

SUMMARY
Autophagy is a conserved, multi-step process of capturing proteolytic cargo in autophagosomes for lysosome degradation. The capacity to remove toxic proteins that accumulate in neurodegenerative disorders attests to the disease-modifying potential of the autophagy pathway. However, neurons respond only marginally to conventional methods for inducing autophagy, limiting efforts to develop therapeutic autophagy modulators for neurodegenerative diseases. The determinants underlying poor autophagy induction in neurons and the degree to which neurons and other cell types are differentially sensitive to autophagy stimuli are incompletely defined. Accordingly, we sampled nascent transcript synthesis and stabilities in fibroblasts, induced pluripotent stem cells (iPSCs) and iPSC-derived neurons (iNeurons), thereby uncovering a neuron-specific stability of transcripts encoding myotubularin-related phosphatase 5 (MTMR5). MTMR5 is an autophagy suppressor that acts with its binding partner, MTMR2, to dephosphorylate phosphoinositides critical for autophagy initiation and autophagosome maturation. We found that MTMR5 is necessary and sufficient to suppress autophagy in iNeurons and undifferentiated iPSCs. Using optical pulse labeling to visualize the turnover of endogenouslyencoded proteins in live cells, we observed that knockdown of MTMR5 or MTMR2, but not the unrelated phosphatase MTMR9, significantly enhances neuronal degradation of TDP-43, an autophagy substrate implicated in several neurodegenerative diseases. Our findings thus establish a regulatory mechanism of autophagy intrinsic to neurons and targetable for clearing disease-related proteins in a cell type-specific manner. In so doing, our results not only unravel novel aspects of neuronal biology and proteostasis, but also elucidate a strategy for modulating neuronal autophagy that could be of high therapeutic potential for multiple neurodegenerative diseases.
Keywords: Macroautophagy, neuronal autophagy, autophagosome, myotubularin, phosphoinositide, induced pluripotent stem cells (iPSC), RNA stability, TDP-43, optical pulse labeling, dSTORM
eTOC Blurb
Chua et al. establish MTMR5 as a key suppressor of autophagy in neurons. They show that MTMR5 is selectively enriched in neurons compared to other cell types. Furthermore, MTMR5 is necessary and sufficient to inhibit autophagy induction, and knockdown of MTMR5 accelerates degradation of autophagy substrates in neurons, including TDP-43.
INTRODUCTION
Neurodegenerative diseases are a heterogeneous group of sporadic and familial disorders with typical onset in mid- to late-life. In spite of diverse clinical manifestations and atrophy in distinct neuroanatomical regions particular for each disease, neurodegenerative disorders harbor overlapping histopathologic features, including the abnormal accumulation of misfolded and aggregated proteins1. Although the relationship between such aggregates and the pathogenesis of each disease is incompletely understood, these shared pathologic characteristics implicate age-related or genetic dysfunction of protein quality control mechanisms as convergent pathways to neurodegeneration.
One such mechanism of protein quality control is macroautophagy, here referred to as autophagy. From Greek etymologic roots meaning “self-digestion,” autophagy is a highly conserved, multi-step process of capturing protein and organelle substrates, both selectively and in bulk, into specialized autophagosome vesicles for trafficking to lysosomes, within which such cargo is degraded by proteolytic enzymes2. Autophagy operates at a constitutive level but can be stimulated as part of the adaptive response to stress and nutrient deprivation to maintain protein homeostasis2. Precise and multifaceted regulatory machinery is required for coordinating autophagy induction. Such regulation is accomplished by (i) signaling cascades inhibiting the autophagy-suppressive mTOR pathway3, (ii) kinases synthesizing phosphoinositide scaffolds upon which autophagy initiation complexes are assembled4,5, and (iii) additional multimeric proteins directing autophagosome membrane elongation and autophagosome-lysosome fusion6,7.
Many of the aggregation-prone proteins found in neurodegenerative diseases are autophagy substrates. Furthermore, genetic ablation of autophagy components produces neurodegeneration in mice8,9, and inherited forms of neurodegenerative disorders are caused by, or associated with, mutations in key autophagy-related genes, including SQSTM1 (sequestosome-1 or p62)10, TBK1 (TANK binding kinase 1)11, and OPTN (optineurin)12 in familial amyotrophic lateral sclerosis; PRKN (Parkin)13 and PINK1 (PTEN-induced kinase 1)14,15 in Parkinson disease; VCP (valosin containing protein)16 in multisystem proteinopathy; WDR45 (WD-repeat domain 45, also known as WIPI4)17 in β-propeller protein-associated neurodegeneration (BPAN); and EPG5 (Ectopic p-granules autophagy protein 5 homolog)18 in Vici syndrome. Collectively, these observations suggest that intact autophagy is required for maintaining neuronal proteostasis and preventing neurodegeneration. The susceptibility of pathogenic proteins to autophagy and the neuroprotection provided by normal autophagy function, together with growing evidence demonstrating that autophagic degradation of these proteins rescues cellular toxicity19–21, implies that the autophagy pathway is a promising therapeutic target for the broad category of neurodegenerative disease. Despite this, however, autophagy modulators have largely failed to provide clinically meaningful benefit for those with neurodegenerative disorders22–24. Additionally, the use of currently available modulators of autophagy is limited by narrow therapeutic indices, dose-dependent toxicity, and wide-ranging adverse effects due to pleiotropic target engagement25,26.
The inherent resistance of neurons to methods of autophagy induction may also contribute to the inefficacy of autophagy stimulators in clinical trials. Starvation and mTOR inhibition are potent inducers of autophagy in most cell types but are largely ineffective in neurons, despite successful target engagement27,28. Negative results in clinical trials thus far may therefore relate to inadequate induction of neuronal autophagy, and alternative strategies for augmenting autophagy in neurons that overcome the prevailing barriers against autophagy-based therapies are sorely needed. Nevertheless, the critical mechanisms that (1) underlie the insensitivity of neurons to autophagy stimuli and (2) are amenable for therapeutic targeting have yet to be identified.
Here, we sought to uncover neuronal determinants governing autophagy and leading to the relative resistance of neurons to autophagy inducers. Using unbiased, genome-wide assessments of cell type-specific gene expression, we identified a selective enrichment of myotubularin-related phosphatase 5 (MTMR5) in iNeurons, or neurons derived from human induced pluripotent stem cells (iPSCs). MTMR5, also known as SET binding factor 1 (SBF1), belongs to a 14-member family of myotubularin-related phosphatases (MTMRs) that catalyze the removal of phosphate groups from the third and fifth positions on membrane phosphoinositides, including phosphatidylinositol-3-phosphate and phosphatidylinositol-3,5-bisphosphate (PtdIns3P and PtdIns(3,5)P2)29. Since these enzymatic events prevent recruitment of autophagy initiation complexes and reduce autophagy induction, MTMRs are considered autophagy suppressors30. Notably, MTMR5 is catalytically inactive30, and instead associates with its paralog and active phosphatase, MTMR2, as a heterodimer to regulate MTMR2’s localization and enhance its phosphatase activity31. Similar to MTMR232 and MTMR1333, MTMR5 is essential for maintaining peripheral nerves, and loss-of-function mutations lead to dysmyelination and a subtype of Charco-Marie-Tooth disease, CMT4B34. However, the precise role of MTMR5 in the central nervous system, and whether MTMR5 regulates autophagy to similar extents in different cell types, is unknown. In this study, our identification of MTMR5’s selective enrichment in neurons indicated a determinative role of MTMR5 in regulating neuronal autophagy. We therefore sought to manipulate MTMR5 expression and found that reductions in MTMR5 robustly enhanced neuronal autophagy, while neuron-like insensitivity to autophagy induction was recapitulated by MTMR5 overexpression in non-neuronal cells. In so doing, our results unravel a pivotal function of myotubularins in neuronal proteostasis and establish a novel target for potentiating autophagy in neurons for therapy design in neurodegenerative diseases.
RESULTS
Neurons are resistant to autophagy induction by Torin1.
We first assessed whether neurons generate autophagosomes to a similar degree as non-neuronal cell types. To monitor autophagosome biogenesis non-invasively, we used an iPSC line edited with CRISPR/Cas9 to tag endogenous LC3 with mEGFP at the N-terminus (Figure 1A). mEGFP-positive vesicles in this iPSC line represent autophagosomes, as indicated by the presence of immunocytochemical markers such as LC3, p62, and ATG5 (Figure S1A–C). We subsequently edited mEGFP-LC3 iPSCs further to allow doxycycline-inducible, rapid differentiation of nearly 100% efficiency35 into glutamatergic forebrain-like neurons using TALENS (iNeurons; Figure 1B–C), or a piggybac/transposase system for doxycycline-inducible differentiation into skeletal muscle36 (iMuscle; Figure 1D, S1D). Lastly, we differentiated mEGFP-LC3 iPSCs into astrocytes using dual-SMAD inhibition, as previously described37 (iAstrocytes; Figure 1C). Treatment with 250nM Torin1 for 4h produced large numbers of mEGFP-positive autophagosomes in iAstrocytes, iMuscle, and undifferentiated iPSCs, but significantly fewer autophagosomes in iNeurons (Figure 1E–G). Arguing against intrinsic resistance of iNeurons to Torin1, we observed reduced phosphorylation of mTOR pathway components in treated iNeurons, indicating successful Torin1-mediated inhibition of the mTOR pathway in these cells (Figure S2). These results together demonstrate that, compared to non-neuronal cells, neurons are less sensitive to the autophagy-inducing effects of the mTOR inhibitor Torin1.
Figure 1. Neurons are resistant to Torin1-mediated induction of autophagy.
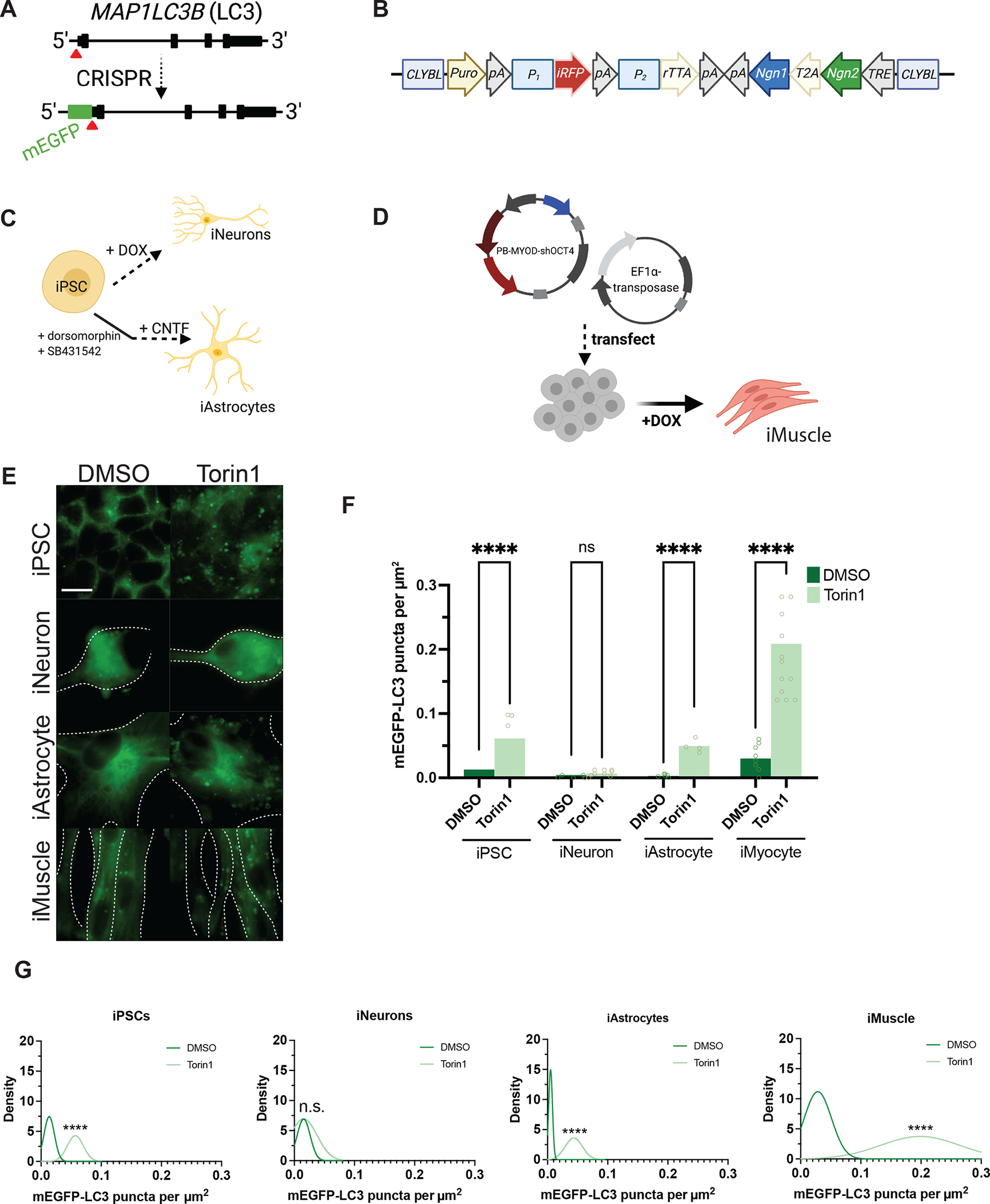
(A) Targeting strategy using CRISPR/Cas9 to knock-in mEGFP immediately 5’ to exon1 in the MAP1LC3B gene. (B) Schematic of the cassette used to integrate NGN1 and NGN2 at the CLYBL safe harbor locus under the control of a Tet-ON system. Puro, puromycin-resistance gene; pA, poly-A tail; P1, P2, promotors; iRFP, near-infrared fluorescent protein; rTTA, reverse tetracycline-controlled transactivator; NGN1 and NGN2, neurogenin-1 and -2; T2A, self-cleaving peptide; TRE, tetracycline response element. (C) Protocols used to differentiate iPSCs into iNeurons using doxycycline-mediated, forced expression of differentiation factors, or into iAstrocytes using dual-SMAD inhibition followed by terminal differentiation by culturing in CNTF. (D) Protocol for differentiating iPSCs into iMuscle using a piggybac/transposase system to integrate doxycycline-inducible MYOD and OCT4 shRNA. (E) Representative 100X images of mEGFP-LC3-positive vesicles in the cell types indicated after treatment with DMSO vehicle or 250nM Torin1 for 4h. Dotted lines indicate cell borders, within which cell area in μm2 was calculated using Fiji. (F) Scatterplots of blinded manual quantifications of mEGFP-LC3-positive vesicles imaged as in (E), normalized to cell area in μm2. Data are from three independent experiments. n.s., not significant; *p<0.05; ****p<0.0001; one-way ANOVA with Šídák’s multiple comparisons test. (G) Density plots of data from (F), which visualize the distribution of quantification data over a continuous interval with kernel smoothing to reduce noise and facilitate visualization of the distribution shape of data and peaks in which data are concentrated. n.s., not significant; ****p<0.0001, two-sample Kolmogorov-Smirnov test. See also Figure S1, Figure S2, Figure S5, and the STAR Methods.
MTMR5 expression is selectively enhanced in neurons.
To uncover differences across the human genome in the expression of autophagy-related genes that may account for the relative insensitivity of neurons to mTOR inhibition, we analyzed the transcriptome of iNeurons compared against isogenic iPSCs and the fibroblasts from which these iPSCs were reprogrammed. Steady-state measurements of mRNA transcripts via RNA-seq or PCR-based methods do not capture key aspects of gene expression such as rates of synthesis and turnover of RNA38. Therefore, we took advantage of Bru-seq and BruChase-seq to assess the relative synthesis and stabilities of RNA transcripts genome-wide. Briefly, these methods consist of the incorporation of bromouridine (BrU) into synthesizing RNA, followed with or without a chase using excess uridine. BrU-labeled transcripts are pulled down with anti-bromouridine antibodies, synthesized into cDNA, then analyzed by deep sequencing (Figure 2A)38.
Figure 2. RNA expression of autophagy factors in different cell types.
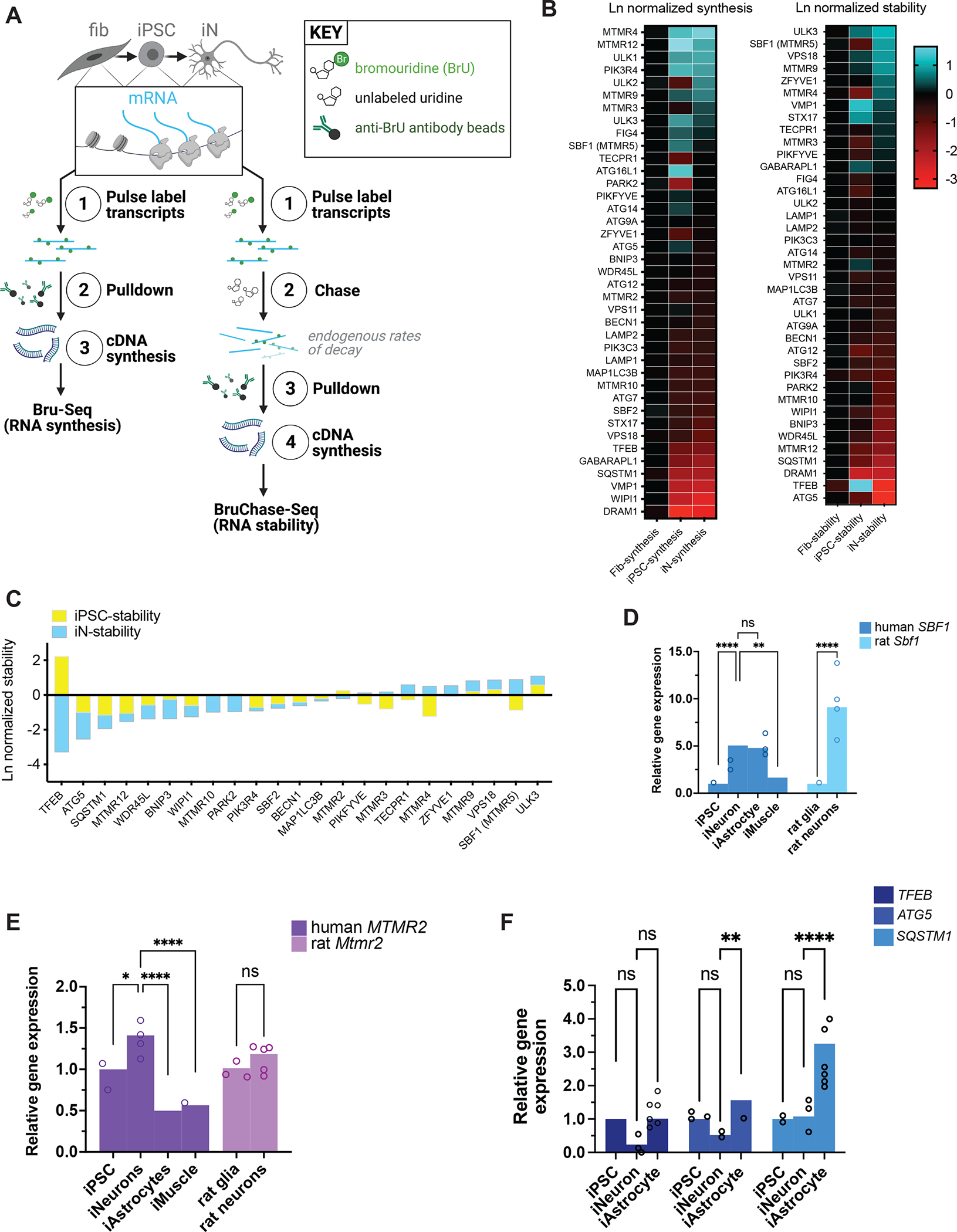
(A) Schematic of Bru-Seq and BruChase-Seq. RNA transcripts from fibroblasts, iPSCs reprogrammed from the fibroblasts, and iNeurons differentiated from the same iPSCs were pulse-labeled with bromouridine with and without chasing with unlabeled uridine, followed by pulldown with anti-bromouridine antibody beads, then subjected to RNA-Seq. (B) Heatmaps of RNA synthesis data from Bru-Seq (left) and RNA stability data from BruChase-Seq (right), plotted for each cell type and natural log (Ln)-normalized to fibroblasts. Fib, fibroblast; iPSC, induced pluripotent stem cell; iN, iNeuron. (C) Bar graph of RNA stability data from BruChase-Seq, Ln-normalized to fibroblasts. SBF1 was among the most stable RNA transcripts. (D) RT-PCR measurements of total steady-state human SBF1 or rat Sbf1 RNA in each cell type indicated. **p<0.01, ****p<0.0001, one-way ANOVA. (E) RT-PCR measurements of human MTMR2 or rat Mtmr2 RNA in the indicated cell types. ns, not significant; *p<0.05; ****p<0.0001, one-way ANOVA. (F) RT-PCR measurements of human TFEB, ATG5, or SQSTM1 RNA in each cell type indicated. ns, not significant; **p<0.01; ****p<0.0001, one-way ANOVA. See also Figure S5.
Among several candidate transcripts, SBF1—encoding the protein MTMR5—exhibited significantly greater stability in iNeurons (Figure 2B–C) compared to fibroblasts and undifferentiated iPSCs (p=0.0279 and p=0.0003, respectively; one-way ANOVA). SBF1 transcript synthesis rates were similar in iNeurons compared to iPSCs and fibroblasts (p=0.1242 and p=0.6363, respectively; one-way ANOVA), indicating that differences in SBF1 mRNA were exclusive to transcript stabilities among the different cell types. In contrast, neither the synthesis nor stability of MTMR2 mRNA in iNeurons (Figure 2B–C) was significantly different compared to those in fibroblasts (p=0.1040 and p=0.0846 for synthesis and stability differences, respectively; one-way ANOVA) or iPSCs (p=0.8170 and p=0.1209, respectively; one-way ANOVA). We then asked if the heightened stability of SBF1 RNA correlated with its steady state levels. Consistent with our Bru-seq data, measurements of SBF1 RNA by RT-PCR revealed a selective, nearly five-fold enrichment in iNeurons compared to their iPSCs of origin (Figure 2D). This expression pattern was conserved across species, as rat cortical neurons exhibited a similar, higher abundance of orthologous Sbf1 RNA compared to cortical glia (Figure 2D). When comparing against additional cell types, neuronal SBF1 was also more abundant compared to iMuscle SBF1, though similarly enriched in iAstrocytes (Figure 2D). For MTMR2, steady-state RNA levels were only slightly elevated in iNeurons and rat cortical neurons, but approximately 50% lower in iAstrocytes and iMuscle compared to undifferentiated iPSCs (Figure 2E).
In addition, our Bru-seq and RT-PCR analyses revealed that in iPSCs, the expression of TFEB, a master transcriptional regulator of autophagy and lysosomal genes39, and ATG5, a critical component of the ATG5-ATG12-ATG16L1 complex which regulates phagophore elongation2, trended toward slightly higher levels compared to iNeurons, but these were not significant (Figure 2B–C, 2F). iAstrocytes also express higher levels of TFEB and ATG5 (Figure 2F), but also SQSTM1, which encodes the autophagy adaptor p62/sequestosome-1 protein that recruits ubiquitinated substrates to autophagosomes40 (Figure 2F). Overall, iNeurons tended to exhibit lower expression of these three, autophagy-promoting factors.
Importantly, for SBF1, neuronal enrichment was not limited to RNA alone—immunoblotting also demonstrated higher levels of MTMR5 protein in iNeurons compared to iPSCs, iAstrocytes, and iMuscle (Figure 3A–B). Except for a selective enrichment in iMuscle, MTMR2 protein was not significantly different among the different cell types (Figure 3A–B). We also performed immunohistochemical staining of MTMR5 in human brain tissue, which revealed strong MTMR5 signal in neurons within the frontal cortex (Figure 3C) and within the deep cerebellar nuclei (Figure 3D, iii–v). Consistent with the putative localization of this protein41, the staining pattern of MTMR5 in frontal cortex was primarily cytoplasmic, prominent in both neuronal somata (Figure 3C, iv–v) and processes (Figure 3C, vi), and punctate or vesicular in morphology (Figure 3C, iv–v). In contrast, there was scant detection of MTMR5 in non-neuronal cells of the surrounding neuropil (Figure 3C). In the cerebellum, and similar to neurons of the frontal cortex, neurons of the deep cerebellar nuclei (Figure 3D, iii–v), stellate and basket cells of the molecular layer (Figure 3D, ii), and Golgi cells of the granular layer (Figure 3D, ii) showed marked MTMR5 staining compared to the surrounding neuropil (Figure 3D, v). Taken together, our findings from iPSC-derived cells and human brain tissue corroborate our Bru-seq and BruChase-seq analyses and validate a neuron-specific excess of the autophagy suppressor protein, MTMR5, due to enhanced stability of the SBF1 transcript.
Figure 3. MTMR5 protein is enriched in neurons.
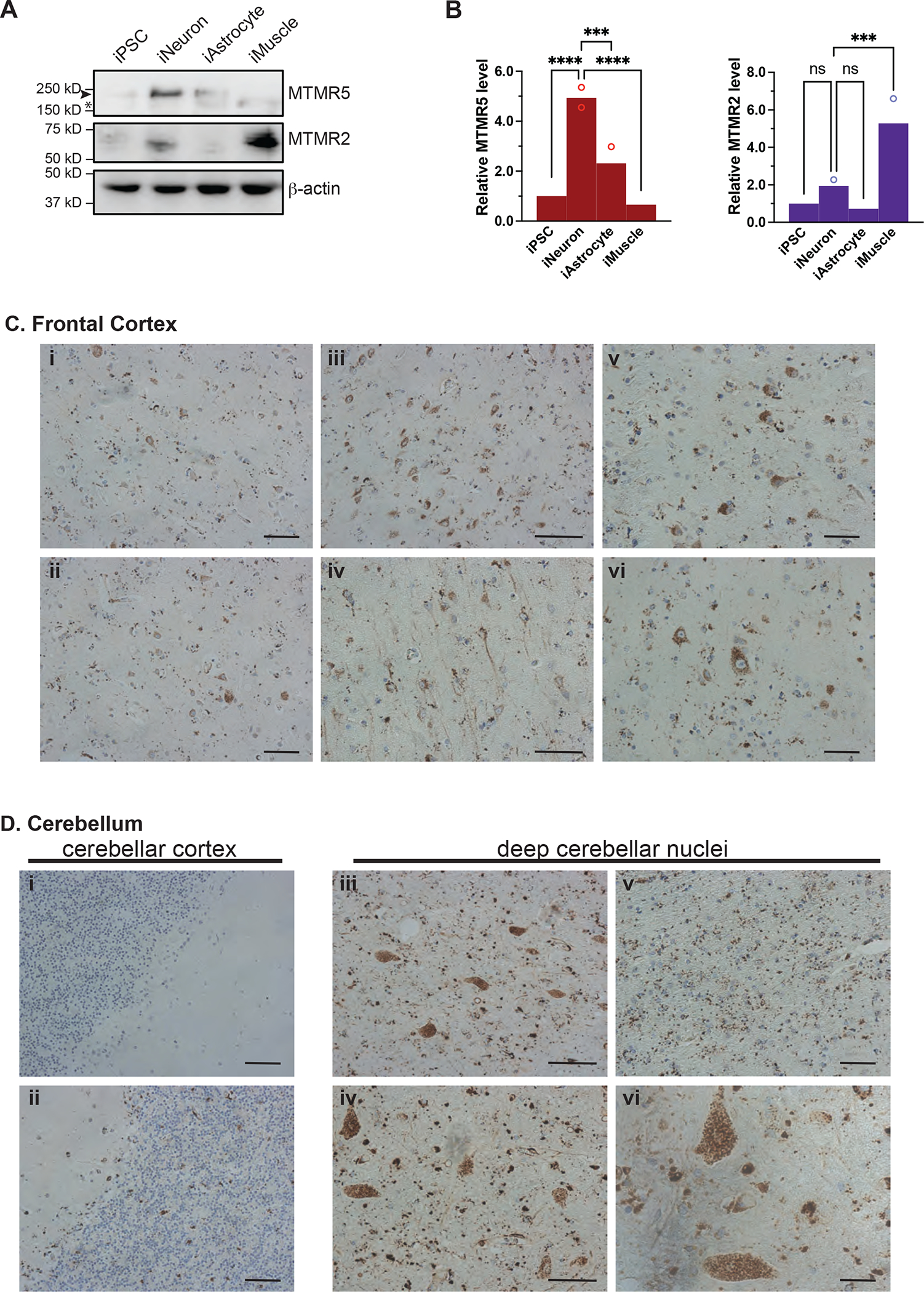
(A) Representative Western analysis of MTMR5, MTMR2, and actin loading control in each of the cell types indicated. Arrowhead, MTMR5, 208kD; asterisk, non-specific band. (B) Band intensity quantifications of MTMR5 and MTMR2 as depicted in (A), normalized to actin band intensity. Data are from three independent experiments. ns, not significant; ***p<0.001; ****p<0.0001, one-way ANOVA. (C) Immunohistochemical staining of MTMR5 in sections of human frontal cortex (i–iv, 10x magnification, scale bar 40μm; v–vi, 20x magnification, scale bar 20μm), and in (D) cerebellum (i–ii, cortex; iii–v deep cerebellar nuclear nuclei, 10x, scale bar 30μ; vi deep cerebellar nuclei, 20x; scale bar 10μm).
MTMR5 is sufficient for suppressing autophagy.
We next wondered if enhancing MTMR5 Expression leads to a neuron-like blunting of autophagy induction in non-neuronal cells. We chose to increase endogenous MTMR5 expression through CRISPR activation (CRISPRA), in which catalytically inactive Cas9 (dCas9) fused to transcriptional activators (VP64, MS2, p65, HSF1) is directed to a locus of interest by a single guide RNA (sgRNA) to enhance native gene expression (Figure 4A)42. Using the CRISPRA system and a sgRNA specific for the SBF1 locus, we increased expression of endogenous MTMR5 in undifferentiated iPSCs (Figure 4B). We then measured the effect of MTMR5 overexpression on autophagosome number after Torin1-mediated induction of autophagy. iPSCs expressing non-targeted sgRNA showed no change in autophagosome biogenesis after treatment with Torin1 (Figure 4C–E). However, cells expressing sgRNA targeting SBF1 (Figure 4C–E) demonstrated a significant attenuation of autophagosome induction after Torin1 treatment, similar to that observed in iNeurons (Figure 1E–G). These results confirm that MTMR5 is sufficient for restricting autophagosome biogenesis, as its overexpression in non-neuronal cells recapitulates the insensitivity of neurons to Torin1-mediated induction of autophagy.
Figure 4. MTMR5 is sufficient to desensitize iPSCs to Torin1 induction of autophagy.
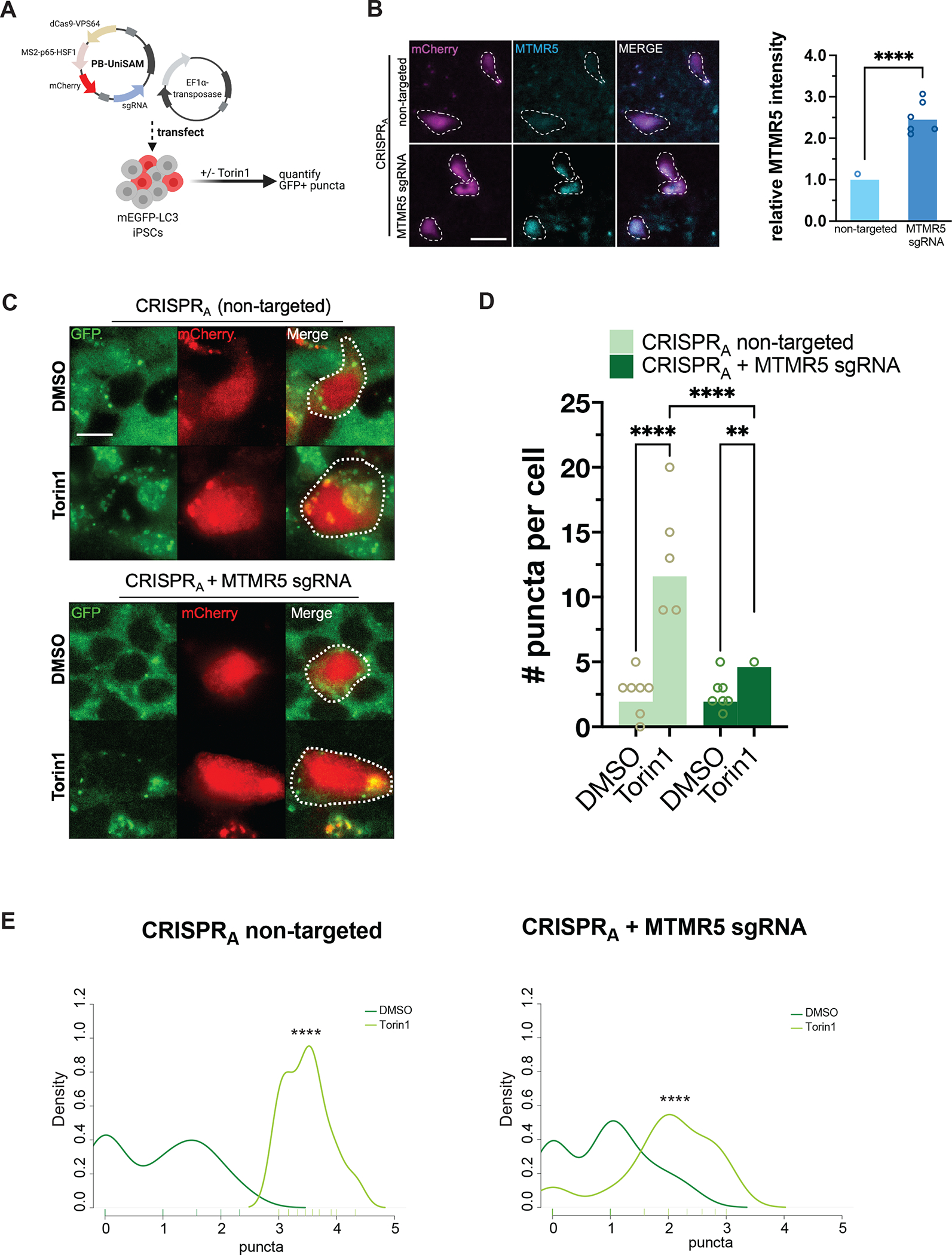
(A) Schematic of CRISPRA experimental workflow using piggybac/transposase system to express dCas9 fused with VPS64-MS2-p65 transactivators. Cells were treated with 250nM Torin1 or vehicle control for 4h then fixed, stained, and imaged. (B) Representative images of iPSCs transfected with CRISPRA vectors without a targeting sgRNA (top) or with sgRNA targeting the native SBF1 locus, followed by immunocytochemistry staining for MTMR5 to confirm protein overexpression. Cells harboring the piggybac/transposase vectors are indicated by the mCherry expression marker. Scale bar, 15μm. ****p<0.0001, Student’s t test. (C) Representative images of mEGFP-LC3-positive vesicles visualized in mCherry-positive cells after treatment with DMSO vehicle or Torin1. Scale bar, 10μm. (D) Scatterplot of blinded manual quantifications of mEGFP-LC3-positive vesicles imaged as in (C). Data are from three independent experiments. **p<0.01; ****p<0.0001, one-way ANOVA. (E) Density plots of data from (D). ****p<0.0001, two-sample Kolmogorov-Smirnov test.
MTMR5 is necessary for neuronal resistance to autophagy stimuli.
We next asked whether reducing MTMR5 levels augments autophagy induction in neurons. To address this, we knocked down MTMR5 by transducing mEGFP-LC3 iNeurons with shRNA directed against SBF1 or a non-targeted control (Figure 5A, top), and visualized mEGFP-LC3 puncta after application of Torin1. We did not observe any changes to autophagosome formation in iNeurons transduced with non-targeted shRNA (Figure 5B–D). However, knockdown of SBF1 in iNeurons significantly increased the number of mEGFP-LC3-positive puncta after Torin1 treatment (Figure 5C–D). Importantly, depleting pools of phosphoinositides with the PI3K inhibitor, VPS34-IN1 (Figure 5E–F), abrogated Torin1-induced biogenesis of autophagosomes in these cells, demonstrating that the potentiation of autophagy induction by MTMR5 knockdown is dependent on phosphoinositides.
Figure 5. MTMR5 is necessary for suppressing autophagy in neurons.
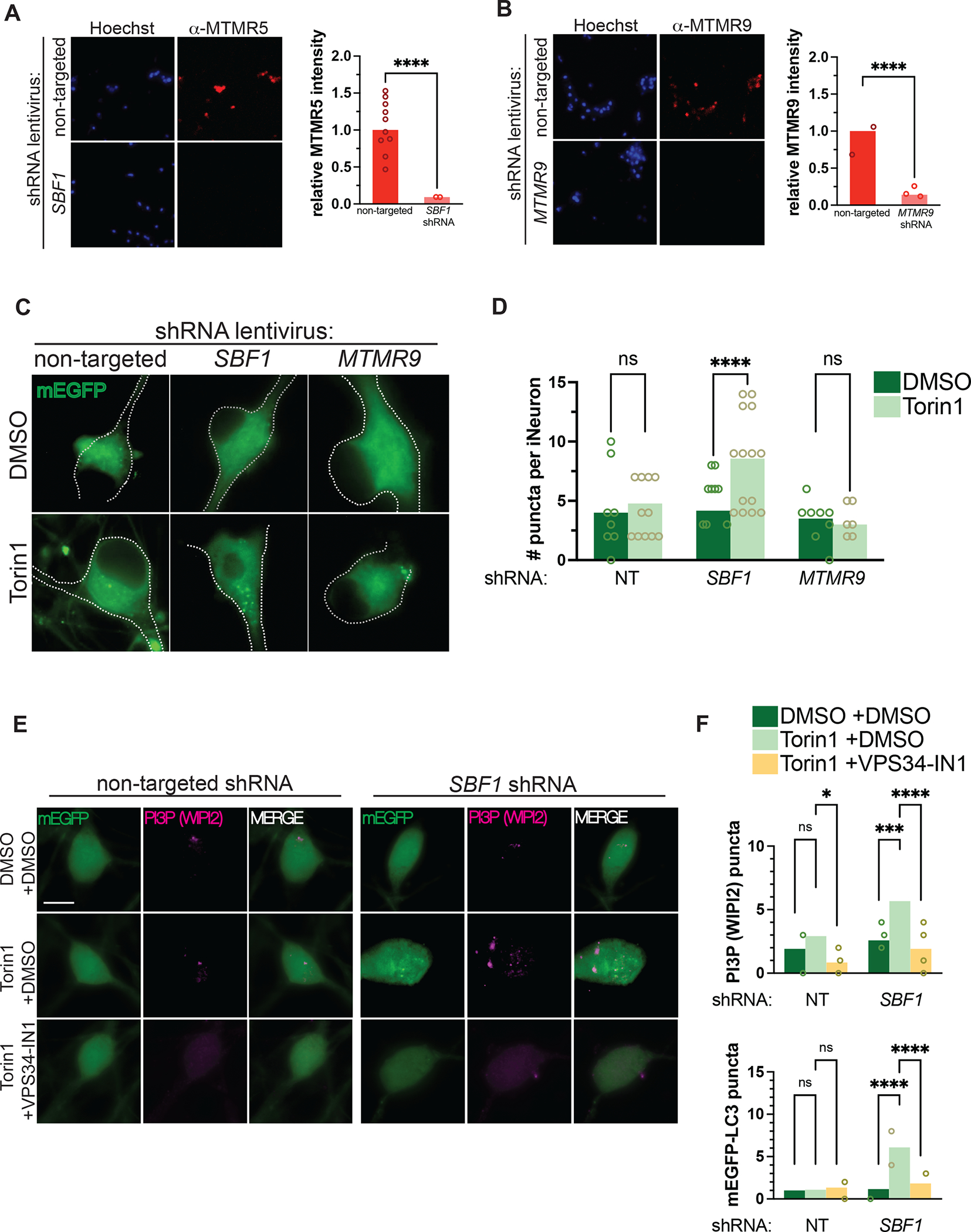
(A–B) Representative immunocytochemical staining against MTMR5 (A) and MTMR9 (B) in iNeurons transduced with non-targeted shRNA lentivirus (top rows), SBF1 shRNA (bottom left row), or MTMR9 shRNA lentivirus (bottom right row), respectively. (C) Representative images of DIV14 iNeurons transduced with non-targeted, SBF1, or MTMR9 shRNA and treated with DMSO vehicle or 250nM Torin1 for 4h. (D) Scatterplots of blinded manual quantifications of mEGFP-LC3-positive puncta imaged in iNeurons as treated in (C). Data are from 3 independent experiments. ns, not significant; ****p<0.0001, one-way ANOVA. (E) Representative images of mEGFP-LC3 iNeurons, transduced with non-targeted (left panels) or SBF1 shRNA lentivirus (right panels), and treated with or without Torin1 (240nM, 1 hour) and with or without VPS34-IN1 (10μM, 1 hour), a Class III PI3K inhibitor. Cells were fixed and immunostained for WIPI2 to visualize PtdIns3P (PI3P). (F) Quantifications of punctate WIPI2 immunostaining (top panel) or mEGFP-LC3 vesicles (bottom panel). ns, not significant; ****p<0.0001, one-way ANOVA. See also Figure S3 and Figure S4.
Prior studies report that MTMR5 associates with MTMR2 as a heterodimer complex31, a phenomenon that we confirmed using two-color direct stochastic optical reconstruction microscopy (dSTORM, Figure S3A)43. Given the physical interaction between MTMR5 and MTMR2, we next asked whether genetically depleting MTMR2 would abrogate the effects on autophagy observed after MTMR5 knockdown (Fig 5). Consistent with a functional interaction between the two myotubularin orthologs, MTMR2 knockdown also led to enhanced numbers of puncta after Torin1 treatment (Figure S4A–B). This enhancement also resulted in augmented autophagic flux after knockdown of SBF1 or MTMR2, as indicated by slight decreases in the levels of autophagy substrate p62 after Torin1 treatment (Figure S4C–D), and the greater accumulation of p62 after lysosomal inhibition with bafilomycin (Figure S4E–F). In contrast to SBF1 and MTMR2, MTMR9 knockdown failed to produce a similar induction of mEGFP-LC3 puncta in response to Torin1 (Figure 5B–D). These results indicate that MTMR5 is necessary for repression of autophagy induction in neurons, and the sensitization of neurons to autophagy stimuli is specific to the MTMR5-MTMR2 axis.
MTMR5 knockdown enhances degradation through autophagy.
Since the accumulation of mEGFP-LC3-positive autophagosomes could indicate autophagy induction or a late-stage block in the pathway, we next asked whether increases in mEGFP-LC3 number upon SBF1 knockdown are associated with a corresponding enhancement in autophagic degradation. To address this, we used CRIPSR/Cas9 to label the endogenous protein TDP-43 at the carboxy-terminus with the photoswitchable fluorophore, Dendra244 in iPSCs (Figure 6A). TDP-43 is not only a degradative substrate of autophagy45, but also integrally involved in the pathogenesis of amyotrophic lateral sclerosis, frontotemporal dementia, and other neurodegenerative proteinopathies46,47. Fusion with the Dendra2 allows for non-invasive tracking of endogenous TDP-43 protein levels in live cells via optical pulse labeling (OPL), a technique we previously used to track the turnover of overexpressed TDP-43 in primary neurons48. Briefly, in OPL we exploit the property of Dendra2 to irreversibly switch emission maxima from green to red wavelengths upon exposure to short-wavelength light49 (termed “photoconversion,” Figure 6B). After pulsing the total pool of TDP-43-Dendra2 at the beginning of an experiment, we then use automated fluorescence microscopy50 to track the pool of photoconverted “red” Dendra2 signal over time (Figure 6B).
Figure 6. Knockdown of the MTMR5-MTMR2 axis enhances the proteolytic clearance of TDP-43-Dendra2.
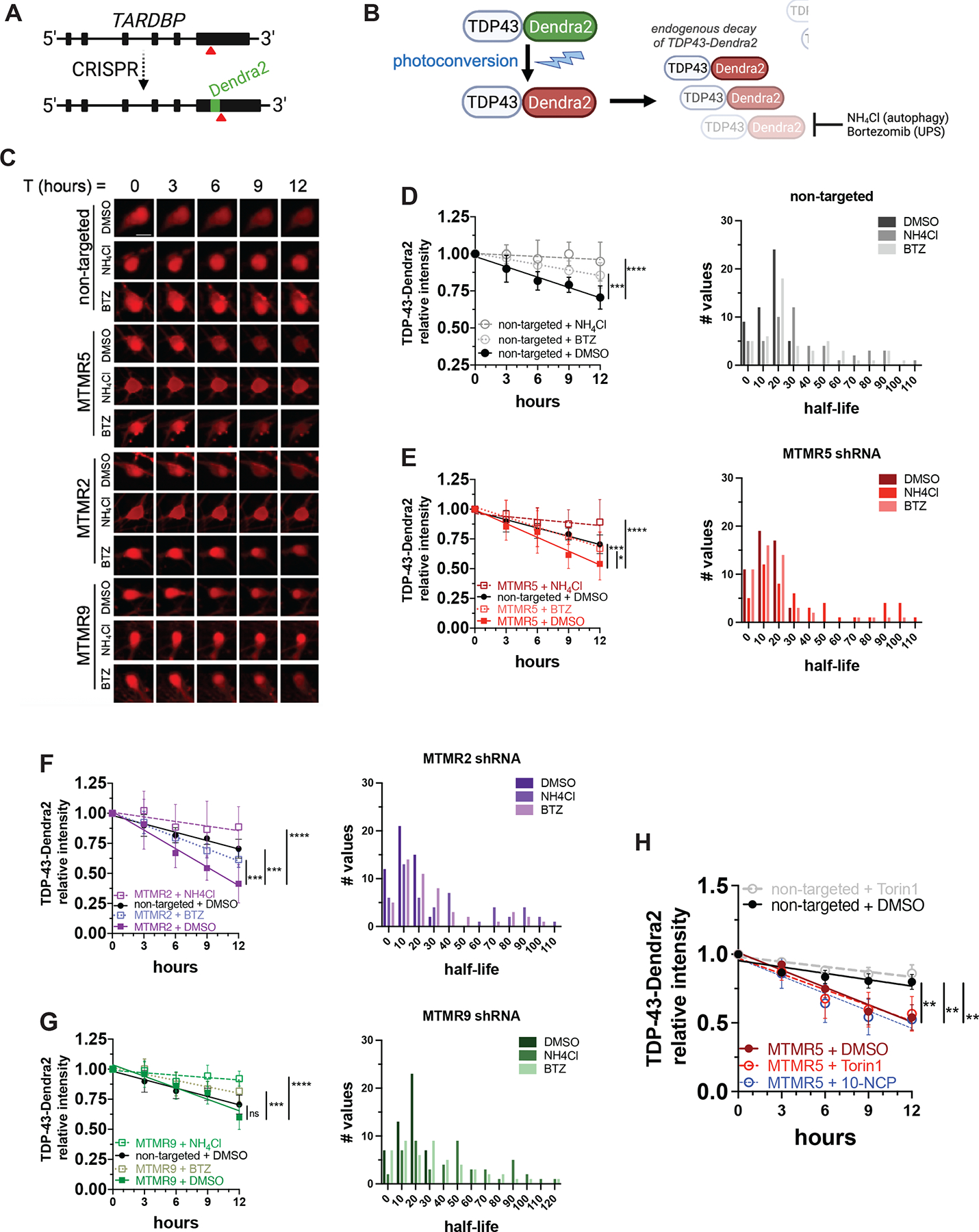
(A) Strategy to label endogenous TDP-43 by inserting Dendra2 immediately upstream of the TARDBP stop codon using CRISPR/Cas9. (B) Schematic of optical pulse labeling (OPL) to photoconvert Dendra2 emission maxima from green to red, followed by tracking red fluorescence decay to monitor TDP-43-Dendra2 degradation, which can be blocked with NH4Cl to inhibit autophagy or bortezomib to inhibit the ubiquitin-proteasome system (UPS). (C) Representative OPL of TDP-43-Dendra2 iNeurons imaged by automated fluorescence microscopy and after treatment with DMSO, NH4Cl, or bortezomib and transduction with the indicated shRNA lentiviruses; scale bar, 10μm. (D–G) TDP-43-Dendra2 fluorescence measured in iNeurons after knockdown of each indicated MTMR by transduction with non-targeted (D), SBF1 (E), MTMR2 (F), or MTMR9 (G) shRNA lentivirus and after the indicated drug treatments (left panels), and histogram plot of the half-lives of each measured iNeuron (right panels). Data are represented at each time point as mean ± SD; *p<0.05; ***p<0.001; ****p<0.0001, one-way ANCOVA. (H) TDP-43-Dendra2 fluorescence measured in iNeurons transduced with non-targeted or SBF1 shRNA and after the treatment with DMSO vehicle or autophagy inducing agents Torin1 or 10-NCP. Data are represented at each time point as mean ±SD; **p<0.01, one-way ANCOVA. See also Figure S3 and Figure S6.
After transduction with non-targeted shRNA lentivirus, we measured a baseline TDP-43-Dendra2 half-life of approximately 16h in iNeurons via OPL (Figure 6C, D). TDP-43-Dendra2 half-life was significantly prolonged by inhibiting autophagic flux through treatment with ammonium chloride, and to a lesser extent after proteasomal inhibition with bortezomib (Figure 6C, D). These observations suggest that TDP-43 undergoes constitutive proteolytic breakdown in iNeurons, primarily through autophagy but also to some degree through the ubiquitin-proteasome pathway. SBF1 knockdown significantly accelerated TDP-43-Dendra2 decay in iNeurons, resulting in an attenuated half-life of approximately 12h (Figure 6C, E). In the setting of SBF1 knockdown, treatment with either Torin1 or a neuron-specific inducer of autophagy, 10-NCP48, had no further effect on TDP-43-Dendra2 half-life (Figure 6H), suggesting that SBF1 knockdown achieved a maximal degree of autophagy stimulation. Consistent with autophagy-mediated degradation of TDP-43-Dendra2 upon SBF1 knockdown, the protein was effectively stabilized by ammonium chloride treatment (Figure 6C, E).
Given that MTMR5, a catalytically inactive myotubularin51, binds with the active phosphatase MTMR2 to enhance its activity31, we wondered whether MTMR2 serves as a functional effector of MTMR5’s suppressive effect, and if MTMR2 and SBF1 knockdown have similar effects on TDP-43-Dendra2 degradation. We performed OPL after transduction with MTMR2 shRNA and found that, indeed, MTMR2 knockdown also augmented the degradation of TDP-43-Dendra2 (Figure 6C, F), resulting in a half-life of about 10h. As before, this effect was blocked by ammonium chloride more so than bortezomib (Figure 6C, F). In contrast, and in line with our autophagosome quantification experiments (Figure 5C,D), MTMR9 knockdown did not significantly change TDP-43-Dendra2 turnover (Figure 6C, G), indicating that MTMR9 is dispensable for regulating the degradation of autophagy substrates in neurons. Together, these findings confirm that MTMR5 and MTMR2, but not MTMR9, are primary determinants of autophagy inhibition in iNeurons, and knockdown of either myotubularin increases neuronal degradation of autophagy substrates (Figure 7F).
Figure 7. Knockdown of the MTMR5-MTMR2 axis enhances the proteolytic clearance of Dendra2.
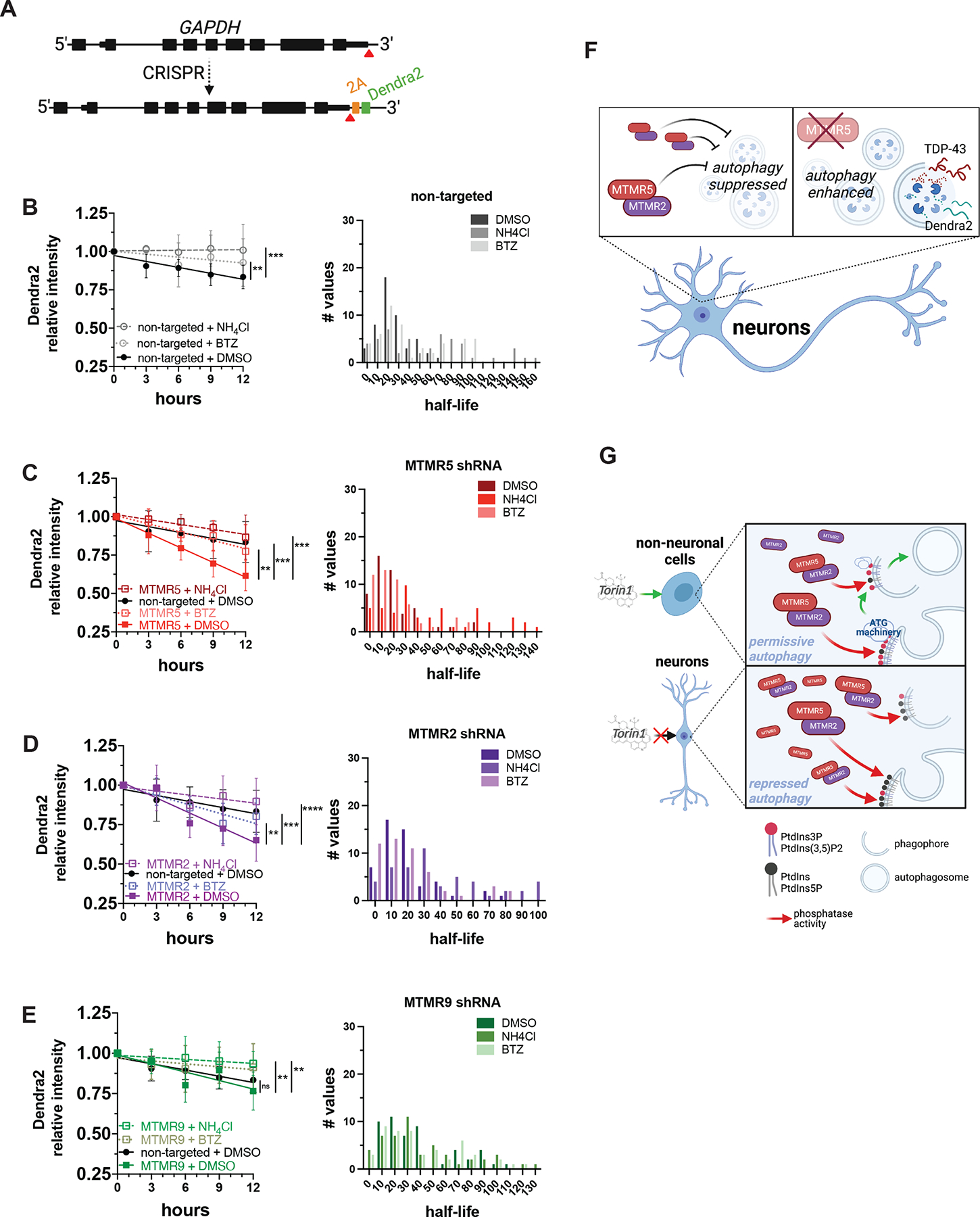
(A) Strategy for inserting Dendra2 at the native GAPDH locus using CRISPR/Cas9. 2A, self-cleaving peptide. (B–E) Dendra2 fluorescence measured in GAPDH-2A-Dendra2 iNeurons after knockdown of each indicated MTMR by transduction with non-targeted (B), SBF1 (C), MTMR2 (D), or MTMR9 (E) shRNA lentivirus and after the indicated drug treatments (left panels), and histogram plot of the half-lives of each measured iNeuron (right panels). Data are represented at each time point as mean ± SD; **p<0.01; ***p<0.001, one-way ANCOVA. (F) Schematic summary of MTMR5 influence on the proteolysis of autophagy substrates. In neurons, autophagy induction is suppressed by native levels of MTMR5 (left), but opposing MTMR5 activity (e.g., through shRNA-mediated knockdown) disinhibits autophagy and accelerates autophagic degradation of substrates, such as TDP-43 and Dendra2 (right). (G) Working model of cell type-specific regulation of autophagy. In non-neuronal cells (left), autophagy operates under permissive conditions due to relatively lower levels of MTMR5, enabling sufficient levels of PtdIns3P to recruit autophagy-related protein complexes (ATG machinery) necessary for autophagosome biogenesis and in response to stimuli (e.g., Torin1). However, in neurons (right), higher levels of MTMR5 impair autophagy induction by potentiating MTMR2, depleting PtdIns3P scaffolds necessary for assembling ATG machinery, and leading to a repressed state of autophagy induction (right panel). See also Figure S6.
We also wondered if the observed changes in protein turnover ascribed to MTMR2/5 knockdown are specific to TDP-43, or if these manipulations affect the autophagic degradation of other proteins. To resolve this, we created a separate iPSC line in which sequences encoding Dendra2 and a self-cleavable 2A peptide were inserted immediately 5’ to the GAPDH stop codon (Figure 7A). These cells, which we termed GAPDH-2A-Dendra2 iPSCs, express untagged Dendra2 that is diffusely distributed throughout the cell, as expected. As measured by OPL, the half-life of Dendra2 in iNeurons was approximately 25h (Figure 7B). As with TDP-43-Dendra2, treatment with ammonium chloride and bortezomib prolonged the half-life of Dendra2 (Figure 7B), implying combined autophagic and proteasomal clearance of Dendra2. Knockdown of MTMR5 accelerated Dendra2 turnover, reducing the half-life to approximately 13h (Figure 7C). The enhanced decay of Dendra2 was largely abolished with lysosomal inhibition, and less so with proteasomal inhibition (Figure 7C). Similar to the effects seen with TDP-43-Dendra2, clearance of Dendra2 in GAPDH-2A-Dendra2 iNeurons was also enhanced by knockdown of MTMR2 (Figure 7D), but not MTMR9 (Figure 7E).
Although MTMR5 knockdown was sufficient to accelerate the degradation of TDP-43-Dendra2 in glutamatergic iNeurons, this may not be the case for other neuronal subtypes. In addition to forebrain-like glutamatergic neurons, we differentiated iPSCs into motor neurons (iMotor Neurons) through doxycycline-induced expression of NGN2, LHX3, and ISL1, which were integrated at the CLYBL safe harbor locus35 (Figure S5A). These cells are cultured in different media and exhibit markers of cholinergic lower motor neurons (Figure S5B). Although iMotor Neurons harbor high levels of SBF1 (Figure S5C), they induce autophagosome formation to a greater extent than iNeurons upon Torin1 treatment (Figure S5D–E). Intriguingly and in line with expression patterns in iPSCs and iAstrocytes (Figure 2), iMotor Neurons express markedly higher levels of TFEB and ATG5 (Figure S5F). The abundance of these pro-autophagy factors may account in part for their differential sensitivity and response to Torin1 compared to iNeurons.
We then asked whether manipulating MTMR5 in iMotor Neurons similarly affected TDP-43-Dendra2 levels. Following transduction of iMotor Neurons with shRNA lentivirus targeting SBF1, we measured TDP-43-Dendra2 turnover by OPL. In cells transduced with non-targeted control, TDP-43-Dendra2 half-life was approximately 12h (Figure S6A), suggesting a baseline difference in TDP-43-Dendra2 clearance between iNeurons and iMotor Neurons. In contrast to what we observed in iNeurons, knockdown of SBF1, MTMR2, or MTMR9 in iMotor Neurons had little effect on TDP-43-Dendra2 decay rates (Figure S6B–D). Together with the relatively rapid turnover of TDP-43-Dendra2 in iMotor Neurons, these results suggest autophagic clearance of TDP-43-Dendra2 is not regulated by MTMR5 in iMotor Neurons, as occurs in iNeurons.
We also created GADPH-2A-Dendra2 iMotor Neurons to examine the turnover of Dendra2 alone by OPL in these cells. As with iNeurons, Dendra2 showed a half-life of about 24h in iMotor Neurons, and this was extended to similar degrees by ammonium chloride and bortezomib (Figure S7E). SBF1 and MTMR2 knockdown both accelerated Dendra2 turnover in iMotor Neurons (Figure S7F,G) while MTMR9 knockdown had little effect (Figure S7H), testifying to the specificity of MTMR2/5 in regulating protein turnover. These findings demonstrate not only proteolytic profiles unique to each individual degradative substrate of autophagy and specific for each neuronal subtype, but also cell type- and substrate-specific enhancement of autophagy after knocking down SBF1 or MTMR2.
DISCUSSION
In the present study, we identify a neuron-specific determinant of autophagy inhibition, MTMR5, the suppressive effects of which are dependent on MTMR2 (Figure 7, S4, S6). Not only is SBF1 mRNA stabilized in iNeurons compared to isogenic fibroblasts and iPSCs, but MTMR5 protein is also elevated in iNeurons and neurons in human brain tissue (Figure 3). Overexpression of MTMR5 recapitulates neuron-like resistance to autophagy inducers in iPSCs (Figure 4), while reduction of MTMR5 or MTMR2 leads to enhanced autophagosome biogenesis (Figure 5, S4), as well as accelerated clearance of two autophagy substrates, Dendra2-tagged TDP-43 and Dendra2 itself (Figure 6–7, S6). This effect is dependent on pools of phosphoinositides involved in autophagy initiation (Figure 5E–F). Accordingly, we propose a working model of regulating autophagy in neurons, in which non-neuronal cells exhibit a permissive state of autophagy induction by dint of low levels of MTMR5 and sufficient levels of phosphoinositides, but neurons are restrained from efficient autophagy induction machinery assembly due to an excess of MTMR5 (Figure 7G). Our results and working model highlight the MTMR5-MTMR2 axis as a novel target for selectively augmenting autophagy in neurons and enhancing the efficacy of autophagy-based therapies for neurodegenerative diseases.
Our results also illustrate different profiles of autophagic activity in distinct cell types, correlating with each cell’s unique complement of autophagy-related factors. For instance, undifferentiated iPSCs exhibit low levels of SBF1/MTMR5, and higher levels of the autophagy-promoting factors and ATG5 (Figure 2,3), in keeping with the relative sensitivity of iPSCs to Torin1 (Figure 1E–G). Interestingly, and to our surprise, iAstrocytes robustly generate autophagosomes after Torin1 treatment (Figure 1E–G) despite expressing a similar level of SBF1 mRNA as iNeurons (Figure 2E) and are moderately enriched with MTMR5 protein (Figure 3A–B). One possible explanation for this observation is that iAstrocytes express nearly undetectable levels of MTMR2 (Figure 3A), but also higher levels of TFEB, ATG5, and SQSTM1 (Figure 2H). Thus, the combination of multiple, abundant autophagy-promoting factors, together with a relative paucity of MTMR2, may counteract the inhibitory influence of SBF1/MTMR5 in glia, though the necessity of this balance of autophagy factors for preventing neuron-like resistance to Torin1 remains to be determined. In contrast, iMuscle cells express the highest levels of MTMR2 (Figure 3A–B), yet are robustly responsive to Torin1 (Figure 1E–G). Although MTMR2 is the nominal effector arm of the MTMR5-MTMR2 axis as both an active phosphatase and autophagy suppressor, we found marked asymmetry in MTMR5 and MTMR2 expression due to iMuscle harboring minimal levels of MTMR5 protein (Figure 3A–B). This suggests that low MTMR5 has a greater determinative and permissive influence on autophagy induction in Torin1-sensitive iMuscle cells. Even among neurons, in our immunohistochemical analyses of human brain tissue, MTMR5 demonstrated marked variability among different neuronal subtypes in the cerebellum (Figure 3D). In contrast to neurons in the deep cerebellar nuclei, Purkinje neurons and granule neurons had minimal to no staining for MTMR5 (Figure 3D, i–ii). Altogether, our observations suggest that modifying therapy strategies to provide optimal autophagy modulation in different neurodegenerative diseases, each exhibiting predilections for divergent subpopulations of neurons, glia, and muscle, may require consideration of the relative stoichiometries of autophagy-promoting and -suppressive factors, both relative to MTMR5 and unique to each cell type.
Separately, even within each cell type, we found evidence of differential handling of TDP-43 and Dendra2 by the autophagy pathway as degradative substrates (Figure 6–7, S6), showing approximate half-lives of 16h and 24h, respectively, at baseline, and 12h and 13h, respectively, after MTMR5 knockdown in iNeurons. Corresponding baseline half-lives for TDP-43 and Dendra2 in iMotor Neurons were 12h (Figure S6A) and 24h (Figure S7E), respectively, and the rate of decay for Dendra2, but not TDP-43 (Figure S6B–C), was augmented by MTMR2/5 knockdown (Figure S7F–G). These observations hint at substrate-specific differences in mechanisms for cargo recognition and targeting to the autophagosome-lysosome system, including substrate-specific ligases and rates for ubiquitination52,53 modifying access to ubiquitin-to-LC3 adaptors in selective autophagy54, varying degrees of post-translational modifications on substrates55 and autophagy receptors56,57 modifying proteolytic susceptibility, and substrate-specific autophagy machinery required for efficient protein clearance58. Based on the specialized function of TDP-43 in autoregulating its RNA expression59, greater autophagic degradation of TDP-43 could affect total steady-state levels, but this was not captured by our OPL studies which only tracked a single pool of TDP-43-Dendra2 in each cell after photoconversion. Overall, the extent to which these biological aspects apply to TDP-43 or Dendra2 in cell types from our iPSC-based model system, and whether and how these mechanisms contrast among each cell type and for other protein substrates, remains to be determined.
Maintaining intact, robust autophagy appears to be necessary for skeletal muscle health. For instance, pharmacologically or genetically abrogating autophagy leads to numerous skeletal muscle pathologies. After autophagy inhibition with 3-methyladenine, skeletal muscle in mice fails to recover and regrow following cardiotoxin injections60. Blocking autophagy through global knock-out of ulk1, an essential component of autophagy initiation machinery, constrains skeletal muscle regrowth after myectomy in zebrafish61. Conditional knock-out of Atg562 or Atg763 in mouse skeletal muscle leads to disruption of ultrastructural organization, accumulation of ubiquitinated inclusions, and dysfunctional mitochondria and sarcoplasmic reticulum. Thus, the abundance of pro-autophagy factors we noted in iMuscle may be necessary for promoting intact and productive autophagy, itself required for maintaining skeletal muscle proteostasis, organelle function, and structural integrity.
Separate from glutamatergic neurons, glia, and skeletal muscle, the regulatory mechanisms intrinsic to motor neurons deserve special mention. Not only are hallmarks of impaired autophagy such as ubiquitinated and p62-positive inclusions64 apparent in motor neuron disease, but also genetically attenuating SBF1 and MTMR2 has little effect in iMotor Neurons as compared to glutamatergic iNeurons (Figure S6). As a potential counter to the high expression (Figure S5C) and suppressive influence of SBF1, iMotor Neurons express the highest levels of TFEB among the cell types analyzed, as well as higher levels of ATG5 compared to iNeurons (Figure S5F). The enhancing effects of these autophagy-promoting factors are reflected by shorter half-lives for TDP-43-Dendra2 and Dendra2, neither of which were further accelerated by SBF1 or MTMR2 knockdown (Figure 7). Other components of the autophagy machinery in motor neurons have a profound influence on proteostasis and neuroprotection, as demonstrated by Atg765 and Tbk166 conditional deletion in motor neurons leading to earlier neurodegeneration in transgenic mouse models of amyotrophic lateral sclerosis. Interestingly, attenuated autophagy in these studies limited glial inflammation and extended lifespan65,66, emphasizing the interplay of cell and non-cell autonomous effects of disrupting autophagy on disease pathogenesis. These effects may also be species-specific, given that murine embryonic stem cell (ESC)-derived cranial motor neurons harbor greater proteasomal capacity and resistance to protein aggregation-related toxicity in comparison to ESC-derived spinal motor neurons67. Regardless, our results not only suggest that human iMotor Neurons operate at a higher basal rate of autophagic flux, but also that individual neuronal subtypes may be unique in their individual complement68 of activating factors and, in turn, intrinsic rates of autophagic activity. Such differences may explain discrepant reports in the literature regarding the extent to which neurons respond to autophagy stimuli69, not just in cell and tissue samples containing multiple subtypes of neurons, but also those containing heterogenous populations of non-neuronal cells.
One function of myotubularins is to modulate steady-state pools of phosphoinositides by reducing PtdIns3P and PtdIns(3,5)P241. Our results imply that targeting MTMR5 is a promising strategy for amplifying autophagic degradation of misfolded proteins in neurodegenerative disorders. However, additional aspects of MTMR5 function and the nature of de-repressed autophagy after MTMR5 knockdown require further clarification. First, although morphologic markers of autophagy induction or flux inhibition are not apparent after MTMR5 knockdown with vehicle treatment (Figure 5, S4), nevertheless there is evidence of enhanced decay of autophagy substrates (Figure 6–7, S6) that is not further enhanced with Torin1 or 10-NCP (Figure 6F). One possible explanation for this apparent discrepancy is that morphologically visible markers of autophagy (e.g., epifluorescence monitoring of LC3 and p62) only reflect steady-state levels of those proteins and do not fully capture nor correlate with the kinetics of other aspects of autophagic flux, such as substrate loading, assembly of autophagosome machinery, and synthesis of autophagy effectors70. To this end and consistent with prior studies48,68,71, monitoring the proteolysis of fluorescent autophagy substrates, including at multiple early time-points and with OPL, is a more sensitive measure of autophagic flux than single time-point quantifications of autophagy markers alone. Adding to the biological complexity of autophagy in different cell types is the asymmetric subcellular distribution of different pathway intermediates in neurons. Previous investigations suggested that autophagosome biogenesis occurs distally in axons, with lysosomal fusion occurring in proximal segments and the soma72. Because puncta within axon terminals were challenging to consistently locate due to random process length and orientation of axons in cultured iNeurons, we focused primarily on LC3-positive puncta distributed within neuronal somata. Although intrasoma autophagosomes likely represent mature proteolytic structures, additional revelatory details about neuronal autophagy may have been missed without evaluating distal compartments. Future studies will require more delineation of whether and how autophagy in neuronal processes differs after manipulating the MTMR2/5 axis72.
Second, given the presence of additional protein-protein interaction motifs in MTMR5, including PH (plekstrin-homology), DENN (differentially expressed in normal and neoplastic cells), and SID (SET-interacting domain) motifs, and other documented MTMR5 binding partners including multiple Rab GTPases73,74 and KMT2A/HRX75, it remains a possibility that MTMR5 modulates autophagy through additional or alternative mechanisms than phosphoinositide metabolism, such as membrane trafficking73 or transcriptional and epigenetic regulation76. Third, it remains unknown if targeting MTMR5 is the optimal strategy for rescuing protein dyshomeostasis in the central nervous system while avoiding peripheral and age-dependent requirements of intact MTMR5. Ideally, newly developed therapies would not reduce peripheral MTMR5 expression, since loss of testicular Sbf1 in mice leads to infertility77, and dysfunction in Schwann cell MTMR5 leads to CMT4B334, a dysmyelinating inherited polyneuropathy. Fourth, the mechanisms by which the SBF1 transcript and MTMR5 protein are stabilized in iNeurons remain undefined. Possibilities include hypomethylation of neuronal SBF1 or neuronal enrichment of enhancers with cognate binding sites in the SBF1 promoter region, including BRD3, NOTCH1, and TCF1278. However, our Bru-Seq data show that synthesis of SBF1 transcripts is similar between iNeurons and iPSCs (Figure 2B, p=0.1242), suggesting that heightened SBF1 stability (Figure 2B–E) may instead be due to neuron-specific attenuation of SBF1 decay. Lastly, what are the teleologic underpinnings of MTMR5 expression and restricted autophagy in neurons? It is possible that limiting excess autophagy induction in neurons affords an evolutionary advantage by preventing depletion of essential cytoplasmic constituents or exacerbating inefficient cargo capture that outstrips the degradative capacity of the lysosomal compartment in neurons79, though these possibilities require further investigation. Nonetheless, our results establish a critical role of MTMR5 in neuronal autophagy and identify a novel and promising mechanistic target for ameliorating proteostatic deficits in neurodegenerative diseases.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources should be directed to the Lead Contact, Jason Chua (jchua6@jhmi.edu).
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and code availability
Bru-seq/BruChase-seq, RT-PCR, Western blot, and microscopy data reported in this paper will be shared by the Lead Contact upon request without restriction. The 2-D dSTORM dataset is available at https://spaces.hightail.com/space/mTwffdRoyB.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
mEGFP-LC3 human induced pluripotent stem cell lines.
Human iPSCs reprogrammed from skin fibroblasts obtained from a healthy adult Asian male (AICS-0030-022) were engineered by the Allen Institute for Cell Science (www.allencell.org) to express fluorescently labeled LC3 using CRISPR/Cas9 to insert mEGFP at the N-terminal exon of the MAP1LC3B gene. Cells were maintained in TeSR-E8 (STEMCELL Technologies) with daily media changes. All studies using this cell line were performed in compliance with approved protocols through the University of Michigan Human Pluripotent Stem Cell Research Oversight Committee (HPSCRO).
TDP-43-Dendra2 human iPSCs.
TDP-43-Dendra2 iPS cells were engineered after reprogramming fibroblasts from a normal healthy adult male (56 years old; CRL-2465, ATCC), as previously described44,48. Cells were maintained in TeSR-E8 with daily media changes. All studies using this cell line were performed in compliance with approved protocols through the University of Michigan Human Pluripotent Stem Cell Research Oversight Committee (HPSCRO).
GAPDH-2A-Dendra2 human iPSCs.
GAPDH-2A-Dendra2 iPS cells were engineered from the same CRL-2465 line (ATCC) as the TDP-43-Dendra2 iPSC line. Cells were maintained in TeSR-E8 with daily media changes. All studies using this cell line were performed in compliance with approved protocols through the University of Michigan Human Pluripotent Stem Cell Research Oversight Committee (HPSCRO).
Rat cortical neurons and cortical glia.
Primary culture cells were maintained with media changes every 2 days. Cells were passaged with 0.25% Trypsin-EDTA, re-plated in new PDL-coated flasks, maintained in culture for 12–14 days.
Rodents.
Rats (Rattus norvegicus) used for primary neuron collection were housed singly in chambers equipped with environmental enrichment. All studies were designed to minimize animal use. Rats were cared for by the Unit for Laboratory Animal Medicine at the University of Michigan; all individuals were trained and approved in the care and long-term maintenance of rodent colonies, in accordance with the NIH-supported Guide for the Care and Use of Laboratory Animals (National Academies Press, 2011). All personnel handling the rats and administering euthanasia were properly trained in accordance with the University of Michigan Policy for Education and Training of Animal Care and Use Personnel. Euthanasia was performed according to the recommendations of the Guidelines on Euthanasia of the American Veterinary Medical Association. All vertebrate animal work was approved by the Committee on the Use and Care of Animals (UCUCA) at the University of Michigan and in accordance with the United Kingdom Animals Act (1986). All experiments were performed in accordance with UCUCA guidelines.
METHOD DETAILS
Plasmids for genomic editing.
To engineer mEGFP-LC3 iPSCs capable of rapid differentiation into iNeurons and iMotor Neurons, TALENs vectors targeting the CLYBL safe harbor locus were obtained from Addgene (#62196 and #62197), and HDR vectors encoding doxycycline-inducible transcription factors NGN1/2 (pUCM-CLYBL-NGN1&2) or LHX3/ISL1/NGN2 (pUCM-CLYBL-hNIL) for knock-in at CLYBL were kindly provided by Michael Ward (NINDS). For rapid differentiation into iMuscle, piggy-bac/transposon vectors encoding doxycycline-inducible MYOD1/shRNA-OCT4 (PB-MYOD-shOCT4) were kindly provided by Michael Ward (NINDS). To generate TDP-43-Dendra2 iPSCs, oligonucleotides encoding sgRNA targeting sequences flanking the stop codon of the endogenous TARDBP gene were annealed, digested, and ligated into the BbsI site in pX335-U6-Chimeric_BB-CBh-hSpCas9n(D10A) (Addgene #42335). For generating GAPDH-2A-Dendra2 iPSCs, oligonucleotides encoding sgRNA targeting sequences flanking the stop codon of the endogenous GAPDH gene were also annealed, digested, and ligated into the BbsI site in into pX335-U6-Chimeric_BB-CBh-hSpCas9n(D10A). HDR vectors encoding the Dendra2 open reading frame (ORF) flanked by 400bp of sequence homologous to sequences flanking the endogenous TARDBP stop codon, or the Dendra2 ORF and 400-bp homology sequences flanking the GAPDH stop codon, were synthesized in pUC-minus(MCS) by Blue Heron, LLC.
Harvesting of primary culture cells.
Rat cortical neurons and cortical glia were dissected from E20-E21 rat pups, fractionated by centrifugation, and plated on poly-D-lysine (PDL)-coated flasks at a density of ~30–50 × 106 cells per flask. After DIV 7–8, glial flasks were shaken with a benchtop cell agitator at 180rpm × 30min to detach and remove microglia, then 240rpm for 6 hours and washed twice with PBS to detach and remove oligodendrocyte precursors. After DIV 12–14, cells were collected for RNA and protein analyses.
Transfection.
Prior to transfection, iPSCs were split with EDTA and seeded onto vitronectin-coated plates. On the day of transfection, cells were switched from TeSR E8 (StemCell Technologies 05990) to mTeSR 1 (StemCell Technologies 85850), and total DNA amounts of 5μg (2.5μg donor vector plus 1.25μg of each TALENS arm plasmid) or 2μg (1μg piggybac vector plus 1μg of transposase plasmid) were combined with Lipofectamine Stem (ThermoFisher, STEM00003) in Opti-MEM (ThermoFisher 11058021), then added dropwise to iPSCs and incubated overnight, followed by media change to TeSR-E8 the next morning. Cells with fluorescent selection markers (iRFP for NGN1/2 iPSCs or Dendra2 for TDP-43-Dendra2 and GAPDH-2A-Dendra2 iPSCs) were manually selected and passaged to generate iPSC colonies with 100% of cells expressing the fluorescent selection marker.
Differentiation protocols.
Rapid differentiation was performed as previously described6. TDP-43-Dendra2 was previously engineered for rapid differentiation by integrating using TALENs. GAPDH-2A-Dendra2 iPSC lines were previously engineered. On DIV 0 iPSCs were split with Accutase and plated on PEI-coated slides for live cell imaging or Matrigel-coated plates for RNA and protein analyses. For iNeurons, on DIV 1–2 media were changed daily from N2 medium (TesR-E8) to transition medium (half TesR-E8, half DMEM/F12), with both media supplemented with doxycycline, N2, NEAA, BDNF, NT3, and laminin. On DIV 3, media were changed to B27 medium (Neurobasal-A supplemented with doxycycline, B27, Glutamax, BDNF, NT3, laminin, and Culture One). From DIV 6–14, equal volumes of B27 medium without Culture One were added every 4 days to each well. For iMotor Neurons, media were changed on DIV 1 to D1 medium (TesR-E8 supplemented with doxycycline, N2, and Compound E) and DIV 3 to D3 medium (DMEM/F12 supplemented with doxycycline, N2, NEAA, Glutamax, and Compound E). On DIV 6, media were changed to B27 medium with Culture One. From DIV 10–14, equal volumes of B27 medium without Culture One were added every 4 days to each well. For iAstrocytes37, iPSCs were differentiated to neuroectoderm by dual-SMAD signaling inhibition in 3N medium (half Neurobasal-A, half DMEM/F12 supplemented with SB431542, dorsomorphin, N2, B27, NEAA, Glutamax, hr-insulin, and pen/strep) for 2–4 weeks. Following this, neurospheres were formed and mechanically chopped, passaged, and cultured in EL20 medium (half Neurobasal-A, half DMEM/F12 supplemented with EGF, LIF, N2, B27, NEAA, Glutamax, and pen/strep) for 2–4 weeks. Cells were then passaged and maintained as monolayers of astrocyte precursors in EF20 medium (half Neurobasal-A, half DMEM/F12 supplemented with EGF, FGF2, N2, B27, NEAA, and Glutamax) for 2–4 weeks. Finally, astrocyte precursors were differentiated to mature astrocytes with AstroMED medium (Neurobasal-A supplemented with CNTF, B27, NEAA, Glutamax, and pen/strep) for 4 weeks. For iMuscle, iPSCs were cultured in myogenic progenitor media (MEMa supplemented with sodium pyruvate, non-essential amino acids, Glutamax, 2-mercaptoethanol, doxycycline, and 5% knock-out serum replacement) for 2 days, followed by myogenic induction medium (DMEM supplemented with Glutamax, IGF-1, 2-mercaptoethanol, non-essential amino acids, doxycycline, and 5% knock-out serum replacement) for an additional 5 days.
Lentiviral transduction.
TRC Lentiviral shRNA plasmids against human SBF1 (Horizon RHS3979-201739167), MTMR2 (Horizon RHS3979-224867040), MTMR9 (Horizon RHS3979-201908246), TFEB (Horizon RHS3979-201744686), ATG5 (Horizon RHS3979-201857832), SQSTM1 (Horizon RHS3979-201739507), or non-targeted control (Horizon RHS6848) were produced through the University of Michigan Vector Core. On DIV7, half the working volume of B27 medium was removed from iNeuron and iMotor Neuron cultures and saved at 4° C, or TeSR E8 from undifferentiated iPSCs, and replaced with equivalent volumes of lentiviral lysate. The following day, the saved conditioned B27 media was re-warmed at room temperature and added back to iNeuron and iMotor Neuron cultures. The remaining seven days of differentiation were continued unchanged and as described above, and cells were imaged on DIV14. For iPSCs, fresh TeSR-E8 was added the following day after addition of lentiviral lysates, and daily media changes continued for 6 more days before treatment with Torin1 and imaging.
Bru-seq and BruChase-seq.
RNA labeled with bromouridine (BrU) was prepared and analyzed as previously described80. Briefly, BrU-labeled RNA was extracted with anti-BrU antibodies from total RNA samples from healthy adult fibroblasts, isogenic iPSCs reprogrammed from these fibroblasts, and isogenic iNeurons differentiated from these iPSCs. Strand-specific DNA libraries were prepared with Illumina TruSeq Kit (Illumina) and sequenced using the Illumina sequencing platform24. Strand-specific sequenced data was aligned to human ribosomal DNA complete repeating unit (U13369.1) using Bowtie (v0.12.8). Remaining unaligned reads were then mapped to the human genome build hg19/GRCh37 using TopHat (v1.4.1)24. Bru-seq data from iPSCs and iNeurons were compared to fibroblasts and fold differences quantified using DESeq (version 1.4.1) in R (version 2.15.1). Genes having a mean RPKM ≥ 0.5, length ≥ 300 bp, false discovery rate (FDR) ≤ 0.1 and a 1.5-fold change were included for downstream bioinformatics analyses. For BruChase-seq, a stability index for each transcript was calculated as a ratio of transcript abundance at 6 hours vs. 0.5 hours, and median values across replicates of the same condition were used. Genes showing greater or less than 1.5-fold change in the stability index were included in final dataset.
Live cell imaging and optical pulse labeling.
Cells were incubated in maintenance medium for each respective cell type supplemented with DMSO vehicle or 250nM Torin1 for four hours. For autophagosome puncta quantification, live cells were imaged at 100X using ONI Nanoimager (www.oni.bio), and images were processed and analyzed using Fiji. For optical pulse labeling, an automated microscopy platform was used as previously described48. Briefly, images were obtained at the indicated time points with a Nikon TE2000 microscope equipped with the PerfectFocus system, a high-numerical aperture 20X objective lens and a 16-bit Andor Clara digital camera with a cooled charge-coupled device. Illumination was provided by a Lambda XL Xenon lamp (Sutter) with a liquid light guide. The ASI 2000 stage was controlled by rotary encoders in all three planes of movement. All components were housed in a custom-designed, climate-controlled environmental chamber (InVivo Scientific) kept at 37° C and 5% CO2. The Semrock BrightLine full-multiband filter set (DAPI, FITC, TRITC, Cy5) was used for fluorophore photoactivation (DAPI), excitation and detection (FITC, TRITC). The illumination, filter wheels, focusing, stage movements and image acquisitions were fully automated and coordinated with a mix of proprietary (ImagePro) and publicly available (μManager) software.
RT-PCR.
RNA was isolated from cell pellets using the RNeasy Mini Kit (QIAgen). cDNA was reverse transcribed from 1μg RNA with the iScript kit (Bio-Rad). Quantitative PCR was then performed on 0.5 μl of cDNA using Power SYBR Green (Applied Biosystems) with primers complementary to human SBF1, MTMR2, TFEB, ATG5, SQSTM1, and GAPDH for iPSCs and differentiated cells, or to rat Sbf1 and Gapdh for primary cultured cells.
Western blot.
Cell pellets were lysed in ice-cold RIPA buffer supplemented with c0mplete protease inhibitors (Roche). Whole-cell lysates were then sonicated and clarified by centrifugation at 14,000×g for 15 minutes at 4°C. Protein concentrations were determined by BCA protein assay. After boiling at 100°C for 10 minutes in loading buffer, 20μg of protein samples were resolved on 10% or 15% SDS-PAGE gels, transferred to 0.2-μm PVDF membranes using the Mini Trans-Blot system (Bio-Rad), and probed by the indicated antibodies. Detection was performed by chemiluminescence.
Immunocytochemistry.
Cells were fixed in 4% paraformaldehyde (Sigma P6148) and permeabilized with 0.1% Triton X-100 (Bio-Rad 161-0407) in 1X phosphate buffered saline (PBS). Fixative was quenched with 10mM glycine (Fisher BP381-1), followed by incubating in blocking solution comprised of 3% bovine serum albumin w/v (BSA, Fisher BP9703-100), 2% fetal calf serum v/v (Sigma F4135), and 0.1% Triton X-100 v/v in 1X PBS for one hour at room temperature. Cells were then incubated with the indicated primary antibodies and diluted in blocking solution at 4° C overnight, washed three times for five minutes each in 1X PBS, then incubated with the indicated secondary antibodies diluted (1:250) in blocking solution at room temperature for one hour. Finally, cells were washed three times for five minutes each in 1X PBS. For visualizing nuclei where indicated, cells were further incubated twice for five minutes each in Hoescht 33258 dye (Invitrogen H3569, 1:10000 in 1X PBS), then mounted for fluorescence imaging.
dSTORM microscopy.
After immunocytochemical staining with the indicated primary and secondary antibodies, cells were incubated in dSTORM buffer: 555mM dextrose (VWR 76346-096), 0.1 M Tris (Fisher 1610716), 25mM NaCl (Fisher S25542), 143mM 2-mercaptoethanol (VWR 97064-880), 80μg/mL catalase (VWR 470300-672), 500μg/mL glucose oxidase (Fisher ICN19519610), pH 8.5. Individual iNeurons were imaged on an ONI Nanoimager, using a light program consisting of 5000 images taken with the 641 laser set at 35%, followed by 5000 images taken with the 488 laser set at 100%. For each iNeuron, all 10,000 images were uploaded to the ONI Collaborative Discovery cloud platform (CODI, http://alto.codi.bio), ROIs were manually drawn around each iNeuron, and images processed with the following filters and parameters to generate localization data: drift correction, p-value localization = 0.2, and localization precision = 20nm. Localization data were clustered using the CODI Clustering Tool with a minimum cluster size of 15nm. Resultant clusters were analyzed for co-localization using the CODI Counting Tool, with a maximum radius set to 150nm and number of circle sizes up to the max radius set to 10. Co-localization was scored when ≥15 localizations of both proteins were present.
QUANTIFICATION AND STATISTICAL ANALYSIS
All live cell imaging, immunocytochemistry, RT-PCR, and Western blot experiments were performed in biological triplicate and quantified using Fiji. Quantification of mEGFP-LC3-positive puncta were blinded to genotype and drug treatment. After thresholding images of PtdIns3P (WIPI2) and p62 immunocytochemistry, puncta were quantified using the Analyze Particles tool (Fiji). Quantifications of image analyses and Western blot band intensities, normalized to actin loading control, are reported as mean ± SEM. Statistical significance of mean differences was determined using Student’s unpaired t test, or multiple comparisons with ANOVA and ANCOVA, and with p<0.05 considered significant. Data were analyzed and plotted using GraphPad Prism.
Supplementary Material
Data S1. Bru-seq and BruChase-seq transcript values, related to Figure 2. Raw synthesis and stability data from Bru-seq and BruChase-seq analyses of fibroblasts, iPSCs, and iNeurons, with each transcript normalized to synthesis and stability values in fibroblasts, and each transcript natural log transformed and normalized to values in fibroblasts.
Data S2. Autophagosome and phosphoinositide measurements, related to Figure 1, Figure 4, Figure 5. Quantifications of mEGFP-LC3 or WIPI2 puncta normalized to cell surface area or per cell in each cell type and treatment condition indicated.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-β-actin | Cell Signaling Technology | Cat#4967S; RRID: AB_330288 |
| Rabbit polyclonal anti-ATG5 | Novus | Cat#NB110-53818; RRID: AB_828587 |
| Rabbit polyclonal anti-Histone 3 (monomethyl K9) | abcam | Cat#ab8896; RRID: AB_732929 |
| Rabbit monoclonal anti-MTMR5 | abcam | Cat#ab181106 |
| Mouse monoclonal anti-MTMR5 | Santa Cruz | Cat#sc-393488 |
| Mouse monoclonal anti-MTMR2 | Santa Cruz | Cat#sc-365184 |
| Mouse monoclonal anti-SQSTM1/p62 | Novus | Cat#H00008878-M01; RRID: AB_437085 |
| Mouse monoclonal anti-TFEB | MyBioSource | Cat#MBS120432; RRID: AB_2271743 |
| Goat anti-mouse Atto488 | Rockland Immunochemicals | Cat#610-152-121; RRID: AB_10893825 |
| Goat anti-mouse Alexa Fluor 594 | ThermoFisher Scientific | Cat#A11032; RRID: AB_2534091 |
| Goat anti-rabbit Alexa Fluor 647 | ThermoFisher Scientific | Cat#A32733; RRID: AB_2633282 |
| Chemicals, peptides, and recombinant proteins | ||
| Dimethyl sulfoxide (DMSO) | Sigma-Aldrich | Cat#D8418 |
| TeSR-E8 | STEMCELL | Cat#05990 |
| Torin1 | Tocris | Cat#4247 |
| Bafilomycin A1 | Cayman | Cat#NC135138 |
| Ammonium chloride (NH4Cl) | Sigma-Aldrich | Cat#A9434 |
| Bortezomib | Sigma-Aldrich | Cat#179324-69-7 |
| Experimental models: Cell lines | ||
| Human: mono-allelic mEGFP-tagged MAP1LC3 WTC iPSC line (tag at N-term) | Coriell Institute | Cat#AICS-0030-022 |
| Human: TDP-43-Dendra2 iPSC line (tag at C-term) | This paper | N/A |
| Human: GAPDH-2A-Dendra2 iPSC line | This paper | N/A |
| Rat: primary cortical neurons | This paper | N/A |
| Rat: primary cortical glia | This paper | N/A |
| Oligonucleotides | ||
| Primer: human ATG5 RT-PCR; Forward: GACTGGACTTGTGGTGCGCT; Reverse: AGCCGCAAAAAGCACCG | This paper | N/A |
| Primer: rat Atg5 RT-PCR; Forward: CTGTGATCCCGGTAGACCCA; Reverse: CCGTCCAAACCACACGTCTC | This paper | N/A |
| Primer: human GAPDH RT-PCR; Forward: AAGGTGAAGGTCGGAGTCAA; Reverse: AATGAAGGGGTCATTGATGG | This paper | N/A |
| Primer: rat Gapdh RT-PCR; Forward: CATCTCCCTCACAATTCCATCC; Reverse: GAGGGTGCAGCGAACTTTAT | This paper | N/A |
| Primer: human MTMR2 RT-PCR; Forward: GCCAGCACGGAGAGTCGAT; Reverse: AGCAGTCTTCGCGGCTACAG | This paper | N/A |
| Primer: rat Mtmr2 RT-PCR; Forward: CCACCTCCACCCCCAAAATG; Reverse: CAGGGAAGAGATCCGTGTGC | This paper | N/A |
| Primer: human MTMR9 RT-PCR; Forward: GAGGCGGCTTTCTCCTACTG; Reverse: CTAGGACTCGGCCACAGAGA | This paper | N/A |
| Primer: rat Mtmr9 RT-PCR; Forward: GCCTTGACCTTGGCTGTAGG; Reverse: CAGCACAGGGCACAGGTATC | This paper | N/A |
| Primer: human SBF1 RT-PCR; Forward: GAGGAGAGCACGGTGTTCAG; Reverse: CGCTCTCCGTGTCATAGCTC | This paper | N/A |
| Primer: rat Sbf1 RT-PCR; Forward: GCTGTAGTGTAGCCCTGCTC; Reverse: GACGTTGGTAGCTCCTGGAC | This paper | N/A |
| Primer: human SQSTM1 RT-PCR; Forward: GTACCAGGACAGCGAGAGGA; Reverse: CTCAAGCCCCATGTTGCACG | This paper | N/A |
| Primer: rat Sqstm1 RT-PCR; Forward: CTGATCCCCGGCTGATTGAG; Reverse: GAGGTGGGTTGTGGGACTTG | This paper | N/A |
| Primer: human TFEB RT-PCR; Forward: GAGAATGATGCCTCCGCACC; Reverse: GCGCAACCCTATGCGTGA | This paper | N/A |
| Primer: rat Tfeb RT-PCR; Forward: CGGCAGTCCTATGATGGGGA; Reverse: CCCCCTTCTCCAAAATGCCC | This paper | N/A |
| Recombinant DNA | ||
| pX335-U6-Chimeric_BB-CBh-hSpCas9n(D10A) plasmid | Addgene | Cat#42335 |
| pUCM-TDP-43-Dendra2 HDR plasmid | Blue Heron, LLC | N/A |
| pUCM-GAPDH-2A-Dendra2 HDR plasmid | Blue Heron, LLC | N/A |
| pUCM-CLYBL-NGN1&2 HDR plasmid | Michael Ward (NINDS) | N/A |
| pUCM-CLYBL-hNIL HDR plasmid | Michael Ward (NINDS) | N/A |
| PB-MYOD-shOCT4 plasmid | Michael Ward (NINDS) | N/A |
| TRC lentiviral human SBF1 shRNA | Horizon | Cat#RHS3979-201739167 |
| TRC lentiviral human MTMR2 shRNA | Horizon | Cat#RHS3979-224867040 |
| TRC lentiviral human MTMR9 shRNA | Horizon | Cat#RHS3979-201908246 |
| TRC lentiviral human TFEB shRNA | Horizon | Cat#RHS3979-201744686 |
| TRC lentiviral human SQSTM1 shRNA | Horizon | Cat#RHS3979-201739507 |
| TRC lentiviral human non-targeted shRNA | Horizon | Cat#RHS6848 |
| Software and algorithms | ||
| Prism | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Fiji/ImageJ | NIH, open-source | https://fiji.sc/ |
| ImagePro | Mediacy | https://www.mediacy.com/imagepro |
| μManager | Vale Lab (UCSF) | https://micro-manager.org |
| Bowtie | SourceForge | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| TopHat | Center for Computational Biology (Johns Hopkins) | https://ccb.jhu.edu/software/tophat/index.shtml |
| DESeq | Huber Group (EMBL) | https://www.huber.embl.de/users/anders/DESeq/ |
| R | R Core Team | https://www.r-project.org |
| Collaborative Discovery platform (CODI) | ONI | http://alto.codi.bio |
Highlights.
Neurons are uniquely resistant to methods of inducing autophagy
iPSC-derived, rat, and human neurons highly express the autophagy suppressor, MTMR5
MTMR5 is necessary and sufficient to inhibit autophagosome biogenesis
Knockdown of MTMR5 enhances autophagic degradation of TDP-43
ACKNOWLEDGEMENTS
We thank Sandra Mojica-Perez and Michael Uhler (University of Michigan Human Stem Cell and Gene Editing Core) and Andrea Serio (The Francis Crick Institute) for their expertise and guidance in iAstrocyte differentiation. Figures 1A–D, 2A, 3A, 5A–B, 5F, and S5 were created using BioRender (www.biorender.com). This research was supported by the NIH National Institute for Neurological Disorders and Stroke (NINDS) R25 NS089450-06, R01 NS099340, and the Intramural Research Program of the NIH/NINDS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
We worked to ensure diversity in experimental samples through the selection of the cell lines. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
REFERENCES
- 1.Dugger BN, and Dickson DW (2017). Pathology of Neurodegenerative Diseases. Cold Spring Harbor perspectives in biology 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parzych KR, and Klionsky DJ (2014). An overview of autophagy: morphology, mechanism, and regulation. Antioxidants & redox signaling 20, 460–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Laplante M, and Sabatini DM (2009). mTOR signaling at a glance. Journal of Cell Science 122, 3589–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palamiuc L, Ravi A, and Emerling BM (2020). Phosphoinositides in autophagy: current roles and future insights. Febs j 287, 222–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Claude-Taupin A, and Morel E (2021). Phosphoinositides: Functions in autophagy-related stress responses. Biochim Biophys Acta Mol Cell Biol Lipids 1866, 158903. [DOI] [PubMed] [Google Scholar]
- 6.Barz S, Kriegenburg F, Sánchez-Martín P, and Kraft C (2021). Small but mighty: Atg8s and Rabs in Membrane Dynamics during Autophagy. Biochim Biophys Acta Mol Cell Res, 119064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gómez-Sánchez R, Tooze SA, and Reggiori F (2021). Membrane supply and remodeling during autophagosome biogenesis. Curr Opin Cell Biol 71, 112–119. [DOI] [PubMed] [Google Scholar]
- 8.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, et al. (2006). Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880–884. [DOI] [PubMed] [Google Scholar]
- 9.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, et al. (2006). Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889. [DOI] [PubMed] [Google Scholar]
- 10.Fecto F, Yan J, Vemula SP, Liu E, Yang Y, Chen W, Zheng JG, Shi Y, Siddique N, Arrat H, et al. (2011). SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Archives of neurology 68, 1440–1446. [DOI] [PubMed] [Google Scholar]
- 11.Freischmidt A, Wieland T, Richter B, Ruf W, Schaeffer V, Muller K, Marroquin N, Nordin F, Hubers A, Weydt P, et al. (2015). Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nature neuroscience 18, 631–636. [DOI] [PubMed] [Google Scholar]
- 12.Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y, Kinoshita Y, Kamada M, Nodera H, Suzuki H, et al. (2010). Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465, 223–226. [DOI] [PubMed] [Google Scholar]
- 13.Nakano T, Nakaso K, Nakashima K, and Ohama E (2004). Expression of ubiquitin-binding protein p62 in ubiquitin-immunoreactive intraneuronal inclusions in amyotrophic lateral sclerosis with dementia: analysis of five autopsy cases with broad clinicopathological spectrum. Acta Neuropathol 107, 359–364. [DOI] [PubMed] [Google Scholar]
- 14.Beilina A, Van Der Brug M, Ahmad R, Kesavapany S, Miller DW, Petsko GA, and Cookson MR (2005). Mutations in PTEN-induced putative kinase 1 associated with recessive parkinsonism have differential effects on protein stability. Proc Natl Acad Sci U S A 102, 5703–5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Silvestri L, Caputo V, Bellacchio E, Atorino L, Dallapiccola B, Valente EM, and Casari G (2005). Mitochondrial import and enzymatic activity of PINK1 mutants associated to recessive parkinsonism. Human molecular genetics 14, 3477–3492. [DOI] [PubMed] [Google Scholar]
- 16.Watts GD, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D, Pestronk A, Whyte MP, and Kimonis VE (2004). Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet 36, 377–381. [DOI] [PubMed] [Google Scholar]
- 17.Haack TB, Hogarth P, Kruer MC, Gregory A, Wieland T, Schwarzmayr T, Graf E, Sanford L, Meyer E, Kara E, et al. (2012). Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X-linked dominant form of NBIA. Am J Hum Genet 91, 1144–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cullup T, Kho AL, Dionisi-Vici C, Brandmeier B, Smith F, Urry Z, Simpson MA, Yau S, Bertini E, McClelland V, et al. (2013). Recessive mutations in EPG5 cause Vici syndrome, a multisystem disorder with defective autophagy. Nat Genet 45, 83–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang IF, Guo BS, Liu YC, Wu CC, Yang CH, Tsai KJ, and Shen CK (2012). Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA-binding protein 43. Proc Natl Acad Sci U S A 109, 15024–15029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kruger U, Wang Y, Kumar S, and Mandelkow EM (2012). Autophagic degradation of tau in primary neurons and its enhancement by trehalose. Neurobiol Aging 33, 2291–2305. [DOI] [PubMed] [Google Scholar]
- 21.Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O’Kane CJ, et al. (2004). Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 36, 585–595. [DOI] [PubMed] [Google Scholar]
- 22.Morrison KE, Dhariwal S, Hornabrook R, Savage L, Burn DJ, Khoo TK, Kelly J, Murphy CL, Al-Chalabi A, Dougherty A, et al. (2013). Lithium in patients with amyotrophic lateral sclerosis (LiCALS): a phase 3 multicentre, randomised, double-blind, placebo-controlled trial. The Lancet. Neurology 12, 339–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mandrioli J, D’Amico R, Zucchi E, Gessani A, Fini N, Fasano A, Caponnetto C, Chio A, Dalla Bella E, Lunetta C, et al. (2018). Rapamycin treatment for amyotrophic lateral sclerosis: Protocol for a phase II randomized, double-blind, placebo-controlled, multicenter, clinical trial (RAP-ALS trial). Medicine 97, e11119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Simuni T, Fiske B, Merchant K, Coffey CS, Klingner E, Caspell-Garcia C, Lafontant DE, Matthews H, Wyse RK, Brundin P, et al. (2021). Efficacy of Nilotinib in Patients With Moderately Advanced Parkinson Disease: A Randomized Clinical Trial. JAMA neurology 78, 312–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pallet N, and Legendre C (2013). Adverse events associated with mTOR inhibitors. Expert opinion on drug safety 12, 177–186. [DOI] [PubMed] [Google Scholar]
- 26.Schmeisser K, and Parker JA (2019). Pleiotropic Effects of mTOR and Autophagy During Development and Aging. Front Cell Dev Biol 7, 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maday S, and Holzbaur EL (2016). Compartment-Specific Regulation of Autophagy in Primary Neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience 36, 5933–5945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsvetkov AS, Miller J, Arrasate M, Wong JS, Pleiss MA, and Finkbeiner S (2010). A small-molecule scaffold induces autophagy in primary neurons and protects against toxicity in a Huntington disease model. Proc Natl Acad Sci U S A 107, 16982–16987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blondeau F, Laporte J, Bodin S, Superti-Furga G, Payrastre B, and Mandel JL (2000). Myotubularin, a phosphatase deficient in myotubular myopathy, acts on phosphatidylinositol 3-kinase and phosphatidylinositol 3-phosphate pathway. Human molecular genetics 9, 2223–2229. [DOI] [PubMed] [Google Scholar]
- 30.Hnia K, Vaccari I, Bolino A, and Laporte J (2012). Myotubularin phosphoinositide phosphatases: cellular functions and disease pathophysiology. Trends in molecular medicine 18, 317–327. [DOI] [PubMed] [Google Scholar]
- 31.Kim SA, Vacratsis PO, Firestein R, Cleary ML, and Dixon JE (2003). Regulation of myotubularin-related (MTMR)2 phosphatidylinositol phosphatase by MTMR5, a catalytically inactive phosphatase. Proc Natl Acad Sci U S A 100, 4492–4497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bolino A, Bolis A, Previtali SC, Dina G, Bussini S, Dati G, Amadio S, Del Carro U, Mruk DD, Feltri ML, et al. (2004). Disruption of Mtmr2 produces CMT4B1-like neuropathy with myelin outfolding and impaired spermatogenesis. J Cell Biol 167, 711–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robinson FL, Niesman IR, Beiswenger KK, and Dixon JE (2008). Loss of the inactive myotubularin-related phosphatase Mtmr13 leads to a Charcot-Marie-Tooth 4B2-like peripheral neuropathy in mice. Proc Natl Acad Sci U S A 105, 4916–4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakhro K, Park JM, Hong YB, Park JH, Nam SH, Yoon BR, Yoo JH, Koo H, Jung SC, Kim HL, et al. (2013). SET binding factor 1 (SBF1) mutation causes Charcot-Marie-Tooth disease type 4B3. Neurology 81, 165–173. [DOI] [PubMed] [Google Scholar]
- 35.Fernandopulle MS, Prestil R, Grunseich C, Wang C, Gan L, and Ward ME (2018). Transcription Factor-Mediated Differentiation of Human iPSCs into Neurons. Curr Protoc Cell Biol 79, e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu W, Hill SE, Nathan WJ, Paiano J, Callen E, Wang D, Shinoda K, van Wietmarschen N, Colón-Mercado JM, Zong D, et al. (2021). Neuronal enhancers are hotspots for DNA single-strand break repair. Nature 593, 440–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Serio A, Bilican B, Barmada SJ, Ando DM, Zhao C, Siller R, Burr K, Haghi G, Story D, Nishimura AL, et al. (2013). Astrocyte pathology and the absence of non-cell autonomy in an induced pluripotent stem cell model of TDP-43 proteinopathy. Proc Natl Acad Sci U S A 110, 4697–4702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paulsen MT, Veloso A, Prasad J, Bedi K, Ljungman EA, Magnuson B, Wilson TE, and Ljungman M (2014). Use of Bru-Seq and BruChase-Seq for genome-wide assessment of the synthesis and stability of RNA. Methods (San Diego, Calif.) 67, 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, et al. (2011). TFEB links autophagy to lysosomal biogenesis. Science (New York, N.Y.) 332, 1429–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, and Johansen T (2007). p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282, 24131–24145. [DOI] [PubMed] [Google Scholar]
- 41.Lorenzo O, Urbe S, and Clague MJ (2006). Systematic analysis of myotubularins: heteromeric interactions, subcellular localisation and endosome related functions. J Cell Sci 119, 2953–2959. [DOI] [PubMed] [Google Scholar]
- 42.Kampmann M (2020). CRISPR-based functional genomics for neurological disease. Nat Rev Neurol 16, 465–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Endesfelder U, and Heilemann M (2015). Direct stochastic optical reconstruction microscopy (dSTORM). Methods Mol Biol 1251, 263–276. [DOI] [PubMed] [Google Scholar]
- 44.Weskamp K, Tank EM, Miguez R, McBride JP, Gómez NB, White M, Lin Z, Gonzalez CM, Serio A, Sreedharan J, et al. (2020). Shortened TDP43 isoforms upregulated by neuronal hyperactivity drive TDP43 pathology in ALS. The Journal of clinical investigation 130, 1139–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang X, Fan H, Ying Z, Li B, Wang H, and Wang G (2010). Degradation of TDP-43 and its pathogenic form by autophagy and the ubiquitin-proteasome system. Neuroscience letters 469, 112–116. [DOI] [PubMed] [Google Scholar]
- 46.McAleese KE, Walker L, Erskine D, Thomas AJ, McKeith IG, and Attems J (2017). TDP-43 pathology in Alzheimer’s disease, dementia with Lewy bodies and ageing. Brain pathology (Zurich, Switzerland) 27, 472–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, et al. (2006). TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochemical and biophysical research communications 351, 602–611. [DOI] [PubMed] [Google Scholar]
- 48.Barmada SJ, Serio A, Arjun A, Bilican B, Daub A, Ando DM, Tsvetkov A, Pleiss M, Li X, Peisach D, et al. (2014). Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nature chemical biology 10, 677–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lešková A, Kusá Z, Labajová M, Krausko M, and Jásik J (2019). The Photoconvertible Fluorescent Protein Dendra2 Tag as a Tool to Investigate Intracellular Protein Dynamics. Methods Mol Biol 1992, 201–214. [DOI] [PubMed] [Google Scholar]
- 50.Weskamp K, Safren N, Miguez R, and Barmada S (2019). Monitoring Neuronal Survival via Longitudinal Fluorescence Microscopy. Journal of visualized experiments : JoVE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mruk DD, and Cheng CY (2011). The myotubularin family of lipid phosphatases in disease and in spermatogenesis. The Biochemical journal 433, 253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee YC, Huang WC, Lin JH, Kao TJ, Lin HC, Lee KH, Lin HC, Shen CJ, Chang WC, and Huang CC (2018). Znf179 E3 ligase-mediated TDP-43 polyubiquitination is involved in TDP-43-ubiquitinated inclusions (UBI) (+)-related neurodegenerative pathology. J Biomed Sci 25, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hebron ML, Lonskaya I, Sharpe K, Weerasinghe PP, Algarzae NK, Shekoyan AR, and Moussa CE (2013). Parkin ubiquitinates Tar-DNA binding protein-43 (TDP-43) and promotes its cytosolic accumulation via interaction with histone deacetylase 6 (HDAC6). J Biol Chem 288, 4103–4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Itakura E, and Mizushima N (2011). p62 Targeting to the autophagosome formation site requires self-oligomerization but not LC3 binding. J Cell Biol 192, 17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.François-Moutal L, Perez-Miller S, Scott DD, Miranda VG, Mollasalehi N, and Khanna M (2019). Structural Insights Into TDP-43 and Effects of Post-translational Modifications. Frontiers in molecular neuroscience 12, 301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chu CT (2019). Mechanisms of selective autophagy and mitophagy: Implications for neurodegenerative diseases. Neurobiol Dis 122, 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Conway O, Akpinar HA, Rogov VV, and Kirkin V (2020). Selective Autophagy Receptors in Neuronal Health and Disease. J Mol Biol 432, 2483–2509. [DOI] [PubMed] [Google Scholar]
- 58.Filimonenko M, Stuffers S, Raiborg C, Yamamoto A, Malerød L, Fisher EM, Isaacs A, Brech A, Stenmark H, and Simonsen A (2007). Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J Cell Biol 179, 485–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ayala YM, De Conti L, Avendaño-Vázquez SE, Dhir A, Romano M, D’Ambrogio A, Tollervey J, Ule J, Baralle M, Buratti E, et al. (2011). TDP-43 regulates its mRNA levels through a negative feedback loop. The EMBO journal 30, 277–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nichenko AS, Southern WM, Atuan M, Luan J, Peissig KB, Foltz SJ, Beedle AM, Warren GL, and Call JA (2016). Mitochondrial maintenance via autophagy contributes to functional skeletal muscle regeneration and remodeling. American journal of physiology. Cell physiology 311, C190–200. [DOI] [PubMed] [Google Scholar]
- 61.Saera-Vila A, Kish PE, Louie KW, Grzegorski SJ, Klionsky DJ, and Kahana A (2016). Autophagy regulates cytoplasmic remodeling during cell reprogramming in a zebrafish model of muscle regeneration. Autophagy 12, 1864–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Raben N, Hill V, Shea L, Takikita S, Baum R, Mizushima N, Ralston E, and Plotz P (2008). Suppression of autophagy in skeletal muscle uncovers the accumulation of ubiquitinated proteins and their potential role in muscle damage in Pompe disease. Human molecular genetics 17, 3897–3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, Metzger D, Reggiani C, Schiaffino S, and Sandri M (2009). Autophagy is required to maintain muscle mass. Cell metabolism 10, 507–515. [DOI] [PubMed] [Google Scholar]
- 64.Leigh PN, Whitwell H, Garofalo O, Buller J, Swash M, Martin JE, Gallo JM, Weller RO, and Anderton BH (1991). Ubiquitin-immunoreactive intraneuronal inclusions in amyotrophic lateral sclerosis. Morphology, distribution, and specificity. Brain 114 (Pt 2), 775–788. [DOI] [PubMed] [Google Scholar]
- 65.Rudnick ND, Griffey CJ, Guarnieri P, Gerbino V, Wang X, Piersaint JA, Tapia JC, Rich MM, and Maniatis T (2017). Distinct roles for motor neuron autophagy early and late in the SOD1(G93A) mouse model of ALS. Proc Natl Acad Sci U S A 114, E8294–e8303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gerbino V, Kaunga E, Ye J, Canzio D, O’Keeffe S, Rudnick ND, Guarnieri P, Lutz CM, and Maniatis T (2020). The Loss of TBK1 Kinase Activity in Motor Neurons or in All Cell Types Differentially Impacts ALS Disease Progression in SOD1 Mice. Neuron 106, 789–805. e785. [DOI] [PubMed] [Google Scholar]
- 67.An D, Fujiki R, Iannitelli DE, Smerdon JW, Maity S, Rose MF, Gelber A, Wanaselja EK, Yagudayeva I, Lee JY, et al. (2019). Stem cell-derived cranial and spinal motor neurons reveal proteostatic differences between ALS resistant and sensitive motor neurons. eLife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tsvetkov AS, Arrasate M, Barmada S, Ando DM, Sharma P, Shaby BA, and Finkbeiner S (2013). Proteostasis of polyglutamine varies among neurons and predicts neurodegeneration. Nature Chemical Biology 9, 586–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, and Ohsumi Y (2004). In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Molecular biology of the cell 15, 1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yoshii SR, and Mizushima N (2017). Monitoring and Measuring Autophagy. Int J Mol Sci 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Loos B, du Toit A, and Hofmeyr JH (2014). Defining and measuring autophagosome flux—concept and reality. Autophagy 10, 2087–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Maday S, and Holzbaur EL (2014). Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway. Dev Cell 30, 71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yoshimura S, Gerondopoulos A, Linford A, Rigden DJ, and Barr FA (2010). Family-wide characterization of the DENN domain Rab GDP-GTP exchange factors. J Cell Biol 191, 367–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jean S, Cox S, Schmidt EJ, Robinson FL, and Kiger A (2012). Sbf/MTMR13 coordinates PI(3)P and Rab21 regulation in endocytic control of cellular remodeling. Molecular biology of the cell 23, 2723–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cui X, De Vivo I, Slany R, Miyamoto A, Firestein R, and Cleary ML (1998). Association of SET domain and myotubularin-related proteins modulates growth control. Nat Genet 18, 331–337. [DOI] [PubMed] [Google Scholar]
- 76.Antunes ETB, and Ottersbach K (2020). The MLL/SET family and haematopoiesis. Biochim Biophys Acta Gene Regul Mech 1863, 194579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Firestein R, Nagy PL, Daly M, Huie P, Conti M, and Cleary ML (2002). Male infertility, impaired spermatogenesis, and azoospermia in mice deficient for the pseudophosphatase Sbf1. The Journal of clinical investigation 109, 1165–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fagerberg L, Hallström BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, Habuka M, Tahmasebpoor S, Danielsson A, Edlund K, et al. (2014). Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Molecular & cellular proteomics : MCP 13, 397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH, and Nixon RA (2008). Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. The Journal of neuroscience : the official journal of the Society for Neuroscience 28, 6926–6937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tank EM, Figueroa-Romero C, Hinder LM, Bedi K, Archbold HC, Li X, Weskamp K, Safren N, Paez-Colasante X, Pacut C, et al. (2018). Abnormal RNA stability in amyotrophic lateral sclerosis. Nature communications 9, 2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Bru-seq and BruChase-seq transcript values, related to Figure 2. Raw synthesis and stability data from Bru-seq and BruChase-seq analyses of fibroblasts, iPSCs, and iNeurons, with each transcript normalized to synthesis and stability values in fibroblasts, and each transcript natural log transformed and normalized to values in fibroblasts.
Data S2. Autophagosome and phosphoinositide measurements, related to Figure 1, Figure 4, Figure 5. Quantifications of mEGFP-LC3 or WIPI2 puncta normalized to cell surface area or per cell in each cell type and treatment condition indicated.
Data Availability Statement
Bru-seq/BruChase-seq, RT-PCR, Western blot, and microscopy data reported in this paper will be shared by the Lead Contact upon request without restriction. The 2-D dSTORM dataset is available at https://spaces.hightail.com/space/mTwffdRoyB.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.