

SUMMARY
Excitotoxicity induced by NMDA receptor (NMDAR) is a major cause of neuronal death in ischemic stroke. However, past efforts of directly targeting NMDARs have unfortunately failed in clinical trials. Here we reveal an unexpected mechanism underlying NMDAR-mediated neurotoxicity, which leads to identification of a novel target and development of an effective therapeutic peptide for ischemic stroke. We show that NMDAR-induced excitotoxicity is enhanced by physical and functional coupling of NMDAR to an ion channel TRPM2 upon ischemic insults. TRPM2-NMDAR association promotes the surface expression of extrasynaptic NMDARs, leading to enhanced NMDAR activity and increased neuronal death. We identified a specific NMDAR-interacting motif on TRPM2, and designed a membrane-permeable peptide to uncouple TRPM2-NMDAR interaction. This disrupting-peptide protects neurons against ischemic injury in vitro and protects mice against ischemic stroke in vivo. These findings provide an unconventional strategy to mitigate excitotoxic neuronal death without directly targeting NMDARs.
Keywords: Ischemic stroke, TRPM2, NMDA receptors (NMDARs), Ca2+ signaling, excitotoxicity, neuronal death, therapeutic peptide
eTOC Blurb
Zong et al. discover an unexpected association between the oxidative stress-sensitive ion channel TRPM2 and the extrasynaptic NMDA receptor (NMDAR) in the neurons, which enhances excitotoxicity during ischemic brain injury. Neuron-specific knockout of TRPM2 or uncoupling of TRPM2-NMDAR association using an interfering peptide protects mice against ischemic stroke.
Graphical Abstract
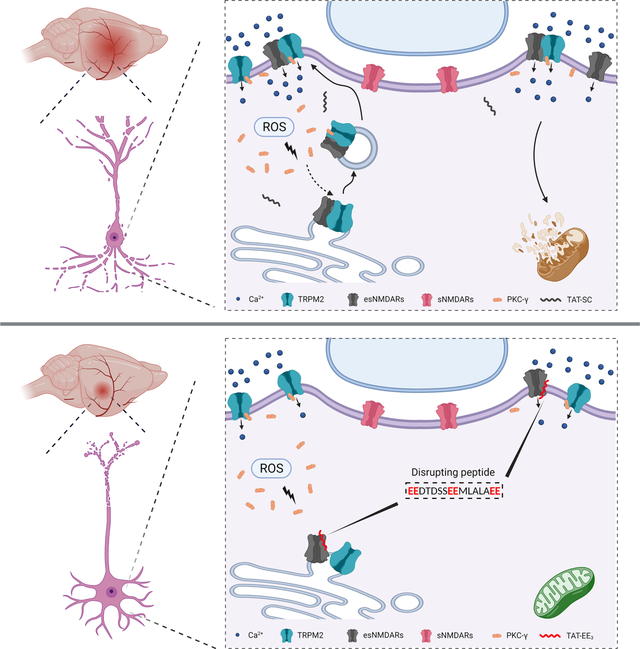
INTRODUCTION
Ischemic stroke is a leading cause of disability and mortality worldwide (Virani et al., 2020). Numerous factors are involved in neuronal damage during ischemic stroke, among which Ca2+ overload plays a key role (Granzotto et al., 2020). Ca2+ overload caused by excitotoxic mechanisms through NMDA receptor (NMDAR) and subsequent activation of non-glutamatergic Ca2+-permeable channels (Tymianski, 2011) triggers a series of downstream cytotoxic events, including reactive oxygen species (ROS) generation, mitochondrial dysfunction, and necrosis/apoptosis cascade activation, which ultimately leads to neuronal death (Choi, 2020). NMDAR-mediated excitotoxicity has been extensively studied since its first discovery fifty years ago (Olney, 1969). However, NMDAR antagonists all failed to attenuate ischemic stroke in human patients (Sena et al., 2007).
Failure of NMDAR antagonists shifted focus of neuroprotection research in ischemic stroke towards identifying the downstream intracellular signaling pathways triggered by NMDARs (Wu and Tymianski, 2018), and the investigation of subtype-dependent (Ge et al., 2020) as well as localization-dependent excitotoxic effects of NMDARs (Hardingham and Bading, 2010). Over a third of surface NMDARs are located extrasynaptically (Petit-Pedrol and Groc, 2021) and closely related to neurotoxicity. In contrast, activation of synaptic NMDARs promotes neuron survival through activation of ERK and CREB signaling (Bading, 2013; Hardingham, 2019). Moreover, many non-glutamatergic Ca2+-permeable channels in neurons are also identified as potential therapeutic targets (Tymianski, 2011), including TRPM2 (Belrose and Jackson, 2018).
TRPM2 was discovered as an oxidative stress sensitive Ca2+-permeable channel (Hara et al., 2002; Perraud et al., 2001; Sano et al., 2001) which is gated by elevated intracellular Ca2+ and ADP ribose (ADPR) (Huang et al., 2019; Wang et al., 2018; Zhang et al., 2018), and can also be regulated by different stress factors such as acidic pH(Du et al., 2009b; Starkus et al., 2010; Yang et al., 2010), glutathione (Belrose et al., 2012), Zn2+ (Mortadza et al., 2017), and heat (Kashio et al., 2012). TRPM2 is abundantly expressed in the brain (Fonfria et al., 2006) and is involved neuronal death caused by oxidative stress (Belrose and Jackson, 2018; Mai et al., 2020). Contribution of TRPM2 to ischemic brain injury was initially indicated by TRPM2 knockdown (Jia et al., 2011), and was later demonstrated in TRPM2 global knockout mice (Alim et al., 2013; Gelderblom et al., 2014; Shimizu et al., 2013). It was also reported that TRPM2 in immunocompetent cells plays a critical role in ischemic stroke (Gelderblom et al., 2014). Thus, the mechanisms by which TRPM2 results in deleterious effects in ischemic stroke remain elusive (Belrose and Jackson, 2018; Mai et al., 2020). More importantly, it was unknown whether TRPM2 is involved in excitotoxicity in ischemic stroke. Given the complexity of excitotoxicity, which includes over-activation of glutamatergic NMDARs and subsequent activation of non-glutamatergic Ca2+-permeable channels, a mechanism which can influence both NMDARs’ excitotoxicity and its downstream pathways may be an ideal candidate target for ischemic stroke to produce better therapeutic outcome. As TRPM2 activation requires elevated intracellular Ca2+/ADPR, which can be produced subsequent to NMDARs’ activation during ischemic stroke, we reasoned that TRPM2 may influence NMDARs’ excitotoxicity.
Here we unveil a previously unknown mechanism by which TRPM2 mediates neuronal death during ischemic stroke. We found that TRPM2 exacerbates NMDARs’ excitotoxicity by physically and functionally interacting with NMDARs. We identified the interacting motifs and designed a disruptive peptide TAT-EE3, which can uncouple TRPM2-NMDARs interaction thereby protecting neurons against ischemic injury in vitro and in vivo. Our findings establish that TRPM2 exacerbates extrasynaptic NMDAR’s excitotoxicity, therefore intervention of TRPM2-NMDAR coupling may represent a promising therapeutic strategy for ischemic stroke.
RESULTS
TRPM2 deletion in neurons protects mice against ischemic stroke
Neuron-specific Trpm2 deletion was achieved by crossing nestin-cre mice with Trpm2fl/fl mice, which was confirmed by PCR, western blot, and current recording (Figure S1A–K). Successful middle cerebral artery occlusion (MCAO) was confirmed by monitoring blood flow reduction by 85% (Figure S1L–M). Similar to global Trpm2 knockout (gM2KO), neuron-specific Trpm2 deletion (Cre+, Trpmfl/fl; nM2KO) exhibited reduced infarct volume and improved neurological performance compared with control (Cre−, Trpm2fl/fl; WT) littermates (Figure 1A–D). Apoptosis is an important cause of neuronal death. In the penumbra after MCAO (Figure S1N), the number of TUNEL-positive neurons was markedly smaller in brain slices from gM2KO and nM2KO than those of control littermates (Figure 1E–H, Figure S1O–P). These results suggest that neuronal TRPM2 plays a key role in causing ischemic brain damage.
Figure 1 |. Neuron-specific Trpm2 deletion protects the brain against ischemic damage.
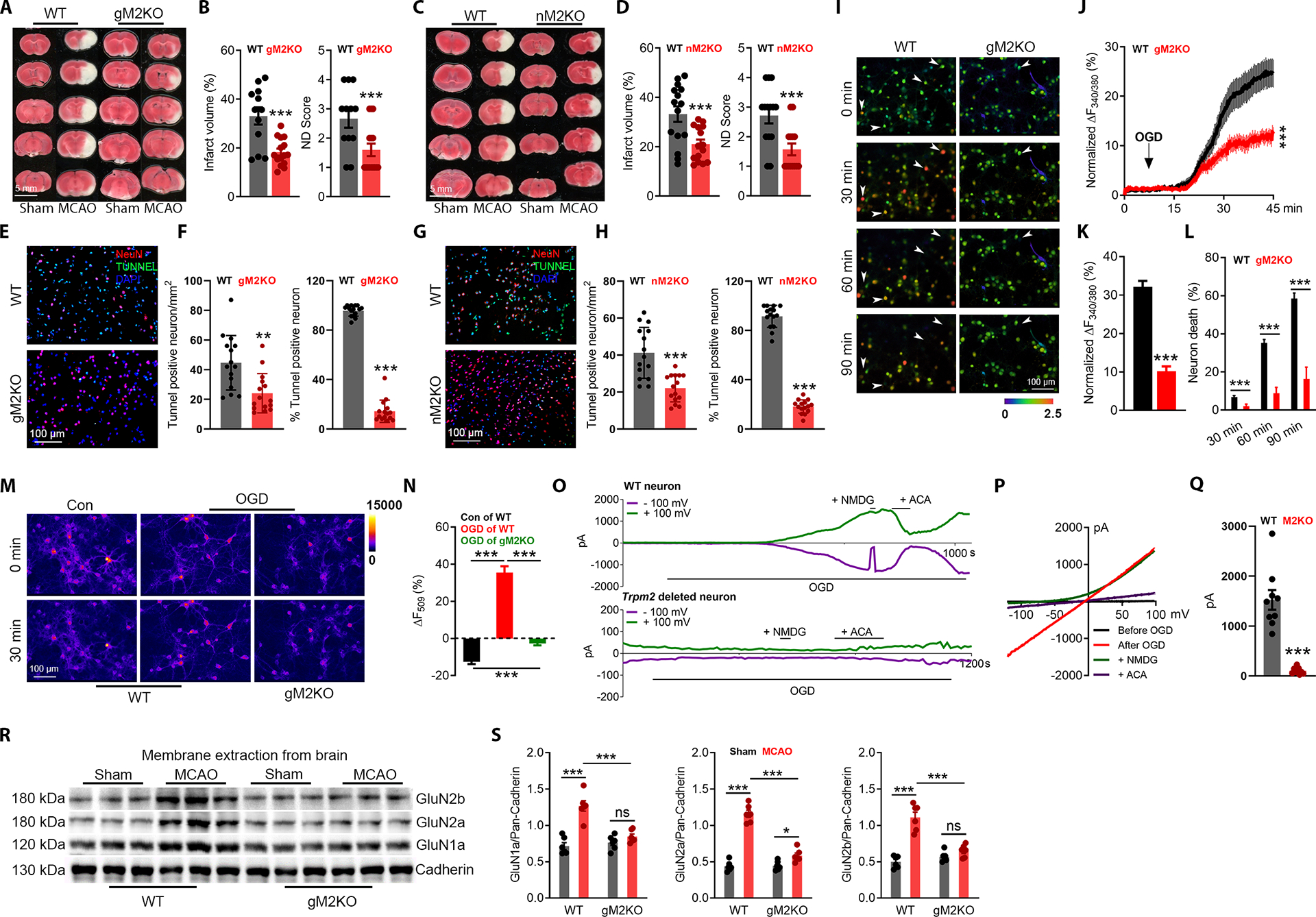
(A-B), Global Trpm2 deletion (gM2KO) attenuates ischemic stroke. (A), TTC staining of brain slices of wild-type (WT) and gM2KO mice 24 hrs after MCAO. Average infarct volume (B-left) and ND score (B-right) (n=12 for WT and 15 for gM2KO).
(C-D), Neuron-specific Trpm2 deletion (nM2KO) produces similar protective effects as that of gM2KO. (C), TTC staining of nM2KO and Cre− control littermates (WT) 24 hrs after surgery. Average infarct volume (D-left) and ND score (D-right) (n=15 for WT and 15 for nM2KO).
(E-H), TUNEL staining of the penumbra (Supplementary Fig. 1N) in brain sections from gM2KO (E-F) and nM2KO (G-H) mice. (E, G), Merged images (Red: NeuN; Blue: DAPI; Green: TUNEL). (F, H), Quantification of TUNEL-positive neurons and mean percentage of TUNNEL positive neurons in all NeuN positive cells (n=5/group).
(I-L), Ratio Ca2+ imaging. (I), Arrows indicate representative lysed neurons with increasing intracellular Ca2+. (J), Averaged Ca2+ imaging traces (n=20/group). (K), Quantification of OGD-induced Fura-2 fluorescence changes (n=238/6 dishes for WT and 233/6 dishes for gM2KO). (L), OGD-induced neuronal death (n=6/group).
(M-N), R123 imaging. (M), R123-labelled mitochondria before and 30 min after OGD. Control group (no OGD treatment) was used to show the rapid photo bleaching of R123. (N), Quantification of R123 change (n=39, 74, n=123, respectively).
(O-Q), OGD-induced TRPM2 activation in WT neurons. (O) Time-dependent current activation and blockade by ACA. NMDG was used to ensure the tight seal. (P), I-V relationship. (Q), Average of current amplitudes.
(R-S), WB for surface expression of NMDARs in the brain (n=12/group).
(ns, no statistical significance, *, p < 0.05, **, p < 0.01, ***, p < 0.001; ANOVA, Bonferroni’s test; mean ± SEM)
Besides apoptosis, Ca2+ overload also necrosis in neurons (Choi, 1995). Using isolated cortical neurons, we performed oxygen-glucose deprivation (OGD) to mimic in vivo ischemic injury. OGD induced a persistent increase of [Ca2+]i (Figure 1I–J), until the lysis of neurons as reflected by a complete loss of Fura-2 fluorescence (Figure 1I; see the arrow-pointed cells). The lysed neurons were counted as dead neurons. Throughout the 90 mins of OGD exposure, a notable number of neurons (6.8%) in the WT group died after 30 mins of OGD, which was increased to 35.2% and 58.5% at 60 and 90 mins (Figure 1I–L). In contrast, there was only 1.9%, 8.7%, and 16.3% of neurons died at 30, 60, and 90 mins in the gM2KO group, respectively (Figure 1L). Also, the increase of [Ca2+]i was remarkably higher in WT than in gM2KO neurons (Figure 1I–L). Similar results were also observed in neurons isolated from nM2KO mice (Figure S2A–D). During OGD, 80% of the Ca2+ entry in neurons is mediated by NMDARs (Goldberg and Choi, 1993; Lipton, 1999). Indeed, NMDAR blockers, AP5 and MK801, inhibited the increase of [Ca2+]i and neuronal death induced by OGD in WT neurons, but there was no further reduce in gM2KO neurons (Figure S2E–H). Mitochondrial dysfunction is an early event promoting neuronal death (Lemasters et al., 2009). We found that OGD induced mitochondrial depolarization in WT neurons as indicated by increased Rh123 fluorescence, but this increase was largely prevented by Trpm2 deletion (Figure 1M–N), NMDARs blockers AP5 or MK-801, removing extracellular Ca2+, or chelating intracellular Ca2+ using BAPTA-AM (Figure SI–L). These results suggest a critical role of TRPM2 in aggregating NMDAR-mediated Ca2+ overload, mitochondrial dysfunction and neuronal death during OGD.
Then we examined whether TRPM2 in neurons can be activated by OGD. Sub-optimal Ca2+ and ADPR concentrations (Du et al., 2009a) in the pipette solution were used for TRPM2 recording (Figure S1Q). During OGD, TRPM2 current was elicited in neurons from WT mice (Figure 1O–Q), but not in the neurons from Trpm2 deletion mice (Figure 1O bottom; Figure 1Q). We also found that TRPM2 expression in the brain was markedly upregulated by MCAO (Figure S1R–S). Moreover, MCAO-induced increase of NMDAR’s surface expression was inhibited by Trpm2 deletion (Figure 1R–S). Importantly, the effects of TRPM2 on NMDAR’s surface expression were specific, as the surface expression of other membrane proteins such as TRPM4 and Pannexin-1 were not influenced by Trpm2 deletion (Figure S2M–N). The above data suggest that the upregulation of TRPM2 expression is important for the increase of NDMAR’s surface expression after ischemic stroke.
TRPM2 interacts with NMDARs
To understand how TRPM2 influences NMDAR’s surface expression, we examined whether TRPM2 interacts with NMDARs, and found that TRPM2 can be co-immunoprecipitated by GluN1a, GluN2a, and GluN2b in HEK293T overexpression system (Luo et al., 2002; Schmitz et al., 2003) (Figure 2A). Reciprocally, GluN1a, GluN2a, and GluN2b can also be co-immunoprecipitated by TRPM2 (Figure 2B). Interestingly, when transfected with GluN1a, GluN2a, or GluN2b separately, TRPM2 can be only co-immunoprecipitated with GluN2a and GluN2b, but not GluN1a, indicating that in NMDAR, GluN2a and GluN2b interact with TRPM2. We further confirmed the presence of TRPM2-NMDAR association in the brain lysates (Figure 2D–E). To determine the consequences of TRPM2-NMDAR association, we found that NMDAR currents were much bigger in WT neurons (Figure 2F–G) and in NMDARs/TRPM2 co-expressing HEK293 cells (Figure 2J–K) than that in the TRPM2-KO neurons and NMDARs/EGFP co-expressing cells, respectively. Similarly, surface expression of NMDAR was higher in NMDARs/TRPM2 than NMDARs/EGFP expressing cells (Figure 2H–I). The increased surface expression and enhanced NMDAR current were also observed when GluN1a/GluN2a or GluN1a/GluN2b were separately transfected with TRPM2 (Figure S3A–H). Moreover, both the current amplitude and surface expression of TRPM2 were also increased when TRPM2 was co-expressed with NMDARs (Figure S3I–L). These results suggest that the physical interaction between TRPM2 and NMDAR produces functional consequences.
Figure 2 |. TRPM2 physically and functionally interacts with NMDARs.
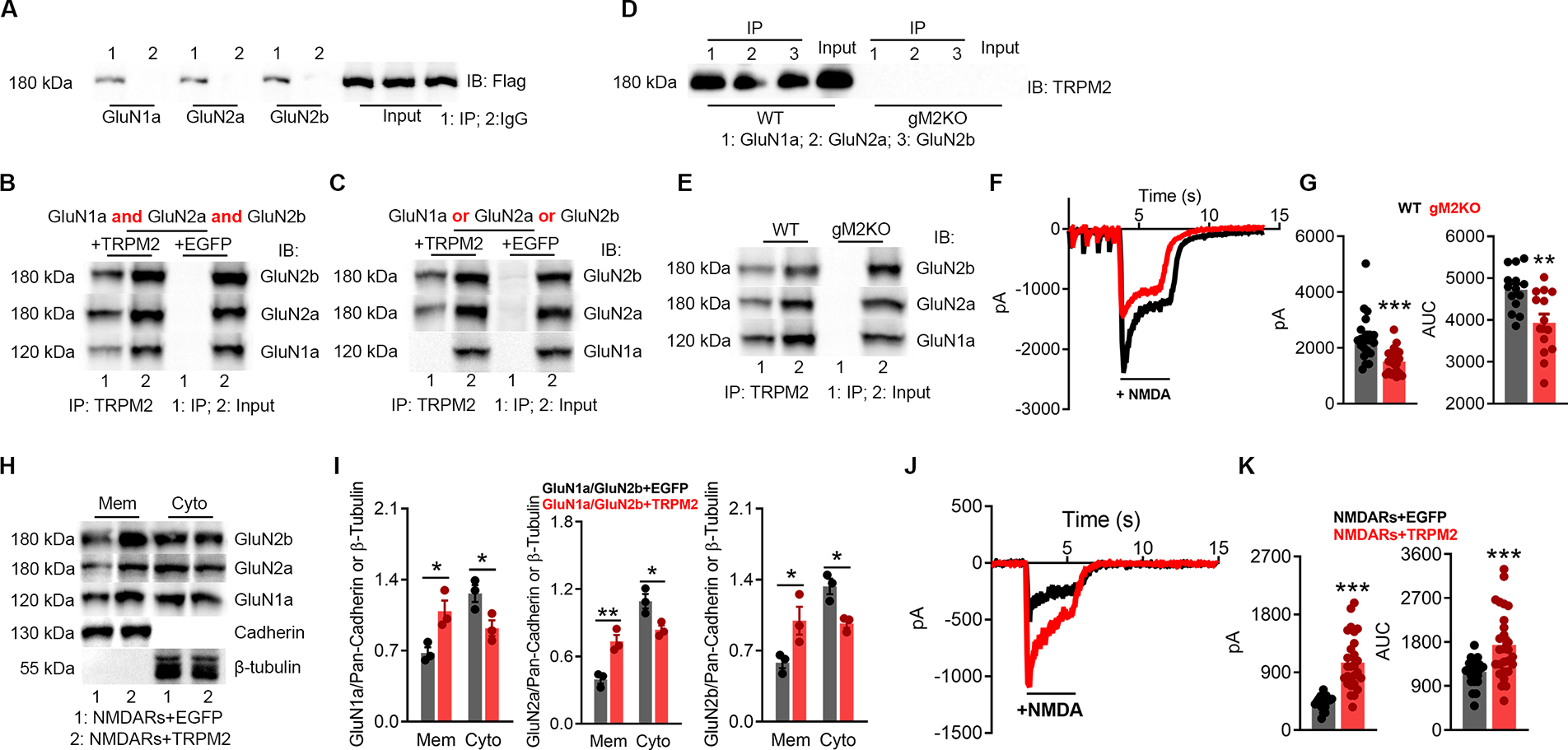
(A-B), Co-IP of NMDARs and TRPM2 expressed in HEK-293T cells. (A), immunoprecipitation (IP) using anti-NMDARs and IB with anti-Flag. (B), IP using anti-Flag and IB with anti-NMDARs.
(C), Co-IP of TRPM2 co-expressed with GluN1a, GluN2a, or GluN2b in HEK-293T cells. IP using anti-Flag and IB with anti-NMDARs.
(D-E), Co-IP of NMDARs and TRPM2 in the brain lysates. (D) IP using anti-NMDARs and IB with anti-TRPM2. (E) IP using anti-TRPM2 and IB with anti-NMDARs.
(F-G), NMDAR current recording from isolated WT and gM2KO neurons. (G), Average peak current amplitude and area under curve (AUC) (n=20/group).
(H-I), Surface expression of NMDARs in HEK-293T cells co-transfected with NMDARs/TRPM2, or NMDARs/EGFP plasmids. Membrane (Mem) and cytosol (Cyto) protein levels assessed with WB.
(J-K), NMDAR current recording from HEK293T cells transfected with NMDARs/TRPM2, or NMDARs/EGFP. (K), Average peak current amplitude and AUC from NMDARs/EGFP group (n=23) and NMDARs/TRPM2 group (n=27).
(*, p < 0.05, **, p < 0.01, ***, p < 0.001; ANOVA, Bonferroni’s test; mean ± SEM,)
Identification of the interaction motifs in NMDARs and TRPM2
N- and C-tail fragments of TRPM2 were subcloned and tagged with Flag and GFP, respectively. When co-expressed with NMDARs, the TRPM2-NT but not the TRPM2-CT was detected in the immunoprecipitates of anti-NMDAR (Figure 3A–B). For NMDARs, the C-tail of GluN2a and GluN2b were generated and tagged with GFP, and showed interaction with TRPM2, whereas the GFP-tagged C-tail deleted GluN2a (GluN2a-ΔCT) and GluN2b (GluN2b-ΔCT) were absent in the immunoprecipitates of anti-TRPM2 (Figure 3C). Moreover, TRPM2-NT produced a similar potentiation on NMDAR current and NMDA-induced Ca2+ entry as the full-length TRPM2 (TRPM2-FL) (Figure 3D–E). These findings suggest that the C-termini of GluN2a and GluN2b interact with TRPM2’s N-terminal domain. To narrow down the NMDAR-interacting domain in TRPM2-NT (Figure 3F), we generated a series of truncation constructs by incrementally deleting about 50 residues. We found that the N-terminal amino acid residues from 631 to 679 are critical for the TRPM2-NMDAR interaction (Figure 3G–I), as both forward and reverse Co-IP experiments confirmed the interaction of NMDARs with the fragments of 1–727 and 1–679, whereas the fragments shorter than 631 residues failed to interact with NMDARs. We further shortened the 1–679 fragment and found that the region between residues 665 and 679 is the minimal sequence required for the TRPM2-NMDARs interaction (Figure 3J–L).
Figure 3 |. EE3 motif in TRPM2 mediates TRPM2-NMDARs coupling.
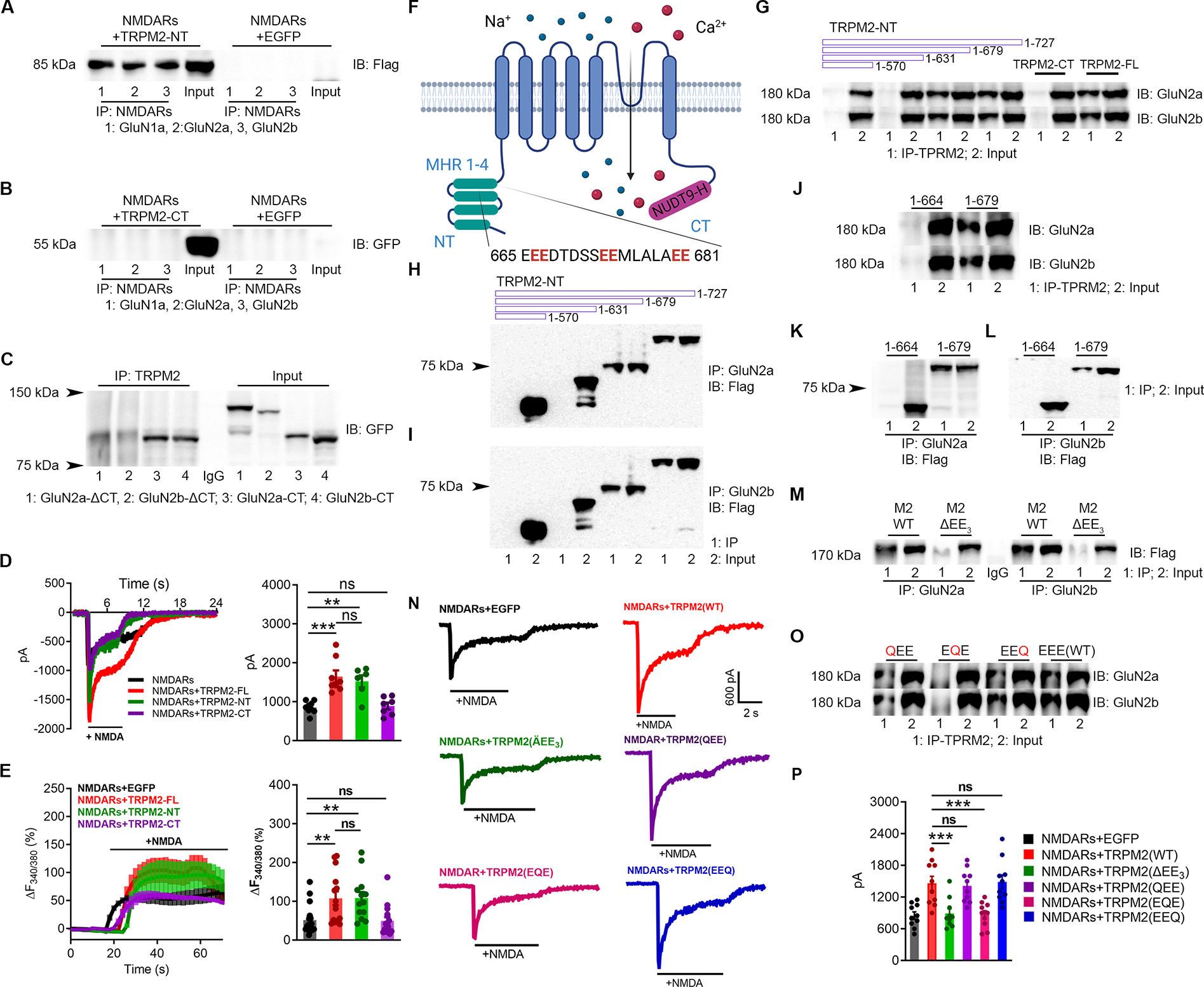
(A-B), Co-IP of N-terminal and C-terminal fragments of TRPM2 (TRPM2-NT (flag-tagged, ~85 kDa), TRPM2-CT (GFP-tagged, ~60 kDa)) with NMDARs. (A), IP using anti-NMDARs and IB with anti-Flag. (B) IP using anti-NMDARs and IB using anti-GFP.
(C), Co-IP of TRPM2 with the C-terminus of GluN2a (GluN2a-CT, 1054–1068), C-terminus deleted GluN2a (GluN2a-ΔC, 1–1053), C-terminus of GluN2b (GluN2b-CT, 1041–1691), and C-terminus deleted GluN2b (GluN2b-ΔC, 1–1047). All the constructs were GFP-tagged. IP using anti-TRPM2 and IB using anti-GFP.
(D), NMDAR current recording in HEK-293T cells transfected with NMDARs and EGFP, TRPM2-full length (FL), TRPM2-NT or TRPM2-CT (left). Average current amplitudes (n=9,8,8,8, respectively).
(E), NMDARs-mediated Ca2+ influx in HEK-293T cells transfected with NMDARs and EGFP, TRPM2-full length (FL), TRPM2-NT or TRPM2-CT (left). Averaged changes of F340/380.
(F), Membrane topology of TRPM2. The EE3 domain is located in the MHR4.
(G-L), Co-IP of Flag-tagged TRPM2 N-terminal segments with different lengths (1–570, 1–631, 1–678 and 1–727) with NMDARs. (G), IP using anti-TRPM2 (anti-GFP for TRPM2-CT) and IB using anti-GluN2a/2b. (H, I) IP using anti-GluN2a (H) or anti-GluN2b (I), and IB using anti-Flag.
(J-L), Co-IP of the TRPM2 N-tail fragments (1–664 and 1–679) with NMDARs. (J), IP using anti-TRPM2 and IB using anti-GluN2a/2b. (K, L), IP using anti-GluN2a (K) or anti-GluN2b (L), and IB using anti-Flag.
(M-P), Physical and functional coupling of TRPM2 and NMDARs through EE3 domain. (M, O), EE3 motif deletion mutant of TRPM2 (TRPM2-ΔEE3), and EE3 mutations of TRPM2, TRPM2-QEE (E666Q, E667Q), TRPM2-EQE (E673Q, E674Q), and TRPM2-EEQ (E680Q, E681Q) were co-expressed with NMDARs in HEK293T cells for co-IP. (M) IP using anti-GluN2a/b, and IB using anti-Flag. (O), IP using anti-TRPM2 and IB using anti-GluN2a/2b. (N), NMDAR current recording in HEK293T cells co-expressed with EGFP, WT-TRPM2, TRPM2-ΔEE3, TRPM2-QEE, TRPM2-EQE, and TRPM2-EEQ. (P), Mean current amplitude (n=10/group)
(ns, no statistical significance, **, p < 0.01, ***, p < 0.001; ANOVA, Bonferroni’s test; mean ± SEM)
Interestingly, the 15 residues between 665 and 679 contain two “glutamate-glutamate” (EE) repeats separated by five residues and followed by another EE repeat (Figure 3F), so we denoted the sequence between 665 to 681 the “EE3” motif for simplicity. This EE3 motif is conserved in TRPM2 from different species, but absent in all other TRPM channels (Figure S4A–B). When the EE3 domain was deleted from the full-length TRPM2 (TRPM2-ΔEE3), interaction between TRPM2 and NMDARs was disappeared (Figure 3M). Intriguingly, when the middle “EE” of the EE3 motif was replaced by two glutamine residues (“QQ”), the resulting TRPM2-EQE mutant failed to interact with NMDARs, whereas the replacements of the first and third EE repeats (TRPM2-QEE, TRPM2-EEQ) did not influence the TRPM2-NMDARs interaction (Figure 3O). Consistent with the disrupted interaction, TRPM2-ΔEE3 and TRPM2-EQE mutants failed to enhance NMDAR currents, whereas TRPM2-QEE and TRPM2-EEQ mutants produces an effect similar to TRPM2-WT (Figure 3N & 3P). Importantly, these mutants exhibited similar electrophysiological properties as TRPM2-WT (Figure S4C–H). These results indicate that the EE3 motif is essential for both the physical and functional coupling between TRPM2 and NMDARs.
The C-tail of GluN2a and GluN2b have diverged substantially in evolution. As the EE3 motif in TRPM2 is largely negatively charged (Figure S4A–B), we focused on a segment of GluN2a/GluN2b regions (GluN2a:1254-1270; GluN2b: 1259-1274; Figure 4C) near the binding site for CaMKII (Bayer et al., 2001; Goodell et al., 2017), which is positively charged by lysine (K) and arginine (R) residues. We designated this region the “KKR” motif (Figure S4I), and KKR motif is highly conserved between GluN2a and GluN2b, and in different species (Figure S4J–K). Deletion of KKR motif abolished the interaction between TRPM2 and GluN2a/GluN2b (Figure 4A, B), indicating KRR motif is required for the TRPM2-NMDAR interaction.
Figure 4 |. KKR region in GluN2a and GluN2b is required for the direct binding to the EE3 motif in TRPM2.
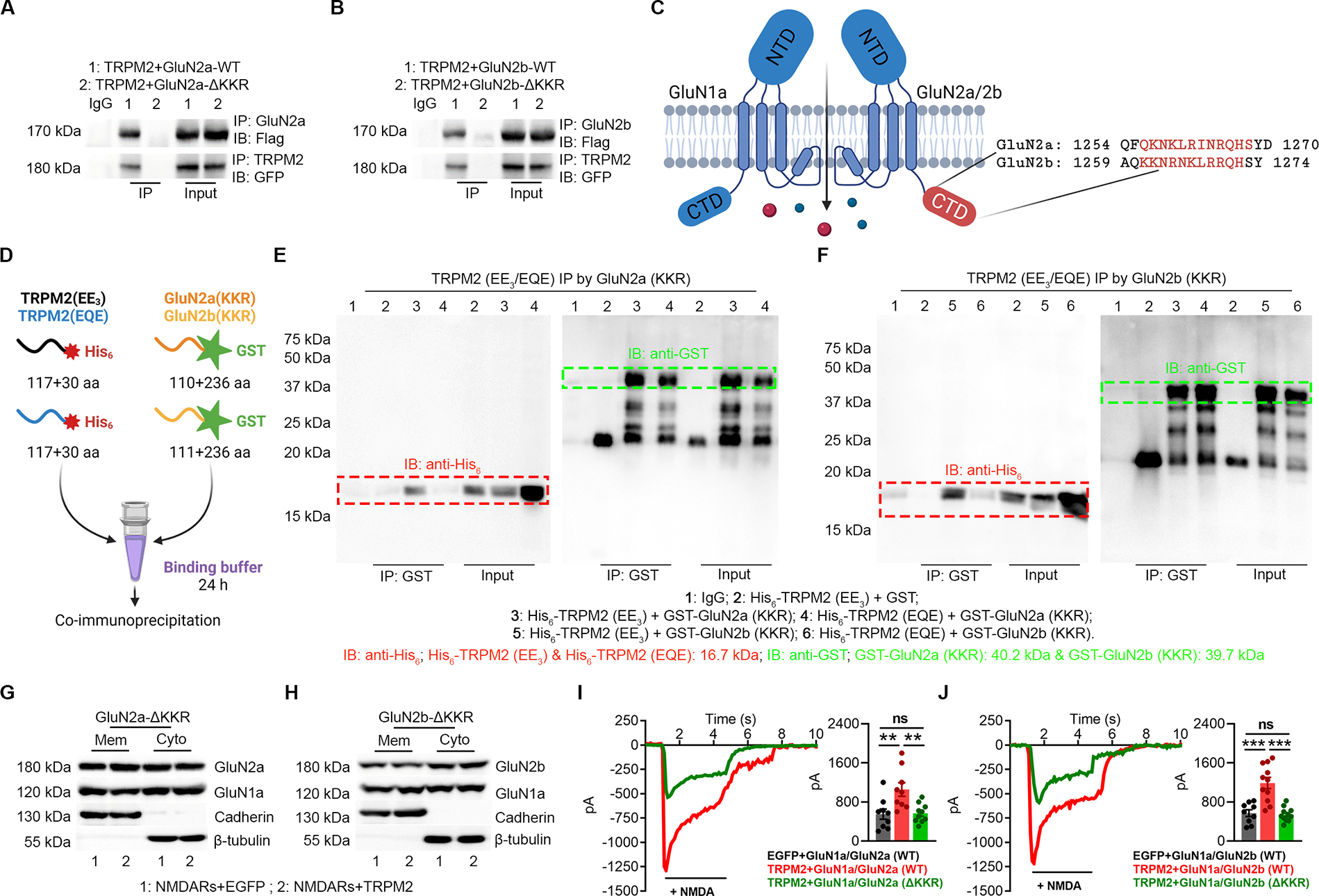
(A-B) Co-IP of KKR region deleted GluN2a (GluN2a-ΔKKR, A) and GluN2b (GluN2b-ΔKKR, B) with TRPM2. TRPM2 is flag-tagged and GluN2a/GluN2b is GFP-tagged. IP using anti-GluN2a/b and IB with anti-Flag (upper). IP using anti-TRPM2 and IB using anti-GFP (lower).
(C) Structure of GluN2a and GluN2b. The KKR domain (red label indicates the deleted sequence (ΔKKR)) is localized within the C terminal domain (CTD).
(D), Schematic diagram of the in vitro binding assay. EE3 and EQE containing fragments were labelled by a N-terminal His6 tag and KKR containing fragments were labelled by a N-terminal GST tag.
(E-F) Co-IP of EE3 and EQE containing fragments with KKR containing fragments from GluN2a (E) and GluN2b (F). IP using anti-GST and IB using anti-His6.
(G-H) Surface expression of NMDARs in HEK-293T cells co-transfected with TRPM2 and GluN2a-ΔKKR (G) and GluN2b-ΔKKR (H).
(I, J) NMDAR current recording in HEK-293T cells co-transfected with TRPM2 and GluN2a-ΔKKR (I) and GluN2b-ΔKKR (J).
(**, p<0.01; ***, p < 0.001; ANOVA, Bonferroni’s test; mean ± SEM)
Then we sought to determine whether EE3 and KKR motifs directly bind to each other. A 117-residue TRPM2 segment containing either the EE3 or EQE motif was subcloned into a modified His6-tag vector (Li and Hao, 2010) (Figure 5–B, Figure S5A–B), and the KKR-containing fragments in GluN2a (110-residues) and GluN2b (111-residues) were subcloned into a modified GST-tag vector (Li and Hao, 2010) for expression and purification (Figure S5C). In vitro co-IP binding assays were performed (Figure 4D). EE3 motif was co-immunoprecipitated by KKR motifs derived from GluN2a/2b, but EQE was not (Figure 4E–F; Figure S5D). Similarly, KKR motifs were effectively co-immunoprecipitated by EE3 motif, but not by EQE motif (Figure S5E). These results indicate that TRPM2-GluN2a/GluN2b association is mediated by direct interaction between their EE3 and KKR motifs, respectively. Moreover, deletion of KKR motif abolished the increased surface expression of NMDAR (Figure 4G–H; Figure S5F–G) and the enhanced NMDAR current (Figure 4 I, J) induced by TRPM2, confirming the importance of KKR motif in the functional coupling of TRPM2-NMDARs.
Figure 5 |. N-tail of TRPM2 Interacts with PKC-γ.
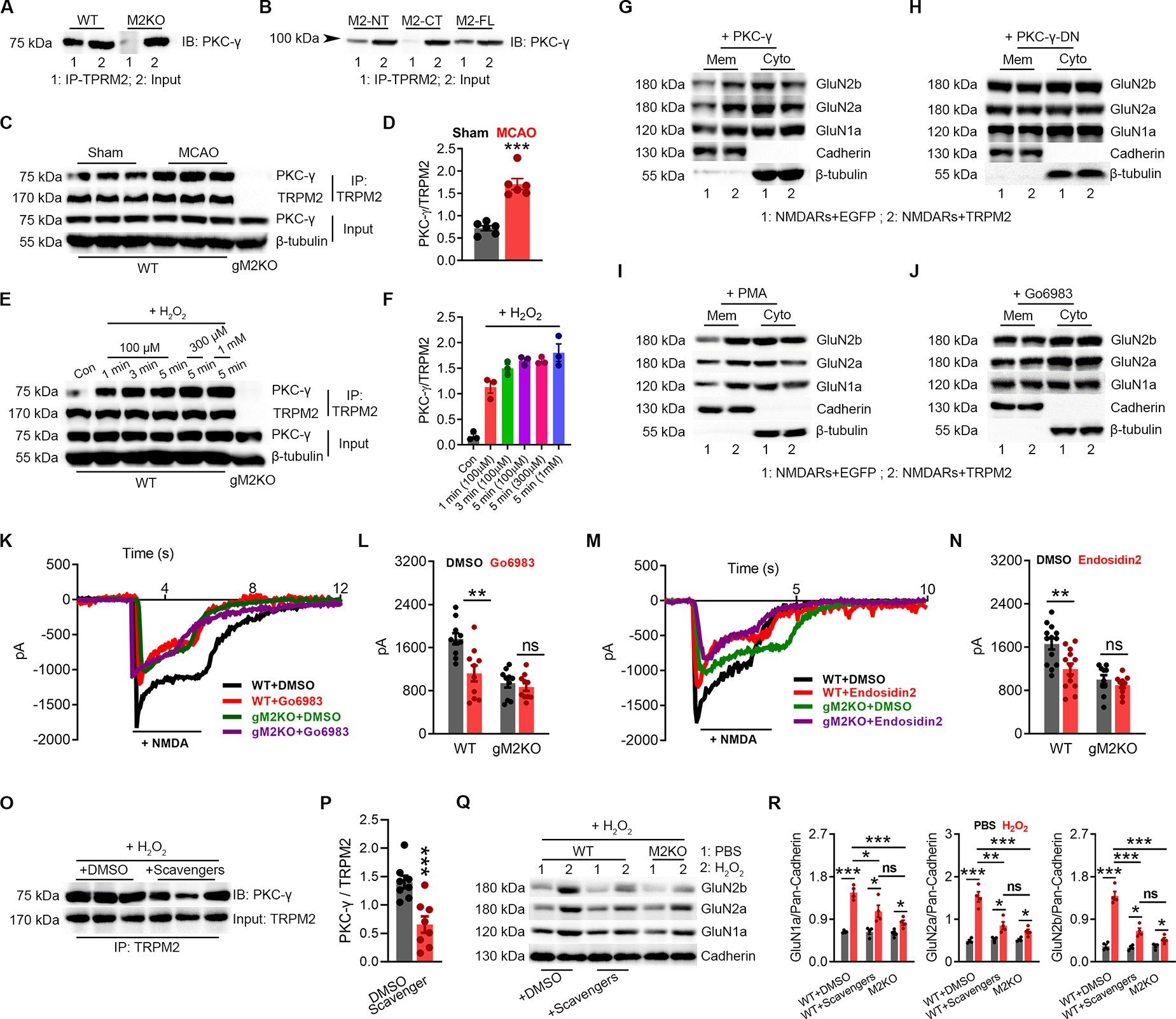
(A-B), Co-IP of PKCγ and TRPM2 using brain lysates (A), and in HEK293T cells expressing PKCγ with TRPM2-FL, TRPM2-NT, or TRPM2-CT (B). IP using anti-TRPM2 ( anti-TRPM2-NT for M2-NT, and anti-TRPM2-CT for M2-CT) and IB using anti- PKCγ.
(C-D), Co-IP of TRPM2 and PKCγ using brain lysates from WT mice subjected to MCAO or sham surgery. IP using anti-TRPM2 and IB using anti- PKCγ. (D) Quantification of PKCγ/TRPM2 (n=6/group).
(E-F), Co-IP of TRPM2 and PKCγ using neuron lysates from WT mice subjected to H2O2 treatment (at 100 μM for 1 min, 3 min, and 5 min, at 300 μM for 5 min and at 1 mM for 5 min) (E), IP using anti-TRPM2 and IB using anti-PKCγ. (F), Quantification of PKCγ/TRPM2 (n=3/group).
(G-H), Surface expression of NMDARs in HEK-293T cells co-transfected with PKC-γ/EGFP and PKC-γ/TRPM2 (G) or PKC-γ-DN/EGFP and PKC-γ-DN/TRPM2 (H).
(I-J), Surface expression of NMDARs in HEK-293T cells co-transfected with TRPM2 or EGFP with the treatment of PKC activator PMA (I) or inhibitor Go6893 (J).
(K-L), NMDAR current recording in WT and gM2KO neurons treated with or without Go6983 at 1 μM for overnight. (L), Mean current amplitude (n=10~15 neurons from 2 mice).
(M-N), NMDAR current recording in WT and gM2KO neurons treated with or without the treatment of exocytosis inhibitor endosidin2 at 1 μM overnight. (N) Mean current amplitude (n=10~15 neurons from 2 mice).
(O, P) Co-IP of TRPM2 and PKCγ using neuron lysates from WT mice subjected to H2O2 treatment at 100 μM for 3 min with the preincubation of DMSO or scavengers. (P), Co-IP using anti-TRPM2 and IB using anti- PKCγ. (D) Quantification of PKCγ/TRPM2 (n=6/group).
(Q, R) Surface expression of NMDARs in isolated WT neurons subjected to H2O2 at 100 μM for 3 min with the preincubation of DMSO or scavengers (n=4 in each group).
(ns, no statistical significance, *, p < 0.05, **, p < 0.01, ***, p < 0.001; ANOVA, Bonferroni’s test; mean ± SEM)
Mechanisms of TRPM2-NMDAR functional coupling
Next, we aimed for understanding how TRPM2 potentiates NMDAR current. PKC regulates NMDAR’s surface trafficking (Lan et al., 2001; Zheng et al., 1999). PKCγ, the neuron-specific PKC, can be readily co-immunoprecipitated by anti-TRPM2 in the brain lysates (Figure 5A). We further narrowed down the binding area for PKC on TRPM2 is located on its N-tail using HEK293 cells co-transfected with PKCγ and TRPM2-FL, TRPM2-CT, or TRPM2-NT (Figure 5B). Moreover, MCAO increased the binding of PKCγ to TRPM2 in the brain (Figure 5C–D). Similarly, cultured neurons treated with H2O2 exhibited markedly increased TRPM2-PKCγ interaction (Figure 5E–F).
Then we sought to determine whether TRPM2-PKCγ association influences the surface expression of NMDARs. NMDARs/TRPM2 were co-expressed with wild-type PKCγ or dominant-negative PKCγ (PKCγ-DN) (Colgan et al., 2018; Soh and Weinstein, 2003) (Figure 5G–H, Figure S6A–B). Over-expression of PKCγ further increased the TRPM2-induced increase of NMDAR’s surface expression, whereas PKCγ-DN abolished this increase, suggesting that a functional PKCγ is required for TRPM2-mediated increase of NMDAR’s surface expression. Indeed, PKC activator PMA also markedly increased the TRPM2-induced increase of NMDAR’s surface expression (Figure 5I; Figure S6C, E), whereas PKC inhibitors Go6983 and staurosporine abolished this increase (Figure 5J & Figure S6D, F–G). Similarly, Go6983 and staurosporine reduced the NMDAR current amplitude in WT neurons to a similar level as in gM2KO neurons (Figure 5K–L, Figure S6H–I).
Surface trafficking of NMDARs involves exocysts (Sans et al., 2003). Endosidin 2, an exocyst inhibitor, prevented the increased surface expression of NMDARs induced by TRPM2 (Figure S6J–K). Also, endothsidin 2 pretreatment inhibited NMDAR currents in WT neurons, but not in gM2KO neurons (Figure 5M–N). CaMKII is also important in NMDAR’s trafficking. CaMKII inhibitor KN93 inhibited the increased surface expression of NMDARs (Figure S6L–M) induced by TRPM2, and reduced NMDAR current amplitude in WT neurons to the similar level in gM2KO neurons (Figure S6N–O). Preincubation with oxidative stress scavengers Mn(III)TBAP together with L-NMA inhibited the H2O2-induced binding of PKCγ to TRPM2 (Figure 5O–P). Moreover, the enhanced surface expression of NMDARs induced by H2O2 in WT neurons was reduced to the similar levels as that in M2KO neurons (Figure 5Q–R).
Taken together, the above results indicate that PKCγ interacts with TRPM2, which can be promoted by oxidative stress in vitro and by MCAO in vivo, and suggest that the TRPM2-PKCγ association may be required for the functional coupling between TRPM2 and NMDAR.
TAT-EE3 eliminates TRPM2-NMDAR coupling
As the TRPM2 N-terminal EE3 motif is critical for the TRPM2-NMDAR interaction, we designed a membrane-permeable interfering peptide, TAT-EE3, and scrambled control TAT-SC or TAT-EQE, to investigate whether disruption of the physical interaction influences their functional coupling. Similar to TRPM2-Δ(EE)3 and TRPM2-EQE mutants, TAT-EE3 treatment completely disrupted the interaction of TRPM2 with GluN2a (Figure 6A) and GluN2b (Figure 6B). Also, the enhancement of NMDAR’s surface expression (Figure 6D, Figure S6Q) (Figure 6C, Figure S6P), and the increase of NMDAR current amplitude (Figure 6E–F) induced by TRPM2 were inhibited by TAT-EE3.
Figure 6|. TAT-EE3 disrupts the physical and functional interaction between TRPM2 and NMDAR.
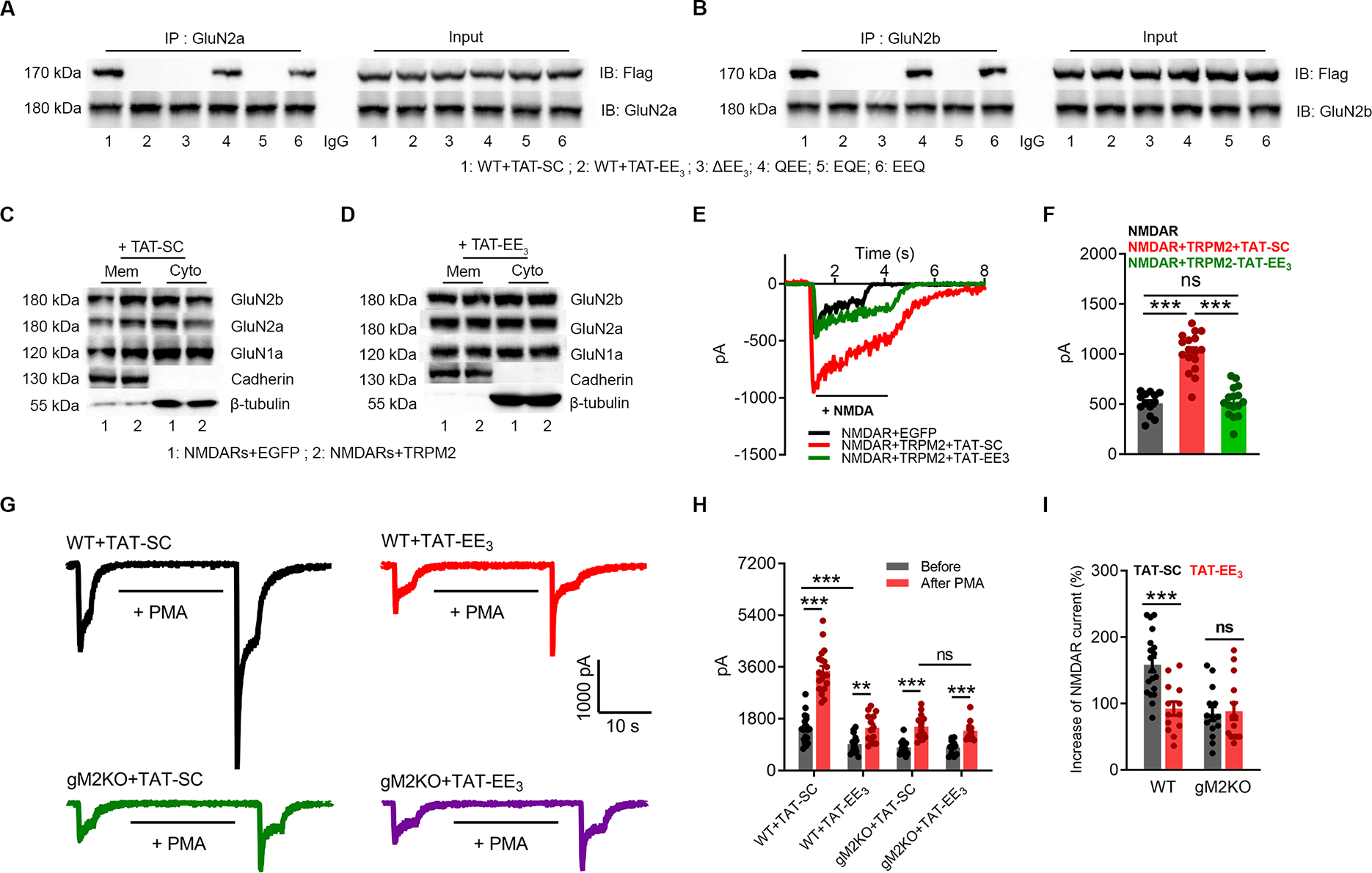
(A-B), Co-IP of NMDARs with WT or EE3-domain mutants of TRPM2 (Flag-tagged). IP using anti-GluN2a (A) or anti-GluN2b (B), and IB using anti-Flag.
(C-D), Surface expression of NMDARs in HEK-293T cells co-transfected with TRPM2 or EGFP with the treatment of 10 μM TAT-SC (C) or TAT-EE3 (D) overnight.
(E-F), NMDAR current recording in HEK293T cells co-transfected with TRPM2 and NMDARs and treated with 10 μM TAT-EE3 or TAT-SC for overnight. (F), Mean current amplitude (n=13,16,17, respectively).
(G-I), Effects of TAT-EE3 on PKC-induced changes of NMDAR currents recorded in WT and gM2KO neurons. Neurons were pre-incubated with 10 μM TAT-SC or TAT-EE3 overnight. (G) NMDAR current recording in WT and gM2KO neurons before and after PMA (1 μM) perfusion for 20 s. (H), Average current amplitude. (I), Average percentage increases of NMDAR currents induced by PMA (n=10~15/group).
(ns, p>0.05; **, p < 0.01; ***, p < 0.001; ANOVA, Bonferroni’s test; mean ± SEM)
PKC can increase both the surface trafficking and channel activity of NMDAR (Lan et al., 2001). In WT neurons treated with TAT-SC, NMDA-induced current was increased from 1542.1±117.1 pA to 3642.1±180.8 pA by a 20-s PMA perfusion (Figure 6G, top left), which is about 1.5-fold increase (Figure 6H–I). However, in WT neurons treated with TAT-EE3, NMDAR currents were substantially reduced before and after PMA perfusion (Figure 6G, top right), and the current potentiation by PMA was also eliminated (Figure 6H–I). In neurons from gM2KO mice, the basal NMDAR current amplitude (Figure 6G, bottom) and PMA-induced increase were much smaller than that of WT neurons, and TAT-EE3 did not cause additional inhibition (Figure 6H–I). These data further support our hypothesis that the TRPM2-PKCγ association is required for the functional coupling between TRPM2 and NMDAR.
Disruption of TRPM2-NMDAR interaction protects neurons against OGD
The functional uncoupling of TRPM2 from NMDARs by TAT-EE3 prompted us to investigate its potential protective effect against ischemic injury. Pre-treatment of TAT-EE3 inhibited the NMDAR currents in WT neurons but not in M2KO neurons (Figure 7A–B), indicating the specificity of TAT-EE3 in disrupting the TRPM2-NMDAR interaction. When neurons were exposed to OGD, the increase of intracellular Ca2+ was markedly inhibited in WT but not in gM2KO neurons pre-treated with TAT-EE3 (Figure 7C–D). Moreover, OGD-mediated neuronal death in WT neurons treated with TAT-SC was 10.6%, 47.6%, and 70.5% at 30, 60, and 90 min, respectively, which were drastically reduced by TAT-EE3 to 3.6%, 9.9%, and 45.2%, similar levels as those in M2KO neurons (Figure 7E). Moreover, TAT-EE3 inhibited OGD-induced mitochondria depolarization in WT neurons to the similar levels as those in neurons from gM2KO or nM2KO mice (Figure 7G–H). The observed mitochondrial dysfunction was supposed to be mediated by Ca2+ entry, as membrane depolarization was inhibited when neurons were perfused with Ca2+-free OGD solution (last column in Figure 7G: micrographs, and Figure 7H). In summary, TAT-EE3 inhibited the exacerbation of excitotoxicity caused by TRPM2-NMDAR coupling, and protects neuron against OGD.
Figure 7|. Uncoupling TRPM2 and NMDARs by TAT-EE3 protects neurons against OGD-induced injury.
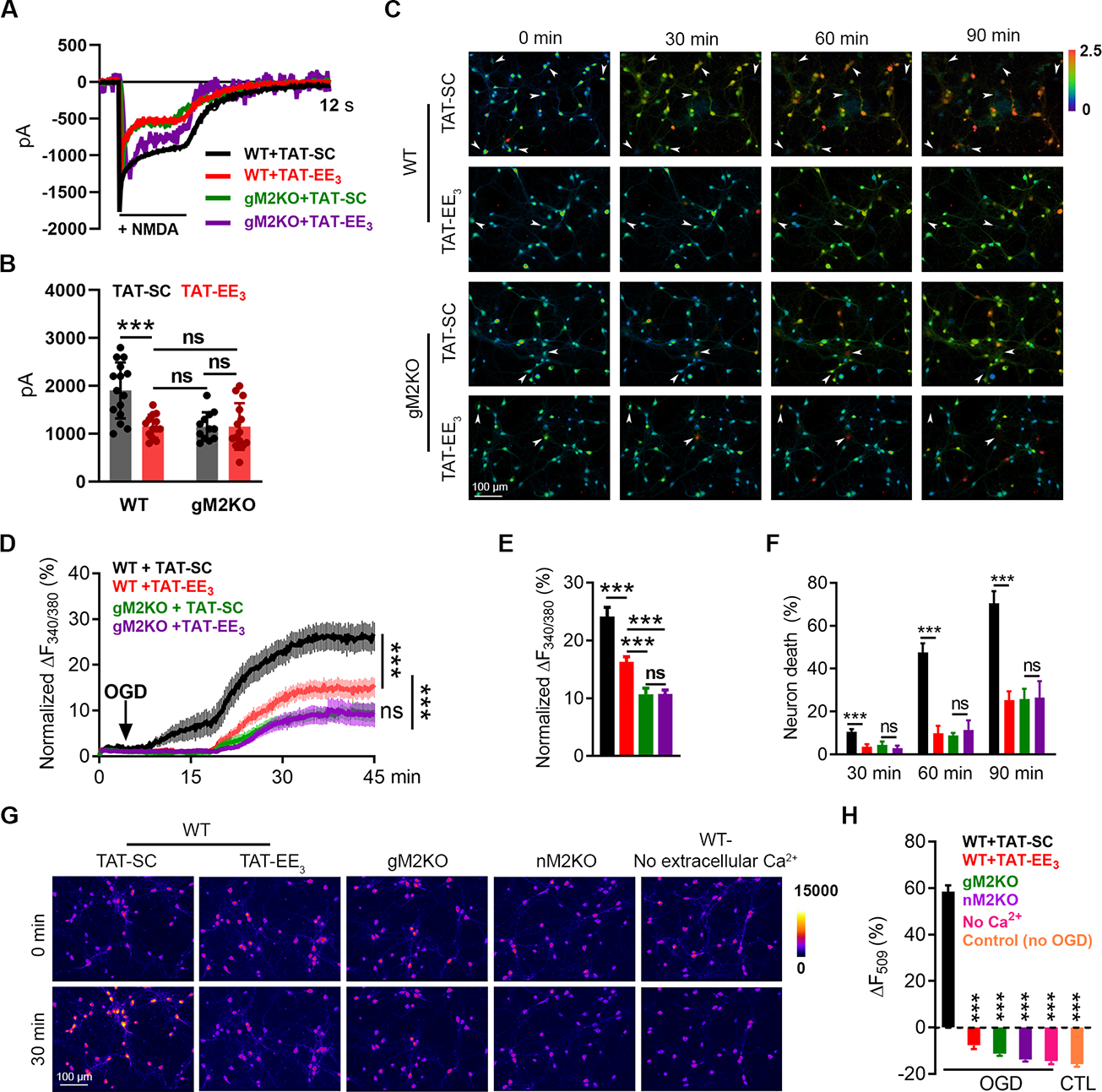
(A-B), NMDAR current recording in WT and global TRPM2 knockout (gM2KO) neurons treated with 10 μM TAT-EE3 or TAT-SC for overnight. (B), Mean current amplitude (n=15, 13, 12, and 14, respectively).
(C-F), Ratio Ca2+ imaging. (C), Arrows indicate representative lysed neurons with increasing intracellular Ca2+. (D), Averaged Ca2+ imaging traces (n=20/group). (E), Quantification of OGD-induced Fura-2 fluorescence changes (n=179,269,232,242/4 dishes, respectively). (F), OGD-induced neuronal death (n=4/group).
(G-H), R123 imaging. (G), R123-labelled mitochondria before and 30 min after OGD. (H), Quantification of R123 change (n=296,262,265,208,145,258/4 dishes, respectively).
(***, p < 0.001; ANOVA, Bonferroni’s test; mean ± SEM)
Uncoupling of TRPM2-NMDAR association attenuates ischemic stroke
We first determined the efficacy of TAT-EE3 in disrupting the TRPM2-NMDAR interaction in vivo. TAT-EE3, TAT-SC and TAT-EQE, were intraperitoneally (i.p.) administrated to WT mice prior to MCAO surgery, and the TRPM2-NMDAR interaction was assessed by Co-IP at 2h, 12h, and 24h after i.p. injection. TAT-EE3 effectively disrupted the TRPM2-NMDAR interaction at 2h and 12h, while at 24h, there was a ~ 40% recover of TRPM2-NMDAR interaction. In contrast, the sequence-specific scramble TAT-EQE did not influence the interaction, similar to that of the random scramble TAT-SC (Figure 8A). We then designed an i.p. injection strategy (Figure 8B). TAT-EE3, TAT-EQE or TAT-SC, were i.p. administrated before MCAO (pre-MCAO), or after MCAO (post-MCAO), followed by re-injection every 12 h till the time for infarction assessment. For pre-MCAO treatment, TAT-EE3 or TAT-SC (100 nmol/kg, estimated to be 2 μM based on 1 ml blood) were i.p. administrated 15 mins before MCAO or sham surgery as previously reported (Weilinger et al., 2016).
Figure 8 |. TAT-EE3 alleviates ischemic stroke by preserving pro-survival signaling.
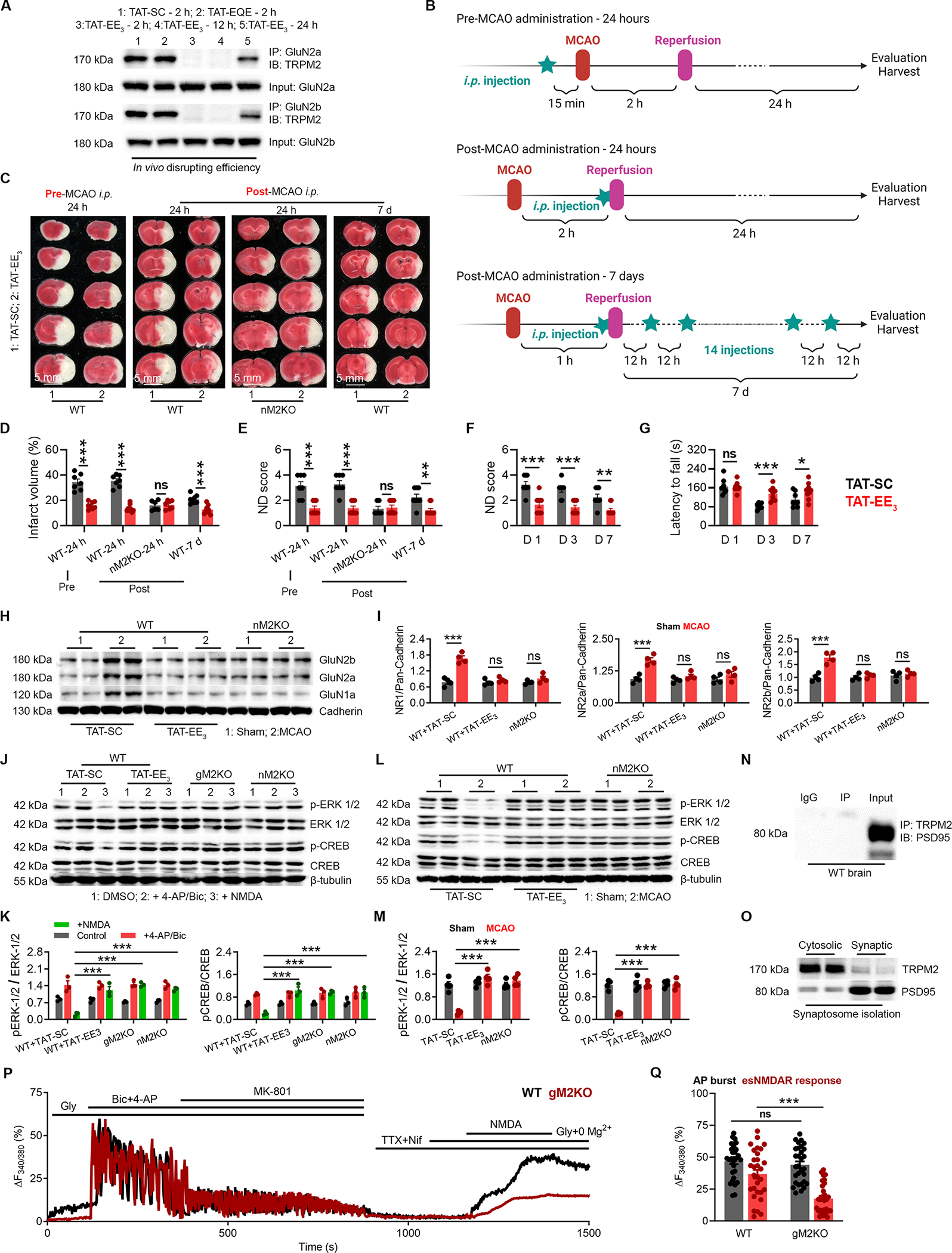
(A) Co-immunoprecipitation (Co-IP) of TRPM2 and GluN2a/2b in the brain lysates from WT mice after MCAO with the treatment of TAT-SC for 2 h, TAT-EQE for 2 h, and TAT-EE3 for 2 h, 12 h and 24 h. IP using anti-GluN2a/2b and IB using anti-TRPM2.
(B) Graphic illustration of injection strategy. For pre-MCAO administration, TAT-EE3 was injected intraperitoneally (i.p.) 30 min before the MCAO. For post-MCAO administration, TAT-EE3 was injected right before the reopening of the occulated MCA to best mimic the treatment of ischemic stroke under clinical situation. For examining the long-term protective effects, mice were subjected to a 1-h MCAO, and TAT-EE3 was administered every 12 hours based on the pre-evaluated in vivo disrupting efficacy (Fig 8A).
(C-G), TAT-EE3 protects WT mice against MCAO. (C), TTC staining of brain slices. (D), Mean infarct volume. (E), Average neurological deficit (ND) score. Average ND score (F) and average latency to fall time (G) for the long-term MCAO experiment (Neuron-specific TRPM2 knockout (nM2KO)).
(H-I), WB analysis of the surface expression of NMDARs in the brain (n=4/group).
(J-K), WBof ERK1/2 and CREB phosphorylation in cultured neurons isolated from global TRPM2 knockout (gM2KO) and nM2KO mice, and WT littermates. WT neurons were pre-treated with TAT-EE3 or TAT-SC overnight (n=3/group).
(L-M), WB of ERK1/2 and CREB phosphorylation in the brains from WT and nM2KO mice subjected to MCAO (n=4/group).
(N) IP using anti-PSD-95 and IB using anti-TRPM2.
(O) WB of TRPM2 expression in synaptosomes.
(P-Q) Isolation of extrasynaptic NMDAR-mediated Ca2+ response in WT and gM2KO neurons. (P) Representative Ca2+ imaging traces (Q) Quantification of AP burst (synaptic NMDAR-mediated response) induced by 4-AP/Bic, and extrasynaptic NMDAR-mediated response by NDMA (n=30/group).
(ns, no statistical significance, *, p < 0.05, **, p < 0.01, ***, p < 0.001; ANOVA, Bonferroni’s test; mean ± SEM).
TAT-EE3 treated mice exhibited reduced infarct volume and improved ND score (Figure 8C–E). For the post-MCAO treatment, we evaluated the protective effects of TAT-EE3 at 24 h, 3 day and 7 day. As shown in Figure 8C–E, post-MCAO TAT-EE3 treatment reduced infarct volume and improved ND score in WT mice, but did not produce further protective effects in the nM2KO mice, indicating that TAT-EE3 specifically targets TRPM2. For the long-term MCAO (7d), we evaluated ND score as well as behavioral changes using rotarod test at D1, D3 and D7. TAT-EE3 improved ND score at D1, D3 and D7 (Figure 8F), and inhibited the reduction of “latency to fall” time (Figure 8G). Moreover, the MCAO-induced increase of the surface expression of NMDARs was markedly inhibited by TAT-EE3 to a similar level as that in gM2KO and nM2KO (Figure 8H–I). These results suggest that TAT-EE3 disrupts TRPM2-NMDAR coupling (Figure 8A) and reduces surface expression of NMDARs (Figure 8H), thereby protecting mice against ischemic stroke.
Synaptic NMDAR (sNMDAR) promote while extrasynaptic NMDAR (sNMDAR) inhibit neuron survival (Figure S7A). sNMDAR and esNMDAR were selectively activated based on a well-established protocol (Hardingham et al., 2002; Nicolai et al., 2010). As shown in Figure 8J–K, activation of esNMDAR by NMDA inhibited the activation of the pro-survival ERK1/2 (pERK1/2) and CREK (pCREB), whereas activation of sNMDAR by 4-AP/Bic increased the phosphorylation of ERK1/2 and pCREB. In contrast, TAT-EE3 treatment markedly reduced the inhibition of NMDA on pERK1/2 and pCREB (Figure 8J,K). Similarly, inhibition of pERK1/2 and pCREB in the brain by MCAO were prevented by TAT-EE3, to a similar level as that in nM2KO (Figure 8L–M) and gM2KO (Figure S7B).
The preferential effects of TRPM2 on esNMDARs is consistent with the observation that TRPM2 is absent in the synaptome databases (Bayes et al., 2012; Yan et al., 2020). We found that TRPM2 was not present in the co-immunoprecipitate by anti-PSD95, a synaptic protein associating with sNMDAR (Figure 8N). Moreover, TRPM2 was barely detected in isolated synaptosomes from the brain (Figure 8O). We also isolated the esNMDAR-mediated Ca2+ response (Bengtson et al., 2008; Hardingham et al., 2002), and found that the esNMDAR-mediated increase of intracellular Ca2+ was inhibited by Trpm2 deletion, whereas the sNMDAR-mediated Ca2+ response was not altered. These results suggest that TRPM2 is located at the extrasynaptic sites and preferentially enhances esNMDARmediated Ca2+ signals.
DISCUSSION
In this study, we revealed a potential therapeutic strategy for ischemic stroke by targeting TRPM2-NMDAR association. Previous studies have shown the protective effects of global Trpm2 deletion (Alim et al., 2013; Belrose and Jackson, 2018; Gelderblom et al., 2014; Mai et al., 2020). Our neuron specific Trpm2 deletion results establish that neuronal TRPM2 plays a key role in aggregating excitotoxic neuronal apoptosis and necrosis in ischemic stroke through a previously unknown mechanism: physical and functional coupling with NMDARs. TRPM2 directly interact with GluN2a/b through the unique EE3 motif in its N-tail, and the KKR motifs in the C-tail of GluN2a/b. The EE3 motif is evolutionally conserved in TRPM2 of different species, but absent in other subfamilies of TRP channels, including TRPM4, which was shown to interact with GluN2a/2b (Yan et al., 2020). We showed that TRPM2 selectively enhances the activity of esNMDAR by increasing its surface expression, which is mediated by recruitment of PKCγ to the TRPM2-NMDAR complex. PKC regulates NMDAR’s surface trafficking via different mechanisms. Some studies demonstrated that PKC phosphorylates serine residues (Ser896 and Ser897) on GluN1 (Horak and Wenthold, 2009; Scott et al., 2001; Standley et al., 2000), whereas others showed the regulation of PKC on NMDAR is not mediated by directly phosphorylation (Zheng et al., 1999), but by triggering auto-phosphorylation of CaMKII which associates with NMDARs (Yan et al., 2011). Interestingly, CaMKII inhibitor can reduce the enhanced surface expression of NMDARs in our study. Moreover, we found that TRPM2-induced increase of NMDAR’s surface expression can be inhibited by the exocyst inhibitor endosidin2 (Sans et al., 2003; Zhang et al., 2016). NMDARs interact with the exocyst (Sans et al., 2003) for PKC-induced surface trafficking. Although we do not know the exact mechanism by which PKCγ mediates trafficking of the TRPM2/NMDAR complex, we propose the following working model. Under oxidative stress conditions, TRPM2 recruits PKCγ to the TRPM2/NMDARs to bring PKCγ to the close proximity of NMDAR interacting partners such as CaMKII, and consequently promotes NMDAR’s surface trafficking, leading to enhanced excitotoxicity. Nonetheless, future investigations are needed to understand where PKCγ begin to bind to TRPM2-NMDAR complex.
We found that TRPM2 exacerbates excitotoxicity by preferentially enhancing the function of esNMDAR. Disruption of TRPM2-NMDAR interaction by TAT-EE3 prevented the inhibition on phosphorylation of ERK1/2 and CREB, the pro-survival signaling that can be shut off by esNMDAR activation during ischemic stroke (Hardingham and Bading, 2010; Hardingham et al., 2002), indicating that disruption of TRPM2-NMDAR coupling largely eliminates the esNMDAR-mediated excitotoxicity. It is not surprising that TRPM2 preferentially influence esNMDARs, since we found that TRPM2 could not be detected in the synaptosomes, nor immunoprecipitated by anti-PSD-95. Moreover, Trpm2 deletion significantly reduced extrasynaptic but not the synaptic NMDA-induced Ca2+ response. Similarly, other studies have demonstrated a predominantly extrasynaptic distribution of TRPM2 in cultured hippocampal neurons (Olah et al., 2009), and that TRPM2 is absent in the synaptic proteins (Bayes et al., 2012). Importantly, TRPM2 only enhances NMDAR function during ischemic stroke, as TRPM2-NMDAR interaction and increase of NMDAR’s surface trafficking are promoted by PKCγ under oxidative stress conditions. Thus, disrupting the TRPM2-NMDARs interaction to specifically target esNMDAR will unlikely generate similar side effects caused by inhibition of synaptic NMDAR by conventional NMDAR antagonists.
The most exciting result in our study is that the TRPM2-derived interfering peptide TAT-EE3 protects mice against ischemic stroke in both short- and long-term MCAO. Cell-permeable peptides such as TAT-fused peptides have been well characterized and are considered as powerful tools for both clinical applications and basic studies (Xie et al., 2020). We found that TAT-EE3 disrupts TRPM2-NMDARs interaction in vitro and in vivo, effectively inhibits excitotoxicity and prevents the reduction of phosphorylated CREB and ERK1/2 levels. Our results indicate that peptide-based uncoupling of TRPM2-NMDAR association is a promising therapeutic strategy for ischemic stroke. NMDARs interact with various proteins (Petit-Pedrol and Groc, 2021). A recent study showed that interaction of GluN2a/b with the Ca2+-impermeable channel TRPM4 through a 57-amino acid domain (TwinF) in the N-tail of TRPM4 enhances excitotoxicity (Yan et al., 2020). Interestingly, interaction of TRPM4 with NMDARs does not influence NDMAR currents nor Ca2+ signaling, but promotes pro-death signaling. The authors demonstrated that the physical coupling of TRPM4 and NMDARs is required to promote excitotoxicity (Yan et al., 2020). Similar to TRPM2, TRPM4 preferentially influences esNMDAR. It would be exciting to speculate that, like scaffolding proteins for synaptic NMDARs’ trafficking, TRPM2 and TRPM4 may represent a family of proteins which are able to form interacting complex with NMDARs, and may exclusively localize NMDARs to the extrasynaptic sites. Moreover, as shown by previous studies that GluN2b is preferentially localized at extrasynaptic sites (Ge et al., 2020), it will be of a great interest to understand how TRPM2 in our study and TRPM4 in the previous study (Yan et al., 2020) only influence esNMDAR but interacting with both GluN2a and GluN2b.
In summary, we found that TRPM2 in neurons promotes neuronal death during ischemic stroke by coupling with esNMDAR via its EE3 motif. Targeting TRPM2-NMDAR interaction could be a promising strategy for developing more effective and safer therapies for ischemic stroke.
STAR * METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact Lixia Yue (lyue@uchc.edu).
Material Availability
This study did not generate new unique reagents.
Data and Code Availability
Data reported in this paper will be shared by the lead contact upon request
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All the experimental mice bred and hosted in the animal facility building of University of Connecticut School of Medicine (UCONN Health) were fed with standard chow diet and water ad libitum. Standard housing conditions were maintained at a controlled temperature with a 12-h light/dark cycle. All experimental procedures and protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of University of Connecticut School of Medicine (animal protocol: AP-200135-0723), and were conducted in accordance with the U.S. National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. Male mice at 8 to 12 weeks old were used in this study.
The global TRPM2 knockout (TRPM2-KO, or gM2KO) mice were generated by Dr. Yasuo Mori’s lab at Kyoto University Japan. The deletion of Trpm2 was developed in C57B6J mouse by replacing the third exon (S5–S6 linker in the pore domain) with a neomycin coding region. The knockout mice exhibited no differences in behavior or impairment in breeding, compared to wild type (WT) C57BJ6 mice (Yamamoto et al., 2008). TRPM2-KO mice were back-crossed to C57BL/6 mice for ≥10 generations before being used for experiments.
The neuron specific knockout of TRPM2 (nM2KO) was generated by breeding TRPM2fl/fl mice with Nestin-Cre ((B6.Cg-Tg)Nes-cre)1kln/J: 003771; JAX laboratory). TRPM2fl/fl mice were generated by Dr. Barbara Miller (Miller et al., 2013) (Penn State University, Pennsylvania). The exons 21 and 22 encoding transmembrane domain 5 and 6 and pore loop were flanked by loxp recombination sites and will be deleted by Cre recombinase (Miller et al., 2013). The mice were backcrossed with C57BL/6 mice for ≥10 generations before being used for experiments. The TRPM2flox/flox (TRPM2fl/fl) with Cre+ mice and TRPM2fl/fl with Cre− mice from the same litters were paired for experiments throughout the manuscript.
The inducible global knockout was also generated by using TRPM2fl/fl mice breeding with global Cre, Rosa26-CreERT2 (B6.129-Gt(ROSA)26Sortm1(cre/ERT2)Tyj/J: 008463; JAX laboratory). Knockout was induced by Tamoxifen treatment and confirmed by genotyping. The mice were backcrossed with C57BL/6 mice for ≥10 generations before being used for experiments.
Cortical neuron isolation and culture
Mice pups at P0 were euthanized based on animal protocol. Whole brain was dissected out immediately and immersed in ice-cold Hank’s Balanced Salt Solution (HBSS). Meninges were removed thoroughly, and tissue of different brain areas was taken based on purposes. Brain tissue was cut into small pieces and digested with 0.25% trypsin (Thermal Fisher Scientific, 15090–046) in HBSS at 37 °C for 20 min. Digestion solution was quickly removed, and tissue pellets are washed with Neurobasal® Medium (Thermal Fisher Scientific, 21103–049) for 3 times. Cells were resuspended with appropriate amount of Neurobasal® Medium supplemented with 2% B27® supplement (Thermal Fisher Scientific, 17504–044), 3% horse serum (Thermal Fisher Scientific, 16050114), 0.25% L-glutamine (Thermal Fisher Scientific, 25030–081) and 1% penicillin/streptomycin (Thermal Fisher Scientific, 15140–122). Isolated cells were counted and plated on coverslips pre-coated with poly-L-lysine (Sigma-Aldrich, P4707) at a density of about 500 × 103 cells/cm2 for OGD and H2O2 treatment, and 100 × 103 cells/cm2 for current recording. Cytosine arabinoside (Sigma-Aldrich, C1768) was added to maintain a concentration at 1μM to inhibit the proliferation of non-neuronal cells. 24 h after plating, culture medium was changed to Neurobasal® Medium supplemented with 2% B27® supplement, 0.25% L-glutamine and 1% penicillin/streptomycin. The concentration of Cytosine arabinoside (araC) was increased to 2 μM. Medium was changed every 3 days. OGD and H2O2 treatment was conducted at 7th day of culture, and current recording was conducted at 7th, 10th and 14th day of culture.
Cell line culture and transfection
HEK293T cells were cultured in Dulbecco’s Modified Eagle’s medium (DMEM) (Thermal Fisher Scientific, 12100–038) supplemented with 10% BGS (HyClone, SH30541.03) and 0.5% penicillin/streptomycin (Thermal Fisher Scientific, 15140–122) at 37 °C and 5% CO2. 8h prior to transfection, culture medium was replaced with DMEM supplemented only with 2.5% BGS. Cells were transfected when at a confluence about 80–90% using Lipofectamine® 3000 Transfection Kit (Thermal Fisher Scientific, 2232162) based on instruction.
METHOD DETAILS
Middle cerebral artery occlusion (MCAO)
Eight- to nine-week-old male mice (~25 g) were subjected to right middle cerebral artery occlusion (MCAO) for 120 min followed by 24 hours of reperfusion, or MCAO for 60 min followed by 7-days reperfusion. The genotype information was blinded to the surgeon who conduct the surgeries. MCAO surgery was performed as previously described (Liu and McCullough, 2014; Wu et al., 2012). In brief, mice were anesthetized with 2% isoflurane (vol/vol) in 100% oxygen and the anesthesia was maintained with 1.5% isoflurane during surgery through nose cone (Harvard Apparatus). The unilateral right middle cerebral artery (MCA) occlusion was carried out by advancing a silicone-coated 6–0 monofilament (Doccol Corporation, Sharon, MA) 10 to 11 mm from internal carotid artery bifurcation via an external carotid artery incision (Chiang et al., 2011). Mouse body temperature was monitored by a rectal temperature probe and maintained at ~ 37°C with an automatic temperature-regulating heating pad connected to animal temperature controller (TCA T-2DF, Physitemp). Cerebral blood flow was monitored after occlusion as well as after reperfusion. The bregma was exposed and the skull bone countersunk at two 3 × 3-mm areas over both MCA supply territories for bilateral monitoring of local cortical blood flow. Successful occlusion was confirmed by 85% reduction of cerebral blood flow monitored using laser Doppler blood FlowMeter (Moor-VMS-LDF1, Moor Instrument, Dever, UK). Sham control mice underwent the same procedure but without insertion of filament to occlude the MCA.
Neurological deficit score evaluation
Neurological deficit was scored based on previously reported criteria (Longa et al., 1989). In brief, score 0 represents no neurological deficit; score 1 represents failure to extend left paw; Score 2 represents circling to the left; score 3 represents falling to the left; score 4 represents inability of spontaneously walking and decreased level of consciousness; and score 5 represents death due to brain ischemia. The observer to score the neurological deficit was an experienced observer and blinded by the group assignment and genotype information. If the animal score was 0 or 5, it was removed from the study.
Rotarod test
For examining the long-term protective effect of TAT-EE3, motor coordination of mice was evaluated by rotarod test right after the evaluation of neurological deficit score at 1st, 3rd, and 7th day after MCAO. Briefly, mice were placed on a rotating rod with the speed range of 6–56 rounds per minute for 5 minutes. Each mouse was tested for 3 times with two 20-min interval in between. The falling from the rotating rod was recorded and the averaged latency of each mouse was used for quantification.
Infarct volume assessment by Triphenyl Tetrazolium chloride (TTC) staining
Tetrazolium chloride (Sigma-Aldrich, T-8877) was dissolved in PBS at a concentration of 20 mg/ml 30 min prior to use. Post-stroke mice were euthanized and brains were frozen at −80°C for 5 min, cut into coronary slices at a thickness of 1 mm. Brain slices were stained with 2% TTC (vol/vol) for 20 min, and then washed using PBS for 3 times, and fixed in 10% Neutral buffered formalin for later scanning. TTC labels non-injured tissue, leaving the infarct area white. The stained slices were scanned for data analysis using ImageJ software. The infarct volume was calculated and presented as a percentage of total brain volume as previously reported (Ren et al., 2003; Schulien et al., 2020).
Antibodies, chemicals and reagents
Rabbit polyclonal antibodies to TRPM2 (Novus, NB110-81601, 1:500 in 5% BSA for WB, 1:50 in protein extraction for IP); Rabbit polyclonal antibodies to GluN1 (Cell Signaling Technology, 5704S, 1:1000 in 5% BSA for WB, 1:50 in protein extraction for IP); Rabbit polyclonal antibodies to GluN2A (Cell Signaling Technology, 4205S, 1:1000 in 5% BSA for WB, 1:50 in protein extraction for IP). Rabbit polyclonal antibodies to GluN2B (Cell Signaling Technology, 4207S, 1:1000 in 5% BSA for WB, 1:50 in protein extraction for IP); Mouse polyclonal antibodies to flag (Sigma-Aldrich, F3165, 1:5000 in 5% BSA for WB): Rabbit polyclonal antibodies to GFP (Cell Signaling Technology, 2956S, 1:2000 in 5% BSA for WB); Rabbit polyclonal antibodies to CREB (Cell Signaling Technology, 4820S, 1:2000 in 5% BSA for WB); Rabbit polyclonal antibodies to phosphor-CREB(Ser133) (Cell Signaling Technology, 9198S, 1:2000 in 5% BSA for WB); Rabbit polyclonal antibodies to p44/42 MAPK (ERK1/2) (Cell Signaling Technology, 9102S, 1:2000 in 5% BSA for WB); Rabbit polyclonal antibodies to phospho-p44/42 MAPK (ERK1/2) (Tyr202/204) (Cell Signaling Technology, 4377T, 1:2000 in 5% BSA for WB); Rabbit polyclonal antibodies to PKC-γ (Cell Signaling Technology, 59090S, 1:5000 in 5% BSA for WB); Rabbit polyclonal antibodies to Pan-cadherin (Cell Signaling Technology, 4068S, 1:5000 in 5% BSA for WB); Rabbit polyclonal antibodies to TRPM4 (abcam, ab123936, 1:5000 in 5% BSA for WB); Rabbit polyclonal antibodies to pannexin-1 (Cell Signaling Technology, 91137S, 1:5000 in 5% BSA for WB); Rabbit polyclonal antibodies to GAPDH (Cell Signaling Technology, 2118S, 1:5000 in 5% BSA for WB); Rabbit polyclonal antibodies to β-tubulin (Cell Signaling Technology, 4820S, 1:5000 in 5% BSA for WB); Mouse monoclonal antibodies to Caspase-3 (Santa Cruz Biotechnology, sc-7272, 1:1000 in 5% BSA for WB, 1:50 in 5% BSA and 15% goat serum for immunofluorescence staining); Rabbit polyclonal antibodies to NeuN (Abcam, ab187477, 1:50 in 5% BSA and 15% goat serum for immunofluorescence staining); Goat anti-rabbit IgG-FITC (Santa Cruz Biotechnology, sc-2012, 1:1000 in 5% BSA and 15% goat serum for immunofluorescence staining); Goat anti-mouse IgG-rhodamine (Thermal Fisher Scientific, 31660, 1:1000 in 5% BSA and 15% goat serum for immunofluorescence staining); Prolong® Gold antifade reagent with DAPI (Life technologies, P36935), HRP-linked anti-rabbit IgG (Cell Signaling Technology, 7074S, 1:10000 in 5% BSA for WB); HRP-linked anti-mouse-IgG (Cell Signaling Technology, 7076S, 1:10000 in 5% BSA for WB); Tetrazolium chloride (Sigma-Aldrich, T-8877); NMDA (Tocris, 0114); Glutamate (Sigma-Aldrich, 49621); Glycine(Sigma-Aldrich, 50046); Bicuculine (TCI, B1890); 4-AP (Sigma-Aldrich, A-0152); MK-801 (Sigma-Aldrich, M107); PMA (Sigma-Aldrich, 524400); 4-α-PMA (Sigma-Aldrich, P128); H2O2 (Thermal Fisher Scientific, 200745); BAPTA (Cayman chemical, 11706); BAPTA-AM (Cayman chemical, 15551); Mn(III)TBAP (chloride) (Cayman chemical, 75850); L-NNA (Cayman chemical, 80220); NP40 (Thermal Fisher Scientific, 28324); Triton™ X-100 (T-9284), Bovine Serum Albumin (Sigma-Aldrich, 9048-46-8), Goat Serum (Thermal Fisher Scientific, 16210–064). All chemicals for making artificial cerebrospinal fluid (aCSF; see below) and recording solution (see below) were purchased from Sigma-Aldrich.
Membrane permeable peptide TAT-EE3 for disrupting TRPM2 and NMDARs coupling and scramble control TAT-SC peptides
TAT-SC (sequence: YGRKKRRQRRR VILLKDHTLEYPVF), TAT-EQE (sequence: YGRKKRRQRRR EEDTDSSQQMLALAEE), TAT-EE3 (sequence: YGRKKRRQRRR EEDTDSSEEMLALAEE) were ordered from GenScript Biotech, and dissolved in PBS to make a stock concentration at 10 mM. HEK-293T cells or isolated neurons were treated with TAT-SC or TAT-EE3 at a concentration of 10 μM for at least 8h prior to use. Mice were intraperitoneal injected (ip) with TAT-SC or TAT-EE3 at a dose of 100 nmol/kg. Detailed injection strategy is described in Figure 8B.
Plasmids and enzymes
GluN1a (Addgene, 17928), GluN2A (Addgene, 17924), GluN2B (Addgene, 17925), PKC-γ (Addgene, 112266), PKC-γ-DN (Addgene, 21239). The pcDNA4/TO-FLAG-hTRPM2 construct was a kind gift from Dr. Sharenberg AM (University of Washington, Seattle)(Perraud et al., 2003).
XbaI (BioLabs, R0145S), BamHI (BioLabs, R3136S), XhoI (BioLabs, R0146S), DpnI (BioLabs, R0176S), EcoRI (BioLabs, R3101S), KpnI (BioLabs, R3142S), NotI (BioLabs, R3189S) and T4 DNA ligase (Thermal Fisher Scientific, 2148085), PfuUltra HF (Agilent, 600380–51), and Q5® High-Fidelity DNA Polymerase (Biolabs, M0491S) were used to generating different deletion or mutation constructs.
Subcloning
For TRPM2, subcloning of N terminus (1–727) was achieved by introducing a stop codon (A2282T) by PCR using PfuUltra HF. C terminal of TRPM2 was amplified by PCR using Q5® High-Fidelity DNA Polymerase, cut by EcoRI and XbaI, and inserted into EGFP-C3 vector. To look for the binding part in N terminus of TRPM2, a series of stop codons were introduced by PCR using PfuUltra HF (C1831A, C1994T, G2138T, A2090T). EE3 motif was deleted by PCR using Q5® High-Fidelity DNA Polymerase. EE was mutated to QQ by PCR using PfuUltra HF (G2093C, G2096C; G2117C, G2120C; G2138C, G2141C). For GluN2A and GluN2B, C terminal was amplified by PCR using Q5® High-Fidelity DNA Polymerase, cut by EcoRI and XbaI, and inserted into EGFP-C3 vector. Deletion of C terminus of GluN2a and GluN2b were achieved by introducing a stop codon by PCR using PfuUltra HF (G4371T for GluN2B and G4518T for GluN2B). Deletion of the KKR region in GluN2a and GluN2b were achieved by PCR using Q5® Site-Directed Mutagenesis Kit based on the instruction.
For E. coli expression system, EE3 containing segment and EQE containing segment In TRPM2 were amplified by PCR using Q5® High-Fidelity DNA Polymerase, cut by KpnI and NotI, and inserted into a homemade His6-tagged vector; and KKR containing segment in GluN2a and GluN2b were amplified by PCR using Q5® High-Fidelity DNA Polymerase, cut by KpnI and NotI, and inserted into a homemade GST-tagged vector. The information of all the primers is listed in the Table S1.
E.coli expression and purification of proteins
The human GluN2a and GluN2b genes were cloned into a modified pGEX vector containing a removable tobacco etch virus (TEV) protease recognition site. The GluN2a (residues 1208 – 1317; MW: 16.7 kDa) and GluN2b (residues 1212 – 1322; MW: 16.7 kDa) proteins were expressed in Escherichia coli BL21(DE3) cells grown to an OD600 (optical density at 600 nm) between 0.8–1.0 at 37°C followed by induction of protein expression at 21°C overnight using 0.5 mM isopropyl-D-1-thiogalactopyranoside (IPTG). Cells were harvested by centrifugation, resuspended by lysis buffer containing 25 mM Tris-HCl (pH 8.0), 200 mM NaCl, 1% phenylmethylsulfonyl fluoride (PMSF) and 2 mM dithiothreitol (DTT), and lysed by using high-pressure homogenization (Avestin EmulsiFlex C3). The lysates were clarified by centrifugation at 30,000 rpm at 4°C for 30 min, and the supernants were applied to a Glutathione Sepharose 4B column (GE Healthcare). After being extensively washed with lysis buffer, the GST-tagged GluN2a and GluN2b fusion proteins were eluted with a buffer containing 25 mM Tris-HCl (pH 8.0), 200 mM NaCl, 15 mM reduced glutathione and 2 mM DTT. The proteins were concentrated using an Amicon stirred ultrafiltration cell unit with a 10-kDa cutoff membrane (EMD Millipore) and stored at −80°C until use. The human wild-type TRPM2 (residues 644 – 760; ~ 16.7 kDa) and its EQE mutant were cloned into a modified pET15b vector containing a removable TEV protease recognition site. The proteins were expressed in E. coli BL21(DE3) as described above. Cell pellets were resuspended in denaturing buffer containing 25 mM Tris-HCl (pH 8.0), 300 mM NaCl, 1% PMSF and 6 M Urea and lysed by high-pressure homogenization. The lysates were clarified by centrifugation at 30,000 rpm at 4 °C for 30 min, and the supernants were applied to a Ni2+-nitrilotriacetic acid (NTA) column (GE Healthcare). After being extensively washed with 25 mM imidazole in the denaturing buffer, the His6-tagged TRPM2 fusion proteins were eluted with a buffer containing 25 mM Tris-HCl (pH 8.0), 300 mM NaCl and 250 mM imidazole. To refold the TRPM2 proteins, the denatured samples were dialyzed at 4 ° C overnight against two changes of a buffer without urea [25 mM Tris-HCl (pH 8.0), 200 mM NaCl and 2 mM DTT]. The protein solutions were concentrated using an Amicon stirred ultrafiltration cell unit with a 10-kDa cutoff membrane and stored at −80°C until use.
In vitro protein-protein direct binding assay
About 1~10 μg of purified proteins were added into 1 mL freshly prepared binding buffer (25 mM HEPES, 100 mM NaCl, 0.01% Triton X-100 and 5% glycerol) for in vitro direct binding at 4 °C for overnight. Then Co-immunoprecipitation assay was performed using either anti-GST or anti-His antibodies at 1:50 dilution as detailed described in the Co-immunoprecipitation section. The precipitated proteins were detected by coommassie blue staining and western blot.
Oxygen-glucose deprivation
Oxygen-glucose deprivation (OGD) was achieved by replace the glucose in aCSF with sucrose, and 95% N2 and 5% CO2 was used to equilibrate sucrose-aCSF to displace oxygen (Povysheva et al., 2019; Weilinger et al., 2016). This condition typically yielded a pO2 of < 5 mm Hg in the imaging chamber (Thompson et al., 2006). At least 10 min was allowed for neurons to adapt to the change from culture medium to aCSF before OGD was applied.
Real-time monitoring of mitochondrial function
Mitochondria function was evaluated using Rhodamine-123 dequenching as previously reported. Rhodamine-123 (Rh123, Thermal Fisher Scientific, R302) was dissolved in DMSO to make a stock concentration at 10 mg/ml. Pre-warmed Neurobasal® Medium was used to dilute Rhodamine-123 to 5 μg/ml as working concentration. Culture medium was removed and cultured neurons on the 25 mm coverslip were washed using prewarmed PBS for 3 times, then 2 ml of Rh123 working solution was added. Cells were incubated with Rh123 at 37 °C for 5 min. Then Rh123 working solution was replaced with culture medium. At least 10 min were allowed to achieve Rh123 equilibration after the transition of culture medium to aCSF before experiments.
Fluorescence intensities at 509 nm with excitation at 488nm was collected every 15 s for 30 min using CoolSNAP HQ2 (Photometrics) and data were analyzed using NIS-Elements (Nikon).
Ratio calcium imaging experiments
Changes of intracellular Ca2+ was measured using ratio Ca2+ imaging as we described previously (Du et al., 2010). In brief, Fura-2 AM (Thermal Fisher Scientific, F1221) was dissolved in DMSO to make a stock concentration at 1 mM. Pre-warmed Neurobasal® Medium (Thermal Fisher Scientific, 21103–049) was used to dilute Fura-2 AM to a working concentration at 2.5 μM, and 0.02% Pluronic™ F-127 (Thermal Fisher Scientific, P3000MP) was added to facilitate loading of Fura-2 AM. Cells plated on 25 mm glass coverslips were washed using pre-warmed PBS for 3 times, and then incubated with 2 ml of Fura-2 AM working solution for 30~45 min at 37 °C. Non-incorporated dye was washed away using HEPES-buffered Saline Solution (HBSS) containing (in mM): 20 HEPES, 10 glucose, 1.2 MgCl2, 1.2 KH2PO4, 4.7 KCl, 140 NaCl, 1.3 Ca2+ (pH 7.4).
Ca2+ influx was measured by perfusing the cells with Tyrode’s solution for transfected HEK293T cells or aCSF for neurons under different conditions. Ionomycin (Iono) at 1 μM was applied at the end of the experiment as an internal control. Fluorescence intensities at 510 nm with 340 nm and 380 nm excitation were collected at a rate of 1 Hz using CoolSNAP HQ2 (Photometrics) and data were analyzed using NIS-Elements (Nikon). The 340:380 nm ratio in the presence of different treatments was normalized to the maximal Ca2+ signal elicited by 1 μM Ionomycin (Iono) as we previously reported (Du et al., 2010).
Recording of extracellular NMDAR mediated calcium influx was performed as reported by Dr. Hilmar Bading previously (Bengtson et al., 2008; Hardingham et al., 2002). Briefly, bicuculine (Bic, 50 μM) and 4-Aminopyridine (4-AP, 2.5 mM) in the presence of glycine (10 μM) were used to promote synaptic glutamate release and to induce the AP burst for 3 min. Then in the background of AP bursting, MK-801 (10μM), an activity-dependent NMDA receptor blocker, was applied to block synaptic NMDA receptor. After 10 min, neurons were washed using aCSF with tetrodotoxin (TTX, 1 μM) to block synaptic activities for 5 min. Then neurons were washed using aCSF for 2 min and NMDA (100 μM) was applied to induce Ca2+ influx mediated by extrasynaptic NMDA receptor in the presence of glycine (10 μM) and in the absence of Mg2+.
Co-immunoprecipitation
NP-40 lysis buffer (10% NP40, 150 mM NaCl, 1 mM EDTA, 50 mM Tris, pH=8.0) containing proteinase inhibitors (Sigma-Aldrich, 539131-10VL) and phosphatase inhibitors (Thermal Fisher Scientific, 78428) was used to lyse both cultured cells and frozen brain tissue. For transfected cells, proteins were extracted 36 hours after transfection. Cell and tissue lysate were lysed by ultrasound using an ultrasonic cleaner (Thermal Fisher Scientific) filled with ice-cold water for 30 min. After incubated on ice for 1 h, lysate was centrifuged at 13000 g for 30 min and supernatant was collected. Protein concentration was measured using Pierce™ Rapid Gold BCA Protein Assay Kit (Thermal Fisher Scientific, A53225). 300 μg of protein was taken and diluted using NP-40 lysis buffer to make a total volume of 500 μl. Unused protein was allocated and frozen at −80 °C for future use. Appropriate amount of antibody was added based on instruction. After protein-antibody mixture was incubated on ice for 2 h, 25 μl of pre-washed Protein A/G PLUS-Agarose (Santa Cruz Biotechnology, sc-2003) was added, and the whole mixture was incubated at 4 °C for overnight. Then the mixture was centrifuged at 2500g for 1min to get agarose beads. Agarose beads was washed using NP-40 lysis buffer for 7 times, mixed with same amount of 2x Laemmli Sample Buffer (BIO-RAD, 1610737), and boiled at 95 °C for 5 min. Then samples were ready for western blotting analysis.
Western blotting
NP-40/Triton lysis buffer (10% NP40, 1% Triton™ X-100, 150 mM NaCl, 1 mM EDTA, 50 mM Tris, pH=8.0) containing proteinase inhibitors and phosphatase inhibitors was used to lyse both cultured cells and frozen brain tissue. Surface protein was extracted using Pierce® Cell Surface Protein Isolation Kit (Thermal Fisher Scientific, 89881) in transfected HEK-293T cells, and using ProteoExtract™ Native Membrane Protein Extraction Kit (Calbiochem, 444810) in brain tissue based on instructions. Synaptosome was isolated using the Syn-PER™ Synaptic Protein Extraction Reagent (Thermal Fisher Scientific, 87793). For transfected cells, proteins were extracted 36 hours after transfection. Cell and tissue lysate were lysed by ultrasound using an ultrasonic cleaner filled with ice-cold water for 30 min. After incubated on ice for 1 h, lysate was centrifuged at 13000 g for 30 min and supernatant was collected. Protein concentration was measured using Pierce™ Rapid Gold BCA Protein Assay Kit.
30–50 μg of total protein was loaded and separated proteins were transferred to Nitrocellulose membranes. Membranes were blocked with 5% BSA and 2.5% goat serum in Tris buffered saline (TBS, pH=7.4) at room temperature for 2 h, and incubated with primary antibodies in TBS with 0.05% Tween (TBS-T) at room temperature for 2 h. Then membranes were incubated with secondary antibodies in TBS-T for 1 h at room temperature for 1 h for detection. Blots were developed with ImageQuant LAS 4000 imaging system. Band intensity was quantified using ImageJ software and normalized with appropriate loading controls.
Electrophysiology
Whole cell currents were recorded using an Axopatch 200B amplifier. Data were digitized at 10 or 20 kHz and digitally filtered offline at 1 kHz. Patch electrodes were pulled from borosilicate glass and fire-polished to a resistance of ~3 MΩ when filled with internal solutions. Series resistance (Rs) was compensated up to 90% to reduce series resistance errors to <5 mV. Cells in which Rs was >10 MΩ were discarded (Du et al., 2009b). For heterologous expression, transfected HEK-293 cells were identified by GFP fluorescence. TRPM2 current recording in transfected HEK-293T cells was performed as we previously reported (Du et al., 2009a; b). TRPM2 and NMDAR currents recordings from cultured neurons were performed using aCSF as extracellular solution as we previously reported (Zeng et al., 2010). In brief, for TRPM2 current recordings, voltage stimuli lasting 250 ms were delivered at 1-s intervals, with voltage ramps ranging from −100 to +100 mV at holding potential of 0 mV to elicited currents. For NMDAR current recordings, a gap-free protocol at holding potential of −80 mV was applied to elicit NMDA currents upon agonist stimulation. A fast perfusion system was used to exchange extracellular solutions and to deliver agonists and antagonists to the cells, with a complete solution exchange achieved in about 1–3 s (Jiang et al., 2005).
Normal Tyrode solution for current recording in HEK-293 cells contained (mM): 145 NaCl, 5 KCl, 2 CaCl2, 10 HEPES, 10 glucose, osmolarity=290–320 mOsm/Kg, and pH=7.4 was adjusted with NaOH. Extracellular solution for current recording in neuron, the aCSF solution contained (mM):124 NaCl, 2.5 KCl, 2 MgSO4, 2 CaCl2, 1.2 NaH2PO4, 24 NaHCO3, 5 HEPES, 12.5 glucose, osmolarity=300–310 mOsm/Kg, with pH=7.4 adjusted with NaOH. For oxygen-glucose-deprivation (OGD) solution, glucose was eliminate from extracellular solution, and the solution was saturated with nitrogen (N2) bubbling for 30 min before the experiments.
The internal pipette solution for whole cell current recordings of TRPM2 overexpressed in HEK293 cells (pipette solution-1: P1) contained (in mM): 135 Cs-methanesulfonate (CsSO3CH3), 8 NaCl, 0.5 CaCl2, 1 EGTA, and 10 HEPES, with pH adjusted to 7.2 with CsOH. Free [Ca2+]i buffered by EGTA was ~ 100 nM calculated using Max chelator (Du et al., 2009b). ADPR 200 μM was included in the pipette solution for most experiments. The intracellular pipette solution to test the effects of OGD on TRPM2 currents in neuron was adjusted to sub-optimal condition (pipette solution-2: P2), containing (in mM) 135 CsSO3CH3, 8 NaCl, 3 MgCl2 and 10 HEPES (pH 7.2) with 5 μM EGTA and 1 μM ADPR. Free [Ca2+]i buffered by EGTA was ~ 500 nM calculated using Max chelator. The intracellular solution for NMDAR current recording (pipette solution-3: P3) contained (mM): 110 K-ASP, 20 KCl, 1 MgSO4, 10 mM BAPTA, 0.1 GTP, 5 ATP-Mg2, 10 HEPES, osmolarity=275–285 mOsm/Kg, pH=7.2 adjusted with KOH. For the experiments using cells pretreated with the disrupting peptides TAT-SC and TAT-EE3, 10 μM TAT-SC or TAT-EE3 or TAT-EQE was included in the pipette solution, and at least 10 min was allowed for achieving intracellular equilibration of TAT-SC or TAT-EE3 before current recording.
For current recordings in neurons, tetrodotoxin (0.5 μM) was included in the external solution to block voltage-gated Na+ current, and 10 μM nifedipine was used to block voltage-gated Ca2+ currents for recording TRPM2 currents.
The above-mentioned pipette solutions and extracellular solutions were specific for TRPM2 or NMDAR current recordings, without “cross-contamination” for each other or from other channel activation. The omitting of ADPR and including high concentration of potent calcium chelator in the pipette solution for NMDAR current recordings eliminated any possibility of TRPM2 channel activation because TRPM2 requires Ca2+ and ADPR to be activated. No Ca2+ pipette solution for NMDAR current recording also prevented other Ca2+-activated currents such as TRPM4. Moreover, using CsSO3CH3 in the pipette solution for TRPM2 current recordings eliminated contamination from any potassium channels for recordings in neurons and in HEK-293 cells.
Immunofluorescence staining
Brains harvested from mice were frozen at −80 °C prior to use, and was mounted in Fisher Healthcare™ Tissue-Plus™ O.C.T. Compound (Thermal Fisher Scientific, 23-730-571) prior to cutting. Brains were cut into sagittal slices at a thickness of 6 to 8 μm, mounted to Superfrost® Plus Microscope Slides (Thermal Fisher Scientific, 12-550-15), and frozen at −80 °C for future use. Prior to staining, slides were taken to room temperature for at least 30 min allowing for dehydration. Slices were fixed in 10% formaldehyde for 15 min following washing using PBS for 3 times, and incubated in blocking solution containing 5% BSA, 15% goat serum and 1% Triton X-100 at room temperature for 2 h. Primary antibodies were diluted as described previously in TBS-T containing 15% goat serum. Slices were incubated with primary antibodies for at least 12 h at 4 °C following washing using PBS for 3 times, and incubated with secondary antibodies at room temperature for 2 h. Then slices were washed using PBS for 3 times, and mounted using Prolong® Gold anti-fade reagent with DAPI. Slices were kept at 4 °C before taking pictures. TUNEL staining was performed based on the instruction of kit at the penumbra areas (Figure S1N).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data analysis was done by experimenters blind to experimental conditions. All data are expressed as mean ± SEM. For two groups’ comparison, statistical significance was determined using Student’s t-test. For multiple groups’ comparison, statistical significance was determined using one-way or two-way analysis of variance (ANOVA) followed by Bonferroni posttest. Distribution of the data was analyzed prior to analysis to determine whether the data met assumptions of the statistical approach. P<0.05 was regarded as significant. The exact sample size and related information can be found in the figure legends.
Supplementary Material
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal antibodies to TRPM2 | Novus | Cat#NB110-81601 |
| Rabbit polyclonal antibodies to TRPM2 N terminal part | Abmart | Cat#634-1-1-R1 |
| Rabbit polyclonal antibodies to TRPM2 C terminal part | Abmart | Cat#634-2-1-R1 |
| Rabbit polyclonal antibodies to GluN1 | Cell Signaling Technology | Cat#5704S |
| Rabbit polyclonal antibodies to GluN2A | Cell Signaling Technology | Cat#4205S |
| Rabbit polyclonal antibodies to GluN2B | Cell Signaling Technology | Cat#4207S |
| Mouse monoclonal antibodies to flag | Sigma-Aldrich | Cat#F3165 |
| Rabbit polyclonal antibodies to phosphor-CREB(Ser133) | Cell Signaling Technology | Cat#9198S |
| Rabbit polyclonal antibodies to p44/42 MAPK (ERK1/2) | Cell Signaling Technology | Cat#9102S |
| Rabbit polyclonal antibodies to phospho-p44/42 MAPK (ERK1/2) (Tyr202/204) | Cell Signaling Technology | Cat#4377T |
| Rabbit polyclonal antibodies to PKC-γ | Cell Signaling Technology | Cat#59090S |
| Rabbit polyclonal antibodies to Pan-cadherin | Cell Signaling Technology | Cat#4068S |
| Pannexin-1 (D9M1C) Rabbit mAb | Cell Signaling Technology | Cat#91137S |
| Anti-TRPM4 antibody | Abcam | Cat#ab123936 |
| Rabbit polyclonal antibodies to GAPDH | Cell Signaling Technology | Cat#2118S |
| Rabbit polyclonal antibodies to β-tubulin | Cell Signaling Technology | Cat#4820S |
| Rabbit polyclonal antibodies to NeuN | Abcam | Cat#ab187477 |
| Goat anti-rabbit IgG-FITC | Santa Cruz Biotechnology | Cat#sc-2012 |
| Goat anti-mouse IgG-rhodamine | Thermal Fisher Scientific | Cat#31160 |
| Bacterial and virus strains | ||
| NEB® 5-alpha Competent E. coli (High Efficiency) | Biolabs | Cat#C2987U |
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| 2,3,5-Triphenyltetrazolium chloride | Sigma-Aldrich | Cat#T-8877 |
| N-Methyl-D-aspartic acid | Tocris | Cat#0114 |
| Glutamate | Sigma-Aldrich | Cat#49621 |
| Glycine | Sigma-Aldrich | Cat#50046 |
| Bicuculine | TCI | Cat#B1890 |
| 4-Aminopyridine | Sigma-Aldrich | Cat#A-0152 |
| MK-801 | Sigma-Aldrich | Cat#M107 |
| Phorbol-12-myristate-13-acetate | Sigma-Aldrich | Cat#524400 |
| 30% Hydrogen Peroxide | Thermal Fisher Scientific | Cat#200745 |
| NP40 | Thermal Fisher Scientific | Cat#28324 |
| Triton™ X-100 | Sigma-Aldrich | Cat#T-9284 |
| Bovine Serum Albumin | Sigma-Aldrich | Cat#9048-46-8 |
| Goat Serum | Thermal Fisher Scientific | Cat#16210-064 |
| Rhodamine-123 | Thermal Fisher Scientific | Cat#R302 |
| Fura-2 AM | Thermal Fisher Scientific | Cat#F1221 |
| Ionomycin | Sigma-Aldrich | Cat#I0634 |
| Pluronic™ F-127 | Thermal Fisher Scientific | Cat#P3000MP |
| Proteinase inhibitors | Sigma-Aldrich | Cat#539131-10VL |
| Phosphatase inhibitors | Thermal Fisher Scientific | Cat#78428 |
| Laemmli Sample Buffer | BIO-RAD | Cat#1610737 |
| Protein A/G PLUS-Agarose | Santa Cruz Biotechnology | Cat#sc-2003 |
| TAT-SC | Genescript | Customized in this study |
| TAT-EQE | Genescript | Customized in this study |
| TAT-EE3 | Genescript | Customized in this study |
| XbaI | BioLabs | Cat#R0145S |
| XhoI | BioLabs | Cat#R0146S |
| BamHI | BioLabs | Cat#R3136S |
| DpnI | BioLabs | Cat#R0176S |
| KpnI | BioLabs | Cat#R3142S |
| NotI | BioLabs | Cat#R3189S |
| EcoRI | BioLabs | Cat#R3101S |
| T4 DNA ligase | Thermal Fisher Scientific | Cat#2148085 |
| PfuUltra HF | Agilent | Cat#600380-51 |
| Q5® High-Fidelity DNA Polymerase | BioLabs | Cat#M0491S |
| Dulbecco’s Modified Eagle’s medium | Thermal Fisher Scientific | Cat#12100-038 |
| Bovine Calf Serum | HyClone | Cat#SH30541.03 |
| Penicillin/streptomycin | Thermal Fisher Scientific | Cat#15140-122 |
| 2.5% trypsin | Thermal Fisher Scientific | Cat#15090-046 |
| Neurobasal® Medium | Thermal Fisher Scientific | Cat#21103-049 |
| B27® supplement | Thermal Fisher Scientific | Cat#17504-044 |
| Horse serum | Thermal Fisher Scientific | Cat#16050114 |
| L-glutamine | Thermal Fisher Scientific | Cat#25030-081 |
| Cytosine arabinoside | Thermal Fisher Scientific | Cat#C1768 |
| Poly-L-lysine | Sigma-Aldrich | Cat#P4707 |
| Critical commercial assays | ||
| Lipofectamine® 3000 Transfection Kit | Thermal Fisher Scientific | Cat#2232162 |
| Pierce™ Rapid Gold BCA Protein Assay Kit | Thermal Fisher Scientific | Cat#A53225 |
| Pierce® Cell Surface Protein Isolation Kit | Thermal Fisher Scientific | Cat#89881 |
| ProteoExtract™ Native Membrane Protein Extraction Kit | Calbiochem | Cat#444810 |
| Fisher Healthcare™ Tissue-Plus™ O.C.T. Compound | Thermal Fisher Scientific | Cat#23-730-571 |
| ProLong™ Gold Antifade Mountant | Thermal Fisher Scientific | Cat#P10144 |
| QIAprep® Spin Miniprep Kit | QIAGEN | Cat#27106 |
| QIAGEN® Plasmid Maxi Kit | QIAGEN | Cat#12163 |
| Qiaquick® PCR Purification Kit | QIAGEN | Cat#28104 |
| Syn-PER™ Synaptic Protein Extraction Reagent | Thermal Fisher Scientific | Cat#87793 |
| Q5® Site-Directed Mutagenesis Kit | BioLabs | Cat#E0554S |
| In Situ Cell Death Detection Kit | Millipore Sigma | Cat#11684795910 |
| Deposited data | ||
| Experimental models: Cell lines | ||
| 293T | ATCC | Cat#CRL-3216™ |
| Experimental models: Organisms/strains | ||
| C57B6J mice | JAX laboratory | https://www.jax.org/strain/000664 |
| Trpm2−/− mice | Yamamoto et al., 2008 | Dr. Yasuo Mori |
| TRPM2fl/fl mice | Miller et al., 2013 | Dr. Barbara Miller |
| Nestin-cre mice | JAX laboratory | https://www.jax.org/strain/003771 |
| Oligonucleotides | ||
| Primers for TRPM2 subcloning and mutagenesis, see Table S1 | This study | N/A |
| Primers for GluN2a and GluN2b subcloning and mutagenesis, see Table S1 | This study | N/A |
| Primers for E. coli expression, see Table S1 | This study | N/A |
| Recombinant DNA | ||
| GluN1a | Luo et al., 2002 | Addgene 17928 |
| GluN2A | Luo et al., 2002 | Addgene 17924 |
| GluN2B | Luo et al., 2002 | Addgene 17925 |
| PKC-γ | Colgan et al., 2018 | Addgene 112266 |
| PKC-γ-DN | Soh et al., 2003 | Addgene 21239 |
| pcDNA4/TO-FLAG-hTRPM2 | Perraud et al., 2003 | Dr. Sharenberg AM (University of Washington, Seattle) |
| Software and algorithms | ||
| GraphPad Prism 6.0 | GraphPad Software | https://www.graphpad.com/updates/ |
| Biorender | Biorender | N/A |
| pClamp 9.2 | Molecular Devices | https://biorender.com/ |
| Adobe Illustrator | Adobe | https://www.adobe.com/products/illustrator/ |
| NIS Elements AR4 | Nikon | https://www.microscope.healthcare.nikon.com/products/software/nis-elements/nis-elements-advanced-research |
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Other | ||
HIGHLIGHTS.
TRPM2 physically and functionally interacts with extrasynaptic NMDAR
TRPM2-NMDAR interaction exacerbates excitotoxicity during ischemic stroke
TRPM2 recruits PKCγ thereby increasing NMDAR’s surface expression
Uncoupling TRPM2-NMDAR interaction attenuates ischemic brain injury
ACKNOWLEDGMENTS
We would like to thank Drs Rajkumar Verma (UCONN Health) and Louise McCullough (UT Health) for constructive discussions about this project. We thank Drs. Rindy Jaffe and Dejian Ren for helpful comments to the manuscript. We thank Dr. Andrew M. Scharenberg (University of Washington) for kindly providing TRPM2 plasmid. This work was partially supported by the National Institute of Health (R01-HL143750) and American Heart Association (19TPA34890022) to LY, and National Institute of Health (R01-GM135592) to BH.
Footnotes
DECLARATION OF INTERESTS
P.Z., J.F., and L.Y. have a pending UCONN provisional patent for TRPM2-based therapies for stroke, filed: July, 2020. The authors declare no other competing financial interests related to this work.
INCLUSION AND DIVERSITY
We worked to ensure sex balance in the selection of non-human subjects. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCE
- Alim I, Teves L, Li R, Mori Y, and Tymianski M (2013). Modulation of NMDAR subunit expression by TRPM2 channels regulates neuronal vulnerability to ischemic cell death. The Journal of neuroscience : the official journal of the Society for Neuroscience 33, 17264–17277. 10.1523/JNEUROSCI.1729-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bading H (2013). Nuclear calcium signalling in the regulation of brain function. Nature reviews. Neuroscience 14, 593–608. 10.1038/nrn3531. [DOI] [PubMed] [Google Scholar]
- Bayer KU, De Koninck P, Leonard AS, Hell JW, and Schulman H (2001). Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature 411, 801–805. 10.1038/35081080. [DOI] [PubMed] [Google Scholar]
- Bayes A, Collins MO, Croning MD, van de Lagemaat LN, Choudhary JS, and Grant SG (2012). Comparative study of human and mouse postsynaptic proteomes finds high compositional conservation and abundance differences for key synaptic proteins. PLoS One 7, e46683. 10.1371/journal.pone.0046683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belrose JC, and Jackson MF (2018). TRPM2: a candidate therapeutic target for treating neurological diseases. Acta pharmacologica Sinica 39, 722–732. 10.1038/aps.2018.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belrose JC, Xie YF, Gierszewski LJ, MacDonald JF, and Jackson MF (2012). Loss of glutathione homeostasis associated with neuronal senescence facilitates TRPM2 channel activation in cultured hippocampal pyramidal neurons. Mol Brain 5, 11. 10.1186/1756-6606-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtson CP, Dick O, and Bading H (2008). A quantitative method to assess extrasynaptic NMDA receptor function in the protective effect of synaptic activity against neurotoxicity. BMC neuroscience 9, 11. 10.1186/1471-2202-9-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang T, Messing RO, and Chou WH (2011). Mouse model of middle cerebral artery occlusion. J Vis Exp. 10.3791/2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW (1995). Calcium: still center-stage in hypoxic-ischemic neuronal death. Trends in neurosciences 18, 58–60. [PubMed] [Google Scholar]
- Choi DW (2020). Excitotoxicity: Still Hammering the Ischemic Brain in 2020. Frontiers in neuroscience 14, 579953. 10.3389/fnins.2020.579953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colgan LA, Hu M, Misler JA, Parra-Bueno P, Moran CM, Leitges M, and Yasuda R (2018). PKCalpha integrates spatiotemporally distinct Ca(2+) and autocrine BDNF signaling to facilitate synaptic plasticity. Nat Neurosci 21, 1027–1037. 10.1038/s41593-018-0184-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Xie J, and Yue L (2009a). Intracellular calcium activates TRPM2 and its alternative spliced isoforms. Proceedings of the National Academy of Sciences 107, 7239–7244. 10.1073/pnas.0811725106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Xie J, and Yue L (2009b). Modulation of TRPM2 by acidic pH and the underlying mechanisms for pH sensitivity. J. Gen. Physiol. 134, 471–488. 10.1085/jgp.200910254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Xie J, Zhang Z, Tsujikawa H, Fusco D, Silverman D, Liang B, and Yue L (2010). TRPM7-mediated Ca2+ signals confer fibrogenesis in human atrial fibrillation. Circulation research 106, 992–1003. CIRCRESAHA.109.206771 [pii] 10.1161/CIRCRESAHA.109.206771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonfria E, Murdock PR, Cusdin FS, Benham CD, Kelsell RE, and McNulty S (2006). Tissue distribution profiles of the human TRPM cation channel family. Journal of receptor and signal transduction research 26, 159–178. 10.1080/10799890600637506. [DOI] [PubMed] [Google Scholar]
- Ge Y, Chen W, Axerio-Cilies P, and Wang YT (2020). NMDARs in Cell Survival and Death: Implications in Stroke Pathogenesis and Treatment. Trends in molecular medicine 26, 533–551. 10.1016/j.molmed.2020.03.001. [DOI] [PubMed] [Google Scholar]
- Gelderblom M, Melzer N, Schattling B, Gob E, Hicking G, Arunachalam P, Bittner S, Ufer F, Herrmann AM, Bernreuther C, et al. (2014). Transient receptor potential melastatin subfamily member 2 cation channel regulates detrimental immune cell invasion in ischemic stroke. Stroke; a journal of cerebral circulation 45, 3395–3402. 10.1161/STROKEAHA.114.005836. [DOI] [PubMed] [Google Scholar]
- Goldberg MP, and Choi DW (1993). Combined oxygen and glucose deprivation in cortical cell culture: calcium-dependent and calcium-independent mechanisms of neuronal injury. J Neurosci 13, 3510–3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodell DJ, Zaegel V, Coultrap SJ, Hell JW, and Bayer KU (2017). DAPK1 Mediates LTD by Making CaMKII/GluN2B Binding LTP Specific. Cell reports 19, 2231–2243. 10.1016/j.celrep.2017.05.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granzotto A, Canzoniero LMT, and Sensi SL (2020). A Neurotoxic Menage-a-trois: Glutamate, Calcium, and Zinc in the Excitotoxic Cascade. Frontiers in molecular neuroscience 13, 600089. 10.3389/fnmol.2020.600089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara Y, Wakamori M, Ishii M, Maeno E, Nishida M, Yoshida T, Yamada H, Shimizu S, Mori E, Kudoh J, et al. (2002). LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Molecular cell 9, 163–173. [DOI] [PubMed] [Google Scholar]
- Hardingham G (2019). NMDA receptor C-terminal signaling in development, plasticity, and disease. F1000Research 8. 10.12688/f1000research.19925.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, and Bading H (2010). Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci 11, 682–696. 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, and Bading H (2002). Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nature neuroscience 5, 405–414. 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Horak M, and Wenthold RJ (2009). Different roles of C-terminal cassettes in the trafficking of full-length NR1 subunits to the cell surface. The Journal of biological chemistry 284, 9683–9691. 10.1074/jbc.M807050200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Roth B, Lu W, and Du J (2019). Ligand recognition and gating mechanism through three ligand-binding sites of human TRPM2 channel. eLife 8. 10.7554/eLife.50175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia J, Verma S, Nakayama S, Quillinan N, Grafe MR, Hurn PD, and Herson PS (2011). Sex differences in neuroprotection provided by inhibition of TRPM2 channels following experimental stroke. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 31, 2160–2168. 10.1038/jcbfm.2011.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Li M, and Yue L (2005). Potentiation of TRPM7 inward currents by protons. J. Gen. Physiol 126, 137–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashio M, Sokabe T, Shintaku K, Uematsu T, Fukuta N, Kobayashi N, Mori Y, and Tominaga M (2012). Redox signal-mediated sensitization of transient receptor potential melastatin 2 (TRPM2) to temperature affects macrophage functions. Proceedings of the National Academy of Sciences of the United States of America 109, 6745–6750. 10.1073/pnas.1114193109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan JY, Skeberdis VA, Jover T, Grooms SY, Lin Y, Araneda RC, Zheng X, Bennett MV, and Zukin RS (2001). Protein kinase C modulates NMDA receptor trafficking and gating. Nature neuroscience 4, 382–390. 10.1038/86028. [DOI] [PubMed] [Google Scholar]
- Lemasters JJ, Theruvath TP, Zhong Z, and Nieminen AL (2009). Mitochondrial calcium and the permeability transition in cell death. Biochimica et biophysica acta 1787, 1395–1401. 10.1016/j.bbabio.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, and Hao B (2010). Structural basis of dimerization-dependent ubiquitination by the SCF(Fbx4) ubiquitin ligase. The Journal of biological chemistry 285, 13896–13906. 10.1074/jbc.M110.111518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton P (1999). Ischemic cell death in brain neurons. Physiol Rev 79, 1431–1568. 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- Liu F, and McCullough LD (2014). The middle cerebral artery occlusion model of transient focal cerebral ischemia. Methods in molecular biology 1135, 81–93. 10.1007/978-1-4939-0320-7_7. [DOI] [PubMed] [Google Scholar]
- Longa EZ, Weinstein PR, Carlson S, and Cummins R (1989). Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke; a journal of cerebral circulation 20, 84–91. [DOI] [PubMed] [Google Scholar]
- Luo JH, Fu ZY, Losi G, Kim BG, Prybylowski K, Vissel B, and Vicini S (2002). Functional expression of distinct NMDA channel subunits tagged with green fluorescent protein in hippocampal neurons in culture. Neuropharmacology 42, 306–318. 10.1016/s0028-3908(01)00188-5. [DOI] [PubMed] [Google Scholar]
- Mai C, Mankoo H, Wei L, An X, Li C, Li D, and Jiang LH (2020). TRPM2 channel: A novel target for alleviating ischaemia-reperfusion, chronic cerebral hypo-perfusion and neonatal hypoxic-ischaemic brain damage. Journal of cellular and molecular medicine 24, 4–12. 10.1111/jcmm.14679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BA, Wang J, Hirschler-Laszkiewicz I, Gao E, Song J, Zhang XQ, Koch WJ, Madesh M, Mallilankaraman K, Gu T, et al. (2013). The second member of transient receptor potential-melastatin channel family protects hearts from ischemia-reperfusion injury. American journal of physiology. Heart and circulatory physiology 304, H1010–1022. 10.1152/ajpheart.00906.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortadza SS, Sim JA, Stacey M, and Jiang LH (2017). Signalling mechanisms mediating Zn(2+)-induced TRPM2 channel activation and cell death in microglial cells. Scientific reports 7, 45032. 10.1038/srep45032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolai J, Burbassi S, Rubin J, and Meucci O (2010). CXCL12 inhibits expression of the NMDA receptor’s NR2B subunit through a histone deacetylase-dependent pathway contributing to neuronal survival. Cell death & disease 1, e33. 10.1038/cddis.2010.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olah ME, Jackson MF, Li H, Perez Y, Sun HS, Kiyonaka S, Mori Y, Tymianski M, and MacDonald JF (2009). Ca2+-dependent induction of TRPM2 currents in hippocampal neurons. J Physiol 587, 965–979. 10.1113/jphysiol.2008.162289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olney JW (1969). Brain lesions, obesity, and other disturbances in mice treated with monosodium glutamate. Science 164, 719–721. 10.1126/science.164.3880.719. [DOI] [PubMed] [Google Scholar]
- Perraud AL, Fleig A, Dunn CA, Bagley LA, Launay P, Schmitz C, Stokes AJ, Zhu Q, Bessman MJ, Penner R, et al. (2001). ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature 411, 595–599. [DOI] [PubMed] [Google Scholar]
- Perraud AL, Schmitz C, and Scharenberg AM (2003). TRPM2 Ca2+ permeable cation channels: from gene to biological function. Cell Calcium 33, 519–531. 10.1016/s0143-4160(03)00057-5. [DOI] [PubMed] [Google Scholar]
- Petit-Pedrol M, and Groc L (2021). Regulation of membrane NMDA receptors by dynamics and protein interactions. The Journal of cell biology 220. 10.1083/jcb.202006101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Povysheva N, Nigam A, Brisbin AK, Johnson JW, and Barrionuevo G (2019). Oxygen-Glucose Deprivation Differentially Affects Neocortical Pyramidal Neurons and Parvalbumin-Positive Interneurons. Neuroscience 412, 72–82. 10.1016/j.neuroscience.2019.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren M, Senatorov VV, Chen RW, and Chuang DM (2003). Postinsult treatment with lithium reduces brain damage and facilitates neurological recovery in a rat ischemia/reperfusion model. Proceedings of the National Academy of Sciences of the United States of America 100, 6210–6215. 10.1073/pnas.0937423100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano Y, Inamura K, Miyake A, Mochizuki S, Yokoi H, Matsushime H, and Furuichi K (2001). Immunocyte Ca2+ influx system mediated by LTRPC2. Science 293, 1327–1330. [DOI] [PubMed] [Google Scholar]
- Sans N, Prybylowski K, Petralia RS, Chang K, Wang YX, Racca C, Vicini S, and Wenthold RJ (2003). NMDA receptor trafficking through an interaction between PDZ proteins and the exocyst complex. Nature cell biology 5, 520–530. 10.1038/ncb990. [DOI] [PubMed] [Google Scholar]
- Schmitz C, Perraud AL, Johnson CO, Inabe K, Smith MK, Penner R, Kurosaki T, Fleig A, and Scharenberg AM (2003). Regulation of vertebrate cellular Mg2+ homeostasis by TRPM7. Cell 114, 191–200. 10.1016/s0092-8674(03)00556-7. [DOI] [PubMed] [Google Scholar]
- Schulien AJ, Yeh CY, Orange BN, Pav OJ, Hopkins MP, Moutal A, Khanna R, Sun D, Justice JA, and Aizenman E (2020). Targeted disruption of Kv2.1-VAPA association provides neuroprotection against ischemic stroke in mice by declustering Kv2.1 channels. Science advances 6. 10.1126/sciadv.aaz8110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DB, Blanpied TA, Swanson GT, Zhang C, and Ehlers MD (2001). An NMDA receptor ER retention signal regulated by phosphorylation and alternative splicing. J Neurosci 21, 3063–3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sena E, van der Worp HB, Howells D, and Macleod M (2007). How can we improve the pre-clinical development of drugs for stroke? Trends in neurosciences 30, 433–439. 10.1016/j.tins.2007.06.009. [DOI] [PubMed] [Google Scholar]
- Shimizu T, Macey TA, Quillinan N, Klawitter J, Perraud AL, Traystman RJ, and Herson PS (2013). Androgen and PARP-1 regulation of TRPM2 channels after ischemic injury. J Cereb Blood Flow Metab 33, 1549–1555. 10.1038/jcbfm.2013.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soh JW, and Weinstein IB (2003). Roles of specific isoforms of protein kinase C in the transcriptional control of cyclin D1 and related genes. J Biol Chem 278, 34709–34716. 10.1074/jbc.M302016200. [DOI] [PubMed] [Google Scholar]
- Standley S, Roche KW, McCallum J, Sans N, and Wenthold RJ (2000). PDZ domain suppression of an ER retention signal in NMDA receptor NR1 splice variants. Neuron 28, 887–898. 10.1016/s0896-6273(00)00161-6. [DOI] [PubMed] [Google Scholar]
- Starkus JG, Fleig A, and Penner R (2010). The calcium-permeable non-selective cation channel TRPM2 is modulated by cellular acidification. The Journal of physiology 588, 1227–1240. 10.1113/jphysiol.2010.187476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RJ, Zhou N, and MacVicar BA (2006). Ischemia opens neuronal gap junction hemichannels. Science 312, 924–927. 10.1126/science.1126241. [DOI] [PubMed] [Google Scholar]
- Tymianski M (2011). Emerging mechanisms of disrupted cellular signaling in brain ischemia. Nature neuroscience 14, 1369–1373. 10.1038/nn.2951. [DOI] [PubMed] [Google Scholar]
- Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Delling FN, et al. (2020). Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation 141, e139–e596. 10.1161/CIR.0000000000000757. [DOI] [PubMed] [Google Scholar]
- Wang L, Fu TM, Zhou Y, Xia S, Greka A, and Wu H (2018). Structures and gating mechanism of human TRPM2. Science 362. 10.1126/science.aav4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weilinger NL, Lohman AW, Rakai BD, Ma EM, Bialecki J, Maslieieva V, Rilea T, Bandet MV, Ikuta NT, Scott L, et al. (2016). Metabotropic NMDA receptor signaling couples Src family kinases to pannexin-1 during excitotoxicity. Nature neuroscience 19, 432–442. 10.1038/nn.4236. [DOI] [PubMed] [Google Scholar]
- Wu LJ, Wu G, Akhavan Sharif MR, Baker A, Jia Y, Fahey FH, Luo HR, Feener EP, and Clapham DE (2012). The voltage-gated proton channel Hv1 enhances brain damage from ischemic stroke. Nature neuroscience 15, 565–573. nn.3059 [pii] 10.1038/nn.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu QJ, and Tymianski M (2018). Targeting NMDA receptors in stroke: new hope in neuroprotection. Mol Brain 11, 15. 10.1186/s13041-018-0357-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, Bi Y, Zhang H, Dong S, Teng L, Lee RJ, and Yang Z (2020). Cell-Penetrating Peptides in Diagnosis and Treatment of Human Diseases: From Preclinical Research to Clinical Application. Front Pharmacol 11, 697. 10.3389/fphar.2020.00697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S, Shimizu S, Kiyonaka S, Takahashi N, Wajima T, Hara Y, Negoro T, Hiroi T, Kiuchi Y, Okada T, et al. (2008). TRPM2-mediated Ca2+influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nature medicine 14, 738–747. 10.1038/nm1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Bengtson CP, Buchthal B, Hagenston AM, and Bading H (2020). Coupling of NMDA receptors and TRPM4 guides discovery of unconventional neuroprotectants. Science 370. 10.1126/science.aay3302. [DOI] [PubMed] [Google Scholar]
- Yan JZ, Xu Z, Ren SQ, Hu B, Yao W, Wang SH, Liu SY, and Lu W (2011). Protein kinase C promotes N-methyl-D-aspartate (NMDA) receptor trafficking by indirectly triggering calcium/calmodulin-dependent protein kinase II (CaMKII) autophosphorylation. The Journal of biological chemistry 286, 25187–25200. 10.1074/jbc.M110.192708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Zou J, Xia R, Vaal ML, Seymour VA, Luo J, Beech DJ, and Jiang LH (2010). State-dependent inhibition of TRPM2 channel by acidic pH. The Journal of biological chemistry 285, 30411–30418. 10.1074/jbc.M110.139774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H, Guo M, Martins-Taylor K, Wang X, Zhang Z, Park JW, Zhan S, Kronenberg MS, Lichtler A, Liu HX, et al. (2010). Specification of region-specific neurons including forebrain glutamatergic neurons from human induced pluripotent stem cells. PloS one 5, e11853. 10.1371/journal.pone.0011853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Brown MQ, van de Ven W, Zhang ZM, Wu B, Young MC, Synek L, Borchardt D, Harrison R, Pan S, et al. (2016). Endosidin2 targets conserved exocyst complex subunit EXO70 to inhibit exocytosis. Proceedings of the National Academy of Sciences of the United States of America 113, E41–50. 10.1073/pnas.1521248112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Toth B, Szollosi A, Chen J, and Csanady L (2018). Structure of a TRPM2 channel in complex with Ca(2+) explains unique gating regulation. eLife 7. 10.7554/eLife.36409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Zhang L, Wang AP, Bennett MV, and Zukin RS (1999). Protein kinase C potentiation of N-methyl-D-aspartate receptor activity is not mediated by phosphorylation of N-methyl-D-aspartate receptor subunits. Proceedings of the National Academy of Sciences of the United States of America 96, 15262–15267. 10.1073/pnas.96.26.15262. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data reported in this paper will be shared by the lead contact upon request
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.