

Abstract
Ariboflavinosis is a pathological condition occurring as a result of riboflavin deficiency. This condition is treatable if detected early enough, but it lacks timely diagnosis. Critical symptoms of ariboflavinosis include neurological and visual manifestations, yet the effects of flavin deficiency on the retina are not well investigated. Here, using a diet induced mouse model of riboflavin deficiency, we provide the first evidence of how retinal function and metabolism are closely intertwined with riboflavin homeostasis. We find that diet induced riboflavin deficiency causes severe decreases in retinal function accompanied by structural changes in the neural retina and retinal pigment epithelium (RPE). This is preceded by increased signs of cellular oxidative stress and metabolic disorder, in particular dysregulation in lipid metabolism, which is essential for both photoreceptors and the RPE. Though many of these deleterious phenotypes can be ameliorated by riboflavin supplementation, our data suggests that some patients may continue to suffer from multiple pathologies at later ages. These studies provide an essential cellular and mechanistic foundation linking defects in cellular flavin levels with the manifestation of functional deficiencies in the visual system and paves the way for a more in-depth understanding of the cellular consequences of ariboflavinosis.
Keywords: Flavins, Retbindin, Ariboflavinosis, Metabolism, Retinal degeneration, RPE dystrophy, Riboflavin
Graphical abstract
Abbreviations
- α-SMA
alpha smooth muscle actin
- CALCRL
calcitonin receptor-like receptor
- COL2A
collagen type II alpha 1 chain
- DEG
differentially expressed genes
- DUSP1
dual specificity phosphatase 1
- ECM2
extracellular matrix protein 2
- EMT
epithelial mesenchymal transition
- EGR1
early growth response 1
- ERG
electroretinography or electroretinograms
- FAD
flavin adenine dinucleotide
- FMN
flavin mononucleotide
- FOS
fos proto-oncogene
- FPKM
fragments per kilobase of transcript per million mapped reads
- Iba-1
Ionized calcium-binding adaptor molecule 1
- IPL
inner plexiform layer
- IS
inner segments
- LAMA2
laminin subunit alpha
- MEPA1
prostate transmembrane protein, androgen induced 1
- MEX-3C
RNA-binding E3 ubiquitin ligase
- MRPL49
mitochondrial ribosomal protein L49
- NR
neural retina
- NR4A1
nuclear receptor subfamily 4 group
- OS
outer segments
- OXL2
lysyl oxidase like 2
- P
postnatal day
- PDGFR
platelet-derived growth factor receptor
- PNA
peanut agglutinin
- PPM
part per million
- Prph2
peripherin 2, aka Rds
- PRRX1
paired related homeobox 1
- RC
regular chow
- RDC
riboflavin deficient chow
- RF
riboflavin, aka, vitamin B2
- RPE
retinal pigment epithelium
- RPE65
retinal pigment epithelium protein 65, aka retinoid isomerohydrolase
- RPE-Ch
retinal pigment epithelium-choroid
- Rtbdn
retbindin
- TCA
tricaboxylic cycle, aka Kreb's cycle or citric acid cycle
- VIM
vimentin
1. Introduction
Riboflavin (Vitamin B2), a necessary nutrient for proper growth and development, is a water-soluble vitamin, excess of which is flushed out of the body [1]. Circulating riboflavin is differentially taken up by various tissues according to their requirements and is quickly converted to its functional forms, flavin adenine mononucleotide (FMN) and flavin adenine dinucleotide (FAD) [2,3]. FMN and FAD are essential co-factors in the metabolism of carbohydrates, proteins and lipids [1].
Riboflavin deficiency (ariboflavinosis) is a systemic condition that can either be a congenital disorder or develop in adults due to malnutrition [1,4]. The development of technologies like exome sequencing [5] has facilitated the identification of causative mutations in cases of congenital ariboflavinosis, known as Brown-Vialetto-Van Laere syndrome. These patients exhibit defects in riboflavin transporters and suffer from multiple organ system failures, including severe neurological disorders, sometimes resulting in death [4]. Early diagnosis coupled with riboflavin supplementation can rescue the disease [5], however, patients may still develop multiple pathologies at later ages [1,6]. One commonly reported pathology in ariboflavinosis is impaired vision, but the link between loss of vision and riboflavin deficiency is unexplored [4,7].
Tissues with established blood barriers express riboflavin binding and carrier proteins, which are essential in taking up flavins and maintaining appropriate flavin levels [8]. Recently, we demonstrated that the novel retinal riboflavin binding protein, retbindin (Rtbdn) [9], helps concentrate FAD and FMN in the neural retina (NR). The retina is one of the most metabolically active and energy consuming tissues [[10], [11], [12]], and consistent with this high-energy demand, retinal tissue has one of the highest flavin levels in the body [13,14]. Underscoring the importance of maintaining sufficient levels of retinal flavins in the retina, and the essential role of retbindin in that process, we found that ablation of Rtbdn resulted in significantly reduced levels of retinal flavins leading to retinal degeneration [15].
The objective of the current investigation is to determine the role of riboflavin in adult retinal homeostasis using a nutritional model of adult-onset ariboflavinosis. Mice were maintained, for varying durations, on a riboflavin deficient diet starting at postnatal day (P) 30. We found that, while the retina maintains levels of flavins longer than the blood, prolonged dietary riboflavin deficiency leads to depletion of retinal flavins and downregulation of Rtbdn. This was accompanied by metabolic abnormalities and alterations in upregulation of oxidative stress markers, and inflammatory pathways gradually leading to impaired retinal function and degeneration of retinal pigment epithelium (RPE).
2. Material and methods
2.1. Animal information
Animal experiments were approved by the University of Houston Institutional Animal Care and Use Committee and adhered to the recommendations in the NIH Guide for the Care and Use of Laboratory Animals and to those from the Association for Research in Vision and Ophthalmology. All mice were on C57BL/6 J-129S1/SvImJ background and were genotyped for and found to be negative for both the rd8 allele [16] and the RPE65 Leu450Met variant [17,18]. Mice were reared under cyclic light conditions (12 h L/D, ∼30 lux) and fed ad libitum with the same nutrients except for either riboflavin-containing regular chow (RC) (AIN-93G; Envigo, Indianapolis, IN) or riboflavin deficient chow (RDC) (RF-Deficient-AIN-93G, Envigo, Indianapolis, IN). Mice of both sexes were used in all experiments and showed no differences in their response to the treatment regimen. The age of mice used is specified in each experiment. Animal weight was not used as a criterion for enrolling animals in the studies, but mice did not exhibit defects in overall weight gain/loss. All enrolled animals had normal immune status and had no issues with breeding healthy litters. To help prevent bias during analysis/experiment, identity of treatments was masked until all analyses (e.g. histological measurements, ERG assessment, etc.) were completed. For each specific experiment, the number of experimental replicates is indicated.
2.2. Antibodies
Antibodies used are summarized in Table S1.
2.3. Tissue collection
Tissue collection for HPLC, immunoblot, metabolomics, and transcriptomics was done as published previously [12,15,19,20]. Briefly, mice were anesthetized by intramuscular injection of 85 mg/kg ketamine and 14 mg/kg xylazine. Using forceps and tweezers, the NR was extracted and the RPE choroid (RPE-Ch) was collected and separated rapidly from the rest of the eyecup, rinsed quickly in phosphate buffer saline (pH = 7.4) and immediately frozen in liquid nitrogen. Blood samples were collected via tail-clip for flavin measurements with HPLC [20]. Following tissue collection, mice were euthanized.
2.4. HPLC
Micro-extraction of flavins from tissues was performed as previously described [20]. Briefly, frozen tissues were homogenized using a handheld motorized pestle in 100 μl of 1X PBS (pH 6.8 unless otherwise indicated) and a 30 μl aliquot was saved for protein assay. The rest was centrifuged at 1,000×g for 10 min at 4 °C and supernatant was separated and incubated at 37 °C in 10% trichloroacetic acid for 15 min to precipitate proteins. The tube was centrifuged at 10,000×g for 10 min at 4 °C; supernatant was carefully collected, filtered through 0.45 μm filter and used for HPLC analysis. To avoid any effect of freeze-thaw on flavin levels, HPLC analysis was performed right after extraction. The chromatographic separation was as previously described [20]. Briefly, the mobile phase was phosphate buffer (50 mM) at pH (3.1). The HPLC setup was composed of Waters binary HPLC pump (1525), Waters auto-sampler (2707), Waters multi wavelength fluorescence detector (2475) and a Waters X-Bridge C18 3.5 μm column with dimensions of 4.6X250mm (Waters, Milford, MA, USA). For HPLC quantification of flavins, a previously described method was followed [20].
2.5. Metabolomics
2.5.1. Tissue extraction and metabolomics analyses
Extraction of the samples was prepared according to Metabolon, Inc. (Morrisville, NC, USA) and several recovery standards were added to the samples prior to the first step in the extraction process for quality control purposes. In order to increase maximum yield, methanol extraction was done prior to analysis. To remove proteins, dissociate small molecules bound to proteins or trapped in the precipitated protein matrix, and to recover chemically diverse metabolites, proteins were precipitated with methanol under vigorous shaking for 2 min followed by centrifugation. The resulting extract was divided into five fractions: two for analysis by two separate reverse phase (RP)/UPLC-MS/MS methods with positive ion mode electrospray ionization (ESI), one for analysis by RP/UPLC-MS/MS with negative ion mode ESI, one for analysis by HILIC/UPLC-MS/MS with negative ion mode ESI, and one sample was reserved for backup. After removal of the organic solvent, the sample extracts were stored overnight under nitrogen before analysis.
2.5.2. Measurements
Metabolomics measurements were performed at Metabolon, Inc (Morrisville, NC, USA). All methods utilized a Waters ACQUITY ultra-performance liquid chromatography (UPLC) and a Thermo Scientific Q-Exactive high resolution/accurate mass spectrometer interfaced with a heated electrospray ionization (HESI-II) source and Orbitrap mass analyzer operated at 35,000 mass resolution. The extracted samples were dried and then reconstituted in solvents, which were compatible with each of the four methods above. Each reconstituted solvent contained a series of standards at fixed concentrations to ensure consistency in injection and chromatography. One aliquot was analyzed using acidic positive ion conditions, and chromatographically optimized for more hydrophilic compounds. In this method, the extract was gradient eluted from the C18 column (Waters UPLC BEH C18–2.1 × 100 mm, 1.7 μm) using water and methanol, containing 0.05% perfluoropentanoic acid (PFPA) and 0.1% formic acid (FA). The other aliquot was also analyzed using acidic positive ion conditions; but it was chromatographically optimized for more hydrophobic compounds. In this method, the extract was eluted in a gradient manner from the same aforementioned C18 column using methanol, acetonitrile, water, 0.05% PFPA and 0.01% FA and then was operated at an overall higher organic content. The third aliquot was analyzed using basic negative ion optimized conditions and a separate dedicated C18 column. The basic extracts were eluted as gradients from the column using methanol and water, however with 6.5 mM ammonium bicarbonate at pH 8. The last and fourth aliquot was then analyzed via negative ionization following elution from a HILIC column (Waters UPLC BEH Amide 2.1 × 150 mm, 1.7 μm) and using a gradient consisting of water and acetonitrile with 10 mM ammonium formate, pH 10.8. The MS analysis alternated between MS and data-dependent MSn scans using dynamic exclusion. The scan range varied slightly between methods but overall covered 70–1000 m/z. Compounds were finally identified by comparing to the respective library entries of purified standards or recurrent unknown entities. Peaks were then quantified by measurement of respective area-under-the-curve. For studies that spanned multiple days, a data normalization step was performed to correct for the variation resulting from instrument inter-day tuning differences. Biochemical data for each metabolite were then normalized to the protein concentration as measured by Bradford assay to account for differences in metabolite levels due to differences in the amount of material present in each sample.
2.5.3. Quality control
Along with the experimental samples (and controls), various types of technical control samples were analyzed, including 1) a pooled matrix sample generated by taking a small volume of each experimental sample, which served as a technical replicate throughout the data set, 2) extracted water samples which served as process blanks, and 3) a cocktail of quality control standards that were carefully chosen not to interfere with the measurement of endogenous compounds which were spiked into every analyzed sample, allowing instrument performance monitoring and aiding chromatographic alignment. Instrument variability was consistently measured by calculating the median relative standard deviation (RSD) for the internal standards that were added to each sample prior to injection into the mass spectrometers. Overall, process variability was measured by calculating the median RSD for all endogenous metabolites (i.e., non-instrument standards) present in 100% of the technical replicates of pooled samples. Internal standards reflected instrument variability of only 6% median RSD, while endogenous metabolites reflected a total process variability of only 2% median RSD. Experimental samples were randomized across the platform run with QC samples spaced evenly between the injections.
2.6. Immunoblotting
NRs and RPE-Ch were harvested as described previously [12,15,19]. Protein lysates were first prepared by homogenizing the retinas using a motorized pestle (VWR, Radnor, PA) in 1 × PBS (pH 7.2) containing 1% Triton X-100 and complete protease inhibitor mixture (Roche, Basel, Switzerland). Following a 1 h incubation at 4 °C, the insoluble material was separated via centrifugation at 4,000×g. Supernatants were incubated for 1 h at room temperature in Laemmli buffer containing β-mercaptoethanol. Equal amounts of total protein (15 μg) for each sample were loaded onto the 10% SDS-PAGE gel, separated, and blotted onto PVDF membrane using standard protocols. Protein detection was performed with the respective antibodies (α-sma:1:1000, e-cadherin: 1:500, n-cadherin:1:1000, vimentin: 1:1000; Proteintech) and membranes were imaged with a ChemiDocTM MP imaging system (Bio-Rad, Hercules, CA). Densitometric analyses was performed on unsaturated bands using Image Lab software version 4.1 (Bio-Rad), and band intensities were normalized to β-actin band intensities in the corresponding lanes on the same immunoblot.
2.7. Histology and morphometric analysis
Histology and associated morphometry was performed as described previously [15,19]. Whole eyes were dissected, and a hot needle was used to mark the superior portion of the cornea to maintain orientation. Then a small corneal slit was made near the ora serrata, and the eyes were fixed for 2h at 4 °C in Davidson fixative (32% ethanol, 11% acetic acid, 2% formaldehyde). Both cornea and lens were removed and the remaining eyecup was returned to fixative overnight at 4 °C, then dehydrated and embedded in paraffin. For light microscopy histology, hematoxylin and eosin stained sections were used from at least three mice per group. To assess photoreceptor loss, nuclei in the ONL were counted in sections, which were passing through the optic nerve head. Measurements were made at 12 different locations along the vertical axis, with six each in the superior and inferior hemispheres. OS length was measured from the tip of the OS to the base from the same regions. Measurements of the thickness of the inner plexiform layer (IPL) were made on the same sections at 500 μm distance from the optic nerve head in the superior and inferior regions by using the line measurement tool in ZEN 3.0 imaging software. All morphometric measurements were taken from the images by an observer masked to sample identity (treatment vs. control group), and then averaged.
2.8. Immunofluorescence
For immunolabeling of paraffin-embedded tissues, 10 μm thick sections were prepared from eyes that were fixed for 2h in Davidson fixative at 4 °C followed by paraffin embedding, sectioning and then processed for immunofluorescence as described previously [9,15]. Briefly, sections were boiled for 30 min with Tris-EDTA pH 9.0 with 0.1% Tween then blocked in 1x PBS, pH 7.4, 2% donkey serum, 0.5% Triton X-100, 50 mg/ml bovine serum albumin. After blocking, sections were incubated overnight in primary antibodies in blocking buffer, followed by washing (4 × 15 min with 1x PBS, pH 7.4), incubation in appropriate fluorescently labeled secondary antibodies in blocking buffer, additional washing, and mounting using ProlongGold mounting media with DAPI (Life Technologies, Carlsbad, CA). Control sections that received no primary antibody were included in each experiment. Cone count was determined using fluorescently tagged peanut agglutinin (PNA) (1:500; Life Technologies) for 1h at room temperature. Images were processed using ZEN Image Analysis software (Zeiss). To ensure reproducibility and accuracy in cone counts, sections that went through the center of the optic nerve were selected. Where confocal images are indicated, they represent an 8 slice stack, at 290 nm per slice, 2.32 μm total, captured with a 63X oil objective (NA1.4) and collapsed to a single projection image. Changes from this standard are indicated in the figure legend. For 3D reconstruction, the 3D create image tool in the ZEN 2.0 software was used following adjustment of orientation in the YX plane or ZY plane, and exported as a tiff file.
2.9. RPE flat-mount and cell count
RPE was dissected away from the NR, immediately fixed in 4% paraformaldehyde for 30 min at room temperature and washed once with 1 × PBS. The flat mount of RPE was prepared by making four incisions and laid flat. Flat mounts were incubated with either anti-β-catenin (1:500; Proteintech) or phalloidin (1:500; Thermo Fisher). The flat-mounts were then washed four times for 5 min each in 1 × PBS. Then using Fluoroshield™ with DAPI (Sigma), flat mounts were mounted on a glass microscope slide. For imaging, two 40x images (160 μm × 160 μm) were taken from the central area of each leaflet for each cohort.
RPE cell counts were performed from flat mounts using ImageJ software and only cells with over half of their body in frame were counted. Number of cells were plotted as per area of 0.0256 mm2 for all cohorts.
2.10. Bipolar endfeet count
To count bipolar endfeet, 40x images were captured from retinal cross sections stained with anti-PKCα. Single-plane (159.7 μm × 159.7 μm) images were generated and rod bipolar endfeet were counted using the events marker feature in Zen 2 (Blue Edition) (Carl Zeiss, Jena, Germany). Four images from each section were captured, and four sections were evaluated per sample, resulting in 16 images per sample. Two to three independent samples were assessed per group.
2.11. Electroretinography
Full field electroretinograms (ERG) were recorded as previously described [15]. Prior to ERG recording, mice were dark adapted overnight in red Optimice cages. Mice were anesthetized by intramuscular injection of 85 mg/kg ketamine and 14 mg/kg xylazine and eyes were dilated using 1% cyclopentolate HCl (Cyclogyl, Pharmaceutical Systems, Tulsa, OK). Light-evoked ERG responses were recorded using a UTAS system (LKC, Gaithersburg, MD) and a platinum wire loop electrode in contact with the cornea through a methyl cellulose layer (Ax10968, Accutome, Malvern, PA). Scotopic ERGs (rod photoreceptor-driven responses) were recorded using a strobe flash stimulus of 157 cd-s/m2. Photopic ERGs (cone photoreceptor-driven responses) were recorded and averaged from 25 flashes at 90 cd-s/m2 for white light, following a 5 min light adaptation with background light at an intensity of 29.03 cd/m2. C-wave ERGs were performed on mice that were dark-adapted overnight prior to the experiment. C-wave ERGs were recorded and averaged from 5 flashes at 79 cd-s/m2 with an extended recording time of 4080 ms. Animals were placed on a warming bed to maintain body temperature during the experiment and were monitored following the experiment until animals were fully recovered from anesthesia, as well as checked the next day.
2.12. RNA-seq
Preparation of RNA library and transcriptome sequencing was conducted by Novogene Corporation Inc. (Sacramento, CA, USA). Briefly, RNA was extracted from frozen NR and RPE, quality control performed to ensure RNA integrity number (RIN) > 7 and then respective cDNA libraries were prepared from ∼500 ng of isolated RNA with the TruSeq Stranded Total RNA Library Prep Kit according to the manufacturer's directions (Illumina, RS-122-2001, San Diego, CA, USA). Paired-end 100-cycle sequencing was performed on HiSeq 2000 or HiSeq 2500 sequencers according to the manufacturer's directions (Illumina, San Diego, CA, USA).
Reference genome and gene model annotation files were downloaded from genome website browser (NCBI/UCSC/Ensembl) directly. Indexes of the reference genome were built using STAR [21] and paired-end clean reads were aligned to the reference genome using STAR (v2.5). STAR used the method of Maximal Mappable Prefix (MMP), which can generate a precise mapping result for junction reads. STAR counts the number of reads per gene while mapping. The counts coincide with those produced by htseq-count with default parameters. Fragments per kilobase of transcript per million mapped reads (FPKM) for each gene was then calculated based on the length of the gene and reads count mapped to this gene. FPKM considers the effect of sequencing depth and gene length for the read counts at the same time, and is currently the most commonly used method for estimating gene expression levels [22]. Gene Ontology, Enrichr [23], and Biojupies [24] were used to assess the effected pathways.
2.12.1. Differential expression analysis
For determining differentially expressed genes between RC and RDC groups for each of NR and RPE, differential expression analysis was performed using the DESeq2 R package (1.14.1). DESeq2 (http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html) provide statistical routines for determining differential expression in digital gene expression data using a model based on the negative binomial distribution. The resulting P-values were adjusted using the Benjamini and Hochberg's approach for controlling the False Discovery Rate (FDR). Genes with an adjusted P-value <0.05 found by DESeq2 were assigned as differentially expressed.
2.13. Statistical analysis
Statistical testing was performed using GraphPad Prism, v.9.2 (GraphPad Software) and post hoc testing in cases with multiple groups was employed. All immunoblotting, immunofluorescence and histology experiments were performed on at least three separate occasions regardless of the number of individual animals used for each study. For comparisons of two groups, t-tests were used. For comparison of multiple groups, one-way ANOVA with Holm-Sidak post-hoc test was used. In cases where two variables were analyzed, two-way ANOVA was used. Two tailed tests were used in all cases. For cases where multiple t-tests were performed (e.g. for analysis of multiple metabolites), false discovery rate (FDR) was also calculated to correct for the multiple comparisons that normally occur in metabolomic-and transcriptomic based studies.
3. Results
3.1. Feeding riboflavin deficient diet leads to progressive depletion of flavins from the blood, followed by the RPE then the NR
Our first goal was to understand the time course over which riboflavin deficiency develops in the blood, NR and RPE following the onset of dietary riboflavin deficiency. Wild-type mouse mating pairs were maintained on regular chow (RC, 6 ppm riboflavin) during pregnancy and post-partum until pups reached postnatal day (P) 30. At that point, half of each litter was transferred to riboflavin deficient chow (RDC) and the other half continued on RC to the specific timepoint listed for each experiment. Flavin measurements were performed on extracts obtained from whole blood, NR, and RPE-Ch every 15 days until P90 using a well-established HPLC method [20]. By 15 days on RDC, blood flavin levels dropped precipitously and significantly to ∼10% of baseline levels (Fig. 1A). In contrast, the NR and RPE-Ch retained riboflavin and its derivatives, FAD and FMN, over a longer period of time, even after circulating levels in blood had significantly diminished (Fig. 1B–D). In the RPE-Ch, flavin levels began declining significantly at P45 and were depleted by P90 while their levels were maintained for a longer time in the NR and with a gradual decline over the course of 3 months (Fig. 1B–D). However, by P90, flavin levels in the NR were diminished. In the NR and RPE-Ch, riboflavin, FAD, and FMN exhibited similar patterns of decline, with FAD being preserved for slightly longer than the others in NR from animals fed RDC (Fig. 1D).
Fig. 1.

Levels of retinal flavins and retbindin are reduced in RDC fed mice. (A) Total blood flavin levels decline with duration on RDC. (B) Riboflavin, (C) FMN and (D) FAD levels in NR and RPE-Ch of RDC mice. Data are presented as percent of values from RC fed P30 mice. (E) Immunoblot showing the disappearance of retbindin (Rtbdn) in P240 RDC neural retinas (210 days on RDC diet). (F) Quantification of Rtbdn levels in P240 RDC retinas. Although Rtbdn protein disappears in retinal extracts from RDC fed mice, (G) the transcript levels were comparable to that of age-matched mice on RC. Rtbdn expression levels were measured in FPKM (Fragments per kilobase of transcript per million mapped reads) from animals fed RDC (blue) and RC (black). (H) Rtbdn labeling (red) in retinal section from P90 RC fed is mainly located at the tips of the outer segment and some around the inner segment (H, left two images) while Prph2 (green) is located at the outer segment. Rtbdn labeling is absent in P90 Rtbdn−/− retina (H, right two images). (I) Rtbdn labeling is reduced in P90 fed RDC when compared to those at P60 and disappeared at P240. Data are presented as an average from n = 3–6 for A–D and plotted as means ± SEM. Statistical analysis was assessed by one-way ANOVA for A-D (* = p < 0.05, ** = p < 0.01, *** = p < 0.001, **** = p < 0.0001 for comparison between each time point and P30. In graphs with two sets of symbols, top symbols refer to NR, bottom to RPE-Ch. Plotted are means ± SEM. NR, neural retina; RPE-Ch, retinal pigment epithelium-choroid; OS, outer segments; IS, inner segments; ONL, outer nuclear layer. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
We recently showed that Rtbdn is a retinal-specific riboflavin binding protein and in the absence of Rtbdn, retinal flavin levels drop [9,15], suggesting that a key function for Rtbdn involved riboflavin binding. The relationship of Rtbdn to riboflavin was confirmed by our experiments from RDC fed animals: Rtbdn was reduced to undetectable levels in retinal extracts from P240 mice that had been on RDC for 120 days (Fig. 1E and F). This decline in Rtbdn protein levels was not accompanied by a change in Rtbdn transcript levels (Fig. 1G), suggesting that the lack of flavins in RDC-fed mice reduced either the stability of Rtbdn protein or the translationability of its transcript. To evaluate the time course of the decrease in Rtbdn protein expression, we labeled retinal sections collected at multiple timepoints from mice on RDC starting at P30 for Rtbdn and the outer segment (OS) marker Prph2 (Fig. 1I). Sections from RC-fed and Rtbdn−/− retinas were included as controls (Fig. 1H). We observed a gradual decline in Rtbdn labeling first detected one month after the start of the RDC diet (P60) and becoming much more pronounced at P90 (60 days on RDC). After seven months on RDC (P240), virtually no Rtbdn protein was visible by immunofluorescence in the retina (Fig. 1I, right) although levels of Rtbdn at P30 were measured (data not shown) and were consistent with previously published levels [9]. These findings suggest that the stability of Rtbdn depends on the availability of riboflavin.
3.2. Flavin deficiency results in a gradual functional decline and thinning of the inner retina
To determine how riboflavin deficiency affects functional competence of the retina, electroretinogram (ERG) responses were recorded longitudinally from mice on RC and RDC up to P300 (270 days on the diet). No differences were observed in scotopic ERG responses at P90 and P120 in mice on RDC vs. RC (Fig. 2B). However, both scotopic a-wave (rod's signal) and scotopic b-wave (second order neurons' signal) were reduced significantly at P180 in RDC mice compared to age-matched RC littermates (Fig. 2A and B). This decline was progressive, and by P300 scotopic a-waves in RDC-fed mice were reduced by 40% compared to RC controls (Fig. 2B). Cone ERG responses in RDC-fed mice also declined progressively compared to RC controls, however at a slower rate, with significant differences in photopic ERG amplitudes first detected at P240 (Fig. 2C and D). By P300, photopic responses were reduced by ∼44% in RDC-fed mice vs. controls (Fig. 2D). To evaluate whether this ERG deficit was accompanied by photoreceptor loss, we counted photoreceptor nuclei (Fig. 2G, I), measured outer segment length (Fig. 2H), and counted cones in PNA labeled sections (Fig. 2E and F) at P240 (210 days on the diet). RDC fed mice did not exhibit a significant reduction in either the number of total photoreceptors, the number of cones, or the mean length of the OS compared to RC fed mice, suggesting that the ERG reduction at this age was attributable to molecular defects in photoreceptors rather than overt cell loss or structural defects.
Fig. 2.

RDC fed mice exhibit a gradual visual loss followed by structural alterations. Full-field scotopic (A) and photopic (C) ERG waveforms obtained for P240 RC (black) and RDC fed (blue) mice. ERG amplitudes (B-D) were measured at different ages showing progressive decline in both scotopic a- and b-wave as well as photopic b-wave amplitudes across the designated ages (n = 6–11 animals/age/group, each symbol represents the averaged value of the left and right eyes from one animal). Retinal cross sections from P240 RC and RDC fed mice labeled with PNA (E) and cone cells were counted (F). Representative light microscopic images from P240 RC and RDC fed retinas (G) showing no change in OS length (H), photoreceptor count (I), or cone count (F), while a significant change is seen in the IPL (black vertical lines in G and quantification in J). Number of samples in B&D, p90, n = 6; P120, n = 10; P180, n = 11; p240, n = 11; P300, n = 7. For H-J, n = 3; I-J, n = 3–4. Between-group differences were assessed by two-way ANOVA with Sidak post-hoc test for B, D, I, and J. T-tests were used for F and H. (* = p < 0.05, ** = p < 0.01, *** = p < 0.001, **** = p < 0.0001). Plots represent mean ± SEM. OS, outer segment; ONL, outer nuclear layer; INL, inner nuclear layer; IPL, inner plexiform layer; ONH, optic nerve head; S, superior; I, inferior. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
In contrast to the preservation of the outer retina, we observed significant pan-retinal thinning in the inner plexiform layer (IPL) (black vertical lines in Fig. 2G and 2J) at P240. The inner plexiform layer contains synapses between second order neurons (bipolar cells), amacrine cells, and ganglion cells. To investigate the cellular basis for the inner retinal thinning observed in RDC-fed animals, we immunolabeled retinal sections from both diet groups with antibodies designed to help evaluate the synaptic connectivity of the rod and cone photoreceptors with second order neurons (Fig. 3A). We first co-labeled for either protein kinase c alpha (PKCα), a marker for rod bipolar cells [25] and vesicular glutamate transporter 1 (VGLUT1), a marker for rod and cone photoreceptor terminals [26,27] or PKCα and the ribbon synapse marker C-terminal binding protein 2 (CtBP2) [28] (Fig. 3B) as markers for photoreceptor synaptic ribbons [29,30]. In the P300 RC fed retina, we observed a thick layer of photoreceptor terminals (VGLUT1 staining) with a similarly thick layer of synaptic ribbons (CtBP2). However, this layer of photoreceptor terminals was demonstrably thinner in the RDC retina (Fig. 3B), and was accompanied by regression of some photoreceptor terminals into the ONL (arrows Fig. 3B). In conjunction with regression of terminals into the ONL in retinas from RDC-fed mice, we also observed rod bipolar dendrites (PKCα) extending further into the OPL than in RC animals, and in some cases, we observed dendritic sprouting extending into the ONL (arrows, Fig. 3B, RDC). In addition to the intermittent sprouting of bipolar dendrites abnormally into the ONL, we also observed overall shortening of bipolar dendritic processes in the OPL (Fig. 3B and C). Assessment of 3D renderings of PKCα-labeled rod bipolar cells in RDC retina showed an overall reduction in cell length of ∼19% when compared to RC retina (Fig. 3C). We also observed significant reduction in rod bipolar endfeet counts from PKCα-labeled retinal cross sections in retinas from the RDC group vs. RC controls (Fig. 3D).
Fig. 3.

RDC fed mice show thinning of the inner retina. (A) Schematic diagrams showing localization of markers involved in photoreceptor-bipolar synapse of rods and cones. (B) Retinal sections from P300 RDC and RC mice immunolabeled for rod bipolar cells (PKCα), photoreceptor synaptic vesicles (VGLUT-1) and ribbon synapses (CtBP2) markers. Bottom images are taken at higher magnification at the OPL/INL areas colabeled with VGLUT-1 and PKCα (left) and PKCα and CtBP2 (right) taken at different regions of the OPL. (C) Length of 3D renderings (generated from PKCα stained retinas) of rod bipolar cells was measured in RC and RDC retinas. (D) Rod bipolar endfeet for both cohorts were counted from samples labeled with PKCα stained retinas. Statistical analysis was assessed by one-way ANOVA **** = p < 0.0001 (N = 3 separate animals, plotted is mean ± SEM). (E) Colabeling with secretagogin (SCGN), a marker that preferentially labels cone bipolar cells, CtBP2, and PNA show the synaptic interaction at the cone pedicle. Insets show magnified images of a single cone synaptic connection. (F) Labeling with tubulin βIII marks axons in the inner plexiform layer. 3D renderings of marked regions (white dashed boxed in the lower magnify cation images, top panel corresponds to top box and bottom panel corresponds to bottom box) show axonal density in the IPL of RC and RDC retinas. All images presented are collapsed confocal stacks, while insets in C are single layer captures. OS, outer segment; ONL, outer nuclear layer; OPL, outer plexiform layer, INL, inner nuclear layer; IPL, inner plexiform layer; GC, ganglion cell layer.
To assess whether these abnormalities also affected cone bipolar cells, we colabeled retinal sections from P300 RC and RDC animals with antibody against secretagogin (SCGN), a calcium binding protein, that labels several subtypes of cone bipolar cells [31,32], CtBP2 and PNA to label flat contacts. As depicted in the schematic drawing shown in Fig. 3A, the synaptic interaction of cones is organized differently from the rods. Cone photoreceptors have multiple ribbon synapses [33], mandating a much larger terminal than rods, and thus interact with multiple bipolar dendrites [34]. The larger synaptic terminals of cones allows further assessment of the synapse by high magnification confocal imaging. The combined labeling of PNA (which labels flat contacts at the base of cone pedicles), SCGN and CtBP2 showed highly organized synaptic connection with multiple ribbons in the cone pedicle in RC retinas (Fig. 3E, side panels and insets). However, in the RDC retinas, the cone pedicles showed fewer synaptic ribbons in each pedicle with more disorganized labeling of CtBP2. Overall, OPL integrity is compromised in the RDC retina, with evidence of synaptic loss as well as reduction in the number and length of bipolar cells.
The IPL houses the reticulum of axons connecting bipolar cells, amacrine cells and ganglion cells [35,36]. Assessment of the axonal meshwork using anti-tubulin βIII antibody allowed for a preliminary understanding of the changes to interactions between second and third order neurons in the IPL. Images at high magnification were captured at the top of the IPL (near the INL, corresponding to the OFF-lamina of the IPL), and in the inner part of the IPL (corresponding to the ON-lamina). Here we observed that dense tubulin labeling was similar in both regions of the IPL in the RC retina (Fig. 3F, RC panels located on the right representing areas boxed in low magnification images). However, in the RDC retina, the meshwork appears less dense (yellow arrows point to gaps in fibril network) in the OFF-lamina, while more normal density is seen in the inner IPL (ON-layer).
3.3. RDC diet induces metabolic dysregulation that precedes the functional decline
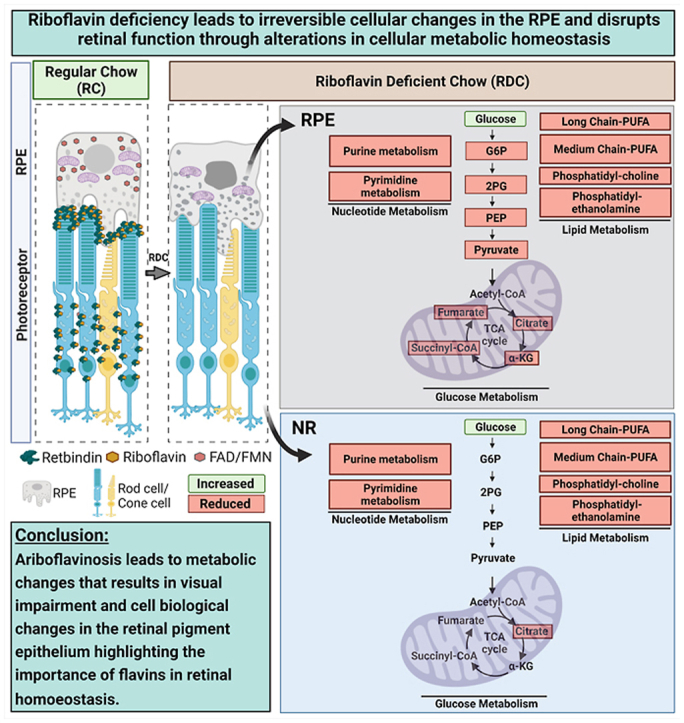
To investigate the potential mechanisms by which riboflavin deficiency leads to a decline in retinal function, we asked how the elimination of riboflavin (and hence its derivatives, FMN and FAD from the tissue) from the diet affects the metabolic processes in the retina. Steady state levels of various metabolites in the NR and RPE-Ch were compared between P120 RDC-fed animals (3 months on RDC) and their age-matched littermates on RC. We selected P120 since it preceded the development of significant declines in retinal function, a strategy designed to ensure that any changes observed were not due to structural alterations [19]. Our goal was to understand what metabolic changes preceded the functional and structural deficits. In the RDC-fed animals, glucose levels were significantly elevated in the NR (∼250%, Fig. 4A, values for RDC-fed animals are plotted as % of RC-fed animals), while all other glycolytic intermediates remained similar to age-matched RC-fed littermates. Among TCA cycle intermediates, only citrate was significantly changed in RDC-fed animals and was reduced to 75% of the levels in animals on RC (Fig. 4B). In the RPE-Ch of RDC-fed animals, glucose levels were also increased (∼120%, Fig. 4C) compared to controls, while all other glycolytic and TCA cycle intermediates were significantly reduced in the RDC-fed group vs. controls. Glycolytic intermediates in the RPE-Ch of RDC-fed animals were reduced by half (<50%), compared to the age matched RC fed animals (Fig. 4C), while TCA cycle intermediates in RDC-fed RPE-Ch were reduced to ≤75% of levels in RC-fed animals (Fig. 4D).
Fig. 4.

Retinal metabolic dysregulation precedes onset of the functional decline in mice on RDC diet. Steady-state levels of metabolites in mice fed on riboflavin-deficient chow (RDC) until P120 are presented as percent change from levels for age-matched littermate controls on regular chow (RC) for both NR and RPE-Ch. Glycolytic (A, C) and TCA cycle (B, D). Dotted red line represent levels in retinas of RC fed animals. Each respective class of metabolites is identified as a header in each table (E–G). Values which were not significantly different are in white boxes and those significantly lower are marked in red. Statistical analysis was assessed by t-tests with FDR correction for multiple tests (*p < 0.05 ** = p < 0.01, *** = p < 0.001, **** = p < 0.0001), with n = 6 samples for each dietary group. Each sample contained at least 6 independent retinas or RPE. Plotted are means ± SEM. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
In addition to glucose and TCA metabolites, we also evaluated metabolites that arise during lipid metabolism since many lipid-processing enzymes utilize flavins as cofactors. We found that steady state levels of multiple lipid metabolites critical for NR and RPE-Ch homeostasis were reduced at P120 in RDC-fed mice compared to RC-fed controls. Multiple long chain fatty acids, including the long chain polyunsaturated fatty acids and the monounsaturated fatty acids, were significantly reduced in both the NR (<60%) and RPE-Ch (≤60%) of RDC mice vs. controls (Fig. 4E). Important membrane lipid components including phosphatidylcholine and phosphatidylethanolamine metabolites are also significantly reduced (<80%) in RDC-fed animals in both the NR and RPE-Ch (Fig. 4F). Furthermore, both purine and pyrimidine metabolism in the NR and RPE-Ch are also compromised, with reductions (<75%) in several key metabolites (Fig. 4G). Combined, these findings suggest that energy generation, lipid metabolism, and nucleotide metabolism are all disrupted in the NR and RPE-Ch in RDC fed mice prior to the onset of functional defects.
3.4. Metabolic impairment is accompanied by homeostatic changes in NR and RPE-Ch of RDC treated mice
Given the significant disruption to metabolic pathways, we proceeded to investigate homeostatic changes in the NR and RPE of RDC animals by RNA-seq. Computational analysis of libraries generated from RDC and RC NR revealed 29 DEGs, listed in Table S2. Volcano plot was used to visualize the distribution of DEGs. We observed 11 genes above 1.5 log2 fold change (Fig. 5A, Fold change of 1.5 was used to show a cluster of genes with highest fold change and p-value.) Enrichment analysis of statistically significant upregulated DEGs resulted in three upregulated pathways (p53, hypoxia, and TNF-α signaling via NF-kβ) listed by increasing p-value (Fig. 5B). These pathways are indicative of a stress response to the RDC diet. Considering that riboflavin bioavailability is important for proper aerobic respiration [37] and antioxidant generation, upregulation of hypoxia-related genes, and cell survival/apoptotic and stress-response pathways (p53 and TNF-α), are consistent with the function of riboflavin.
Fig. 5.

Transcriptomic analysis of NR indicates upregulated stress response. (A) Volcano plot of DEGs determined from pairwise comparison of RDC vs RC neural retina at P120. Statistically significant genes above 1.5 log2 fold changed are marked in blue (upregulated genes) or red (down regulated genes). (B) Log p-value of statistically significant upregulated pathways. (C) Heat map of DEGs involved in TNF-α signaling and oxidative stress responses, presented as log2FC of normalized gene expression. (D) DEGs, Mrpl49 and Mex3C, were statistically significantly upregulated genes involved in energy homeostasis in RDC NR at P120. Genes are presented as FPKM (fragments per kilobase of transcript per million mapped reads). Each group had an n value of 4 samples. Each sample contained 4 pooled neural retinas with selections were intentionally spread over two/three separate litters. Statistical analysis was assessed by multiple t-test using FDR correction for multiple tests (** = p < 0.01). Plotted as means ± SEM. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Many of the highest upregulated genes (according to fold change and p-value), such as early growth response 1 (Egr1), fos proto-oncogene (Fos), dual specificity phosphatase 1 (DUSP1), and nuclear receptor subfamily 4 group 1 (Nr4a1) are involved in responses to oxidative-stress related changes to the environment and are interconnected with signaling in the TNF-α response pathway [[38], [39], [40], [41], [42], [43], [44], [45]]. The heatmap in Fig. 5C clearly depicts a stark difference in the expression of these response genes relative to animals treated with the standard diet.
Considering the changes to metabolism, we expected to see significant differential regulation of genes involved in energy or metabolic homeostasis. However, mitochondrial ribosomal Protein l49 (Mrpl49) and mex-3 RNA binding family member C (Mex3c) were the only two identified genes that played roles in the energy homeostatic pathways (Fig. 5D), where Mrpl49 is involved in regulation of complexes in oxidative phosphorylation and Mex3c regulates energy expenditure. Surprisingly, there were no notable downregulated pathways, or downregulation of metabolic enzymes or riboflavin transporters observed in RDC treated NR (Figs. S1A–C).
Because the RPE is a highly metabolically active junction between the blood supply and the energy-consumptive photoreceptor layer, it is not astonishing that the RPE demonstrated a stronger transcript-level response to ariboflavinosis than the NR. In fact, 256 genes were differentially expressed, and Fig. 6A shows the greater number of up and down regulated genes beyond the 1.5 log2 fold change threshold (Fig. 6A, list of top 50 DEGs in the RPE can be found in Table S3). Gene ontological (GO) analysis of upregulated DEGs indicated that epithelial to mesenchymal transition (EMT), hypoxia, TNF-α signaling, angiogenesis, and other responses related to stress, immune, and inflammatory pathways were significantly upregulated (Fig. 6B). However, EMT had the highest p-value, approximately 5 times that of the next upregulated pathway.
Fig. 6.

Transcriptomic analysis of RPE suggests upregulated stress, inflammation, and EMT pathways. (A) Volcano plot of DEGs determined from pairwise comparison of RDC vs RC RPE at P120. Statistically significant genes above 1.5 log2 fold changed are marked in blue (upregulated genes) or red (downregulated genes). (B) Log p-value of statistically significant upregulated pathways. (C) Heat map of DEGs involved in hypoxia and angiogenesis, presented as log2FC of normalized gene expression. (D) Heat map of DEGs involved in TNF-α signaling, general inflammation, and the complement pathway presented as log2FC of normalized gene expression. (E) Statically significant genes associated with EMT. Genes were separated into broad categories of “ECM remodeling” and “Genes implicated in EMT”. DEGs are presented as FPKM (fragments per kilobase of transcript per million mapped reads). Each group had an n value of 4 samples and each sample contained 4 pooled RPEs with selections were intentionally spread over two/three separate litters. Those RPEs were the corresponding tissue for the neural retina samples analyzed in Fig. 5. Statistical analysis was assessed by t-test with FDR correction for multiple tests (** = p < 0.01, *** = p < 0.001) for comparisons between RC and RDC fed animals. Plotted as means ± SEM. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
The RPE of RDC treated animals appeared to show an upregulation of the same genes previously identified to be differentially expressed in NR of RDC animals. Genes such as DUSP1, Egr1, and Nr4a1, and the Fos family of genes indicate a similar mechanism of hypoxia and oxidative induced stress responses as a result of flavin deprivation (see heatmap, Fig. 6C). In addition to hypoxia, angiogenic promoting genes, such as Stanniocalcin (Stc1) [46,47], are also upregulated in RDC-treated RPE (Fig. 6C). Our analysis also indicated the enrichment of pathways involved in inflammation and immunity, such as complement and IL2/STAT5 signaling. Heatmaps in Fig. 6D list statistically significant genes involved in these pathways. Notable genes include CD36, serpine1 and calcitonin receptor-like receptor gene (Calcrl) [[48], [49], [50], [51]], which may play a role in the homeostatic and immune dysregulation identified in some retinopathies.
EMT is a multiphase cellular reprogramming that involves the loss of cellular identity and is a primary feature of many cancers [52] but has also been observed in end-stage retinal pathologies [53,54]. In RPE of RDC-fed mice, many of the upregulated genes were affiliated with the cellular and extracellular matrix changes in EMT. In Fig. 6E, significantly upregulated genes associated with EMT are presented. Of the large number of DEGs in this pathway, genes were placed into broad categories based on their role in EMT and, the top 3 or 4 genes in each category were presented as normalized gene expression levels in fpkm (fragments per kilobase of exon model per million mapped reads). Laminin subunit alpha (Lama2), secreted protein acidic and cysteine rich (Sparc), extracellular matrix protein 2 (Ecm2), collagen type II alpha 1 chain (Col2A) were amongst the highest statistically significant genes involved in ECM signaling and remodeling, while lysyl oxidase like 2 (Loxl2), paired related homeobox 1 (Prrx1), platelet-derived growth factor receptor (Pdgfr), and prostate transmembrane protein, androgen induced 1 (Pmepa1) have been reported to be primarily associated with signaling or induction of EMT [[55], [56], [57], [58]]. Other prototypic markers of EMT such as vimentin (Vim) were upregulated as well [59]. To determine whether RPEs of RDC-treated animals exhibit any loss in identity, transcript levels of RPE-specific genes were assessed (top panel of Fig. 6E). No changes were observed between cohorts. Furthermore, as is the case with the NR, there were no differential expression of genes related to riboflavin transport or critical metabolic enzymes in the transcriptome of RDC-treated RPEs.
3.5. Riboflavin deficiency induced structural changes in the RPE of RDC fed mice
To investigate the changes resulted from alterations in the expression of genes in the RPE-Ch, we examined the RPE structure at P240, a time point showing a significant reduction in photoreceptor function (Fig. 2A–D). Normally, the RPE cell layer exhibits a regular, hexagonal array structure, but we observed signs of abnormal RPE shape and hypertrophy as well as increased presence of multi-nucleated RPE cells in RDC fed mice (Fig. 7A, asterisks). This phenotype is often associated with RPE atrophy (e.g. see Ref. [60]), and is observed in multiple areas of the RPE flat mounts (Fig. S2). To quantify whether there was significant RPE cell loss in the RDC group, we counted RPE cells and observed that the mean number of RPE cells/field decreased in the RDC group vs. control. We also observed abnormal localization of ezrin, which normally localizes to apical RPE microvilli (Fig. 7C, upper panel). In RDC-fed mice, ezrin localization was not restricted to the microvilli, instead localizing throughout the whole RPE cell (Fig. 7C, lower panels). Despite these structural changes, the RPE cells remained differentiated as evident from their expression of RPE65 (Fig. 7D). To help assess RPE function, we also performed prolonged ERG recordings to capture RPE-generated c-waves at P240 (Fig. S3). We observed a 19% reduction in c-wave amplitudes in the RDC-fed diet (Figs. S3A and B), but no significant changes in c-wave implicit time (Fig. S3C). Since the RPE responses are dependent on light-induced rod photoreceptor activity [61], and thus RPE response are reduced when scotopic a-waves are reduced independent of RPE function. To help understand whether there were changes in RPE function independent from photoreceptor function, we normalized c-waves to scotopic a-waves. After normalization, there was no difference in c-wave amplitude between the RC and RDC groups suggesting that the changes in c-wave amplitude are likely a reflection of reduction in photoreceptor responses (data not shown).
Fig. 7.

Riboflavin deficiency results in RPE dystrophy. (A) RPE flat-mounts collected at P240 from RDC fed mice show significant intracellular localization of β-catenin (green) (arrowheads) away from the plasma membrane, disruption of regular hexagonal cell shape, RPE hypertrophy, and frequent multinucleation (asterisks) compared to RC fed mice. Each experiment was performed three times on independent samples. (B) RPE cells were counted and plotted from flat-mounts stained in (A), **p < 0.01 by t-test, plotted is mean ± SEM. (C-D) Staining of retinal cross sections for Ezrin (green) and Rtbdn (red) (C) and RPE65 (green) (D) indicates that RPE cells are still differentiated in RDC. Notice the redistribution of ezrin in RDC fed RPE (white arrows, bottom of C). Rtbdn disappears in retinas from P240 RDC while it is seen in retinal sections from RC mice, localized basal to ezrin, which labels the microvilli of the RPE (C). RPE, Retinal pigment epithelium; OS, outer segment, IS, inner segment, OS, outer segment; ONL. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Since we identified EMT associated pathways being significantly altered in RPEs from RDC vs. RC fed animals, we investigated markers of EMT in our RPE flat mounts. In the RPE, β-catenin disassociation from cadherin [62] and mislocalization into the cytoplasm has been identified as a hallmark of EMT [63], and we observed pronounced intracellular accumulation of β-catenin in RPE cells from RDC vs. RC fed animals (Fig. 7A, arrowheads).
3.6. Riboflavin supplementation partially rescues the phenotypes associated with RF deficiency
Upon diagnosis, patients with ariboflavinosis are placed on riboflavin supplementation [5]. However, it is not clear to what extent this supplementation could correct structural and functional defects in the neural retina and RPE. We first asked whether a return to riboflavin-containing chow after several months on RDC could rescue flavin and Rtbdn levels. We placed mice on RDC from P30–P120 (by which time our data show that retinal flavins are depleted), then returned them to RC until P210 (called RDC > RC) and assessed for Rtbdn levels in comparison to mice that were kept on RDC or those that were kept on RC from birth until P210. Under these conditions, Rtbdn levels and cellular distribution in the RDC > RC group recovered to control (RC) levels (Fig. 8A–C). Similarly, we observed that in the RDC > RC group, retinal flavin levels were significantly improved compared to the RDC group, and were not significantly different from the RC group (Fig. 8D–F).
Fig. 8.

Flavin and Rtbdn protein levels recover with riboflavin supplementation. Representative immunoblot (A) and quantification (B) from three independent experiments showing the reappearance of Rtbdn following reintroduction of riboflavin in the diet (RDC > RC). (C) representative immunofluorescence images of retinal cross sections from animals fed RC and RDC until P210, and those supplemented after P120 with riboflavin up to P210 (RDC > RC) reflect restoration of Rtbdn protein. Levels of riboflavin (D), FMN (E) and FAD (F) were fully recovered in NR and RPE at P210 to normal levels in mice supplemented with riboflavin after being on RDC until P120. B was quantified from 4 different experiment on 4 independent samples/each. Number of samples for D-F, n = 3 per group. Differences between groups were assessed by one-way ANOVA with Holm-Sidak post-hoc test (****p < 0.0001). RPE, retinal pigment epithelium, OS, outer segment; IS, inner segment, ONL, outer nuclear layer.
To determine whether this rescue of retinal flavin/Rtbdn levels could lead to improvements in retinal structure and function, we designed a second group of RDC > RC animals. In this group, animals were put on RDC from P30–P240 to allow structural and functional phenotypes to develop, and were then returned to RC (labeled in Fig. 9 as the RDC > RC group). We assessed phenotypes 30 and 60 days later. At P270, mice that were returned to RC for 30 days had no improvements in scotopic a-wave, b-wave, or photopic b-wave amplitudes when compared to mice that remained on RDC (Fig. 8A–C). However, by 60 days after returning to RC (RDC > RC at P300), scotopic a-wave, scotopic b-wave, and photopic b-wave in the RDC > RC group were rescued back to levels statistically indistinguishable from RC mice (Fig. 9A–C). In RDC treated mice we observed a significant increase in the implicit time of the scotopic ERG and this increase was slightly corrected in the RDC > RC treated animals (Fig. 9D). Similarly, intensity-response curves (plotting scotopic a-wave and b-wave amplitudes at a range of light intensities) were also suppressed in RDC mice, but scotopic a responses were completely rescued after RDC animals were returned to RC for 60 days (Fig. S4A) while scotopic b-waves were partially rescued (Fig. S4B).
Fig. 9.

Sustained riboflavin supplementation rescue of the functional and structural degenerations of the retina and the RPE. (A-C) Gradual improvements in scotopic-a wave (A) and b-wave (B) and in photopic b-wave (C) responses were observed in RDC mice switched back to RC at P240 for 30 (P270) and 60 (P300) days (RDC > RC group, grey). (D-F) Implicit time (D), OPL (E), and IPL (F) were measured from RC, RDC, and RDC > RC mice at P300. RDC > RC mice exhibit significant recovery in IPL thickness. (G) RPE flatmounts were stained for β-catenin (green). Flatmounts from RDC and RDC > RC groups both exhibited some cytoplasmic accumulation of β-catenin (green, arrowheads) as well as multinucleated enlarged RPE cells (asterisks). (H) RPE cell numbers were quantified from 6 regions each of a 0.0256 mm2 window in any one of 3 independent samples and plotted from three independent animals per cohort. At P300, RDC mice have reduced number of RPE cells, but this is partially rescued in RDC > RC mice. One-way ANOVA (A-C, H) with Holm-Sidak post hoc comparison and two-way ANOVA with Bonferroni's post-hoc test (D) were used to evaluate the statistical significance between groups. In A-C differences at P240 were assessed by t-test since only two groups were present. (* = p < 0.05, ** = p < 0.01, *** = p < 0.001, **** = p < 0.0001). Graphs plot mean ± SEM. Number of samples for A-C, 4–10, each symbol represents the averaged value from one animal (two eyes). Number of samples for E-F, n = 3. OPL, Outer plexiform layer, IPL, inner plexiform layer. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Structural effects of riboflavin deficiency in the NR were also partially corrected at P300 after switching to RC diet for 60 days. Both of the OPL and IPL thickness were significantly reduced in P300 mice on RDC diet (Fig. 9E and F, respectively and Fig. S4C). However, RDC mice that were returned to RC for 60 days (RDC > RC) had OPL and IPL thicknesses that were rescued almost back to those seen in RC mice (Fig. 9E and F and vertical black lines in Fig. S4C). We also observed partial rescue in some RPE phenotypes associated with riboflavin-deficiency in animals that were returned to RC at P240 (Fig. 9G). For example, at P300, RDC-fed mice had a statistically significant reduction in the number of RPE cells per field, and there was a significant improvement in the number of RPE cells per field in RDC > RC group (Fig. 9H). However, accumulation of cytoplasmic β-catenin was similar in RDC > RC and RDC groups (arrowheads in Fig. 9G), and we continued to observe some enlarged, multinucleated RPE cells (asterisks, Fig. 9G), suggesting that some of the changes are non-reversible or longer treatment with riboflavin supplementation may be required.
To understand the effects of riboflavin supplementation on the secondary order neurons in the IPL, we performed analysis on RDC > RC retinas using the same markers used in Fig. 3. This evaluation revealed that, similar to RDC, RDC > RC retinas exhibited thinning of the OPL, as evidenced by the thinner layer of labeling for synaptic ribbons (CtBP2) and photoreceptor terminals (VGLUT-1) compared to RC retinas (Fig. S5, right-most panel). Like RDC, the dendrites of RDC > RC retinas also penetrate into the ONL with some instance of sprouting processes (white arrows). Furthermore, it appears that the reintroduction of riboflavin was not able to prevent rod bipolar cell loss or improve density of axons in the IPL (Figs. S5E and F). Combined, these data indicate that many retinal phenotypes associated with flavin deficiency can be partially rescued by re-introduction of dietary riboflavin.
4. Discussion
To help evaluate visual complications of riboflavin deficiency, we established a model system of diet-induced ariboflavinosis in mice. We found that in the absence of dietary riboflavin, cellular flavin levels were maintained longer in the NR and RPE than in the blood (from which flavins were rapidly depleted). This is likely due to the presence of specific riboflavin binding proteins in the NR and RPE. We have shown previously that one such protein is retbindin [9], elimination of which leads to a reduction of 50% in total NR flavins [15,19], suggesting that it is a key protein for sequestering/maintaining retinal flavin levels. Critically, we here show that Rtbdn protein levels drop as flavins are depleted from the retina and re-appear as flavin is reintroduced into the diet, suggesting that Rtbdn protein stability is likely promoted by riboflavin binding. These findings indicate that Rtbdn's function is linked to its RF-binding abilities and leads us to hypothesize that downstream metabolic and degenerative phenotypes associated with Rtbdn elimination are due to its function as a flavin binding protein [19]. Other riboflavin binding proteins and transporters like solute carrier family 52 members (SLC52) A1, A2 and A3 are also known to be present in blood vessels supplying tissues with their flavin requirements. In fact, RNA-seq analyses showed the presence of SLC52A2 in both NR and RPE while SLC52A3 is present only in the RPE [64,65]. Collectively, our data suggest that Rtbdn binds riboflavin that is provided by the choroidal blood supply through the RPE riboflavin transporters (SLC52A2/3) to supply the outer retina while SLC52A2 riboflavin transporter supplies riboflavin to the inner retinal cells as hypothesized in Fig. 10.
Fig. 10.

Riboflavin delivery to the retina. (A) Schematic diagram displaying riboflavin entry to the retina and the location of Rtbdn and Slc52A2/3 riboflavin transporters. (B) Retinal sections (left images) taken from WT animals on RC and RDC labeled for Rtbdn and cartoons (right) depicting the location of retbindin in RC and its absence in RDC photoreceptor.
We have previously shown that retinal flavin levels decline with age in several retinal degenerative and diabetic models [20], and some diabetic patients suffer from systemic flavin deficiency [1,20]. However, how the adult retina responds and adjusts to flavin deficiency has been heretofore unknown. Here we show that flavin deficiency progressively compromises photoreceptor function with rod responses declining earlier than cone responses. This is clearly due to physiological as opposed to structural changes, since we observe no photoreceptor degeneration at time-points when function is impaired, and photoreceptor function improves with the reintroduction of dietary riboflavin. This functional impairment is consistent with the role of flavins in energy and cellular metabolism as phototransduction is highly energy dependent. Our findings are consistent with clinical data showing that ariboflavinosis can be reversible with timely diagnosis followed by riboflavin supplementation [66,67].
The RPE exhibited signs of RPE atrophy and increased multinucleation after several months of flavin deficiency. RPE cell loss in RDC-fed animals was not significant at P240 but became statistically significant by P300. RPE multinucleation is a common feature of various RPE pathologies, including aging and drusen development in age-related macular degeneration [68,69]. Current studies suggest that RPE multinucleation normally arises as a result of lack of cytokinesis [68,70] but RPE cell multi-nucleation can also be a consequence of oxidative stress (e.g. Refs. [68,70,71]). This has been hypothesized to be a method by which the RPE monolayer can be repaired (and maintained intact) due to damage incurred during aging [68]. Interestingly, we observed improvement in RPE cell number after return to a riboflavin-containing diet. It is possible that this reflects cell proliferation to replace lost cells, however, RPE cells are generally considered post-mitotic. There is evidence for limited mitosis in adult murine RPE cells [68] but this occurs without cytokinesis and is thought to give rise to the multinucleated cells. Given the large number of DEGs in RDC RPEs involved in EMT, there is another possibility that multinucleation and loss of a tight junction integrity could be associated with early stages of EMT, since energy deprivation [72] and hypoxia [73] are drivers of EMT in many cancers. Fundamentally, however, the rescue of RPE cell number at P300 in the RDC > RC group vs. the RDC group likely reflects the fact that RPE cell loss, which is not yet significant by P240 in animals on RDC, is halted by the return to regular chow. This highlights the multifaceted requirements of riboflavin and the necessity of timely re-supplementation of dietary riboflavin to prevent any further cell loss.
We found activation of oxidative stress response pathways and increased expression of oxidative stress markers in the retinas of riboflavin-deficient mice. Many of these markers have been previously reported to associate with prolonged oxidative injury to the retina [74]. More interesting was that genes responsible for elevated energy production and expenditure were also significantly elevated, a further reflection of cellular stress. Glutathione, one of the most potent free radical scavengers, is recycled via the flavin dependent activity of the enzyme glutathione reductase [75], and reduced flavins levels correlate with glutathione depletion [76]. Glutathione depletion induces Fos/Jun expression, consistent with what we find in the flavin deficient retinas. We also find the reactive oxygen species sensor early growth response (Egr1) is upregulated in flavin-deficient retinas. Egr1 has been previously implicated in early stages of multiple retinal pathophysiology and increases during retinal aging and in inherited retinal degenerative disorders [38,77]. These signs of oxidative stress also likely predispose the NR-RPE interdependence towards functional compromise.
β-oxidation of fatty acids is facilitated via essential dehydrogenases and can be a critical part of energy generation in highly metabolically active tissues such as the retina. These dehydrogenases need flavins for their activity [78], and suboptimal flavin levels have been shown both in vitro and in vivo to result in reduced β-oxidation and poor lipid recycling. This can initiate a cascade of metabolic abnormalities [79]. Here, we show that flavin deficiency disrupts steady state lipid metabolism in both the NR and the RPE-Ch. We see significantly reduced levels of essential lipids like phosphatidylethanolamine, phosphatidylcholine, mono and polyunsaturated fatty acids in the NR upon flavin deficiency. Compromised dehydrogenase activity and poor turnover of lipids are known to affect both rod and cone functions [80] consistent with our functional results here. These findings suggest that impaired lipid metabolism may be a key mechanistic link between flavin deficiency and the development of impaired retinal function. It is important to emphasize that this effect is different from that seen for glucose metabolism. Both the NR and the RPE-Ch accumulate glucose in riboflavin-deficient animals. This increase in glucose was associated with impaired glycolysis in the RPE, but not in the NR. This difference in metabolic alterations in response to riboflavin deficiency is consistent with our previous findings in absence of Rtbdn showing separate metabolic priorities for the NR and RPE, especially when measuring steady state levels of metabolites [12]. We did not observe decreased transcript levels for flavin-dependent metabolic enzymes, highlighting the fact that the metabolic defects arise likely due to impaired enzyme function in the absence of critical flavin cofactors rather than any altered transcriptional regulation.
The inner retinal and RPE abnormalities exhibited by RDC and RDC > RC retinas demonstrate a cumulative effect of impaired metabolic processes and reduced antioxidant capacity. In the inner retina specifically, we observed atrophy and retraction of the photoreceptor synaptic terminals, loss of synaptic connectivity at the cone pedicle, and dendritic sprouting and shortening of rod bipolar cells, without photoreceptor cell loss or obvious structural changes to photoreceptor outer segments. Similar to the process of light capture in the outer segment, the propagation of the visual signal through subsequent neurons is very energetically costly [[81], [82], [83]] and is highly susceptible to oxidative damage [84,85]. The metabolic demands and antioxidant requirements of the various cell types in the retina differ greatly [10,86], and are even specific to subcellular components of the cell. Synaptic terminals may be more susceptible to metabolic dysfunction and redox changes introduced by sustained ariboflavinosis than photoreceptor outer segments. This is consistent with our prior studies showing a link between the downregulation of synaptic transmission and elimination of a critical antioxidant [87]. Synaptic remodeling also is known to accompany prolonged reductions in retinal function. Reduced synaptic input from photoreceptors can lead to morphological abnormalities at photoreceptor synapses and the interconnectedness between photoreceptors and bipolar cells is essential for their homeostasis and survival [[88], [89], [90]] Rod bipolar cells are some of the first cells to exhibit aberrant behavior and ectopic connections in retinal degenerative diseases [[90], [91], [92]] such as retinitis pigmentosa. The dendritic sprouting of rod bipolar cells that we observe in RDC and RDC > RC animals has been observed in the mouse aging retina (beyond a year old), in models of retinal detachment [93], in models of CACNA1F mutations, where the function of calcium voltage-gated channels are impaired [94], and in other retinal models where the ribbon synapse is greatly dysregulated or completely absent. Why bipolar cells adopt this sprouting behavior under certain stress or pathological conditions is still uncertain [95], however, some studies suggest that these outgrowths form when synaptic transmission between the rod and bipolar is significantly impaired [96].
Assessment of the IPL's axonal reticulum further substantiates our observation of loss of synaptic connectivity and axonal retraction amongst the cells of the inner retina. Bipolar cell processes synapse onto amacrine cells and ganglion cells in the IPL [97]. In RDC and RDC > RC we observed large voids in the axonal meshwork suggesting loss of bipolar processes or some loss of connectivity to amacrine and ganglion cells. Defects in inner retinal connectivity are interesting because optic nerve atrophy (resulting from RGC loss), has been seen in patients with riboflavin transporter deficiencies (RTDs) [98]. However, our findings regarding the inner retinal changes in this model are more analogous to early inner retinal abnormalities seen in retinopathies and degenerative disease where photoreceptors are primarily affected [92,99]. In these instances, RGCs and amacrine cells are ultimately the last group of cells to succumb to the effects of the pathology [92,[100], [101], [102]]. However, very few investigations have really examined the degeneration of the inner retina as a result of ariboflavinosis, and loss of overall riboflavin, as opposed to loss of riboflavin transporters with specific/restricted distribution. As RGC loss and optic nerve irregularities can be assessed clinically, it is possible that in RTD patients, secondary neuron abnormalities may precede RGC/optic nerve changes but are too subtle for standard clinical observation.
The inner retinal aberrations do, in part, recapitulate the neuropathological features of many RTD patients. Evaluations by imaging and nerve biopsy from RTD2 and RTD3 (mutations in SLC52A2 and SLC52A3 respectively [5,[103], [104], [105], [106]] individuals do show indications of axonal degeneration, neuronal loss [103,107], gliotic events, shortened axonal length [98,108] and variations in brain and spinal cord abnormalities [98,109]. These clinical findings manifest in many central nervous system (CNS) related impairments. Our observations are consistent with clinical findings, as we also demonstrate neuronal cell loss, shorter axons, and atrophy in the neuronal meshwork of the IPL. We have already delineated the reasons for neuronal cell/NR/RPE susceptibility to riboflavin deficiency. However, our understanding of the specific pathomechanisms involved in peripheral and CNS dysfunction as a consequence of ariboflavinosis remains limited. Studies have suggested problems with mitochondrial function and regulation, in oxidative phosphorylation intermediates [98] while others investigation have also implicated neuronal filament aggregation [108], similar to events in amyotrophic lateral sclerosis (ALS) [110]. Because our model has revealed clear biochemical aberrations in synaptic connectivity, and second order neurons, this study demonstrates that retinal models of ariboflavinosis can serve as a vehicle for understanding not only the visual impairment experienced by RTD patients, but findings can also facilitate our understanding of CNS dysfunction, given the homology between the two neuronal systems.
Patients born with mutations in tissue specific riboflavin transporters have severe tissue manifestations of ariboflavinosis even if blood flavin levels are normal [5]. It is likely that the metabolic health of their retina is compromised and likely can be reversed, if identified early enough.
5. Conclusion
In conclusion, we present a new model of ariboflavinosis and share mechanistic data suggesting that visual impairment in riboflavin deficiency occurs as a result of increased cellular oxidative stress and impaired lipid handling. We highlight the importance of flavins for retinal metabolic homeostasis and function and show that disruptions in the uptake and sequestering of flavins lead to severe functional and structural consequences in the NR and RPE. Future studies may utilize this model to determine whether similar mechanisms contribute to the established neurological disorders associated with ariboflavinosis. Visual phenotypes associated with riboflavin transporters’ mutations have not been well investigated. Here, using an animal model for riboflavin deficiency (ariboflavinosis), we show that patients suffering from ariboflavinosis due to mutations in the riboflavin transporters have retinal changes that may not be reversible if they are not identified early and provided with riboflavin supplementation.
Author contributions
TS, LI, MRA and MIN contributed to designing the experiments and writing the manuscript while TS, LI, MSM, RC, MK, XZ performed experiments and associated analyses.
Declaration of competing interest
The authors declare no competing interest.
Acknowledgments
This study was supported by a grant from the National Eye Institute (EY026499 and EY10609 to MIN and MRA). The authors thank Dr. Shannon Conley for reviewing the manuscript and providing critical comments.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2022.102375.
Contributor Information
Muayyad R. Al-Ubaidi, Email: malubaid@central.uh.edu.
Muna I. Naash, Email: mnaash@central.uh.edu.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Powers H.J. Riboflavin (vitamin B-2) and health. Am. J. Clin. Nutr. 2003;77(6):1352–1360. doi: 10.1093/ajcn/77.6.1352. [DOI] [PubMed] [Google Scholar]
- 2.McCormick D.B. The fate of riboflavin in the mammal. Nutr. Rev. 1972;30(4):75–79. doi: 10.1111/j.1753-4887.1972.tb03991.x. [DOI] [PubMed] [Google Scholar]
- 3.Muller F. Flavin radicals: chemistry and biochemistry. Free Radic. Biol. Med. 1987;3(3):215–230. doi: 10.1016/0891-5849(87)90009-8. [DOI] [PubMed] [Google Scholar]
- 4.Haack T.B., Makowski C., Yao Y., Graf E., Hempel M., Wieland T., Tauer U., Ahting U., Mayr J.A., Freisinger P., Yoshimatsu H., Inui K., Strom T.M., Meitinger T., Yonezawa A., Prokisch H. Impaired riboflavin transport due to missense mutations in SLC52A2 causes Brown-Vialetto-Van Laere syndrome. J. Inherit. Metab. Dis. 2012;35(6):943–948. doi: 10.1007/s10545-012-9513-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson J.O., Gibbs J.R., Megarbane A., Urtizberea J.A., Hernandez D.G., Foley A.R., Arepalli S., Pandraud A., Simon-Sanchez J., Clayton P., Reilly M.M., Muntoni F., Abramzon Y., Houlden H., Singleton A.B. Exome sequencing reveals riboflavin transporter mutations as a cause of motor neuron disease. Brain. 2012;135(Pt 9):2875–2882. doi: 10.1093/brain/aws161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Powers H.J., Corfe B.M., Nakano E. Riboflavin in development and cell fate. Subcell. Biochem. 2012;56:229–245. doi: 10.1007/978-94-007-2199-9_12. [DOI] [PubMed] [Google Scholar]
- 7.Kruse H.D., Sydenstricker V.P., Sebrell W.H., Cleckley H.M. U.S. Govt. print. off.; Washington,: 1940. Ocular Manifestations of Ariboflavinosis. [Google Scholar]
- 8.Subramanian V.S., Ghosal A., Kapadia R., Nabokina S.M., Said H.M. Molecular mechanisms mediating the adaptive regulation of intestinal riboflavin uptake process. PLoS One. 2015;10(6) doi: 10.1371/journal.pone.0131698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kelley R.A., Al-Ubaidi M.R., Naash M.I. Retbindin is an extracellular riboflavin-binding protein found at the photoreceptor/retinal pigment epithelium interface. J. Biol. Chem. 2015;290(8):5041–5052. doi: 10.1074/jbc.M114.624189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Linton J.D., Holzhausen L.C., Babai N., Song H., Miyagishima K.J., Stearns G.W., Lindsay K., Wei J., Chertov A.O., Peters T.A., Caffe R., Pluk H., Seeliger M.W., Tanimoto N., Fong K., Bolton L., Kuok D.L., Sweet I.R., Bartoletti T.M., Radu R.A., Travis G.H., Zagotta W.N., Townes-Anderson E., Parker E., Van der Zee C.E., Sampath A.P., Sokolov M., Thoreson W.B., Hurley J.B. Flow of energy in the outer retina in darkness and in light. Proc. Natl. Acad. Sci. U. S. A. 2010;107(19):8599–8604. doi: 10.1073/pnas.1002471107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ames A., Li Y.Y., Heher E.C., Kimble C.R. Energy metabolism of rabbit retina as related to function: high cost of Na+ transport. J. Neurosci. 1992;12(3):840–853. doi: 10.1523/JNEUROSCI.12-03-00840.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sinha T., Naash M.I., Al-Ubaidi M.R. The symbiotic relationship between the neural retina and retinal pigment epithelium is supported by utilizing differential metabolic pathways. iScience. 2020;23(4) doi: 10.1016/j.isci.2020.101004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Batey D.W., Daneshgar K.K., Eckhert C.D. Flavin levels in the rat retina. Exp. Eye Res. 1992;54(4):605–609. doi: 10.1016/0014-4835(92)90139-j. [DOI] [PubMed] [Google Scholar]
- 14.Batey D.W., Eckhert C.D. Identification of FAD, FMN, and riboflavin in the retina by microextraction and high-performance liquid chromatography. Anal. Biochem. 1990;188(1):164–167. doi: 10.1016/0003-2697(90)90546-l. [DOI] [PubMed] [Google Scholar]
- 15.Kelley R.A., Al-Ubaidi M.R., Sinha T., Genc A.M., Makia M.S., Ikelle L., Naash M.I. Ablation of the riboflavin-binding protein retbindin reduces flavin levels and leads to progressive and dose-dependent degeneration of rods and cones. J. Biol. Chem. 2017;292(51):21023–21034. doi: 10.1074/jbc.M117.785105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen X., Kezic J., Bernard C., McMenamin P.G. Rd8 mutation in the Crb1 gene of CD11c-eYFP transgenic reporter mice results in abnormal numbers of CD11c-positive cells in the retina. J. Neuropathol. Exp. Neurol. 2013;72(8):782–790. doi: 10.1097/NEN.0b013e31829e8375. [DOI] [PubMed] [Google Scholar]
- 17.Danciger M., Matthes M.T., Yasamura D., Akhmedov N.B., Rickabaugh T., Gentleman S., Redmond T.M., La Vail M.M., Farber D.B. A QTL on distal chromosome 3 that influences the severity of light-induced damage to mouse photoreceptors. Mamm. Genome. 2000;11(6):422–427. doi: 10.1007/s003350010081. [DOI] [PubMed] [Google Scholar]
- 18.Kim S.R., Fishkin N., Kong J., Nakanishi K., Allikmets R., Sparrow J.R. Rpe65 Leu450Met variant is associated with reduced levels of the retinal pigment epithelium lipofuscin fluorophores A2E and iso-A2E. Proc. Natl. Acad. Sci. U.S.A. 2004;101(32):11668–11672. doi: 10.1073/pnas.0403499101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sinha T., Du J., Makia M.S., Hurley J.B., Naash M.I., Al-Ubaidi M.R. Absence of retbindin blocks glycolytic flux, disrupts metabolic homeostasis, and leads to photoreceptor degeneration. Proc. Natl. Acad. Sci. U. S. A. 2021;118(6) doi: 10.1073/pnas.2018956118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sinha T., Makia M., Du J., Naash M.I., Al-Ubaidi M.R. Flavin homeostasis in the mouse retina during aging and degeneration. J. Nutr. Biochem. 2018;62:123–133. doi: 10.1016/j.jnutbio.2018.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mortazavi A., Williams B.A., McCue K., Schaeffer L., Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008;5(7):621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 23.Kuleshov M.V., Jones M.R., Rouillard A.D., Fernandez N.F., Duan Q., Wang Z., Koplev S., Jenkins S.L., Jagodnik K.M., Lachmann A., McDermott M.G., Monteiro C.D., Gundersen G.W., Ma'ayan A., Enrichr A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44(W1):W90–W97. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Torre D., Lachmann A., Ma'ayan A., BioJupies Automated generation of interactive notebooks for RNA-seq data analysis in the cloud. Cell Syst. 2018;7(5):556–561 e3. doi: 10.1016/j.cels.2018.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Negishi K., Kato S., Teranishi T. Dopamine cells and rod bipolar cells contain protein kinase C-like immunoreactivity in some vertebrate retinas. Neurosci. Lett. 1988;94(3):247–252. doi: 10.1016/0304-3940(88)90025-0. [DOI] [PubMed] [Google Scholar]
- 26.Bellocchio E.E., Reimer R.J., Fremeau R.T., Jr., Edwards R.H. Uptake of glutamate into synaptic vesicles by an inorganic phosphate transporter. Science. 2000;289(5481):957–960. doi: 10.1126/science.289.5481.957. [DOI] [PubMed] [Google Scholar]
- 27.Johnson J., Sherry D.M., Liu X., Fremeau R.T., Jr., Seal R.P., Edwards R.H., Copenhagen D.R. Vesicular glutamate transporter 3 expression identifies glutamatergic amacrine cells in the rodent retina. J. Comp. Neurol. 2004;477(4):386–398. doi: 10.1002/cne.20250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmitz F., Konigstorfer A., Sudhof T.C. RIBEYE, a component of synaptic ribbons: a protein's journey through evolution provides insight into synaptic ribbon function. Neuron. 2000;28(3):857–872. doi: 10.1016/s0896-6273(00)00159-8. [DOI] [PubMed] [Google Scholar]
- 29.Grassmeyer J.J., Cahill A.L., Hays C.L., Barta C., Quadros R.M., Gurumurthy C.B., Thoreson W.B. Ca(2+) sensor synaptotagmin-1 mediates exocytosis in mammalian photoreceptors. Elife. 2019;8 doi: 10.7554/eLife.45946. eLife. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Piatigorsky J. Dual use of the transcriptional repressor (CtBP2)/ribbon synapse (RIBEYE) gene: how prevalent are multifunctional genes? Trends Neurosci. 2001;24(10):555–557. doi: 10.1016/s0166-2236(00)01894-4. [DOI] [PubMed] [Google Scholar]
- 31.Puthussery T., Gayet-Primo J., Taylor W.R. Localization of the calcium-binding protein secretagogin in cone bipolar cells of the mammalian retina. J. Comp. Neurol. 2010;518(4):513–525. doi: 10.1002/cne.22234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wagner L., Oliyarnyk O., Gartner W., Nowotny P., Groeger M., Kaserer K., Waldhausl W., Pasternack M.S. Cloning and expression of secretagogin, a novel neuroendocrine- and pancreatic islet of Langerhans-specific Ca2+-binding protein. J. Biol. Chem. 2000;275(32):24740–24751. doi: 10.1074/jbc.M001974200. [DOI] [PubMed] [Google Scholar]
- 33.Matsui K., von Gersdorff H. The great escape of glutamate from the depth of presynaptic invaginations. Neuron. 2006;50(5):669–671. doi: 10.1016/j.neuron.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 34.Strettoi E., Novelli E., Mazzoni F., Barone I., Damiani D. Complexity of retinal cone bipolar cells. Prog. Retin. Eye Res. 2010;29(4):272–283. doi: 10.1016/j.preteyeres.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kolb H., Linberg K.A., Fisher S.K. Neurons of the human retina: a Golgi study. J. Comp. Neurol. 1992;318(2):147–187. doi: 10.1002/cne.903180204. [DOI] [PubMed] [Google Scholar]
- 36.Kolb H., Nelson R., Fernandez E., Jones B. The organization of the retina and visual system, WEBVISION. https://webvision.med.utah.edu/ [PubMed]
- 37.Udhayabanu T., Manole A., Rajeshwari M., Varalakshmi P., Houlden H., Ashokkumar B. Riboflavin responsive mitochondrial dysfunction in neurodegenerative diseases. J. Clin. Med. 2017;6(5) doi: 10.3390/jcm6050052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pagel J.I., Deindl E. Disease progression mediated by egr-1 associated signaling in response to oxidative stress. Int. J. Mol. Sci. 2012;13(10):13104–13117. doi: 10.3390/ijms131013104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cheng J.P., Hu W.X., Liu X.J., Zheng M., Shi W., Wang W.H. Expression of c-fos and oxidative stress on brain of rats reared on food from mercury-selenium coexisting mining area. J. Environ. Sci. (China) 2006;18(4):788–792. [PubMed] [Google Scholar]
- 40.Tormos C., Javier Chaves F., Garcia M.J., Garrido F., Jover R., O'Connor J.E., Iradi A., Oltra A., Oliva M.R., Saez G.T. Role of glutathione in the induction of apoptosis and c-fos and c-jun mRNAs by oxidative stress in tumor cells. Cancer Lett. 2004;208(1):103–113. doi: 10.1016/j.canlet.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 41.Ozolins T.R., Hales B.F. Oxidative stress regulates the expression and activity of transcription factor activator protein-1 in rat conceptus. J. Pharmacol. Exp. Therapeut. 1997;280(2):1085–1093. [PubMed] [Google Scholar]
- 42.Liu C., Shi Y., Du Y., Ning X., Liu N., Huang D., Liang J., Xue Y., Fan D. Dual-specificity phosphatase DUSP1 protects overactivation of hypoxia-inducible factor 1 through inactivating ERK MAPK. Exp. Cell Res. 2005;309(2):410–418. doi: 10.1016/j.yexcr.2005.06.022. [DOI] [PubMed] [Google Scholar]
- 43.Liu Y.X., Wang J., Guo J., Wu J., Lieberman H.B., Yin Y. DUSP1 is controlled by p53 during the cellular response to oxidative stress. Mol. Cancer Res. 2008;6(4):624–633. doi: 10.1158/1541-7786.MCR-07-2019. [DOI] [PubMed] [Google Scholar]
- 44.Zong C., Qin D., Yu C., Gao P., Chen J., Lu S., Zhang Y., Liu Y., Yang Y., Pu Z., Li X., Fu Y., Guan Q., Wang X. The stress-response molecule NR4A1 resists ROS-induced pancreatic beta-cells apoptosis via WT1. Cell. Signal. 2017;35:129–139. doi: 10.1016/j.cellsig.2017.03.012. [DOI] [PubMed] [Google Scholar]
- 45.Ipseiz N., Uderhardt S., Scholtysek C., Steffen M., Schabbauer G., Bozec A., Schett G., Kronke G. The nuclear receptor Nr4a1 mediates anti-inflammatory effects of apoptotic cells. J. Immunol. 2014;192(10):4852–4858. doi: 10.4049/jimmunol.1303377. [DOI] [PubMed] [Google Scholar]
- 46.He L.F., Wang T.T., Gao Q.Y., Zhao G.F., Huang Y.H., Yu L.K., Hou Y.Y. Stanniocalcin-1 promotes tumor angiogenesis through up-regulation of VEGF in gastric cancer cells. J. Biomed. Sci. 2011;18:39. doi: 10.1186/1423-0127-18-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao M., Xie W., Tsai S.H., Hein T.W., Rocke B.A., Kuo L., Rosa R.H., Jr. Intravitreal stanniocalcin-1 enhances new blood vessel growth in a rat model of laser-induced choroidal neovascularization. Invest. Ophthalmol. Vis. Sci. 2018;59(2):1125–1133. doi: 10.1167/iovs.17-23083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Picard E., Houssier M., Bujold K., Sapieha P., Lubell W., Dorfman A., Racine J., Hardy P., Febbraio M., Lachapelle P., Ong H., Sennlaub F., Chemtob S. CD36 plays an important role in the clearance of oxLDL and associated age-dependent sub-retinal deposits. Aging. 2010;2(12):981–989. doi: 10.18632/aging.100218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Awadalla M.S., Burdon K.P., Thapa S.S., Hewitt A.W., Craig J.E. A cross-ethnicity investigation of genes previously implicated in primary angle closure glaucoma. Mol. Vis. 2012;18:2247–2254. [PMC free article] [PubMed] [Google Scholar]
- 50.Shastry B.S. Genetic susceptibility to primary angle closure glaucoma (PACG) Discov. Med. 2013;15(80):17–22. [PubMed] [Google Scholar]
- 51.Higgins P.J. Balancing AhR-dependent pro-oxidant and nrf2-responsive anti-oxidant pathways in age-related retinopathy: is SERPINE1 expression a therapeutic target in disease onset and progression? J. Mol. Genet. Med. 2014;8(2):101. doi: 10.4172/1747-0862.1000101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brabletz T., Kalluri R., Nieto M.A., Weinberg R.A. EMT in cancer. Nat. Rev. Cancer. 2018;18(2):128–134. doi: 10.1038/nrc.2017.118. [DOI] [PubMed] [Google Scholar]
- 53.Zhou M., Geathers J.S., Grillo S.L., Weber S.R., Wang W., Zhao Y., Sundstrom J.M. Role of epithelial-mesenchymal transition in retinal pigment epithelium dysfunction. Front. Cell Dev. Biol. 2020;8:501. doi: 10.3389/fcell.2020.00501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sripathi S.R., Hu M.W., Turaga R.C., Mertz J., Liu M.M., Wan J., Maruotti J., Wahlin K.J., Berlinicke C.A., Qian J., Zack D.J. Proteome landscape of epithelial-to-mesenchymal transition (EMT) of retinal pigment epithelium shares commonalities with malignancy-associated EMT. Mol. Cell. Proteomics. 2021;20 doi: 10.1016/j.mcpro.2021.100131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cuevas E.P., Eraso P., Mazon M.J., Santos V., Moreno-Bueno G., Cano A., Portillo F. LOXL2 drives epithelial-mesenchymal transition via activation of IRE1-XBP1 signalling pathway. Sci. Rep. 2017;7 doi: 10.1038/srep44988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Steller E.J., Raats D.A., Koster J., Rutten B., Govaert K.M., Emmink B.L., Snoeren N., van Hooff S.R., Holstege F.C., Maas C., Borel Rinkes I.H., Kranenburg O. PDGFRB promotes liver metastasis formation of mesenchymal-like colorectal tumor cells. Neoplasia. 2013;15(2):204–217. doi: 10.1593/neo.121726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guo J., Fu Z., Wei J., Lu W., Feng J., Zhang S. PRRX1 promotes epithelial-mesenchymal transition through the Wnt/beta-catenin pathway in gastric cancer. Med. Oncol. 2015;32(1):393. doi: 10.1007/s12032-014-0393-x. [DOI] [PubMed] [Google Scholar]
- 58.Zhang L., Wang X., Lai C., Zhang H., Lai M. PMEPA1 induces EMT via a non-canonical TGF-beta signalling in colorectal cancer. J. Cell Mol. Med. 2019;23(5):3603–3615. doi: 10.1111/jcmm.14261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu S., Du Y., Beckford J., Alachkar H. Upregulation of the EMT marker vimentin is associated with poor clinical outcome in acute myeloid leukemia. J. Transl. Med. 2018;16(1):170. doi: 10.1186/s12967-018-1539-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang X., Zhao L., Campos M.M., Abu-Asab M., Ortolan D., Hotaling N., Bharti K., Wong W.T. CSF1R blockade induces macrophage ablation and results in mouse choroidal vascular atrophy and RPE disorganization. Elife. 2020;9 doi: 10.7554/eLife.55564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Samuels I.S., Sturgill G.M., Grossman G.H., Rayborn M.E., Hollyfield J.G., Peachey N.S. Light-evoked responses of the retinal pigment epithelium: changes accompanying photoreceptor loss in the mouse. J. Neurophysiol. 2010;104(1):391–402. doi: 10.1152/jn.00088.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen Y.T., Stewart D.B., Nelson W.J. Coupling assembly of the E-cadherin/beta-catenin complex to efficient endoplasmic reticulum exit and basal-lateral membrane targeting of E-cadherin in polarized MDCK cells. J. Cell Biol. 1999;144(4):687–699. doi: 10.1083/jcb.144.4.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhao C., Yasumura D., Li X., Matthes M., Lloyd M., Nielsen G., Ahern K., Snyder M., Bok D., Dunaief J.L., LaVail M.M., Vollrath D. mTOR-mediated dedifferentiation of the retinal pigment epithelium initiates photoreceptor degeneration in mice. J. Clin. Invest. 2011;121(1):369–383. doi: 10.1172/JCI44303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kubo Y., Miki S., Akanuma S.I., Hosoya K.I. Riboflavin transport mediated by riboflavin transporters (RFVTs/SLC52A) at the rat outer blood-retinal barrier. Drug Metabol. Pharmacokinet. 2019;34(6):380–386. doi: 10.1016/j.dmpk.2019.08.002. [DOI] [PubMed] [Google Scholar]
- 65.Kubo Y., Yahata S., Miki S., Akanuma S.I., Hosoya K.I. Blood-to-retina transport of riboflavin via RFVTs at the inner blood-retinal barrier. Drug Metabol. Pharmacokinet. 2017;32(1):92–99. doi: 10.1016/j.dmpk.2016.09.006. [DOI] [PubMed] [Google Scholar]
- 66.Suwannasom N., Kao I., Pruss A., Georgieva R., Baumler H. Riboflavin: the health benefits of a forgotten natural vitamin. Int. J. Mol. Sci. 2020;21(3) doi: 10.3390/ijms21030950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thakur K., Tomar S.K., Singh A.K., Mandal S., Arora S. Riboflavin and health: a review of recent human research. Crit. Rev. Food Sci. Nutr. 2017;57(17):3650–3660. doi: 10.1080/10408398.2016.1145104. [DOI] [PubMed] [Google Scholar]
- 68.Chen M., Rajapakse D., Fraczek M., Luo C., Forrester J.V., Xu H. Retinal pigment epithelial cell multinucleation in the aging eye - a mechanism to repair damage and maintain homoeostasis. Aging Cell. 2016;15(3):436–445. doi: 10.1111/acel.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Al-Hussaini H., Schneiders M., Lundh P., Jeffery G. Drusen are associated with local and distant disruptions to human retinal pigment epithelium cells. Exp. Eye Res. 2009;88(3):610–612. doi: 10.1016/j.exer.2008.09.021. [DOI] [PubMed] [Google Scholar]
- 70.Rajapakse D., Chen M., Curtis T.M., Xu H. PKCzeta-dependent upregulation of p27kip1 contributes to oxidative stress induced retinal pigment epithelial cell multinucleation. Aging. 2017;9(10):2052–2068. doi: 10.18632/aging.101299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Telegina D.V., Kozhevnikova O.S., Bayborodin S.I., Kolosova N.G. Contributions of age-related alterations of the retinal pigment epithelium and of glia to the AMD-like pathology in OXYS rats. Sci. Rep. 2017;7 doi: 10.1038/srep41533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen G., Liu H., Zhang Y., Liang J., Zhu Y., Zhang M., Yu D., Wang C., Hou J. Silencing PFKP inhibits starvation-induced autophagy, glycolysis, and epithelial mesenchymal transition in oral squamous cell carcinoma. Exp. Cell Res. 2018;370(1):46–57. doi: 10.1016/j.yexcr.2018.06.007. [DOI] [PubMed] [Google Scholar]
- 73.Tam S.Y., Wu V.W.C., Law H.K.W. Hypoxia-induced epithelial-mesenchymal transition in cancers: HIF-1alpha and beyond. Front. Oncol. 2020;10:486. doi: 10.3389/fonc.2020.00486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Strunnikova N., Zhang C., Teichberg D., Cousins S.W., Baffi J., Becker K.G., Csaky K.G. Survival of retinal pigment epithelium after exposure to prolonged oxidative injury: a detailed gene expression and cellular analysis. Invest. Ophthalmol. Vis. Sci. 2004;45(10):3767–3777. doi: 10.1167/iovs.04-0311. [DOI] [PubMed] [Google Scholar]
- 75.Beutler E. Effect of flavin compounds on glutathione reductase activity: in vivo and in vitro studies. J. Clin. Invest. 1969;48(10):1957–1966. doi: 10.1172/JCI106162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bergelson S., Pinkus R., Daniel V. Intracellular glutathione levels regulate Fos/Jun induction and activation of glutathione S-transferase gene expression. Cancer Res. 1994;54(1):36–40. [PubMed] [Google Scholar]
- 77.Chaum E., Yin J., Lang J.C. Molecular responses transduced by serial oxidative stress in the retinal pigment epithelium: feedback control modeling of gene expression. Neurochem. Res. 2011;36(4):574–582. doi: 10.1007/s11064-010-0370-5. [DOI] [PubMed] [Google Scholar]
- 78.Wang Y., Palmfeldt J., Gregersen N., Makhov A.M., Conway J.F., Wang M., McCalley S.P., Basu S., Alharbi H., St Croix C., Calderon M.J., Watkins S., Vockley J. Mitochondrial fatty acid oxidation and the electron transport chain comprise a multifunctional mitochondrial protein complex. J. Biol. Chem. 2019;294(33):12380–12391. doi: 10.1074/jbc.RA119.008680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Houten S.M., Violante S., Ventura F.V., Wanders R.J. The biochemistry and physiology of mitochondrial fatty acid beta-oxidation and its genetic disorders. Annu. Rev. Physiol. 2016;78:23–44. doi: 10.1146/annurev-physiol-021115-105045. [DOI] [PubMed] [Google Scholar]
- 80.Tyni T., Paetau A., Strauss A.W., Middleton B., Kivela T. Mitochondrial fatty acid beta-oxidation in the human eye and brain: implications for the retinopathy of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Pediatr. Res. 2004;56(5):744–750. doi: 10.1203/01.PDR.0000141967.52759.83. [DOI] [PubMed] [Google Scholar]
- 81.Viegas F.O., Neuhauss S.C.F. A metabolic landscape for maintaining retina integrity and function. Front. Mol. Neurosci. 2021;14 doi: 10.3389/fnmol.2021.656000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wong-Riley M.T. Energy metabolism of the visual system. Eye Brain. 2010;2:99–116. doi: 10.2147/EB.S9078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Okawa H., Sampath A.P., Laughlin S.B., Fain G.L. ATP consumption by mammalian rod photoreceptors in darkness and in light. Curr. Biol. 2008;18(24):1917–1921. doi: 10.1016/j.cub.2008.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mattson M.P., Liu D. Energetics and oxidative stress in synaptic plasticity and neurodegenerative disorders. NeuroMolecular Med. 2002;2(2):215–231. doi: 10.1385/NMM:2:2:215. [DOI] [PubMed] [Google Scholar]
- 85.Urano S., Asai Y., Makabe S., Matsuo M., Izumiyama N., Ohtsubo K., Endo T. Oxidative injury of synapse and alteration of antioxidative defense systems in rats, and its prevention by vitamin E. Eur. J. Biochem. 1997;245(1):64–70. doi: 10.1111/j.1432-1033.1997.00064.x. [DOI] [PubMed] [Google Scholar]
- 86.Hurley J.B., Lindsay K.J., Du J. Glucose, lactate, and shuttling of metabolites in vertebrate retinas. J. Neurosci. Res. 2015;93(7):1079–1092. doi: 10.1002/jnr.23583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ikelle L., Naash M.I., Al-Ubaidi M.R. Modulation of SOD3 levels is detrimental to retinal homeostasis. Antioxidants. 2021;10(10) doi: 10.3390/antiox10101595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Varela C., Igartua I., De la Rosa E.J., De la Villa P. Functional modifications in rod bipolar cells in a mouse model of retinitis pigmentosa. Vision Res. 2003;43(8):879–885. doi: 10.1016/s0042-6989(02)00493-5. [DOI] [PubMed] [Google Scholar]
- 89.Beier C., Hovhannisyan A., Weiser S., Kung J., Lee S., Lee D.Y., Huie P., Dalal R., Palanker D., Sher A. Deafferented adult rod bipolar cells create new synapses with photoreceptors to restore vision. J. Neurosci. 2017;37(17):4635–4644. doi: 10.1523/JNEUROSCI.2570-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Strettoi E., Pignatelli V. Modifications of retinal neurons in a mouse model of retinitis pigmentosa. Proc. Natl. Acad. Sci. U. S. A. 2000;97(20):11020–11025. doi: 10.1073/pnas.190291097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Strettoi E., Pignatelli V., Rossi C., Porciatti V., Falsini B. Remodeling of second-order neurons in the retina of rd/rd mutant mice. Vision Res. 2003;43(8):867–877. doi: 10.1016/s0042-6989(02)00594-1. [DOI] [PubMed] [Google Scholar]
- 92.Strettoi E. A survey of retinal remodeling. Front. Cell. Neurosci. 2015;9:494. doi: 10.3389/fncel.2015.00494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lewis G.P., Linberg K.A., Fisher S.K. Neurite outgrowth from bipolar and horizontal cells after experimental retinal detachment. Invest. Ophthalmol. Vis. Sci. 1998;39(2):424–434. [PubMed] [Google Scholar]
- 94.Mansergh F., Orton N.C., Vessey J.P., Lalonde M.R., Stell W.K., Tremblay F., Barnes S., Rancourt D.E., Bech-Hansen N.T. Mutation of the calcium channel gene Cacna1f disrupts calcium signaling, synaptic transmission and cellular organization in mouse retina. Hum. Mol. Genet. 2005;14(20):3035–3046. doi: 10.1093/hmg/ddi336. [DOI] [PubMed] [Google Scholar]
- 95.Liets L.C., Eliasieh K., van der List D.A., Chalupa L.M. Dendrites of rod bipolar cells sprout in normal aging retina. Proc. Natl. Acad. Sci. U. S. A. 2006;103(32):12156–12160. doi: 10.1073/pnas.0605211103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dick O., tom Dieck S., Altrock W.D., Ammermuller J., Weiler R., Garner C.C., Gundelfinger E.D., Brandstatter J.H. The presynaptic active zone protein bassoon is essential for photoreceptor ribbon synapse formation in the retina. Neuron. 2003;37(5):775–786. doi: 10.1016/s0896-6273(03)00086-2. [DOI] [PubMed] [Google Scholar]
- 97.Morgan J.L., Dhingra A., Vardi N., Wong R.O. Axons and dendrites originate from neuroepithelial-like processes of retinal bipolar cells. Nat. Neurosci. 2006;9(1):85–92. doi: 10.1038/nn1615. [DOI] [PubMed] [Google Scholar]
- 98.Manole A., Jaunmuktane Z., Hargreaves I., Ludtmann M.H.R., Salpietro V., Bello O.D., Pope S., Pandraud A., Horga A., Scalco R.S., Li A., Ashokkumar B., Lourenco C.M., Heales S., Horvath R., Chinnery P.F., Toro C., Singleton A.B., Jacques T.S., Abramov A.Y., Muntoni F., Hanna M.G., Reilly M.M., Revesz T., Kullmann D.M., Jepson J.E.C., Houlden H. Clinical, pathological and functional characterization of riboflavin-responsive neuropathy. Brain. 2017;140(11):2820–2837. doi: 10.1093/brain/awx231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Soto F., Kerschensteiner D. Synaptic remodeling of neuronal circuits in early retinal degeneration. Front. Cell. Neurosci. 2015;9:395. doi: 10.3389/fncel.2015.00395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mazzoni F., Novelli E., Strettoi E. Retinal ganglion cells survive and maintain normal dendritic morphology in a mouse model of inherited photoreceptor degeneration. J. Neurosci. 2008;28(52):14282–14292. doi: 10.1523/JNEUROSCI.4968-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Damiani D., Novelli E., Mazzoni F., Strettoi E. Undersized dendritic arborizations in retinal ganglion cells of the rd1 mutant mouse: a paradigm of early onset photoreceptor degeneration. J. Comp. Neurol. 2012;520(7):1406–1423. doi: 10.1002/cne.22802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.O'Brien E.E., Greferath U., Fletcher E.L. The effect of photoreceptor degeneration on ganglion cell morphology. J. Comp. Neurol. 2014;522(5):1155–1170. doi: 10.1002/cne.23487. [DOI] [PubMed] [Google Scholar]
- 103.Foley A.R., Menezes M.P., Pandraud A., Gonzalez M.A., Al-Odaib A., Abrams A.J., Sugano K., Yonezawa A., Manzur A.Y., Burns J., Hughes I., McCullagh B.G., Jungbluth H., Lim M.J., Lin J.P., Megarbane A., Urtizberea J.A., Shah A.H., Antony J., Webster R., Broomfield A., Ng J., Mathew A.A., O'Byrne J.J., Forman E., Scoto M., Prasad M., O'Brien K., Olpin S., Oppenheim M., Hargreaves I., Land J.M., Wang M.X., Carpenter K., Horvath R., Straub V., Lek M., Gold W., Farrell M.O., Brandner S., Phadke R., Matsubara K., McGarvey M.L., Scherer S.S., Baxter P.S., King M.D., Clayton P., Rahman S., Reilly M.M., Ouvrier R.A., Christodoulou J., Zuchner S., Muntoni F., Houlden H. Treatable childhood neuronopathy caused by mutations in riboflavin transporter RFVT2. Brain. 2014;137(Pt 1):44–56. doi: 10.1093/brain/awt315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Green P., Wiseman M., Crow Y.J., Houlden H., Riphagen S., Lin J.P., Raymond F.L., Childs A.M., Sheridan E., Edwards S., Josifova D.J. Brown-Vialetto-Van Laere syndrome, a ponto-bulbar palsy with deafness, is caused by mutations in c20orf54. Am. J. Hum. Genet. 2010;86(3):485–489. doi: 10.1016/j.ajhg.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cali E., Dominik N., Manole A., Houlden H. In: GeneReviews((R)), Seattle (WA) Adam M.P., Ardinger H.H., Pagon R.A., Wallace S.E., Bean L.J.H., Gripp K.W., Mirzaa G.M., Amemiya A., editors. 1993. Riboflavin transporter deficiency. [PubMed] [Google Scholar]
- 106.Bosch A.M., Stroek K., Abeling N.G., Waterham H.R., Ijlst L., Wanders R.J. The Brown-Vialetto-Van Laere and Fazio Londe syndrome revisited: natural history, genetics, treatment and future perspectives. Orphanet J. Rare Dis. 2012;7:83. doi: 10.1186/1750-1172-7-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chaya S., Zampoli M., Gray D., Booth J., Riordan G., Ndondo A., Fieggen K., Rusch J., van der Watt G., Pillay K., van der Westhuizen F., Menezes M., Wilmshurst J. The first case of riboflavin transporter deficiency in sub-saharan Africa. Semin. Pediatr. Neurol. 2018;26:10–14. doi: 10.1016/j.spen.2017.03.002. [DOI] [PubMed] [Google Scholar]
- 108.Rizzo F., Ramirez A., Compagnucci C., Salani S., Melzi V., Bordoni A., Fortunato F., Niceforo A., Bresolin N., Comi G.P., Bertini E., Nizzardo M., Corti S. Genome-wide RNA-seq of iPSC-derived motor neurons indicates selective cytoskeletal perturbation in Brown-Vialetto disease that is partially rescued by riboflavin. Sci. Rep. 2017;7 doi: 10.1038/srep46271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Carey G., Kuchcinski G., Gauvrit F., Defebvre L., Nguyen S., Dhaenens C.M., Dessein A.F., Vianey-Saban C., Acquaviva C., Tard C. Three cases of adult-onset Brown-Vialetto-Van Laere syndrome: novel variants in SLC52A3 gene and MRI abnormalities. Neuromuscul. Disord. 2021;31(8):752–755. doi: 10.1016/j.nmd.2021.06.009. [DOI] [PubMed] [Google Scholar]
- 110.Didonna A., Opal P. The role of neurofilament aggregation in neurodegeneration: lessons from rare inherited neurological disorders. Mol. Neurodegener. 2019;14(1):19. doi: 10.1186/s13024-019-0318-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.