

SUMMARY
Ferroptosis, a newly emerged form of regulated necrotic cell death, has been demonstrated to play an important role in multiple diseases including cancer, neurodegeneration, and ischemic organ injury. Mounting evidence also suggests its potential physiological function in tumor suppression and immunity. Execution of ferroptosis is driven by iron-dependent phospholipid peroxidation. As such, metabolism of biological lipids regulates ferroptosis via controlling phospholipid peroxidation, as well as various other cellular processes relevant to phospholipid peroxidation. In this review, we will provide a comprehensive analysis by focusing on how lipid metabolism impacts the initiation, propagation, and termination of phospholipid peroxidation, how multiple signal transduction pathways communicate with ferroptosis via modulating lipid metabolism, and how such intimate crosstalk of ferroptosis with lipid metabolism and related signaling pathways can be exploited for the development of rational therapeutic strategies.
In this review, Liang et al. describe the recent development on the pivotal role of lipid metabolism, as well as various signaling pathways that modulate lipid metabolism, in ferroptosis; they also highlight some unanswered questions in the field and suggest potential ferroptosis induction-based cancer therapeutic strategies.
INTRODUCTION
Programmed cell death, such as apoptosis, plays important physiological and pathological roles (Bedoui et al., 2020; Green, 2019; Kist and Vucic, 2021). There are other modes of programmed or regulated cell death distinct from apoptosis mechanistically and functionally (Green, 2019; Kist and Vucic, 2021). Among them, ferroptosis is a death process driven by iron-dependent phospholipid (PL) peroxidation (Jiang et al., 2021). Conceptually, ferroptosis can be considered as a byproduct of cellular metabolism: oxygen and iron are essential drivers of metabolism, leading to the production of reactive species (ROS) as an inevitable byproduct; if a specific class of ROS, phospholipid peroxides (PLOOH), cannot be neutralized efficiently and thus accumulate to disrupt plasma membrane integrity, cells will succumb to ferroptotic death (Figure 1). In the cell, the substrates of PL peroxidation are PLs containing polyunsaturated fatty acyl (PUFA) chains at the sn2 position; in presence of bioactive iron, PUFA-PLs can be converted to PLOOH through both enzymatic and non-enzymatic lipid peroxidation reactions. Since all mammalian cells contain certain levels of PUFA-PLs and bioactive iron, specific surveillance or protection mechanisms need to be in place to protect them from unwanted ferroptosis (Figure 1). The canonical surveillance mechanism is mediated by glutathione peroxidase-4 (GPX4), the only mammalian enzyme known to catalyze the reduction of phospholipid hydroperoxides into phospholipid alcohols (Seiler et al., 2008; Ursini et al., 1982). There are also GPX4-independent mechanisms that can mitigate ferroptosis. For example, enzymes such as FSP1, GCH1, and DHODH produce metabolites possessing free radical-trapping activity (Bersuker et al., 2019; Doll et al., 2019; Kraft et al., 2020; Mao et al., 2021; Soula et al., 2020), which can terminate the Fenton radical chain reaction and prevent the amplification of lipid peroxidation.
Figure 1. A summary of ferroptosis.
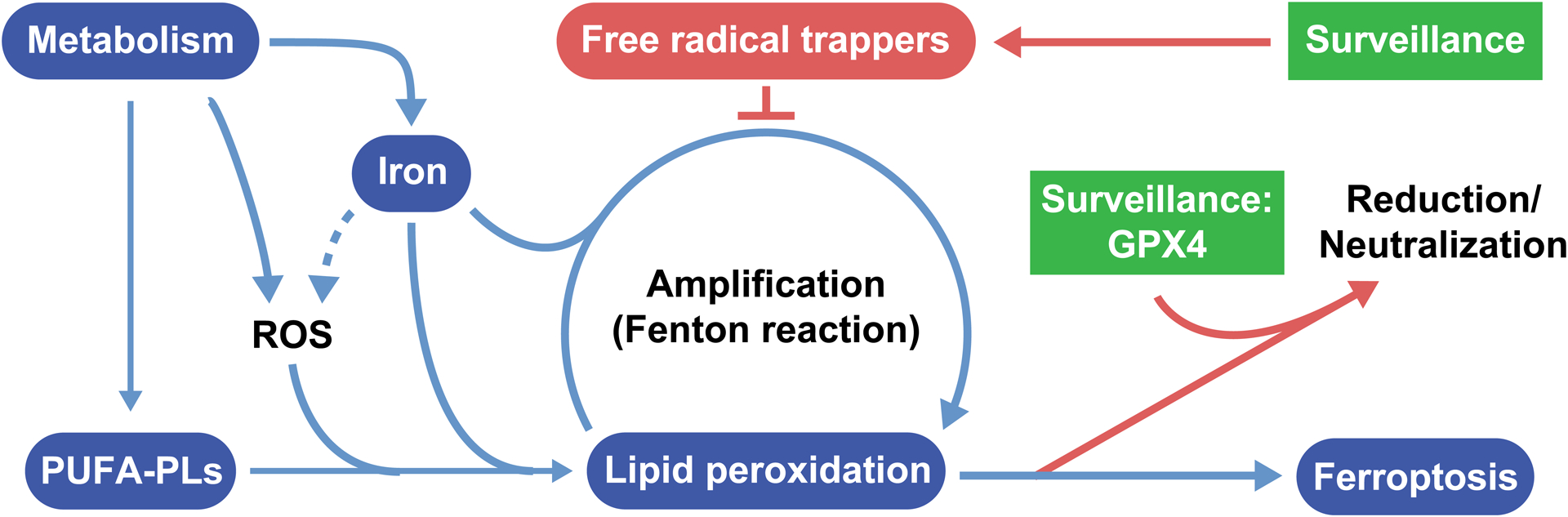
The figure shows the core features of ferroptosis, including metabolic production of PUFA-PLs, which are the substrates of phospholipid peroxidation; iron-dependent reactions leading to the initiation and propagation of phospholipid peroxidation; and surveillance mechanisms that can keep phospholipid peroxidation in check.
Unsurprisingly, the decision of ferroptosis is determined by which of these cellular processes – PL peroxidation or ferroptosis surveillance – can overcome the other. While PL peroxidation can be considered a specific process of lipid metabolism, various ferroptosis surveillance mechanisms have also been shown to be highly regulated by cellular lipids and lipid metabolism (discussed later). Therefore, cellular lipids and lipid metabolism play a central role for the regulation of ferroptosis.
Among the various classes of cellular lipids, phospholipids (PLs), along with sphingolipids and cholesterol, are major components of cellular membranes. The chemical diversity of PLs arises from combinations of two fatty acyl chains (designated sn1 and sn2) and the head group. The sn1 position tends to be occupied by saturated fatty acids (SFA) or monounsaturated fatty acids (MUFA), while the sn2 position can be a SFA, MUFA, or PUFA. Membrane lipid composition is highly diverse among organisms, cell types, organelles, and even membrane subdomains, and is dynamically regulated (reviewed in (Harayama and Riezman, 2018)). Generally, SFA-PLs make cell membranes more rigid, while PLs with a higher degree of unsaturation render membranes more pliable. PL content is regulated by multiple mechanisms, including the synthesis, uptake, storage, release, and beta oxidation of fatty acids, as well as by PL synthesis and PL remodeling process known as the Lands’ Cycle (Snaebjornsson et al., 2020) (Figure 2). Intriguingly, although increasing PUFA-PL content in the cell membrane can fulfill numerous cellular functions through improving the fluidity of the cell, it also increases the intrinsic susceptibility to ferroptosis (PUFA-PLs are the substrates of PL peroxidation) (Doll et al., 2017; Kagan et al., 2017). Under physiological conditions, such alteration of ferroptosis susceptibility is likely under close regulation; but this same property might be exploited therapeutically for the treatment of human diseases such as cancer. In this review, we will discuss in detail the myriad roles lipids play in ferroptosis, especially how lipid metabolism dynamically regulates the balance of cellular function and ferroptosis surveillance, and how cellular signaling pathways intersect with lipid metabolism to regulate ferroptosis, in the contexts of normal biology and disease.
Figure 2. Lipid metabolic pathways across different cellular compartments.
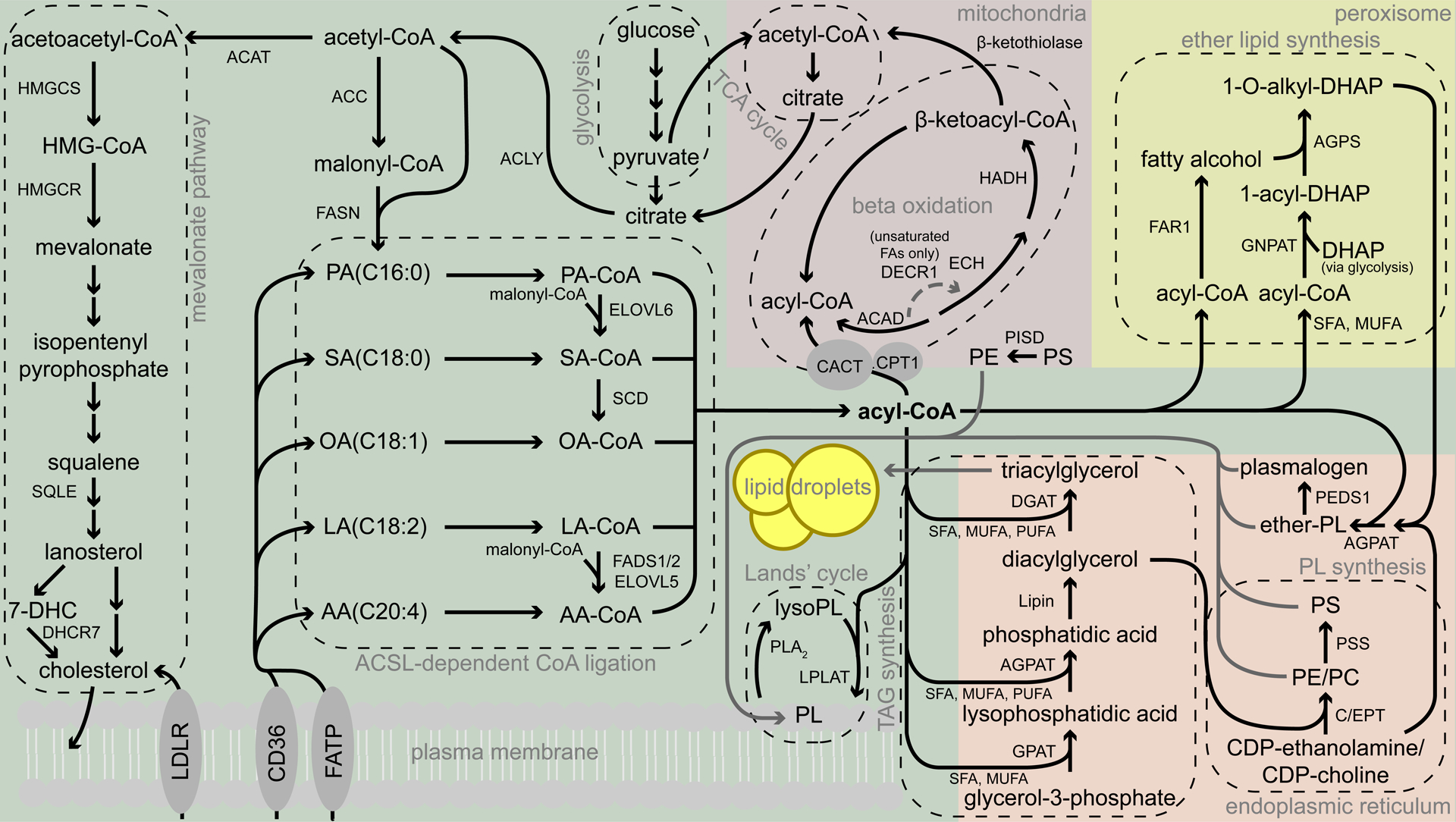
Fatty acids can either be taken up by the cell, through transporters including CD36 or the FATP family, or synthesized de novo from glucose via conversion to acetyl-CoA. Acetyl-CoA can be used for cholesterol synthesis via the mevalonate pathway or for the production or extension of new fatty acids. The ACSL enzymes ligate coenzyme A to fatty acids for elongation, beta oxidation, ether lipid synthesis, triglyceride synthesis, or phospholipid synthesis. Phospholipids are transported to the membrane, where they can then be remodeled by the Lands’ cycle.
ACAT, acetyl-CoA acetyltransferase; HMGCS, hydroxymethylglutaryl-CoA synthase; HMGCR, hydroxymethylglutaryl-CoA reductase; SQLE, squalene monooxygenase; 7-DHC, 7-dehydrocholesterol; DHCR7, 7-dehydrocholesterol reductase; LDLR, low density lipoprotein receptor; CD36, fatty acid translocase; FATP, fatty acid transport protein; ACLY, ATP-citrate lyase; ACC, acetyl-CoA carboxylase; FASN, fatty acid synthase; PA, palmitic acid; SA, stearic acid; OA, oleic acid; LA, linoleic acid; AA, arachidonic acid; ACSL, acyl-CoA synthetase, long chain; ELOVL, elongation of very long chain fatty acid protein; SCD, stearoyl-CoA desaturase; FADS, fatty acid desaturase; CPT1, carnitine palmitoyltransferase 1; CACT, carnitine-acylcarnitine translocase; ACAD, acyl-CoA dehydrogenase; DECR1, 2,4-dienoyl-CoA reductase 1; ECH, enoyl-CoA hydratase; HADH, hydroxyacyl-CoA dehydrogenase; PISD, phosphatidylserine decarboxylase proenzyme; GPAT, glycerol-3-phosphate acyltransferase; AGPAT, acylglycerol-3-phosphate O-acyltransferase; DGAT, diacylglycerol acyltransferase; C/EPT, choline/ethanolamine phosphotransferase; PE, phosphatidylethanolamine, PC, phosphatidylcholine; PSS, phosphatidylserine synthase; PEDS1, plasmanylethanolamine desaturase 1; FAR1, fatty acyl-CoA reductase 1; GNPAT, glyceronephosphate O-acyltransferase; DHAP, dihydroxyacetone phosphate; AGPS, alkylglycerone phosphate synthase.
PHOSPHOLIPID PEROXIDATION
As the committing event of ferroptosis, PL peroxidation occurs in three steps: initiation, propagation, and termination (Figure 3). The peroxidation of fatty acyl chains of PLs is facilitated by a bisallylic group (−CH=CH−CH2−CH=CH−) due to the weak bonds connecting the hydrogens in the central methylene group. These bonds are weak enough that a strong oxidant can cleave it, forming a radical. For this reason, PUFA-PLs, which contain bisallylic groups in PUFA chains, are most susceptible to oxidation. Due to the surrounding double bonds, the radical can be isomerized to a more thermodynamically favorable state: a conjugated diene (Figure 3A). Finally, the radical reacts with molecular oxygen to form a hydroperoxide. Lipid peroxidation can be propagated when one lipid radical reacts with the methylene hydrogen in another bisallylic group on a nearby lipid. Interestingly, it appears that multiple bisallylic groups within a single fatty acid chain can also propagate lipid peroxidation (Else and Kraffe, 2015). Finally, termination occurs when the propagation can no longer continue, either when there are no more available lipid substrates, or when cellular antioxidants reduce the oxidized lipids.
Figure 3. Lipid peroxidation.
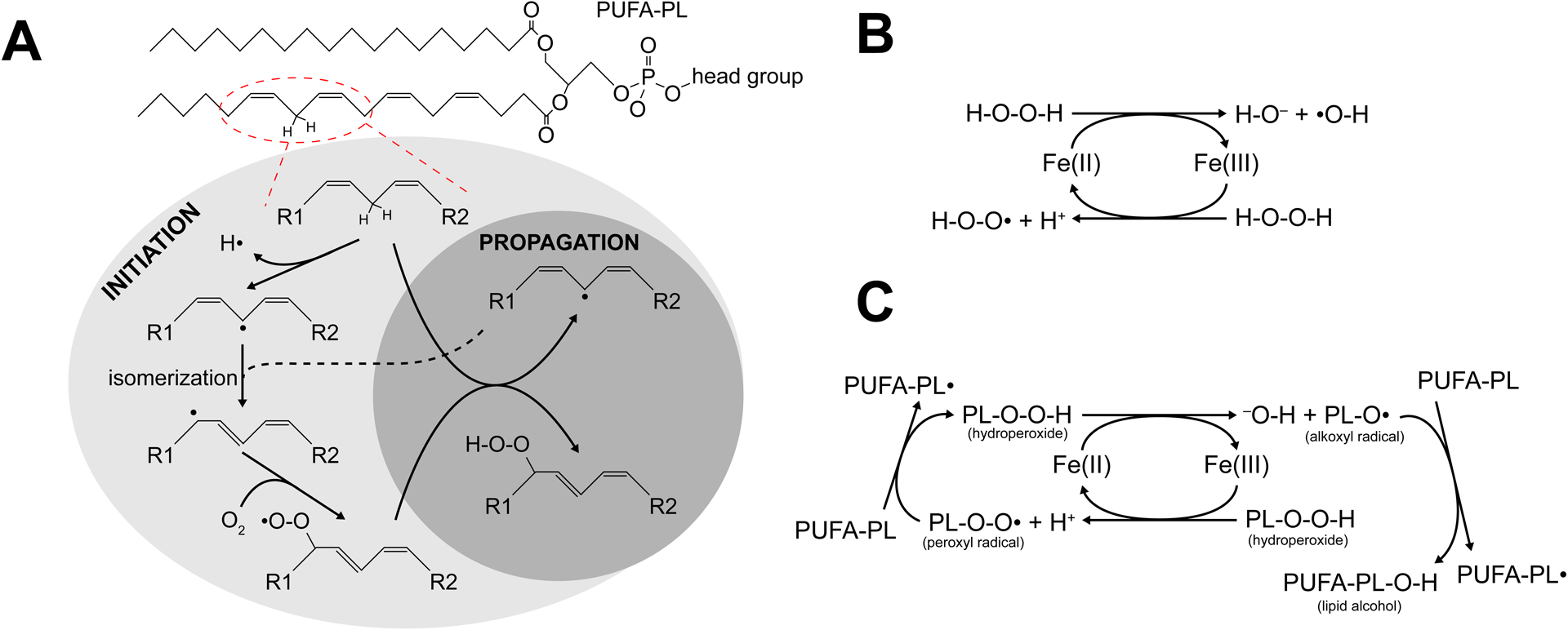
(A) An example phospholipid with a PUFA chain in the sn2 position can be oxidized at the encircled position, either enzymatically or non-enzymatically. The resulting radical can be isomerized to a more energetically favorable conformation and react with molecular oxygen, forming a peroxyl radical. This radical can then react with another PUFA chain, creating another radical on that chain and forming a hydroperoxide. (B) The basic Fenton reaction. Iron can shift between the ferrous and ferric states, and both transition states can react with hydrogen peroxide to generate a radical. (C) The Fenton reaction can also utilize phospholipid hydroperoxides as substrates. The phospholipid radicals formed by this process can react with other PUFAs to propagate a chain reaction of oxidation.
Initiation and Propagation of PL Peroxidation
PL peroxidation can be initiated in cells both enzymatically and nonenzymatically. Nonenzymatic peroxidation of lipids is catalyzed by redox active metals, particularly iron. Within cells, iron is typically bound within a complex, such as heme (a prosthetic group of numerous proteins, such as hemoglobin), iron-sulfur clusters (a cofactor for many enzymes), and ferritin (a dedicated iron storage protein). A small amount of iron within cells is not bound in such a way, called the labile iron pool. Labile iron is highly reactive, particularly with hydrogen peroxide (H2O2). This reaction, known as the Fenton reaction, cycles between ferrous (Fe2+) and ferric (Fe3+) ions, converting H2O2 to either a hydroxide ion (OH−) and a hydroxyl (HO•) radical or a proton and a peroxyl (HOO•) radical (Figure 3B). Both hydroxyl and peroxyl radicals can initiate lipid peroxidation. Like H2O2, PLOOH can also undergo iron-catalyzed Fenton reaction, generating lipid hydroxyl radical PLO• and lipid peroxyl radical PLOO• (Figure 3C). If PLOOH is formed and is not neutralized quickly enough, it can propagate peroxidation to neighboring PUFA-PLs in the presence of labile iron. As such, cellular processes resulting in an increase of the cellular labile iron pool, such as autophagic degradation of ferritin (Gao et al., 2016; Hou et al., 2016), uptake of transferrin (Gao et al., 2015), or inhibition of iron exporter ferroportin (Bao et al., 2021; Chen et al., 2020; Geng et al., 2018) can all sensitize the cell to ferroptosis.
Multiple enzymes have been shown to drive lipid peroxidation. Importantly, these enzymes are all iron-dependent. Lipoxygenases, a family of enzymes that are critical for several signaling pathways and contains non-heme iron for their activity, directly catalyze the formation of lipid radicals. In humans, lipoxygenases are named for their preferred oxidation site relative to arachidonic acid (AA) and differ in substrate selectivity (both target site and substrate localization). For instance, ALOX5 converts AA into 5-HPETE, a precursor for leukotrienes, a family of inflammatory signaling lipids. It should be noted that although the enzymatic activity of ALOXs can sensitize cells to ferroptosis, they are not indispensable due to the existence of other enzymatic and nonenzymatic mechanisms for PL peroxidation. Furthermore, pharmacological inhibitors of ALOXs should be used with caution, as many of them are strong radical trapping agents and thus inhibit ferroptosis independently of ALOXs (Shah et al., 2018; Yang et al., 2016). Expression of ALOX genes is hardly detectable in some of the commonly used cell lines in ferroptosis research (Ghandi et al., 2019), suggesting a limited role of the ALOX family members in ferroptosis, likely only in tissues expressing ALOXs. The ER-residing P450 oxidoreductase (POR), has also been shown to be critical for the initiation of PL peroxidation and ferroptosis (Yan et al., 2021; Zou et al., 2020b). The P450 system is a heme-containing enzyme family critical for the metabolism and detoxification of many drugs, proteins, or xenobiotics via oxidation in the endoplasmic reticulum (Pandey and Sproll, 2014). POR can donate electrons to a member of the cytochrome p450 family (CYP), allowing the CYP to oxidize and metabolize another molecule. CYP family members, particularly the CYP4A family, have been demonstrated to directly oxidize arachidonic acid as a substrate (Schwartzman et al., 1990). While POR expression is ubiquitous, expression of CYP isoforms is highly tissue-specific; for example, many CYP4A isoforms are only expressed in the liver or kidney (Simpson, 1997). Recent studies suggest that POR can enhance lipid peroxidation without affecting the lipidome of cells, and that POR does not need CYP to induce ferroptosis. Instead, POR, as well as another ER-residing oxidoreductase CYB5R1, directly generate H2O2 to induce lipid peroxidation through the Fenton reaction (Yan et al., 2021). Importantly, unlike the tightly regulated ALOX enzymes, POR is ubiquitously expressed, suggesting POR is a constitutive catalyst for PL peroxidation.
Termination of PL Peroxidation and Ferroptosis Surveillance
PL peroxidation can only propagate for so long; eventually, cells die of ferroptosis or survive because the chain reaction will either run out of substrates, or something will stop it in its tracks. GPX4 can catalyze the reduction of toxic PLOOH to a nontoxic PL alcohol (PLOH) (Ursini et al., 1982), thus representing the canonical mode of ferroptosis surveillance (Figure 4). Inhibition of GPX4 enzymatic activity directly by targeting its selenocysteine site (Yang et al., 2014), or indirectly through targeting the glutamate/cystine transporter system xc− and thus glutathione synthesis (Dixon et al., 2012) leads to the propagation of PL peroxidation, the accumulation of PLOOH, and ferroptosis. GPX4 is an essential gene, as systemic GPX4 knockout is embryonic lethal in mice and conditional GPX4 knockout triggered diseases such as acute renal failure, likely due to ferroptosis (Friedmann Angeli et al., 2014).
Figure 4. Ferroptosis surveillance.

Ferroptosis is a death process driven by iron-dependent phospholipid peroxidation. The canonical ferroptosis surveillance mechanism is mediated by GPX4, which catalyzes the reduction of lipid peroxides into lipid alcohols. In parallel, enzymes such as FSP1, DHODH, GCH1, and NOS2, which produce metabolites with free radical-trapping activity to terminate the phospholipid Fenton reaction, are classified as the second surveillance mechanism. PUFA-PLs render cells more prone to ferroptosis, whereas MUFAs, likely though MUFA-PLs, can inhibit ferroptosis. As such, the balance between MUFA-PLs and PUFA-PLs may determine the susceptibility of the cell to ferroptosis. The balance between MUFA-PLs and PUFA-PLs is dictated by the supply and activation of MUFAs and PUFAs, as well as by the PL remodeling process (Lands’ cycle). These processes in combination form another potential mechanism for ferroptosis surveillance.
Besides enzymatic reduction of PLOOH by GPX4, lipophilic radical-trapping antioxidants (RTA) can terminate the propagation of PL peroxidation, thus blocking ferroptosis caused by GPX4 deficiency (Dixon et al., 2012). Recent studies indicate that various enzymes inhibit ferroptosis by catalyzing the production of endogenous RTAs, representing another surveillance mechanism (Figure 4). CoQ10 is an important component of the oxidative phosphorylation machinery for its ability to carry two electrons, owing to its three redox states: ubiquinone (fully oxidized), semiquinone, and ubiquinol (fully reduced). CoQ10 is highly lipophilic and can (in the ubiquinol form) act as a radical trapping agent. FSP1 (also known as AIFM2), a cytosolic NADH:ubiquinone oxidoreductase was identified as having the capability of terminating PL peroxidation by generating ubiquinol (Bersuker et al., 2019; Doll et al., 2019). FSP1 can also reduce α-tocopherol, likely through ubiquinol, which may explain the strong anti-ferroptotic activity of FSP1 despite the relatively weak biological activity of ubiquinol. Similarly, the mitochondrial protein DHODH has been shown to inhibit ferroptosis by reducing ubiquinone to ubiquinol inside the mitochondrial inner membrane (Mao et al., 2021). Tetrahydrobiopterin, also known as BH4, is another endogenous RTA that acts similarly to α-tocopherol. Two CRISPR screens for novel regulators of ferroptosis identified a handful of genes in the tetrahydrobiopterin synthesis pathway, including GCH1, PTS, and SPR, for promoting survival upon GPX4 inhibition (Kraft et al., 2020; Soula et al., 2020).
Yet another surprising GPX4-independent mechanism cells can use to defend against ferroptosis is through nitric oxide (NO). NO is an important signaling molecule for a number of cellular processes (Martínez-Ruiz et al., 2011) and is produced by nitric oxide synthases (NOS), which are expressed in specific tissues and contexts. NO can react with superoxide and form reactive nitrogen species (RNS), which can cause damage to lipids and proteins, binding to transition metals or altering their structure by S-nitrosylating cysteine residues (Gow et al., 2002). However, nitric oxide is also a potent RTA, and NO concentrations as low as 30 nM can outcompete physiological pools of α-tocopherol, around 20 μM (O’Donnell et al., 1997). Recent studies have identified role of NO in ferroptosis. M1 macrophages, which express the iNOS member of the NOS family, have elevated NO concentrations. Stimulation with lipopolysaccharide and IFN-γ efficiently suppressed ferroptosis in M1 macrophages, and knockdown of iNOS sensitized these cells to RSL3-induced ferroptosis (Kapralov et al., 2020). Further studies found that elevated iNOS activity in macrophages following infection with the bacteria P. aeruginosa could stave off lipid peroxidation that damages airway epithelial cells (Dar et al., 2021). Another study identified that the NO donor NOC 18 could inhibit ferroptosis through this mechanism as well (Homma et al., 2021). Although NO appears to be a potent ferroptosis inhibitor in certain contexts, this potential mechanism needs to be accessed cautiously because of the damaging effects of reactive nitrogen species and myriad signaling roles of NO.
The reactions of RTAs with PL radicals are rather complicated and not fully understood, and it is not clear how the remaining non-radical PLOOHs or their by-products are detoxified, especially when GPX4 activity is insufficient. Recently, Ca2+-independent phospholipase A2β (iPLA2β) was identified as a suppressor of ferroptosis due to its ability to preferentially remove oxidized fatty acyl chains of PLs (Beharier et al., 2020; Chen et al., 2021; Sun et al., 2021). This potentially represents yet another surveillance mechanism in addition to those mediated by GPX4 and RTAs (Figure 4). However, whether such a mechanism is sufficient to protect cells from ferroptosis in the absence of the function of GPX4 and RTA-producing enzymes is not clear.
VERSATILE ROLES OF LIPIDS IN FERROPTOSIS
Polyunsaturated fatty acids
The composition of cellular membranes is an important determinant of whether a cell will survive or die under ferroptosis-triggering conditions. As discussed, PUFA-PLs can propagate lipid peroxidation, which is central to the execution of ferroptosis. In animal cells, arachidonic acid (AA) is one of the most abundant PUFA species. As an ω-6 fatty acid, AA has a 20-carbon chain and 4 unsaturated bonds (termed 20:4). PLs with AA or adrenic acid (AdA, 22:4) at the sn2 position have been identified as being particularly sensitive to PL peroxidation, likely because both AA and AdA have two bisallylic groups. In mammals, these species are preferentially oxidized over other PUFA-PLs, such as those containing linoleic acid (LA, 18:2), which only has a single bisallylic group (Kagan et al., 2017). However, LA supplementation can enhance ferroptosis in mammalian cells (Yang et al., 2016), and Arabidopsis thaliana, which contains LA as its primary PUFA species, can undergo ferroptosis under heat stress (Distéfano et al., 2017; He et al., 2020). Further, docosahexanoic acid (22:6), a PUFA with three biallylic groups, may be more readily oxidizable than AA or AdA, although it is less abundant in PLs in most tissues.
While AA is readily available to be absorbed from meat in the diet, it can also be produced from LA, another ω-6 PUFA, through a series of elongation and desaturation reactions. LA, the primary PUFA obtained from plant food sources, can be converted to AA first by the fatty acid desaturase FADS2, which converts LA to γ-LA (18:3). ELOVL5, a long chain fatty acid elongase, is primarily responsible for the elongation of 18 carbon fatty acids to 20 carbons (Robichaud et al., 2018). ELOVL5 converts γ-LA to dihomo-γ-LA (DGLA, 20:3). Finally, FADS1 desaturates DGLA to AA (Figure 2). Conceivably, FADS1, FADS2, and ELOVL5 have all been identified as enhancers of ferroptosis (Lee et al., 2020b; Yamane et al., 2021).
For a free fatty acid (FA) to be esterified into a PL, the fatty acid must first be linked to Coenzyme A (CoA). The Acyl-CoA Synthetase Long Chain (ACSL) family of ligases catalyze this reaction, in a substrate-specific manner (Figure 2). For example, in vitro studies have identified AA and eicosapentanoic acid (20:5) as the primary substrates of ACSL4 (Kang et al., 1997). Mice with an adipocyte-specific knockout of Acsl4 have a sharp decrease in PLs containing arachidonic or docosapentanoic (22:5) fatty acid chains, with a concomitant increase in LA (Killion et al., 2018). Unsurprisingly, ACSL4 is central to ferroptosis (Dixon et al., 2015; Doll et al., 2017; Kagan et al., 2017). Loss of ACSL4 severely tempers ferroptotic cell death in many cell lines, but does not totally block ferroptosis (Zou et al., 2020a), suggesting complementary mechanism(s) for the loss of ACSL4.
Loss of ACSL4 results in an increase in unesterified arachidonate. Although free AA is still sensitive to peroxidation, such peroxidation is insufficient to induce ferroptosis. Therefore, peroxidation of PLs but not of fatty acids appears to be crucial for ferroptosis (Kagan et al., 2017). This highlights a limitation of one of the most important tools used in the field: fluorescent probes of lipid peroxidation. Proxy measurements with probes such as BODIPY 581/591 C11 are used widely to monitor PL peroxidation. However, this method cannot distinguish PL peroxidation from peroxidation of fatty acids or other membrane lipids.
Fatty acyl-CoA can be esterified to the sn2 position of a lysophospholipid (lysoPL), resulting in a phospholipid (PL). This reaction is catalyzed by the lysophosphatidyl CoA acyltransferases, or LPCAT family. As ACSLs, individual LPCAT family members have different substrate preference (Kazachkov et al., 2008). LPCAT3 is primarily responsible for esterifying AA into lysoPLs, and thus is also an important player in ferroptosis (Dixon et al., 2015). Potential role of other LPCAT family members in ferroptosis has not been systematically investigated.
Monounsaturated fatty acids
Unlike PUFAs, monounsaturated fatty acids (MUFAs) have been demonstrated to be potent inhibitors of ferroptosis (Magtanong et al., 2019; Yang et al., 2016). In an ACSL3-dependent manner, exogenous MUFAs were able to efficiently block ferroptosis and limit lipid peroxidation at the plasma membrane. While this phenotype is quite clear, the underlying mechanism has not been defined. One possible explanation is that MUFAs compete with PUFAs for PL synthesis. However, this mechanism needs excessive amount of MUFAs over PUFAs; it also suggests that saturated fatty acids (SFAs) would have similar inhibitory activity, which is not the case. Another possibility is that exogenous MUFAs can restructure lipid metabolism within the cell. Excess lipids can be stored in cells within specialized organelles, lipid droplets, in the form of triacylglycerol (TAG), a particularly non-reactive form of lipid (Figure 2). However, inhibition of DGAT1/2, the enzymes that convert fatty acyl-CoA into TAG, was unable to restore cell death when oleic acid (18:1) was added to cells (Magtanong et al., 2019). Therefore, how MUFAs inhibit ferroptosis remains to be an important yet unanswered question.
Ether-linked phospholipids
Recent research has demonstrated that the sn2 position of the lipid is not the only determinant of sensitivity to ferroptosis. The sn1 fatty acid, typically an SFA, can be linked to the glycerol moiety either via an ester linkage (as the sn2 fatty acid) or via an ether linkage. Ether-linked PLs, though less common in animals than the ester-linked PLs, comprise a significant percentage of the PL pool. While most PLs are wholly produced at the endoplasmic reticulum, ether-linked PLs start in the peroxisome (Dean and Lodhi, 2018). Recently, a CRISPR-Cas9 screen identified multiple peroxisomal genes involved in the ether lipid synthesis pathway as promoters of ferroptosis (Zou et al., 2020a). This is the first evidence suggesting that the sn1 fatty acyl chains can also influence ferroptosis sensitivity. Importantly, this suggests that two separate synthesis pathways converge on PL production to control ferroptosis. In support of this, ACSL4 knockout further suppressed sensitivity to ferroptosis over individual knockout of genes involved in peroxisomal ether PL synthesis.
A subclass of ether PLs called plasmalogens are identifiable by a vinyl ether linkage. After the ether linkage is complete, the nascent PL is shuttled to the endoplasmic reticulum, where the sn2 lipid is added. An additional step catalyzed by PEDS1 (also known as TMEM189) desaturates the first C-C bond after the ether group, creating the vinyl ether (Werner et al., 2020). This vinyl ether moiety may be critical; it may function as an antioxidant that can halt the propagation step of lipid peroxidation (Sindelar et al., 1999). Zou et al did not observe a difference with or without PEDS1. However, a later study reported that although the peroxisomal synthesis of ether lipids was critical for ferroptosis, PEDS1-dependent plasmalogen synthesis suppressed ferroptosis (Cui et al., 2021). These contradiction warrants further investigation to reconcile the seemingly paradoxical regulation of ferroptosis by PEDS1 and plasmalogens.
Cholesterol
Cholesterol (Ch) is another essential lipid component of mammalian membranes and is required to maintain membrane integrity, fluidity, and membrane microstructures. Most mammalian cells obtain cholesterol via both endogenous synthesis and exogenous uptake (Figure 2). Ch is synthesized through the mevalonate pathway, which generates its precursor squalene via a series of isoprenoid intermediates: isopentenyl pyrophosphate (IPP), geranyl-PP, and farnesyl-PP. IPP is also required for protein prenylation, CoQ10 and Dolichol synthesis (reviewed in (Snaebjornsson et al., 2020)). The mevalonate pathway has been suggested to affect ferroptosis in three distinct ways: (1) As a selenoprotein, GPX4 synthesis requires a unique selenocysteine tRNA (Sec-tRNA). IPP regulates Sec-tRNA maturation through adenosine isopentenylation at position 37 (i6A), and therefore is required for GPX4 synthesis (Warner et al., 2000). (2) The radical trapping antioxidant CoQ10 is produced by conjugating ten isoprene units from IPP to a hydroxybenzoic acid backbone, thus inhibition of mevalonate pathway will decrease CoQ synthesis (Littarru and Langsjoen, 2007). (3) Some metabolic intermediates in the cholesterol synthesis pathway can act as RTAs (Garcia-Bermudez et al., 2019). Indeed, due to the unique connection of the mevalonate pathway to GPX4 and CoQ10, inhibiting HMGCR, a rate limiting enzyme in mevalonate pathway, can induce ferroptosis in some cancer cells (Viswanathan et al., 2017). Further, it was shown recently that 7-dehydrocholesterol reductase (DHCR7), which catalyzes the final step in distal cholesterol biosynthesis, played an unexpected pro-survival function against ferroptosis (Angeli et al., 2021). Inhibition of DHCR7 leads to the accumulation of 7-dehydrocholesterol (7-DHC), which has superior reactivity towards peroxyl radicals. Similarly, another study showed that loss of squalene monooxygenase (SQLE), a rate-limiting enzyme in the cholesterol biosynthetic pathway, leads to the accumulation of squalene and confers protection against ferroptosis in cholesterol auxotrophic cells (Garcia-Bermudez et al., 2019).
Interestingly, Ch is susceptible to peroxidation to generate various Ch-OOH products, and GPX4 is also capable of reducing Ch-OOH to Ch-OH (Thomas et al., 1990). In theory, in the presence of iron, Ch-OOH can propagate peroxidation to adjacent lipids, presumably including PUFA-PLs (reviewed in (Girotti and Korytowski, 2019)). As such, a highly relevant question is, how is Ch peroxidation induced and can it lead to ferroptosis? Furthermore, Ch and its metabolites can act as signaling molecules in the tumor microenvironment (TME) and have been reported to regulate cancer cell ferroptosis. Ch in the TME upregulates CD36 (encoding the fatty acid transporter) expression in tumor-infiltrating CD8+ T cells. Consequently, CD36-mediated uptake of fatty acids induces lipid peroxidation and ferroptosis, leading to the exhaustion of CD8+ T cells and impairment of antitumor functionality, which could be reversed by blocking CD36 or inhibiting ferroptosis (Ma et al., 2021). 27-hydroxycholesterol (27HC) is the most abundant oxysterol in the plasma membrane and blood. Long-term treatment of estrogen receptor (ER)-negative breast cancer cells with 27HC selects for more aggressive cells that exhibit elevated cellular uptake of lipids or increased lipid biosynthesis (Liu et al., 2021). However, this will also increase cellular dependence on GPX4 to suppress ferroptosis in the 27HC-resistant cells, as GPX4 knockdown can attenuate the enhanced tumorigenic and metastatic activity of 27HC-resistant cells (Liu et al., 2021).
LIPID METABOLISM AND FERROPTOSIS
The composition of cellular lipids is dynamically regulated to fulfill various cellular functions. While a given cellular function may require a specific lipid composition, the same lipid composition might also make the cell more vulnerable to certain stressors. Therefore, cells may employ sophisticated mechanisms to sense and modulate PL composition (reviewed in (Harayama and Riezman, 2018)). An example relevant to this review: as PUFA-PLs make cell membranes more flexible, they facilitate processes that involve frequent membrane bending and change of morphology, such as endocytosis and cell migration; however, excessive PUFA-PLs predispose the cell to ferroptosis. Therefore, cells respond to elevated PUFAs by altering relevant lipid metabolism, such as increasing saturated phosphatidylcholine (PC) synthesis. Indeed, lipid metabolism can profoundly affect PL composition by adjusting the balance between MUFA-PL and PUFA-PL, thus impacting the susceptibility of the cell to ferroptosis. Lipid metabolism controls PL composition through two major mechanisms: (1) modulating the supply of FA through synthesis, uptake, storage, and β oxidation; (2) the expression and regulation of enzymes in PL synthesis and remodeling.
FA Synthesis
SFA and MUFA can be synthesized de novo in the cell. Briefly, acetyl CoA is carboxylated to form malonyl CoA by acetyl CoA carboxylase (ACC), then malonyl-CoA is further converted to palmitic acid, an SFA, by fatty acid synthase (FASN). Palmitoyl-CoA or stearoyl-CoA can then be desaturated by SCD1 to form palmitoleic acid and oleic acid, both MUFAs, respectively. Expression of ACC, FASN and SCD1 are under the control of the transcription factors LXR and the sterol regulatory element-binding protein-1 (SREBP1) (Snaebjornsson et al., 2020). ACC, FASN and SCD1 are overexpressed in some cancer cells which are dependent on FA synthesis for energy. Knockdown of SREBP1 or SCD1 will sensitize cancer cells to ferroptosis (Tesfay et al., 2019; Yi et al., 2020). Unlike SFA or MUFA, long chain ω-3 or ω-6 PUFAs cannot be synthesized de novo by mammalian cells. As a result, upregulated lipid synthesis increases MUFA-PL contents and tends to confer resistance to ferroptosis. Mammals require the essential PUFAs αlinolenic acid (ALA; 18:3n-3) and linoleic acid (LA; 18:2n-6) through diet, and can then synthesize other long chain PUFAs through a chain of desaturation and elongation reactions (using malonyl-CoA as a source of carbon) that are catalyzed by various desaturase (FADS1/2) and elongation enzymes (e.g., ELOVL5) (Figure 2). FADS1, FADS2, and ELOVL5 have all been identified as enhancers of ferroptosis (Lee et al., 2020b).
Lipid uptake
Dietary FA is another important source of FA pool, especially for cells deficient of lipid synthesis. Free FA or lipoprotein are absorbed through CD36, FA transport proteins (FATPs) and FA-binding proteins (FABPs) (Nagarajan et al., 2021). The content of dietary FAs (MUFA vs PUFA) significantly affects the cellular pool of FAs, which ultimately changes the cellular profiles of PL and sensitivity to ferroptosis. Melanoma cells metastasized via blood vessels have been shown to be sensitive to ferroptosis, and administration of a ferroptosis inhibitor significantly enhanced metastasis of these cells through the vasculature (Ubellacker et al., 2020). Intriguingly, these melanoma cells absorbed MUFA-enriched lipoproteins from the lymphatic vessel, reshaping the plasma membrane to have elevated MUFA-PLs and protecting the cells from ferroptosis (Ubellacker et al., 2020).
In some instances, lipid uptake can promote PUFA content and ferroptosis sensitivity in the cell. For example, CD36-mediated fatty acid uptake promotes ferroptosis in both B1 and marginal zone B (MZB) cells (Muri et al., 2019) as well as in tumor-infiltrating CD8+ T cells, which negatively regulates antitumor immunity (Ma et al., 2021). Supplementation of ω-3 or ω-6 PUFAs was reported to selectively induce ferroptosis in cancer cells under ambient acidosis (Dierge et al., 2021). Acidic pH promotes autocrine TGFβ2 signaling, which induces CD36 to facilitate FA uptake and stimulates the formation of lipid droplets. Upon exceeding the buffering capacity of triglyceride storage into lipid droplets, ω-3 and ω-6 PUFA peroxidation led to cytotoxic effects, exacerbated in the presence of diacylglycerol acyltransferase inhibitors, which inhibit lipid droplet formation. A long-chain ω-3 PUFA-rich diet significantly delayed mouse tumor growth compared with a MUFA-rich diet, an effect further accentuated by administration of DGAT inhibitors or ferroptosis inducers (Dierge et al., 2021). Since dietary PUFA shapes PL profiles in both cancer cells and immune cells, further studies are required to test whether dietary PUFA is complement to pharmacological approaches to inhibit tumor growth by ferroptosis.
Beta-oxidation
Beta-oxidation is generally believed to have a suppressive effect on ferroptosis by decreasing the availability of unesterified PUFAs. Acyl-CoA is linked to carnitine, a reaction catalyzed by carnitine palmitoyltransferase 1 (CPT1), allowing it to enter the mitochondria through carnitine-acylcarnitine translocase (CACT). Acyl-CoA is released from carnitine by CPT2 in the mitochondria, allowing it to enter the beta-oxidation cycle. A molecule of acetyl-CoA is released each cycle as two carbons are cleaved from the acyl chain (cleavage occurs at the beta carbon). An additional step is required for some cycles when unsaturated fatty acids are reduced, because these bonds must be saturated prior to oxidation. 2,4-dienoyl-CoA-reductase 1 (DECR1) is the rate limiting enzyme for PUFA reduction, as it catalyzes this step in the mitochondria (Figure 2). In human prostate cancer (PCa), beta-oxidation is the main bioenergetic pathway and a promising therapeutic vulnerability. DECR1 is frequently overexpressed in PCa tissues and is associated with poor survival. DECR1 knockout induces ER stress and sensitizes castration resistant-PCa cells to ferroptosis in vitro and in vivo, and inhibition of beta-oxidation enhanced ferroptosis in cancer cells (Blomme et al., 2020; Nassar et al., 2020).
Phospholipid synthesis and remodeling
Ferroptosis sensitivity is not only influenced by enzymes catalyzing FA synthesis, but also by enzymes that catalyze the esterification of FAs into PLs. The enzymes ACSL3 and ACSL4 were shown to regulate the composition of PLs and ferroptosis sensitivity due to their distinct substrate preference towards MUFAs and PUFAs, respectively. Activated FAs are incorporated into sn-2 position of glycerol backbone through two distinct pathways: the de novo synthesis pathway and remodeling pathway. In the de novo synthesis pathway, acyl-CoA and glycerol-3-phosphate (a byproduct of glycolysis) are combined to form lysophosphatidic acid. Another acyl-CoA, typically a MUFA-CoA or PUFA-CoA, is esterified by lysophosphatidic acid acyltransferases (LPAATs, also known as AGPATs) to form phosphatidic acid. Phosphatidic acid is converted to diacylglycerol (DAG) by a phosphatidyl phosphatase, known as Lipin in mammals. DAG can either be converted into a triacylglycerol (TAG) by the DGAT proteins or be linked to a phospholipid head group (Figure 2). In the PL remodeling pathway (Lands’ cycle), phospholipase A2 (PLA2) hydrolyzes acyl chains at the sn-2 position of PLs, generating a 1-acyl-lysoPL, which then can be re-acylated by lysoPL acyltransferases (LPLATs) (Figures 2, 4). As individual LPAAT or LPLAT enzymes differ in their preference for FA substrates and lysoPL targets, this process thus generates diverse PL species. LPCAT3 (LysoPC acyltransferase 3), and AGPAT3 (1-acylglycerol-3-phosphate O-acyltransferase 3, also called LPAAT3), were shown to promote ferroptosis (Dixon et al., 2015). Both enzymes prefer PUFA-CoA as a substrate and sensitize cancer cells to ferroptosis by increasing the proportion of PUFA-PLs. Interestingly, LPCAT3 is essential for SREBP1 maturation in hepatocytes. LPCAT3 depletion reduced PUFA-phosphatidyl choline (PC) and impaired SREBP1 maturation (Rong et al., 2017). Further study is required to better understand the substrate preferences of LPLATs, and whether any of them are uniquely important for the incorporation of MUFAs into PLs.
Lipid storage and release
Cellular FAs can be stored in lipid droplets in the form of TAG (Figure 2). When it is needed, FA can be released from lipid droplets through lipolysis, a process mediated by lipases or by lipophagy, the autophagic degradation of lipid droplets (Cohen, 2018). The relationship between lipid droplets and ferroptosis is complicated, and context appears to play an important role. For example, PUFAs can be redirected from PLs to lipid droplets in the form of PUFA-TAG, which will prevent PUFA oxidation on the membrane and thus make the cell less vulnerable to ferroptosis. Indeed, it has been reported that lipid droplets protect drosophila glial cell niche and neural stem cells from damaging PUFA peroxidation (Bailey et al., 2015). Conversely, PUFA-TAG can serve as a source of PUFA for PUFA-PL synthesis. For example, in clear cell renal cell carcinoma (ccRCC), which typically have a high basal level of lipid droplets, it was shown that PUFAs are enriched in both PLs and lipid droplets (Zou et al., 2019); in hepatocytes, it was reported that selective degradation of lipid droplets by RAB7A-related lipophagy increases the production of free FAs for subsequent lipid peroxidation and ferroptosis (Bai et al., 2019). Further, it is intriguing to ask whether MUFA-TAGs can interconvert with MUFA-PLs and therefore inhibit ferroptosis under some circumstance. ccRCC cells amass MUFAs (particularly oleic acid) in the form of TAG and stored them within lipid droplets when there is abundant supply of exogenous lipids and/or oxygen. However, once extracellular lipids and oxygen become limiting, oleic acid is released and incorporated into PLs (Ackerman et al., 2018). This explains, at least partially, how hypoxia inhibit ferroptosis through PL remodeling in ccRCC.
FERROPTOSIS, LIPID METABOLISM, AND CANCER SIGNALING
Given the central role of lipids and lipid metabolism in ferroptosis, it is conceivable that signaling pathways that directly or indirectly modulate lipid metabolism may exert important function in ferroptosis. Although ferroptosis has been associated with multiple diseases, including cancer, neurodegeneration, and ischemic organ injury (see (Jiang et al., 2021) as a recent review), the signaling pathways governing lipid metabolism and ferroptosis have only been extensively studied in cancer. As discussed below, oncogenic mutations of these pathways may enhance the synthesis of MUFA-PLs or PUFA-PLs, among other relevant alterations, making cancer cells more resistant or sensitive to ferroptosis induction (Figure 5). Therefore, therapeutic approaches can be designed accordingly to specifically induce cancer cell ferroptosis.
Figure 5. Selected ferroptosis-regulatory signaling pathways.
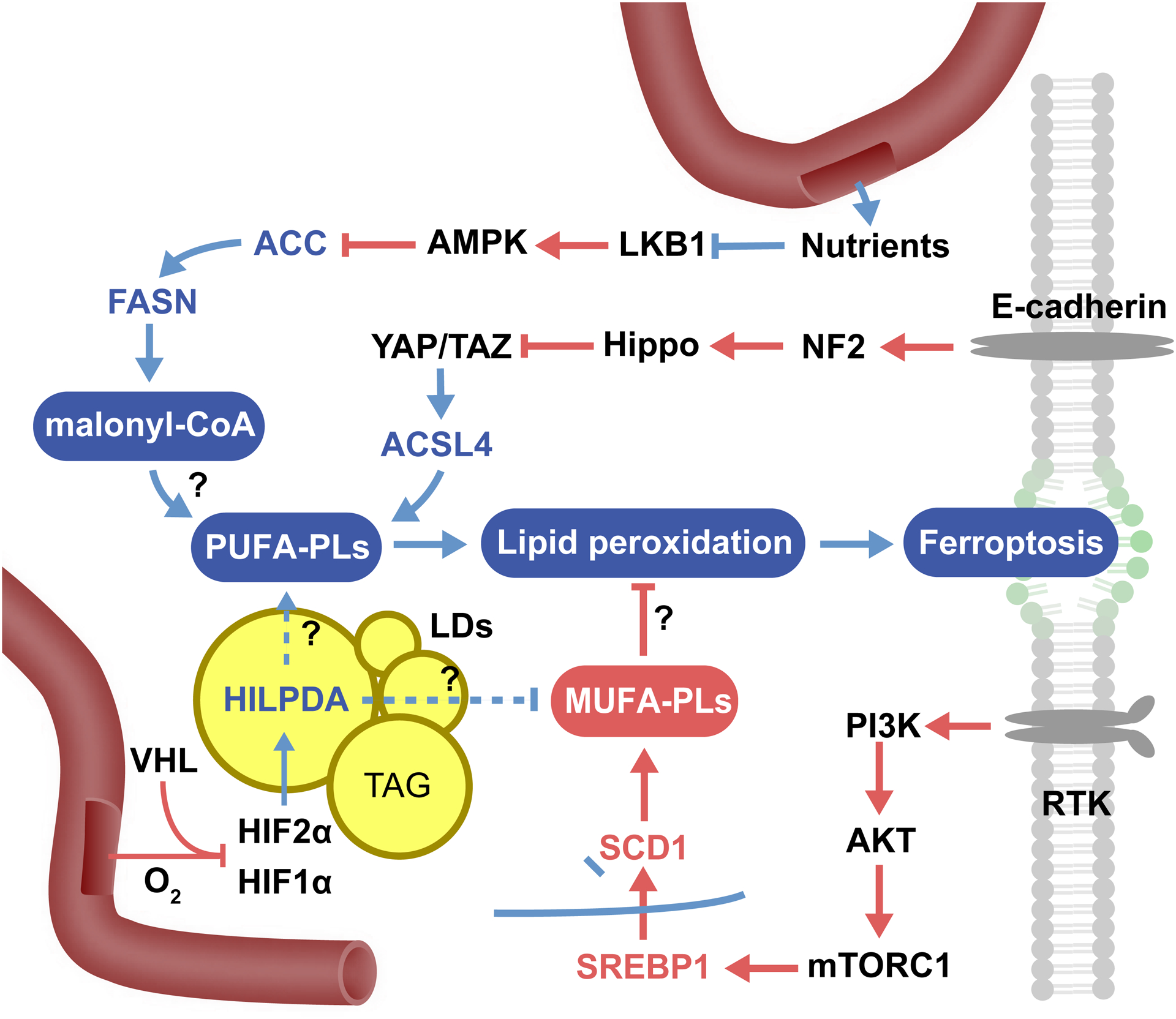
Lipid metabolism is a complex process regulated by many signaling pathways, and the activation or suppression of these pathways can therefore modulate sensitivity to ferroptosis. The pathways outlined here, not meant to be exhaustive, are a sampling of some of those regulating ferroptosis. Question marks indicate processes that have not been clearly defined mechanistically. For example, it is unclear how HILPDA selectively releases PUFA from lipid droplets, how exactly MUFAs efficiently block ferroptosis, or the exact mechanism underlying how inhibition of ACC (and thus de novo lipogenesis) can inhibit ferroptosis.
PI3K-AKT-mTOR pathway
mTOR is a central signaling hub responsible for myriad processes within the cell, including protein synthesis, transcription, and the cell cycle, and it is an important target for cancer therapy (Guertin and Sabatini, 2007). Lipogenesis is an important function of the PI3K-AKT-mTOR axis, and it supports the growth of cancer cells (Porstmann et al., 2008). mTORC1 activates the transcription factor SREBP1, a master regulator of lipogenesis through transcriptional control over multiple lipogenic factors. Included in this is SCD1, which converts SFAs into MUFAs. Recently, it was found that cancer cells with activating mutations to PIK3CA, which encodes the catalytic subunit p110α of PI3K, or deleterious mutations to PTEN (which counteracts the activity of PI3K) develop potent resistance to ferroptosis due to SREBP1-dependent SCD1 upregulation (Yi et al., 2020). Notably, mTORC1 was also shown to regulate ferroptosis through the promotion of GPX4 synthesis (Zhang et al., 2021). Further, pharmacological inhibition of PI3K, AKT, or mTORC1 all sensitized these cells to ferroptosis, suggesting a novel combination therapy for the treatment of cancers harboring oncogenic activation of PI3K-AKT-mTOR pathway (Yi et al., 2020; Zhang et al., 2021).
In addition to oncogenic PI3K-mTOR signaling, cancer cells may acquire SCD1-mediated ferroptosis suppression through other mutations. For example, ~10% of lung adenocarcinoma patients present mutations in both LKB1 and KEAP1. These patients have a remarkably poor prognosis (Shen et al., 2019). KEAP1 is an inhibitor of the master antioxidant transcription factor, NRF2, which can also upregulate SCD1 expression. It was reported recently that lung tumors with mutations in LKB1 and KEAP1 develop a strong dependence on SCD1 activity (Wohlhieter et al., 2020). Therefore, in cancers harboring either PI3K-mTOR activation or LKB1/KEAP1 inactivation, therapies of same mechanism might be effective: a combination of ferroptosis induction with pharmacological agents leading to SCD1 inhibition either directly (SCD1 inhibitor) or indirectly (PI3K/AKT/mTORC1 inhibitors).
LKB1-AMPK pathway
Ferroptosis is tightly tied to cellular metabolism, and thus it may not be a surprise that AMPK, a major sensor of cellular energy stress, has been reported to play a role in suppressing ferroptosis (Lee et al., 2020a; Li et al., 2020). AMPK is activated upon the reduction of ATP and elevation of AMP in the cell. One of AMPK’s many roles is to shut down anabolic processes; in this capacity, AMPK had a profound effect on lipid metabolism. AMPK phosphorylates and inhibits activity of ACC1 (acetyl CoA carboxylase 1) and ACC2 (Fullerton et al., 2013), which convert acetyl-CoA into malonyl-CoA and are crucial in de novo lipogenesis by fatty acid synthase (FASN). Moreover, ACC2 suppresses fatty acid oxidation through inhibiting CPT1 (Abu-Elheiga et al., 2001). Therefore, activation of AMPK will simultaneously suppress fatty acid synthesis and increases fatty acid oxidation. It has been shown that both activation of AMPK and inhibition of ACC or FASN could efficiently block ferroptosis (Lee et al., 2020a; Li et al., 2020), and that LKB1/STK11, a tumor suppressor and protein kinase that activates AMPK , could also inhibit ferroptosis (Li et al., 2020). Intriguingly, mammals cannot produce PUFAs through de novo lipogenesis. Why then does inhibition of ACC-FASN pathway and downstream SFA/MUFA synthesis suppress ferroptosis? Through untargeted lipidomic analysis, Lee, et al. showed that activation of AMPK led to an overall decrease of fatty acids (including SFA, MUFA, PUFA, and various PLs), and AMPK knockout led to overall increase of fatty acids in the cell. One possible mechanism proposed by Lee, et al. is that malonyl-CoA (which is produced by ACC) was not only required in SFA/MUFA synthesis but was also required in the elongation step of long chain PUFA synthesis, e.g., from γ-linolenic acid (C18:3) to DGLA (20:3). Another possible mechanism is that decreased fatty acid oxidation by AMPK inhibition leads to an overall increase of fatty acid abundance, including PUFA and PUFA-PL. Alternatively, could it be that a protein involved in ferroptosis requires palmitoylation, or do SFAs play any active role in ferroptosis? These questions warrant further investigation. Lastly, evidence suggests the therapeutic efficacy of AMPK activation for many types of cancer (Kishton et al., 2016; Li et al., 2015; Sengupta et al., 2007), thus it will be of interest to determine whether combined induction of AMPK activation and ferroptosis is an effective therapy.
E-cadherin-Hippo-YAP/TAZ pathway
The Hippo pathway senses cell-cell contacts and is activated as cells begin to butt up against one another, activating a signaling cascade to inhibit the activity of protooncogenic transcriptional co-activators YAP and TAZ (Piccolo et al., 2014). At least one of the ways Hippo senses cell-cell contact is through the adhesion protein E-cadherin. Knockout of E-cadherin or inactivation of Hippo pathway all sensitized these densely cultured cells to ferroptosis (Wu et al., 2019). Overexpression of constitutively active YAP or TAZ mutants could also sensitize these cells to ferroptosis (Wu et al., 2019; Yang et al., 2019). It appears that there are multiple effectors downstream of YAP and TAZ responsible for the increase in sensitivity to ferroptosis, including transferrin receptor, NADPH oxidases NOX2 and NOX4, and ACSL4 (Wu et al., 2019; Yang et al., 2019; Yang et al., 2020). This may result in an overall ferroptosis-sensitive cell state. Transferrin receptor imports labile iron to the cell, NOX2/4 generate ROS, and ACSL4 mediates the synthesis of PUFA-PLs. Together, they provide the kindling, fuel, and match for ferroptosis. Importantly, as loss of function mutations of the Hippo pathway is a strong malignant event rending cancer cells resistant to common therapies, increased ferroptosis susceptibility caused by these same mutants suggests that these mutants can be used as biomarkers to predict the responsiveness of cancers to ferroptosis-inducing treatments.
Notably, although E-cadherin is a bona fide tumor suppressor in cancers including gastric and breast lobular carcinoma and whose loss of function has been considered as a critical step in metastasis (Berx et al., 1995; Liu and Chu, 2014; Schmalhofer et al., 2009), a recent study showed that in breast ductal carcinoma, loss of E-cadherin may be instead an obstacle for metastasis: circulating cancer cells lacking E-cadherin had higher levels of ROS and formed fewer metastatic sites (Padmanaban et al., 2019). It is enticing to speculate that the function of E-cadherin in suppressing ferroptosis is behind this phenomenon, a therapeutically relevant scenario easily testable.
VHL-HIF pathway
A great deal of research has been conducted on the role of ferroptosis in ischemia-reperfusion injury, and mounting evidence suggests that the inhibition of ferroptosis can improve outcomes in ischemic stroke, heart attack, and acute kidney injury (Gao et al., 2015; Guan et al., 2019; Linkermann et al., 2014). This is likely due to the massive burst of ROS that occurs with reperfusion, fueling uncontrollable lipid peroxidation and killing cells. However, less attention has been paid to the molecular mechanisms surrounding ferroptosis in hypoxic cells. Lipid metabolism under hypoxia is substantially altered to accommodate the oxygen-poor environment (Munir et al., 2019). One of these changes is the accumulation of lipid droplets, which are organelles storing excessive lipids primarily in the form of very non-reactive neutral lipids: triacylglycerol and cholesterol esters.
Lipid droplets are abundant not only in various normal tissues or cell types such as adipocytes and hepatocytes, but also in certain cancers, such as clear cell renal cell carcinoma (ccRCC). ccRCC is a form of kidney cancer identifiable by the presence of large lipid droplets. This is a result of a high rate of mutation to VHL, a tumor suppressor that suppresses stability of the hypoxia inducible factors 1 and 2 alpha (HIF1/2α). Constitutive activation of HIF1/2α alters a plethora of cellular processes, including metabolism of sugar and lipids. VHL mutation has been reported to enhance sensitivity to ferroptosis through the alterations to lipid storage (Miess et al., 2018). Moreover, HIF2α was found to increase incorporation of PUFAs into triacylglycerols and phospholipids through direct upregulation of HILPDA, a lipid droplet associated protein. HILPDA can bind and inhibit adipose triglyceride lipase, a protein responsible for breaking down triacylglycerols and appears to stimulate the specific conversion of PUFA-linked TAGs to PLs (Zou et al., 2019). HIF1α has also been identified as an enhancer of ferroptosis through epigenetic suppression of SCD1 (Jiang et al., 2017). However, HIF1α also suppressed FADS2, which promotes the desaturation of linoleic acid into arachidonic acid. Therefore, the impact of hypoxia on lipid metabolism and ferroptosis is complex and likely to be context dependent.
CONCLUDING REMARKS
In recent years, we have witnessed substantial progress in ferroptosis research, including the understanding of the mechanisms and pathological functions of this unique form of cell death. Although lipid metabolism and related signaling pathways have been established as central players of ferroptosis, even in this specific territory, many fundamental questions remain to be answered and will be the focus of future investigation. To name a few: First, as distribution and metabolism of various lipid species have exquisite tissue specificity, investigation of their interplay with ferroptosis in a tissue-specific manner holds better opportunity for the identification of novel physiological function of ferroptosis. This is obviously important, considering that a physiological role for ferroptosis, if any, has not been defined. Second and technically, fluorescent probes widely used for the measurement of lipid peroxidation cannot distinguish peroxidation of phospholipids versus free fatty acids, a major technical hurdle in the field. Therefore, there is an urgent need to develop new technology to accurately measure phospholipid peroxidation in cells, or to make mass spectrometry-based method more feasible for routine measurement of the cellular (oxi)lipidome. A lipidomics-based approach is advantageous as it can determine the specific identity of oxidized phospholipids (head groups), helping to answer a pressing question in the field: whether ferroptosis is triggered by the peroxidation of certain specific phospholipids (phosphatidylethanolamine, instead of other phospholipids) as suggested previously (Kagan et al., 2017); and if so, what are the mechanisms underlying the selectivity of peroxidation and the role of these specific oxidized phospholipids in ferroptosis. Related and conceptually important, although phospholipid peroxidation has been considered the ultimate executioner and point of no-return of ferroptosis, it is formally possible that there exists a specific molecular mediator downstream of phospholipid peroxidation (such a mechanism can explain why phosphatidyl ethanolamine peroxidation might be specifically crucial for ferroptosis (Kagan et al., 2017)). If so, this specific molecule might be used as a precise biomarker for ferroptosis in vivo and in patient samples, better than all currently used markers that cannot unambiguously distinguish ferroptosis from certain other cellular events such as general oxidative stress. Therapeutically, we foresee that further mechanistic dissection of the role of lipid metabolism and related signaling in ferroptosis will nominate novel drug targets, biomarkers, and therapeutic strategies – ferroptosis inducers as single agents or in combination with other targeted agents for cancer treatment, and ferroptosis inhibitors as potential agents for the treatment of neurodegeneration and ischemic organ injuries.
Acknowledgment:
The authors thank members of the Jiang lab for critical reading and suggestions. This work is supported by NIH F31CA247112 (to A.M.M.), NIH R01CA204232, NIH R01CA258622, and NIH R01CA244581 (to X.J.), and NCI cancer centre core grant P30 CA008748 to MSKCC.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- Abu-Elheiga L, Matzuk MM, Abo-Hashema KA, and Wakil SJ (2001). Continuous fatty acid oxidation and reduced fat storage in mice lacking acetyl-CoA carboxylase 2. Science 291, 2613–2616. [DOI] [PubMed] [Google Scholar]
- Ackerman D, Tumanov S, Qiu B, Michalopoulou E, Spata M, Azzam A, Xie H, Simon MC, and Kamphorst JJ (2018). Triglycerides Promote Lipid Homeostasis during Hypoxic Stress by Balancing Fatty Acid Saturation. Cell Rep 24, 2596–2605 e2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angeli JPF, Freitas FP, Nepachalovich P, Puentes L, Zilka O, Inague A, Lorenz S, Kunz V, Nehring H, Thamara Nishida Xavier da S, et al. (2021). Nature Portfolio.
- Bai Y, Meng L, Han L, Jia Y, Zhao Y, Gao H, Kang R, Wang X, Tang D, and Dai E (2019). Lipid storage and lipophagy regulates ferroptosis. Biochem Biophys Res Commun 508, 997–1003. [DOI] [PubMed] [Google Scholar]
- Bailey AP, Koster G, Guillermier C, Hirst EM, MacRae JI, Lechene CP, Postle AD, and Gould AP (2015). Antioxidant Role for Lipid Droplets in a Stem Cell Niche of Drosophila. Cell 163, 340–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao WD, Pang P, Zhou XT, Hu F, Xiong W, Chen K, Wang J, Wang F, Xie D, Hu YZ, et al. (2021). Loss of ferroportin induces memory impairment by promoting ferroptosis in Alzheimer’s disease. Cell Death Differ 28, 1548–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedoui S, Herold MJ, and Strasser A (2020). Emerging connectivity of programmed cell death pathways and its physiological implications. Nat Rev Mol Cell Biol 21, 678–695. [DOI] [PubMed] [Google Scholar]
- Beharier O, Tyurin VA, Goff JP, Guerrero-Santoro J, Kajiwara K, Chu T, Tyurina YY, St Croix CM, Wallace CT, Parry S, et al. (2020). PLA2G6 guards placental trophoblasts against ferroptotic injury. Proc Natl Acad Sci U S A 117, 27319–27328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R, et al. (2019). The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berx G, Cleton-Jansen AM, Nollet F, de Leeuw WJ, van de Vijver M, Cornelisse C, and van Roy F (1995). E-cadherin is a tumour/invasion suppressor gene mutated in human lobular breast cancers. EMBO J 14, 6107–6115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomme A, Ford CA, Mui E, Patel R, Ntala C, Jamieson LE, Planque M, McGregor GH, Peixoto P, Hervouet E, et al. (2020). 2,4-dienoyl-CoA reductase regulates lipid homeostasis in treatment-resistant prostate cancer. Nat Commun 11, 2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Chu B, Yang X, Liu Z, Jin Y, Kon N, Rabadan R, Jiang X, Stockwell BR, and Gu W (2021). iPLA2beta-mediated lipid detoxification controls p53-driven ferroptosis independent of GPX4. Nat Commun 12, 3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PH, Wu J, Ding CC, Lin CC, Pan S, Bossa N, Xu Y, Yang WH, Mathey-Prevot B, and Chi JT (2020). Kinome screen of ferroptosis reveals a novel role of ATM in regulating iron metabolism. Cell Death Differ 27, 1008–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S (2018). Lipid Droplets as Organelles. Int Rev Cell Mol Biol 337, 83–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui W, Liu D, Gu W, and Chu B (2021). Peroxisome-driven ether-linked phospholipids biosynthesis is essential for ferroptosis. Cell Death Differ 28, 2536–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dar HH, Anthonymuthu TS, Ponomareva LA, Souryavong AB, Shurin GV, Kapralov AO, Tyurin VA, Lee JS, Mallampalli RK, Wenzel SE, et al. (2021). A new thiol-independent mechanism of epithelial host defense against Pseudomonas aeruginosa: iNOS/NO. Redox Biol 45, 102045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean JM, and Lodhi IJ (2018). Structural and functional roles of ether lipids. Protein Cell 9, 196–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dierge E, Debock E, Guilbaud C, Corbet C, Mignolet E, Mignard L, Bastien E, Dessy C, Larondelle Y, and Feron O (2021). Peroxidation of n-3 and n-6 polyunsaturated fatty acids in the acidic tumor environment leads to ferroptosis-mediated anticancer effects. Cell Metab 33, 1701–1715 e1705. [DOI] [PubMed] [Google Scholar]
- Distéfano AM, Martin MV, Córdoba JP, Bellido AM, D’Ippólito S, Colman SL, Soto D, Roldán JA, Bartoli CG, Zabaleta EJ, et al. (2017). Heat stress induces ferroptosis-like cell death in plants. J Cell Biol 216, 463–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, Superti-Furga G, and Stockwell BR (2015). Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chemical Biology 10, 1604–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, Grocin AG, Xavier da Silva TN, Panzilius E, Scheel CH, et al. (2019). FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693–698. [DOI] [PubMed] [Google Scholar]
- Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A, et al. (2017). ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 13, 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Else PL, and Kraffe E (2015). Docosahexaenoic and arachidonic acid peroxidation: It’s a within molecule cascade. Biochim Biophys Acta 1848, 417–421. [DOI] [PubMed] [Google Scholar]
- Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch A, Eggenhofer E, et al. (2014). Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol 16, 1180–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullerton MD, Galic S, Marcinko K, Sikkema S, Pulinilkunnil T, Chen ZP, O’Neill HM, Ford RJ, Palanivel R, O’Brien M, et al. (2013). Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat Med 19, 1649–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Monian P, Pan Q, Zhang W, Xiang J, and Jiang X (2016). Ferroptosis is an autophagic cell death process. Cell Res 26, 1021–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Monian P, Quadri N, Ramasamy R, and Jiang X (2015). Glutaminolysis and Transferrin Regulate Ferroptosis. Mol Cell 59, 298–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Bermudez J, Baudrier L, Bayraktar EC, Shen Y, La K, Guarecuco R, Yucel B, Fiore D, Tavora B, Freinkman E, et al. (2019). Squalene accumulation in cholesterol auxotrophic lymphomas prevents oxidative cell death. Nature 567, 118–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng N, Shi BJ, Li SL, Zhong ZY, Li YC, Xua WL, Zhou H, and Cai JH (2018). Knockdown of ferroportin accelerates erastin-induced ferroptosis in neuroblastoma cells. Eur Rev Med Pharmacol Sci 22, 3826–3836. [DOI] [PubMed] [Google Scholar]
- Ghandi M, Huang FW, Jane-Valbuena J, Kryukov GV, Lo CC, McDonald ER 3rd, Barretina J, Gelfand ET, Bielski CM, Li H, et al. (2019). Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girotti AW, and Korytowski W (2019). Cholesterol Peroxidation as a Special Type of Lipid Oxidation in Photodynamic Systems. Photochem Photobiol 95, 73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gow AJ, Chen Q, Hess DT, Day BJ, Ischiropoulos H, and Stamler JS (2002). Basal and stimulated protein S-nitrosylation in multiple cell types and tissues. J Biol Chem 277, 9637–9640. [DOI] [PubMed] [Google Scholar]
- Green DR (2019). The Coming Decade of Cell Death Research: Five Riddles. Cell 177, 1094–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan X, Li X, Yang X, Yan J, Shi P, Ba L, Cao Y, and Wang P (2019). The neuroprotective effects of carvacrol on ischemia/reperfusion-induced hippocampal neuronal impairment by ferroptosis mitigation. Life Sci 235, 116795. [DOI] [PubMed] [Google Scholar]
- Guertin DA, and Sabatini DM (2007). Defining the role of mTOR in cancer. Cancer Cell 12, 9–22. [DOI] [PubMed] [Google Scholar]
- Harayama T, and Riezman H (2018). Understanding the diversity of membrane lipid composition. Nat Rev Mol Cell Biol 19, 281–296. [DOI] [PubMed] [Google Scholar]
- He M, Qin CX, Wang X, and Ding NZ (2020). Plant Unsaturated Fatty Acids: Biosynthesis and Regulation. Front Plant Sci 11, 390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homma T, Kobayashi S, Conrad M, Konno H, Yokoyama C, and Fujii J (2021). Nitric oxide protects against ferroptosis by aborting the lipid peroxidation chain reaction. Nitric Oxide 115, 34–43. [DOI] [PubMed] [Google Scholar]
- Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ, Kang R, and Tang D (2016). Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12, 1425–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Stockwell BR, and Conrad M (2021). Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol 22, 266–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Mao C, Yang R, Yan B, Shi Y, Liu X, Lai W, Liu Y, Wang X, Xiao D, et al. (2017). EGLN1/c-Myc Induced Lymphoid-Specific Helicase Inhibits Ferroptosis through Lipid Metabolic Gene Expression Changes. Theranostics 7, 3293–3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan VE, Mao G, Qu F, Angeli JPF, Doll S, Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, et al. (2017). Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol 13, 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang MJ, Fujino T, Sasano H, Minekura H, Yabuki N, Nagura H, Iijima H, and Yamamoto TT (1997). A novel arachidonate-preferring acyl-CoA synthetase is present in steroidogenic cells of the rat adrenal, ovary, and testis. Proc Natl Acad Sci USA 94, 2880–2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapralov AA, Yang Q, Dar HH, Tyurina YY, Anthonymuthu TS, Kim R, St Croix CM, Mikulska-Ruminska K, Liu B, Shrivastava IH, et al. (2020). Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat Chem Biol 16, 278–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazachkov M, Chen Q, Wang L, and Zou J (2008). Substrate preferences of a lysophosphatidylcholine acyltransferase highlight its role in phospholipid remodeling. Lipids 43, 895–902. [DOI] [PubMed] [Google Scholar]
- Killion EA, Reeves AR, El Azzouny MA, Yan Q-W, Surujon D, Griffin JD, Bowman TA, Wang C, Matthan NR, Klett EL, et al. (2018). A role for long-chain acyl-CoA synthetase-4 (ACSL4) in diet-induced phospholipid remodeling and obesity-associated adipocyte dysfunction. Mol Metab 9, 43–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishton RJ, Barnes CE, Nichols AG, Cohen S, Gerriets VA, Siska PJ, Macintyre AN, Goraksha-Hicks P, de Cubas AA, Liu T, et al. (2016). AMPK Is Essential to Balance Glycolysis and Mitochondrial Metabolism to Control T-ALL Cell Stress and Survival. Cell Metabolism 23, 649–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kist M, and Vucic D (2021). Cell death pathways: intricate connections and disease implications. EMBO J 40, e106700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Muller C, Zandkarimi F, Merl-Pham J, Bao X, Anastasov N, Kossl J, et al. (2020). GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent Sci 6, 41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Zandkarimi F, Zhang Y, Meena JK, Kim J, Zhuang L, Tyagi S, Ma L, Westbrook TF, Steinberg GR, et al. (2020a). Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat Cell Biol 22, 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J-Y, Nam M, Son HY, Hyun K, Jang SY, Kim JW, Kim MW, Jung Y, Jang E, Yoon S-J, et al. (2020b). Polyunsaturated fatty acid biosynthesis pathway determines ferroptosis sensitivity in gastric cancer. Proc Natl Acad Sci USA 117, 32433–32442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Dong X, Du W, Shi X, Chen K, Zhang W, and Gao M (2020). LKB1-AMPK axis negatively regulates ferroptosis by inhibiting fatty acid synthesis. Signal Transduction and Targeted Therapy 5, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Saud SM, Young MR, Chen G, and Hua B (2015). Targeting AMPK for cancer prevention and treatment. Oncotarget 6, 7365–7378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkermann A, Skouta R, Himmerkus N, Mulay SR, Dewitz C, De Zen F, Prokai A, Zuchtriegel G, Krombach F, Welz PS, et al. (2014). Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci U S A 111, 16836–16841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littarru GP, and Langsjoen P (2007). Coenzyme Q10 and statins: biochemical and clinical implications. Mitochondrion 7 Suppl, S168–174. [DOI] [PubMed] [Google Scholar]
- Liu W, Chakraborty B, Safi R, Kazmin D, Chang CY, and McDonnell DP (2021). Dysregulated cholesterol homeostasis results in resistance to ferroptosis increasing tumorigenicity and metastasis in cancer. Nat Commun 12, 5103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, and Chu K-M (2014). E-cadherin and gastric cancer: cause, consequence, and applications. Biomed Res Int 2014, 637308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Xiao L, Liu L, Ye L, Su P, Bi E, Wang Q, Yang M, Qian J, and Yi Q (2021). CD36-mediated ferroptosis dampens intratumoral CD8(+) T cell effector function and impairs their antitumor ability. Cell Metab 33, 1001–1012 e1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magtanong L, Ko P-J, To M, Cao JY, Forcina GC, Tarangelo A, Ward CC, Cho K, Patti GJ, Nomura DK, et al. (2019). Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem Biol 26, 420–432.e429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, Koppula P, Wu S, Zhuang L, Fang B, et al. (2021). DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 593, 586–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Ruiz A, Cadenas S, and Lamas S (2011). Nitric oxide signaling: classical, less classical, and nonclassical mechanisms. Free Radic Biol Med 51, 17–29. [DOI] [PubMed] [Google Scholar]
- Miess H, Dankworth B, Gouw AM, Rosenfeldt M, Schmitz W, Jiang M, Saunders B, Howell M, Downward J, Felsher DW, et al. (2018). The glutathione redox system is essential to prevent ferroptosis caused by impaired lipid metabolism in clear cell renal cell carcinoma. Oncogene 37, 5435–5450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munir R, Lisec J, Swinnen JV, and Zaidi N (2019). Lipid metabolism in cancer cells under metabolic stress. Br J Cancer 120, 1090–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muri J, Thut H, Bornkamm GW, and Kopf M (2019). B1 and Marginal Zone B Cells but Not Follicular B2 Cells Require Gpx4 to Prevent Lipid Peroxidation and Ferroptosis. Cell Rep 29, 2731–2744 e2734. [DOI] [PubMed] [Google Scholar]
- Nagarajan SR, Butler LM, and Hoy AJ (2021). The diversity and breadth of cancer cell fatty acid metabolism. Cancer Metab 9, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassar ZD, Mah CY, Dehairs J, Burvenich IJ, Irani S, Centenera MM, Helm M, Shrestha RK, Moldovan M, Don AS, et al. (2020). Human DECR1 is an androgen-repressed survival factor that regulates PUFA oxidation to protect prostate tumor cells from ferroptosis. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell VB, Chumley PH, Hogg N, Bloodsworth A, Darley-Usmar VM, and Freeman BA (1997). Nitric oxide inhibition of lipid peroxidation: kinetics of reaction with lipid peroxyl radicals and comparison with alpha-tocopherol. Biochemistry 36, 15216–15223. [DOI] [PubMed] [Google Scholar]
- Padmanaban V, Krol I, Suhail Y, Szczerba BM, Aceto N, Bader JS, and Ewald AJ (2019). E-cadherin is required for metastasis in multiple models of breast cancer. [DOI] [PMC free article] [PubMed]
- Pandey AV, and Sproll P (2014). Pharmacogenomics of human P450 oxidoreductase. Front Pharmacol 5, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccolo S, Dupont S, and Cordenonsi M (2014). The biology of YAP/TAZ: hippo signaling and beyond. Physiol Rev 94, 1287–1312. [DOI] [PubMed] [Google Scholar]
- Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S, Griffiths JR, Chung Y-L, and Schulze A (2008). SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metabolism 8, 224–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robichaud P-P, Munganyiki JE, Boilard E, and Surette ME (2018). Polyunsaturated fatty acid elongation and desaturation in activated human T-cells: ELOVL5 is the key elongase. J Lipid Res 59, 2383–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmalhofer O, Brabletz S, and Brabletz T (2009). E-cadherin, beta-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev 28, 151–166. [DOI] [PubMed] [Google Scholar]
- Schwartzman ML, Martasek P, Rios AR, Levere RD, Solangi K, Goodman AI, and Abraham NG (1990). Cytochrome P450-dependent arachidonic acid metabolism in human kidney. Kidney Int 37, 94–99. [DOI] [PubMed] [Google Scholar]
- Seiler A, Schneider M, Förster H, Roth S, Wirth EK, Culmsee C, Plesnila N, Kremmer E, Rådmark O, Wurst W, et al. (2008). Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab 8, 237–248. [DOI] [PubMed] [Google Scholar]
- Sengupta TK, Leclerc GM, Hsieh-Kinser TT, Leclerc GJ, Singh I, and Barredo JC (2007). Cytotoxic effect of 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside (AICAR) on childhood acute lymphoblastic leukemia (ALL) cells: implication for targeted therapy. Mol Cancer 6, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah R, Shchepinov MS, and Pratt DA (2018). Resolving the role of lipoxygenases in the initiation and execution of ferroptosis. ACS Cent Sci 4, 387–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen R, Martin A, Ni A, Hellmann M, Arbour KC, Jordan E, Arora A, Ptashkin R, Zehir A, Kris MG, et al. (2019). Harnessing Clinical Sequencing Data for Survival Stratification of Patients with Metastatic Lung Adenocarcinomas. JCO Precis Oncol 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson AE (1997). The cytochrome P450 4 (CYP4) family. Gen Pharmacol 28, 351–359. [DOI] [PubMed] [Google Scholar]
- Sindelar PJ, Guan Z, Dallner G, and Ernster L (1999). The protective role of plasmalogens in iron-induced lipid peroxidation. Free Radic Biol Med 26, 318–324. [DOI] [PubMed] [Google Scholar]
- Snaebjornsson MT, Janaki-Raman S, and Schulze A (2020). Greasing the Wheels of the Cancer Machine: The Role of Lipid Metabolism in Cancer. Cell Metab 31, 62–76. [DOI] [PubMed] [Google Scholar]
- Soula M, Weber RA, Zilka O, Alwaseem H, La K, Yen F, Molina H, Garcia-Bermudez J, Pratt DA, and Birsoy K (2020). Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat Chem Biol 16, 1351–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun WY, Tyurin VA, Mikulska-Ruminska K, Shrivastava IH, Anthonymuthu TS, Zhai YJ, Pan MH, Gong HB, Lu DH, Sun J, et al. (2021). Phospholipase iPLA2beta averts ferroptosis by eliminating a redox lipid death signal. Nat Chem Biol 17, 465–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesfay L, Paul BT, Konstorum A, Deng Z, Cox AO, Lee J, Furdui CM, Hegde P, Torti FM, and Torti SV (2019). Stearoyl-CoA Desaturase 1 Protects Ovarian Cancer Cells from Ferroptotic Cell Death. Cancer Res 79, 5355–5366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JP, Geiger PG, Maiorino M, Ursini F, and Girotti AW (1990). Enzymatic reduction of phospholipid and cholesterol hydroperoxides in artificial bilayers and lipoproteins. Biochim Biophys Acta 1045, 252–260. [DOI] [PubMed] [Google Scholar]
- Ubellacker JM, Tasdogan A, Ramesh V, Shen B, Mitchell EC, Martin-Sandoval MS, Gu Z, McCormick ML, Durham AB, Spitz DR, et al. (2020). Lymph protects metastasizing melanoma cells from ferroptosis. Nature 585, 113–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ursini F, Maiorino M, Valente M, Ferri L, and Gregolin C (1982). Purification from pig liver of a protein which protects liposomes and biomembranes from peroxidative degradation and exhibits glutathione peroxidase activity on phosphatidylcholine hydroperoxides. Biochim Biophys Acta 710, 197–211. [DOI] [PubMed] [Google Scholar]
- Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, Kaffenberger SD, Eaton JK, Shimada K, Aguirre AJ, et al. (2017). Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner GJ, Berry MJ, Moustafa ME, Carlson BA, Hatfield DL, and Faust JR (2000). Inhibition of selenoprotein synthesis by selenocysteine tRNA[Ser]Sec lacking isopentenyladenosine. J Biol Chem 275, 28110–28119. [DOI] [PubMed] [Google Scholar]
- Werner ER, Keller MA, Sailer S, Lackner K, Koch J, Hermann M, Coassin S, Golderer G, Werner-Felmayer G, Zoeller RA, et al. (2020). The TMEM189 gene encodes plasmanylethanolamine desaturase which introduces the characteristic vinyl ether double bond into plasmalogens. Proc Natl Acad Sci U S A 117, 7792–7798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlhieter CA, Richards AL, Uddin F, Hulton CH, Quintanal-Villalonga À, Martin A, de Stanchina E, Bhanot U, Asher M, Shah NS, et al. (2020). Concurrent mutations in STK11 and KEAP1 promote ferroptosis protection and SCD1 dependence in lung cancer. Cell reports 33, 108444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Minikes AM, Gao M, Bian H, Li Y, Stockwell BR, Chen ZN, and Jiang X (2019). Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature 572, 402–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamane D, Hayashi Y, Matsumoto M, Nakanishi H, Imagawa H, Kohara M, Lemon SM, and Ichi I (2021). FADS2-dependent fatty acid desaturation dictates cellular sensitivity to ferroptosis and permissiveness for hepatitis C virus replication. Cell Chem Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan B, Ai Y, Sun Q, Ma Y, Cao Y, Wang J, Zhang Z, and Wang X (2021). Membrane Damage during Ferroptosis Is Caused by Oxidation of Phospholipids Catalyzed by the Oxidoreductases POR and CYB5R1. Molecular Cell 81, 355–369.e310. [DOI] [PubMed] [Google Scholar]
- Yang WH, Ding CC, Sun T, Rupprecht G, Lin CC, Hsu D, and Chi JT (2019). The Hippo Pathway Effector TAZ Regulates Ferroptosis in Renal Cell Carcinoma. Cell Rep 28, 2501–2508.e2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WH, Huang Z, Wu J, Ding CC, Murphy SK, and Chi JT (2020). A TAZ-ANGPTL4-NOX2 Axis Regulates Ferroptotic Cell Death and Chemoresistance in Epithelial Ovarian Cancer. Mol Cancer Res 18, 79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, and Stockwell BR (2016). Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci USA 113, E4966–4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, et al. (2014). Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi J, Zhu J, Wu J, Thompson CB, and Jiang X (2020). Oncogenic activation of PI3K-AKT-mTOR signaling suppresses ferroptosis via SREBP-mediated lipogenesis. Proceedings of the National Academy of Sciences of the United States of America 117, 31189–31197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Swanda RV, Nie L, Liu X, Wang C, Lee H, Lei G, Mao C, Koppula P, Cheng W, et al. (2021). mTORC1 couples cyst(e)ine availability with GPX4 protein synthesis and ferroptosis regulation. Nat Commun 12, 1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y, Henry WS, Ricq EL, Graham ET, Phadnis VV, Maretich P, Paradkar S, Boehnke N, Deik AA, Reinhardt F, et al. (2020a). Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature 585, 603–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y, Li H, Graham ET, Deik AA, Eaton JK, Wang W, Sandoval-Gomez G, Clish CB, Doench JG, and Schreiber SL (2020b). Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol 16, 302–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W, Tseng Y-Y, Deasy R, Kost-Alimova M, Dančík V, et al. (2019). A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nature Communications 10, 1617. [DOI] [PMC free article] [PubMed] [Google Scholar]