and elicit anti-cancer effects were emphasized. NaB regulates the epigenomic via rewiring metabolomics and regulating epigenetic genes, consequently, modulate the tumor promote/suppress genes and KEAP1/NRF2 signaling to exhibit anti-cancer effects.
Introduction
Colorectal cancer (CRC) is one of the most diagnosed and important cause of cancer-related deaths in the US and worldwide [1]. Accumulation of genetic and epigenetic/epigenomic alterations in colon epithelial cells transform them into adenocarcinomas and also serves as a crucial driving factor in CRC progression and metastasis [2]. There are three epigenetic mechanisms including histone acetylation, DNA methylation, and non-coding microRNAs that are responsible for modifying the expression of critical genes associated with physiologic and pathologic processes [3]. Specifically, DNA methylation, with the addition of a methyl group to 5-cytosine in the CpG dinucleotide, is one of the most common epigenetic mechanisms associated with aberrant gene expression in cancer. For instance, CpG hyper-/hypo-methylation in DNA promoter regions is believed to play a crucial role in regulating gene expression, perhaps by interfering transcription factor binding [4]. In addition, histone tail acetylation enhances the accessibility of a gene to the transcription machinery, whereas deacetylated tails are highly un-charged and tightly associated with the DNA backbone, thus limiting accessibility of genes to transcription factors [5]. Interestingly, increasing evidences suggest that cellular metabolism could play an important role in cancers and that the cellular metabolism/metabolites are tightly linked to the basic epigenetic machinery [6]. Previous studies reported the epigenetic modifications such as DNA methylation and histone acetylation are sensitive to cellular metabolic status [7]. Strong molecular link between metabolic rewiring and epigenetic modifications through key metabolic intermediates, such as α-ketoglutarate (aKG), acetyl-CoA (AcCoA), nicotinamide adenine dinucleotide (NAD) of the tricarboxylic acid (TCA) cycle and S-adenosyl methionine (SAM) of methionine cycle which are co-factors for the epigenetic enzymes and work as hubs between epigenetic processes and therapeutic modalities [8-10]. So, modulation of epigenetic such as DNA de-/methylation, histone de-/acetylation as well as transcriptomics through regulating metabolomics may prevent diseases and protective against CRC development and metastasis [11].
The Kelch-like-ECH-associated protein 1 (KEAP1)-Nuclear Factor Erythroid-2 like 2 (NFE2L2, also called NRF2)-Antioxidant Response Element (ARE) signaling axis mediates cellular defense against oxidative stresses via induction of cellular defense and multiple downstream cytoprotective genes [12]. In this KEAP1/NRF2 axis, NRF2 is sequestered by the inhibitor KEAP1 and is transcriptionally inactive until KEAP1 is modified by sulfhydryl modification such as reactive oxygen species (ROS) and dissociated from NRF2. This dissociation allows NRF2 to form transcriptionally active complexes with other proteins, such as v-maf avian musculoaponeurotic fibrosarcoma oncogene homolog (MAFs) in the nucleus, transcriptionally activates cytoprotective and antioxidative genes, to remove oxidative insults like ROS [13]. However, recent accumulating evidence suggests that NRF2 has a contradictory role in cancers [14]. Aberrant activation of NRF2 is highly associated with poor prognosis. The constitutive activation of NRF2 in various cancers including CRC [15, 16] induces pro-survival genes and promotes cancer cell proliferation by repression of cancer cell apoptosis, and enhancement of self-renewal capacity of cancer stem cells. In addition, NRF2 was proven to contribute to the chemoresistance of cancer cells as well as inflammation-induced carcinogenesis dramatically [14]. For example, the in vitro and in vivo studies from Pawel et al. reported that the NRF2 KO cancer cells have significantly reduced proliferation phenotype and more sensitive to chemotherapeutic agents, such as cisplatin and carboplatin [17]. More importantly, NRF2 has been reported to induce metabolic rewiring via modulating the cellular intermediary metabolisms and mitochondrial functions [18]. It is either involved directly in the regulation of several key metabolic genes, or it affects their expression indirectly through crosstalk with other transcription factors [18]. For example, NRF2 has been shown to inhibits lipogenesis, supports β-oxidation of fatty acids, facilitates flux through the pentose phosphate pathway (PPP), and increases nicotinamide adenine dinucleotide phosphate (NADPH) regeneration and purine biosynthesis, therefore, these findings suggest NRF2 directs metabolic rewiring which may further influence the process of epigenetic associated metabolisms or the synthesis of the relevant metabolites such as TCA cycle, methionine metabolism and their respective metabolites [19].
As an important dietary constituent, dietary fiber ensures the potential carcinogens are removed from the colon and the microbiota within the colon converts the fiber into short chain fatty acids (SCFAs) by the process of fermentation [20]. These SCFAs serve as a major source of energy for the colon cells. Butyrate (B), constitutes about 20–30% of the SCFAs, is the predominant energy providing source [21, 22]. B has received particular attention for its cancer prevention effects including inhibit tumor-cell growth as well as its biological activity in many human CRC cells including HCT116 cell line [23]. The CRC microenvironment induced by colorectal tumor lesions shows significantly different from the normal intestinal environment [24]. CRC cells are more sensitive to SCFAs than normal intestinal epithelial cells, suggesting that SCFAs affect tumor cells through some certain pathways [25]. Previous studies have demonstrated that the elevated colonic concentration of B works as an important mediator in the observed protective effect of fermentable dietary fibers against CRC. And B is also recognized for its potential to act on secondary chemoprevention by slowing cell growth and activating apoptosis in colon cancer cells, but it can also act as primary chemopreventive agent [26, 27]. The anti-cancer and cancer prevention mechanisms of B are multiple, but many of these are related to its regulation effects on epigenetics as histone deacetylase (HDAC) inhibitor (HDACi) with subsequent regulatory effects on gene expression [28]. There is a growing interest in dietary HDACi, in particular B because its impact on epigenetic mechanisms will lead to more specific and efficacious therapeutic strategies in the prevention and treatment of cancers [28]. In terms of the metabolomic regulations, B has been reported to play a crucial role in maintaining the homeostasis of host metabolism and gut microbiome diversity [29]. For instance, previous study investigated the effect of NaB on TCA cycle enzymes activity in the brain of rats subjected to an animal model of mania induced by D-amphetamine and found B exerts protective effects against the D-amphetamine-induced TCA cycle enzymes' dysfunction [30].
However, the role of the B in regulating NRF2 signaling as well as metabolic rewiring, CpG methylomic reprogramming, and transcriptomic network in blocking pro-tumorigenic signaling and elicit cancer prevention effects in CRC remains unknown. So, in the current study we aimed to reveal the underlying intricate biological connectivity between metabolomic, epigenomic and transcriptomic regulation as well as the critical role of NRF2 in B-mediated cancer prevention effect in human CRC HCT116 cells.
Materials and methods
Cell culture and reagents
Sodium butyrate (NaB) was purchased from Sigma-Aldrich, Inc. (St. Louis, MO, USA). Versene, trypsin-EDTA, fetal bovine serum (FBS), penicillin-streptomycin (10,000 U/ml), puromycin, Dulbecco′s Modified Eagle′s Medium (DMEM) were purchased from Gibco Laboratories (Grand Island, NY, USA). Dimethyl sulfoxide (DMSO) was purchased from Sigma-Aldrich (St. Louis, MO, USA). 5-azadeoxycytidine (5-aza) and trichostain A (TSA) were obtained from Sigma-Aldrich (St. Louis, MO, USA). The MTS reagent ((3-(4,5-Dimethylthiazol-2-yl)-5-(3-Carboxymethoxyphenyl)-2-(4-Sulfophenyl)-2H-Tetrazolium) CellTiter 96® AQueous One Solution was obtained from Promega (Madison, WI, USA). Methanol (99%), and formic acid (98%) were purchased from Sigma-Aldrich (St. Louis, MO). Acetonitrile and pure water were purchased from Honeywell Burdick & Jackson (Muskegon, MI). Isotope labeled L-methionine (Methyl-13C) was purchased from Cambridge Isotope Laboratories, Inc (Tewksbury, MA). Bicinchoninic acid (BCA) protein assay kit was purchased from Pierce Biotech (Rockford, IL, USA).
The human hepatocellular HepG2-C8 cell line was previously established by stable transfection with the pARE-TI-luciferase construct and was utilized in our previous studies [31, 32]. Human colorectal carcinoma HCT116 cells were purchased from the American Type Culture Collection (ATCC; Manassas, VA, USA). HepG2-C8 cells and HCT116 cells were routinely cultured in DMEM supplemented with 10% FBS at 37°C in a humidified 5 % CO2 atmosphere.
Cell Viability Test
HCT116 cells were seeded in a 96-well plate at a density of 4 × 103 cells/well for overnight and then treated with either 0.1 % DMSO as vehicle control or various concentrations of NaB for 1-, 3-, 5-day in DMEM medium supplemented with 1% FBS. The cell culture medium dissolved with NaB was changed every other day. To test cell viability, the MTS assay was performed followed by the manufacturer’s instructions.
Dosage information/Dosage regimen
Previous studies reported that the physiological range of B concentrations in the intestinal lumen is between 1 and 10 mmol/l of the food content [33], which, assuming a daily production of 9 l of the intestinal content, corresponds to 9–90 mmol/day, i.e. 1–10 g/day. Specifically, to assess the effects of B on inflammation and oxidative stress, previous clinical trial (NCT00696098) recruited 35 patients with ulcerative colitis in clinical remission daily administered 60 ml rectal enemas containing 100 mM NaB (n = 17) which can achieve around 10 mM of NaB in human intestinal lumen [34]. In addition, Jahns et al. [35] treated the normal, adenoma and cancerous human colon tissues with 10 mM B to investigate their chemoprevention potential in CRC. So, all of the doses of B used in current study are within the range covering the physiology range in human and achievable through regular diet (low concentration) or via consuming supplements (moderate and high concentrations).
Luciferase Reporter Activity Assay
Previous established HepG2-C8 ARE-luciferase reporter transfected cells were utilized to examine the effects of NaB on the NRF2-ARE pathway. The NRF2 luciferase reporter stable HepG2 cells have been well developed for studying the NRF2 associated pathway and were also well validated by various antioxidant molecules in previous studies [36, 37]. HepG2-C8 cells were plated in 12-well plates at a density of 1 × 105 cells/well for 24 h, and then treated with 0.1% DMSO or various concentrations of NaB in DMEM supplemented with 1% FBS for 24 h. ARE-luciferase activity was determined using the luciferase activity assay kit (Promega, Madison, WI, USA). The reporter lysis buffer was used to lysate the cells and 10 μL of the cell lysate supernatant were measured for NRF2-ARE activity using a Sirius luminometer (Berthold Detection System GmbH, Pforzheim, Germany). The results were normalized against the protein concentration as determined by the Bicinchoninic Acid (BCA) protein assay. The results are expressed as an inducible fold change compared to the control group.
Quantitative Real-time Polymerase Chain Reaction (RT-PCR)
HCT116 cells were plated in 6-well plate at a density of 3 x 105 cells/well overnight and then treated with either vehicle control, 1 mM, 5 mM and 10mM NaB for 24h. mRNA was extracted using mRNA extraction kit from Thermo Fisher Scientific (Cat No. K0732). The first-strand cDNA was synthesized from 1 μg extracted RNA using SuperScript III First-Strand cDNA Synthesis System (Invitrogen, Grand Island, NY, USA). To determine the RNA expression of specific genes, the cDNA was used as the template for real time PCR using Power SYBR Green PCR Master Mix (Applied Biosystem, Carlsbads, CA). The mRNA expression was calculated as the fold change with normalization to the expression of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) using the 2−ΔΔCT method while GAPDH was used as an internal loading control. The primers were designed and ordered from Integrated DNA Technologies (IDT, Coralville, Iowa, USA) (Supplemental Table 1).
Protein Lyses Preparation and Western Blotting
HCT116 cells (1x106) treated with vehicle control and various concentrations of NaB for 24h and cells were harvested using RIPA buffer supplemented with protein inhibitor cocktail (Sigma, St. Louis, MO). Protein concentrations of each cleared lysates were measured using the BCA method (Pierce, Rockford, IL). Total 45μg proteins of each sample were separated by 4-15% SDS-polyacrylamide gel electrophoresis (Bio-Rad, Hercules, CA). Then the proteins were transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore, Bedford, MA) followed by blocking with 5% BSA in Tris-buffered saline-0.1% Tween 20 (TBST) buffer. Then the membrane was sequentially incubated with specific primary antibodies and then HRP-conjugated secondary antibodies. The blots were visualized by SuperSignal enhanced chemiluminiscence (ECL) detection system and recorded using a Gel Documentation 2000 system (Bio-Rad, Hercules, CA). Primary antibodies were purchased from different resources: anti-DNA Methyltransferase 1 (DNMT1) and anti-GAPDH were purchased from Cell Signaling (Boston, MA); anti-NRF2, anti-KEAP1, anti-Heme Oxygenase 1 (HO1), anti-MYCL proto-oncogene (c-MYC), and anti-NAD(P)H dehydrogenase [quinone] 1 (NQO1) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Isolation of Nucleic Acids and Next-generation Sequencing (NGS)
HCT116 cells were plated in 10cm cell culture plates at a density of 2 × 105 cells/dish for overnight and then treated with the vehicle control, 1mM NaB for 5 days. The cell culture medium dissolved with 1mM NaB was changed every other day and cells were harvest at day 5. RNA and DNA were extracted using an AllPrep DNA/RNA Mini Kit (Qiagen, Valencia, CA, USA). Total four RNA samples (n=2) and two DNA samples (n=1) were subject to RNA-seq and SureSelect Methyl-seq respectively. Each RNA or DNA sample was pooled by 3 independent biological replicates. Previous NGS studies from our lab showed that the pooled DNA and RNA collected from 3 plates of cells and combined into one DNA-seq and RNA-seq sample were not much variation between different plates of cells [38, 39]. 3 μg RNA and 3 μg DNA of each sample was used for NGS. The library preparation and sequencing were performed by RUCDR Infinite Biologics. Briefly, the RNA library was built with the Illumina TruSeq RNA preparation kit (Illumina, San Diego, CA, USA) and then sequenced on an Illumina NextSeq 500 instrument with 75 bp single-end reads, generating 30-40 million reads per sample. The DNA samples were further processed with the Agilent Mouse SureSelect Methyl-seq Target Enrichment System (Agilent Technologies Inc., Santa Clara, CA, USA). Bisulfite conversion was performed using an EZ DNA Methylation-Gold Kit (Zymo Research, USA). And then DNA sequencing was also performed on the Illumina NextSeq 500 instrument with 76-bp single-end reads, generating 30-40 million reads per sample [40].
Bioinformatics Analyses
RNA-seq
Cutadapt, a command-line program produced by second-generation sequencers [41], was used to remove the Illumina Universal Adapter sequence. Hierarchical indexing for spliced alignment of transcripts (HISAT2) was adapted for aligning the reads to the mouse genome (mm10) [42], and remove PCR duplicates. Genomic features with overlapping reads were counted by featureCounts (version 1.5.1) [43] and then data were further analyzed for differential expression with DEGSeq (version 1.36.0) in R (version 3.4.0) [44].
DNA SureSelect Methyl-seq
Bismark (version 0.15.0) alignment algorithm was applied to align the DNA reads to the in silico C-T converted mouse genome (mm10). And the DMRfinder, an efficient tool to identify differentially methylated regions from Methyl-seq data, was used to extract methylation counts and cluster CpG sites into differentially methylated regions (DMRs) [45]. Methylation differences > 10% with P-values < 0.05 were considered as significant. Genomic annotation was performed with ChIPseeker (version 1.10.3) in R (version 3.4.0) [46].
Ingenuity Pathway Analysis (IPA)
Isoforms with false discovery rates (FDR) adjusted P value (q value) < 0.05 and log2 fold changes > 2.0 or < −2.0 were applied for the IPA analysis (IPA 4.0, Ingenuity Systems, www.Ingenuity.com). The input genes were mapped to the IPA knowledge base, and the signaling pathways mediated by NaB were identified.
LC-MS Metabolomic Analysis
LC-MS metabolomic analysis was performed in Metabolomics Shared Resources, Rutgers Cancer Institute of New Jersey (CINJ) as our previously reported [47, 48]. 3 ×106 HCT116 cells were seeded in 10-cm cell culture dishes (n=3) and cultured in dialyzed FBS DMEM medium overnight. For the stable isotope labelling of methyl group, methionine free DMEM media will be used, and supplemented with 2 mM 13C-methionine as we have performed with other isotopes previously [49]. Cells were then be treated with vehicle control (DMSO) vs various concentrations of NaB for 24 hrs. The cells were scraped after PBS wash for 3 times and centrifuged at 4 °C, 15,000 g for 10 min. Then metabolites were extracted with 1 ml cold 40:40:20 methanol:acetonitrile:water solution with 0.5% formic acid, and then followed by 5 min incubation on ice and sequentially neutralized with 50 μL 15% NH4HCO3. The cleared supernatant was then used for LC-MS analysis. LC separation was performed on a XBridge BEH Amide column (2.1 mm × 150 mm, 2.5 μm particle size, 130 Å pore size; Waters) coupled with a Waters XBridge BEH XP VanGuard cartridge (2.1 mm x 5 mm, 2.5 μm particle size, 130 Å pore size) guard column. The solvent A prepared by water/acetonitrile (95:5, v/v) with 20 mM NH3AC and 20 mM NH3OH at pH 9; and solvent B prepared by acetonitrile/water (80:20, v/v) with 20 mM NH3AC and 20 mM NH3OH at pH 9 in the following solvent B percentages over time: 0 min, 100%; 3 min, 100%; 3.2 min, 90%; 6.2 min, 90%; 6.5 min, 80%; 10.5 min, 80%; 10.7 min, 70%; 13.5 min, 70%; 13.7 min, 45%; 16 min, 45%; 16.5 min, 100%. The flow rate was set to 300 μL/min with an injection volume 5 μL. The column temperature was set at 25°C. MS scans were obtained in both positive and negative ion modes with a resolution of 70,000 at m/z 200, in addition to an automatic gain control target of 3 × 106 and m/z scan range of 72 to 1000.
DNA Extraction and Bisulfite Genomic Sequencing TA Cloning
HCT116 cells were treated with vehicle control, 1mM NaB and 5mM NaB treatment for 5 days and the medium dissolved with the compounds were changed every 2 days. For the positive control group, HCT116 cells were treated with 500 nM 5-aza in every other day. On day 4, for the 5-aza and TSA combination treatment, 100 nM TSA was added to the 5-aza containing medium. Cells were then harvested on day 5 for genomic DNA isolated with the QIAamp® DNA mini Kit (Qiagen, Valencia, CA). Then 500 ng DNA was denatured and utilized to bisulfite conversion using EZ DNA Methylation Gold Kits (Zymo Research Corp., Orange, CA) following the manufacturer's instructions. The converted DNA was amplified by PCR using Platinum Taq DNA polymerase (Invitrogen, Grand Island, NY) with primers (Forward: 5’-GAAAGAAAGAAAGAAAAGAAAAG-3’, Reverse: 5’-CACCAAAAATAAAATAAACACCC-3’) that amplify the 62 CpGs located between −291 and 337 of human KEAP1 gene [50] with the translation start site defined as +1. The first 12 CpGs located between −291 to −89 in KEAP1 promoter region were counted as the methylation ratio of these 12 CpGs have been proven to regulate the KEAP1 gene expression in CRC [51]. PCR products were cloned into pCR4 TOPO vector using a TOPO™ TA Cloning Kit (Invitrogen, Carlsbad, CA). Plasmids from at least ten colonies of each treatment group were selected using QIAprep Spin Miniprep Kit (Qiagen, Valencia, CA) and sequenced (Genwiz, Piscataway, NJ).
Statistical analysis
The results are presented as the mean ± SEM or SD and P-value ≤ 0.05 was considered statistically significant. Statistical analysis was carried out using Student’s t test (two-tailed unpaired) for two groups and one-way ANOVA followed by Dunnett’s post hoc test for multiple groups within GraphPad Prism. Metabolomic pathways analysis and pathway enrichment analysis were performed within the Web-based inference MetaboAnalystR 5.0 (https://www.metaboanalyst.ca/).
Results
Cytotoxicity of NaB in HCT116 Cells
Treatment with NaB showed a time- and dose-dependent effect on the HCT116 cell viability (Figure 1a). Determination of an ideal treatment duration and concentration involves a trade-off between toxicity and efficacy. The viability of HCT116 cells that were treated with 1mM NaB for 1-, 3- and 5-day were 97.4%, 93.2% and 89.5%, respectively, therefore 1mM of NaB was used in the subsequent 5-day next generation sequencing (NGS) so that maximal cell viability was achieved; 1mM and 5mM of NaB were used for the 5-day bisulfite sequencing TA cloning studies. In addition, the cell viability of HCT116 after low (1mM), moderate (5mM) and high (10mM) doses treatment for 24 h were 97.4%, 81.5% and 73.2%, respectively. These doses were further utilized for the metabolomic, RT-PCR, western blot analysis, as stated accordingly. In addition, as aforementioned the physiological range of B concentrations in the intestinal lumen is between 1 and 10 mM [33, 52] which can further convince our doses.
Figure 1. NaB-mediated cell viability and regulation of NRF2/NRF2-target genes in HCT116 cells.
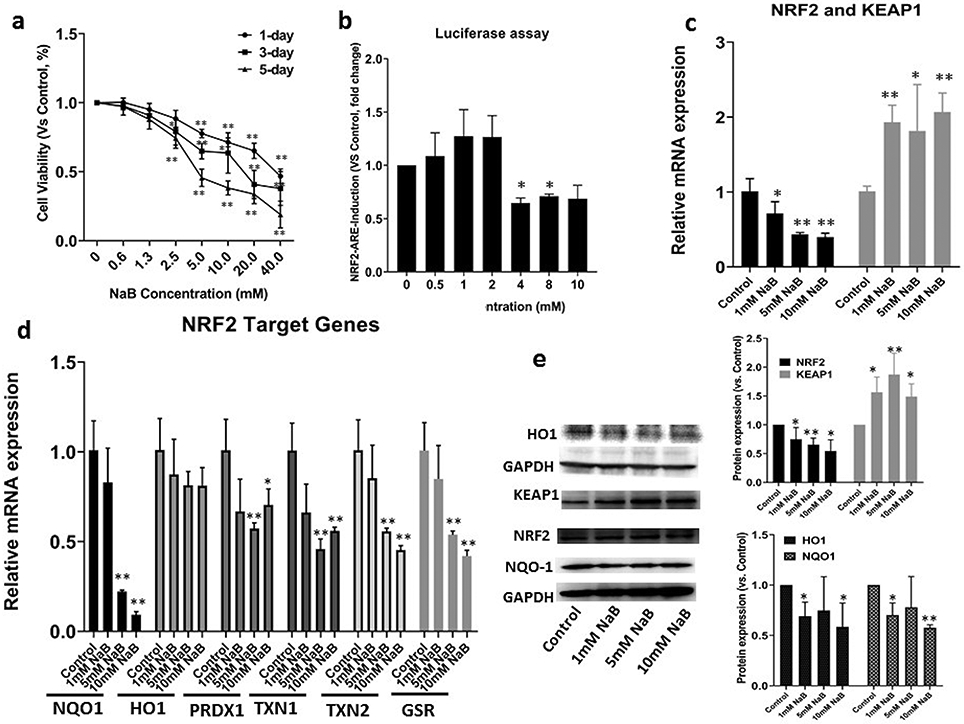
(a) Cell viability of the HCT116 cells after various concentrations of NaB treatment for 1-, 3- and 5-day. Cell viability was determined by the MTS assay; (b) The luciferase activity after NaB treatment in HepG2 cells transfected with the ARE-luciferase reporter vector. The activity was normalized based on the protein concentrations in the BCA protein assay and shown as fold change compare to control group; (c) NRF2 and KEAP1 mRNA expression measured by RT-PCR. The expressions were shown as fold change compare to control group; (d) NRF2-target genes mRNA expression measured by RT-PCR. The expression levels were shown as fold change compare to control group; (e) NRF2, KEAP1, HO1 and NQO1 protein expression quantified by Western Blot after NaB treatment. The protein expressions were shown as fold change compare to control group and GAPDH was used as the housekeeping protein for the normalization. All the data are presented as the means ± SEM of three independent experiments. *, P < 0.05 and **, P < 0.01 indicate significant differences between the treatment groups and the control group (0.1% DMSO). Student’s t test was used to calculate the significance of the differences compared with the control.
NaB Regulates NRF2 signaling and NRF2-target Gene Expression
To investigate the mechanism underlying therapeutic efficacy of NaB, we used HepG2-C8 cells that were stably transfected with the ARE-luciferase reporter. NaB inhibits NRF2-ARE luciferase activity at the NaB concentration range from 4 to 10 mM (Figure 1b) while lower NaB concentration (<2mM) slightly increased the luciferase activities but not significant (these low concentrations are easily achievable in in vivo situation, including human) [53]. In addition, RT-PCR revealed NaB significantly decreased the mRNA expression levels of NRF2 and NRF2-target genes including NQO1, HO1, PRDX1, TXN1 and TXN2, while increased the NRF2 negative regulator KEAP1 mRNA levels compared to control (Figure 1c and 1d). The protein levels measured by Western Blot were further validated showing that NaB decreased NRF2, NOQ1 and HO1, while increased KEAP1 protein expression (Figure 1e). These results indicate that NaB may be act as a potent NRF2 inhibitor in CRC cells and could be a promising therapeutic and chemopreventive agent to against cancers.
NaB Drives Epigenetic CpG Methylation Reprogramming
DNA Methyl-seq profiled differentially methylated regions (DMRs) were used to show the epigenomic modulations by NaB in HCT116 cells as we have reported previously in other systems [38, 48]. More than 50% DMRs were detected in the promoters and distal intergenic regions (Figure 2a). The methylation differences > 10% of specific genes were applied for the cutoff of DMRs. Briefly, 1,100 DMRs located in the promoter and intron regions were filtered which included 493 hypermethylated and 607 hypomethylated after NaB treatment (Figure 2b and Supplemental file 1 (DMRs)). Interestingly, DNA methyl-seq analysis also revealed that NaB decreased the CpG methylation ratio in the KEAP1 promoter region from 94% to 85% indicating NaB may regulates the KEAP1 gene expression through modulating it’s promoter methylation. The methylation of KEAP1 promoter will be validated through bisulfite genomic sequencing TA cloning as below.
Figure 2. DNA Methyl-seq and RAN-seq profiles in HCT116 cells regulated by NaB.

(a) Distribution of annotated DMRs by genomic features including Distal Intergenic, Promoter (<=1kb, 1-2kb and 2-3kb), 1st Intron and other regions as shown in the Fig. 2a legend. Each DMR has at least three CpG sites; (b) Overall DMRs in response to 1mM NaB with cutoffs of methylation difference ≥ 10% or ≤ −10%. “+” represents Intron, “•” represents Promoter, “0” represents the absolute methylation difference <10%, “1” represents the methylation increased by >10% and “2” represents the methylation decreased by >10%; (c) Principal component analysis (PCA) of transcriptomic profiles in control and NaB treatment groups; (d) MA plots showing overall DEGs in response to 1mM NaB with cutoffs of p<0.01 and log2(fold change) ≥ 2.0 or ≤ −2.0; (e) NRF2 and NRF2-target genes mRNA expression reported from RNA-seq analysis. The mRNA expressions were shown as fold changes compare to control group; (f) c-MYC mRNA expression reported from RNA-seq and protein expression quantified by western blot after NaB treatment. GAPDH was used as the housekeeping protein for the protein normalization. The RNA-seq data are presented as the means ± SEM of two independent biological replicates. The western blot protein levels are presented as the means ± SEM of three independent experiments. *, P < 0.05 and **, P < 0.01 indicate significant differences between the treatment groups and the control group (0.1% DMSO). Student’s t test was used to calculate the significance of the differences compared with the control group.
NaB Regulates Differentially Expressed Genes (DEGs)
Differentially Expressed Genes (DEGs) obtained from RNA-seq analysis were utilized to show the transcriptomic regulations of NaB in HCT116 cells. The principal component analysis (PCA) (Figure 2c) showed that the NaB treatment group to be clearly separated from the vehicle control indicating that exposure of HCT116 cells to NaB significantly induced gene expression alterations. The DEG profile was further analyzed and plotted with the cutoff p-value < 0.01 coupled to log2 fold change ≥ 2.0 or ≤ −2.0 (Figure 2d and Supplemental File 2 (DEGs)). The MA plot showing that 3,917 and 783 genes were significantly up- and down-regulated after NaB treatment, respectively, compared with control group. Besides the overall mapping, RNA-seq analysis also further validated that NaB inhibited the NRF2 and significantly induced the KEAP1 transcription levels which are highly in agreement with the RT-PCR and Western Blot results above. RNAseq also shows that NaB significantly inhibited NRF2 target genes HO1 and GCLM gene expression by 92% and 69% respectively (Figure 2e). Moreover, NaB also significantly decreased the cancer marker MYCL proto-oncogene (c-MYC) gene expression by 64%. The c-MYC protein expression level was also validated by western blot (Figure 2f).
NaB Regulates Signaling Pathways of HCT116 Cells
A total of 4,700 genes (3,917 up- and 783 down-regulated) filtered with the thresholds p-value < 0.01 coupled to log2 fold change ≥ 2.0 or ≤ −2.0 from the RNAseq dataset were further used to conduct the Ingenuity Pathway Analysis (IPA). These highly regulated genes (Supplemental File 1) covered 83 significantly modulated signaling pathways (P ≤ 0.05). The top regulated signaling pathways with −log(p-value) > 2.0 and an absolute values z-score >2.0 were further plotted as shown in the Figure 3. Among these 83 highly regulated pathways, 77 were significantly activated by NaB while 6 pathways were inhibited by NaB (Supplemental File 3). Interestingly, IPA shows NaB significantly activated AMP-activated protein kinase (AMPK: one of the central regulators of cellular and organismal metabolism) signaling pathway indicating that NaB may play critical roles in reprogramming energetic metabolism as well as cellular processes such as autophagy and cellular polarity [54]. Other caner- and cell cycle-related signaling pathways including Tumor Microenvironment Pathway and Cell Cycle Control of Chromosomal Replication pathways were also highly regulated NaB. The totality of these modulated signaling pathways may contribute to the biological effects of B and provide new windows of therapeutic opportunity against CRC.
Figure 3. Ingenuity Pathway Analysis (IPA) and Correlation of DEGs and DMRs.

(a) Top 30 regulated pathways in HCT116 cells after NaB treatment; (b) Correlation analysis between gene expression and DNA methylation. Total 43 DMRs (located in the upper left and bottom right corners) were negatively correlated with their corresponding gene expression with the cutoff of methylation difference ≥ 15% or ≤ −15% coupled to gene expression log2(fold change) ≥ 5.0 or ≤ −5.0. 9 of 43 DMRs were located in the promoter region. The DMR locations (gene features: Region, Body, Downstream and Promoter) are indicated by the colors.
Integrated Methyl-seq and RNA-seq Analysis Identifies DNA Methylation and Gene Expression Patterns
The current dogma of DNA methylation implicates regulation of transcription of many downstream target genes in mammalian cells [5]. The correlation between DEGs and DMRs in the promoter regions were conducted to discover the underlying linkage between DNA methylation and transcriptomic gene expression. A cutoff threshold methylation difference ≥ 10% coupled to log2-fold change of gene expression difference ≥ 2 or ≤ −2 was applied to the selection. A total 444 DMRs in the promoter regions were negatively correlated (85 genes promoter methylation increased with downregulated mRNA expression; 359 gene promoter methylation decreased with upregulated mRNA expression) with their corresponding gene expression after NaB treatment (Supplemental File 4). To have a better visualization, the negatively correlated genes filtered with threshold methylation difference ≥ 15% coupled to log2-fold change of gene expression ≥ 5 or ≤ −5 were used for the starburst plot (Figure 3b). Interestingly, this associative analysis revealed that NaB decreased the tumor suppressor gene ATP binding cassette A1 (ABCA1)’s promoter methylation by 11% and increased the gene expression by 6.5-fold compare to control. In addition, the promoter methylation and gene expression of Early growth response protein 3 (EGR3) were increased by 11% and decreased by 94% respectively, compare to control (Supplemental File 4). These results indicated that NaB may regulate the tumor promoter/suppressor genes through modulating the it’s promoter methylation.
NaB Drives Cellular Metabolomic Rewiring
To unravel the potential underlying molecular links between metabolic reprogramming and epigenetic modifications as well as the impact of NaB on epigenetics through regulating mitochondrial metabolic pathways and metabolites, the samples collected from HCT116 cells treated with vehicle control or 1mM, 5mM and 10mM of NaB for 24h were used to perform the metabolomic analysis. PCA revealed a clear separation for each treatment, showing NaB induced significant differential metabolic profile with a dose-dependent manner, with the most significant difference between control and 10mM NaB groups (Figure 4a). A total of 111 metabolites were identified under positive and negative ion modes of which top 30 regulated metabolites (one-way ANOVA computed P value) across 4 groups were further plotted as in figure 4b. In addition, the metabolites regulated by 10 mM NaB were filtered with the thresholds p-value < 0.05 and log2 fold changes > 2.0 or < −2.0. Pathway analysis (Figure 4c) combined with pathway enrichment analysis (Figure 4d) with these filtered metabolites from control vs. 10mM NaB comparison group revealed that the epigenetic associated metabolism such as TCA cycle and methionine metabolism were significantly altered in 10 mM NaB treated HCT116 cells. These results suggested that the alterations of mitochondrial metabolism/metabolites are impacted by NaB and also pointing to the down-stream impact on epigenetic reprogramming, to be discussed below.
Figure 4. Metabolomic profiles of HCT116 cells treated with NaB.

(a) PCA plot of metabolomic profiles in HCT116 cells after control and 1mM, 5mM and 10mM NaB treatment; (b) Top 30 regulated metabolites after NaB treatment; (c) Metabolism pathway analysis and (d) pathway enrichment analysis of HCT116 cells after 10mM NaB treatment.
NaB Regulates Epigenetics via Cellular Metabolism
Mitochondrial TCA cycle and metabolites are tightly linked to the basic epigenetic machinery DNA/histone modifications, chromatin remodeling and modulating phenotypic gene expression [8, 55]. Some of the TCA metabolites are cofactors utilized by the epigenetic enzymes that catalyze the post-translational epigenetic modification including AcCoA, the acetyl (-COH) donor for histone acetylation with the histone acetyltransferases (HATs) catalyzation; α-ketoglutarate (α-KG), the cofactor along with the histone lysine demethylase (KDMs) and ten eleven translocation (TETs) enzymes for histones and DNA demethylation respectively [55], participate in the biological function of the basic epigenetic machinery (Figure 5a). NaB significantly activated the TCA cycle and elevated the metabolites levels in a dose-dependent manner (Figure 5b). The AcCoA, α-KG and citrate were increased by ~ 1.9-, 4.7- and 12.5-fold after 10 mM NaB treatment respectively (Figure 5c). Besides the NaB-induced metabolic regulation, RNAseq also revealed that the transcription levels of epigenetic genes HAT1 (Histone Acetyltransferase 1), KDM1A (Lysine Demethylase 1A), KDM1B (Lysine Demethylase 1B) and TET1 (Tet Methylcytosine Dioxygenase 1), which couple to the above cofactors for the epigenetic regulation, were significantly activated (Figure 5d) after NaB treatment. The metabolomic along with the transcriptomic results from RNAseq indicated that NaB would potentially promote histone acetylation, histone demethylation and DNA demethylation via the contribution of NaB-induced metabolic rewiring.
Figure 5. NaB upregulates epigenetic associated TCA metabolites and epigenetic genes.

(a) Schematic of connectivity between TCA cycle and epigenetic enzymes. The enzymes involved are highlighted in red; (b) Heatmap showing metabolites of TCA cycle in HCT116 cells after treated with NaB for 24h (n=3); (c) Quantified TCA metabolites shown as fold changes compare to control group (n=3); (d) Epigenetic gene expression reported from RNA-seq analysis (n=2). *, P < 0.05 and **, P < 0.01 indicate significant differences between the treatment groups and the control group (0.1% DMSO). Student’s t test was used to calculate the significance of the differences compared with the control group.
In addition to the TCA cycle, the epigenetic associated methionine cycle metabolites like SAM which was also highly regulated by NaB (Figure 6a). SAM, as the universal donor of methyl groups to both DNA and histone methyltransferase enzymes, coupled to DNMTs are tightly linked with basic epigenetic methylation reactions like CpG and histone methylation driving epigenetic reprogramming [56]. To monitor the methyl group transfer and synthesis of SAM after NaB treatment, the stable isotope tracing with 13C-methionine (methyl group donor) experiment was performed. The labeled SAM (m+1) and methionine (m+1)/SAM (m+1) ratio were significantly decreased and increased respectively, after NaB treatment (Figure 6b) indicating the decreased transfer potential of the methyl groups to DNA and histones. Furthermore, the intermediary metabolite S-adenosylhomocysteine (SAH) which is the byproduct of SAM during methyltransferase reactions [57] is a potent inhibitor of DNMTs. The SAM/SAH ratio serves as a biosensor of the cellular metabolic state influencing the activity of methyltransferase enzymes that culminate in chromatin changes in response to cellular challenges [58]. NaB inhibited SAM/SAH ratios indicated the reduced cellular methylation potential [59]. Moreover, both mRNA and protein expression of DNMT1 (Figure 6c) were significantly decreased indicating that the intermediary metabolites coupled to epigenetic transcription enzymes mediated by NaB may contribute to the regulation of epigenetics.
Figure 6. NaB regulates the methionine metabolism and decreases transfer potential of the methyl groups to DNA and histones.

(a) Schematic of connectivity between methionine metabolism and epigenetic enzymes and the regulation effect of NaB on methionine cycle metabolites (n=3). The enzyme involved is highlighted in red; (b) Quantified methionine metabolism metabolites and isotope labeling of methionine to trace the methyl group transfer in HCT116 cells after NaB treatment; (c) Quantified DNMT1 gene and protein expression by RT-PCR and western blot respectively. *, P < 0.05 and **, P < 0.01 indicate significant differences between the treatment groups and the control group (0.1% DMSO). Student’s t test was used to calculate the significance of the differences compared with the control group.
NaB Regulates CpG Methylation in Keap1 Promoter Region
Previous study showed that KEAP1 gene silencing is highly associated with its promoter hypermethylation of the first 12 CpG (−291 to −89) and the KEAP1 promoter regions also have significant different methylation status between CRC and normal cells [51]. Therefore, bisulfite sequencing was conducted to further investigate if NaB treatment would demethylate the first 12 CpGs in KEAP1 promoter region and contribute to the gene and protein expression. Figure 7 shows that the 12 CpGs were hypermethylated in HCT116 cells in the control group (methylation ratio 91.7%). Compared to control, treatment with NaB (1.0mM, 5.0 mM) and the combination of 5-aza (500 nM)/TSA (100 nM) (Positive control) significantly decreased the methylation ratio to 80.0%, 77.7% and 62.5%, respectively. This suggests that NaB has demethylation potential on the promoter of KEAP1 gene and results in the increase of KEAP1 gene expression, consequently, attenuates NRF2 biological functions. Furthermore, the bisulfite sequencing result also aligned with the DNA Methyl-seq data which further validated the regulatory effect of NaB on KEAP1-NRF2 signaling axis.
Figure 7. Effect of NaB on KEAP1 promoter methylation in HCT116 cells.
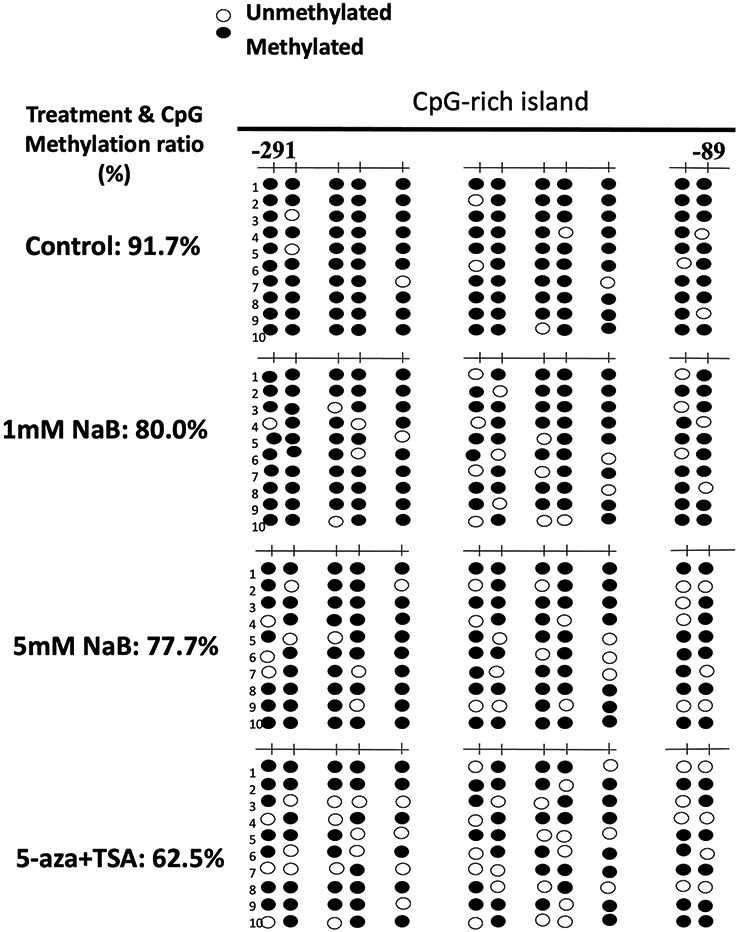
Cells were treated with 1.0 or 5 mM NaB for 5 days, and then genomic DNA was extracted from the treated cells. A combination treatment of 5-aza (500 nM)/TSA (100 nM) was used as positive control, while TSA was added 24 h before cell collection. The methylation pattern of the first 12 CpGs located at −291 and 337 in KEAP1 promoter region, where the translational starting site is considered as +1 was determined. Black dots indicated methylated CpGs while open circles indicate unmethylated CpGs. At least ten clones were picked randomly and sequenced from each of the three independent experiments.
Discussion
NaB has been used as dietary cancer chemopreventive phytochemicals blocking chemical-induced carcinogenesis through regulating NRF2 anti-oxidative signaling in normal cells [60]. However, the molecular mechanisms by which NaB regulate the mitochondrial metabolism, epigenomic/CpG methylation and transcriptomic as well as the role of NRF2 in regulating the CRC cell activities remain unclear. So, in our current study, we performed studies with metabolomics, epigenomic and transcriptomic to uncover underlying intricated biological connectivity between NaB-induced metabolomic, epigenomic and transcriptomic regulations, as well as the role of NRF2 signaling in NaB-mediated anti-cancer and cancer prevention effect in human CRC cells.
The KEAP1-NRF2 signaling is one of primary pathway in maintaining cellular homeostasis in order to respond adaptively to oxidative stress. Under basal condition, NRF2 interacts with two molecules of KEAP1 to activate Cullin 3 (Cul3)-based E3 ligase complex-mediated NRF2 ubiquitination reaction. Once NRF2 is ubiquitinated, it will be degraded and maintained at a low level in the cytoplasm [61]. When cells are exposed to oxidative stress or chemopreventive agents, the cysteine residues of KEAP1 are modified which can promote the detachment of NRF2 from KEAP1 and allow the NRF2 to translocate into nucleus and binds to ARE in the upstream promoter region of multiple genes [14]. In cancer cells, aberrant activation of NRF2 have been widely reported especially in CRCs and it’s highly associate with KEAP1 expression levels. NRF2 overexpression in cancers further promote cell proliferation and enhance the chemoresistance of cancer cells [14]. Previous study reported 8 of 10 CRC cell lines had hypermethylated CpG islands in the KEAP1 promoter region. HT29 CRC cells with a hypermethylated KEAP1 promoter resulted in decreased mRNA level. These results suggested that methylation of the KEAP1 promoter regulates its mRNA level. Furthermore, aberrant KEAP1 promoter methylation was detected in 53% of tumor tissues from 40 surgical CRC specimens, indicating that cancerous tissue showed increased methylation of the KEAP1 promoter region, conferring a protective effect against cytotoxic anticancer drugs [51]. In this context, NaB significantly decreased the KEAP1 promoter methylation (Figure 7) which can promote the KEAP1 mRNA and protein expression levels and then further decrease the NRF2 mRNA and protein levels as well as the NRF2-ARE signaling pathway (Figure 1), and contribute to the NaB-mediated cancer prevention/therapy. Based on these, NaB (as a NRF2 inhibitor in advance cancers) also can be used in combination with other chemotherapy agents to alleviate the chemoresistance and strengthen the anti-cancer effects. Interestingly, NRF2 also has been reported to modulate glutathione (GSH) to glutathione disulfide (GSSG) conversion by regulating glutathione reductase (GSR) to maintain the redox state [62]. Previous clinical trial [34] treated100 mM of NaB (daily intestinal content is ~9-10L, so the NaB concentration is ~10 mM in human intestinal lumen which is covered by our in vitro experiments) in human subject showed NaB decreased the GSH/GSSG ratio, an oxidative stress index, by 26% which are comparable with our metabolic results that 5mM and 10mM NaB decreased the GSH/GSGS ratios by 39% and 48% respectively (Supplemental Figure S1). Beside the dosage, these results also indicate the potential translation of the research data from in vitro to in vivo in current study.
In addition to the regulation effect on NRF2 signaling, NaB also upregulates the tumor suppressor gene ABCA1 and downregulates the tumor promote gene EGR3 potentially through modulating their corresponding promote methylation (Supplemental File 4). Previous studies showed ABCA1 is a known tumor suppressor and downregulation of ABCA1 expression causes high intracellular cholesterol levels, which creates an environment conducive to tumor progression [63]. ABCA1 transporter is directly suppressed by miR-200b-3p and upregulation of miR-200b-3p was also observed in cancers. Therefore, it is proposed that one of the mechanisms of cancer cell proliferation and metastasis in cancers is via miR-200-3p-directed inhibition of the ABCA1 transporter [64]. Moreover, Arnon et al, reported EGR3 has been implicated in cancer cell migration which makes it highly associated with tumor progression and it’s expression level also have been used as a prognostic marker in various cancers including CRC [65]. In colon cancer cell lines, EGR3 has binding sites in several genes related to the cancer therapy agent 5-fluorouracil resistance which support that EGR3 may have a protective function against chemotherapy [66]. Moreover, previous studies indicated that that c-MYC amplification and overexpression was showed in approximately 10 and 70 % in CRC, respectively [67, 68]. And c-MYC gene can promote tumorigenesis in CRC, mediate the critical role in the CRC progression and highly involve in chemotherapy resistance [69]. Deregulation of c-MYC by NaB (Figure 2f) would be promising to against CRC and alleviat chemoresistance. So, the findings in current study may indicate that NaB can be used as a potential cancer prevention therapeutics or in combination with other anti-cancer drugs for cancer therapy, via regulation of the cancer markers.
Recent evidence suggests strong molecular link between metabolic rewiring and epigenetic reprogramming through key metabolic intermediates, such as SAM, aKG and AcCoA, which can be used as targets for cancer prevention/therapy [70]. In cancer cells, aberrant mutations activate oncogenes or inactivate tumor suppressor genes may affect multiple signaling pathways that results in mitochondrial metabolic reprogramming and altered bioenergetics [71]. Moreover, reactive oxygen species (ROS) are known as an important progenitors in carcinogenesis, including CRC [72]. ROS are commonly higher in CRC cells than their normal counterpart cells [73] which has been reported to induced cellular oxidative stress and regulate multiple redox signaling pathways that ultimately impacts on cellular metabolic homeostasis [74]. In current study, NaB regulates metabolic pathways such as TCA cycle and methionine metabolism as well as the metabolites like SAM, SAM/SAH, AcCoA in HCT116 cells (Figure 4-6). These metabolites are tightly linked to the basic epigenetic machinery DNA/histone modifications, chromatin remodeling and modulating phenotypic gene expression with high connectivity between mitochondrial metabolites, and epigenetics [6, 8, 75]. Specifically, SAM is the universal donor of methyl groups to both DNA and histone methyltransferase enzymes, and the changes in methionine metabolism altering levels of SAM can directly influence trimethylation of H3K4 (Histone H3 lysine K4) and consequently regulates gene expression [76]. The intermediary metabolite SAH is the byproduct of SAM during methyltransferase reactions [57], which is a potent inhibitor of both DNA and histone methyltransferases. Thereby, the SAM/SAH ratio serves as a biosensor of the cellular metabolic state influencing the activity of methyltransferase enzymes that culminate in chromatin changes in response to cellular alternations [58]. The methyl group isotope labeling study showed that NaB decreased transfer potential of methyl groups to DNA and histones (Figure 6b). Besides the metabolic regulation, NaB also modulates the epigenetic regulatory enzymes/genes such as DNMT1 (Figure 6c), HAT1, KDM1A, KDM1B and TET1 (Figure 5d). These results indicated that NaB coordinately regulates epigenetic associated genes and metabolites which can further reveal the underlying mechanism of NaB-mediated cancer prevention/treatment effect.
In conclusion, our study integrates the latest Methyl-seq, RNA-seq and isotope labeling LC/MS/MS technologies in dissecting the potential underlying mechanism of epigenomic CpG methylation, mRNA transcriptomic gene expression and mitochondrial metabolic rewiring in HCT116 CRC cells after exposure to NaB. The results provide the experimental evidence for the metabolomic, epigenomic, and transcriptomic effects of NaB in CRC cells. We observed that NaB rewires metabolic profile, reprograms epigenetic CpG methylation, driving alterations of phenotypic gene expression, de-/activated signaling pathways in HCT116 cells. NaB decreased NRF2 gene expression through regulating the KEAP1 promoter methylation, which further regulates NRF2-target genes and contribute the NaB-mediated cancer prevention/therapy. Thus, the anticancer potential of NaB is exhibited through regulation of KEAP1/NRF2-ARE pathways, metabolic rewiring and epigenetic reprogramming in HCT116 cells, making it a promising drug candidate for the prevention and treatment of CRC.
Supplementary Material
Supplemental File 1 NaB regulated DMRs filtered with cutoff methylation difference ≥ 10%.
Supplemental Figure S1: GSH/GSSG ratios after treated with different concentrations of NaB in HCT116 cells. The GSH and GSSH were quantified by LCMS in metabolic experiments. The data is presented as the means ± SEM of three independent experiments. *, P < 0.05 and **, P < 0.01 indicate significant differences between the treatment groups and the control group (0.1% DMSO). Student’s t test was used to calculate the significance of the differences compared with the control.
{kind=link}
Supplemental File 2 NaB regulated DEGs filtered with cutoff p-value < 0.01 coupled to log2 fold change ≥ 2.0 or ≤ −2.0
Supplemental File 3 Top regulated signaling pathways in HCT116 cells after NaB treatment exported from Ingenuity Pathway Analysis (IPA).
Supplemental File 4 Correlated genes which possess negatively DNA methylation and gene expression patterns after NaB treatment filtered by integration of RNA-seq and Methyl-seq analysis.
Acknowledgements:
We thank all the members of Professor Ah-Ng Kong’s laboratory for their invaluable discussion and technical support for preparation of this manuscript.
Funding:
This work was supported in part by institutional funds and by R01 AT009152 from the National Center for Complementary and Integrative Health (NCCIH), and P30 ES005022 from the National Institute of Environmental Health (NIEHS).
Abbreviations:
- B
Butyrate
- NaB
Sodium Butyrate
- CRC
colorectal cancer
- NGS
next generation sequencing
- DMRs
differentially methylated regions
- DEGs
differentially expressed genes
- TCA
tricarboxylic acid
- aKG
α-ketoglutarate
- AcCoA
acetyl-CoA
- SAM
S-adenosyl methionine
- NAD
nicotinamide adenine dinucleotide
- HDACs
histone deacetylases
- HDACi
HDAC inhibitor
- NRF2
Nuclear Factor Erythroid-2 like 2
- KEAP1
Kelch-like-ECH-associated protein 1
- HO1
Heme Oxygenase 1
- NQO1
NAD(P)H dehydrogenase [quinone] 1
- ARE
Antioxidant Response Element
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- DNMT1
DNA Methyltransferase 1
- HAT1
Histone Acetyltransferase 1
- KDM1A
Lysine Demethylase 1A
- KDM1B
Lysine Demethylase 1B
- TET1
Tet Methylcytosine Dioxygenase 1
- ABCA1
ATP binding cassette A1
- EGR3
Early growth response protein 3
Footnotes
Conflict of Interest: The authors declared no competing interests for this work.
Reference
- [1].Wu R, Wang L, Yin R, Hudlikar R, Li S, Kuo HD, Peter R, Sargsyan D, Guo Y, Liu X, Kong AN, Epigenetics/epigenomics and prevention by curcumin of early stages of inflammatory-driven colon cancer. Mol Carcinog 2020, 59, 227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Wang T, Maden SK, Luebeck GE, Li CI, Newcomb PA, Ulrich CM, Joo J-HE, Buchanan DD, Milne RL, Southey MC, Carter KT, Willbanks AR, Luo Y, Yu M, Grady WM, Dysfunctional epigenetic aging of the normal colon and colorectal cancer risk. Clinical Epigenetics 2020, 12, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].McKay J, Mathers JC J. A. p, Diet induced epigenetic changes and their implications for health. 2011, 202, 103–118. [DOI] [PubMed] [Google Scholar]
- [4].Robertson KD, DNA methylation and human disease. Nat Rev Genet 2005, 6, 597–610. [DOI] [PubMed] [Google Scholar]
- [5].Anastasiadi D, Esteve-Codina A, Piferrer F, Consistent inverse correlation between DNA methylation of the first intron and gene expression across tissues and species. Epigenetics & Chromatin 2018, 11, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Janke R, Dodson AE, Rine J, Metabolism and Epigenetics. Annual Review of Cell and Developmental Biology 2015, 31, 473–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Su X, Wellen KE, Rabinowitz JD, Metabolic control of methylation and acetylation. Curr Opin Chem Biol 2016, 30, 52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cyr AR, Domann FE, The redox basis of epigenetic modifications: from mechanisms to functional consequences. Antioxid Redox Signal 2011, 15, 551–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wu R, Li S, Hudlikar R, Wang L, Shannar A, Peter R, Chou PJ, Kuo H-CD, Liu Z, Kong A-N, Redox signaling, mitochondrial metabolism, epigenetics and redox active phytochemicals. Free Radical Biology and Medicine 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Locato V, Cimini S, De Gara L, ROS and redox balance as multifaceted players of cross-tolerance: Epigenetic and retrograde control of gene expression. Journal of experimental botany 2018, 69. [DOI] [PubMed] [Google Scholar]
- [11].Wu R, Wang L, Yin R, Hudlikar R, Li S, Kuo H-CD, Peter R, Sargsyan D, Guo Y, Liu X, Kong AN, Epigenetics/epigenomics and prevention by curcumin of early stages of inflammatory-driven colon cancer. Molecular Carcinogenesis 2020, 59, 227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Baird L, Yamamoto M, The Molecular Mechanisms Regulating the KEAP1-NRF2 Pathway. Mol Cell Biol 2020, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gonzalez-Donquiles C, Alonso-Molero J, Fernandez-Villa T, Vilorio-Marques L, Molina AJ, Martin V, The NRF2 transcription factor plays a dual role in colorectal cancer: A systematic review. PLoS One 2017, 12, e0177549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wu S, Lu H, Bai Y, Nrf2 in cancers: A double-edged sword. Cancer Med 2019, 8, 2252–2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sadeghi MR, Jeddi F, Soozangar N, Somi MH, Samadi N, The role of Nrf2-Keap1 axis in colorectal cancer, progression, and chemoresistance. 2017, 39, 1010428317705510. [DOI] [PubMed] [Google Scholar]
- [16].Lee YJ, Kim WI, Bae JH, Cho MK, Lee SH, Nam HS, Choi IH, Cho SW, Overexpression of Nrf2 promotes colon cancer progression via ERK and AKT signaling pathways. Ann Surg Treat Res 2020, 98, 159–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bialk P, Wang Y, Banas K, Kmiec EB, Functional Gene Knockout of NRF2 Increases Chemosensitivity of Human Lung Cancer A549 Cells In Vitro and in a Xenograft Mouse Model. Mol Ther Oncolytics 2018, 11, 75–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hayes JD, Dinkova-Kostova AT, The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends in Biochemical Sciences 2014, 39, 199–218. [DOI] [PubMed] [Google Scholar]
- [19].Ting-Yu Lin, L. C. C. a. G. M. D., NRF2 Rewires Cellular Metabolism to Support the Antioxidant Response, A Master Regulator of Oxidative Stress - The Transcription Factor Nrf2. Jose Antonio Morales-Gonzalez, Angel Morales-Gonzalez and Eduardo Osiris Madrigal-Santillan December 21st 2016. [Google Scholar]
- [20].den Besten G, van Eunen K, Groen AK, Venema K, Reijngoud DJ, Bakker BM, The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J Lipid Res 2013, 54, 2325–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Louis P, Flint HJ, Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol Lett 2009, 294, 1–8. [DOI] [PubMed] [Google Scholar]
- [22].Silva YP, Bernardi A, Frozza RL, The Role of Short-Chain Fatty Acids From Gut Microbiota in Gut-Brain Communication. 2020, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wang W, Fang D, Zhang H, Xue J, Wangchuk D, Du J, Jiang L, Sodium Butyrate Selectively Kills Cancer Cells and Inhibits Migration in Colorectal Cancer by Targeting Thioredoxin-1. Onco Targets Ther 2020, 13, 4691–4704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Azcarate-Peril MA, Sikes M, Bruno-Barcena JM, The intestinal microbiota, gastrointestinal environment and colorectal cancer: a putative role for probiotics in prevention of colorectal cancer? Am J Physiol Gastrointest Liver Physiol 2011, 301, G401–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Canani RB, Costanzo MD, Leone L, Pedata M, Meli R, Calignano A, Potential beneficial effects of butyrate in intestinal and extraintestinal diseases. World J Gastroenterol 2011, 17, 1519–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Scheppach W, Weiler F, The butyrate story: old wine in new bottles? Curr Opin Clin Nutr Metab Care 2004, 7, 563–567. [DOI] [PubMed] [Google Scholar]
- [27].Hajjar R, Richard CS, Santos MM, The role of butyrate in surgical and oncological outcomes in colorectal cancer. 2021, 320, G601–G608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Berni Canani R, Di Costanzo M, Leone L, The epigenetic effects of butyrate: potential therapeutic implications for clinical practice. Clinical Epigenetics 2012, 4, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kasubuchi M, Hasegawa S, Hiramatsu T, Ichimura A, Kimura I, Dietary gut microbial metabolites, short-chain fatty acids, and host metabolic regulation. Nutrients 2015, 7, 2839–2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Valvassori S, Calixto K, Budni J, Resende W, Varela R, Freitas K, Gonçalves C, Streck E, Quevedo J, Sodium butyrate reverses the inhibition of Krebs cycle enzymes induced by amphetamine in the rat brain. Journal of neural transmission (Vienna, Austria : 1996) 2013, 120. [DOI] [PubMed] [Google Scholar]
- [31].Yu R, Mandlekar S, Lei W, Fahl WE, Tan TH, Kong AN, p38 mitogen-activated protein kinase negatively regulates the induction of phase II drug-metabolizing enzymes that detoxify carcinogens. J Biol Chem 2000, 275, 2322–2327. [DOI] [PubMed] [Google Scholar]
- [32].Wang L, Wu R, Sargsyan D, Su S, Kuo HC, Li S, Chou P, Sarwar MS, Phadnis A, Wang Y, Su X, Kong AN, Nfe2l2 Regulates Metabolic Rewiring and Epigenetic Reprogramming in Mediating Cancer Protective Effect by Fucoxanthin. AAPS J 2022, 24, 30. [DOI] [PubMed] [Google Scholar]
- [33].Banasiewicz T, Domagalska D, Borycka-Kiciak K, Rydzewska G, Determination of butyric acid dosage based on clinical and experimental studies - a literature review. Prz Gastroenterol 2020, 15, 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hamer HM, Jonkers DMAE, Vanhoutvin SALW, Troost FJ, Rijkers G, de Bruïne A, Bast A, Venema K, Brummer R-JM, Effect of butyrate enemas on inflammation and antioxidant status in the colonic mucosa of patients with ulcerative colitis in remission. Clinical Nutrition 2010, 29, 738–744. [DOI] [PubMed] [Google Scholar]
- [35].Jahns F, Wilhelm A, Jablonowski N, Mothes H, Radeva M, Wolfert A, Greulich KO, Glei M, Butyrate suppresses mRNA increase of osteopontin and cyclooxygenase-2 in human colon tumor tissue. Carcinogenesis 2011, 32, 913–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Li S, Li W, Wang C, Wu R, Yin R, Kuo HC, Wang L, Kong AN, Pelargonidin reduces the TPA induced transformation of mouse epidermal cells -potential involvement of Nrf2 promoter demethylation. Chem Biol Interact 2019, 309, 108701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Prawan A, Keum YS, Khor TO, Yu S, Nair S, Li W, Hu L, Kong AN, Structural influence of isothiocyanates on the antioxidant response element (ARE)-mediated heme oxygenase-1 (HO-1) expression. Pharm Res 2008, 25, 836–844. [DOI] [PubMed] [Google Scholar]
- [38].Wang C, Wu R, Sargsyan D, Zheng M, Li S, Yin R, Su S, Raskin I, Kong A-N, CpG methyl-seq and RNA-seq epigenomic and transcriptomic studies on the preventive effects of Moringa isothiocyanate in mouse epidermal JB6 cells induced by the tumor promoter TPA. The Journal of Nutritional Biochemistry 2019, 68, 69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hudlikar RR, Sargsyan D, Li W, Wu R, Zheng M, Kong A-N, Epigenomic, Transcriptomic, and Protective Effect of Carotenoid Fucoxanthin in High Glucose-Induced Oxidative Stress in Mes13 Kidney Mesangial Cells. Chemical Research in Toxicology 2021, 34, 713–722. [DOI] [PubMed] [Google Scholar]
- [40].Guo Y, Su ZY, Zhang C, Gaspar JM, Wang R, Hart RP, Verzi MP, Kong AN, Mechanisms of colitis-accelerated colon carcinogenesis and its prevention with the combination of aspirin and curcumin: Transcriptomic analysis using RNA-seq. Biochem Pharmacol 2017, 135, 22–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Martin M, Cutadapt removes adapter sequences from high-throughput sequencing reads. 2011 2011, 17, 3 %J EMBnet.journal. [Google Scholar]
- [42].Kim D, Langmead B, Salzberg SL, HISAT: a fast spliced aligner with low memory requirements. Nature Methods 2015, 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Liao Y, Smyth GK, Shi W, featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [DOI] [PubMed] [Google Scholar]
- [44].Wang L, Feng Z, Wang X, Wang X, Zhang X, DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [DOI] [PubMed] [Google Scholar]
- [45].Gaspar JM, Hart RP, DMRfinder: efficiently identifying differentially methylated regions from MethylC-seq data. BMC Bioinformatics 2017, 18, 528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Yu G, Wang L-G, He Q-Y, ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015, 31, 2382–2383. [DOI] [PubMed] [Google Scholar]
- [47].Hudlikar RR, Sargsyan D, Cheng D, Kuo HD, Wu R, Su X, Kong AN, Tobacco carcinogen 4-[Methyl(nitroso)amino]-1-(3-pyridinyl)-1-butanone (NNK) drives metabolic rewiring and epigenetic reprograming in A/J mice lung cancer model and prevention with Diallyl Sulphide (DAS). Carcinogenesis 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Li S, Wu R, Wang L, Dina Kuo HC, Sargsyan D, Zheng X, Wang Y, Su X, Kong AN, Triterpenoid ursolic acid drives metabolic rewiring and epigenetic reprogramming in treatment/prevention of human prostate cancer. Mol Carcinog 2022, 61, 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Dragan M, Nguyen M-U, Guzman S, Goertzen C, Brackstone M, Dhillo WS, Bech PR, Clarke S, Abbara A, Tuck AB, Hess DA, Pine SR, Zong W-X, Wondisford FE, Su X, Babwah AV, Bhattacharya M, G protein-coupled kisspeptin receptor induces metabolic reprograming and tumorigenesis in estrogen receptor-negative breast cancer. Cell Death & Disease 2020, 11, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Wang R, An J, Ji F, Jiao H, Sun H, Zhou D, Hypermethylation of the Keap1 gene in human lung cancer cell lines and lung cancer tissues. Biochem Biophys Res Commun 2008, 373, 151–154. [DOI] [PubMed] [Google Scholar]
- [51].Hanada N, Takahata T, Zhou Q, Ye X, Sun R, Itoh J, Ishiguro A, Kijima H, Mimura J, Itoh K, Fukuda S, Saijo Y, Methylation of the KEAP1 gene promoter region in human colorectal cancer. BMC Cancer 2012, 12, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Jiminez Janelle A, Uwiera Trina C, Abbott DW, Uwiera Richard RE, Inglis GD, Suen G, Butyrate Supplementation at High Concentrations Alters Enteric Bacterial Communities and Reduces Intestinal Inflammation in Mice Infected with Citrobacter rodentium. mSphere, 2, e00243–00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Korsten SGPJ, Smits EAW, Garssen J, Vromans H, Modeling of the luminal butyrate concentration to design an oral formulation capable of achieving a pharmaceutical response. PharmaNutrition 2019, 10, 100166. [Google Scholar]
- [54].Tamargo-Gomez I, Marino G, AMPK: Regulation of Metabolic Dynamics in the Context of Autophagy. Int J Mol Sci 2018, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Janke R, Dodson AE, Rine J, Metabolism and epigenetics. Annu Rev Cell Dev Biol 2015, 31, 473–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Feinberg AP, The Key Role of Epigenetics in Human Disease Prevention and Mitigation. 2018, 378, 1323–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Gut P, Verdin E, The nexus of chromatin regulation and intermediary metabolism. Nature 2013, 502, 489–498. [DOI] [PubMed] [Google Scholar]
- [58].Shyh-Chang N, Locasale JW, Lyssiotis CA, Zheng Y, Teo RY, Ratanasirintrawoot S, Zhang J, Onder T, Unternaehrer JJ, Zhu H, Asara JM, Daley GQ, Cantley LC, Influence of threonine metabolism on S-adenosylmethionine and histone methylation. Science 2013, 339, 222–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Zhang H, Liu Z, Ma S, Zhang H, Kong F, He Y, Yang X, Wang Y, Xu H, Yang A, Tian J, Zhang M, Cao J, Jiang Y, Guo X, Ratio of S-adenosylmethionine to S-adenosylhomocysteine as a sensitive indicator of atherosclerosis. Mol Med Rep 2016, 14, 289–300. [DOI] [PubMed] [Google Scholar]
- [60].Dong W, Jia Y, Liu X, Zhang H, Li T, Huang W, Chen X, Wang F, Sun W, Wu H, Sodium butyrate activates NRF2 to ameliorate diabetic nephropathy possibly via inhibition of HDAC. J Endocrinol 2017, 232, 71–83. [DOI] [PubMed] [Google Scholar]
- [61].Bellezza I, Giambanco I, Minelli A, Donato R, Nrf2-Keap1 signaling in oxidative and reductive stress. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 2018, 1865, 721–733. [DOI] [PubMed] [Google Scholar]
- [62].Harvey CJ, Thimmulappa RK, Singh A, Blake DJ, Ling G, Wakabayashi N, Fujii J, Myers A, Biswal S, Nrf2-regulated glutathione recycling independent of biosynthesis is critical for cell survival during oxidative stress. Free Radic Biol Med 2009, 46, 443–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Solomon KR, Allott EH, Freeman MR, Freedland SJ, Words of wisdom. Re: Dysregulation of cholesterol homeostasis in human prostate cancer through loss of ABCA1. Eur Urol 2013, 63, 1128–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Liu K, Zhang W, Tan J, Ma J, Zhao J, MiR-200b-3p Functions as an Oncogene by Targeting ABCA1 in Lung Adenocarcinoma. Technol Cancer Res Treat 2019, 18, 1533033819892590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Knudsen AM, Eilertsen I, Kielland S, Pedersen MW, Sørensen MD, Dahlrot RH, Boldt HB, Munthe S, Poulsen FR, Kristensen BW, Expression and prognostic value of the transcription factors EGR1 and EGR3 in gliomas. Scientific Reports 2020, 10, 9285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Szoke D, Gyorffy A, Surowiak P, Tulassay Z, Dietel M, Gyorffy B, Identification of consensus genes and key regulatory elements in 5-fluorouracil resistance in gastric and colon cancer. Onkologie 2007, 30, 421–426. [DOI] [PubMed] [Google Scholar]
- [67].Rochlitz CF, Herrmann R, de Kant E, Overexpression and amplification of c-myc during progression of human colorectal cancer. Oncology 1996, 53, 448–454. [DOI] [PubMed] [Google Scholar]
- [68].Lee KS, Kwak Y, Nam KH, Kim D-W, Kang S-B, Choe G, Kim WH, Lee HS, Favorable prognosis in colorectal cancer patients with co-expression of c-MYC and ß-catenin. BMC Cancer 2016, 16, 730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Strippoli A, Cocomazzi A, Basso M, Cenci T, Ricci R, Pierconti F, Cassano A, Fiorentino V, Barone C, Bria E, Ricci-Vitiani L, Tortora G, Larocca LM, Martini M, c-MYC Expression Is a Possible Keystone in the Colorectal Cancer Resistance to EGFR Inhibitors. Cancers (Basel) 2020, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Locato V, Cimini S, De Gara L, ROS and redox balance as multifaceted players of cross-tolerance: epigenetic and retrograde control of gene expression. J Exp Bot 2018, 69, 3373–3391. [DOI] [PubMed] [Google Scholar]
- [71].Sun X, Wang M, Wang M, Yu X, Guo J, Sun T, Li X, Yao L, Dong H, Xu Y, Metabolic Reprogramming in Triple-Negative Breast Cancer. 2020, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Basak D, Uddin MN, Hancock J, The Role of Oxidative Stress and Its Counteractive Utility in Colorectal Cancer (CRC). Cancers (Basel) 2020, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Lin S, Li Y, Zamyatnin AA Jr., Werner J, Bazhin AV, Reactive oxygen species and colorectal cancer. J Cell Physiol 2018, 233, 5119–5132. [DOI] [PubMed] [Google Scholar]
- [74].Marengo B, Nitti M, Furfaro AL, Colla R, Ciucis CD, Marinari UM, Pronzato MA, Traverso N, Domenicotti C, Redox Homeostasis and Cellular Antioxidant Systems: Crucial Players in Cancer Growth and Therapy. Oxid Med Cell Longev 2016, 2016, 6235641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Hudlikar R, Wang L, Wu R, Li S, Peter R, Shannar A, Chou PJ, Liu X, Liu Z, Kuo HD, Kong AN, Epigenetics/epigenomics and prevention of early stages of cancer by isothiocyanates. Cancer Prev Res (Phila) 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Mentch SJ, Mehrmohamadi M, Huang L, Liu X, Gupta D, Mattocks D, Gomez Padilla P, Ables G, Bamman MM, Thalacker-Mercer AE, Nichenametla SN, Locasale JW, Histone Methylation Dynamics and Gene Regulation Occur through the Sensing of One-Carbon Metabolism. Cell Metab 2015, 22, 861–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental File 1 NaB regulated DMRs filtered with cutoff methylation difference ≥ 10%.
Supplemental Figure S1: GSH/GSSG ratios after treated with different concentrations of NaB in HCT116 cells. The GSH and GSSH were quantified by LCMS in metabolic experiments. The data is presented as the means ± SEM of three independent experiments. *, P < 0.05 and **, P < 0.01 indicate significant differences between the treatment groups and the control group (0.1% DMSO). Student’s t test was used to calculate the significance of the differences compared with the control.
Supplemental File 2 NaB regulated DEGs filtered with cutoff p-value < 0.01 coupled to log2 fold change ≥ 2.0 or ≤ −2.0
Supplemental File 3 Top regulated signaling pathways in HCT116 cells after NaB treatment exported from Ingenuity Pathway Analysis (IPA).
Supplemental File 4 Correlated genes which possess negatively DNA methylation and gene expression patterns after NaB treatment filtered by integration of RNA-seq and Methyl-seq analysis.