

Abstract
Therapies for genetic disorders caused by mutated mitochondrial DNA are an unmet need, in large part due barriers in delivering DNA to the organelle and the absence of relevant animal models. We injected into mouse eyes a mitochondrially targeted Adeno-Associated-Virus (MTS-AAV) to deliver the mutant human NADH ubiquinone oxidoreductase subunit I (hND1/m.3460G>A) responsible for Leber’s hereditary optic neuropathy, the most common primary mitochondrial genetic disease. We show that the expression of the mutant hND1 delivered to retinal ganglion cells (RGC) layer colocalizes with the mitochondrial marker PORIN and the assembly of the expressed hND1 protein into host respiration complex I. The hND1 injected eyes exhibit hallmarks of the human disease with progressive loss of RGC function and number, as well as optic nerve degeneration. We also show that gene therapy in the hND1 eyes by means of an injection of a second MTS-AAV vector carrying wild type human ND1 restores mitochondrial respiratory complex I activity, the rate of ATP synthesis and protects RGCs and their axons from dysfunction and degeneration. These results prove that MTS-AAV is a highly efficient gene delivery approach with the ability to create mito-animal models and has the therapeutic potential to treat mitochondrial genetic diseases.
Keywords: mitochondrial- targeted AAV, m.G3460A ND1, Leber’s Hereditary Optic Neuropathy
Introduction
Mutations in mitochondrial DNA lead to a spectrum of neurodegenerative diseases for which no effective treatment exists. This is a group of untreatable disorders affecting the eye, nervous system and heart, some clinically characterized over a century ago, but they are now known to be a spectrum of molecularly defined diseases1. Leber’s hereditary optic neuropathy (LHON) is the most common primary mitochondrial genetic disease, leading to rapid blindness in children and young adults. More than 90% of LHON cases are caused by one of three point mutations in the mitochondrial DNA (mtDNA) (m.11778 G>A, m.3460G>A, and m.14484 T>C) encoding for subunits ND4, ND1, and ND6 of the respiration complex I, respectively2, 3. Virtually all mammalian cells contain numerous mitochondria, and correspondingly numerous copies of mtDNA. The levels of mutated mtDNA can vary from 0 to 100%, a situation known as heteroplasmy (coexistence of mutant and wildtype mtDNA), or homoplasmy (all mtDNA are either mutant or wildtype). In LHON, over 85% of the cases are homoplasmic mutations4–6. Clinically, LHON is typically a monosymptomatic disease, characterized by a painless loss of central vision and swelling of retinal nerve fibers followed by optic atrophy and progressive loss of retinal ganglion cells (RGCs)7, 8. To date, no-FDA approved treatment is available for LHON or any other mitochondrial disorders, although several treatment modalities have been proposed, including palliative pharmacological treatments, molecular genetic therapies, and stem cell therapies9–12.
Numerous challenges hamper the development of novel treatments for mitochondrial diseases, including LHON, not least are difficulties targeting drugs and /or nucleic acids inside mitochondria and lack of model systems13. Tools to manipulate mtDNA are limited but several strategies have been developed. One such technique is called allotopic expression, which recodes a mitochondrial gene into the nuclear codon thereby allowing for protein expression in the cytoplasm and transportation into the mitochondria.
Guy et al14 was the first group to employ the allotopic expression of wildtype ND4 in cybrid cells, complementing the ND4/ m.11778G>A mutant protein. We15, 16 and Corral-Debrinski et al17, 18 generated an LHON animal model with loss of visual function and RGC degeneration using allotopic expression of mutant human ND4(R340H). Vision loss and RGC degeneration in this LHON model could be rescued using a second AAV carrying allotopic wildtype ND415–18. This work formed the basis for launching gene therapy trials designed to test the safety and therapeutic efficacy of allotopically expressed human ND4 on patients carrying m.11778G>A mutation10, 11, 19–23.
Another technique is to import specific restriction endonucleases or transcription activator-like effector nucleases (TALENs) into the mitochondria to degrade mtDNA. This strategy has proved successful in inducing mtDNA heteroplasmic shift through the selective elimination of the mutated mtDNA in cybrid cells or germlines24. A recently developed technology based on mitochondrial-targeted TALEN guides deaminase facilitating a C to T or A to G base conversions25, 26.
In our earlier studies27, 28, we pioneered a novel approach to redirect the adeno-associated virus (AAV) virion into mitochondria through the addition of a mitochondrial targeting sequence (MTS) to capsid VP2 (MTS-AAV). Using this MTS-AAV, we delivered the wild type human ND4 gene to LHON cybrids homoplasmic for m.11778G>A mutation and rescued the LHON cybrid cells from defective cellular respiration. We also reported delivery of the wild type human ND4 into the adult rodent visual system, where vision loss and atrophy of the optic nerve induced by mutant R340H ND427, 28 was prevented.
In later studies29, we used this approach to introduce the mutant hND4 gene into mouse zygotes and generated a bona fide LHON mitomice carrying the human m.11778G>A mutation. Our results demonstrated that the translated hND4 protein assembled into host respiratory complexes, decreased respiratory chain function, and increased oxidative stress. The mitomice recapitulate the progressive RGC loss and optic nerve degeneration observed in the human form of the disease29, 30. In a more recent study31, we used the same approach to deliver human ND6/m.14484T>C into mouse eyes, which induced visual and RGC loss.
Given the biochemical and genetic heterogeneity of the disease, no single therapy is likely to exist for all LHON mutations. Currently, no animal model for ND1/m. 3460G>A exists and no gene therapy trials for LHON caused by ND1 or ND6 mutations are available. The studies presented here are part of our continuing efforts to study LHON disease mechanisms and pathophysiology and to develop possible treatment strategies. The results presented in this study further validate the technology’s applicability to facilitate a novel gene therapy for LHON and other mitochondrial genetic diseases.
Materials and Methods
Plasmids and AAVs.
sc-HSPCSB-hND1G3460A-mCherry was constructed as previously described27, 29. In brief, human ND1/m.3460G>A gene was fused in frame with Myc, and mitochondrial-encoded Cherry(mCherry) were cloned into scAAV backbones under the control of the mitochondrial heavy strand promoter including 3 upstream conserved sequence blocks (HSPCSB), where ND1G3460AMyc is followed by mCherry with a stop codon between two genes. mCherry cloned in the same scAAV backbone was used as a control (sc-HSPCSB-mCherry). Wild-type human ND1fused in frame with Myc was cloned into the same AAV backbone used as positive control and the treatment for the disease caused by mutant ND1 (sc-HSPCSB-hND1). The plasmids were purified using Qiagen endotoxin free maxiprep and then packaged with the VP2COX8 plus VP1, VP3 and helper plasmid PXX6 into recombinant virus: MTSAAV/sc-HSPCSB-hND1G3460A-mCherry, MTSAAV/sc-HSPCSB-mCherry, and MTSAAV/sc-HSPCSB-hND1.
Animals.
All animal procedures were performed in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals and the ARVO Statement for the use of Animals in Ophthalmic and Vision Research. For the intraocular injection of recombinant AAV, three months old DBA/1j mice were sedated by inhalation of 1.5% to 2% isoflurane. A local anesthetic (proparacaine HCl) was applied topically to the cornea, and a 32-gauge needle attached to a Hamilton syringe was inserted through the pars plana. 1 μl of mitochondria targeting AAV carrying the mutant human ND1 (MTSAAV/sc-HSPCSB-hND1G3460A-mCherry), or the mCherry (MTSAAV/sc-HSPCSB-mCherry ), or the wild type ND1 (MTSAAV/sc-HSPCSB-hND1) was injected into the eyes. For the treatment, the mice were first injected with mutant human ND1 and two days later were injected with wildtype hND1 (treated group) or mCherry (untreated group). PERGs and OCTs were performed at baseline, and 1, 3/4, 6/8 and 12/14 months after intravitreal AAV injections.
Based on one-way ANOVA, a sample size of N=9 would provide 80% power to detect a 20% effect size in RGC survival at a significance of p=0.05 in one (or more) of the groups. To reflect the male predominance of LHON, all animals used in this study were male DBA/1j mice started from 3 months old and randomly assigned to each study group. Data collection and analysis were done automatically or blinded by different individuals.
PERG and OCT.
Pattern electroretinogram (PERG) was performed as previously described.32, 33 In brief, mice were weighed and anesthetized with intraperitoneal (IP) injections of a mixture of ketamine (80mg/kg body weight) and xylazine (10mg/kg body weight) and were restrained by using a bite bar and a nose holder that allowed unobstructed vision and ocular stability. The animals were maintained at a constant body temperature of 37.6°C with a feedback-controlled heating pad. Under these conditions, the eyes of mice were wide open and in a stable position with undilated pupils pointing laterally and upward. The ERG electrode had a diameter of 0.25mm (made of silver wire configured to a semicircular loop of 2-mm radius) and was placed on the corneal surface by means of a micromanipulator and positioned to encircle the pupil without limiting the field of view. Reference and ground electrodes were stainless-steel needles inserted under the skin of the scalp and tail, respectively. After setting the mice on the stage and before recording, a small drop of balanced saline solution was topically applied on the cornea to prevent drying. A visual stimulus of contrast-reversing bars (field area, 50° × 58°; mean luminance, 50 cd/m2; spatial frequency, 0.05 cycles/deg; contrast, 100%; and temporal frequency, 1 Hz) was aligned with the projection of the pupil at 20 cm distance. Eyes were not refracted for the viewing distance, because the mouse eye has a large depth of focus due to the pinhole pupil. Retinal signals were amplified (10,000-fold) and band-pass filtered (1–30 Hz). Three consecutive responses to 600 contrast reversals each were recorded. The responses were superimposed to check for consistency and then averaged. The PERG is a light-adapted response. To have a corresponding index of outer retinal function, a light-adapted flash ERG (FERG) was also recorded with undilated pupils in response to strobe flashes of 20 cd/m2/s superimposed on a steady background light of 12 cd/m2 and presented within a Ganzfeld bowl. Under these conditions, rod activity is largely suppressed while cone activity is minimally suppressed. Averaged PERGs were automatically analyzed to evaluate the major positive and negative waves by Sigma Plot (Systat software Inc., San Jose, CA). Retinal images were visualized with in vivo spectral-domain (SD) OCT (Bioptigen, Inc. Durham, NC) and were analyzed with semiautomated custom software written using MATLAB software (MathWorks, Inc. Natick, MA)34.
Laser microdissection and PCR.
The injected mouse retinas were fixed overnight at 4°C in 4% PFA/PBS and equilibrated in 30% sucrose/PBS overnight at 4°C. After embedding into optimal cutting temperature (O.C.T.) compound (Sakura, Torrance, CA ), the retinas were cut into 8 μm sections and placed onto director slides (Expression Pathology, Rockville MD). Laser capture micro-dissection (LCM) was performed using a Leica LMD6500 (Leica Microsystems Inc, Buffalo Grove, IL). Cells from the RGC, inner and outer nuclear layers were excised and collected respectively for DNA extraction (DNeasy blood and tissue kit, Qiagen, Hilden, Germany). PCR was performed with primers F 5’ AATTTCCACCAAACCCCCCC3’ and R 5’ GCCTGCGGCGTATTCGATGT3’, and qPCR was performed with primers: hND1 F 5’ AGAACACCTCTGATTACTCCT3’ and R 5’ TTCGGTTGGTCTCTGCTA 3’; mND1 F5’ CATTGTTGGTCCATACGG3’ and R 5’ TGCTAGTGTGAGTGATAGG3’.
Immunohistochemistry.
Mice were anesthetized with isoflurane/oxygen and perfused transcardially with PBS followed by 4% paraformaldehyde in PBS. Longitudinal retinal sections were used for staining. The following antibodies were used: anti-Porin (ab15895 Abcam, 1:400, Waltham, MA), anti-mCherry [1C51] (ab125096, Abcam, 1:500), donkey anti-rabbit IgG 488 (A21206, Invitrogen, 1:600, Waltham, MA), and donkey anti-mouse IgG 546 (A10036, Invitrogen, 1:600). Images were taken with a Leica TCS SP5 confocal microscope (Leica Microsystems Inc, Buffalo Grove, IL).
Blue Native/PAGE and Immunoblotting.
Blue Native (BN)/PAGE was performed using the Invitrogen NativePAGE Gel system with minor modifications. In brief, mitochondria from pooled retina were enriched and used for BN-PAGE gel as described27. Electrophoresis was performed according to the manufacturer’s specifications (Invitrogen). BN-PAGE strips were equilibrated and applied to the 2D SDS gel as described by Invitrogen. Samples were separated in the second dimension and transferred to PVDF membranes (Immobilon; Millipore, Burlington, MA) using the semitransfer system (Bio-Rad, Hercules, CA). The following antibodies were used: anti-Myc [Y69] (ab32072, abcam, 1:500), anti-NDUFS4 [2C7CD4AG3] (ab87399, abcam, 1:1000) and anti-NDUFB6 [21C11BC11] (ab 110244, abcam, 1:1000). Secondary probing with anti-mouse (32230, ThermoFisher, 1:5000) or anti-rabbit HRP-conjugated antibodies (32260, ThermoFisher, 1:5000) was performed for 1 h, followed by detection using ECL reagents (ThermoFisher) and a FUJI Film Imaging system.
Respiratory chain function.
Mice were euthanized two months after injection, and the optic nerves and retinas were collected. Assays for complex I activity were performed in triplicate using a complex I enzyme activity microplate assay kit (Abcam). The rate of ATP synthesis was measured in triplicate by chemiluminescence with a modified luciferinluciferase assay in homogenized optic nerves or retinas digitonin-permeabilized with the complex I substrates malate and pyruvate in real time with an Optocom I luminometer (MGM Instruments, Hamden, CT). In brief, the optic nerves or retinas were put in 1.5mL Buffer A (150 mM KCL, 25 mM Tris-HCL, 2 mM EDTA, 0.1% BSA, 10mM K3PO4, 0.1mM MgCL2, PH 7.4) and homogenized for 10 seconds (optic nerve) or 1min (retina) with Omni TH at 19500rpm. A 100 μL aliquot of the cell suspension was used for each assay. Protein concentration of each suspension was quantitated using the protein assay kit (Bio-Rad).
Optic Nerve Diameters and Axons Counts.
Fifteen months after intraocular injections, optic nerves were dissected from 1 mm behind the globe to the optic chiasm. For axon counting, eight transmission electron micrographs were photographed by a masked observer at a magnification of 1500 X for each optic nerve specimen. The diameter and number of axons were then manually counted by a masked observer. Axons were identified by a clear axoplasmic cytoskeleton or surrounding electron dense myelin sheath. A total of 3,889 axons were counted for mCherry-injected controls, 5,238 axons for mutant hND1 injected mice, and 6,947 axons for treated mice.
Statistical Analysis.
Statistical analyses were performed using the Graphpad Prism software. Two groups were compared using two-tailed t-tests. Multiple groups were compared using one-way ANOVA analysis of variance; Tukey’s multiple comparisons test, Generalized Estimating Equation (GEE) with two main effects Age and Group, and the interaction between Age and Group. P values of <0.05 were considered significant. Values were expressed as means ± standard deviation (SD).
Results
Expression of human ND1/m.3460G>A in mouse retina following intravitreal injection of MTS-AAV2
Even though the mitochondrial double membrane has proven to be a formidable barrier in targeting nucleic acids to the organelle, viruses, naked DNA, and RNA are observed to traverse the membranes35–38. To generate a mouse model carrying human mutated ND1, we cloned the human ND1/m.3460G>A gene linked to a Myc epitope followed by a stop codon (without a tRNA) and mitochondrial encoded mCherry into a self-complementary AAV serotype 2 (AAV2) backbone (mut-hND1)( Fig 1A), as described in the Materials and Methods section. The same AAV construct without ND1 was used as a control (mCherry). All resultant constructs were packaged into mitochondrial targeting AAVs (MTS-AAVs), and the generated AAVs were injected intravitreally into mouse eyes. To reflect the male predominance of LHON, all mice used in this study were male. Ten days after injection, PCR analysis of mtDNA extracted from the retina using hND1 specific primers revealed the presence of the expected amplicon only in mutant hND1 injected mice but not in control mice (Fig1B). Sanger DNA sequencing and alignment to mutant and wild-type human ND1 confirmed that the PCR fragments were human ND1/m.3460 G>A (Fig1C, D).
Figure 1. Delivery and expression of hND1/m.3460G>A in mouse retinal ganglion cells (RGC).
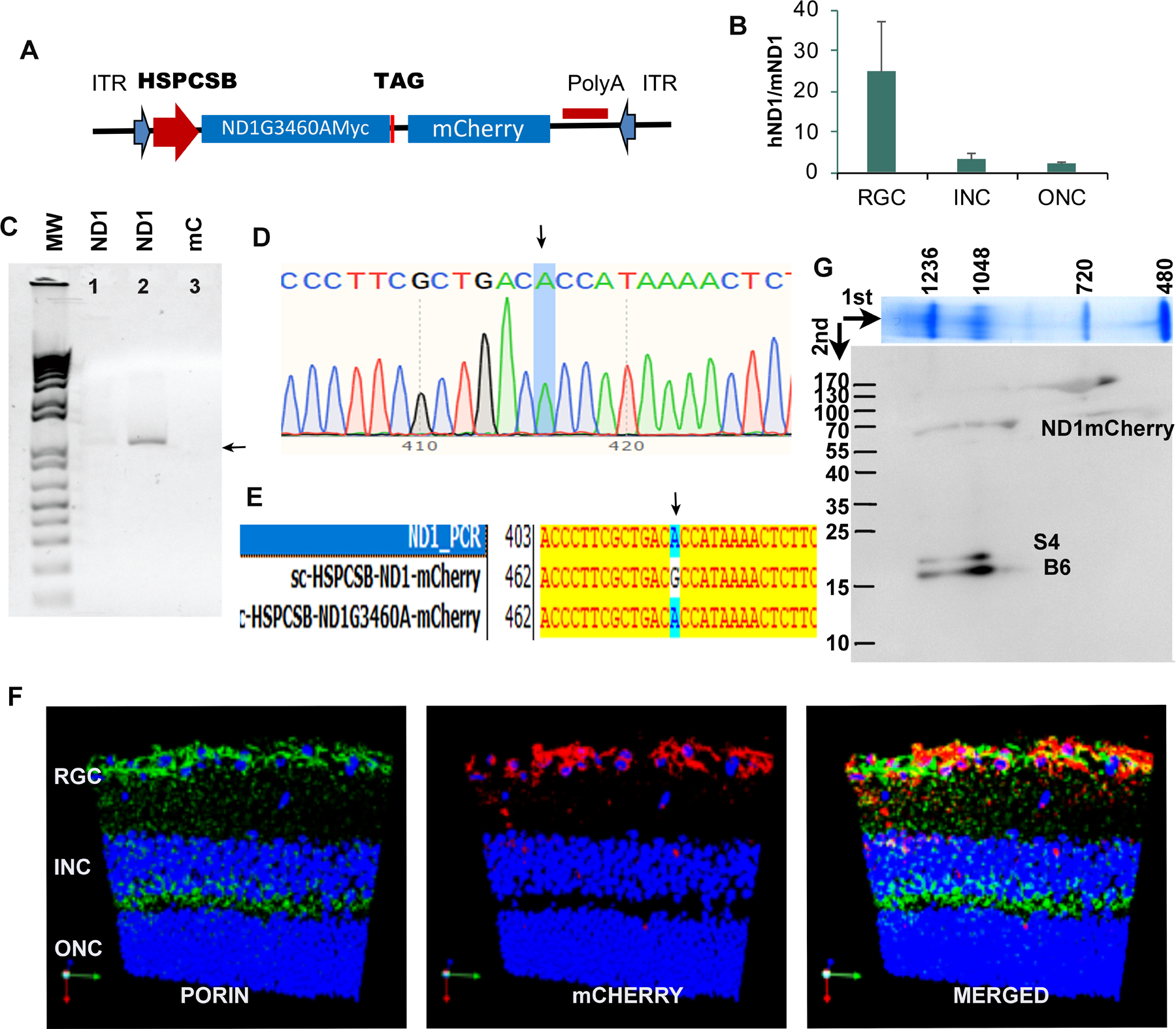
(A). Scheme of an AAV construct where hND1/m.3460G>A fused with the Myc epitope tag was cloned into a self-complementary AAV backbone, under the control of the mitochondrial heavy strand promoter, including three upstream conserved sequence blocks (HSPCSB) of the D-loop responsible for replication. A fluorescence marker, mitochondrial encoded mCherry, was cloned downstream of the mutant gene with a stop codon TAG in between (B). qPCR on microdissected cells showed that hND1 load was 24 times of endogenous mouse ND1 in cells of the RGC layer (RGC), 3 times in the inner nuclear (INC), and 2 times in the outer nuclear (ONC) layer. (C). Using the forward primer targeted to HSPCSB and reverse primer targeted to human ND1, the PCR showed the band with expected size in the whole retina of mutant hND1 injected mice( Panel 1,2), not in mCherry injected control mice (panel 3). (D) Sequencing confirmed the amplified band was hND1/m.3460G>A (arrow). (E) PCR sequence aligned to plasmids carrying wild-type and mutant hND1 respectively further confirmed that the PCR product was the hND1/m.3460G>A (arrow). (F). Representative immunostaining of a retinal longitudinal section showed that mCherry was expressed in the cells in the RGC layer, and colocalized with PORIN, a mitochondrial marker. (G) 2D BN-PAGE reacted against MYC, NDUFS4 (S4), and NDUFB6 (B6) subunits of complex I.
Quantitative PCR performed on micro-dissected cells revealed that the ratio of human ND1 level to that of endogenous mouse counterpart was 24:1 in cells of the ganglion cell layer (RGC). This ratio, however, was only 3:1 in cells of the inner nuclear layer (INC) and 2:1 in cells of the outer nuclear layer (ONC) in the injected mice (Fig. 1E). Immunostaining revealed that mCHERRY was only expressed in the RGC layer and colocalized with the mitochondrial marker, PORIN (Fig1F). These results are consistent with previous reports that AAV2 efficiently infects RGCs after intravitreal injection of adult rodent eyes, as these cells have high expression of heparan sulphate proteoglycans (the primary receptor of AAV2 in their membranes39–42).
To further determine whether the expressed mutant human ND1 protein integrates into the murine respiration complex I, the retinal mitochondria from 20 injected eyes was pooled and two-dimensional blue native PAGE western blots were run. The blots probed with an anti-MYC antibody, identified a band around 64kD that represents ND1MYC-mCHERRY (Fig S.1). The ND1MYC-mCHERRY fusion may have been generated by the lack of the correct secondary structure required by mitochondrial RNase P and nucleases for precise endonucleolytic excision of mitochondrial mRNAs by processing tRNAs from the primary transcript43–46. We then labeled the MYC-stained membrane with antibodies against NDUFS4 (S4) and NDUFB6 (B6) subunits of complex I and observed that they migrated in the same vertical plane as signals seen with the MYC antibody, suggesting that human ND1/m. 3460G>A protein assembles into the murine complex I (Fig 1G).
Mutant hND1 induces progressive retinal degeneration in mice eyes
To determine whether the expressed mutant hND1 can induce retinal atrophy in the mice, a phenotype observed in human LHON, we injected 30 mice (54 eyes) randomly with MTS-AAV delivered mutant hND1(n=27) or mCherry (n=27) and used spectral domain-optical coherence tomography (SD-OCT) to measure the thickness of the ganglion cell plus inner plexiform layers (RGC+IPL) in live animals. We observed swelling of the optic nerve head and adjacent RGC + IPL 4 months after injection of mutant hND1, which is characteristic of early human LHON. The optic nerve head swelling diminished, and progressive thinning of RGC+IPL was seen from 8 months to 14 months after the injection. (Fig 2a Right two panels). These finding are characteristic of human LHON in later atrophic stages. Semi-automated quantification showed that the thickness of RGC+IPL increased from 79.9 +/−10.6um at baseline to 84.5+/−24.2um at 4 months after injection, then dropped to 67.6+/−3.7um at 8 months(p<0.05), and continually dropped to 64.8+/−2.3um at 14 months (p<0.05) after injection (Fig 2C). We did not find any qualitative change in the thickness of RGC+IPL from baseline to 4-, 8- and 14-month after the injection of mCherry. (Fig 2A Left two panels, Fig 2B).
Figure 2. Delivered hND1/m.3460G>A induces retinal degeneration.
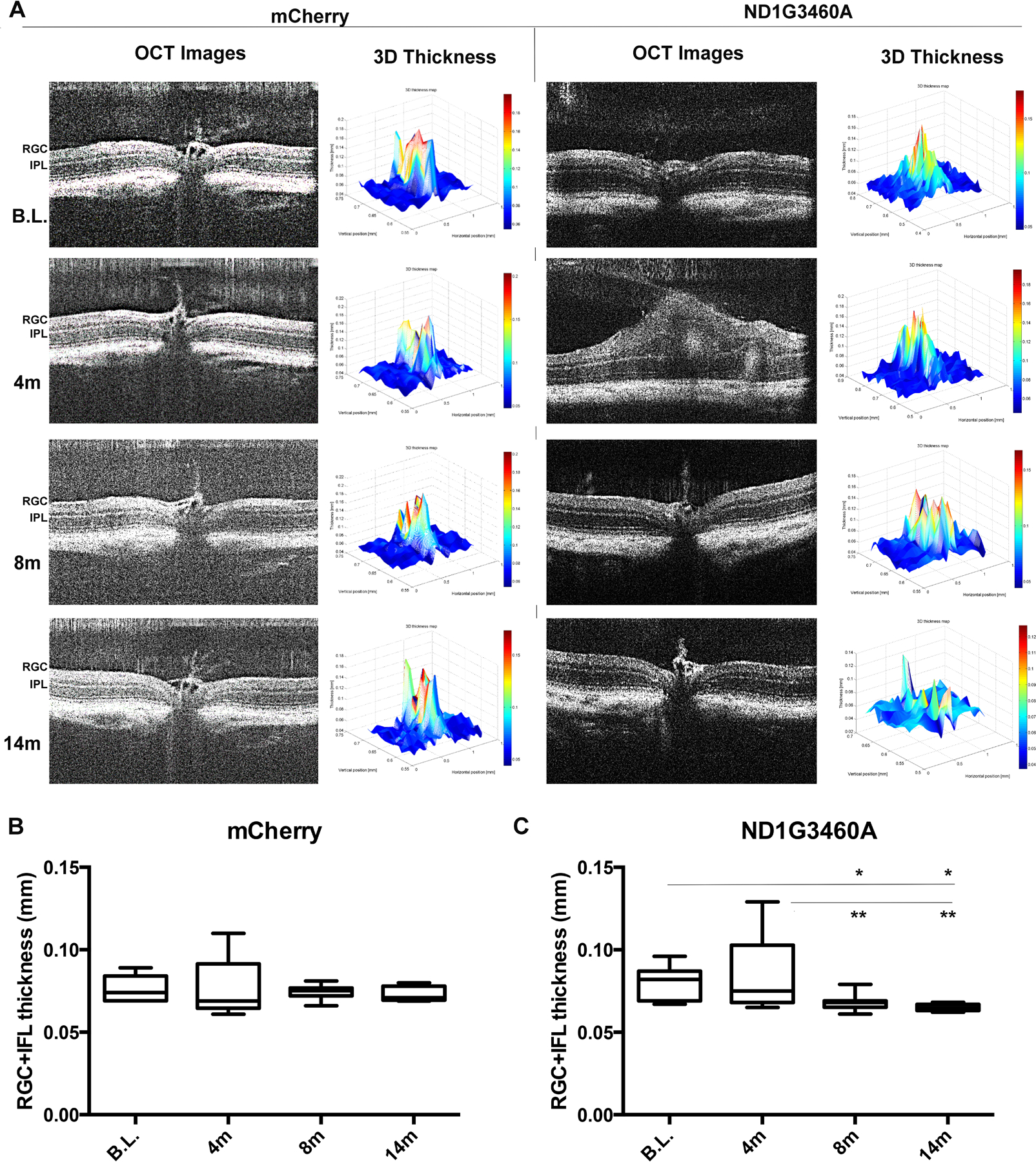
(A). Representative SD-OCT images and the corresponding 3D thickness of RGC-IPL maps of the same animal injected with mCherry (Left two panels) or mutant hND1 (right two panels). Each SD-OCT image (OCT image) represents one scan of a retina, and the corresponding 3D map of the RGC+IPL thickness (3D Thickness) was generated from100 OCT images covering one whole retina using a semiautomated custom software developed from MATLAB software (MathWorks). In mutant ND1-injected mice, optic nerve head swelling was detected at 4 months (4m) post-injection followed by a progressive loss of the RGC+IPL layer’s thickness. No abnormality was seen in the mCherry-injected control mice during the entire experimental period. (B-C) Semiautomatic quantification (n=10 in each group, one-way ANOVA analysis of variance; Tukey’s multiple comparisons test) showed an increase followed by a progressive thinning of the RGC+IPL thickness in mutant hND1-injected mice (C) but not in mCherry-injected control mice (B).
Mutant hND1 induces vision loss in the mice
Given that RGCs are the most vulnerable cell type in patients with LHON mutations, we next determined whether intravitreal injection of mutant hND1 in mouse eyes induced RGC dysfunction using pattern electroretinograms (PERGs), a sensitive electrophysiologic measure for RGC function.
No differences were observed in PERG amplitude between the two groups of mice prior to intravitreal injections (Fig 3 A, B); however, we found a statistically significant decrease in the PERG amplitude at 1 month (1m) after the injection of mutant hND1 relative to the age-matched control mice injected with mCherry (p=0.011, Fig 3 C, D). This decrement persisted at 3m (p=0.019, Fig3 E, F), 6m (p=0.020, Fig3 G, H), and 12m (p=0.010, Fig 3 I, J) after mutant hND1 injection. Using the Generalized Estimating Equation, an unbiased non-parametric method to analyze longitudinal correlated data, we found the mean PERG amplitude significantly decreased with age in both groups (p<0.000). However, mutant hND1-injected mice showed a larger (p<0.001) and faster (p<0.034) decrease in PERG amplitude compared to mCherry-injected control mice (Fig 3K).
Figure 3. Vision loss induced by hND1/m.3460G>A is rescued by gene therapy allele.
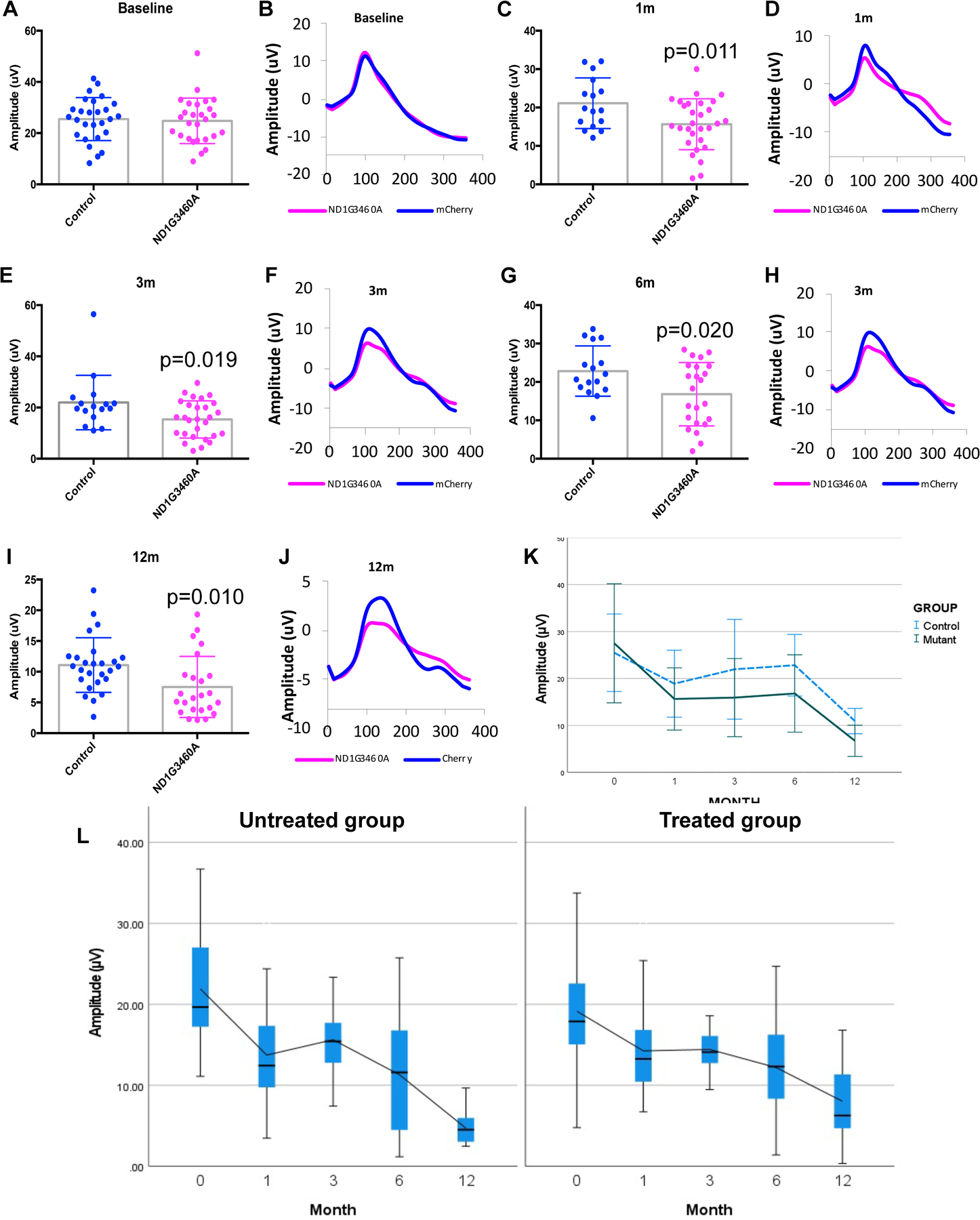
Scatterplot (A) of PERG amplitudes and averaged waveforms (B) of all mice tested in each group showed no significant difference at the baseline between the mice injected with mCherry and hND1/m.3460G>A. however, the difference became statistically significant at 1 month (1m, p=0.011, C, D, two-tailed t-tests ), 3 months (3m, p=0.019, E, F, two-tailed t-tests), 6 months (6m, p=0.020, G, H, two-tailed t-tests), and 12 months (12m, p=0.010, I,J, two-tailed t-tests) after injection. (K). A histogram generated using Generalized Estimating Equation (GEE) analysis showed the mean PERG amplitude significantly decreased with age (p<0.000) but was on average smaller in mutant hND1-injected mice (p<0.001). The time course of age-related PERG changes was different in the two groups (Interaction month × group, p=0.034), indicating the decrease rate was faster in mutant hND1-injected mice than mCherry-injected controls. (L) A box plot using Generalized Estimating Equation (GEE) analysis showed that mice rescued with wildtype hND1(Treated) significantly alleviated the decline in PERG amplitude compared to unrescued control mice (UnTreated) (p=0.028). Month 0 is the baseline, and the following are post injection.
Suppression of the vision loss by mito-targeted wild type ND1
To detect the amelioration of LHON symptoms by intravitreal delivery of a second MTS-AAV2 carrying wild-type hND1, we injected another 32 mice (64 eyes) with MTSAAV delivered mutant hND1 and two days later randomly divided them into treated and untreated groups. The treated group (n=34) received MTS-AAV delivered wildtype hND1, and the untreated group (n=30) received MTS-AAV delivered mCherry. Using the Generalized Estimating Equation, we found a progressive decline in PERG amplitude in both groups; however, the treated mice showed a significant slower reduction in the amplitude than the untreated mice (p=0.028), and the difference between the two groups became statistically significant at 12m after injection (p=0.005)(Fig3L).
Rescue of local energy deficiency induced in the mice by mito-targeted wild type ND1
Although the genetic defects associated with LHON are well characterized, the pathogenesis remains poorly understood. One of the most common hypotheses about the disease is that mtDNA mutations disrupt essential polypeptide subunits of complex I and lead to ATP production impairment, resulting in apoptosis of the retinal ganglion cells (RGCs)8, 47–53. To determine whether delivered mutant hND1 induced local ATP deficits in the mice, we measured complex I activity and the rate of ATP synthesis driven by complex I substrates malate and pyruvate in the retina and the optic nerve two months after injection. Complex I activity and the rate of ATP synthesis were not significantly reduced in the retina of mutant hND1 injected mice compared to that of mCherry injected control mice (Fig S. 2). This insignificance might be due to a small fraction of RGCs in the retina (<6%). However, the complex I activity in the optic nerve of mutant hND1 injected mice decreased significantly to 0.06±0.05 OD450/min/mg relative to 0.17±0.06 OD450/min/mg of mCherry injected control mice (p<0.001) (Fig 4A). Additionally, the rate of complex I-dependent ATP synthesis in the optic nerves of mutant hND1 injected mice was also significantly decreased to 325±192 nmol/min/mg relative to 623±153nmol/min/mg of mCherry injected mice (p<0.05) and relative to 763±9 nmol/min/mg of wildtype hND1 injected positive control mice (p<0.05) (Fig 4B). Intravitreal injection of a second MTS-AAV carrying wild type hND1 significantly increased complex I activity to 0.15±0.02 OD450/min/mg (p<0.01) and the rate of ATP synthesis to 734±337 nmol/min/mg (p<0.05) in the optic nerve relative to untreated mice (Fig 4A, B).
Figure 4. hND1/m.3460G>A causes loss of axons and RGCs.
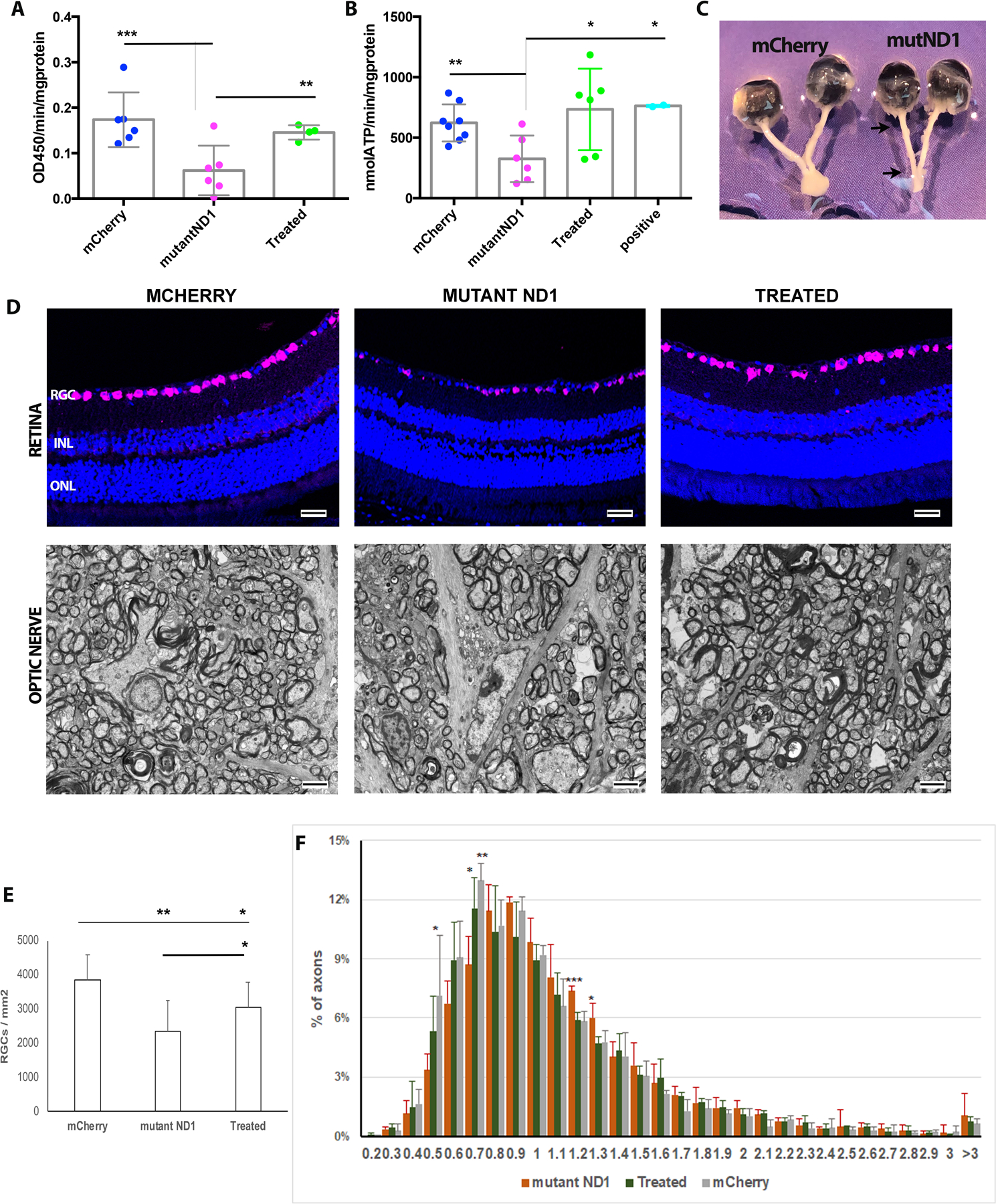
(A). Scatterplots showed that mice injected with hND1/m.3460G>A (mutant ND1) significantly decreased the complex I activity in the optic nerve compared to age-matched mCherry-injected control mice (mCherry). Using a second MTSAAV delivered wildtype hND1 (Treated) rescued the phenotype (one-way ANOVA analysis of variance; Tukey’s multiple comparisons test). (B). Scatterplots showed that the rate of complex I-depended ATP synthesis decreased significantly in the mice injected with hND1/m.3460G>A (mutant ND1) compared to control mice injected with mCherry (mCherry) or wildtype hND1 (positive). Gene therapy using a second MTSAAV delivered wildtype hND1 significantly increased ATP synthesis (Treated) (one-way ANOVA analysis of variance; Tukey’s multiple comparisons test). (C). Gross specimen of a mouse dissected 8 months after injection of mutant hND1 revealed significant thinning of the entire optic nerve from the globe to the optic chiasm (mutND1); whereas the optic nerve in an age-matched mouse injected with mCherry was much thicker (mCherry). (D) Representative immunostaining of retinal longitudinal sections using RBPMS (a pan-RGC marker) showed RGCs loss (top), and Transmission electron micrographs of the retrobulbar optic nerve showed the axon loss (bottom) in a mouse injected with hND1/m.3460G>A (MUTANT ND1) compared to a control mouse injected with mCherry (MCHERRY). Gene therapy using a second MTSAAV delivered wildtype hND1 (TREATED) significantly preserved RGCs and their axons. (E) Quantitation of RGCs in the mice injected with hND1/m.3460G>A(n=4) revealed significant loss relative to control mice injected with mCherry (n=6) (p<0.01) and mice rescued with wildtype hND1 (n=4) (p<0.05, one-way ANOVA analysis of variance; Tukey’s multiple comparisons test). (F). A histogram of the distribution of optic nerve axon diameters in control (mCherry), untreated (mutant ND1), and treated mice. The distribution of axonal diameters in the MTSAAV-hND1 -treated mice shows preservation of small diameter axons with a shift to larger diameter axons in the mice injected with mutant hND1(untreated mice).
Mutant hND1 causes loss of axons and RGCs
To better characterize the pathophysiological mechanism linked to the defects in mutant hND1 injected mice, we performed post-mortem histological analysis 15 months after intravitreal injections. Marked atrophy of the entire optic nerve from the globe to the optic chiasm was found prominent in eyes injected with mutant hND1 but not in eyes injected with mCherry from 8 (Fig 4C) and became more pronounced at 15 months after injection (Fig S3). This result is consistent with the observation in SD-OCT. Light microscopy of the retina performed 15 months after injection demonstrated that mutant hND1 injected mice had fewer cells expressing RBPMS, a pan-RGC marker, than age-matched mCherry-injected control mice. Gene therapy with wildtype alleles preserved more RGCs in the retina than untreated mice (Fig 4D upper panel). RGC quantification revealed a 39% RGC loss in mutant hND1-injected mice relative to the mCherry-injected control mice (2360 ±887 vs. 3856±743 cells /mm2, P< 0.01). Treatment with wildtype hND1 preserved 29% more RGCs than untreated mice (3052±748 vs. 2360 ±887 cells/mm2, p<0.05), although the cell number is still significantly lower than that of the mCherry-injected control mice (P<0.05, Fig 4E). Ultrastructural analysis revealed that the optic nerves of the mutant hND1 injected mice had many cystic spaces and electron-dense debris where axons were degraded. In contrast, age-matched control and treated mice exhibited numerous axons (Fig. 4D lower panel). Quantitative analysis revealed 52% more demyelinated optic nerve axons in untreated mice than in controls, while wildtype hND1 treatment decreased the number of demyelinated axons by 27% compared to untreated mice (Fig S4). However, the difference between groups was not statistically significant (p=0.0800), due to the small sample size and considerable variation. To further analyze the distribution of axons, we manually measured the diameter of every axon that was imaged as described in the materials and methods. The proportion of axons in every bin of 0.1μm was calculated relative to the total existing axons counted of each eye. Compared to age-matched mCherry injected control mice, mutant hND1 injected untreated mice displayed a shift toward larger axons with the highest distribution around 0.9μm. In contrast, wildtype hND1 treatment shifted the curve back to a similar position as the control mice (with the highest distribution around 0.7μm) (Fig 4F). Ordinary one-way ANOVA and multiple comparisons analysis revealed that untreated mice had a significantly lower number of axons with diameters of 0.7–0.8 μm compared to control and wildtype ND1 treated mice (p=0.008, Fig S. 5A). Instead, those mice had significantly more axons with diameters of 1.2–1.4 μm than that of treated and control mice (p=0.001, Fig S. 5B, C). Together, these data suggest that delivered wildtype hND1 prevented the demise of small axons that are preferentially lost in human LHON54.
Discussion
The development of effective treatment for mitochondrial diseases, including LHON, has been hindered by the absence of animal models that bear the same genotypes and corresponding disease phenotypes. We injected a mito-targeted AAV2 vector into mouse eyes to generate a murine model carrying mitochondrially encoded human NADH dehydrogenase 1 gene mutation m. 3460G>A. The injected mice displayed the most common features of LHON, including loss of visual function, progressive demise of RGCs, and loss of axons comprising the optic nerve.
Among the three LHON primary mutations, namely m.11778G>A, m.3460G>A, and m. 14484T>C55, 56, the ND1/m.3460G>A mutation has been generally accepted as the most severe pathogenic variant having the worst visual recovery rate compared to others. ND4/m.11778G>A eyes have an intermediate but still highly significant impairment in visual function and poor visual prognosis, and ND6/ m.14484T>C has the greatest likelihood of spontaneous visual recovery and better prognosis57 58. However, visual recovery for LHON with any mitochondrial mutation is usually incomplete, if it even occurs58.
Data generated here as well as in our previous studies28, 31 demonstrate that each of these three pathogenic mitochondrial variants can induce a progressive loss of RGC functions in intravitreally injected mice, as measured longitudinally with pattern electroretinogram (PERG). However, the onset of symptoms varied between different mitochondrial mutations. Mice injected with human ND1/m.3460G>A showed a 26% reduction in PERG amplitude 1 month after injection compared to age-matched control mice (P = 0.011). Similarly, mice injected with human ND4/m.11778G>A also showed a significant decrease (23%) in PERG amplitude (P < 0.04) at the same time point28. However, mice injected with human ND6/m.14484T>C showed only a slight reduction (14%) in PERG amplitude that was not statistically significant at 1-month post-injection (P = 0.089), but became statistically significant at 3 months post-injection (p=0.0023)31.
As LHON mutations disrupt key subunits of complex I, RGC dysfunction and loss have been interpreted as secondary to changes in the mitochondrial respiratory chain, although biochemical studies show that respiratory chain defects in the disease are more subtle than those seen in other mitochondrial genetic disorders58. The m.3460G>A mutation changes an alanine to threonine within a highly conserved region of ND1 with a predicted alpha-helical secondary structure. This region is directly involved in the ubiquinone reductase activity of Complex I. Mice injected with human ND1/ m. 3460G>A showed severe deficiency in the cellular respiration function in the optic nerve compared to those injected with human ND4/m.11778 G>A28 or ND6/m. 14484 T>C31. We found that mutant hND1 induced a 65% reduction in complex I activity relative to the age-matched controls, while mutant ND4 only induced a decrease of 52%28. Mutant ND6 caused a slight but not statistically significant reduction in activity31. These results are strikingly similar to what has been reported in most in vitro biochemical studies, where ND1/m.3460G>A showed a severe reduction of 60–80% in complex I activity compared to the other two mutations, ND4/m.11778G>A and ND6/m.14484T>C, which had milder (20–50%) or inconsistent reduced activities (0–60%) respectively59–61. Meanwhile, the rate of ATP synthesis was sharply reduced in mutant ND1 cybrids with nicotinamide adenine dinucleotide-dependent substrates, and a severe defect of brain bioenergetics was found in both heteroplasmic and homoplasmic individuals for the ND1 mutation62. However, an in vivo assay using magnetic resonance spectroscopy (MRS) showed that carriers with the ND4/m.11778G>A mutation showed a more severe reduction in ATP production in the brain and skeletal muscle than ND1/m.3460 G>A mutation58, 59, 63–65. Consistently, we found that mutant ND1 injected mice showed a lower reduction of 48% in the rate of complex I-dependent ATP synthesis in the optic nerve, compared with a 55% reduction in mutant ND4 injected mice.
Using a second MTS AVV as gene therapy, we delivered wildtype alleles into mutant hND1-injected eyes. Our biochemical analysis of respiration function in the optic nerve of the rescued mice confirmed significantly improved mitochondrial biogenesis as assessed by complex I activity and the rate of ATP synthesis. Rescued mice showed improved visual function compared to non-rescued mice, as evaluated by PERG. In addition, histopathological and ultrastructural analysis confirmed the preservation of the retinal ganglion cells and small axons in the optic nerve of the treated mice.
Therapies for LHON, in common with most mitochondrial diseases, are inadequate. To date, only the allotopic expression approach has reached human testing for ND4/m. 11778G>A mutation in the USA (NCT02161380)10, 20–23, China (NCT01267422)66, France (NCT02064569)11, 19. The French GenSight has moved into phase 3 trials in the USA and Europe (NCT02652767, NCT02652780, NCT03293524)11. This strategy involves recoding a full-length mitochondrial gene (the ND4) in standard genetic codes and importing the expressed protein from cytoplasm back to mitochondria with the help of mitochondrial targeting sequencing. Given the high hydrophobicity of the protein, its ability to cross the mitochondrial membrane and maintain long-term stable gene expression remains a limitation. In the clinical trial we performed on LHON using allotopically expressed ND4, gene therapy showed limited visual function improvement for patients with during of vision loss more than 12 months10, 20. Based on a similar experience in their phase I-II trial, the French Gensight study group excluded patients with more than a year of visual loss for inclusion into their phase 3 clinical trials11.Therefore, those patients with more than a year of visual loss may have no option for treatment using the allotopic approach. Although improved, most patients in the study cohorts remained with low vision and continued to be legally blind10, 11, 22, 66. Mitochondrial targeting AAV (MTSAAV) delivers exogenous genes directly into mitochondria, likely resulting in more efficient gene expression that may provide more gain in the vision for LHON patients. We are performing preclinical testing of MTSAAV-delivered ND4, under the support of a NIH grant, to gain regulatory approval of this therapy for human testing.
Results from this study demonstrate MTSAAV as a highly efficient gene delivery approach and will significantly benefit the translation of this mitochondrial targeted approach into future clinical trials for LHON patients carrying ND1/m.3460G>A, ND6/m.14484T>C mutation or other mitochondrial genetic diseases.
In conclusion, we delivered human ND1/m.3460G>A into the mouse eyes using mitochondrial targeting AAV, and the delivered DNA induced RGC loss, optic nerve atrophy, and local bioenergetic defects. Our demonstration that the mitochondrial disease phenotype was rescued by a second MTSAAV delivery with wild type ND1 gene supports the link between the disease phenotypes and mutant ND1 load. This study provides a promising basis for the treatment of LHON or other mitochondrial genetic diseases by gene therapy.
Supplementary Material
Acknowledgments
The authors acknowledge the defining contributions of John Guy, MD (deceased), who invented mito-targeting AAV, designed and performed the research. His tireless efforts made this study possible. We also thank Dr. Marco Ruggeri and the Ophthalmic Biophysics Center of the University of Miami for providing OCT analysis techniques; Dr. Alfred S. Lewin and Mr. Vince A. Chiodo at the University of Florida for the MTSAAV package. The authors declare no competing interests.
Funding:
This study is supported by the National Eye Institute R01 EY 027414, R01 EY017141, R24 EY028785, P30 EY014801 Bascom Palmer Eye Institute Core Grant, and institutional support from the Retina Research Foundation (GR015009). R.K. Lee was partially supported by the Walter G. Ross Foundation, the Camiener Foundation Glaucoma Research Fund, and the Guiterrez Family Research Fund.
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its supplementary information files.
References
- 1.Kauppila JHK, Baines HL, Bratic A, Simard ML, Freyer C, Mourier A et al. A Phenotype-Driven Approach to Generate Mouse Models with Pathogenic mtDNA Mutations Causing Mitochondrial Disease. Cell Rep 2016; 16(11): 2980–2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jurkute N, Yu-Wai-Man P. Leber hereditary optic neuropathy: bridging the translational gap. Curr Opin Ophthalmol 2017; 28(5): 403–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yu-Wai-Man P, Griffiths PG, Hudson G, Chinnery PF. Inherited mitochondrial optic neuropathies. Journal of medical genetics 2009; 46(3): 145–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rasool N, Lessell S, Cestari DM. Leber Hereditary Optic Neuropathy: Bringing the Lab to the Clinic. Seminars in ophthalmology 2016; 31(1–2): 107–16. [DOI] [PubMed] [Google Scholar]
- 5.Harding AE, Sweeney MG, Govan GG, Riordan-Eva P. Pedigree analysis in Leber hereditary optic neuropathy families with a pathogenic mtDNA mutation. Am J Hum Genet 1995; 57(1): 77–86. [PMC free article] [PubMed] [Google Scholar]
- 6.Yu-Wai-Man P, Votruba M, Moore AT, Chinnery PF. Treatment strategies for inherited optic neuropathies: past, present and future. Eye 2014; 28(5): 521–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Newman NJ, Biousse V. Hereditary optic neuropathies. Eye 2004; 18(11): 1144–60. [DOI] [PubMed] [Google Scholar]
- 8.Jankauskaite E, Bartnik E, Kodron A. Investigating Leber’s hereditary optic neuropathy: Cell models and future perspectives. Mitochondrion 2017; 32: 19–26. [DOI] [PubMed] [Google Scholar]
- 9.Klopstock T, Metz G, Yu-Wai-Man P, Buchner B, Gallenmuller C, Bailie M et al. Persistence of the treatment effect of idebenone in Leber’s hereditary optic neuropathy. Brain 2013; 136(Pt 2): e230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guy J, Feuer WJ, Davis JL, Porciatti V, Gonzalez PJ, Koilkonda RD et al. Gene Therapy for Leber Hereditary Optic Neuropathy: Low- and Medium-Dose Visual Results. Ophthalmology 2017; 124(11): 1621–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu-Wai-Man P, Newman NJ, Carelli V, Moster ML, Biousse V, Sadun AA et al. Bilateral visual improvement with unilateral gene therapy injection for Leber hereditary optic neuropathy. Sci Transl Med 2020; 12(573). [DOI] [PubMed] [Google Scholar]
- 12.Weiss JN, Levy S, Benes SC. Stem Cell Ophthalmology Treatment Study (SCOTS): bone marrow-derived stem cells in the treatment of Leber’s hereditary optic neuropathy. Neural Regen Res 2016; 11(10): 1685–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rahman S Emerging aspects of treatment in mitochondrial disorders. J Inherit Metab Dis 2015; 38(4): 641–53. [DOI] [PubMed] [Google Scholar]
- 14.Guy J, Qi X, Pallotti F, Schon EA, Manfredi G, Carelli V et al. Rescue of a mitochondrial deficiency causing Leber Hereditary Optic Neuropathy. Ann Neurol 2002; 52(5): 534–42. [DOI] [PubMed] [Google Scholar]
- 15.Qi X, Sun L, Lewin AS, Hauswirth WW, Guy J. The mutant human ND4 subunit of complex I induces optic neuropathy in the mouse. Invest Ophthalmol Vis Sci 2007; 48(1): 1–10. [DOI] [PubMed] [Google Scholar]
- 16.Koilkonda R, Yu H, Talla V, Porciatti V, Feuer WJ, Hauswirth WW et al. LHON gene therapy vector prevents visual loss and optic neuropathy induced by G11778A mutant mitochondrial DNA: biodistribution and toxicology profile. Invest Ophthalmol Vis Sci 2014; 55(12): 7739–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cwerman-Thibault H, Augustin S, Lechauve C, Ayache J, Ellouze S, Sahel JA et al. Nuclear expression of mitochondrial ND4 leads to the protein assembling in complex I and prevents optic atrophy and visual loss. Mol Ther Methods Clin Dev 2015; 2: 15003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baracca A, Solaini G, Sgarbi G, Lenaz G, Baruzzi A, Schapira AH et al. Severe impairment of complex I-driven adenosine triphosphate synthesis in leber hereditary optic neuropathy cybrids. Arch Neurol 2005; 62(5): 730–6. [DOI] [PubMed] [Google Scholar]
- 19.Cwerman-Thibault H, Augustin S, Ellouze S, Sahel JA, Corral-Debrinski M. Gene therapy for mitochondrial diseases: Leber Hereditary Optic Neuropathy as the first candidate for a clinical trial. C R Biol 2014; 337(3): 193–206. [DOI] [PubMed] [Google Scholar]
- 20.Feuer WJ, Schiffman JC, Davis JL, Porciatti V, Gonzalez P, Koilkonda RD et al. Gene Therapy for Leber Hereditary Optic Neuropathy: Initial Results. Ophthalmology 2016; 123(3): 558–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koilkonda RD, Yu H, Chou TH, Feuer WJ, Ruggeri M, Porciatti V et al. Safety and effects of the vector for the Leber hereditary optic neuropathy gene therapy clinical trial. JAMA Ophthalmol 2014; 132(4): 409–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guy J, Feuer WJ, Porciatti V, Schiffman J, Abukhalil F, Vandenbroucke R et al. Retinal ganglion cell dysfunction in asymptomatic G11778A: Leber hereditary optic neuropathy. Invest Ophthalmol Vis Sci 2014; 55(2): 841–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koilkonda RD, Guy J. Leber’s Hereditary Optic Neuropathy-Gene Therapy: From Benchtop to Bedside. J Ophthalmol 2011; 2011: 179412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bacman SR, Williams SL, Pinto M, Peralta S, Moraes CT. Specific elimination of mutant mitochondrial genomes in patient-derived cells by mitoTALENs. Nat Med 2013; 19(9): 1111–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mok BY, de Moraes MH, Zeng J, Bosch DE, Kotrys AV, Raguram A et al. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature 2020; 583(7817): 631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee H, Lee S, Baek G, Kim A, Kang BC, Seo H et al. Mitochondrial DNA editing in mice with DddA-TALE fusion deaminases. Nat Commun 2021; 12(1): 1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu H, Koilkonda RD, Chou TH, Porciatti V, Ozdemir SS, Chiodo V et al. Gene delivery to mitochondria by targeting modified adenoassociated virus suppresses Leber’s hereditary optic neuropathy in a mouse model. Proc Natl Acad Sci U S A 2012; 109(20): E1238–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu H, Ozdemir SS, Koilkonda RD, Chou TH, Porciatti V, Chiodo V et al. Mutant NADH dehydrogenase subunit 4 gene delivery to mitochondria by targeting sequence-modified adeno-associated virus induces visual loss and optic atrophy in mice. Mol Vis 2012; 18: 1668–83. [PMC free article] [PubMed] [Google Scholar]
- 29.Yu H, Koilkonda RD, Chou TH, Porciatti V, Mehta A, Hentall ID et al. Consequences of zygote injection and germline transfer of mutant human mitochondrial DNA in mice. Proc Natl Acad Sci U S A 2015; 112(42): E5689–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu H, Porciatti V, Lewin A, Hauswirth W, Guy J. Longterm Reversal of Severe Visual Loss by Mitochondrial Gene Transfer in a Mouse Model of Leber Hereditary Optic Neuropathy. Sci Rep 2018; 8(1): 5587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu H, Sant DW, Wang G, Guy J. Mitochondrial Transfer of the Mutant Human ND6T14484C Gene Causes Visual Loss and Optic Neuropathy. Transl Vis Sci Technol 2020; 9(11): 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Porciatti V The mouse pattern electroretinogram. Doc Ophthalmol 2007; 115(3): 145–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang X, Chou TH, Ruggeri M, Porciatti V. A new mouse model of inducible, chronic retinal ganglion cell dysfunction not associated with cell death. Invest Ophthalmol Vis Sci 2013; 54(3): 1898–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruggeri M, Wehbe H, Jiao S, Gregori G, Jockovich ME, Hackam A et al. In vivo three-dimensional high-resolution imaging of rodent retina with spectral-domain optical coherence tomography. Invest Ophthalmol Vis Sci 2007; 48(4): 1808–14. [DOI] [PubMed] [Google Scholar]
- 35.Weber-Lotfi F, Ibrahim N, Boesch P, Cosset A, Konstantinov Y, Lightowlers RN et al. Developing a genetic approach to investigate the mechanism of mitochondrial competence for DNA import. Biochim Biophys Acta 2009; 1787(5): 320–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koulintchenko M, Temperley RJ, Mason PA, Dietrich A, Lightowlers RN. Natural competence of mammalian mitochondria allows the molecular investigation of mitochondrial gene expression. Hum Mol Genet 2006; 15(1): 143–54. [DOI] [PubMed] [Google Scholar]
- 37.Hussain SA, Yalvac ME, Khoo B, Eckardt S, McLaughlin KJ. Adapting CRISPR/Cas9 System for Targeting Mitochondrial Genome. Front Genet 2021; 12: 627050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Y, Hu LF, Cui PF, Qi LY, Xing L, Jiang HL. Pathologically Responsive Mitochondrial Gene Therapy in an Allotopic Expression-Independent Manner Cures Leber’s Hereditary Optic Neuropathy. Adv Mater 2021: e2103307. [DOI] [PubMed] [Google Scholar]
- 39.Harvey AR, Kamphuis W, Eggers R, Symons NA, Blits B, Niclou S et al. Intravitreal injection of adeno-associated viral vectors results in the transduction of different types of retinal neurons in neonatal and adult rats: a comparison with lentiviral vectors. Mol Cell Neurosci 2002; 21(1): 141–57. [DOI] [PubMed] [Google Scholar]
- 40.Martin KR, Quigley HA, Zack DJ, Levkovitch-Verbin H, Kielczewski J, Valenta D et al. Gene therapy with brain-derived neurotrophic factor as a protection: retinal ganglion cells in a rat glaucoma model. Invest Ophthalmol Vis Sci 2003; 44(10): 4357–65. [DOI] [PubMed] [Google Scholar]
- 41.Nickells RW, Schmitt HM, Maes ME, Schlamp CL. AAV2-Mediated Transduction of the Mouse Retina After Optic Nerve Injury. Invest Ophthalmol Vis Sci 2017; 58(14): 6091–6104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shevtsova Z, Malik JM, Michel U, Bahr M, Kugler S. Promoters and serotypes: targeting of adeno-associated virus vectors for gene transfer in the rat central nervous system in vitro and in vivo. Exp Physiol 2005; 90(1): 53–9. [DOI] [PubMed] [Google Scholar]
- 43.Ojala D, Montoya J, Attardi G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981; 290(5806): 470–4. [DOI] [PubMed] [Google Scholar]
- 44.Falkenberg M, Larsson NG, Gustafsson CM. DNA replication and transcription in mammalian mitochondria. Annu Rev Biochem 2007; 76: 679–99. [DOI] [PubMed] [Google Scholar]
- 45.Facucho-Oliveira JM, St John JC. The relationship between pluripotency and mitochondrial DNA proliferation during early embryo development and embryonic stem cell differentiation. Stem Cell Rev Rep 2009; 5(2): 140–58. [DOI] [PubMed] [Google Scholar]
- 46.Montoya J, Gaines GL, Attardi G. The pattern of transcription of the human mitochondrial rRNA genes reveals two overlapping transcription units. Cell 1983; 34(1): 151–9. [DOI] [PubMed] [Google Scholar]
- 47.Yu-Wai-Man P, Votruba M, Burte F, La Morgia C, Barboni P, Carelli V. A neurodegenerative perspective on mitochondrial optic neuropathies. Acta neuropathologica 2016; 132(6): 789–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Finsterer J, Zarrouk-Mahjoub S. Leber’s hereditary optic neuropathy is multiorgan not mono-organ. Clinical ophthalmology 2016; 10: 2187–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pilz YL, Bass SJ, Sherman J. A Review of Mitochondrial Optic Neuropathies: From Inherited to Acquired Forms. Journal of optometry 2017; 10(4): 205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sadun AA, La Morgia C, Carelli V. Mitochondrial optic neuropathies: our travels from bench to bedside and back again. Clinical & experimental ophthalmology 2013; 41(7): 702–12. [DOI] [PubMed] [Google Scholar]
- 51.Wang MY, Sadun AA. Drug-related mitochondrial optic neuropathies. Journal of neuro-ophthalmology : the official journal of the North American Neuro-Ophthalmology Society 2013; 33(2): 172–8. [DOI] [PubMed] [Google Scholar]
- 52.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiological reviews 2007; 87(1): 99–163. [DOI] [PubMed] [Google Scholar]
- 53.Sadun AA. Mitochondrial optic neuropathies. Journal of neurology, neurosurgery, and psychiatry 2002; 72(4): 423–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sadun AA, Win PH, Ross-Cisneros FN, Walker SO, Carelli V. Leber’s hereditary optic neuropathy differentially affects smaller axons in the optic nerve. Trans Am Ophthalmol Soc 2000; 98: 223–32; discussion 232–5. [PMC free article] [PubMed] [Google Scholar]
- 55.Wallace DC, Lott MT. Leber Hereditary Optic Neuropathy: Exemplar of an mtDNA Disease. Handb Exp Pharmacol 2017; 240: 339–376. [DOI] [PubMed] [Google Scholar]
- 56.Theodorou-Kanakari A, Karampitianis S, Karageorgou V, Kampourelli E, Kapasakis E, Theodossiadis P et al. Current and Emerging Treatment Modalities for Leber’s Hereditary Optic Neuropathy: A Review of the Literature. Adv Ther 2018; 35(10): 1510–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Asanad S, Meer E, Tian JJ, Fantini M, Nassisi M, Sadun AA. Leber’s hereditary optic neuropathy: Severe vascular pathology in a severe primary mutation. Intractable Rare Dis Res 2019; 8(1): 52–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yu-Wai-Man P, Chinnery PF. Leber Hereditary Optic Neuropathy. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G et al. (eds). GeneReviews((R)): Seattle (WA), 2021. [PubMed] [Google Scholar]
- 59.Yu-Wai-Man P, Turnbull DM, Chinnery PF. Leber hereditary optic neuropathy. J Med Genet 2002; 39(3): 162–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yu-Wai-Man P, Griffiths PG, Chinnery PF. Mitochondrial optic neuropathies - disease mechanisms and therapeutic strategies. Prog Retin Eye Res 2011; 30(2): 81–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Carelli V, Ross-Cisneros FN, Sadun AA. Mitochondrial dysfunction as a cause of optic neuropathies. Prog Retin Eye Res 2004; 23(1): 53–89. [DOI] [PubMed] [Google Scholar]
- 62.Carelli V, Ghelli A, Ratta M, Bacchilega E, Sangiorgi S, Mancini R et al. Leber’s hereditary optic neuropathy: biochemical effect of 11778/ND4 and 3460/ND1 mutations and correlation with the mitochondrial genotype. Neurology 1997; 48(6): 1623–32. [DOI] [PubMed] [Google Scholar]
- 63.Lodi R, Taylor DJ, Tabrizi SJ, Kumar S, Sweeney M, Wood NW et al. In vivo skeletal muscle mitochondrial function in Leber’s hereditary optic neuropathy assessed by 31P magnetic resonance spectroscopy. Ann Neurol 1997; 42(4): 573–9. [DOI] [PubMed] [Google Scholar]
- 64.Lodi R, Carelli V, Cortelli P, Iotti S, Valentino ML, Barboni P et al. Phosphorus MR spectroscopy shows a tissue specific in vivo distribution of biochemical expression of the G3460A mutation in Leber’s hereditary optic neuropathy. J Neurol Neurosurg Psychiatry 2002; 72(6): 805–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yu-Wai-Man P Therapeutic Approaches to Inherited Optic Neuropathies. Semin Neurol 2015; 35(5): 578–86. [DOI] [PubMed] [Google Scholar]
- 66.Yuan J, Zhang Y, Liu H, Wang D, Du Y, Tian Z et al. Seven-Year Follow-up of Gene Therapy for Leber’s Hereditary Optic Neuropathy. Ophthalmology 2020; 127(8): 1125–1127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its supplementary information files.