

Abstract
Background:
The vascular endothelium maintains tissue-fluid homeostasis by controlling the passage of large molecules and fluid between the blood and interstitial space. The interaction of catenins and the actin cytoskeleton with vascular endothelial (VE)-cadherin is the primary mechanism for stabilizing adherens junctions (AJ), thereby preventing lung vascular barrier disruption. Here, we evaluated the role of DOCK4, an atypical Rho family GTPase guanine exchange factor (GEF) in vascular function.
Methods:
We generated mice deficient in DOCK4 and used DOCK4 silencing and reconstitution approaches in human pulmonary artery endothelial cells and used assays to evaluate protein localization, endothelial cell permeability and small GTPase activation.
Results:
Our data show that DOCK4 deficient mice are viable. However, these mice have hemorrhage selectively in the lung, incomplete smooth muscle cell coverage in pulmonary vessels, increased basal microvascular permeability, and impaired response to sphingosine 1-phosphate (S1P) induced reversal of thrombin-induced permeability. Consistent with this, DOCK4 rapidly translocates to the cell periphery and associates with the detergent insoluble fraction following S1P treatment and its absence prevents S1P induced Rac-1 activation and enhancement of barrier function. Moreover, DOCK4-silenced pulmonary artery endothelial cells exhibit enhanced basal permeability in vitro that is associated with enhanced Rho GTPase activation.
Conclusions:
Our findings indicate that DOCK4 maintains AJs necessary for lung vascular barrier function by establishing the normal balance between RhoA and Rac1-mediated actin cytoskeleton remodeling, a previously unappreciated function for the atypical GEF family of molecules. Our studies also identify S1P as a potential upstream regulator of DOCK4 activity.
Keywords: Endothelial cells, lung, permeability, Rho GTPases, guanine exchange factors
Classification: Biological Sciences
BACKGROUND
Vascular permeability is actively regulated to maintain water and protein balance between the extra- and intra- vascular compartments while enhanced leakage in post-capillary venules and capillaries is a hallmark of sterile and pathogen mediated inflammation that can lead to multi-organ failure 1,2. The endothelium plays a vital role in maintaining barrier function. The predominant pathway of endothelial permeability to macromolecules is through adherens junctions (AJs) that are present at cell-cell contacts. Regulation of vascular permeability through this paracellular route depends on actin-based systems that communicate with junctional molecules, such as VE-cadherin via intracellular linker proteins (e.g. catenins and adaptor molecules). Changes in the actin cytoskeleton are primarily controlled by small GTPases 3. Members of the Rho family of small GTPases regulate junction dynamics: Rac is required for maintaining basal permeability, Rho has been linked to gap formation associated with permeability-inducing proinflammatory agonists, thrombin, bradykinin or histamine and cdc42 is required for junctional re-closure following agonist induced permeability 1 .
The small GTPases cycle between an inactive GDP-bound and an active GTP-bound state 4 . Diverse cell surface receptors control this cycle through the regulation of guanine nucleotide exchange factors (GEFs) and GTPase activating proteins (GAPs), which activate or inactivate the small GTPases, respectively. Conventional GEFs, characterized by Dbl homology-pleckstrin homology domains that are critical for GDP/GTP exchange activity are the largest family of direct activators of Rho GTPases numbering up to 70 members 5 and have been implicated in vascular development 6–8. For example, Tiam, Prex and Vav2 promote endothelial barrier function by activating Rac and maintaining vascular barrier function after PAF-induced inflammation 6,9–12. The CDM (CED-5, Dock180, Myoblast city) family of GEFs, which is comprised of 11 members in humans, contain a catalytic DHR2 domain in their C-terminal region 13,14 that is divergent within the family. The more common DHR1 domain located in the N-terminus interacts with the membrane phospholipid, phosphatidylinositol (3,4,5)-trisphosphate to mediate signaling and membrane localization 15,16 of DOCK proteins, important for their effects on the cytoskeleton 17–19. DOCK proteins interact with ELMO proteins to form functional complexes for GTPase activation with the distinctive feature of activating Rac and/or Cdc42 but not Rho family members 20. These atypical GEFs regulate multiple processes, including lymphocyte migration, cell migration, apoptotic-cell engulfment, and tumor invasion 21–25. Roles for CDM family members in endothelial and vascular function are beginning to emerge 20. Dock180/Elmo1 facilitates angiogenesis by protecting endothelial cells from apoptosis via a Rac1/PAK/AKT signaling cascade 26 and promoting endothelial cell migration 27. A recent paper reports that DOCK4 deficiency results in embryonic lethality and heterozygous mice have altered vessel lumen size in tumor models that is attributed to a role for DOCK4 in interacting with the cdc42 GEF DOCK9 to promote filopodial protrusions 28.
Different GEFs coordinate the activation of endothelial small GTPases downstream of several extracellular stimuli, including sphingosine-1-phosphate (S1P), Angiopoietin1 and β-adrenergic receptor agonists, which regulate tonic endothelial barrier properties 29,30. S1P, a metabolic product of sphingomyelin that is present at high levels in the blood mediates barrier function via Rac GTPases. It signals through widely expressed G protein-coupled receptors (GPCR) (S1PR1- S1PR5) to regulate diverse cellular functions in the vascular system. Knockdown of S1P1 in cultured endothelial cells reduces expression of adhesion molecules such as CD31 and VE-cadherin 31. Disruption of the S1P/S1PR1 axis in mice results in increased vascular leakiness and mortality 32,33. Endothelial-specific S1PR1 knockout mice exhibit vessel leakiness in several organs 34 including the lung 35,36. On the other hand, S1P induced S1PR1 activation rescues endothelial cell-barrier function after thrombin treatment 37,38.
Here, we generated mice deficient in Dock4 and used silencing approaches in human cultured endothelial cells to show that DOCK4 is dispensable for embryo viability and for tumor induced angiogenesis. On the other hand, we demonstrate that DOCK4 is critical for basal and S1P induced endothelial barrier function in the lung and identify dysregulation of Rho GTPases in the absence of DOCK4 as the underlying mechanism.
METHODS
The authors declare that all supporting data are available within the article.
Mouse strains.
All animal studies were approved by the Institutional Animal Care and Use Committee. Briefly, Dock4 deficient mice were generated using a standard Cre-LoxP recombination strategy. The targeting vector included Exons 3 and 4 of Dock4, a 1.6 kb homology region upstream of Exon 3 and a 1.7 kb homology region downstream of Exon 4. A Neo cassette flanked by FRT sites was inserted 3′ of Exon 4 and LoxP sites were placed 5′ of Exon 3 and 3′ of the Neo cassette. Cre recombination results in a frameshift that generates a 56-amino acid truncated N-terminal sequence including 15 amino acids of nonsense sequence before a stop codon. A PGK-diphtheria toxin cassette was used for negative selection. The construct was electroporated into JM8.N4 embryonic stem cells. A PCR strategy was used to screen for correct integration of the transgene cassette in embryonic stem cells and a clone with the desired homologous recombination was used for pronuclear injection into C57 Albino (B6(Cg)-Tyrc-2J/J) blastocysts. Chimeric animals were bred to C57BL/6J wild-type (WT) mice to determine germline transmission. Founder lines carrying the Dock4 floxed allele were bred to VE-cadherin:tetracycline-regulated transactivator (tTA)/C57Bl/6J mice 39 and tet-O-cre/C57Bl/6J mice (The Jackson Laboratory, Bar Harbor, ME) for recombination of LoxP sites. Although VE-cadherin based CRE recombinase strategies are frequently used for endothelial restricted deletion 40, VE-cadherin exhibits epithelial cycle stage–specific expression in the Sertoli cells and in differentiating spermatids at stage II and elongated spermatids of mouse testes 41,42. This leads to expression of CRE-recombinase in the spermatids of VE-Cad/Tet-O-Cre mice and thus the generation of global knock-out alleles that are passed onto the offspring as we also describe in another conditional knock-out animal 43. Dock4 knockout (Dock4−/−) animals were viable, born at the expected mendelian ratios and grew without gross developmental defects. The PCR strategy to determine correct location of targeted allele and WT, Dock4 floxed and Dock4 knockout mice is as follows: P1- aggccaaggaaggaaaactc, P2- cagcctgcggagaaataatc where P1 and P2 recognizes WT and floxed animal, 5’Rec- gctggccaggcacttgatgc and 3’Rec- ctgctggccacaggcttccc recognizes knock-out allele after cre, 3’F-aggaacttcatcagtcaggtaca and 3’R-agacaatgtgcctcctctgg recognizes correct 3’ location of targeted allele and 5’F-gatggtgcccctgaagttgagtagc and 5’R- tacgaagttatcccgggtgtgc recognizes correct 5’ location of targeted allele. WT and Dock4−/− mouse colonies were generated from independent breedings and maintained on a C57Bl/6J background. Age matched males and females between 12–30 weeks old were used for all experiments except for pulmonary hemorrhage, where initial observations made in both sexes were confirmed and analyzed histologically only in females due to their availability.
Pulmonary hemorrhage.
Four-micrometer sections of formalin fixed FFPE tissue were stained with hematoxylin-eosin by standard techniques. Individual H&E-stained sections from one intact lung of WT and Dock4−/− female mice were imaged at 0.23 µm/pixel (40X-equivalent resolution) using a Hamamatsu S360 bright field whole slide imager. Peribronchial hemorrhage was determined by quantifying the fraction of small bronchioles (~50–150µm in luminal diameter) with red blood cells within their lumen or in adjacent alveolar space. Diffuse hemorrhage into intra-alveolar space not adjacent to bronchi was also estimated on a scale of trace, 1+, 2+ and 3+.
Lung vascular permeability.
WT and Dock4−/− mice were anesthetized with an i.p. injection of ketamine (100 mg/kg) and xylazine (2.5 g/kg). S1P and PAR-1 treatments were as described 38. Briefly, PAR1 agonist peptide (TFLLRN-NH2) (1 mg/kg body weight) or control (PBS) were administered via the retroorbital route. After 15 minutes, 50μl of S1P (final concentration 1 μmol/L in 4% BSA solution) or vehicle (4% BSA) was given i.v. After an additional 45 min, the left lobe was excised, weighed, completely dried in a 60 °C oven overnight and then weighed again to determine the lung wet–dry ratio.
Lung microvessel permeability.
Evans blue–conjugated albumin (20 mg/kg) was injected retro-orbitally 30 minutes before sacrificing the mice to assess vascular leak. Mice from WT and Dock4−/− were perfused with 50mM sodium citrate, pH 3.5 to preserve Evans blue binding to albumin. Relevant organs were excised and rinsed with phosphate-buffered saline (PBS; 1.44 g of Na2HPO4, 0.24 g of KH2PO4, 8.0 g of NaCl, and 0.20 g of KCl in 1 L, pH 7.4). Organs were blotted with tissue, and each organ was cut in half and weighed (wet weights, in g). One half of the tissue was dried in the oven at 60 °C overnight. The other tissue half was placed in a consistent volume (up to 200 µL) of formamide in a microfuge tube for 48 h (and up to 72 h) to extract the Evans blue. Absorbance max of Evans blue was read at OD620.
Tracer Injection Experiments.
Lung and brain tracer leakage experiments were performed as described 44,45. Mice were injected i.v. with Alexa Fluor 555–cadaverine (6 μg/g; Invitrogen). After 2 h, mice were anesthetized and perfused for 7 min with ice-cold PBS (pH 7.4), and lungs and brains were removed. After dissection, the left lobe and the cerebral hemispheres were weighed and homogenized with 1% Triton X-100 in PBS. Lysates were centrifuged at 12,000 × g for 20 min at 4 °C, and the supernatant was used to quantify fluorescence (excitation/emission 540/590nm; SpectraMax M2e; Molecular Devices). The relative fluorescence values were normalized by the lung and brain weights.
Colloidal Carbon deposition, Image acquisition and Analysis.
Lung permeability was evaluated using colloidal carbon (ink) 46. Mice were injected i.v. with 100 µL of waterproof ink solution (1:4 in PBS, Higgins) and 20 minutes later animals were sacrificed and perfused through the right ventricle with PBS containing 10mM EDTA. Lungs were then inflated via the trachea with 300ul of an OCT-PBS solution (1:1) and lung lobes were excised, embedded in OCT and snap frozen. 10µm cryostat sections were obtained and imaged to assess ink deposition. One unstained section of lung from each Dock4−/− and wild-type mouse was imaged using an Olympus BX51 brightfield microscope with Nomarski interference optics and a 40X objective. Colloidal carbon deposits less than 50 µm in greatest dimension were counted in 5 randomly selected fields in each section.
Lung immunostaining.
10µm-sections of lung specimens were cut with a cryostat (Leica Microsystems) and fixed in cold acetone for 10 min. Sections were stained with goat anti-mouse CD31 antibody (1:100, Novus Biologicals, AF3628) and FITC-conjugated alpha-smooth muscle actin antibody (1:100, Sigma, F3777) followed by secondary Alexa Fluor 594-conjugated antibody (1:100, Life Technologies, A11058) to detect CD31 antibody staining. Slides were covered with mounting medium containing DAPI (Vector Laboratories). One section of immunostained lung from each of three Dock4−/− and three wild type mice was imaged (DAPI, FITC, TRITC) using MiraxScan 150 (3DHISTECH) (0.46 µm/pixel) and Olympus VS120 (0.32 µm/pixel) whole slide imagers. Blood vessels of 20–200µm in greatest cross-sectional dimension that stained with CD31 (Alexa-594) were selected and the coverage of each vessel’s perimeter by SMA (Alexa-488) was assigned visually to one of five 5 categories: 1) 0–25%, 2) 26–50%, 3) 51–75%, 4) more than 76% but less than 100% and 5) 100%. Between 7 and 22 vessels were quantified from one lung of each mouse.
B16F10 tumor implants and immunostaining.
B16F10 cells were cultured in vitro in DMEM/high glucose supplemented with 10% fetal calf serum and maintained at sub-confluent density. Mice were anesthetized with an intraperitoneal injection of a ketamine/xylazine cocktail (90 mg/kg ketamine, 10 mg/kg xylazine), shaved and injected in the flank subcutaneously with 2×105 tumor cells in 100 ul HBSS. Tumors were measured with a caliper every 1–2 days and tumor volume was calculated using an ellipsoid volume formula (1/2xDxd2; height is difficult to measure) where “D” and “d” are the longer and shorter diameter respectively47,48. After 14 days, tumor implants were dissected from subcutaneous tissue and skin and paraffin blocks were prepared as above. Prior to immunostaining 5-μm sections on glass slides were immersed in a modified citrate buffer, pH 6.1 (DAKO Cat. No. S1699) in a steam environment for epitope retrieval. Tissue sections were permeabilized with 0.5% Tween 20 in PBS, blocked with 10% donkey serum in PBS and immunostained using goat anti-CD31 primary antibody (R & D Systems, AF3628) followed by Alexa-488-conjugated donkey anti-goat secondary antibody (Invitrogen A-11055). DNA was visualized with DAPI. Images (512 × 512-pixel single image planes) of DAPI and 488 fluorescence were obtained using an Olympus FV1000 confocal microscope with a 20X objective (0.248 µm/pixel). One image was obtained from randomly selected regions of four separate tissue sections of each tumor implant in WT mice and Dock4 deficient mice. Fiji (2.0.0-rc-68/1.52E Build: bad6864455) was used to extract Tiff images from Olympus oib files and to perform the following steps using a macro applied to the 488 channel image data: run(“Subtract Background...”, “rolling=50”); run(“Median...”, “radius=2”); setAutoThreshold(“Otsu dark”); run(“Convert to Mask”); run(“Fill Holes”); run(“Set Measurements...”, “area perimeter shape skewness area_fraction redirect=None decimal=3”); run(“Analyze Particles...”, “display clear add”); saveAs(“Results”);
CD31-positive areas representing vascular spaces up to 500 µm2 from WT and DOCK4-deficient mice were compared.
Plasma S1P analysis.
Plasma S1P was extracted according to Engelbrecht et al 49. Briefly, Plasma aliquots (5 or 10 µL) were first diluted to 100 mL with TBS Buffer (50 mM Tris-HCl pH 7.5, 0.15 M NaCl). Extraction of S1P was done by adding 100 mL precipitation solution (20 nM D7-S1P in methanol) followed by 30 s of vortexing. Precipitated samples were centrifuged at 18,000 rpm for 5 min and supernatants were transferred to vials for LC-MS/MS analysis using Q Exactive mass spectrometer coupled to a Vanquish UHPLC System (Thermo Fisher Scientific).
Endothelial cell culture.
Human pulmonary artery endothelial cells (HPAEC) (LONZA) were cultured in a T-75 flask coated with 0.1% gelatin in EBM-2 medium supplemented with 10% fetal bovine serum and maintained at 37°C in a humidified atmosphere of 5% CO2 and 95% air until they formed a confluent monolayer as described 50.
Cell transfection and Lentiviral transduction.
DOCK4 siRNA sense 5’ GGAUGCAUACUAAUAUUUtt 3’ and anti-sense 5’ AAAUAUUUAGUAUGCAUCCca or control siRNA sequences were transduced into cells as described 50. Briefly, HPAEC grown to 70% confluence were trypsinized and mixed with 300nM of siRNA along with 100 μl of HCAEC nucleofector solution. Cells were electroporated with an Amaxa (LONZA) nucleofector device in accordance with the manufacturer’s recommended program (S-05), mixed in EBM-2 and plated on 60-mm dishes or coverslips for indicated experiments. HPAEC plated onto gold electrodes were transfected with the indicated siRNA using Santa Cruz transfection reagent in accordance with the manufacturer’s protocol. The cells were used after 48 h of transfection, when there was clear evidence of the expression of protein. Similarly, HPAEC were transduced with lentivirus Control or with 2 independent DOCK4 shRNA-expressing lentiviruses for 48hrs. Cells were allowed to rest for an additional 24hrs, and experiments were performed thereafter.
Immunoblot Analysis.
Total extracts of protein samples were separated in polyacrylamide gel and transferred on nitrocellulose membrane. Transferred proteins were blocked with 5% (wt/vol) skim milk and probed with primary antibodies against Dock4 (1:500; Bethyl, catalog no. IHC-00647), RhoA (1:500; Santa Cruz, sc-418), actin (1:4000; Sigma, A3853), tubulin (1:5000; Sigma, T5168), Phospho-myosin light chain (MLC) (Ser19) (1:1000; Cell Signaling Technologies, 3671) and Total MLC (1:1000; Cell Signaling Technologies, 8505). Lung and Brain tissue samples from wt and Dock4−/− mice were homogenized in RIPA buffer using QIAGEN tissue homogenizer. Samples were spun for 10 min at max speed and supernatant was estimated for equal loading. For analysis of cell fractions, HPAEC were processed using the Subcellular Protein Fractionation Kit (ThermoFischer) following the manufacturer protocol. Briefly, HPAEC were treated with vehicle (Control) or 1 µM S1P for 1 min, lysed and spun to obtain cytoplasmic soluble proteins (supernatant). The resulting pellet was solubilized and centrifuged to obtain plasma, mitochondria and ER/Golgi membranes (supernatant). The pelleted insoluble material, containing the cytoskeleton and nuclei, was dissolved in SDS-containing gel loading buffer. Equal amounts of proteins from control and S1P-treated cells in each of the fractions were subjected to western blot analysis. Densitometric analysis was performed using ImageJ (https://imagej.nih.gov/).
GTPase activation assays.
Confluent monolayers of HPAEC transduced with lentivirus control or lentiviruses expressing 2 independent DOCK4 shRNAs for 72 hours, or HPAEC transfected with Control or Dock4 siRNA for 48 hours, were starved in EBM-2 medium without supplements for 4 hours and assayed for active Rac or RhoA using G-Lisa kits (Cytoskeleton, Inc., Denver, CO). Active Rac and Rho (GTP-Rac and GTP-RhoA ) were used as positive controls.
qPCR Analysis.
Total RNA was isolated from cells using RNeasy Mini Kit (Qiagen) according to manufacturer’s instructions. RNA was quantified using Nanodrop and reverse transcribed using Applied biosystem’s universal cDNA synthesis kit. Real-Time PCR was performed using SYBR green master mix (Applied Biosystems). Primers used for gene expression are listed below (F, forward; R, reverse):
Dock4 F: 5’- GGA TAC CTA CGG AGC ACG AG -3’
R: 5’- AGC CAT CAC ACT TCT CCA GG -3’
Tiam1 F: 5’- GAT CCA CAG GAA CTC CGA AGT -3’
R: 5’- GCT CCC GAA GTC TTC TAG GGT -3’
Prex1 F: 5’- CCT TCT TCC TCT TCG ACA AC -3’
R: 5’- CCA TCT TCC ACA TTC TCC AC -3’
Prex2 F: 5’- TGG GAG GGG TCC AAC ATC A -3’
R: 5’- TCT TCA ACC GTC TGT GTT TTC TT -3’
GAPDH F: 5’- TGA TGA CAT CAA GAA GGT GG -3’
R: 5’- TTT CTT ACT CCT TGG AGG CC -3’
Trans-Endothelial Electrical Resistance.
HPAEC seeded on eight-well gold-plated electrodes (Applied Biosciences, Carlsbad, CA) were transfected with the indicated siRNA for 48 h. Cells were serum-deprived for 1 h, basal resistances were recorded, and then the cells were stimulated with 1 µM S1P as described previously 51. Similarly, HPAEC were infected with both control and DOCK4 shRNA for 48 h, trypsinized and plated on electrodes. Cells were starved for an hour and stimulated with1 µM S1P
Endothelial cell immunostaining and analysis.
HPAEC were electroporated using nucleofector kit (LONZA) with ctl and DOCK4 siRNA (300nM) and plated on 0.1% gelatin coated coverslips for 24hrs. Complete EBM2 (lonza) media was added for another 24 hours. Cells were starved for 6 hours and stimulated with 1µM S1P for the indicated times. Cells were washed and fixed with 2% paraformaldehyde in PBS and stained with Dock4 antibody (Bethyl) for 1h room temperature. Anti-rabbit 568 secondary antibody was used to detect Dock4. Similarly, cells were either stained with VE-cad (sc-9989) or phalloidin for 45min at room temperature. Anti-mouse secondary antibody was used to detect VE-cad. Coverslips were mounted using Invitrogen mounting solution containing DAPI (Prolong antifade) and observed under the microscope. CellProfiler (www.cellprofiler.org) 52 was used to quantify Phalloidin staining. Nuclei identified by DAPI staining were used as seeds for identifying cells by analyzing Phalloidin staining. Analysis used a modification of the ExampleFly pipeline that is available online (using an adaptive strategy, a propagation segmentation algorithm and the Otsu thresholding method). The mean Phalloidin staining intensity per µm2 in each cell was calculated and compared between experimental conditions. Representative images and the CellProfiler pipeline used for analysis are included in online Supplemental Information.
Statistical analysis.
Graphs were generated using GraphPad Prism (GraphPad Software, La Jolla, CA). Graphical presentation of the data shown in Figure 1G was done using the ggplot2 package v3.3.3 53. Comparisons between experimental groups were made using either one-way ANOVA or two-way ANOVA with multiple comparison t-test, using Mann Whitney test, or using Student’s t-test as described in the figure legends. Normality tests were performed using GraphPad Prism Software. Significance values are shown by *p < 0.05, **p < 0.01, ***p < 0.001, ****p<0.0001. Comparisons of CD31 immunostaining results in B16F10 tumors between experimental groups were made using the Wilcoxon Rank Sum Test and the Clustered Wilcoxon Rank Sum Test 54. Variance was not tested to determine whether the applied parametric tests were appropriate.
Figure 1. Generation and Characterization of DOCK4 deficient mice.
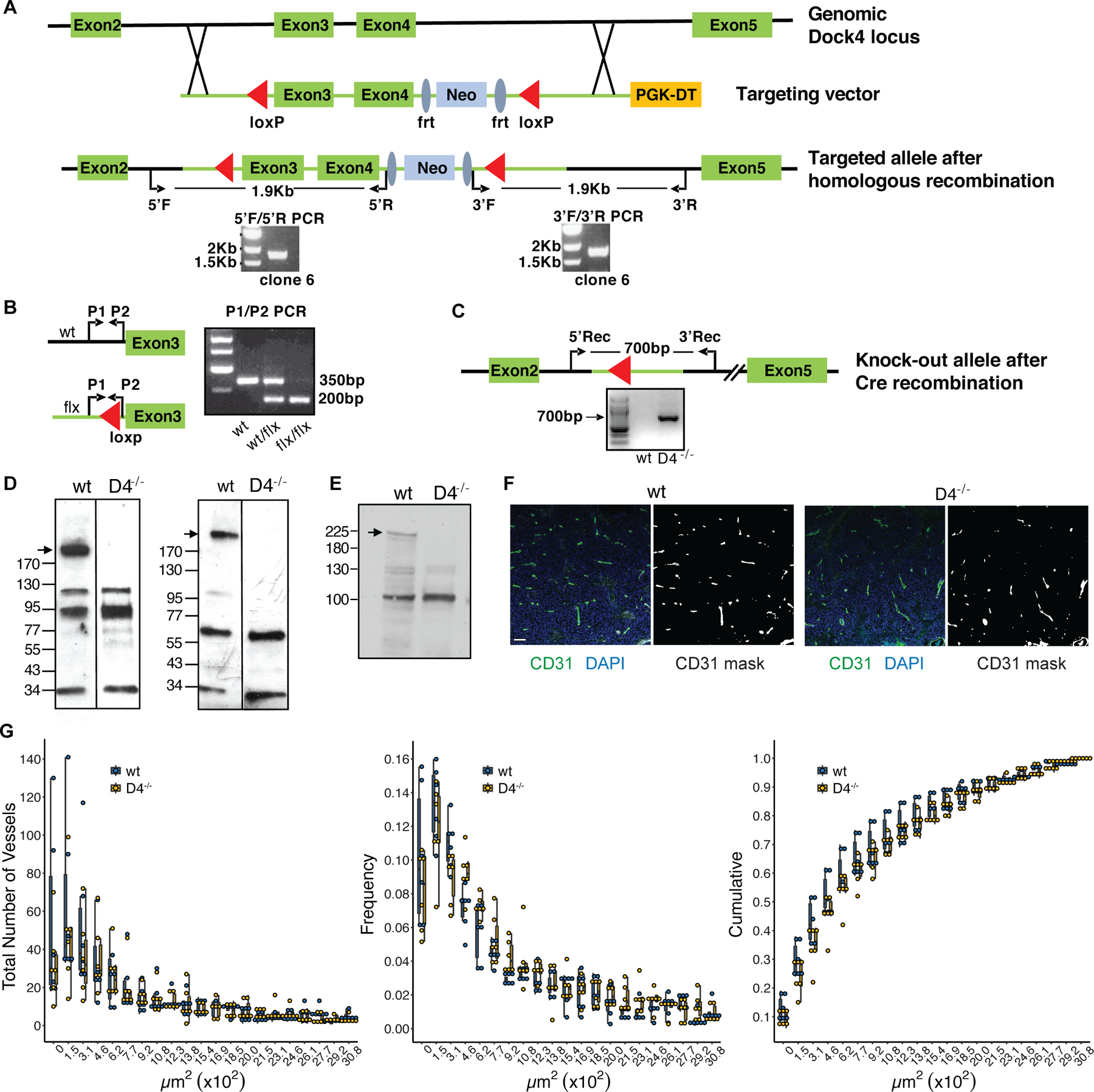
A. Schematic of Dock4 genomic locus and targeting vector. Red triangles: LoxP sites. Blue ellipses: Frt sites. PCR with primer sets 5’F/5’R and 3’F/3’R was used to evaluate integration in ES cells. PCR reactions showing the expected DNA fragments in one positive ES cell clone are shown. B. Genotyping of Dock4 deficient mice using P1/P2 primer set. Primer locations are indicated. Expected DNA fragment sizes are shown for Wild-type (wt), heterozygous (wt/flx) and floxed animals (flx/flx). C. Schematic of Dock4 targeted allele after Cre recombination. PCR using 5’Rec/3’Rec primer set shows the presence of the recombined allele in a Dock4 deficient mouse. D. Western blot showing the absence of Dock4 band in Dock4−/− (D4−/−) mouse brain using N (left) and C (right) terminus antibodies against Dock4. E. Western blot analysis of Dock4−/− (D4−/−) lungs. F&G. CD31 staining and vessel quantification of tumors from wt and Dock4−/− (D4−/−) mice. F. B16F10 tumor implants were immunostained for CD31 (visualized with Alexa 488) and for nuclei (DAPI) (left panels). CD31 masks (right panels) representing vascular structures were identified by image analysis as described in Methods. The scale bar shown as a white rectangle in the left-most panel represents 100 µm and applies to all panels. G. The cross-sectional area of each CD31-expressing vessel in B16F10 tumors was quantified from CD31 masks, such as those illustrated in Fig. 1F, by image analysis as described in Methods. Frequency: the frequency of vessels in bins of 103 µm2 identified in each mouse relative to the total number of vessels identified in the same mouse. Cumulative: the fraction of vessels identified in each mouse with cross-sectional areas that were smaller than or equal to the bin values on the X axis. Individual data points are shown for each animal studied and box plots represent the 25th, 50th and 75th percentiles for the vessels in each bin from mice of each genotype.
RESULTS
DOCK4 is required to maintain vascular barrier function in the lung
To evaluate the physiological role of DOCK4, we generated C57Bl/6J Dock4 knockout (Dock4−/−) mice using a standard Cre-loxP targeted homologous recombination strategy that removes Exon 3 and Exon 4, which encode for 31 amino acids including 26 residues of the N-terminal SH3 domain (Fig. 1A-C). Cre recombination is predicted to result in a frameshift that generates a 56-amino acid truncated N-terminal SH3 domain that includes 16 amino acid nonsense sequence before a stop codon. Dock4−/− mice were viable and fertile and DOCK4 deficiency was confirmed at the protein level using antibodies to the N- and C-terminal ends of the protein (Fig. 1D and E). A recent paper that generated a Dock4 knockout using a gene-trap approach in mice on a mixed strain, reported embryonic lethality in these mice and a role for DOCK4 in lumen formation in heterozygous mice that were implanted intracranially with a syngeneic breast cancer cell line and in a xenograft tumor model following DOCK4 shRNA transduction of the vascular compartment 28. We found that the subcutaneous implantation of B16F10 cells in adult wild-type and Dock4−/− mice resulted in the appearance of tumors in both WT and Dock4−/− mice with similar size (data not shown) and no significant differences in vessel density or size in these highly vascularized tumors (Fig. 1F and G).
Despite the absence of gross abnormalities in the vasculature of Dock4 null animals, the lungs of 12-week old Dock4−/− mice revealed hemorrhage (Fig. 2A) both in peribronchial regions and diffusely within alveoli (Fig. 2B). To determine if this might reflect blood vessel fragility secondary to diminished interaction with perivascular support cells in Dock4−/− mice we determined the anatomic relationship between CD31-positive blood vessels and alpha smooth muscle actin (SMA)-expressing cells that include vascular smooth muscle cells and pericytes 55. Dock4−/− mice showed a smaller fraction of pulmonary blood vessels with 100% SMA coverage compared to wild type mice (Fig. 2C), which could result in vessel fragility and contribute to the observed hemorrhage (Fig. 2A, B). The fractions of these blood vessels with lesser degrees of SMA coverage were not statistically different between Dock4−/− and wild type mice (Fig. 2C). Next, we used 3 approaches to assess whether DOCK4 plays a role in regulating basal permeability in the lung. First, we i.v.-injected 1-Kd fluorescent tracer into WT and Dock4−/− mice and quantified the extravasated dye. Dock4−/− mice exhibited increased dye extravasation compared to control animals (Fig. 2D). Second, we examined lung edema by determining transvascular albumin influx and lung wet-to-dry weight ratio (Fig. 2E). Both were significantly increased in Dock4−/− mouse lungs compared to wild-type counterparts. Third, we assessed extravasation of i.v.-injected Evans Blue, which rapidly binds serum albumin, followed by lung perfusion to remove intravascular dye. The lungs of Dock4−/− animals showed extravasated Evans Blue indicative of barrier disruption that was not observed in the lungs of control animals (Fig. 2F). A similar analysis in the brain of Dock4−/− animals demonstrated barrier disruption as assessed by extravasation of a 1-Kd fluorescent tracer while Evans Blue leakage and wet-to-dry weight ratio analyses did not reveal significant differences (Fig. 2G). Vascular permeability was also not evident in the kidneys of Dock4−/− animals (Fig. 2H). The reduction in SMA coverage (Fig. 2C) known to support endothelial integrity and the observed hemorrhage in larger vessels could contribute to the overall higher Evan’s blue leakage in the lung of DOCK4 versus wild-type mice (Fig. 2F). Therefore, to specifically elucidate whether lung microvascular permeability occurs in the absence of DOCK4, we perfused mice with colloidal carbon that accumulates in open intercellular junctions and is retained within the walls of leaky vessels 56, and evaluated its deposition microscopically. Significantly higher deposition of colloidal carbon was observed in lung capillaries and the smallest other pulmonary vessels of DOCK4 deficient mice compared to wild-type controls (Fig. 2I), suggesting that DOCK4 deficiency specifically leads to impaired microvascular endothelial barrier function. Together, these data suggest that DOCK4 deficiency results in barrier breach for small-molecular-mass tracers in the brain, and in the lung, is important in maintaining large vessel integrity and microvascular barrier function.
Figure 2. Enhanced vascular permeability in Dock4 deficient mice.
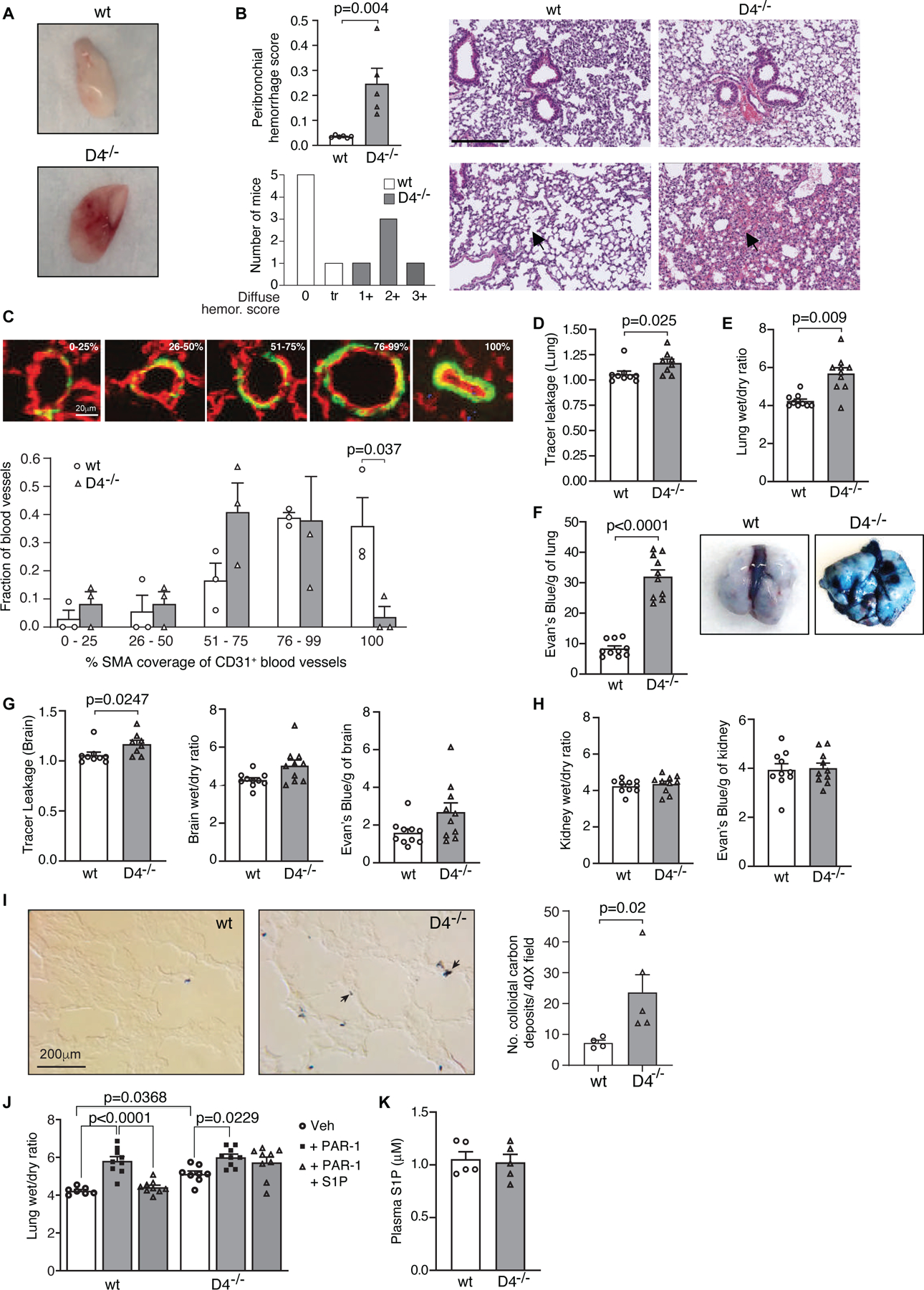
A. A representative image of buffered perfused lungs harvested from wild-type (wt) and Dock4 deficient (D4−/−) mice. B. Assessment of lung hemorrhage score (left). Plot shows fraction of bronchioles with peribronchial hemorrhage (top) and diffuse hemorrhage (bottom) score. Data are mean ± SEM; Student t-test. Representative images of hematoxylin and eosin (H&E)-stained lung sections from wt and D4−/− mice showing peribronchial hemorrhage (top panels) and diffuse hemorrhage (bottom panels). Bar= 250μm. C. CD31 and SMA immunofluorescent staining of wild-type (wt) and Dock4 deficient (D4−/−) lung to assess pericyte coverage. The fractional coverage by SMA of CD31-expressing blood vessels between 20–200μm in greatest diameter in lungs from D4−/− and wt mice was determined. Representative images are shown of pulmonary blood vessels with 0–25%, 26–50%, 51–75% 76–99% and 100% coverage by SMA-expressing cells (green), of CD31-expressing blood vessels (red) in lung. The fraction of blood vessels in each category was calculated for each mouse and these values were compared between D4−/− and wt mice using a Student’s t-test. The white scale bar in the 0–25% panel is 20μm and applies to all panels. D. Quantification of extravasated 1-kDa Alexa Fluor 555–cadaverine shows increased tracer extravasation into the lung of D4−/− compared to wt mice. Data are mean ± SEM; Mann Whitney t-test. E. Lung edema assessed by wet-to- dry ratio shows that Dock4 deletion increases micro- vascular permeability in the lungs. Plot is mean ± SEM; n=10mice/group (unpaired Welch’s t test), F. Permeability in lung as assessed by Evan’s blue dye leakage (left). Data are mean ± SEM; Welch’s t test. Representative image of Evan’s blue in lungs of wt and D4−/− mice (right). G. Quantification of extravasated 1-kDa Alexa Fluor 555–cadaverine (left), wet-dry-ratio (middle) and Evan’s blue dye leak (right) in brains of wt and D4−/− mice. Plot shows mean ± SEM; Welch’s t-test. H. Similar analysis as in G. in kidneys of wt and D4−/− mice. I. Assessment of colloidal carbon deposition in wt and D4−/− lung. Deposits of colloidal carbon, injected i.v. into mice were examined in lungs. Representative images are shown. The black scale bar in the wt panel is 20 μm and applies to both panels. Data are mean ± SEM of the number of colloidal carbon deposits counted/40 X field of lung tissue from each mouse; Student t-test. J. Mice were treated with either PBS or PAR-1 peptide followed 15 mins later by S1P. Lungs were removed after an additional 45 minutes, and vascular permeability was determined by quantifying lung wet/dry weight ratio. S1P failed to prevent PAR-1–induced lung microvascular permeability in D4−/− mice. Data are mean ± SEM; Two-way ANOVA with Tukey’s multiple comparison test. K. Assessment of plasma S1P levels in wt and D4−/− mice. Data are mean ± SEM; Mann Whitney t-test.
Sphingosine 1-phosphate (S1P), a lysosphingolipid has been shown to contribute to maintenance of baseline permeability in vitro and in vivo 29,38. The majority of plasma S1P is bound to circulating macromolecules 57–59. The interactions are complex and varied 59–61 with the result that most plasma S1P is not directly available for cell surface receptor binding. Under homeostasis, S1PR1 signaling is minimal, as determined by the GFP signal in reporter mice. However, a bolus injection of an S1PR1 agonist results in a significant increase in the concentration of free agonist in blood that binds and activates the receptor in vascular endothelial cells of organs, including the lung 62,63. Thus, a bolus intravenous injection of 1 µM S1P (50 µl) is expected to lead to significant activation of the receptor in the lung endothelium. Endothelial treatment with the protease-activated receptor-1 (PAR1)-activating peptide, which is a surrogate for thrombin, disrupts endothelial barrier integrity that can be reversed by the barrier protective action of S1P 38. The i.v. injection of PAR-1 agonist peptide induced an increase in lung permeability, as previously described 38,50, in both WT and Dock4−/− mice as assessed by lung wet/dry weight ratio. 1 µM S1P treatment of WT mice prevented PAR-1 induced endothelial disruption while this was not observed in Dock4−/− mice (Fig. 2J). Differences in the levels of circulating, endogenous S1P were not responsible for the observed increase in baseline permeability as Dock4 deficiency did not affect S1P generation (Fig. 2K). Together, these data indicate that DOCK4 is essential for regulating basal lung barrier function and for S1P mediated reversal of thrombin-induced barrier disruption.
DOCK4 promotes S1P induced enhancement of endothelial barrier properties and Rac1 activation.
To evaluate the role of DOCK4 in human endothelial cell permeability and gain a mechanistic understanding of DOCK4’s role in barrier function, we silenced DOCK4 in HPAEC with siRNAs. DOCK4 silencing in HPAEC resulted in decreased transendothelial electrical resistance (TEER) across the monolayer compared to Control siRNA cells indicating a defect in the maintenance of basal barrier function (Fig. 3A and B). Importantly, S1P failed to increase TEER in DOCK4 compared to control siRNA cells. The defects in the response to S1P were not due to decreased expression of S1PR1 in DOCK4 knockdown cells (Fig. 3C) and occurred despite the presence of canonical GEFs (Fig. 3D) that have been well-described to link S1P to Rac activation in endothelial cells 64. Thus, as observed in vivo, DOCK4 plays an important role in maintaining endothelial barrier function in vitro and is required for its enhancement after S1P stimulation.
Figure 3. DOCK4 silencing in human lung endothelial cells increases basal permeability.
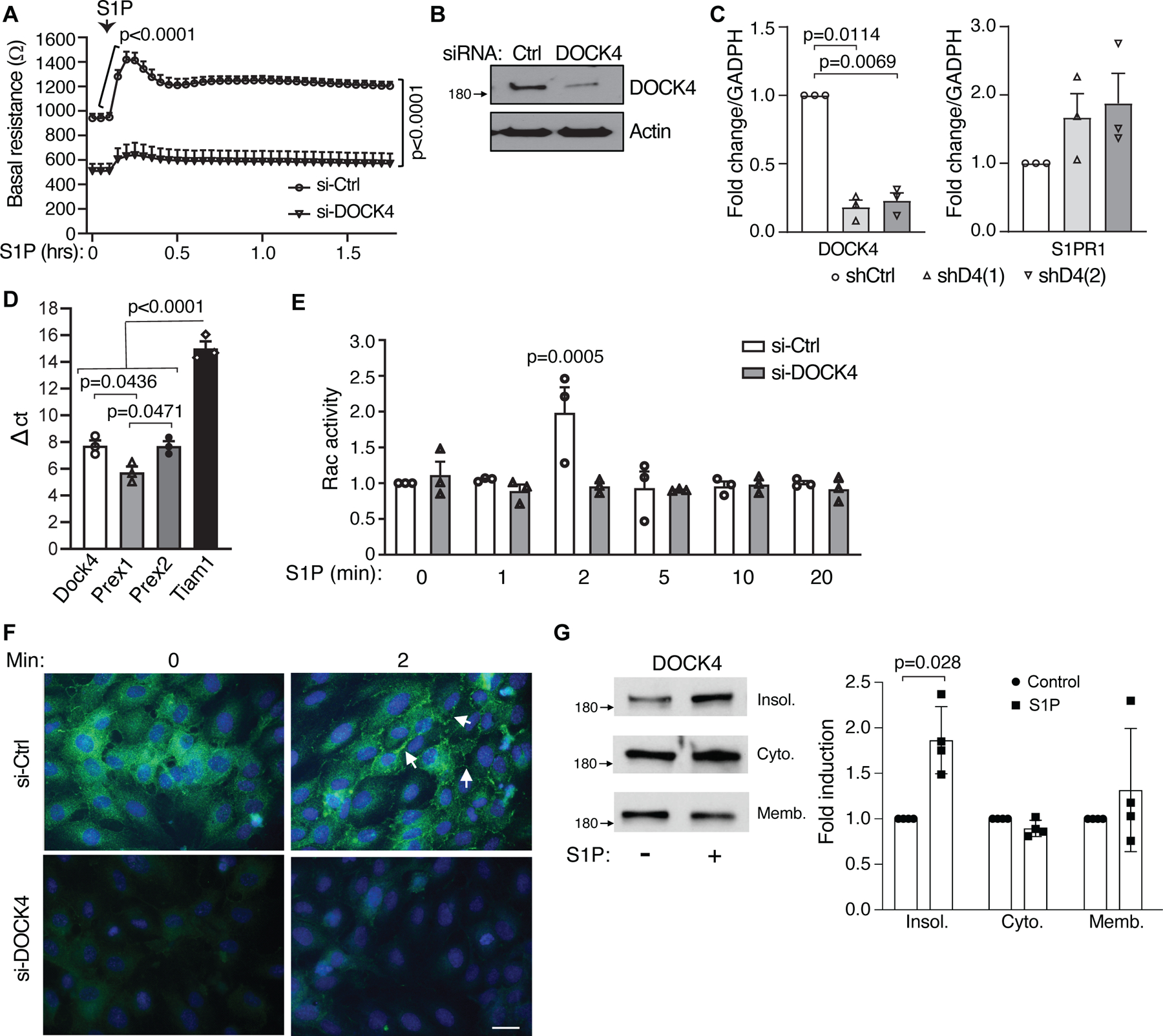
A. HPAEC were electroporated with control (Ctrl) or DOCK4 siRNA and seeded on gold-plated electrodes for 48 hrs. Cells were serum starved for 2 h after which endothelial barrier function was determined by measuring transendothelial electrical resistance (TEER) over time (in hours) before and after addition of 1 µM S1P. Data are mean ± SEM; Two-way ANOVA multiple comparison (Bonferroni). B. Western blot of DOCK4 in siRNA cells. C. mRNA levels of S1PR1 (left) and DOCK4 (right) in DOCK4 knockdown HPAEC using two independent DOCK4 shRNAs. Data are mean fold increase versus levels in HPAEC transduced with shRNA control lentivirus (Ctrl); Welch’s t test. D. mRNA levels of indicated guanine exchange factors in HPAEC. Data are mean ± SEM.; one-way ANOVA, Tukey’s multiple comparisons. E. HPAEC electroporated with Ctrl or DOCK4 siRNA were plated on 6 well plate. After 48 hrs, cells were starved for 5 hrs, stimulated with 1µM S1P for different times and lysates were used to assess Rac1 activity. Data are mean ± SEM; two-way ANOVA, Tukey’s multiple comparisons. F. HPAEC transfected with Ctrl and DOCK4 siRNA were plated on glass coverslips for 48hrs. Cells were serum starved for 4 hrs followed by treatment with 1 µM S1P for the indicated times in minutes. Cells were fixed and stained for DOCK4 (Green) (n=3). Bar = 20 µm. G. HPAEC were treated with S1P or vehicle control for 1 min and DOCK4 levels in equal amounts of protein from each of the obtained fractions (cytosolic, membrane and insoluble cytoskeletal/nuclear) were analyzed by western blot (left). Western blot quantification of DOCK4 levels after S1P treatment (right). Data are mean fold increase in S1P treated vs. control cells ± SD; Mann-Whitney test.
S1P induced Rac activation increases actin polymerization in the periphery of endothelial cells and lamellipodia formation, both of which contribute to barrier enhancement 65,66. S1P induced GTP loading on Rac was observed within 2 min of treatment in Control siRNA cells albeit this was modest as reported by others 67. Notably, this increase failed to occur in similarly treated DOCK4 siRNA cells (Fig. 3E). Immunofluorescence analysis of DOCK4 in endothelial cells showed cytoplasmic staining in untreated cells while S1P stimulation resulted in DOCK4 staining in the cell periphery (Fig. 3F). The submembranous cytoskeleton, together with the peripheral actin ring, are important determinants of the endothelial barrier integrity 68. S1P treatment of HPAEC resulted in an increase in DOCK4 protein in the detergent-insoluble compartment that includes the cytoskeleton (Fig. 3G). Since the cytosolic and membrane fractions contained the majority of DOCK4 (>90%), the increase in the insoluble compartment is not reflected as a significant decrease in the other fractions This mobilization of DOCK4 may facilitate its interaction with adaptor molecules necessary for its function and could localize Rac activation to mediate S1P effects at cortical sites 69.
A role for DOCK4 in regulating Rho GTPases activation in endothelial cells
Endothelial cytoskeleton dynamics play an important role in maintenance of barrier function with the formation of stress fibers and activation of the myosin contraction system resulting in increased endothelial permeability 70,71. To evaluate the role of DOCK4 in regulating the endothelial cytoskeleton, we performed immunofluorescence analysis of F-actin in HPAEC transduced with control lentivirus or two independent DOCK4 targeting shRNA lentiviruses. DOCK4shRNA resulted in a significant increase in actin stress fibers, cell retraction with increased intercellular gap formation and reduced VE-cadherin at cell-cell junctions compared to control cells (Fig. 4A). Activation of Rho GTPases promote stress fiber formation leading to endothelial barrier disruption 1. Knock-down of DOCK4 (Fig. 4B) resulted in an increase in RhoA GTP loading (Fig. 4C) compared to control cells despite comparable total Rho A levels (Fig. 4B) while a 10 min treatment of the DOCK shRNA cells with an inhibitor of Rho associated protein kinase (ROCK), a key Rho effector and regulator of the cytoskeleton 72–74, decreased actin stress fibers to levels observed in control cells (Fig. 4D). Inactivation of myosin light-chain phosphatase through phosphorylation by Rho kinase results in increased net phosphorylation of myosin-light chain thus enhancing actomyosin contractility and endothelial permeability 75. DOCK4 shRNA cells have increased levels of MLC phosphorylation compared to control cells (Fig. 4E), which is consistent with the enhanced RhoA GTP loading and actin stress fibers in these cells. The effect of Dock4 knockdown on Rac activation downstream of S1P was also evaluated using the two independent DOCK4 shRNA lentiviruses. With this independent approach, DOCK4 deficiency prevented S1P-induced Rac activation (Fig. 4F). This recapitulates the results using siRNA (Fig. 3A), and more importantly, together with the demonstrated activation of Rho and its downstream pathway, the imbalance of Rac and Rho in the absence of DOCK4.
Figure 4. DOCK4 maintains the balance between Rho/Rac1 GTPases.
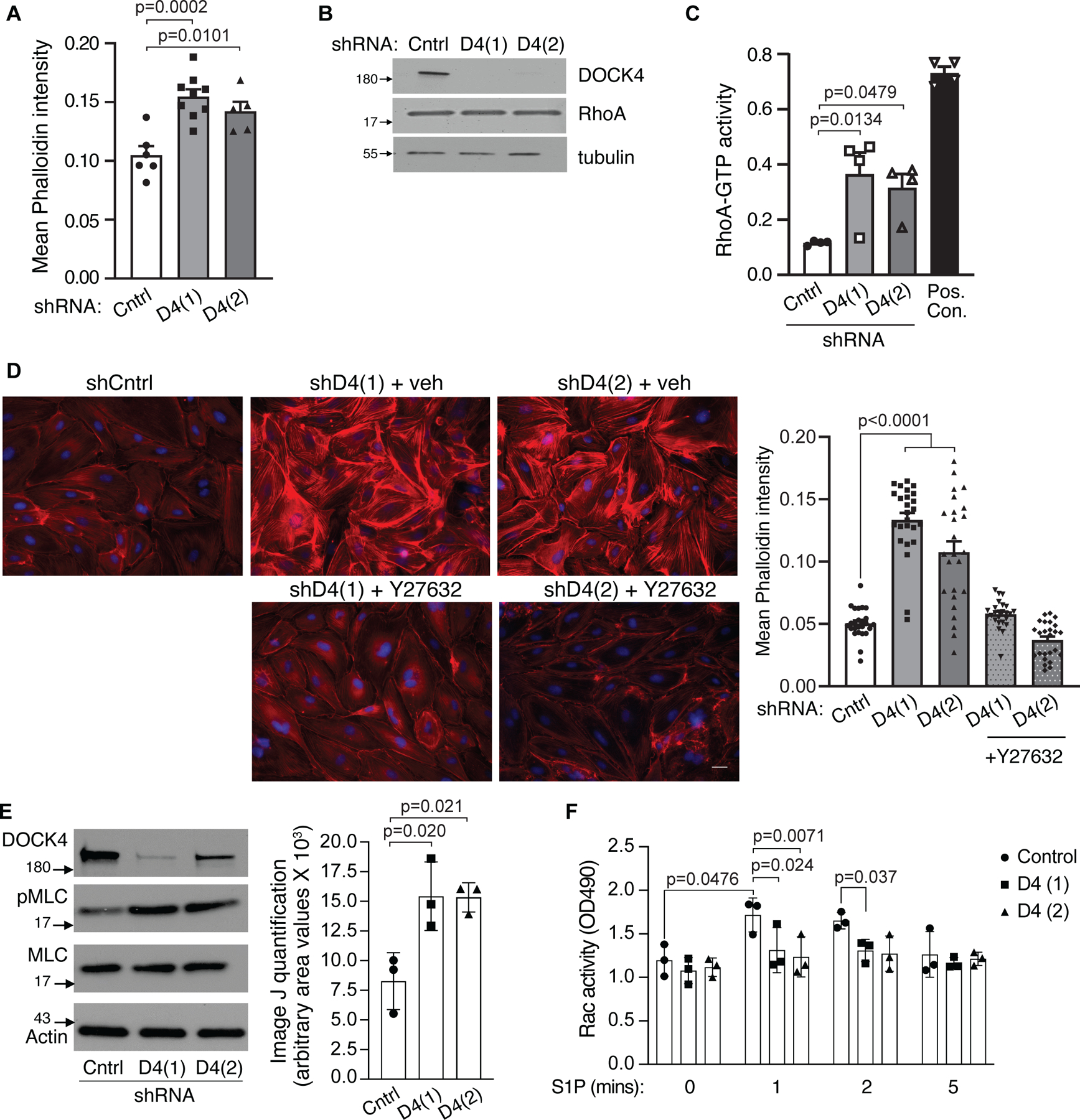
A. DOCK4 depletion induces actin stress fibers. HPAEC were transduced with Control (Cntrl) shRNA or two DOCK4 D4(1) and D4(2) shRNA clones for 72hrs. Cells were fixed and stained with phalloidin 568 and mean phalloidin intensity was measured. Data are mean ± SEM; One-way ANOVA, Tukey’s multiple comparisons. B. Representative western blot of DOCK4 and RhoA in shRNA cells. C. HPAE cells transduced with Cntrl, and D4 shRNAs for 72hrs were starved for 4–5 hours and lysates were used to assess RhoA GTPase activity using an ELISA assay. Data are mean ± SEM; One-way ANOVA, Tukey’s multiple comparisons (n=4). D. Cntrl and DOCK4 shRNA HPAEC treated with vehicle or ROCK inhibitor (Y27632) 10µM for 10 min were fixed, stained with Phalloidin Alexa 568 and quantified for mean phalloidin intensity. Data are mean ± SEM; Two-way ANOVA, Tukey’s multiple comparisons. E. Dock4 deficiency results in increased MLC phosphorylation. HPAEC were transduced with control or DOCK4 shRNA lentiviruses as in A. After 72 hours, the levels of phosphorylated MLC were assessed by western blot analysis. Total MLC and actin served as loading controls. Data are mean ± SD; One-way ANOVA and Tukey’s multiple comparisons. F. HPAEC were transduced with control or DOCK4 ShRNA lentiviruses as in A. After 72 hrs, cells were starved for 4 hours and Rac activity was measured after treatment with 1 uM S1P at the indicated times using an ELISA assay. Data are mean ± SD; Two-way ANOVA and Tukey’s multiple comparisons.
To exclude the possibility of off-target effects of the shRNAs, we re-expressed wild-type DOCK4 or LacZ in DOCK4 shRNA cells using an adenoviral approach that reconstituted DOCK4 (Fig. 5A) due to the transient increase in DOCK4 transcription that escapes the shRNA-mediated mRNA degradation. DOCK4 reconstitution was associated with a dramatic reduction in the inter-endothelial gap formation and the restoration of VE-cadherin to cell-cell contacts that was comparable to that observed in control shRNA cells (Fig. 5B). Similarly, while LacZ adenovirus transduction did not rescue the observed increase in basal permeability in DOCK4 deficient cells, WT DOCK4 reconstitution of DOCK4 shRNA cells restored the basal barrier function in these cells (Fig. 5C) and, importantly, the ability of S1P to enhance barrier function (Fig. 5D).
Figure 5. DOCK4 expression in DOCK4 depleted endothelial cells restores barrier function.
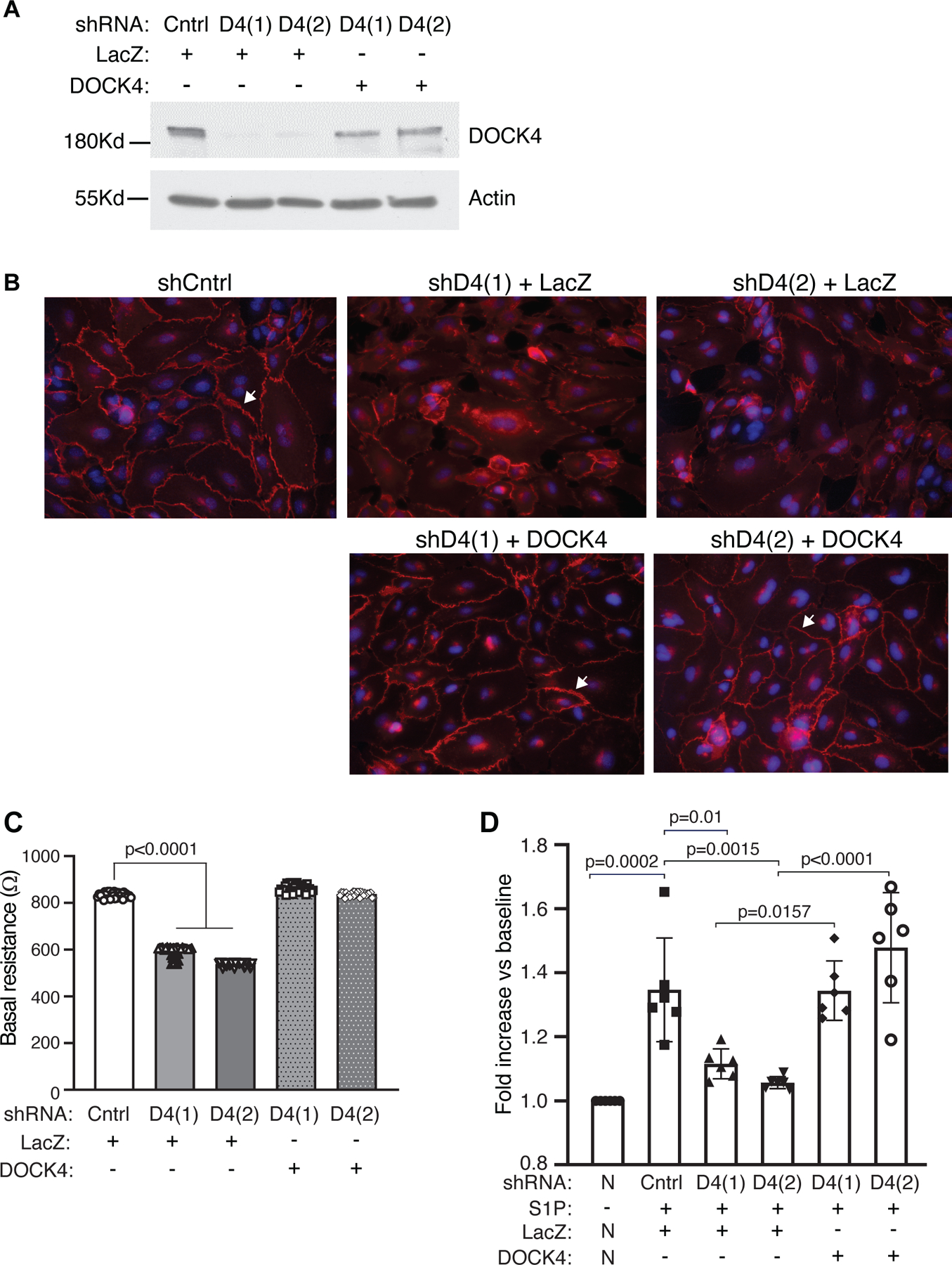
A. Transduction of Control (Cntrl) or D4 shRNA endothelial monolayers with wild-type DOCK4 or LacZ adenovirus as indicated. Monolayers transduced with DOCK4 resulted in DOCK4 protein expression. Representative western blot is shown B. VE-cadherin staining (arrow) of Cntrl and D4 shRNA cells transduced with LacZ or DOCK4 adenovirus as indicated. C. HPAEC were transduced with Cntrl or DOCK4 shRNA clones for 48hrs. Cells were trypsinized and plated on electrodes to allow monolayers to form. Wells were then transduced with LacZ or DOCK4 adenovirus for 24hrs. Basal resistance was measured after 72hrs. Data are mean ± SEM; Two-way ANOVA with Tukey’s multiple comparisons. D. Cntrl or DOCK4 shRNA cells, as in Fig. 5C, were transduced with LacZ or DOCK4 adenovirus respectively. After 24hrs, cells were starved for 2 hrs and TEER was examined before and after stimulation with 1 µM S1P. Each experimental sample was normalized (N) to its own baseline (-S1P). Data are mean ± SEM; Two-way ANOVA with Tukey’s multiple comparisons.
CONCLUSIONS
Rho GTPases play a major role in coordinating signals from cell surface receptors that regulate focal adhesion and cytoskeletal dynamics, thereby controlling cell-cell and cell-matrix interactions in response to external signals. Endothelial barrier integrity critically relies on the cooperative signaling balance between Rac1 and RhoA GTPases. In vivo, endothelial deletion of Rac1 results in lethality at E9.5, due to defects in vasculogenesis and sprouting angiogenesis 76, deletion at E10 results in a reduction in vascular density and hemorrhage in the skin and post-natal deletion results in abnormal retinal angiogenesis 77. Rac1 deletion in primary endothelial cells in vitro interferes with endothelial cell migration, tubulogenesis, adhesion, and permeability in response to sphingosine‐1‐phosphate (S1P), which is attributed to the inability of Rac1‐deficient endothelial cells to form lamellipodial structures and focal adhesions, and to remodel their cell‐cell contacts 77,78. Global deficiency of RhoA results in embryonic lethality 79 while conditional, endothelial restricted deletion of RhoA does not lead to gross vascular abnormalities 80. Here, we demonstrate that global deletion of DOCK4 does not impair embryo viability but does disrupt tonic barrier function in vessels in vivo and in endothelial cells in vitro that is associated with a large induction of Rho activation and associated stress fibers and unresponsiveness to a Rac activator, S1P. Together, these data suggest that an atypical GEF, DOCK4 plays an important role in balanced signaling through Rac and Rho GTPases. The surface receptors that regulate DOCK4 activity and therefore the role of this protein in specific endothelial signaling pathways remain largely unknown 81. Our study demonstrating that S1P induces the relocation of DOCK4 to the cell periphery with increased translocation to a detergent-insoluble compartment and that DOCK4 is required for S1P induced Rac activation and enhancement of barrier function suggests that DOCK4 participates in endothelial S1PRs’ signaling to Rac GTPases, a fruitful area for additional studies.
The hemorrhage observed in the lungs of DOCK4 deficient mice was associated with fewer pulmonary blood vessels showing complete coverage by SMA-expressing cells than in wild type mice. Interrupted support by perivascular cells is associated with blood vessel fragility and hemorrhage and is observed as a result of numerous mutations that affect blood vessel development and in pathophysiologic settings including Hereditary Haemorrhagic Telangiectasia, CADASIL and cerebral cavernous malformations 82,83. The endothelium promotes SMC recruitment and proliferation during tubulogenesis with SMC reciprocating with cues that promote endothelial and vascular function 84. S1PR1-deficient mice die around embryonic 13.5 with incomplete vascular smooth muscle coverage around the developing dorsal aorta, which results in ectopic sprouting 85,86. A similar phenotype was reported in endothelial cell-specific S1PR1 knock-out mice 87. Analysis of post-natal retinal vascular development did not observe the incomplete vascular smooth muscle and pericyte recruitment defects 34,88,89. However, in tumor vessels, endothelial cell S1PR1 is needed for pericyte and vascular smooth muscle coverage 90. Thus, S1PR1 regulation of mural cell coverage is vessel type and context dependent. As DOCK4 contributes to endothelial S1P signaling, we speculate that DOCK4 deficiency in the endothelium contributes to defects in SMC coverage in a pathophysiological context specific manner.
DOCK4 deficient mice exhibit microvascular permeability specifically in lung while only permeability to small molecular weight tracers was observed in the brain, and kidneys showed no increase in albumin leakage. The hemorrhage was associated with decreased perivascular coverage in vessels of DOCK4 deficient mice. Increased microvascular permeability in the lung in DOCK4 deficient animals was also detected by increased deposition of colloidal carbon in the pulmonary microvasculature, which delineates leaky vessels. The selectivity in permeability may be due to differences in vascular beds. For instance, coronary, pulmonary, brain, splanchnic, and skeletal muscle are composed of continuous non-fenestrated endothelium that forms a restrictive barrier 91. Organs such as liver, kidney, and lymphatics are formed from discontinuous and highly permeable endothelial monolayers 92. The increased permeability to only small molecular weight tracers in the brain and the observed trend of vascular edema in this organ may be interesting to follow-up as these results partially recapitulate the phenotypes of S1PR1 deficient mice 93. Interestingly, even knockout of the major TJ molecule Claudin 5 results in a selective defect in small but not large molecular tracers 94. Despite the high levels of Dock4 mRNA in the lung and brain, particularly in the endothelium in these organs (proteinatlas.org, bgee.org), it is plausible that the observed effects in vivo arise from DOCK4 deficiency in more than one cell type as DOCK4 mRNA is widely expressed. Nonetheless, the phenotypes in DOCK4 null mouse lungs were recapitulated in endothelial cells in vitro. Depletion of DOCK4 in endothelial cells, the well-recognized gatekeepers of vascular permeability in vivo 95, both reduced basal barrier function and rendered the endothelium resistant to S1P induced barrier enhancement. The lack of recovery of S1P induced barrier function after acute PAR1 challenge supports the involvement of DOCK4 in regulating endothelial barrier properties in vivo. The viability and normal angiogenesis observed in DOCK4 deficient mice suggest that DOCK4 may be redundant with other GEFs required for S1PR1 functions. For example, the GEF Tiam1 has been shown to mediate tumor angiogenesis downstream of S1PR1 in cooperation with VEGFR2 96.
Several studies have shown that cooperation among the Rho GTPases is required for balancing signaling and related cytoskeletal reorganization, which are essential for maintaining the stability of the mature endothelium including the integrity of AJs. Activation of RhoA by permeability-increasing mediators regulates stress fiber formation and disruption of AJs 97–99. Activation of Rac1 can promote AJ assembly by suppressing RhoA activity at the junction level 100 as the structural basis for Rho regulation is the sequestration of GEFs for RhoA by AJ and TJ. For example, Rac1 inhibition of p190RhoGAP, which localizes to AJs via p120-catenin leads to inhibition of RhoA 101. Active Rac1 also promotes cortical actin assembly and re-annealing of endothelial AJ in part by inhibiting VE-cadherin disassembly from junctions 100,102. In addition to AJs, integrin mediated adhesion and focal adhesion complexes at the cell-substrate interface can lead to Rac activation, and cross-talk between AJ and integrins is known to coordinate barrier function 3,103. Interestingly, the conditional loss of endothelial focal adhesion kinase results in RhoA hyperactivation, which in turn antagonizes Rac1 activity and subsequent loss of lung vascular barrier function 103. The concept that RhoA can downregulate Rac1 activity is supported by recent findings that RhoA suppresses Rac1 activity by activating FILGAP, a specific GTPase for Rac1 104. Our studies suggest that in the resting endothelium, the absence of DOCK4 leads to insufficient Rac activation-induced AJ stabilization that in turn causes RhoA activation or defective integrin mediated adhesion and subsequent RhoA hyperactivation. This not only has direct consequences for basal barrier maintenance through stress fiber-mediated cell retraction but, coupled with the resulting inadequate Rac activation provides a feed-forward loop with deleterious effects on endothelial barrier homeostasis. The restoration of VE-cadherin at contact sites and the normalization of the actin cytoskeleton after inhibition of the Rho effector Rho Kinase indicates that the interruption of this loop is sufficient to restore basal barrier properties in DOCK4 deficient cells.
Upstream regulators of DOCK4 remain largely unknown. The binary DOCK−ELMO complexes adopt a closed auto-inhibited conformation, while in the ternary complex of DOCK-ELMO-Rac proteins, the DOCK-ELMO complex adopts an active state that is induced by conformational changes of ELMO. This allows for the interaction of DOCK4’s DHR2 domain with Rac to promote GTP loading 81. Interaction of upstream regulators with ELMO, including brain-specific angiogenesis inhibitor (BAI) and RHOG could contribute to the open conformation 105. Alternatively, phosphorylation of DOCK proteins 106,107 and/or ELMO 108 in the binary complex could promote the active/open state while subsequent interactions of ELMO with membrane proteins, such as RHOG, could recruit the complex to membrane compartments that allow Rac interaction and GTP loading 81. Several ELMO interacting proteins have been shown to regulate DOCK4-mediated Rac activation in different cell types. These include BOC, the Shh receptor, that regulates axonal growth turning 109 and RhoG which recruits ELMO2 and Dock4 to form a complex with EphAs in migrating breast cancer cells 110. In endothelial cells, a role for DOCK4 has been demonstrated downstream of the scavenger receptor class B type 1 (SR-B1) that mediates the delivery of LDL into arteries thereby promoting atherosclerosis 111. Endothelial DOCK4 has also been placed downstream of VEGF to activate Rac and formation of filopodia that is important for lumen morphogenesis while in the same study, targeted DOCK4 deletion resulted in embryonic lethality that is associated with alterations in vessel density and lumen size 28. However, in our viable DOCK4 deficient mice, vessel size and density under basal conditions and in a melanoma model showed no significant differences compared to WT animals. The discrepancy in results with Abraham et al. 28 may potentially be related to the different genetic backgrounds studied (C57BL/6J vs. CD-1/C57Bl/6) and/or the different genetic interventions used to generate mutant DOCK4 alleles (gene targeting by homologous recombination vs. gene trap). Mutant alleles may inadvertently produce DOCK4 protein fragments, which cannot be evaluated in Abraham et al. 28 as DOCK4 protein levels were not reported.
Our studies suggest that S1P may link to Rac via DOCK4 as the absence of DOCK4 prevents S1P induced Rac activation and barrier enhancement and S1P treatment leads to DOCK4 translocation to cell borders. S1P signals through widely expressed GPCRs (S1PR1- S1PR5). Of these, S1PR1 is primarily implicated in barrier strengthening by stimulating the G-protein Gαi, which in turn induces intracellular Ca2+ transients, activation of the small GTPases Rac1 and Cdc42, phosphatidylinositol 3-kinase (PI3K), and ERK 64,102,112,113. DOCK family members have been implicated downstream of GPCRs, as ELMO proteins directly interact with the receptors, such as the brain-specific angiogenesis inhibitor (BAI), or the associated G proteins 114. In other cases, indirect activation of DOCK proteins by GPCRs has been documented. Engagement of the Met-Leu-Phe receptor in neutrophils activates RHOG that in turn recruits DOCK2 to the membrane leading to Rac activation 115 while GPCR activation of PI3K results in the recruitment of DOCK2 via its DHR1 domain to PIP3 at the membrane 116. Our data indicating translocation of DOCK4 to the cell periphery and increased association with a cytoskeletal-rich insoluble compartment places DOCK4 downstream of SIPR activation. Elucidating the molecular mechanism of DOCK4 activation by S1PR remains an interesting area for future studies.
In summary, we show that DOCK4 maintains lung vascular barrier function by balancing the activation of RhoA and Rac1GTPases. Loss of pulmonary endothelial barrier function that occurs as a consequence of various types lung injury is associated with an overall ~40% mortality in affected patients and pulmonary hypertension 2,117. It would be of interest to determine whether DOCK4 downregulation is associated with enhanced susceptibility to lung edema in the context of sterile and pathogenic stimuli.
Supplementary Material
{kind=link}
HIGHLIGHTS.
Mice deficient in DOCK4, an unconventional guanine exchange factor for Rho GTPases have increased basal microvascular permeability.
DOCK4 deficiency leads to hemorrhage in the lung and decreased smooth muscle cell coverage of large pulmonary vessels
DOCK4 deficiency prevents sphingosine-1-phosphate-mediated reversal of thrombin-induced barrier leakage in the lung
DOCK4 silencing in endothelial cells prevents sphingosine-1-phosphate induced Rac-1 GTPase activation and enhancement of barrier function
DOCK4 silencing in endothelial cells increases basal permeability that is associated with enhanced Rho GTPase activation
SIGNIFICANCE STATEMENT.
The endothelial monolayer plays a critical role in regulating vascular permeability both under homeostasis and following exposure to edemagenic agonists. Members of the Rho family of GTPases and conventional guanine exchange factors of these GTPases have been demonstrated to play important roles in regulating endothelial permeability. We provide in vitro and in vivo evidence that DOCK4, a member of the CDM (CED-5, Dock180, Myoblast city) family of unconventional guanine exchange factors for Rho GTPases, plays a key role in regulating vascular permeability in the lung and identify an imbalance of endothelial Rho and Rac GTPase activation in the absence of DOCK4 as the primary mechanism.
ACKNOWLEDGEMENTS
a) The statistical analysis was conducted with support from Harvard Catalyst | The Harvard Clinical and Translational Science Center.
b) The work was supported by National Institutes of Health R01HL065095, R01DK099507 and P01HL036028 (TM), and K01AR054984 (XC). The Harvard Clinical and Translational Science Center was supported by the National Center for Advancing Translational Sciences, National Institutes of Health Award UL 1TR002541 and financial contributions from Harvard University and its affiliated academic healthcare centers.
Non-standard Abbreviations and Acronyms
- AJs
adherens Junctions
- Dock4−/−, D4−/−
Dock4 knock-out
- GPCR
G-protein coupled receptor
- GEF
guanine-Exchange Factor
- HPAEC
human pulmonary artery endothelial cells
- MLC
myosin light chain
- S1P
sphingosine 1-phosphate
- VE-cadherin
vascular-endothelial cadherin
- WT, wt
wild-type
Footnotes
c) The authors have no conflicts of interest to disclose.
REFERENCES
- 1.Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev 2006;86:279–367. doi: 10.1152/physrev.00012.2005 [DOI] [PubMed] [Google Scholar]
- 2.Matthay MA, Zemans RL. The acute respiratory distress syndrome: pathogenesis and treatment. Annu Rev Pathol 2011;6:147–163. doi: 10.1146/annurev-pathol-011110-130158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sukriti S, Tauseef M, Yazbeck P, Mehta D. Mechanisms regulating endothelial permeability. Pulm Circ 2014;4:535–551. doi: 10.1086/677356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bos JL, Rehmann H, Wittinghofer A. GEFs and GAPs: critical elements in the control of small G proteins. Cell 2007;129:865–877. doi: 10.1016/j.cell.2007.05.018 [DOI] [PubMed] [Google Scholar]
- 5.Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol 2005;6:167–180. doi: 10.1038/nrm1587 [DOI] [PubMed] [Google Scholar]
- 6.van Buul JD, Geerts D, Huveneers S. Rho GAPs and GEFs: controling switches in endothelial cell adhesion. Cell Adh Migr 2014;8:108–124. doi: 10.4161/cam.27599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barry DM, Xu K, Meadows SM, Zheng Y, Norden PR, Davis GE, Cleaver O. Cdc42 is required for cytoskeletal support of endothelial cell adhesion during blood vessel formation in mice. Development 2015;142:3058–3070. doi: 10.1242/dev.125260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yuan L, Sacharidou A, Stratman AN, Le Bras A, Zwiers PJ, Spokes K, Bhasin M, Shih SC, Nagy JA, Molema G, et al. RhoJ is an endothelial cell-restricted Rho GTPase that mediates vascular morphogenesis and is regulated by the transcription factor ERG. Blood 2011;118:1145–1153. doi: 10.1182/blood-2010-10-315275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hunter SG, Zhuang G, Brantley-Sieders D, Swat W, Cowan CW, Chen J. Essential role of Vav family guanine nucleotide exchange factors in EphA receptor-mediated angiogenesis. Mol Cell Biol 2006;26:4830–4842. doi: 10.1128/MCB.02215-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mertens AE, Roovers RC, Collard JG. Regulation of Tiam1-Rac signalling. FEBS Lett 2003;546:11–16. doi: 10.1016/s0014-5793(03)00435-6 [DOI] [PubMed] [Google Scholar]
- 11.Amado-Azevedo J, de Menezes RX, van Nieuw Amerongen GP, van Hinsbergh VWM, Hordijk PL. A functional siRNA screen identifies RhoGTPase-associated genes involved in thrombin-induced endothelial permeability. PLoS One 2018;13:e0201231. doi: 10.1371/journal.pone.0201231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kather JN, Kroll J. Rho guanine exchange factors in blood vessels: fine-tuners of angiogenesis and vascular function. Exp Cell Res 2013;319:1289–1297. doi: 10.1016/j.yexcr.2012.12.015 [DOI] [PubMed] [Google Scholar]
- 13.Cote JF, Vuori K. Identification of an evolutionarily conserved superfamily of DOCK180-related proteins with guanine nucleotide exchange activity. J Cell Sci 2002;115:4901–4913. doi: 10.1242/jcs.00219 [DOI] [PubMed] [Google Scholar]
- 14.Meller N, Irani-Tehrani M, Kiosses WB, Del Pozo MA, Schwartz MA. Zizimin1, a novel Cdc42 activator, reveals a new GEF domain for Rho proteins. Nat Cell Biol 2002;4:639–647. doi: 10.1038/ncb835 [DOI] [PubMed] [Google Scholar]
- 15.Cote JF, Motoyama AB, Bush JA, Vuori K. A novel and evolutionarily conserved PtdIns(3,4,5)P3-binding domain is necessary for DOCK180 signalling. Nat Cell Biol 2005;7:797–807. doi: 10.1038/ncb1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Premkumar L, Bobkov AA, Patel M, Jaroszewski L, Bankston LA, Stec B, Vuori K, Cote JF, Liddington RC. Structural basis of membrane targeting by the Dock180 family of Rho family guanine exchange factors (Rho-GEFs). J Biol Chem 2010;285:13211–13222. doi: 10.1074/jbc.M110.102517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hasegawa H, Kiyokawa E, Tanaka S, Nagashima K, Gotoh N, Shibuya M, Kurata T, Matsuda M. DOCK180, a major CRK-binding protein, alters cell morphology upon translocation to the cell membrane. Mol Cell Biol 1996;16:1770–1776. doi: 10.1128/mcb.16.4.1770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kiyokawa E, Hashimoto Y, Kobayashi S, Sugimura H, Kurata T, Matsuda M. Activation of Rac1 by a Crk SH3-binding protein, DOCK180. Genes Dev 1998;12:3331–3336. doi: 10.1101/gad.12.21.3331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gumienny TL, Brugnera E, Tosello-Trampont AC, Kinchen JM, Haney LB, Nishiwaki K, Walk SF, Nemergut ME, Macara IG, Francis R, et al. CED-12/ELMO, a novel member of the CrkII/Dock180/Rac pathway, is required for phagocytosis and cell migration. Cell 2001;107:27–41. doi: 10.1016/s0092-8674(01)00520-7 [DOI] [PubMed] [Google Scholar]
- 20.Laurin M, Cote JF. Insights into the biological functions of Dock family guanine nucleotide exchange factors. Genes Dev 2014;28:533–547. doi: 10.1101/gad.236349.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gadea G, Blangy A. Dock-family exchange factors in cell migration and disease. Eur J Cell Biol 2014;93:466–477. doi: 10.1016/j.ejcb.2014.06.003 [DOI] [PubMed] [Google Scholar]
- 22.Kunimura K, Uruno T, Fukui Y. DOCK family proteins: key players in immune surveillance mechanisms. Int Immunol 2020;32:5–15. doi: 10.1093/intimm/dxz067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nishikimi A, Kukimoto-Niino M, Yokoyama S, Fukui Y. Immune regulatory functions of DOCK family proteins in health and disease. Exp Cell Res 2013;319:2343–2349. doi: 10.1016/j.yexcr.2013.07.024 [DOI] [PubMed] [Google Scholar]
- 24.Pakes NK, Veltman DM, Williams RS. Zizimin and Dock guanine nucleotide exchange factors in cell function and disease. Small GTPases 2013;4:22–27. doi: 10.4161/sgtp.22087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hernandez-Vasquez MN, Adame-Garcia SR, Hamoud N, Chidiac R, Reyes-Cruz G, Gratton JP, Cote JF, Vazquez-Prado J. Cell adhesion controlled by adhesion G protein-coupled receptor GPR124/ADGRA2 is mediated by a protein complex comprising intersectins and Elmo-Dock. J Biol Chem 2017;292:12178–12191. doi: 10.1074/jbc.M117.780304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schaker K, Bartsch S, Patry C, Stoll SJ, Hillebrands JL, Wieland T, Kroll J. The bipartite rac1 Guanine nucleotide exchange factor engulfment and cell motility 1/dedicator of cytokinesis 180 (elmo1/dock180) protects endothelial cells from apoptosis in blood vessel development. J Biol Chem 2015;290:6408–6418. doi: 10.1074/jbc.M114.633701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sanematsu F, Hirashima M, Laurin M, Takii R, Nishikimi A, Kitajima K, Ding G, Noda M, Murata Y, Tanaka Y, et al. DOCK180 is a Rac activator that regulates cardiovascular development by acting downstream of CXCR4. Circ Res 2010;107:1102–1105. doi: 10.1161/CIRCRESAHA.110.223388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abraham S, Scarcia M, Bagshaw RD, McMahon K, Grant G, Harvey T, Yeo M, Esteves FOG, Thygesen HH, Jones PF, et al. A Rac/Cdc42 exchange factor complex promotes formation of lateral filopodia and blood vessel lumen morphogenesis. Nat Commun 2015;6:7286. doi: 10.1038/ncomms8286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Curry FR, Adamson RH. Tonic regulation of vascular permeability. Acta Physiol (Oxf) 2013;207:628–649. doi: 10.1111/apha.12076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Radeva MY, Waschke J. Mind the gap: mechanisms regulating the endothelial barrier. Acta Physiol (Oxf) 2018;222. doi: 10.1111/apha.12860 [DOI] [PubMed] [Google Scholar]
- 31.Krump-Konvalinkova V, Yasuda S, Rubic T, Makarova N, Mages J, Erl W, Vosseler C, Kirkpatrick CJ, Tigyi G, Siess W. Stable knock-down of the sphingosine 1-phosphate receptor S1P1 influences multiple functions of human endothelial cells. Arterioscler Thromb Vasc Biol 2005;25:546–552. doi: 10.1161/01.ATV.0000154360.36106.d9 [DOI] [PubMed] [Google Scholar]
- 32.Camerer E, Regard JB, Cornelissen I, Srinivasan Y, Duong DN, Palmer D, Pham TH, Wong JS, Pappu R, Coughlin SR. Sphingosine-1-phosphate in the plasma compartment regulates basal and inflammation-induced vascular leak in mice. J Clin Invest 2009;119:1871–1879. doi: 10.1172/jci38575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oo ML, Chang SH, Thangada S, Wu MT, Rezaul K, Blaho V, Hwang SI, Han DK, Hla T. Engagement of S1P(1)-degradative mechanisms leads to vascular leak in mice. J Clin Invest 2011;121:2290–2300. doi: 10.1172/JCI45403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jung B, Obinata H, Galvani S, Mendelson K, Ding BS, Skoura A, Kinzel B, Brinkmann V, Rafii S, Evans T, et al. Flow-regulated endothelial S1P receptor-1 signaling sustains vascular development. Dev Cell 2012;23:600–610. doi: 10.1016/j.devcel.2012.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burg N, Swendeman S, Worgall S, Hla T, Salmon JE. Sphingosine 1-Phosphate Receptor 1 Signaling Maintains Endothelial Cell Barrier Function and Protects Against Immune Complex-Induced Vascular Injury. Arthritis Rheumatol 2018;70:1879–1889. doi: 10.1002/art.40558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Christensen PM, Liu CH, Swendeman SL, Obinata H, Qvortrup K, Nielsen LB, Hla T, Di Lorenzo A, Christoffersen C. Impaired endothelial barrier function in apolipoprotein M-deficient mice is dependent on sphingosine-1-phosphate receptor 1. FASEB J 2016;30:2351–2359. doi: 10.1096/fj.201500064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McVerry BJ, Garcia JG. Endothelial cell barrier regulation by sphingosine 1-phosphate. J Cell Biochem 2004;92:1075–1085. doi: 10.1002/jcb.20088 [DOI] [PubMed] [Google Scholar]
- 38.Tauseef M, Kini V, Knezevic N, Brannan M, Ramchandaran R, Fyrst H, Saba J, Vogel SM, Malik AB, Mehta D. Activation of sphingosine kinase-1 reverses the increase in lung vascular permeability through sphingosine-1-phosphate receptor signaling in endothelial cells. Circ Res 2008;103:1164–1172. doi: 10.1161/01.RES.0000338501.84810.51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun JF, Phung T, Shiojima I, Felske T, Upalakalin JN, Feng D, Kornaga T, Dor T, Dvorak AM, Walsh K, et al. Microvascular patterning is controlled by fine-tuning the Akt signal. Proc Natl Acad Sci U S A 2005;102:128–133. doi: 10.1073/pnas.0403198102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Payne S, De Val S, Neal A. Endothelial-Specific Cre Mouse Models. Arterioscler Thromb Vasc Biol 2018;38:2550–2561. doi: 10.1161/ATVBAHA.118.309669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu N, Sargent KM, Clopton DT, Pohlmeier WE, Brauer VM, McFee RM, Weber JS, Ferrara N, Silversides DW, Cupp AS. Loss of vascular endothelial growth factor A (VEGFA) isoforms in the testes of male mice causes subfertility, reduces sperm numbers, and alters expression of genes that regulate undifferentiated spermatogonia. Endocrinology 2013;154:4790–4802. doi: 10.1210/en.2013-1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aivatiadou E, Mattei E, Ceriani M, Tilia L, Berruti G. Impaired fertility and spermiogenetic disorders with loss of cell adhesion in male mice expressing an interfering Rap1 mutant. Mol Biol Cell 2007;18:1530–1542. doi: 10.1091/mbc.e06-10-0902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Venkatesh D, Mruk D, Herter JM, Cullere X, Chojnacka K, Cheng CY, Mayadas TN. AKAP9, a Regulator of Microtubule Dynamics, Contributes to Blood-Testis Barrier Function. Am J Pathol 2016;186:270–284. doi: 10.1016/j.ajpath.2015.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Niaudet C, Hofmann JJ, Mae MA, Jung B, Gaengel K, Vanlandewijck M, Ekvarn E, Salvado MD, Mehlem A, Al Sayegh S, et al. Gpr116 Receptor Regulates Distinctive Functions in Pneumocytes and Vascular Endothelium. PLoS One 2015;10:e0137949. doi: 10.1371/journal.pone.0137949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Armulik A, Genove G, Mae M, Nisancioglu MH, Wallgard E, Niaudet C, He L, Norlin J, Lindblom P, Strittmatter K, et al. Pericytes regulate the blood-brain barrier. Nature 2010;468:557–561. doi: 10.1038/nature09522 [DOI] [PubMed] [Google Scholar]
- 46.Majno G, Palade GE, Schoefl GI. Studies on inflammation. II. The site of action of histamine and serotonin along the vascular tree: a topographic study. J Biophys Biochem Cytol 1961;11:607–626. doi: 10.1083/jcb.11.3.607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kersemans V, Cornelissen B, Allen PD, Beech JS, Smart SC. Subcutaneous tumor volume measurement in the awake, manually restrained mouse using MRI. J Magn Reson Imaging 2013;37:1499–1504. doi: 10.1002/jmri.23829 [DOI] [PubMed] [Google Scholar]
- 48.Tomayko MM, Reynolds CP. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother Pharmacol 1989;24:148–154. doi: 10.1007/BF00300234 [DOI] [PubMed] [Google Scholar]
- 49.Engelbrecht E, Levesque MV, He L, Vanlandewijck M, Nitzsche A, Niazi H, Kuo A, Singh SA, Aikawa M, Holton K, et al. Sphingosine 1-phosphate-regulated transcriptomes in heterogenous arterial and lymphatic endothelium of the aorta. Elife 2020;9. doi: 10.7554/eLife.52690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yazbeck P, Tauseef M, Kruse K, Amin MR, Sheikh R, Feske S, Komarova Y, Mehta D. STIM1 Phosphorylation at Y361 Recruits Orai1 to STIM1 Puncta and Induces Ca(2+) Entry. Sci Rep 2017;7:42758. doi: 10.1038/srep42758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chavez A, Schmidt TT, Yazbeck P, Rajput C, Desai B, Sukriti S, Giantsos-Adams K, Knezevic N, Malik AB, Mehta D. S1PR1 Tyr143 phosphorylation downregulates endothelial cell surface S1PR1 expression and responsiveness. J Cell Sci 2015;128:878–887. doi: 10.1242/jcs.154476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McQuin C, Goodman A, Chernyshev V, Kamentsky L, Cimini BA, Karhohs KW, Doan M, Ding L, Rafelski SM, Thirstrup D, et al. CellProfiler 3.0: Next-generation image processing for biology. PLoS Biol 2018;16:e2005970. doi: 10.1371/journal.pbio.2005970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wickham H. ggplot2: Elegant Graphics for Data Analysis https://ggplot2.tidyverse.org; 2016. [Google Scholar]
- 54.Rosner B, Glynn RJ, Lee ML. Extension of the rank sum test for clustered data: two-group comparisons with group membership defined at the subunit level. Biometrics 2006;62:1251–1259. doi: 10.1111/j.1541-0420.2006.00582.x [DOI] [PubMed] [Google Scholar]
- 55.Alarcon-Martinez L, Yilmaz-Ozcan S, Yemisci M, Schallek J, Kilic K, Can A, Di Polo A, Dalkara T. Capillary pericytes express alpha-smooth muscle actin, which requires prevention of filamentous-actin depolymerization for detection. Elife 2018;7. doi: 10.7554/eLife.34861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marchesi VT. The passage of colloidal carbon through the inflamed endothelium. Proc Roay Soc B 1962;156:550–552. [Google Scholar]
- 57.Christoffersen C, Obinata H, Kumaraswamy SB, Galvani S, Ahnstrom J, Sevvana M, Egerer-Sieber C, Muller YA, Hla T, Nielsen LB, et al. Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc Natl Acad Sci U S A 2011;108:9613–9618. doi: 10.1073/pnas.1103187108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Obinata H, Kuo A, Wada Y, Swendeman S, Liu CH, Blaho VA, Nagumo R, Satoh K, Izumi T, Hla T. Identification of ApoA4 as a sphingosine 1-phosphate chaperone in ApoM- and albumin-deficient mice. J Lipid Res 2019;60:1912–1921. doi: 10.1194/jlr.RA119000277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cartier A, Hla T. Sphingosine 1-phosphate: Lipid signaling in pathology and therapy. Science 2019;366. doi: 10.1126/science.aar5551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee MH, Appleton KM, El-Shewy HM, Sorci-Thomas MG, Thomas MJ, Lopes-Virella MF, Luttrell LM, Hammad SM, Klein RL. S1P in HDL promotes interaction between SR-BI and S1PR1 and activates S1PR1-mediated biological functions: calcium flux and S1PR1 internalization. J Lipid Res 2017;58:325–338. doi: 10.1194/jlr.M070706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Venkataraman K, Lee YM, Michaud J, Thangada S, Ai Y, Bonkovsky HL, Parikh NS, Habrukowich C, Hla T. Vascular endothelium as a contributor of plasma sphingosine 1-phosphate. Circ Res 2008;102:669–676. doi: 10.1161/CIRCRESAHA.107.165845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kono M, Tucker AE, Tran J, Bergner JB, Turner EM, Proia RL. Sphingosine-1-phosphate receptor 1 reporter mice reveal receptor activation sites in vivo. J Clin Invest 2014;124:2076–2086. doi: 10.1172/JCI71194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kono M, Conlon EG, Lux SY, Yanagida K, Hla T, Proia RL. Bioluminescence imaging of G protein-coupled receptor activation in living mice. Nat Commun 2017;8:1163. doi: 10.1038/s41467-017-01340-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reinhard NR, Mastop M, Yin T, Wu Y, Bosma EK, Gadella TWJ Jr., Goedhart J, Hordijk PL. The balance between Galphai-Cdc42/Rac and Galpha12/13-RhoA pathways determines endothelial barrier regulation by sphingosine-1-phosphate. Mol Biol Cell 2017;28:3371–3382. doi: 10.1091/mbc.E17-03-0136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wilkerson BA, Argraves KM. The role of sphingosine-1-phosphate in endothelial barrier function. Biochim Biophys Acta 2014;1841:1403–1412. doi: 10.1016/j.bbalip.2014.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xiong Y, Hla T. S1P control of endothelial integrity. Curr Top Microbiol Immunol 2014;378:85–105. doi: 10.1007/978-3-319-05879-5_4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shikata Y, Birukov KG, Garcia JG. S1P induces FA remodeling in human pulmonary endothelial cells: role of Rac, GIT1, FAK, and paxillin. J Appl Physiol (1985) 2003;94:1193–1203. doi: 10.1152/japplphysiol.00690.2002 [DOI] [PubMed] [Google Scholar]
- 68.Belvitch P, Htwe YM, Brown ME, Dudek S. Cortical Actin Dynamics in Endothelial Permeability. Curr Top Membr 2018;82:141–195. doi: 10.1016/bs.ctm.2018.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Abbasi T, Garcia JG. Sphingolipids in lung endothelial biology and regulation of vascular integrity. Handb Exp Pharmacol 2013:201–226. doi: 10.1007/978-3-7091-1511-4_10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Carbajal JM, Schaeffer RC Jr. RhoA inactivation enhances endothelial barrier function. Am J Physiol 1999;277:C955–964. doi: 10.1152/ajpcell.1999.277.5.C955 [DOI] [PubMed] [Google Scholar]
- 71.Wojciak-Stothard B, Potempa S, Eichholtz T, Ridley AJ. Rho and Rac but not Cdc42 regulate endothelial cell permeability. J Cell Sci 2001;114:1343–1355. [DOI] [PubMed] [Google Scholar]
- 72.van Nieuw Amerongen GP, Beckers CM, Achekar ID, Zeeman S, Musters RJ, van Hinsbergh VW. Involvement of Rho kinase in endothelial barrier maintenance. Arterioscler Thromb Vasc Biol 2007;27:2332–2339. doi: 10.1161/ATVBAHA.107.152322 [DOI] [PubMed] [Google Scholar]
- 73.Vouret-Craviari V, Boquet P, Pouyssegur J, Van Obberghen-Schilling E. Regulation of the actin cytoskeleton by thrombin in human endothelial cells: role of Rho proteins in endothelial barrier function. Mol Biol Cell 1998;9:2639–2653. doi: 10.1091/mbc.9.9.2639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cao J, Ehling M, Marz S, Seebach J, Tarbashevich K, Sixta T, Pitulescu ME, Werner AC, Flach B, Montanez E, et al. Polarized actin and VE-cadherin dynamics regulate junctional remodelling and cell migration during sprouting angiogenesis. Nat Commun 2017;8:2210. doi: 10.1038/s41467-017-02373-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shen Q, Rigor RR, Pivetti CD, Wu MH, Yuan SY. Myosin light chain kinase in microvascular endothelial barrier function. Cardiovasc Res 2010;87:272–280. doi: 10.1093/cvr/cvq144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tan W, Palmby TR, Gavard J, Amornphimoltham P, Zheng Y, Gutkind JS. An essential role for Rac1 in endothelial cell function and vascular development. FASEB J 2008;22:1829–1838. doi: 10.1096/fj.07-096438 [DOI] [PubMed] [Google Scholar]
- 77.Nohata N, Uchida Y, Stratman AN, Adams RH, Zheng Y, Weinstein BM, Mukouyama YS, Gutkind JS. Temporal-specific roles of Rac1 during vascular development and retinal angiogenesis. Dev Biol 2016;411:183–194. doi: 10.1016/j.ydbio.2016.02.005 [DOI] [PubMed] [Google Scholar]
- 78.Fiedler LR. Rac1 regulates cardiovascular development and postnatal function of endothelium. Cell Adh Migr 2009;3:143–145. doi: 10.4161/cam.3.2.8279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol 2008;9:690–701. doi: 10.1038/nrm2476 [DOI] [PubMed] [Google Scholar]
- 80.Zahra FT, Sajib MS, Ichiyama Y, Akwii RG, Tullar PE, Cobos C, Minchew SA, Doci CL, Zheng Y, Kubota Y, et al. Endothelial RhoA GTPase is essential for in vitro endothelial functions but dispensable for physiological in vivo angiogenesis. Sci Rep 2019;9:11666. doi: 10.1038/s41598-019-48053-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chang L, Yang J, Jo CH, Boland A, Zhang Z, McLaughlin SH, Abu-Thuraia A, Killoran RC, Smith MJ, Cote JF, et al. Structure of the DOCK2-ELMO1 complex provides insights into regulation of the auto-inhibited state. Nat Commun 2020;11:3464. doi: 10.1038/s41467-020-17271-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sweeney M, Foldes G. It Takes Two: Endothelial-Perivascular Cell Cross-Talk in Vascular Development and Disease. Front Cardiovasc Med 2018;5:154. doi: 10.3389/fcvm.2018.00154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Su VL, Calderwood DA. Signalling through cerebral cavernous malformation protein networks. Open Biol 2020;10:200263. doi: 10.1098/rsob.200263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mendez-Barbero N, Gutierrez-Munoz C, Blanco-Colio LM. Cellular Crosstalk between Endothelial and Smooth Muscle Cells in Vascular Wall Remodeling. Int J Mol Sci 2021;22. doi: 10.3390/ijms22147284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liu Y, Wada R, Yamashita T, Mi Y, Deng CX, Hobson JP, Rosenfeldt HM, Nava VE, Chae SS, Lee MJ, et al. Edg-1, the G protein-coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. J Clin Invest 2000;106:951–961. doi: 10.1172/JCI10905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Paik JH, Skoura A, Chae SS, Cowan AE, Han DK, Proia RL, Hla T. Sphingosine 1-phosphate receptor regulation of N-cadherin mediates vascular stabilization. Genes Dev 2004;18:2392–2403. doi: 10.1101/gad.1227804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Allende ML, Yamashita T, Proia RL. G-protein-coupled receptor S1P1 acts within endothelial cells to regulate vascular maturation. Blood 2003;102:3665–3667. doi: 10.1182/blood-2003-02-0460 [DOI] [PubMed] [Google Scholar]
- 88.Gaengel K, Niaudet C, Hagikura K, Lavina B, Muhl L, Hofmann JJ, Ebarasi L, Nystrom S, Rymo S, Chen LL, et al. The sphingosine-1-phosphate receptor S1PR1 restricts sprouting angiogenesis by regulating the interplay between VE-cadherin and VEGFR2. Dev Cell 2012;23:587–599. doi: 10.1016/j.devcel.2012.08.005 [DOI] [PubMed] [Google Scholar]
- 89.Yanagida K, Engelbrecht E, Niaudet C, Jung B, Gaengel K, Holton K, Swendeman S, Liu CH, Levesque MV, Kuo A, et al. Sphingosine 1-Phosphate Receptor Signaling Establishes AP-1 Gradients to Allow for Retinal Endothelial Cell Specialization. Dev Cell 2020;52:779–793 e777. doi: 10.1016/j.devcel.2020.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cartier A, Leigh T, Liu CH, Hla T. Endothelial sphingosine 1-phosphate receptors promote vascular normalization and antitumor therapy. Proc Natl Acad Sci U S A 2020;117:3157–3166. doi: 10.1073/pnas.1906246117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Stevens T, Phan S, Frid MG, Alvarez D, Herzog E, Stenmark KR. Lung vascular cell heterogeneity: endothelium, smooth muscle, and fibroblasts. Proc Am Thorac Soc 2008;5:783–791. doi: 10.1513/pats.200803-027HR [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Aird WC. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ Res 2007;100:158–173. doi: 10.1161/01.RES.0000255691.76142.4a [DOI] [PubMed] [Google Scholar]
- 93.Yanagida K, Liu CH, Faraco G, Galvani S, Smith HK, Burg N, Anrather J, Sanchez T, Iadecola C, Hla T. Size-selective opening of the blood-brain barrier by targeting endothelial sphingosine 1-phosphate receptor 1. Proc Natl Acad Sci U S A 2017;114:4531–4536. doi: 10.1073/pnas.1618659114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nitta T, Hata M, Gotoh S, Seo Y, Sasaki H, Hashimoto N, Furuse M, Tsukita S. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol 2003;161:653–660. doi: 10.1083/jcb.200302070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Claesson-Welsh L, Dejana E, McDonald DM. Permeability of the Endothelial Barrier: Identifying and Reconciling Controversies. Trends Mol Med 2021;27:314–331. doi: 10.1016/j.molmed.2020.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Balaji Ragunathrao VA, Anwar M, Akhter MZ, Chavez A, Mao Y, Natarajan V, Lakshmikanthan S, Chrzanowska-Wodnicka M, Dudek AZ, Claesson-Welsh L, et al. Sphingosine-1-Phosphate Receptor 1 Activity Promotes Tumor Growth by Amplifying VEGF-VEGFR2 Angiogenic Signaling. Cell Rep 2019;29:3472–3487 e3474. doi: 10.1016/j.celrep.2019.11.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Knezevic N, Roy A, Timblin B, Konstantoulaki M, Sharma T, Malik AB, Mehta D. GDI-1 phosphorylation switch at serine 96 induces RhoA activation and increased endothelial permeability. Mol Cell Biol 2007;27:6323–6333. doi: 10.1128/MCB.00523-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Singh I, Knezevic N, Ahmmed GU, Kini V, Malik AB, Mehta D. Galphaq-TRPC6-mediated Ca2+ entry induces RhoA activation and resultant endothelial cell shape change in response to thrombin. J Biol Chem 2007;282:7833–7843. doi: 10.1074/jbc.M608288200 [DOI] [PubMed] [Google Scholar]
- 99.Gorovoy M, Neamu R, Niu J, Vogel S, Predescu D, Miyoshi J, Takai Y, Kini V, Mehta D, Malik AB, et al. RhoGDI-1 modulation of the activity of monomeric RhoGTPase RhoA regulates endothelial barrier function in mouse lungs. Circ Res 2007;101:50–58. doi: 10.1161/CIRCRESAHA.106.145847 [DOI] [PubMed] [Google Scholar]
- 100.Daneshjou N, Sieracki N, van Nieuw Amerongen GP, Conway DE, Schwartz MA, Komarova YA, Malik AB. Rac1 functions as a reversible tension modulator to stabilize VE-cadherin trans-interaction. J Cell Biol 2015;208:23–32. doi: 10.1083/jcb.201409108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zebda N, Tian Y, Tian X, Gawlak G, Higginbotham K, Reynolds AB, Birukova AA, Birukov KG. Interaction of p190RhoGAP with C-terminal domain of p120-catenin modulates endothelial cytoskeleton and permeability. J Biol Chem 2013;288:18290–18299. doi: 10.1074/jbc.M112.432757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mehta D, Konstantoulaki M, Ahmmed GU, Malik AB. Sphingosine 1-phosphate-induced mobilization of intracellular Ca2+ mediates rac activation and adherens junction assembly in endothelial cells. J Biol Chem 2005;280:17320–17328. doi: 10.1074/jbc.M411674200 [DOI] [PubMed] [Google Scholar]
- 103.Schmidt TT, Tauseef M, Yue L, Bonini MG, Gothert J, Shen TL, Guan JL, Predescu S, Sadikot R, Mehta D. Conditional deletion of FAK in mice endothelium disrupts lung vascular barrier function due to destabilization of RhoA and Rac1 activities. Am J Physiol Lung Cell Mol Physiol 2013;305:L291–300. doi: 10.1152/ajplung.00094.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ohta Y, Hartwig JH, Stossel TP. FilGAP, a Rho- and ROCK-regulated GAP for Rac binds filamin A to control actin remodelling. Nat Cell Biol 2006;8:803–814. doi: 10.1038/ncb1437 [DOI] [PubMed] [Google Scholar]
- 105.Weng Z, Situ C, Lin L, Wu Z, Zhu J, Zhang R. Structure of BAI1/ELMO2 complex reveals an action mechanism of adhesion GPCRs via ELMO family scaffolds. Nat Commun 2019;10:51. doi: 10.1038/s41467-018-07938-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Feng H, Hu B, Liu KW, Li Y, Lu X, Cheng T, Yiin JJ, Lu S, Keezer S, Fenton T, et al. Activation of Rac1 by Src-dependent phosphorylation of Dock180(Y1811) mediates PDGFRalpha-stimulated glioma tumorigenesis in mice and humans. J Clin Invest 2011;121:4670–4684. doi: 10.1172/JCI58559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Laurin M, Huber J, Pelletier A, Houalla T, Park M, Fukui Y, Haibe-Kains B, Muller WJ, Cote JF. Rac-specific guanine nucleotide exchange factor DOCK1 is a critical regulator of HER2-mediated breast cancer metastasis. Proc Natl Acad Sci U S A 2013;110:7434–7439. doi: 10.1073/pnas.1213050110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Abu-Thuraia A, Gauthier R, Chidiac R, Fukui Y, Screaton RA, Gratton JP, Cote JF. Axl phosphorylates Elmo scaffold proteins to promote Rac activation and cell invasion. Mol Cell Biol 2015;35:76–87. doi: 10.1128/MCB.00764-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Makihara S, Morin S, Ferent J, Cote JF, Yam PT, Charron F. Polarized Dock Activity Drives Shh-Mediated Axon Guidance. Dev Cell 2018;46:410–425 e417. doi: 10.1016/j.devcel.2018.07.007 [DOI] [PubMed] [Google Scholar]
- 110.Hiramoto-Yamaki N, Takeuchi S, Ueda S, Harada K, Fujimoto S, Negishi M, Katoh H. Ephexin4 and EphA2 mediate cell migration through a RhoG-dependent mechanism. J Cell Biol 2010;190:461–477. doi: 10.1083/jcb.201005141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Huang L, Chambliss KL, Gao X, Yuhanna IS, Behling-Kelly E, Bergaya S, Ahmed M, Michaely P, Luby-Phelps K, Darehshouri A, et al. SR-B1 drives endothelial cell LDL transcytosis via DOCK4 to promote atherosclerosis. Nature 2019;569:565–569. doi: 10.1038/s41586-019-1140-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Li Q, Chen B, Zeng C, Fan A, Yuan Y, Guo X, Huang X, Huang Q. Differential activation of receptors and signal pathways upon stimulation by different doses of sphingosine-1-phosphate in endothelial cells. Exp Physiol 2015;100:95–107. doi: 10.1113/expphysiol.2014.082149 [DOI] [PubMed] [Google Scholar]
- 113.Fu P, Shaaya M, Harijith A, Jacobson JR, Karginov A, Natarajan V. Sphingolipids Signaling in Lamellipodia Formation and Enhancement of Endothelial Barrier Function. Curr Top Membr 2018;82:1–31. doi: 10.1016/bs.ctm.2018.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Xu X, Jin T. ELMO proteins transduce G protein-coupled receptor signal to control reorganization of actin cytoskeleton in chemotaxis of eukaryotic cells. Small GTPases 2019;10:271–279. doi: 10.1080/21541248.2017.1318816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Damoulakis G, Gambardella L, Rossman KL, Lawson CD, Anderson KE, Fukui Y, Welch HC, Der CJ, Stephens LR, Hawkins PT. P-Rex1 directly activates RhoG to regulate GPCR-driven Rac signalling and actin polarity in neutrophils. J Cell Sci 2014;127:2589–2600. doi: 10.1242/jcs.153049 [DOI] [PubMed] [Google Scholar]
- 116.Nishikimi A, Fukuhara H, Su W, Hongu T, Takasuga S, Mihara H, Cao Q, Sanematsu F, Kanai M, Hasegawa H, et al. Sequential regulation of DOCK2 dynamics by two phospholipids during neutrophil chemotaxis. Science 2009;324:384–387. doi: 10.1126/science.1170179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and outcomes of acute lung injury. N Engl J Med 2005;353:1685–1693. doi: 10.1056/NEJMoa050333 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.