

Abstract
The mouse is the most commonly used mammalian model to study disease, including kidney disease. However, close attention needs to be paid to the differences and effects of genetic background. The default choice of most investigators is to use C57BL/6 mice, but not all C57BL/6 mice are the same. Ever since the C57BL/6 line was first established, differences in the genetic background have risen between substrains, which have major implications in the phenotypes expressed in kidney disease. Furthermore, considering that C57BL/6 substrains are relatively resistant to kidney damage, there can be major benefits in selecting other mouse inbred strains when studying kidney disease. These strains can show more similar responses regarding kidney damage as in humans and results may therefore translate better to human application. Genetically diverse mice, such as the Diversity Outbred mice, allow investigators to study kidney phenotypes with comparable levels of genetic diversity as seen in humans, which yield results that more closely reflect the variation in human disease outcomes due to genetic variation. Hence, embracing the genetic diversity that is present in mice can lead to better translational research methods. Investigators need to always take into consideration that genetic background is a variable that can alter results significantly, and optimization of translational research asks for careful strain selection and more rigorous reporting of the genetic background that is being used in experiment.
Keywords: Genetic background, mouse model, kidney disease, translational research, genetic diversity
Introduction
Genetic research is important in understanding disease and developing new therapies. There are obvious limitations for conducting genetic research in humans, therefore using animal models is arguably the best option to study the genetics underlying diseases. The mouse is the most commonly used mammalian model, but regarding kidney disease and kidney-related phenotypes, many are concerned that results obtained from mouse experiments do not translate well to the human population. We argue that the use of the mouse in the kidney field has been too narrow (using mostly young male mice from a single inbred strain that is very resistant to insults to the kidney) and demonstrate that genetic background has a major impact on the phenotype of kidney disease. Different mouse strains and substrains exhibit a range of severities in kidney disease and we call for better characterization of strains so that investigators can choose the appropriate strain with the genetic background suitable for the phenotype they wish to study. We also argue that embracing the genetic diversity that is present in the mouse leads to more applicable results that can be translated into humans as well. We encourage investigators to pay more attention to the specific differences between strains and substrains before designing experiments and to better report husbandry details (diet, health status, housing) that can have large impacts on the kidney phenotype and will allow for better translational research.
Researchers use B6, but not all B6 mice are the same
The development of the C57BL/6 (B6) dates to the 1920s when Clarence Cook Little established the strain by inbreeding mice over many generations1. B6 rapidly became the standard inbred strain to conduct biomedical research and its use is still increasing. While there are more than 450 classically derived mouse inbred strains, as of 2017, 60% of all published mouse studies used B6, compared to 36% in 19902. Since 1990, citations in the scientific literature using B6 mice have risen by over 800%2.
Every B6 mouse used today is a direct descendant from the original line established by Little, though over time many B6 substrains have developed. Although these substrains are related to each other, they are genetically and phenotypically different. The two major substrains of B6 mice are C57BL/6J (B6J), from the originally B6 mice created and maintained at The Jackson Laboratory, and C57BL/6N (B6N), first maintained at the National Institutes of Health (NIH)3. The B6N substrain was later sent to Charles River Laboratories (B6NCrl)4, Harlan (now Envigo) (B6NHsd)5, and Taconic (B6NTac)5 (Figure 1)2. In addition, a B6N colony is maintained at The Jackson Laboratory (B6NJ).
Figure 1.
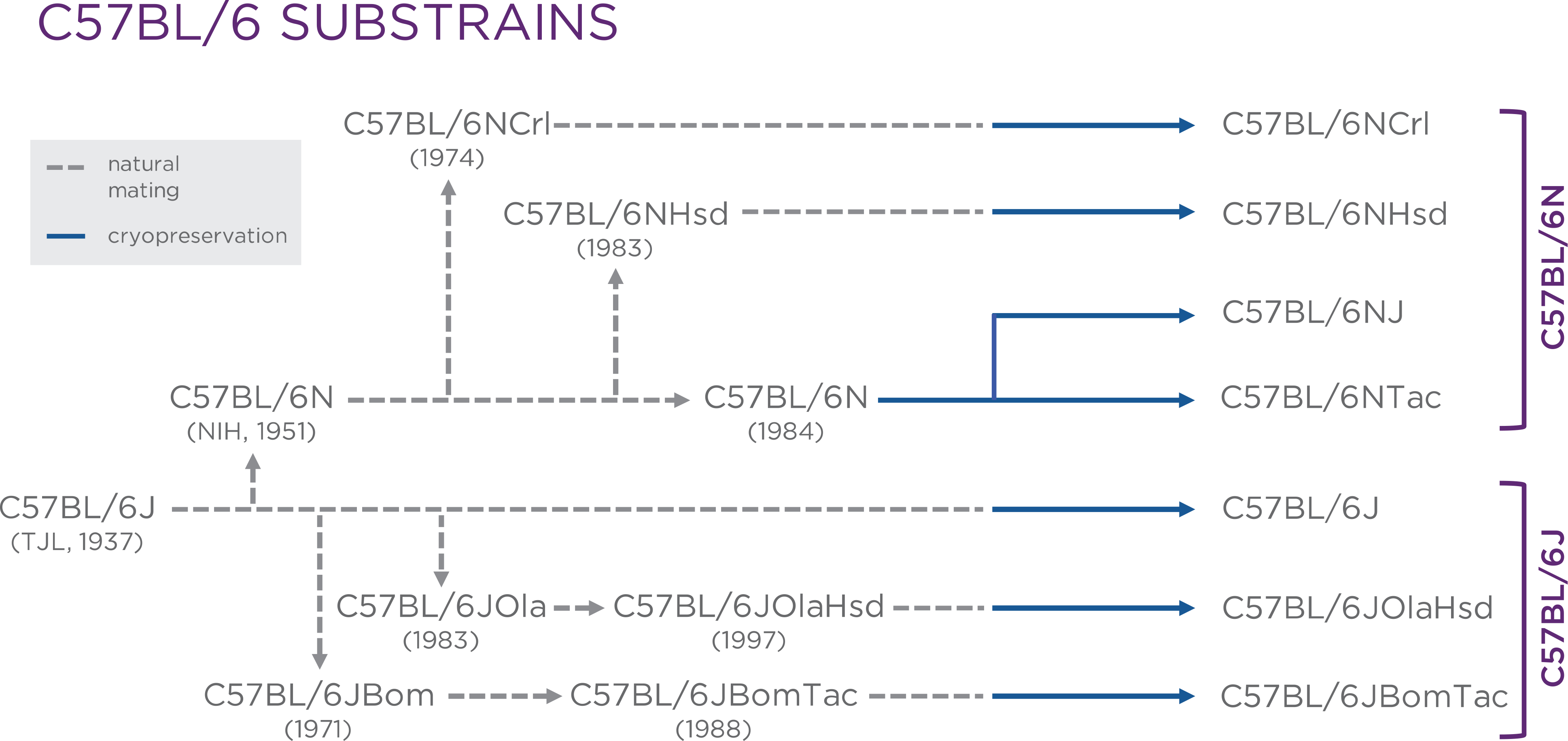
All C57BL/6 substrains are direct descendants of the originally C57BL/6J mice created and maintained at The Jackson Laboratory. New substrains were established due to genetic drift as the original C57BL/6J mice were sent to different institutes. (Figure was provided by Dr. Philip Dubé, and used with permission from Drug Discovery World (DDW).2
Studies have revealed differences between substrains caused by genetic drift between the different colonies over time. Well-known differences include: i) The absence of the nicotinamine nucleotide transhydrogenase protein in B6J mice compared to B6N due to a deletion in the Nnt gene6, ii) the stronger preferences for alcohol of B6J compared to B6NCrl mice7, iii) the expression of the retinal degeneration allele Crb1rd8 in B6N while B6J mice express the wildtype8, iv) B6JOlaHsd, a substrain established in 1983 and maintained at Harlan (now Envigo)9, mice are homozygous for a spontaneous deletion in the genes encoding alpha-synuclein and multimerin-110, v) B6NHsd mice carry a Dock2 mutation affecting B cell signaling and immune tolerance11.
Genetic differences between substrains emphasize the necessity for denoting the correct nomenclature in studies, ensuring the correct substrains are used in a study, and when comparing studies. One example that shows that not doing this leads to wrong conclusions and many years of wasted effort is when Cariappa et al. concluded that the gene Siae contributed to B cell development and signaling without denoting the specific B6 substrain used for the experiments12. Mahajan et al. failed to reproduce the results when backcrossing the Siae mutation with B6J mice11. Further analysis revealed that a copy number mutation in Dock2 had arisen in B6NHsd mice, which were used in the original study. Dock2 was discovered to be the cause of the differences in B cell functions between the B6 substrains used in the different studies and not Siae.
Differences between B6 substrains also have implications in kidney disease phenotype. For example, Usami et al. describes that numerous kidney stones were detected in both B6J and B6N substrains, however the stone count was 16.2-fold higher in B6J mice. Expression of genes related to kidney stones and six genes known to have genetic differences between the two substrains, were evaluated. The expression level of Nnt was significantly reduced in B6N mice. These findings may suggest that the Nnt gene can suppress kidney stone formation13. Bozic et al. concluded that alpha synuclein (SNCA) is significantly downregulated in dilated renal tubules of both human and mouse kidney affected by fibrosis14. Conditional gene silencing of Snca in renal proximal tubular epithelial cells (RPTECs) prompts tubulointerstitial fibrosis through two unrelated pathways. This provides evidence that disruption of SNCA signaling in RPTECs contributes to the pathogenesis of renal tubulointerstitial fibrosis by facilitating partial epithelial-to-mesenchymal transition and extracellular matrix accumulation. The study emphasizes the importance of preserving basal SNCA levels in the kidney as a therapeutic strategy to attenuate the progression of kidney fibrosis14. Furthermore, in ageing mice, Schmitt et al. showed that 19–22 months old B6J mice purchased from Charles River Laboratories in France exhibit significant nodular glomerulosclerosis with characteristics of amyloidosis, and enhanced interstitial fibrosis compared to B6J mice purchased from the NIA in the US, Elevage Janvier in France, and the University of Heidelberg in Germany15 (Figure 2). Since all animals were kept under the same conditions during the experiment, it is likely that a mutation has occurred in the mice from Charles River due to genetic drift and may explain the differences seen amongst the groups in the study.
Figure 2.

Evaluation of glomerular morphology of 19–22 months old B6J mice purchased from four different vendors. Representative examples of periodic acid-Schiff-stained renal sections from mice of (A) group 1 from Charles River Laboratories, (B) group 2 from the NIA, (C) group 3 from Elevage Janvier and (D) group 4 from the University of Heidelberg. (E) Glomerulosclerosis score. (F) Representative picture of Congo red-stained section from mouse of group 1 in normal light and (G) in polarized light. Data in (E) are presented as the mean ± SEM. **P < 0.002 versus group 1. (Reproduced from Schmitt R, Jacobi C, Susnik N, Broecker V, Haller H, Melk A. Ageing mouse kidney—not always the same Old story. Nephrology Dialysis Transplantation. 2009;24(10):3002–3005 by permission of the European Renal Association.
These examples highlight the influence that small differences in genetic background can have on experimental results. A founding principle of scientific research is the ability to reproduce results, which requires the consistent use of materials and methods across experiments. The genetic background in mice is a critical factor in experiment methods, and when disregarded, compromises scientific reproducibility. Many publications still lack the annotation of the exact genetic background and the source used. We urge authors (and journals) to pay more attention to these important details.
Strain differences and benefits for considering other strains
The variation of genetic background among other inbred strains has even more significant implications on the phenotypes in mice compared to B6 substrains. Like the variety in disease susceptibility in humans, various strains of mice exhibit differences in phenotypes as modulated by their genetic background. The comparison of various inbred strains for kidney phenotypes led to the identification of candidate genes for albuminuria16, mesangial matrix expansion17,18, and lithiopathies19. Additionally, various inbred strains are better models compared to B6 to induce kidney diseases as seen in humans. For example, multiple genetic factors regulate the risk for developing diabetic nephropathy (DN) in humans and mice20. DN is a common complication for patients with diabetes, and its severity is heavily influenced by genetic background. Injection of streptozotocin (STZ) and introduction of the Ins2+/C96Y (Akita) mutation are well established methods for inducing type I diabetes in mouse models21. Levels of albuminuria are an important outcome to assess the disease severity as they are a central manifestation of DN 22, 23. The B6 genetic background is relatively resistant to developing albuminuria under diabetic conditions despite showing moderate mesangial expansion and glomerular basement membrane (GBM) thickening21. DBA/2J mice with an Akita mutation exhibit significantly higher albuminuria levels than B6J mice with the same mutation (Figure 3). Similar results are seen when inducing diabetes through low dose STZ-induced diabetes21. Therefore, the B6 genetic background is not a good strain for studying DN. Other strains, like DBA/2J, show a much better DN phenotype, and can lead to a better understanding of the disease. The genome of DBA/2J harbors alleles that predispose mice to kidney dysfunction24. Several genomic regions have been identified25, but the causal genes remain unknown.
Figure 3.
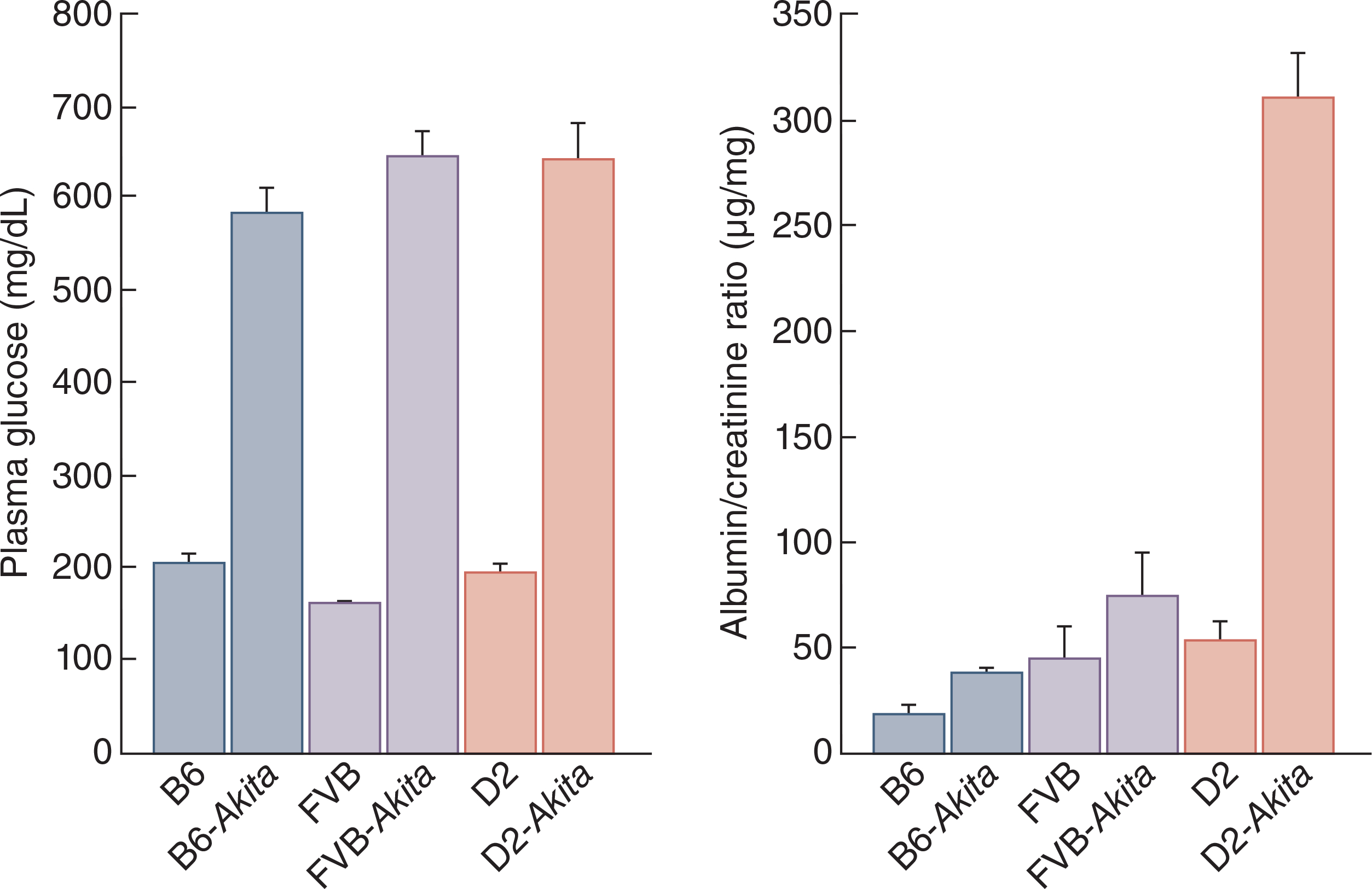
Phenotypic comparison of the Ins2Akita mutation in males of three inbred strains show comparable levels in hyperglycemia in Akita/+ males in all three genetic backgrounds (A). Following necropsy at 6 months of age, development of diabetic nephropathy was only limited to the DBA/2J strain as measured by the increased albumin/creatinine ratio (data from the Diabetes Complications Consortium)
For mouse models of type II diabetes, differences in genetic background also show variation in disease susceptibility. Diet-induced and inherited deficiency in leptin (db/db mice) are well established models for type II diabetes25. The renal manifestations in the db/db mice have been characterized primarily in the C57BLKS/J strain, which shares 84% of its alleles with B6J and 16% of its alleles with the DBA/2J strain26. However, GBM thickening associated with increased albuminuria, and the lack of the progressive increase in albuminuria cannot be easily altered in the C57BLKS-db/db mouse as in the human condition27. In contrast, renal lesions in KK/Ta mice, an inbred strain derived from Japanese native mice28, closely resemble those in human DN. Glomeruli of KK/Ta mice show diffuse hyperplasia of mesangial areas with mild mesangial cell proliferation29. Moreover, the KK-Ay/Ta mouse was established by the transfer of the yellow obese gene (Ay allele) into the KK/Ta mouse with the purpose of inducing a more severe DN phenotype. KK-Ay/Ta mice demonstrate type II diabetes and DN at a younger age compared to KK/Ta mice29. When compared to the C57BLKS-db/db and KK/Ta strains, the KK-Ay/Ta strain is a better model for more severe phenotypes associated with DN (Table 1). Using KK-Ay/Ta mice is advantageous because it exhibits diabetes in a polygenic way (analogous to humans) and not solely due to a defect in leptin receptor binding30,31.
Table 1.
Glomerular changes in different strains of mice
| Age (weeks) | DME | GBMT | SS | |
|---|---|---|---|---|
|
| ||||
| C57BLKS-db/db | 21 | ++ | ++ | − |
| KK/Ta | 16–20 | + | + | − |
| KK-Ay/Ta | 20 | ++ | ++ | + |
Note. +, Present; −, Absent; DME, diffuse mesangial expansion; GBMT, glomerular basement membrane thickening; SS, segmental sclerosis. Data obtained from Ito et al (2006).
Genetic background also modulates the response to cisplatin nephrotoxicity. Cisplatin is a chemotherapeutic used in the treatment of many solid cancers32. Studies have shown approximately 30% of patients will develop kidney damage in the form of acute kidney injury (AKI) that ultimately leads to chronic kidney disease (CKD)33,34. Again, B6J mice are resistant to developing glomerular changes associated with kidney fibrosis and CKD, and to developing tubulointerstitial fibrosis, which makes B6J mice less useful for studying the response to cisplatin. Low dose cisplatin treatment of FVB/n mice shows increased transforming growth factor-beta and fibronectin, suggesting a robust fibrotic phenotype, and increased progressive loss of kidney function35. Therefore, the FVB/n strain is a better model for studying the nephrotoxic effect of cisplatin and its long-term kidney outcomes36.
The remnant kidney model (RKM) – produced by unilateral nephrectomy and partial perfusion of the remaining kidney – of progressive renal disease has been used extensively to study glomerulosclerosis37. In this model, B6 mice (authors did not specify substrain) are resistant to developing glomerulosclerosis after RKM, whereas 129/Sv mice (authors did not specify substrain, while there are several 129/Sv substrains with known differences in renal phenotypes) develop robust induction of marked glomerulosclerosis, which is linked to increased blood pressure38. Amongst other differences between the two strains, B6 mice have one copy of the renin gene (Ren-1c), whereas 129/Sv mice have two active copies (Ren-1d and Ren-2). Polymorphisms in the renin gene may influence susceptibility to the development of hypertension and further glomerulosclerosis in 129/Sv and B6 strains39. Mice with two renin genes have a 10-fold renal hypertrophic response to salt, higher plasma renin activity, and increased blood pressure compared to mice with one renin gene40. 129/Sv mice have increased renin level and activated renin-angiotensin-aldosterone system, which accelerates glomerulosclerosis, while B6 mice maintain normal renin levels and blood pressure, which lead to resistance against glomerulosclerosis38. While there have been studies that have rejected the hypothesized relationship between renin genes and elevations in blood pressure or development of renal lesions41, the 129/Sv strain remains a better model for studying glomerulosclerosis and renal lesions compared to B6.
Considering the above, it is critical to better understand the differences in genetic background that lead to variations in phenotypes when studying kidney phenotypes and must always be considered when choosing the best model for studying phenotypes that most closely represent what is observed in humans. In general, the B6 substrains are very resistant to kidney damage and many other inbred strains are often a better choice to study kidney disease.
Embracing genetic diversity
Although inbred mice are widely used, the generalizability of findings is limited with results obtained from a single strain. Findings from genetically diverse populations are more robust and generalizable to a genetically diverse human population. Resources such as the Diversity Outbred (DO) mice allow investigators to overcome these limitations. The DO mice are a heterogeneous stock derived from eight inbred founder strains, selected to maximize genetic diversity, and maintained by randomized breeding42. Each DO mouse is a unique individual with a high level of allelic heterozygosity. This heterogenous stock provides an advantageous opportunity to study medically relevant traits in a powerful model system that more closely reflect the genetic mechanisms of human disease43.
DO mice also provides a better model for studying kidney phenotypes. For example, DO mice were used to study renal function in the context of varying genetic determination of Cst3 levels as associated with levels of plasma Cystatin C (CysC), similar to what occurs in humans44. Increased plasma levels of CysC are a sensitive indicator for early stages of AKI45, and studies have shown there is a positive correlation between Cst3 expression and plasma CysC levels44. While future investigation on the cause-effect relationship is needed, the results obtained through DO mice indicate a relationship between Cst3 expression levels, kidney inflammation, filtration function, and their implications for AKI and CKD progression44.
DO mice also give investigators the power to complete high resolution mapping of loci associated with kidney phenotypes. For example, Takemon et al. identified novel modifier genes for X-linked Alport Syndrome (XLAS) by introducing a mutated Col4a5 allele into a cohort of DO mice and measure the variation in albuminuria and glomerular filtration rate as a consequence of the genetic variation in the population46. In this study, females heterozygous for the Col4a5 mutation (located on the X-chromosome) in an otherwise B6J genetic background were crossed with male DO mice, and only the offspring with the mutation (hemizygous mutant males and heterozygous mutant females) were selected to generate the experimental cohort. Since the DO male provides eight possible haplotypes and the Col4a5 mutant female only B6J haplotypes, each locus for animal in the DO-XLAS population was either homozygous for the B6J alleles or heterozygous for B6J and one of seven heterozygous states46. Several candidate modifier genes, including Fmn1, were identified and follow-up experiments confirmed that a decrease in Fmn1 expression leads to lower albuminuria in mice with Alport disease46.
Similar to the DO-XLAS strategy which works for dominant alleles or alleles located on the X-chromosome, studying the effect of recessive mutations in a genetically diverse mouse population is also possible. For example, in mouse models, polycystic kidney disease is mostly transmitted as a recessive trait47. There are numerous recessive alleles that lead to PKD in mice, such as the cpk mutation in the Cys1 gene48,49, the b2b1585 mutation in the Pkd1 gene (JAX strain 018189), or the RC allele in Pkd150. Generation of DO experimental cohorts with either mutation is possible by crossing homozygous mutant mice with DO mice and crossing the F1 progeny again to generate homozygous mutant offspring in a genetically diverse background.
Other outbred mouse populations are available51 and have been used to genetically map kidney-related phenotypes52 but are less tractable with regards to their genetics and have lower genetic variation compared to the DO. Embracing genetic diversity in mouse models can lead to novel findings and better understanding of the genetic underpinnings of kidney disease and the identification of resilience genes and new therapeutic targets that are not possible in inbred strains.
Optimizing translational mouse studies
Ideally, we would like to have kidney phenotypes, pathology, and expression profiles of many different inbred strains and genetically diverse populations, such as the DO, under various disease conditions so that we can compare those with human patient data and in this way select the best strain to study a particular disease or condition, but this would require investment and effort. Some data is available for kidney disease and can at least move us in the right direction. For example, the Mouse Phenome Database (phenome.jax.org) has more than 60 kidney-related phenotyping measurements for many different inbred strains and has convenient tools that allow an investigator to explore these and help select the best mouse strain for their project.
Another important part is to educate investigators better so that they do not immediately order B6 when they start a mouse experiment, but instead carefully look at the alternatives, maybe from direct knowledge in different strains or by extrapolation from other (associated) phenotypes in these strains. In addition, often the control strain used in studies is not identical to that used for backcrossing. The control strain is selected from a parental colony from commercial suppliers, and the selected strain may only have in common the genealogic name with the rest of the animals used in the experimental cohort. Using strains from other colonies means that mice within the same cohort differ in environmental upbringing among other possible genetic variations. For example, The Jackson Laboratory lacks the segmented filamentous bacteria (SFB) compared with some other animal houses, which in turn affects an IL-17-associated phenotype53. Purchasing control mice from different vendors may introduce unwanted confounding variables to experiments. Using littermates as controls ensures not only that the genetic background is comparable but also the environment. We strongly urge journals to set this as a quality standard of reporting data.
Age is also another overlooked component in animal studies. The age at which rodents are commonly used for research is 8–12 weeks54. In this age range many developmental processes are ongoing, and changes in physiology with age may have a large impact on experimental variables54. Choosing an age of rodent suitable to the human disease being studied can have a critical impact on the performance of the model. Older animals entering senescence may respond differently to their young counterparts. By emphasizing the importance of the age of the rodent used in biomedical research, the number of animals used can be reduced while maintaining or ameliorating data quality. Accurate and consistent reporting age in peer-reviewed literature will allow comparison of experimental methodology.
Furthermore, there is evidence that paternal age can influence behavioral traits and is associated with a reduced life span in the offspring animals indicating paternal age influences age-related pathologies intergenerationally55. In addition, metabolic status of dams can also influence age-dependent phenotypes in offspring56. For example, insulin secretion was impaired in male offspring of obese dams between 3 and 6 months of age57, suggesting metabolic phenotypes of parents have an impact on phenotypes of offspring. Similarly, both male and female mouse offspring of dams fed high-fed diets from 2 weeks pre-conception developed glucose intolerance from 36 until 42 weeks, despite normal glucose tolerance for the first 3 months of life58. Thus, stressing the importance of evaluating parental phenotypes prior to selecting offspring for experimental cohorts. The provided evidence calls for careful consideration of possible confounding variables for data variation when designing experiments.
Finally, more rigorous reporting of experimental conditions is needed as it will help us understand differences in outcome between studies and identify the factors that cause these differences. Every nephrologist will agree that factors such as genetic background, sex, age, diet, and health status are important factors in human patients, yet most publications fail to report these factors for mouse studies. The mention of “standard rodent chow” is not adequate when there can be a 10% difference in fat content between these “standard” diets. The composition of diets varies from batch to batch, for example, in grain-based nutritional contents fluctuate between batches, which creates inconsistencies in interventions even though investigators are attempting to choose the same diet59. Mouse room health status can also have a profound impact on a phenotype. For example, atherosclerotic plaques were measured in a large number of inbred strains that were kept in a ‘dirty’ room and a ‘clean’ room. For some strains there was no difference, but for most strains the changes were dramatic60. It is not difficult to imagine that a similar effect can be expected for various forms of kidney disease and a mouse room health room report as supplemental data to a publication would be useful. Many publishers of scientific journals have adopted the checklist of recommendations of National Centre for the Replacement, Reduction and Refinement (NC3R) in the form of the Animal Research: Reporting of In Vivo Experiments (ARRIVE) (www.arriveguidelines.org), which helps to better evaluate and reproduce the experiments described in publications. However, among the nephrology-related publications, only AJP-Renal Physiology is using these guidelines.
We urge investigators to consider genetic background in their mouse studies and to be thorough with the reporting of exact genetic background, diet composition, and health status in study designs, grant applications, and study methods sections. It will increase reproducibility and make the mouse a better translational model for kidney disease.
Acknowledgements
RK is supported by grants from the National Institutes of Health (ES29916, AG038070, DK131019, and DK131061) and the Alport Syndrome Foundation. As a Post-baccalaureate Fellow, RB gratefully acknowledges funding support from JAX Genomic Education.
Footnotes
Disclosure
The authors report no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rader KA. Experiment and Change: Institutionalizing Inbred Mice (1922–30). In: Making Mice: Standardizing Animals for American Biomedical Research, 1900–1955. Princeton, New Jersey: Princeton University Press; 2004:59–96. [Google Scholar]
- 2.Dube P How the B6 mouse strain is revolutionizing drug discovery. Drug Discovery World (DDW). https://www.ddw-online.com/how-the-b6-mouse-strain-is-revolutionising-drug-discovery-1764-201710/. Published October 2, 2017. Accessed November 30, 2021. [Google Scholar]
- 3.Matsuo N Behavioral profiles of three C57BL/6 substrains. Frontiers in Behavioral Neuroscience. 2010. doi: 10.3389/fnbeh.2010.00029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bankstahl M, Müller CJ, Wilk E, et al. Generation and characterization of pilocarpine-sensitive C57BL/6 mice as a model of temporal lobe epilepsy. Behavioural Brain Research. 2012;230(1):182–191. doi: 10.1016/j.bbr.2012.02.004 [DOI] [PubMed] [Google Scholar]
- 5.Bryant CD, Zhang NN, Sokoloff G, et al. Behavioral differences among C57BL/6 substrains: Implications for transgenic and knockout studies. Journal of Neurogenetics. 2008;22(4):315–331. doi: 10.1080/01677060802357388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mekada K, Abe K, Murakami A, et al. Genetic differences among C57BL/6 substrains. Experimental Animals. 2009;58(2):141–149. doi: 10.1538/expanim.58.141 [DOI] [PubMed] [Google Scholar]
- 7.Mulligan MK, Ponomarev I, Boehm SL, et al. Alcohol Trait and transcriptional genomic analysis of C57BL/6 substrains. Genes, Brain and Behavior. 2008;7(6):677–689. doi: 10.1111/j.1601-183x.2008.00405.x [DOI] [PubMed] [Google Scholar]
- 8.Mattapallil MJ, Wawrousek EF, Chan C-C, et al. Therd8mutation of thecrb1gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Investigative Opthalmology & Visual Science. 2012;53(6):2921. doi: 10.1167/iovs.12-9662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liron T, Raphael B, Hiram-Bab S, et al. Bone loss in C57BL/6J-OlaHsd mice, a substrain of C57BL/6J carrying mutated alpha-synuclein and multimerin-1 genes. J Cell Physiol. 2017;233(1):371–377. doi: 10.1002/jcp.25895 [DOI] [PubMed] [Google Scholar]
- 10.Specht CG, Schoepfer R. Deletion of the alpha-synuclein locus in a subpopulation of C57BL/6J inbred mice. BMC Neuroscience. 2001;2(1):11. doi: 10.1186/1471-2202-2-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mahajan VS, Demissie E, Mattoo H, et al. Striking immune phenotypes in gene-targeted mice are driven by a copy-number variant originating from a commercially available C57BL/6 strain. Cell Reports. 2016;15(9):1901–1909. doi: 10.1016/j.celrep.2016.04.080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cariappa A, Takematsu H, Liu H, et al. B cell antigen receptor signal strength and peripheral B cell development are regulated by a 9-O-acetyl sialic acid esterase. Journal of Experimental Medicine. 2008;206(1):125–138. doi: 10.1084/jem.20081399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Usami M, Unno R, Iwatsuki S, et al. MP34–11 expression of NNT appears to suppress kidney stone formation in C57BL/6 mouse substrains. Journal of Urology. 2015;193(4S). doi: 10.1016/j.juro.2015.02.1307 [DOI] [Google Scholar]
- 14.Bozic M, Caus M, Rodrigues-Diez RR, et al. Protective role of renal proximal tubular alpha-synuclein in the pathogenesis of Kidney Fibrosis. Nature Communications. 2020;11(1). doi: 10.1038/s41467-020-15732-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmitt R, Jacobi C, Susnik N, Broecker V, Haller H, Melk A. Ageing mouse kidney—not always the same Old story. Nephrology Dialysis Transplantation. 2009;24(10):3002–3005. doi: 10.1093/ndt/gfp232 [DOI] [PubMed] [Google Scholar]
- 16.Tsaih S-W, Pezzolesi MG, Yuan R, Warram JH, Krolewski AS, Korstanje R. Genetic analysis of Albuminuria in aging mice and concordance with loci for human diabetic nephropathy found in a genome-wide association scan. Kidney International. 2010;77(3):201–210. doi: 10.1038/ki.2009.434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Noordmans GA, Caputo CR, Huang Y, et al. Genetic analysis of mesangial matrix expansion in aging mice and identification of FAR2 as a candidate gene. Journal of the American Society of Nephrology. 2013;24(12):1995–2001. doi: 10.1681/asn.2012080838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Backer G, Eddy S, Sheehan SM, et al. FAR2 is associated with kidney disease in mice and humans. Physiological Genomics. 2018;50(8):543–552. doi: 10.1152/physiolgenomics.00118.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Groot T, Ebert LK, Christensen BM, et al. Identification of ACER2 as a first susceptibility gene for lithium-induced nephrogenic diabetes insipidus in mice. Journal of the American Society of Nephrology. 2019;30(12):2322–2336. doi: 10.1681/asn.2018050549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gurley SB, Clare SE, Snow KP, et al. Impact of genetic background on nephropathy in diabetic mice. American Journal of Physiology-Renal Physiology. 2006;290(1). doi: 10.1152/ajprenal.00204.2005 [DOI] [PubMed] [Google Scholar]
- 21.Østergaard MV, Pinto V, Stevenson K, et al. DBA2J db/DB mice are susceptible to early albuminuria and glomerulosclerosis that correlate with systemic insulin resistance. American Journal of Physiology-Renal Physiology. 2017;312(2). doi: 10.1152/ajprenal.00451.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krolewski AS. Genetics of diabetic nephropathy: Evidence for major and minor gene effects. Kidney International. 1999;55(4):1582–1596. doi: 10.1046/j.1523-1755.1999.00371.x [DOI] [PubMed] [Google Scholar]
- 23.Wang J, Takeuchi T, Tanaka S, et al. A mutation in the insulin 2 gene induces diabetes with severe pancreatic β-cell dysfunction in the Mody Mouse. Journal of Clinical Investigation. 1999;103(1):27–37. doi: 10.1172/jci4431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Soto I, Howell G, John C, et al. DBA/2J Mice Are Susceptible to Diabetic Nephropathy and Diabetic Exacerbation of IOP Elevation. PLoS One. 2014;9(9):e107291. doi: 10.1371/journal.pone.0107291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sheehan S, Tsaih S, King B et al. Genetic analysis of albuminuria in a cross between C57BL/6J and DBA/2J mice. American Journal of Physiology-Renal Physiology. 2007;293(5):F1649–F1656. doi: 10.1152/ajprenal.00233.2007 [DOI] [PubMed] [Google Scholar]
- 26.Qi Z, Fujita H, Jin J, et al. Characterization of susceptibility of inbred mouse strains to diabetic nephropathy. Diabetes. 2005;54(9):2628–2637. doi: 10.2337/diabetes.54.9.2628 [DOI] [PubMed] [Google Scholar]
- 27.Sharma K, McCue P, Dunn SR. Diabetic kidney disease in thedb/dbmouse. American Journal of Physiology-Renal Physiology. 2003;284(6). doi: 10.1152/ajprenal.00315.2002 [DOI] [PubMed] [Google Scholar]
- 28.Kondo K, Nozawa K, Tomita T, et al. Inbred strains resulting from Japanese mice. Bull Exp Animals. 1957; 6:107–12. [Google Scholar]
- 29.Ito T, Tanimoto M, Yamada K, et al. Glomerular changes in the KK-ay/TA mouse: A possible model for human type 2 diabetic nephropathy. Nephrology. 2006;11(1):29–35. doi: 10.1111/j.1440-1797.2006.00543.x [DOI] [PubMed] [Google Scholar]
- 30.Breyer M, Böttinger E, Brosius F et al. Mouse Models of Diabetic Nephropathy. Journal of the American Society of Nephrology. 2004;16(1):27–45. doi: 10.1681/asn.2004080648 [DOI] [PubMed] [Google Scholar]
- 31.Chakraborty G, Thumpayil S, Lafontant D, et al. Age dependence of glucose tolerance in adult KK-Ay mice, a model of non–insulin dependent diabetes mellitus. Lab Anim (NY). 2009;38(11):364–368. doi: 10.1038/laban1109-364 [DOI] [PubMed] [Google Scholar]
- 32.Dasari S, Bernard Tchounwou P. Cisplatin in cancer therapy: Molecular mechanisms of action. European Journal of Pharmacology. 2014;740:364–378. doi: 10.1016/j.ejphar.2014.07.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hsu RK, C-yuan Hsu. The role of acute kidney injury in chronic kidney disease. Seminars in Nephrology. 2016;36(4):283–292. doi: 10.1016/j.semnephrol.2016.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Latcha S, Jaimes EA, Patil S, Glezerman IG, Mehta S, Flombaum CD. Long–term renal outcomes after cisplatin treatment. Clinical Journal of the American Society of Nephrology. 2016;11(7):1173–1179. doi: 10.2215/cjn.08070715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharp CN, Doll MA, Dupre TV, et al. Repeated administration of low-dose cisplatin in mice induces fibrosis. American Journal of Physiology-Renal Physiology. 2016;310(6). doi: 10.1152/ajprenal.00512.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sears S, Sharp C, Krueger A et al. C57BL/6 mice require a higher dose of cisplatin to induce renal fibrosis and CCL2 correlates with cisplatin-induced kidney injury. American Journal of Physiology-Renal Physiology. 2020;319(4):F674–F685. doi: 10.1152/ajprenal.00196.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kren S, Hostetter TH. The course of the remnant kidney model in mice. Kidney International. 1999;56(1):333–337. doi: 10.1046/j.1523-1755.1999.00527.x [DOI] [PubMed] [Google Scholar]
- 38.Ma L-J, Fogo AB. Model of robust induction of glomerulosclerosis in mice: Importance of genetic background. Kidney International. 2003;64(1):350–355. doi: 10.1046/j.1523-1755.2003.00058.x [DOI] [PubMed] [Google Scholar]
- 39.Lu X, Li N, Shushakova N, et al. C57BL/6 and 129/sv mice: Genetic difference to renal ischemia-reperfusion. Journal of Nephrology. 2011;25(5):738–743. doi: 10.5301/jn.5000053 [DOI] [PubMed] [Google Scholar]
- 40.Wang Q, Hummler E, Nussberger J, et al. Blood pressure, cardiac, and renal responses to salt and deoxycorticosterone acetate in mice: Role of renin genes. Journal of the American Society of Nephrology. 2002;13(6):1509–1516. doi: 10.1097/01.asn.0000017902.77985.84 [DOI] [PubMed] [Google Scholar]
- 41.Hartner A Strain differences in the development of hypertension and glomerular lesions induced by deoxycorticosterone acetate salt in mice. Nephrology Dialysis Transplantation. 2003;18(10):1999–2004. doi: 10.1093/ndt/gfg299 [DOI] [PubMed] [Google Scholar]
- 42.Svenson KL, Gatti DM, Valdar W, et al. High-resolution genetic mapping using the mouse diversity outbred population. Genetics. 2012;190(2):437–447. doi: 10.1534/genetics.111.132597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gatti DM, Svenson KL, Shabalin A, et al. Quantitative trait locus mapping methods for diversity outbred mice. G3 Genes|Genomes|Genetics. 2014;4(9):1623–1633. doi: 10.1534/g3.114.013748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huda MN, VerHague M, Albright J, et al. Dissecting the genetic architecture of cystatin C in diversity outbred mice. G3 Genes|Genomes|Genetics. 2020;10(7):2529–2541. doi: 10.1534/g3.120.401275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu X, Guan Y, Xu S, et al. Early predictors of Acute Kidney Injury: A narrative review. Kidney and Blood Pressure Research. 2016;41(5):680–700. doi: 10.1159/000447937 [DOI] [PubMed] [Google Scholar]
- 46.Takemon Y, Wright V, Davenport B, et al. Uncovering modifier genes of X-linked Alport Syndrome using a novel Multiparent Mouse model. Journal of the American Society of Nephrology. 2021;32(8):1961–1973. doi: 10.1681/asn.2020060777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guay-Woodford L Murine models of polycystic kidney disease. Am J Physiol Renal Physiol. 2003;(285):1034–1049. doi: 10.1152/ajprenal.00195.2003. [DOI] [PubMed] [Google Scholar]
- 48.Fry J, Koch W, Jennette J, Mcfarland E, et al. A Genetically Determined Murine Model of Infantile Polycystic Kidney Disease. Journal of Urology. 1985;134(4):828–833. doi: 10.1016/s0022-5347(17)47448-9 [DOI] [PubMed] [Google Scholar]
- 49.Simon E, Cook S, Davisson M, D’Eustachio P, Guay-Woodford L. The Mouse Congenital Polycystic Kidney (cpk) Locus Maps within 1.3 cM of the Chromosome 12 Marker D12Nyu2. Genomics. 1994;21(2):415–418. doi: 10.1006/geno.1994.1285 [DOI] [PubMed] [Google Scholar]
- 50.Arroyo J, Escobar-Zarate D, Wells H et al. The genetic background significantly impacts the severity of kidney cystic disease in the Pkd1RC/RC mouse model of autosomal dominant polycystic kidney disease. Kidney Int. 2021;99(6):1392–1407. doi: 10.1016/j.kint.2021.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yalcin B, Nicod J, Bhomra A, et al. Commercially available outbred mice for genome-wide association studies. PLoS Genetics. 2010;6(9). doi: 10.1371/journal.pgen.1001085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang W, Korstanje R, Thaisz J, et al. Genome-wide association mapping of quantitative traits in outbred mice. G3 Genes|Genomes|Genetics. 2012;2(2):167–174. doi: 10.1534/g3.111.001792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Holmdahl R, Malissen B. The need for littermate controls. Eur J Immunol. 2011;42(1):45–47. doi: 10.1002/eji.201142048 [DOI] [PubMed] [Google Scholar]
- 54.Jackson S, Andrews N, Ball D et al. Does age matter? The impact of rodent age on study outcomes. Lab Anim. 2016;51(2):160–169. doi: 10.1177/0023677216653984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xie K, Ryan DP, Pearson BL, et al. Epigenetic alterations in longevity regulators, reduced life span, and exacerbated aging-related pathology in old father offspring mice. Proceedings of the National Academy of Sciences. 2018;115(10). doi: 10.1073/pnas.1707337115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schoonejans JM, Ozanne SE. Developmental programming by maternal obesity: Lessons from animal models. Diabetic Medicine. 2021;38(12). doi: 10.1111/dme.14694 [DOI] [PubMed] [Google Scholar]
- 57.Samuelsson A-M, Matthews PA, Argenton M, et al. Diet-induced obesity in female mice leads to offspring hyperphagia, adiposity, hypertension, and insulin resistance. Hypertension. 2008;51(2):383–392. doi: 10.1161/hypertensionaha.107.101477 [DOI] [PubMed] [Google Scholar]
- 58.Khan IY, Taylor PD, Dekou V, et al. Gender-linked hypertension in offspring of lard-fed pregnant rats. Hypertension. 2003;41(1):168–175. doi: 10.1161/01.hyp.0000047511.97879.fc [DOI] [PubMed] [Google Scholar]
- 59.Pellizzon MA, Ricci MR. Choice of laboratory rodent diet may confound data interpretation and reproducibility. Current Developments in Nutrition. 2020;4(4). doi: 10.1093/cdn/nzaa031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Srivastava U, Paigen B, Korstanje R. Differences in Health Status Affect Susceptibility and Mapping of Genetic Loci for Atherosclerosis (Fatty Streak) in Inbred Mice. Arterioscler Thromb Vasc Biol. 2012;32(10):2380–2386. doi: 10.1161/atvbaha.112.255703 [DOI] [PMC free article] [PubMed] [Google Scholar]