

Abstract
Background
Arteriovenous fistulae (AVF) are the gold standard for vascular access for hemodialysis. Although the vein must thicken and dilate for successful hemodialysis, excessive wall thickness leads to stenosis causing AVF failure. Since transforming growth factor-β (TGF-β) regulates extracellular matrix deposition and smooth muscle cell (SMC) proliferation, critical components of wall thickness, we hypothesized that disruption of TGF-β signaling prevents excessive wall thickening during venous remodeling.
Methods
A mouse aortocaval fistula model was used. SB431542, an inhibitor of TGF-β receptor I, was encapsulated in nanoparticles and applied to the AVF adventitia in C57BL/6J mice. Alternatively, AVF were created in mice with conditional disruption of TGF-β receptors in either SMC or endothelial cells. Doppler ultrasound was performed serially to confirm patency and to measure vessel diameters. AVF were harvested at predetermined time points for histological and immunofluorescence analyses.
Results
Inhibition of TGF-β signaling with SB431542-containing nanoparticles significantly reduced p-Smad2-positive cells in the AVF wall during the early maturation phase (days 7-21) and was associated with decreased AVF wall thickness that showed both decreased collagen density and decreased SMC proliferation. SMC-specific TGF-β signaling disruption decreased collagen density but not SMC proliferation or wall thickness. Endothelial cell-specific TGF-β signaling disruption decreased both collagen density and SMC proliferation in the AVF wall and was associated with reduced wall thickness, increased outward remodeling and improved AVF patency.
Conclusions
Endothelial cell-targeted TGF-β inhibition may be a translational strategy to improve AVF patency.
Keywords: arteriovenous fistula, transforming growth factor beta, extracellular matrix, smooth muscle cell, endothelial cell, nanoparticle
Subject terms: Animal Models of Human Disease, Basic Science Research, Vascular Biology, Vascular Disease
Graphical Abstract
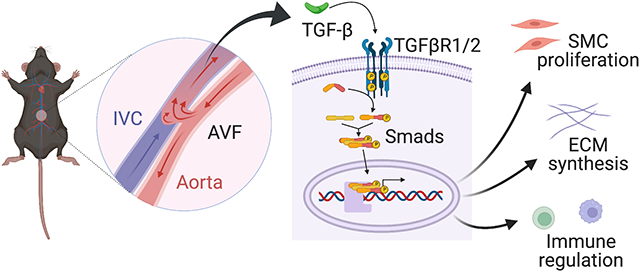
Introduction
There are approximately 750,000 patients with end-stage renal disease in the United States and these patient numbers continue to increase worldwide, requiring a large amount of resources for clinical care.1 Arteriovenous fistulae (AVF) currently remain the gold standard for hemodialysis vascular access owing to their superior patency and fewer complications compared to grafts or catheters.2,3 In order for AVF to be functional, the outflow vein must remodel to support the higher magnitude and pressure of arterial blood flow; the vein must thicken and dilate to adapt to the new fistula environment that includes repetitive punctures with large-bore needles.4 However, excessive wall thickness leads to stenosis and causes low rates of fistula utilization.5,6 Nonetheless, the mechanisms that regulate the venous remodeling to the fistula environment are not well defined.7-9
Transforming growth factor-β (TGF-β) is a multifunctional cytokine that modulates both extracellular matrix (ECM) deposition and smooth muscle cell (SMC) proliferation, both of which are important components of neointimal hyperplasia that lead to AVF stenosis.10-18 TGF-β expression is elevated in stenoses in human AVF,14 and genetic polymorphisms that express high amounts of TGF-β protein correlate with lower AVF patency;19 these data suggest that ‘excessive’ signaling by TGF-β leads to AVF stenosis. TGF-β is a major stimulator of synthesis of multiple components of the ECM via both Smad-dependent and independent pathways.10-12 TGF-β is also a context-dependent cytokine with differential effects on cell proliferation.16,17 As such, TGF-β may serve as a critical regulator of venous remodeling such as occurs during AVF maturation.
We have recently used a mouse aortocaval model that recapitulates human AVF maturation to show that TGF-β expression is significantly increased in the AVF wall during early remodeling.20,21 In addition, expression of TGF-β–activated kinase 1 (TAK1) is elevated in the AVF wall and correlates with wall thickness,13 suggesting that TGF-β signaling may regulate venous wall thickness. We hypothesized that the TGF-β pathway regulates wall thickening during AVF remodeling via regulation of both matrix components as well as cell composition; we directly tested this hypothesis using both pharmacological and cell type-specific genetic approaches.
Materials and Methods
The authors declare that all supporting data are available within the article.
Animals
Mice used for this study included wild type female and male C57BL/6J, male Myh11-CreERT2; Tgfbr2fl/fl; mT/mG or male Cdh5-CreERT2; Tgfbr1fl/fl; Tgfbr2fl/fl; mT/mG. C57BL/6J were purchased from The Jackson Laboratory (Bar Harbor, ME). Myh11-CreERT2; Tgfbr2fl/fl; mT/mG and Cdh5-CreERT2; Tgfbr1fl/fl; Tgfbr2fl/fl; mT/mG were generated as previously described.22,23 The bacterial artificial chromosome containing Myh11-CreERT2 inserts on the Y chromosome and female mice do not express the construct. Based on prior studies, female mice have similar fistula dilation and wall thickening as male mice during days 0-21 in this aorta-caval fistula model despite different AVF inflow shear stress.24
Cre-Lox recombination
For mutant strains, Cre-Lox recombination was induced by tamoxifen (Sigma-Aldrich) at 2 mg/d (~80 mg/kg/d) i.p. for 5 consecutive days starting at 2 weeks prior to surgery, while controls were treated with vehicle (100% corn oil) alone (Figure S1). Induction at a younger age was avoided due to the known risk of spontaneous aortic disease in Myh11-CreERT2; Tgfbr2fl/fl; mT/mG mice.22 These mice possess the mT/mG double-fluorescent reporter that ubiquitously expresses membrane-targeted tdTomato, a variant red fluorescent protein (RFP), except where excised by Cre to express membrane-targeted GFP. To confirm adequate Cre recombination with tamoxifen, the inferior vena cava (IVC) was harvested prior to injection or a day after the 1st, 3rd or 5th injection. Additionally, the IVC was harvested 14 days from the start of the injections when the mice would undergo AVF creation.
Animal Model
All animal experiments were performed in strict compliance with federal guidelines and with approval from the Institutional Animal Care and Use Committee of Yale University. Mice were 9–11 weeks of age when the surgeries were performed. Anesthesia was administered using 2% to 2.5% isoflurane. Briefly, after exposing the IVC and aorta, an aortocaval fistula was created by puncturing the distal aorta into the IVC using a 25-gauge needle, as previously described.20,25 Visualization of pulsatile arterial blood flow in the IVC was used to assess initial technical success of AVF creation.
Ultrasound measurement
Doppler ultrasound (Vevo 770 High-Resolution Imaging System; Fujifilm Visual Sonics Inc., Toronto, Canada) using probe RMV704 (40MHz) was performed at baseline and serially postoperatively to confirm the patency of the AVF, to obtain blood flow velocities and to measure the inner diameter of the aorta and AVF.20 Shear stress and blood flow were calculated from the values obtained by ultrasound using the Hagen-Poiseuille formula: T=4ηV/r, where T is shear stress, η is blood viscosity, V is flow velocity, and r is the radius. Blood viscosity was assumed to be constant at 0.035 poise.
Synthesis and characterization of nanoparticles
Poly(lactic-co-glycolic acid) nanoparticles were made encapsulating Rhodamine B (Sigma-Aldrich) or SB431542 (Abcam); see Data Supplement for details. SB431542-containing nanoparticles are hereafter referred to as NP-SB.
SB431542 treatment
C57BL/6J mice were treated with a single periadventitial application of NP-SB in pluronic gel (20 mg/kg of SB431542) immediately after AVF creation, prior to abdomen closure (Figure S1A, B); control groups were treated with unloaded nanoparticles in pluronic gel (NP-control). All groups received equal volume of the solution at 200 μl. Mice were randomly assigned to the groups.
Histology
Animals were euthanized and perfused with normal saline followed by 10% formalin via the left ventricle under physiological pressure and the AVF was extracted en bloc. The tissue was then embedded in paraffin and cut in 5-μm cross sections. Elastin Van Gieson staining was used to measure intima-media thickness, hematoxylin and eosin staining was used to count the number of medial cells, and trichrome and picrosirius red staining was used to measure collagen density in 5-μm cross sections of the IVC using sections obtained 50–200 μm cranial to the fistula. Eight equidistant points around the IVC wall were averaged in each cross section to obtain the mean AVF wall thickness which refers to both the intima and media, since in the veins of mice and rats there is only one elastic lamina that separates the media and the adventitia.26 Since this elastic lamina is not always discernable throughout the cross-sections, we considered the dark-stained layer alongside the vessel lumen, using Van Gieson’s staining, the intima-media layer.13,20,21 Additional unstained cross sections in this same region were used for immunofluorescence (IF) microscopy.
Immunofluorescence
Tissue sections were de-paraffined using xylene and rehydrated in a graded series of alcohols. Sections were heated in citric acid buffer (pH 6.0) at 100°C for 10 min for antigen retrieval. The sections were then blocked with 2% bovine serum albumin for 1 hour at room temperature, prior to incubation overnight at 4°C with the primary antibodies (Major resources Table). For negative controls, IgG isotype controls and endogenous tissue background controls were used. Sections were then treated with secondary antibodies at room temperature for 1 hour using Alexa Fluor 488-, 568- or 647-conjugated IgG (Life Technologies, Eugene, OR) and stained with 4′,6-diamidino-2-phenylindole (P36935, Invitrogen). For mice possessing the reporter gene, although mT/mG fluorescence was quenched and undetectable in paraffin sections with the aforementioned process (Figure S10C, D), Alexa Fluor 647 was carefully chosen as the secondary antibody for detection of the protein of interest. Positively staining cells were counted in the presence of well-defined nuclei per high power field or the mean intensity in the vessel wall was measured using ImageJ software (National Institutes of Health, Bethesda, MD). Four to eight high power field images were used to acquire the mean value per animal. For studies detecting mT/mG expression, animals were perfused with cold 4% paraformaldehyde, and the vessels were fixed overnight in 4% paraformaldehyde at 4°C, cryoprotected in 15% sucrose for 6 to 8 hours at 4°C, embedded in OCT, and 5-μm–thick sections were obtained.
Western Blot
Total protein in tissues or endothelial cells (EC) was extracted using radioimmunoprecipitation assay lysis buffer; protein concentrations were assessed using a colorimetric assay (Bio-Rad; Hercules, CA). For immunoblotting, proteins (20–40 μg) were separated on an 8% to 10% SDS-PAGE gel and electrophoretically transferred to a polyvinylidene difluoride microporous membrane (0.45 μm pore size; Immobilon, Millipore) and blocked with 5% bovine serum albumin for 1 hour at room temperature, then blotted with primary antibody (Major Resources Table) of targeted proteins overnight at 4°C. After incubation with horseradish peroxidase conjugated secondary antibody for 1 hour at room temperature, the membranes were developed using the Western Lightning Plus ECL reagent (PerkinElmer, Waltham, MA). Where needed, membranes were stripped with Restore Western Blot Stripping Buffer (Pierce Biotechnology, Rockford, IL) and reprobed as described above.
Human AVF Sample Collection
The principles outlined in the Declaration of Helsinki were followed. This study was approved by the Human Investigation Committee of VA Connecticut, and patients gave informed consent to use the samples. Deidentified vein and AVF samples were collected from patients with end-stage renal disease undergoing AVF creation or revision as previously described.13 Control veins and AVF were unmatched samples from different patients. The control veins were veins that did not have an AVF but were discarded as part of the operative procedure; the AVF were small segments of mature AVF that were harvested during surgical revision procedures for focal stenoses but did not include the actual lesion itself. Part of the samples were formalin fixed and paraffin embedded, and another part was frozen at −80°C.
Statistics
All data were analyzed using Prism 9 software (GraphPad Software, Inc., La Jolla, CA). Error bars represent the standard error of the mean. The Shapiro-Wilk test was performed to test for normality; the F test was performed to evaluate homogeneity of variances. For 2-group comparisons, the unpaired Student t test was used for normally distributed data. Welch’s correction was used for data with unequal variances. For multiple group comparisons with normally distributed data, the 1-way ANOVA followed by the Tukey post hoc test was used. For comparisons involving 2 factors, the 2-way ANOVA followed by the Sidak post hoc test was used. For data that were not normally distributed or when the sample sizes were smaller than 6, the nonparametric Mann-Whitney U test was used for 2-group comparisons, and the Kruskal-Wallis test followed by the Dunn multiple comparisons test was used for multiple groups. For 2-group comparisons of categorical values, Fischer’s exact test was used. Patency and survival were analyzed with Kaplan–Meier analysis. P values of ≤0.05 were considered to indicate statistical significance.
Results
Increased p-Smad2 in human and mouse AVF
To determine if canonical Smad-dependent signaling is present in mature AVF, we examined mature AVF specimens from human patients. Both p-Smad2 and Smad2 expression were significantly increased in the AVF of patients undergoing surgical revision compared to the control veins with no difference in the phosphorylated to total ratio (Figure 1A). This increased expression was confirmed using IF showing significantly more p-Smad2 positive cells throughout the AVF wall (Figure 1B).
Figure 1. Increased p-Smad2 in mature human and mouse arteriovenous fistula (AVF).

(A) Representative Western blot (WB) of p-Smad2 and Smad2 in human control vein and AVF with densitometry; p-Smad2: *P=0.0026 (t test), P=0.1000 (U test), Smad2: **P=0.0015 (t test), P=0.1000 (U test), p:t Smad2: P=0.0574 (t test), P=0.1000 (U test); n=3 per group. (B) Representative photomicrographs showing immunofluorescence (IF) of p-Smad2 (red), αSMA (green) and DAPI (blue) in human control vein and AVF wall, quantified as percentage of p-Smad2 positive cells in the AVF wall, * P=0.0020 (t test), P=0.1000 (U test); n=3 per group. (C) Representative WB of p-Smad2 and Smad2 in the AVF of mice harvested at day 21 after sham operation or AVF creation with densitometry. p-Smad2: *P=0.0011 (t test), P=0.1000 (U test), Smad2: **P=0.0122 (t test), P=0.1000 (U test), ***p:t Smad2: P=0.0348 (t test), P=0.1000 (U test); n=3 per group. (D) Representative photomicrographs showing IF of p-Smad2 (red), αSMA (green) and DAPI (blue) in the AVF wall at days 0 to 21 quantified as percentage of p-Smad2 positive cells in the AVF wall; P=0.0194 (ANOVA), P=0.0403 (Kruskal-Wallis), * P=0.0205 (day 0 vs 21, Tukey’s post hoc); n=4 per group. Four high power field images were used to acquire the mean value per animal (B, D). All data are represented as mean value±SEM. L, lumen. Scale bars, 20 μm.
Similar analyses were performed in a mouse AVF model that recapitulates human AVF maturation, with early vessel remodeling characterized by wall thickening and vessel dilation by day 21.20 Both p-Smad2 and Smad2 expression were significantly increased in the AVF at day 21 compared to the IVC of sham-operated male mice with significantly increased phosphorylated to total ratio (Figure 1C). TGF-β1 immunoreactivity showed a trend towards an increase postoperatively in the AVF wall (Figure S2A). Similarly, p-Smad2 immunoreactivity was present in several cell types in the AVF wall and the number of p-Smad2 positive cells significantly increased (Figure 1D) with no change in total Smad2 immunoreactivity postoperatively (Figure S2B). There was a similar trend in the immunoreactivity of these proteins in the AVF wall of female mice (Figure S3A-C). These results show that Smad2 is phosphorylated during both human and mouse venous remodeling.
TGF-β regulates venous remodeling
Since TGF-β expression and Smad2 phosphorylation increases in the remodeling AVF wall in mice (Figure 1, Figure S2-3), we utilized SB431542, an inhibitor of TGF-β receptor I, to inhibit TGF-β/Smad signaling. The efficacy of SB431542 to inhibit Smad2 phosphorylation was validated in vitro using mouse EC (Figure S4). To allow local drug delivery directly to the AVF, drugs were encapsuled in nanoparticles, dissolved in pluronic gel and were applied to the AVF adventitia once at the time of surgery; nanoparticle-based drug delivery enables sustained drug release in vivo avoiding repetitive administration and minimizing the required dose.27-29 The enhanced retention of the drug with this delivery system was validated using Rhodamine B-containing nanoparticles (Figure S5-7).
Mice received a single periadventitial application of either NP-SB (20 mg/kg of SB431542) or an equivalent amount of control nanoparticles (NP-control) immediately after AVF creation (Figure S1A, B) and were followed through postoperative day 42. There was no significant difference in body weight (Figure 2A) or survival (Figure 2B) between the groups, suggesting lack of toxicity of NP-SB. To assess the impact of NP-SB on TGF-β signaling, phosphorylation of Smad2/3 in the AVF wall was also assessed; as expected, the percentage of p-Smad2 (Figure 2C) and p-Smad3 (Figure 2D) immunoreactive cells showed a significant decrease with NP-SB treatment at days 7 and 21. However, there was no significant difference in the immunoreactivity of TAK1 (Figure 2E). These results suggest that inhibition of TGF-β signaling with SB431542 predominantly affects the canonical Smad-dependent pathway.
Figure 2. SB431542-containing nanoparticles (NP-SB) is associated with diminished arteriovenous fistula (AVF) wall thickness.

C57BL/6J mice were treated with NP-SB in pluronic gel once, immediately after AVF creation (day 0). Control mice were treated with unloaded nanoparticles in the same manner (NP-control). (A) Line graph showing the relative body weight. Values are normalized by preoperative measurement. P=0.2866 (ANOVA); n=15 for NP-control, n=13 for NP-SB. (B) Line graph showing survival, P=0.9969 (Log-rank); n=17 for NP-control, n=15 for NP-SB at day 0. (C) Representative photomicrographs showing immunofluorescence (IF) of p-Smad2 (red) merged with αSMA (green) and DAPI (blue) in AVF sections at days 7 and 21, quantified as percentage of p-Smad2-positive cells in the AVF wall; P<0.0001 (ANOVA); *P=0.0001 (day 7, Sidak’s post hoc), **P=0.0335 (day21, Sidak’s post hoc); n=4 per group. (D) Representative photomicrographs showing IF of p-Smad3 (red) merged with αSMA (green) and DAPI (blue) in AVF sections at days 7 and 21, quantified as percentage of p-Smad3-positive cells in the AVF wall; P=0.0010 (ANOVA); *P=0.0431 (day 7, Sidak’s post hoc); n=4 per group. (E) Representative photomicrographs showing IF of TAK1 (red) merged with αSMA (green) and DAPI (blue) in AVF sections at days 7 and 21, quantified as IF intensity of TAK1 in the AVF wall; P=0.3418 (ANOVA); n=4 per group. (F) Line graph showing the outward remodeling of the AVF and aorta. Values are normalized by preoperative measurement; AVF: P=0.9305; aorta: P=0.0977 (ANOVA); n=15 for NP-control, n=13 for NP-SB . (G) Representative photomicrographs of Van Gieson staining showing wall thickness (denoted by yellow arrowheads) of the AVF (day 21), and quantification of AVF wall thickness shown with baseline (day 0), P=0.0524 (t test), P=0.1061 (U test); n=5 for day 0 control, n=7 for NP-control, n=5 for NP-SB. (H) Line graph showing AVF patency. P= 0.7265 (Log-rank); n=17 for NP-control, n=15 for NP-SB at day 0. Four (C-E) or 8 (G) high power field images were used to acquire the mean value per animal. All data are represented as mean value±SEM. L, lumen; A.U., arbitrary unit. Scale bars, 20 μm.
There was no significant difference in outward remodeling of the fistula and the aorta (Figure 2F, Figure S8A-C) without any difference in aortic flow velocities, shear stress and blood flow (Figure S8D-G). The AVF wall thickness increased from baseline to day 21 in both groups (Figure 2G), compatible with the adaptive remodeling process of the AVF; however, there was a trend towards decreased wall thickness in mice treated with NP-SB compared to control (Figure 2G). There were no AVF failures from day 21 to 42 in mice treated with NP-SB (NP-SB: 0%, NP-control: 14.3%; P>0.9999, Figure S8H), without any significant difference in patency at day 42 (Figure 2H). These results show that TGF-β signaling disruption with NP-SB is associated with diminished AVF wall thickness.
TGF-β regulates SMC proliferation and ECM deposition
Since NP-SB treatment was associated with decreased AVF wall thickness (Figure 2G) and venous remodeling may be due to altered composition of the vascular wall, we determined whether the decreased wall thickness was attributable to changes in the vascular cells and/or ECM, both of which are regulated by the TGF-β pathway.10,11,14-18 The number of medial cells in the AVF wall increased from day 7 to 21 in both groups with a trend towards fewer cells in mice treated with NP-SB (Figure 3A). There was also a significant decrease of proliferative SMC at day 7 with NP-SB treatment (Figure 3B) but no difference in apoptotic SMC (Figure 3C). Since TGF-β stimulates PDGF-B production by endothelial cells and also causes FGF-2 release from subendothelial matrix,30 these potent mitogens of SMC were assessed; surprisingly, no change in immunoreactivity of PDGF-B or FGF-2 was detected with NP-SB (Figure S9). Since the TGF-β pathway regulates SMC differentiation,15-18 phenotypes of SMC were also assessed. Although there was no difference in the immunoreactivity of αSMA and Myh11 (contractile SMC markers) in the media, there was a trend towards decreased Myh10 and vimentin (proliferative SMC markers) immunoreactivity with NP-SB treatment (Figure 3D). These results suggest that TGF-β regulates cell proliferation in the remodeling AVF wall.
Figure 3. SB431542-containing nanoparticles (NP-SB) regulate SMC proliferation and ECM composition.

C57BL/6J mice were administered with NP-SB or unloaded nanoparticles (NP-control) in pluronic gel once, immediately after arteriovenous fistula (AVF) creation (day0). (A) Representative photomicrographs of hematoxylin and eosin staining of the AVF wall at days 7 and 21, and the total number of medial cells in the AVF wall per cross section (n/cs). Dashed lines denote the elastic lamina; P=0.0797 (ANOVA); n=4 (day 7) and 8 (day 21) for NP-control, n=4 (day 7) and 5 (day 21) for NP-SB. (B) Representative photomicrographs showing immunofluorescence (IF) of PCNA (red) merged with αSMA (green) and DAPI (blue) in AVF wall at days 7 and 21, quantified as percentage of PCNA-αSMA dual-positive cells in the AVF wall; P=0.0010 (ANOVA); *P=0.0062 (day 7, Sidak’s post hoc); n=4 each. (C) Representative photomicrographs showing IF of cleaved caspase-3 (red) merged with αSMA (green) and DAPI (blue) in AVF wall at days 7 and 21, quantified as percentage of cleaved caspase-3-positive cells in the AVF wall; P=0.7744 (ANOVA); n=4 each. (D) Representative photomicrographs showing IF of αSMA (red), Myh11 (red), Myh10 (red) or vimentin (red) merged with ICAM-1 (green) and DAPI (blue) in AVF wall (day 21), quantified as IF intensity in the medial layer; αSMA: P=0.7480 (t test with Welch’s correction), P>0.9999 (U test); Myh11: P>0.9999 (U test); Myh10: P=0.0593 (t test), P=0.2000 (U test); vimentin: P=0.1238 (t test with Welch’s correction), P=0.3429 (U test); n=4 each. (E) Representative photomicrographs of Masson’s trichrome staining of the AVF wall (day 21), quantified as percentage of collagen area within the AVF wall, *P=0.0496 (t test), P=0.0653 (U test); n=8 for NP-control, n=5 for NP-SB. (F) Representative photomicrographs of picrosirius red staining of the AVF wall (days 7 and 21) examined under polarized light. Mature collagens appear as strongly birefringent red-orange fibers and immature collagens as weakly birefringent green fibers. Bar graphs show percentage of immature or mature collagen area within the AVF wall; immature collagen: P=0.0188 (ANOVA), *P=0.0040 (day 7, Sidak post hoc); mature collagen: P=0.7491 (ANOVA); n=4 (day 7) and 8 (day 21) for NP-control, n=4 (day 7) and 5 (day 21) for NP-SB. (G) Representative photomicrographs showing IF of collagen type I (top, green), collagen type III (middle, green) or fibronectin (bottom, green) merged with DAPI (blue) in the AVF wall (day 7), quantified as IF intensity in the AVF wall; collagen type I : *P=0.0205 (t test), P=0.0571 (U test) ; collagen type III: P=0.4323 (t test), P=0.4857 (U test) ; fibronectin: P=0.5595 (t test with Welch’s correction), P=0.3429 (U test) ; n=4 each. High-magnification images of the AVF wall shown in boxed areas are shown to the lower-right. Four (B-F) or 8 (G) high power field images were used to acquire the mean value per animal. All data are represented as mean value±SEM. L, lumen; A.U., arbitrary unit. Scale bars, 20 μm.
We next determined whether NP-SB regulates deposition of several components of the ECM present in the remodeling AVF wall. Mice treated with NP-SB showed significantly decreased collagen density in the AVF wall (Figure 3E). Picrosirius red staining showed that immature collagens decreased whereas mature collagens increased from day 7 to 21, compatible with venous adaptive remodeling after AVF creation (Figure 3F); however, mice treated with NP-SB had increased percentage of immature collagens compared to control mice at day 7 (Figure 3F). Similarly, immunoreactivity of collagen type I was reduced in the AVF walls of mice treated with NP-SB (Figure 3G). These data suggest that the conversion of immature to mature collagen fibers in the AVF wall are inhibited with NP-SB early postoperatively, followed by a decrease in total collagenous fibers.
TGF-β regulates inflammatory cells
Since TGF-β modulates inflammatory responses via inhibition of T cells and regulation of macrophages,31,32 and we have previously shown that inflammatory cells regulate wall thickening during venous remodeling,21,33-35 we determined the effect of NP-SB on accumulation of several inflammatory cell types within the remodeling AVF wall. In mice treated with NP-control or NP-SB, the numbers of CD3-positive cells in the AVF wall increased from baseline to day 7 and then decreased by day 21 (Figure 4A). However, the numbers of CD3-positive cells were significantly increased with NP-SB treatment at day 7 (Figure 4A). The numbers of CD68-positive cells also increased from baseline to day 7 and then decreased by day 21 in both groups, compatible with our previous report;21 however, there were no difference in the numbers of CD68-positive cells between the groups (Figure 4A). Since TGF-β inhibits proliferation and differentiation of both helper T cells (Th1, Th2) and cytotoxic lymphocytes (CTL),36-38 these subtypes of CD3-positive cells were assessed at day 7. Both CD3/CD4 dual-positive (Th1, Th2) and CD3/CD8 dual-positive (CTL) cells were significantly increased in the AVF wall with NP-SB treatment (Figure 4B). Since TGF-β also stimulates regulatory T cell (Treg) polarization and Treg T cells inhibit Th1 and Th2 T cells,36-38 Treg T cells in the AVF wall were assessed (day 7). Although the number of CD4/Foxp3 dual-positive (Treg) cells did not differ between mice treated with NP-SB or NP-control, there was a trend towards a diminished ratio of Foxp3 positive- to negative- CD4-positive cells with NP-SB (NP-SB: 0.85, NP-control: 2.17; P=0.1122, Figure 4C).
Figure 4. SB431542-containing nanoparticles (NP-SB) regulate inflammatory cells.

C57BL/6J mice were administered with NP-SB or unloaded nanoparticles (NP-control) in pluronic gel once, immediately after arteriovenous fistula (AVF) creation (day0). (A) Representative photomicrographs showing immunofluorescence (IF) of CD3 (green) or CD68 (green) merged with αSMA (red) and DAPI (blue) in the AVF wall (day 7), quantified as the number of CD3 or CD68 positive cells in the AVF wall (days 0–21); CD3: P=0.0029 (ANOVA), *P=0.0025 (day 7, Sidak’s post hoc); CD68: P=0.9018 (ANOVA); n=4 each. (B) Representative photomicrographs showing IF of CD4 (red) or CD8 (red) merged with CD3 (green) and DAPI (blue) in the AVF wall (day 7), quantified as the number of dual-positive cells in the AVF wall; CD3+ CD4+: *P=0.0066 (t test), P=0.0286 (U test); CD3+ CD8+: *P=0.0072 (t test), P=0.0286 (U test); n=4 each. (C) Representative photomicrographs showing IF of Foxp3 (red) merged with CD4 (green) and DAPI (blue) in fistula wall (day 7), quantified as the number of Foxp3-positive (black square dot) or Foxp3-negative (white square dot), CD4-positive cells as well as the ratio of Foxp3-positive to Foxp3-negative CD4-positive cells in the AVF wall; CD4+ Foxp3+: P=0.4949 (t test), P=0.4857 (U test); CD4+ Foxp3−: * P=0.0280 (t test), P=0.0286 (U test); Foxp3-positive to negative ratio: P=0.1122 (t test), P=0.2000 (U test); n=4 each. (D, E) Representative photomicrographs showing IF of (D) IFNγR (red), iNOS (red), (E) CD206 (red), IL-10 (red) or TGM2 (red) merged with CD68 (green) and DAPI (blue) in the AVF wall (day 7), quantified as the number of dual-positive cells in the AVF wall; CD68+ IFNγR+: P=0.1662 (t test), P=0.2000 (U test); CD68+ iNOS+: P=0.2206 (t test), P=0.3429 (U test); CD68+ CD206+: *P=0.0024 (t test), P=0.0286 (U test); CD68+ IL-10+: P=0.0795 (t test), P=0.0571 (U test); CD68+ TGM2+: P=0.1256 (t test), P=0.1143 (U test); n=4 each. Four high power field images were used to acquire the mean value per animal. All data are represented as mean value±SEM. L, lumen; n/HPF, number per high power field. Scale bars, 20 μm.
T cells temporally precede macrophage accumulation in the AVF wall in our model,21 and since TGF-β signaling plays an important role in anti-inflammatory macrophage polarization,39,40 we also examined if there were changes in macrophage phenotypes. Although the numbers of CD68/IFNγ-receptor (pro-inflammatory) and CD68/inducible nitric oxide synthase (iNOS, pro-inflammatory) dual-positive cells were similar in the AVF wall (Figure 4D), there was a significant decrease of CD68/CD206 (anti-inflammatory) dual-positive cells, a trend towards a decrease of CD68/IL-10 (anti-inflammatory) and similar CD68/transglutaminase 2 (TGM2, anti-inflammatory) dual-positive cells with NP-SB (Figure 4E). In toto, these data suggest that the inhibitory effect of TGF-β with NP-SB treatment resulted in decreased polarization of Treg cells, leading to increased numbers of CD3-positive T cells as well as decreased polarization of anti-inflammatory macrophages, suggesting that TGF-β regulates inflammation in the remodeling AVF wall.
Inhibition of TGF-β signaling in SMC has minimal impact on AVF maturation
TGF-β is a context-dependent cytokine and its effect on SMC proliferation may depend on cell type.15 Since medial SMC are one of the largest constituent cell types of the vascular wall (Figure 3A), we investigated the effect of SMC-specific TGF-β inhibition in the AVF. Myh11-CreERT2; Tgfbr2fl/fl; mT/mG mice were treated with tamoxifen, while control litters were treated with corn oil vehicle, 2 weeks prior to AVF creation (Figure S1C). Adequate Cre recombination was confirmed with the mT/mG reporter (Figure S10A).
Mice with SMC-specific Tgfbr2 depletion showed similar body weight change and survival after surgery as compared to the control mice (Figure S11A, B). We next examined how downstream effectors of the TGF-β pathway were affected in the medial SMC of the AVF at day 7. There were significantly decreased numbers of p-Smad2/αSMA dual-positive cells (Figure 5A), p-Smad3/αSMA dual-positive cells (Figure 5B), no difference in TAK1 immunoreactivity (Figure S11C) and a trend towards increased p-Akt1 immunoreactivity in the media of the AVF (Figure S11D). There was no difference in outward remodeling of the AVF and the aorta (Figure 5C, Figure S11E-G) with no difference in aortic flow velocities, shear stress and blood flow (Figure S11H-K). There was no difference in the AVF wall thickness throughout time (Figure 5D) as well as no difference in AVF patency at day 42 (Figure 5E).
Figure 5. Inhibition of TGF-β signaling in smooth muscle cells has minimal impact on arteriovenous fistula (AVF) maturation in mice.

Myh11-CreERT2; Tgfbr2fl/fl; mT/mG mice were treated with vehicle (Control) or tamoxifen (TGF-βRiSMC) and underwent AVF creation (day0). (A) Representative photomicrographs showing immunofluorescence (IF) of p-Smad2 (white) merged with ICAM-1 (green), αSMA (red) and DAPI (blue) in the AVF wall at day 7 (yellow arrowheads denote p-Smad2-αSMA dual positive cells), quantified as percentage of p-Smad2-αSMA dual positive cells in the AVF wall; *P=0.0210 (t test), P=0.0317 (U test); n=5 for control, n=4 for TGF-βRiSMC. (B) Representative photomicrographs showing IF of p-Smad3 (white) merged with αSMA (red) and DAPI (blue) in the AVF wall at day 7 (yellow arrowheads denote p-Smad3-αSMA dual positive cells), quantified as percentage of p-Smad3-αSMA dual positive cells in the AVF wall; *P=0.0279 (t test), P=0.0635 (U test); n=5 for control, n=4 for TGF-βRiSMC. (C) Outward remodeling of the AVF and aorta up to day 21. Values are normalized by preoperative measurement; AVF: P=0.6830 (ANOVA); aorta: P=0.3200 (ANOVA); n=16 for control, n=17 for TGF-βRiSMC. (D) Representative photomicrographs of Van Gieson’s staining showing wall thickness (yellow arrowheads) of the AVF at day 21, and quantification of AVF wall thickness at days 7-42; P=0.6321 (ANOVA); n=5 (day 7), 7 (day 21) and 3 (day 42) for control, n=4 (day 7), 8 (day 21) and 5 (day 42) for TGF-βRiSMC. (E) Line graph showing AVF patency up to day 42, P=0.2378 (Log-rank); n=17 each at day 0. (F) Representative photomicrographs showing IF of PCNA (red, top) or cleaved caspase-3 (red, bottom) merged with αSMA (green) and DAPI (blue) in the AVF at day 7, quantified as percentage of PCNA-αSMA and cleaved caspase-3-αSMA dual-positive cells; PCNA: P=0.3283 (t test), P=0.5556 (U test), cleaved caspase-3: P=0.5535 (t test), P=0.6984 (U test); n=5 for control, n=4 for TGF-βRiSMC. (G) Representative photomicrographs of Masson’s trichrome staining of the AVF wall at day 21, quantified as percentage of collagen area within the AVF wall, *P=0.0204 (t test); n=7 for control, n=8 for TGF-βRiSMC. (H) Representative photomicrographs showing IF of PDGF-B (red, top) or FGF-2 (red, bottom) merged with ICAM-1 (green) and DAPI (blue) in the AVF at day 7, quantified as IF intensity in the AVF wall; PDGF-B: P=0.3782 (t test), P=0.2857 (U test); FGF-2: P=1515 (t test), P=0.1111 (U test); n=5 for control, n=4 for TGF-βRiSMC. (I) Representative photomicrographs showing IF of CD3 (green) or CD68 (green) merged with αSMA (red) and DAPI (blue) in the AVF wall (day 7), quantified as the number of CD3 or CD68 positive cells in the AVF wall (day 7); CD3: P=0.2198 (t test), P=0.4127 (U test); CD68: P=0.3367 (t test), P=0.6746 (U test); n=5 for control, n=4 for TGF-βRiSMC. Four (A, B, F, G, I) or 8 (D, H) high power field images were used to acquire the mean value per animal. All data are represented as mean value±SEM. L, lumen; A.U., arbitrary unit; n/HPF, number per high power field. Scale bars, 20 μm.
Since SMC-specific Tgfbr2 depletion did not significantly impact AVF remodeling (Figure 5C, D), we assessed whether there were changes in SMC proliferation and ECM deposition. There was no difference in the percentage of proliferative or apoptotic SMC at day7 (Figure 5F). However, mice with SMC-specific Tgfbr2 depletion showed significantly decreased collagen density in the AVF wall (Figure 5G) with no difference in the ratio of immature collagen but a trend towards deceased mature collagen (Figure S12A). Similarly, there was a trend towards reduced immunoreactivity of collagen type I with no change in collagen type III or fibronectin (Figure S12B). Since TGF-β promotes endothelial-to-mesenchymal transition (EndMT),16 mesenchymal markers N-cadherin and CD90 (Thy-1) as well as matrix marker vimentin were assessed. As expected, in mice with SMC-specific inhibition of TGF-β signaling, the immunoreactivity of these proteins in the AVF wall was similar to that of control mice (Figure S12C). In addition, in mice with SMC-specific inhibition of TGF-β signaling, the immunoreactivity of PDGF-B and FGF-2 in the AVF wall was similar to that of control mice (Figure 5H).
Since TGF-β inhibition with NP-SB increased T cell population and decreased anti-inflammatory macrophage polarization (Figure 4A, E), we determined whether mice with SMC-specific Tgfbr2 depletion had altered numbers of inflammatory cells. In these mice, there were similar numbers of CD3-positive or CD68-positive cells in the AVF wall compared to control mice (Figure 5I). There were also similar numbers of CD68/IFNγ-receptor, CD68/iNOS, CD68/CD206 and CD68/TGM2 dual-positive cells (Figure S12D). In toto, these data suggest that TGF-β inhibition in SMC attenuates ECM deposition, but does not inhibit SMC proliferation nor impact inflammatory cell accumulation, and is associated with no change in AVF wall thickness.
Inhibition of TGF-β signaling in EC impacts AVF maturation
Since inhibition of TGF-β signaling in SMC had minimal impact on AVF maturation, we next investigated how EC-specific TGF-β inhibition affects AVF maturation. Cdh5-CreERT2; Tgfbr1fl/fl; Tgfbr2fl/fl; mT/mG mice were treated with tamoxifen, while control litters were treated with corn oil vehicle, 2 weeks prior to AVF creation (Figure S1C). Adequate Cre recombination was confirmed with the mT/mG reporter (Figure S10B).
Mice with EC-specific Tgfbr1/Tgfbr2 depletion showed similar body weight change and survival after surgery compared to control mice (Figure S13A, B). There were significantly decreased numbers of p-Smad2/ICAM-1 dual-positive cells in the AVF wall (Figure 6A) but no difference in the percentage of p-Smad3/ICAM-1 dual-positive cells (Figure 6B) or immunoreactivity of TAK1 (Figure S13C) and p-Akt1 (Figure S13D) in the intima of the AVF. Surprisingly, there was a significant increase in outward remodeling of the AVF without any change in the relative diameter of the aorta (Figure 6C, Figure S13E-G) albeit with increased aortic flow velocities, shear stress and blood flow (Figure S13H-K). Furthermore, there was a trend towards decreased AVF wall thickness (Figure 6D) and there was improved AVF patency at day 42 (Figure 6E).
Figure 6. Inhibition of TGF-β signaling in endothelial cells impact arteriovenous fistula (AVF) maturation in mice.

Cdh5-CreERT2; Tgfbr1fl/fl; Tgfbr2fl/fl; mT/mG mice were treated with vehicle (Control) or tamoxifen (TGF-βRiEC) and underwent AVF creation (day0). (A) Representative photomicrographs showing immunofluorescence (IF) of p-Smad2 (white) merged with ICAM-1 (green), αSMA (red) and DAPI (blue) in the AVF wall at day 7 (yellow arrowheads denote p-Smad2-ICAM1 dual positive cells), quantified as percentage of p-Smad2-ICAM1 dual positive cells in the AVF wall; *P=0.0312 (t test), P=0.0317 (U test); n=5 each. (B) Representative photomicrographs showing IF of p-Smad3 (white) merged with ICAM-1 (green) and DAPI (blue) in the AVF wall at day 7 (yellow arrowheads denote p-Smad3-ICAM1 dual positive cells), quantified as percentage of p-Smad3-ICAM1 dual positive cells in the AVF wall; P=0.1002 (t test with Welch’s correction), P=0.1349 (U test); n=5 each. (C) Outward remodeling of the AVF and aorta up to day 21. Values are normalized by preoperative measurement; AVF: P=0.0010 (ANOVA), *P=0.0014 (day14, Sidak’s post hoc), **P=0.0170 (day21, Sidak’s post hoc); aorta: P=0.4837 (ANOVA); n=18 for control, n=17 for TGF-βRiEC. (D) Representative photomicrographs of Van Gieson’s staining showing wall thickness (yellow arrowheads) of the AVF at day 21, and quantification of AVF wall thickness at days 7-42; P= 0.0665 (ANOVA); n=5 (day 7), 11 (day 21) and 4 (day 42) for control, n=5 (day 7), 11 (day 21) and 4 (day 42) for TGF-βRiEC. (E) Line graph showing AVF patency up to day 42, *P=0.0239 (Log-rank); n=19 for control, n=20 for TGF-βRiEC at day 0. (F) Representative photomicrographs showing IF of PCNA (red, top) or cleaved caspase-3 (red, bottom) merged with αSMA (green) and DAPI (blue) in the AVF at day 7, quantified as percentage of PCNA-αSMA and cleaved caspase-3-αSMA dual-positive cells; PCNA: P=0.0957 (t test), P=0.2222 (U test), cleaved caspase-3: P=0.2857 (U test); n=5 each. (G) Representative photomicrographs of Masson’s trichrome staining of the AVF wall at day 21, quantified as percentage of collagen area within the AVF wall, *P=0.0323 (t test); n=10 each. (H) Representative photomicrographs showing IF of PDGF-B (red, top) or FGF-2 (red, bottom) merged with ICAM-1 (green) and DAPI (blue) in the AVF at day 7, quantified as IF intensity in the AVF wall; PDGF-B: P=0.9010 (t test), P=0.8413 (U test); FGF-2: *P= 0.0445 (t test), P=0.0952 (U test); n=5 each. (I) Representative photomicrographs showing IF of CD3 (green) or CD68 (green) merged with αSMA (red) and DAPI (blue) in the AVF wall (day 7), quantified as the number of CD3 or CD68 positive cells in the AVF wall (day 7); CD3: P=0.7484 (t test), P=0.7937 (U test); CD68: P=0.7200 (t test), P>0.9999 (U test); n=5 each. Four (A, B, F, G, I) or 8 (D, H) high power field images were used to acquire the mean value per animal. All data are represented as mean value±SEM. L, lumen; A.U., arbitrary unit; n/HPF, number per high power field. Scale bars, 20 μm.
Since EC-specific Tgfbr1/Tgfbr2 depletion reduced AVF wall thickness, we determined if this was attributable to changes in SMC proliferation or ECM deposition. There was a trend towards decreased proliferative SMC with no change in apoptotic SMC at day7 in mice with EC-specific Tgfbr1/Tgfbr2 depletion (Figure 6F). These mice also showed significantly decreased collagen density in the AVF wall (Figure 6G) with no difference in the ratio of immature collagen but a trend towards deceased mature collagen (Figure S14A). There was a trend towards reduced immunoreactivity of collagen type III with no change in collagen type I and fibronectin (Figure S14B). These mice also showed similar immunoreactivity of EndMT markers with control mice (Figure S14C). On the other hand, there was significantly decreased FGF-2 immunoreactivity in mice with EC-specific inhibition of TGF-β signaling with no change in PDGF-B (Figure 6H).
Since SMC-specific TGF-β inhibition did not show overt effect on inflammatory cells, we determined whether there were changes in inflammatory cells during AVF remodeling with EC-specific TGF-β inhibition. In mice with EC-specific Tgfbr1/Tgfbr2 depletion, there were similar numbers of CD3-positive or CD68-positive cells in the AVF wall compared to control mice at day 7 (Figure 6I). There were also similar numbers of CD68/IFNγ-receptor, CD68/iNOS, CD68/CD206 and CD68/TGM2 dual-positive cells (Figure S14D). In toto, these data suggest that TGF-β inhibition in EC attenuates ECM deposition and SMC proliferation with minimal effect on inflammatory cell accumulation, and is associated with reduced wall thickness, increased outward remodeling and superior patency of the AVF.
Discussion
This study shows increased phosphorylation of the canonical TGF-β/Smad2 signaling pathway in mature human AVF as well as in a mouse AVF model (Figure 1). Pan-inhibition of TGF-β signaling using SB431542 showed attenuated AVF wall thickening (Figure 2) via reduction of SMC proliferation and ECM deposition (Figure 3) as well as altered inflammatory cells (Figure 4). A genetic approach showed that conditional disruption of TGF-β signaling in the SMC attenuated ECM deposition but was not associated with reduced wall thickness (Figure 5); however, TGF-β inhibition in the EC attenuated ECM deposition and SMC proliferation and was associated with reduced wall thickness, increased outward remodeling and superior AVF patency without any effects on inflammatory cell accumulation (Figure 6). These data show that inhibition of the TGF-β pathway, specifically in the EC, results in desirable AVF remodeling with larger vessel diameter and without excessive wall thickening.
TGF-β is a context-dependent cytokine and its effect on SMC proliferation may differ between cell types.15 In murine arterial and venous grafts, TGF-β promotes EndMT leading to increased cells possessing both EC and SMC identity in the neointima.16 In SMC, TGF-β may show both anti-proliferative or pro-proliferative effects depending on the experimental conditions.17,18 In our mouse AVF model, disruption of SMC-specific TGF-β signaling resulted in no change of SMC proliferation whereas disruption of EC-specific TGF-β signaling attenuated SMC proliferation, compatible with previous studies.16-18 Since FGF-2 was decreased in mice with disruption of EC-specific but not SMC-specific TGF-β signaling, crosstalk with the FGF signaling pathway may be one of the mechanisms leading to the different effects on SMC proliferation. Interestingly, pan-inhibition with SB431542 showed anti-proliferative effects in the venous SMC, potentially due to crosstalk between different cell types and pathways affecting SMC proliferation. Although TGF-β is a major promoter of EndMT,16 neither SMC nor EC-specific inhibition of TGF-β signaling altered EndMT markers in the AVF wall. Since increased laminar shear stress suppresses EndMT and laminar shear stress is significantly increased in the IVC of our mouse model,20,41 this may have prevented detection of an effect of TGF-β signaling disruption on EndMT. Also of importance, our mouse model does not recapitulate the environment of chronic kidney disease which may induce EndMT via inflammation or oxidative stress.42 Both cell-specific and pan-inhibition of TGF-β signaling showed inhibitory effects on ECM deposition. These data suggest that differences in AVF wall thickness are attributable to the effects of TGF-β on SMC proliferation. However, AVF diameter did not increase when TGF-β signaling was disrupted in the SMC, possibly associated with vessel hypercontractility that is mediated by endothelial dysfunction such as observed in aortas with SMC-specific TGF-β inhibition in a different study.43 Of note, since wall thickness of the AVF (~10 μm) is about 1% of the lumen diameter (~1-2 mm), we used the vessel diameter as an indicator for outward remodeling.35
Another interesting finding is that inhibition of the TGF-β pathway is relatively specific to the Smad-dependent pathway in our model. Downstream members of the non-canonical pathway, such as TAK1 or Akt1, were minimally affected with pharmacological pan-inhibition or EC-specific TGF-β signaling disruption and even showed a trend towards increased immunoreactivity with SMC-specific TGF-β signaling disruption. Although Smad proteins are primarily activated by TGF-β,44 TAK1 can be activated by various signals,45 and PI3K/Akt signaling can be both activated or deactivated by TGF-β.46,47 Hence, our results on members of the non-canonical pathway may possibly be due to compensatory effects for the disrupted Smad-dependent pathway. Pan-inhibition with SB431542 showed pro-inflammatory effects in the AVF wall with increased numbers of T cells and decreased anti-inflammatory macrophage polarization; on the other hand, SMC or EC-specific inhibition of the TGF-β pathway resulted in no change in the number of T cells or macrophage polarization confirming the specificity of our cell type-specific inhibition. Regarding the role of T cells on AVF maturation, we have previously used cyclosporine A to inhibit CD4+ and CD8+ T cells, which resulted in promotion of outward remodeling and reduction of AVF wall thickening via reduction of collagen.21 Since treatment with NP-SB increased both CD4+ and CD8+ T cells, this may have offset the effect on decreased ECM synthesis resulting in no change in vessel diameter with NP-SB treatment. It is also possible that NP-induced inflammation could mask some effects of NP-SB and prevent detection of altered remodeling,48 whereas the genetically modified mice did not receive NP and showed altered remodeling (Figure S15); however, since we observed fewer T cells in the NP-control groups (~15/HPF, day 7; Figure 4A) compared to the genetically modified groups (~25/HPF; Figure 5I, 6I), we believe that the NP did not induce much inflammation.
TGF-βR1 and TGF-βR2 associate as interdependent components of a heteromeric complex, requiring each other for TGF-β binding and subsequent signaling.49 However, aberrant signaling after depletion of a single receptor is possible and it has been reported that depletion of both TGF-βR1 and TGF-βR2 more effectively diminishes TGF-β-driven Smad2/3 phosphorylation than depletion of either receptor alone.23 SB431542 selectively inhibits TGF-βR1. SMC-specific disruption of TGF-β signaling was based on conditional knockout of TGF-βR2 alone, whereas EC-specific disruption was based on conditional knockout of both TGF-βR1 and TGF-βR2. These differences may have contributed to our finding that EC-specific disruption of TGF-β signaling was associated with the most robust results with reduced AVF wall thickness and increased outward remodeling.
The use of SB431542 was limited to male mice; female and male mice have similar fistula dilation and wall thickening at day 21 in this model,24 as well as increased TGF-β/Smad2 signaling in the early remodeling phase. However, female mice have decreased patency by day 42, preceded by lower flow velocity and shear stress in comparison to male mice,24 confounding interpretation of patency in female mice. Since NP-SB treatment was used through day 42 (Figure 2), NP-SB was not explored in female mice; similarly, the genetic models were followed through day 42 (Figure 5, 6) and therefore male mice were used in these experiments. Studies with female mice are warranted because female sex is an important variable that predicts reduced maturation in human AVF.50 Additional clarification of the effects of TGF-β inhibition on AVF remodeling could also determine whether other cell type-specific TGF-β disruption, such as in fibroblasts or leukocytes, as these cells are essential during AVF remodeling;21,51 these transgenic mice were not available at the time of this study.
In summary, EC-specific and SMC-specific disruption of TGF-β signaling shows differential effects on AVF remodeling. EC-targeted TGF-β inhibition reduces excessive wall thickening and promotes outward remodeling via reduction of ECM deposition and SMC proliferation. Local delivery of pharmacological TGF-β inhibitors to the endothelium, such as via endovascular devices or antigen-coated nanoparticles, may be a translational strategy to improve AVF patency.
Supplementary Material
Highlights.
In a mouse model that recapitulates human AVF maturation, pharmacological inhibition of TGF-β signaling decreased AVF wall thickness but did not improve patency.
SMC-specific TGF-β inhibition via genetic approaches had minimal impact, but endothelial cell-specific TGF-β inhibition was associated with reduced AVF wall thickness, increased outward remodeling and improved patency.
Endothelial-targeted TGF-β inhibition may be a translational strategy to improve AVF patency.
Sources of Funding
This work was supported by US National Institute of Health (NIH) grants R01-HL128406 and R01-HL144476 [to A.D.]; the Uehara Memorial Foundation postdoctoral fellowship [to Y.M.]; as well as with the resources and the use of facilities at the VA Connecticut Healthcare System, West Haven, CT.
Abbreviations
- AVF
arteriovenous fistula
- EC
endothelial cell
- ECM
extracellular matrix
- FGF-2
fibroblast growth factor-2
- iNOS
inducible nitric oxide synthase
- NP
nanoparticle
- PDGF-B
platelet derived growth factor-B
- SMC
smooth muscle cell
- TAK1
TGF-β–activated kinase 1
- TGF-β
transforming growth factor-beta
- TGM2
transglutaminase 2
- Th1
T helper type 1 cells
- Th2
T helper type 2 cells
- Treg
regulatory T cells
Footnotes
Disclosures
None
References
- 1.Saran R, Robinson B, Abbott KC, Bragg-Gresham J, Chen X, Gipson D, Gu H, Hirth RA, Hutton D, Jin Y, et al. US Renal Data System 2019 Annual Data Report: Epidemiology of Kidney Disease in the United States. Am J Kidney Dis. 2020;75:A6–A7. doi: 10.1053/j.ajkd.2019.09.003 [DOI] [PubMed] [Google Scholar]
- 2.Almasri J, Alsawas M, Mainou M, Mustafa RA, Wang Z, Woo K, Cull DL, Murad MH. Outcomes of vascular access for hemodialysis: A systematic review and meta-analysis. J Vasc Surg. 2016;64:236–243. doi: 10.1016/j.jvs.2016.01.053 [DOI] [PubMed] [Google Scholar]
- 3.Murad MH, Elamin MB, Sidawy AN, Malaga G, Rizvi AZ, Flynn DN, Casey ET, McCausland FR, McGrath MM, Vo DH, et al. Autogenous versus prosthetic vascular access for hemodialysis: a systematic review and meta-analysis. J Vasc Surg. 2008;48:34S–47S. doi: 10.1016/j.jvs.2008.08.044 [DOI] [PubMed] [Google Scholar]
- 4.Hu H, Patel S, Hanisch JJ, Santana JM, Hashimoto T, Bai H, Kudze T, Foster TR, Guo J, Yatsula B, et al. Future research directions to improve fistula maturation and reduce access failure. Semin Vasc Surg. 2016;29:153–171. doi: 10.1053/j.semvascsurg.2016.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huijbregts HJ, Bots ML, Wittens CH, Schrama YC, Moll FL, Blankestijn PJ, group Cs. Hemodialysis arteriovenous fistula patency revisited: results of a prospective, multicenter initiative. Clin J Am Soc Nephrol. 2008;3:714–719. doi: 10.2215/CJN.02950707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Al-Jaishi AA, Oliver MJ, Thomas SM, Lok CE, Zhang JC, Garg AX, Kosa SD, Quinn RR, Moist LM. Patency rates of the arteriovenous fistula for hemodialysis: a systematic review and meta-analysis. Am J Kidney Dis. 2014;63:464–478. doi: 10.1053/j.ajkd.2013.08.023 [DOI] [PubMed] [Google Scholar]
- 7.Dember LM, Beck GJ, Allon M, Delmez JA, Dixon BS, Greenberg A, Himmelfarb J, Vazquez MA, Gassman JJ, Greene T, et al. Effect of clopidogrel on early failure of arteriovenous fistulas for hemodialysis: a randomized controlled trial. JAMA. 2008;299:2164–2171. doi: 10.1001/jama.299.18.2164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Katsanos K, Karnabatidis D, Kitrou P, Spiliopoulos S, Christeas N, Siablis D. Paclitaxel-coated balloon angioplasty vs. plain balloon dilation for the treatment of failing dialysis access: 6-month interim results from a prospective randomized controlled trial. J Endovasc Ther. 2012;19:263–272. doi: 10.1583/11-3690.1 [DOI] [PubMed] [Google Scholar]
- 9.Hye RJ, Peden EK, O'Connor TP, Browne BJ, Dixon BS, Schanzer AS, Jensik SC, Dember LM, Jaff MR, Burke SK. Human type I pancreatic elastase treatment of arteriovenous fistulas in patients with chronic kidney disease. J Vasc Surg. 2014;60:454–461.e451. doi: 10.1016/j.jvs.2014.02.037 [DOI] [PubMed] [Google Scholar]
- 10.Ricard-Blum S, Baffet G, Théret N. Molecular and tissue alterations of collagens in fibrosis. Matrix Biol. 2018;68-69:122–149. doi: 10.1016/j.matbio.2018.02.004 [DOI] [PubMed] [Google Scholar]
- 11.Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-beta: the master regulator of fibrosis. Nat Rev Nephrol. 2016;12:325–338. doi: 10.1038/nrneph.2016.48 [DOI] [PubMed] [Google Scholar]
- 12.Ruiz-Ortega M, Rodríguez-Vita J, Sanchez-Lopez E, Carvajal G, Egido J. TGF-beta signaling in vascular fibrosis. Cardiovasc Res. 2007;74:196–206. doi: 10.1016/j.cardiores.2007.02.008 [DOI] [PubMed] [Google Scholar]
- 13.Hu H, Lee SR, Bai H, Guo J, Hashimoto T, Isaji T, Guo X, Wang T, Wolf K, Liu S, et al. TGFβ (Transforming Growth Factor-Beta)-Activated Kinase 1 Regulates Arteriovenous Fistula Maturation. Arterioscler Thromb Vasc Biol. 2020;40:e203–e213. doi: 10.1161/ATVBAHA.119.313848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stracke S, Konner K, Kostlin I, Friedl R, Jehle PM, Hombach V, Keller F, Waltenberger J. Increased expression of TGF-beta1 and IGF-I in inflammatory stenotic lesions of hemodialysis fistulas. Kidney Int. 2002;61:1011–1019. doi: 10.1046/j.1523-1755.2002.00191.x [DOI] [PubMed] [Google Scholar]
- 15.Chen PY, Qin L, Li G, Tellides G, Simons M. Smooth muscle FGF/TGFβ cross talk regulates atherosclerosis progression. EMBO Mol Med. 2016;8:712–728. doi: 10.15252/emmm.201506181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen PY, Qin L, Barnes C, Charisse K, Yi T, Zhang X, Ali R, Medina PP, Yu J, Slack FJ, et al. FGF regulates TGF-β signaling and endothelial-to-mesenchymal transition via control of let-7 miRNA expression. Cell Rep. 2012;2:1684–1696. doi: 10.1016/j.celrep.2012.10.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kobayashi K, Yokote K, Fujimoto M, Yamashita K, Sakamoto A, Kitahara M, Kawamura H, Maezawa Y, Asaumi S, Tokuhisa T, et al. Targeted disruption of TGF-beta-Smad3 signaling leads to enhanced neointimal hyperplasia with diminished matrix deposition in response to vascular injury. Circ Res. 2005;96:904–912. doi: 10.1161/01.RES.0000163980.55495.44 [DOI] [PubMed] [Google Scholar]
- 18.Tsai S, Hollenbeck ST, Ryer EJ, Edlin R, Yamanouchi D, Kundi R, Wang C, Liu B, Kent KC. TGF-beta through Smad3 signaling stimulates vascular smooth muscle cell proliferation and neointimal formation. Am J Physiol Heart Circ Physiol. 2009;297:H540–549. doi: 10.1152/ajpheart.91478.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heine GH, Ulrich C, Sester U, Sester M, Kohler H, Girndt M. Transforming growth factor beta1 genotype polymorphisms determine AV fistula patency in hemodialysis patients. Kidney Int. 2003;64:1101–1107. doi: 10.1046/j.1523-1755.2003.00176.x [DOI] [PubMed] [Google Scholar]
- 20.Yamamoto K, Protack CD, Tsuneki M, Hall MR, Wong DJ, Lu DY, Assi R, Williams WT, Sadaghianloo N, Bai H, et al. The mouse aortocaval fistula recapitulates human arteriovenous fistula maturation. Am J Physiol Heart Circ Physiol. 2013;305:H1718–1725. doi: 10.1152/ajpheart.00590.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsubara Y, Kiwan G, Liu J, Gonzalez L, Langford J, Gao M, Gao X, Taniguchi R, Yatsula B, Furuyama T, et al. Inhibition of T-Cells by Cyclosporine A Reduces Macrophage Accumulation to Regulate Venous Adaptive Remodeling and Increase Arteriovenous Fistula Maturation. Arterioscler Thromb Vasc Biol. 2021;41:e160–e174. doi: 10.1161/ATVBAHA.120.315875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li W, Li Q, Jiao Y, Qin L, Ali R, Zhou J, Ferruzzi J, Kim RW, Geirsson A, Dietz HC, et al. Tgfbr2 disruption in postnatal smooth muscle impairs aortic wall homeostasis. J Clin Invest. 2014;124:755–767. doi: 10.1172/jci69942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen PY, Qin L, Li G, Wang Z, Dahlman JE, Malagon-Lopez J, Gujja S, Cilfone NA, Kauffman KJ, Sun L, et al. Endothelial TGF-β signalling drives vascular inflammation and atherosclerosis. Nat Metab. 2019;1:912–926. doi: 10.1038/s42255-019-0102-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kudze T, Ono S, Fereydooni A, Gonzalez L, Isaji T, Hu H, Yatsula B, Taniguchi R, Koizumi J, Nishibe T, et al. Altered Hemodynamics During Arteriovenous Fistula Remodeling Leads to Reduced Fistula Patency in Female Mice. JVS-Vasc Sci. 2020;1:42–56. doi: 10.1016/j.jvssci.2020.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamamoto K, Li X, Shu C, Miyata T, Dardik A. Technical aspects of the mouse aortocaval fistula. J Vis Exp. 2013:e50449. doi: 10.3791/50449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kudo FA, Muto A, Maloney SP, Pimiento JM, Bergaya S, Fitzgerald TN, Westvik TS, Frattini JC, Breuer CK, Cha CH, et al. Venous identity is lost but arterial identity is not gained during vein graft adaptation. Arterioscler Thromb Vasc Biol. 2007;27:1562–1571. doi: 10.1161/ATVBAHA.107.143032 [DOI] [PubMed] [Google Scholar]
- 27.Taniguchi R, Miura Y, Koyama H, Chida T, Anraku Y, Kishimura A, Shigematsu K, Kataoka K, Watanabe T. Adequately-Sized Nanocarriers Allow Sustained Targeted Drug Delivery to Neointimal Lesions in Rat Arteries. Mol Pharm. 2016;13:2108–2116. doi: 10.1021/acs.molpharmaceut.6b00219 [DOI] [PubMed] [Google Scholar]
- 28.Bai H, Lee JS, Hu H, Wang T, Isaji T, Liu S, Guo J, Liu H, Wolf K, Ono S, et al. Transforming Growth Factor-β1 Inhibits Pseudoaneurysm Formation After Aortic Patch Angioplasty. Arterioscler Thromb Vasc Biol. 2018;38:195–205. doi: 10.1161/ATVBAHA.117.310372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kataoka K, Kwon GS, Yokoyama M, Okano T, Sakurai Y. Block copolymer micelles as vehicles for drug delivery. J Control Release. 1993;24:119–132. [Google Scholar]
- 30.Pintavorn P, Ballermann BJ. TGF-beta and the endothelium during immune injury. Kidney Int. 1997;51:1401–1412. doi: 10.1038/ki.1997.192 [DOI] [PubMed] [Google Scholar]
- 31.Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, Roberts AB, Sporn MB, Ward JM, Karlsson S. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci U S A. 1993;90:770–774. doi: 10.1073/pnas.90.2.770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wahl SM. Transforming growth factor beta (TGF-beta) in inflammation: a cause and a cure. J Clin Immunol. 1992;12:61–74. doi: 10.1007/BF00918135 [DOI] [PubMed] [Google Scholar]
- 33.Kuwahara G, Hashimoto T, Tsuneki M, Yamamoto K, Assi R, Foster TR, Hanisch JJ, Bai H, Hu H, Protack CD, et al. CD44 Promotes Inflammation and Extracellular Matrix Production During Arteriovenous Fistula Maturation. Arterioscler Thromb Vasc Biol. 2017;37:1147–1156. doi: 10.1161/ATVBAHA.117.309385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guo X, Fereydooni A, Isaji T, Gorecka J, Liu S, Hu H, Ono S, Alozie M, Lee SR, Taniguchi R, et al. Inhibition of the Akt1-mTORC1 Axis Alters Venous Remodeling to Improve Arteriovenous Fistula Patency. Sci Rep. 2019;9:11046. doi: 10.1038/s41598-019-47542-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matsubara Y, Kiwan G, Fereydooni A, Langford J, Dardik A. Distinct subsets of T cells and macrophages impact venous remodeling during arteriovenous fistula maturation. JVS-Vasc Sci. 2020;1:207–218. doi: 10.1016/j.jvssci.2020.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yamagiwa S, Gray JD, Hashimoto S, Horwitz DA. A role for TGF-beta in the generation and expansion of CD4+CD25+ regulatory T cells from human peripheral blood. J Immunol. 2001;166:7282–7289. doi: 10.4049/jimmunol.166.12.7282 [DOI] [PubMed] [Google Scholar]
- 37.Gorelik L, Constant S, Flavell RA. Mechanism of transforming growth factor beta-induced inhibition of T helper type 1 differentiation. J Exp Med. 2002;195:1499–1505. doi: 10.1084/jem.20012076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Green EA, Gorelik L, McGregor CM, Tran EH, Flavell RA. CD4+CD25+ T regulatory cells control anti-islet CD8+ T cells through TGF-beta-TGF-beta receptor interactions in type 1 diabetes. Proc Natl Acad Sci U S A. 2003;100:10878–10883. doi: 10.1073/pnas.1834400100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gong D, Shi W, Yi SJ, Chen H, Groffen J, Heisterkamp N. TGFβ signaling plays a critical role in promoting alternative macrophage activation. BMC Immunol. 2012;13:31. doi: 10.1186/1471-2172-13-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang F, Wang H, Wang X, Jiang G, Liu H, Zhang G, Fang R, Bu X, Cai S, Du J. TGF-β induces M2-like macrophage polarization via SNAIL-mediated suppression of a pro-inflammatory phenotype. Oncotarget. 2016;7:52294–52306. doi: 10.18632/oncotarget.10561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alvandi Z, Bischoff J. Endothelial-Mesenchymal Transition in Cardiovascular Disease. Arterioscler Thromb Vasc Biol. 2021;41:2357–2369. doi: 10.1161/ATVBAHA.121.313788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang YX, Tang RN, Wang LT, Liu BC. Role of crosstalk between endothelial cells and smooth muscle cells in vascular calcification in chronic kidney disease. Cell Prolif. 2021;54:e12980. doi: 10.1111/cpr.12980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhu J, Angelov S, Yildirim IA, Wei H, Hu JH, Majesky MW, Brozovich FV, Kim F, Dichek DA. Loss of Transforming Growth Factor Beta Signaling in Aortic Smooth Muscle Cells Causes Endothelial Dysfunction and Aortic Hypercontractility. Arterioscler Thromb Vasc Biol. 2021:ATVBAHA121315878. doi: 10.1161/ATVBAHA.121.315878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Massagué J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19:2783–2810. doi: 10.1101/gad.1350705 [DOI] [PubMed] [Google Scholar]
- 45.Ajibade AA, Wang HY, Wang RF. Cell type-specific function of TAK1 in innate immune signaling. Trends Immunol. 2013;34:307–316. doi: 10.1016/j.it.2013.03.007 [DOI] [PubMed] [Google Scholar]
- 46.Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J Biol Chem. 2000;275:36803–36810. doi: 10.1074/jbc.M005912200 [DOI] [PubMed] [Google Scholar]
- 47.Valderrama-Carvajal H, Cocolakis E, Lacerte A, Lee EH, Krystal G, Ali S, Lebrun JJ. Activin/TGF-beta induce apoptosis through Smad-dependent expression of the lipid phosphatase SHIP. Nat Cell Biol. 2002;4:963–969. doi: 10.1038/ncb885 [DOI] [PubMed] [Google Scholar]
- 48.Nicolete R, dos Santos DF, Faccioli LH. The uptake of PLGA micro or nanoparticles by macrophages provokes distinct in vitro inflammatory response. Int Immunopharmacol. 2011;11:1557–1563. doi: 10.1016/j.intimp.2011.05.014 [DOI] [PubMed] [Google Scholar]
- 49.Wrana JL, Attisano L, Cárcamo J, Zentella A, Doody J, Laiho M, Wang XF, Massagué J. TGF beta signals through a heteromeric protein kinase receptor complex. Cell. 1992;71:1003–1014. doi: 10.1016/0092-8674(92)90395-s [DOI] [PubMed] [Google Scholar]
- 50.Miller CD, Robbin ML, Allon M. Gender differences in outcomes of arteriovenous fistulas in hemodialysis patients. Kidney Int. 2003;63:346–352. doi: 10.1046/j.1523-1755.2003.00740.x [DOI] [PubMed] [Google Scholar]
- 51.Kilari S, Cai C, Zhao C, Sharma A, Chernogubova E, Simeon M, Wu CC, Song HL, Maegdefessel L, Misra S. The Role of MicroRNA-21 in Venous Neointimal Hyperplasia: Implications for Targeting miR-21 for VNH Treatment. Mol Ther. 2019;27:1681–1693. doi: 10.1016/j.ymthe.2019.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Protack CD, Foster TR, Hashimoto T, Yamamoto K, Lee MY, Kraehling JR, Bai H, Hu H, Isaji T, Santana JM, et al. Eph-B4 regulates adaptive venous remodeling to improve arteriovenous fistula patency. Sci Rep. 2017;7:15386. doi: 10.1038/s41598-017-13071-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Almería B, Fahmy TM, Gomez A. A multiplexed electrospray process for single-step synthesis of stabilized polymer particles for drug delivery. J Control Release. 2011;154:203–210. doi: 10.1016/j.jconrel.2011.05.018 [DOI] [PubMed] [Google Scholar]
- 54.Park J, Mattessich T, Jay SM, Agawu A, Saltzman WM, Fahmy TM. Enhancement of surface ligand display on PLGA nanoparticles with amphiphilic ligand conjugates. J Control Release. 2011;156:109–115. doi: 10.1016/j.jconrel.2011.06.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Steenblock ER, Fadel T, Labowsky M, Pober JS, Fahmy TM. An artificial antigen-presenting cell with paracrine delivery of IL-2 impacts the magnitude and direction of the T cell response. J Biol Chem. 2011;286:34883–34892. doi: 10.1074/jbc.M111.276329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lemson MS, Tordoir JH, Daemen MJ, Kitslaar PJ. Intimal hyperplasia in vascular grafts. Eur J Vasc Endovasc Surg. 2000;19:336–350. doi: 10.1053/ejvs.1999.1040 [DOI] [PubMed] [Google Scholar]
- 57.McGah PM, Leotta DF, Beach KW, Eugene Zierler R, Aliseda A. Incomplete restoration of homeostatic shear stress within arteriovenous fistulae. J Biomech Eng. 2013;135:011005. doi: 10.1115/1.4023133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yamamoto K, Protack CD, Kuwahara G, Tsuneki M, Hashimoto T, Hall MR, Assi R, Brownson KE, Foster TR, Bai H, et al. Disturbed shear stress reduces Klf2 expression in arterial-venous fistulae in vivo. Physiol Rep. 2015;3. doi: 10.14814/phy2.12348 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.