

Abstract
Increasing loss of structure and function of neurons and decline in cognitive function is commonly seen during the progression of neurologic diseases, although the causes and initial symptoms of individual diseases are distinct. This observation suggests a convergence of common degenerative features. In myotonic dystrophy type 1 (DM1), the expression of expanded CUG RNA induces neurotransmission dysfunction before axon and dendrite degeneration and reduced MBNL2 expression associated with aberrant alternative splicing. The role of loss of function of MBNL2 in the pathogenesis of neurodegeneration and the causal mechanism of neurodegeneration-reduced expression of MBNL2 remain elusive. Here, we show that increased MBNL2 expression is associated with neuronal maturation and required for neuronal morphogenesis and the fetal to adult developmental transition of RNA processing. Neurodegenerative conditions including NMDA receptor (NMDAR)-mediated excitotoxicity and dysregulated calcium homeostasis triggered nuclear translocation of calpain-2, thus resulting in MBNL2 degradation and reversal of MBNL2-regulated RNA processing to developmental patterns. Nuclear expression of calpain-2 resembled its developmental pattern and was associated with MBNL2 degradation. Knock-down of calpain-2 expression or inhibition of calpain-2 nuclear translocation prevented neurodegeneration-reduced MBNL2 expression and dysregulated RNA processing. Increased calpain-2 nuclear translocation associated with reduced MBNL2 expression and aberrant RNA processing occurred in models for DM1 and Alzheimer's disease (AD) including EpA960/CaMKII-Cre mice of either sex and female APP/PS1 and THY-Tau22 mice. Our results identify a regulatory mechanism for MBNL2 downregulation and suggest that calpain-2-mediated MBNL2 degradation accompanied by re-induction of a developmental RNA processing program may be a converging pathway to neurodegeneration.
SIGNIFICANCE STATEMENT Neurologic diseases share many features during disease progression, such as cognitive decline and brain atrophy, which suggests a common pathway for developing degenerative features. Here, we show that the neurodegenerative conditions glutamate-induced excitotoxicity and dysregulated calcium homeostasis induced translocation of the cysteine protease calpain-2 into the nucleus, resulting in MBNL2 degradation and reversal of MBNL2-regulated RNA processing to an embryonic pattern. Knock-down or inhibition of nuclear translocation of calpain-2 prevented MBNL2 degradation and maintained MBNL2-regulated RNA processing in the adult pattern. Models of myotonic dystrophy and Alzheimer's disease (AD) also showed calpain-2-mediated MBNL2 degradation and a developmental RNA processing program. Our studies suggest MBNL2 function disrupted by calpain-2 as a common pathway, thus providing an alternative therapeutic strategy for neurodegeneration.
Keywords: Alzheimer's disease, calpain-2, excitotoxicity, MBNL2, myotonic dystrophy, RNA-processing
Introduction
Cognitive deficits are prevalent among individuals with myotonic dystrophy type 1 (DM1). The cognitive dysfunctions include impaired executive functions, memory tasks and attention-demanding tasks. Progressive cognitive decline with age is frequently seen in individuals with adult-onset DM1 (Gallais et al., 2017). Neuropathological lesions including neurofibrillary degeneration and amyloid deposition, which are observed in Alzheimer's disease (AD), are also reported in the brain of individuals with DM1 (Maurage et al., 2005; Jimenez-Marin et al., 2021). DM1 is caused by an expansion of CTG repeats in the 3′ untranslated region (UTR) of the dystrophia myotonica protein kinase (DMPK) gene. The primary mechanism of pathogenesis is a gain of function of the mutant DMPK mRNA, which contains expanded CUG repeats, accumulates in nuclear foci and affects cytoplasmic and nuclear functions of RNA binding proteins such as the muscleblind-like (MBNL) protein family (Thornton, 2014). The pathogenic CUG RNA binds and sequesters MBNL proteins resulting in loss of function that has been implicated in DM1 neural pathogenesis.
The MBNL protein family regulates alternative splicing and polyadenylation, translation, mRNA localization and miRNA processing (Rau et al., 2011; Charizanis et al., 2012; Wang et al., 2012). The cytoplasmic fraction of MBNL1 promotes and maintains neuronal morphology, although the underlying mechanism remains elusive (Wang et al., 2018). MBNL2 regulates the developmental transition of alternative splicing and polyadenylation in the brain (Charizanis et al., 2012; Batra et al., 2014), but how this function is associated with neuronal maturation remains unknown. In a brain-specific DM1 mouse model, which expresses expanded CUG RNA in the postnatal brain, MBNL1 and MBNL2 exhibited a sequential response to expanded CUG RNA (Wang et al., 2017). Learning deficits associated with reduced cytoplasmic MBNL1 level occurred before axon and dendrite degeneration, followed by reduced MBNL2 level and aberrant alternative splicing. Reduced cytoplasmic MBNL1 level was because of de-ubiquitination-mediated nuclear translocation, but the causal mechanism of degeneration-reduced MBNL2 level remains undetermined.
The order of phenotype occurrence seen in the DM1 mouse model resembles other neurodegenerative disorders in that morphologic changes associated with neurodegeneration is a relatively later event during disease progression (Oddo et al., 2003; Shahidullah et al., 2013). The fundamental characteristic that distinguishes neurodegenerative disorders such as AD and amyotrophic lateral sclerosis (ALS)/frontotemporal dementia (FTD) is different, which results in distinct pathologies and onset of symptoms at the early stage. In AD, development of amyloid pathology and neurofibrillary tangles favor synaptic dysfunction (Oddo et al., 2003). In C9orf72 ALS/FTD, the formation of hexanucleotide RNA foci and dipeptide repeat aggregates occurs before neurodegeneration and neuronal loss (Liu et al., 2016). Similarly, formation of nuclear expanded CUG RNA foci and neurotransmission dysfunction precedes neuronal degeneration in DM1 (Wang et al., 2017). However, with disease progression, the clinical manifestations and pathologies such as cognitive impairment, structure changes and neuronal loss become similar among neurodegenerative disorders, which suggests a shared mechanism for development of common neurodegenerative features at a later stage.
In addition to DM, deregulation of RNA metabolism also occurs in numerous neurologic disorders such as lobar frontotemporal degeneration and AD (Lester et al., 2021). Although expanded CUG RNA reducing MBNL2 level may be the feature of DM1, whether neurodegeneration reducing MBNL2 level and dysregulation of MBNL2-mediated function is a common molecular feature of neurodegenerative diseases has not been investigated. In the present study, we used cultured hippocampal neuron as an in vitro model to determine the role of MBNL2 during neuronal maturation and how neurodegeneration conditions reduce MBNL2 expression. We used DM1 and AD mouse models to test the hypothesis that dysregulated MBNL2-mediated function may be a common feature across models of neurodegenerative diseases.
Materials and Methods
Animals and tissues
EpA960/CaMKII-Cre mice were described previously (Wang et al., 2017). F1 offspring from EpA960 mice crossed with CaMKII-Cre mice were used in experiments. Both male and female EpA960/CaMKII-Cre mice at age 12–16 months were used. APP/PS1 mice were originally from the Jackson Laboratory [B6;C3-Tg (APPswe, PSEN1dE9) 85Dbo/Mmjax]. The THY-Tau22 mouse model was established as described (Schindowski et al., 2006). Female APP/PS1 mice at 10 or 12 months old and female THY-Tau22 mice at 15 months old, and their age-matched non-Tg control littermates were used in this study. All animals were maintained on a standard 12/12 h light/dark cycle with light on at 8 A.M. Food and water were available ad libitum. All animal experiments were performed with the approval of the Academia Sinica Institutional Animal Care and Utilization Committee in strict accordance with its guidelines and those of the Council of Agriculture Guidebook for the Care and Use of Laboratory Animals.
Primary hippocampal neuron culture, drug treatment, virus infection, and transfection
Hippocampal neurons were cultured from rat embryos at embryonic day (E)18–E19 as described (Wang et al., 2018). In total, 200,000 neurons per well were plated in 12-well plates containing coverslips coated with poly-L-lysine (1 mg/ml). Neurons at 19 d of in vitro culture (DIV) were pretreated with tetrodotoxin (TTX; 1 µm) for 12–16 h to reduce endogenous synaptic activity. Cells were stimulated with glutamate (20 µm), NMDA (50 µm) or A23187 (4 µm) at 20 DIV for 1–3 h (2 h for A23187). MG132 (10 µm), lactacystin (10 µm), calpain inhibitor I (40 µm), calpain inhibitor III (40 µm), EGTA (2 mm) or ivermectin (1 µm) was added 30 min before stimulation for 3 h or the indicated times. After drug treatment, neurons were lysed for immunoblotting or fixed for immunocytochemistry. For genetic-knock-down experiments, hippocampal neurons at 10 DIV were infected with lentivirus-expressing target-specific short hairpin RNA (shRNA) for 24 h and harvested at 16–17 DIV for protein or RNA extraction. To study the knock-down effect on NMDA stimulation, virus-infected cells at 16–17 DIV were pretreated with TTX for 12–16 h before NMDA, then lysed for protein or RNA extraction. Transfection with calcium phosphate precipitation was performed at 11 DIV, followed by fixation at 14 DIV and immunofluorescence staining.
Subcellular fractionation, immunoblotting, and quantification
Subcellular fractionation for cytoplasmic and nuclear protein from cultured hippocampal neurons was performed as described (Chang et al., 2017). Briefly, cells were lysed on ice by using gentle lysis buffer (10 mm HEPES, pH 7.5, 3 mm MgCl2, 10 mm NaCl, and 0.5% NP-40) and centrifuged at 2000 × g for 8 min. The supernatant was collected as the cytoplasmic fraction and pellets were suspended by using gentle lysis buffer containing 0.1% SDS. The suspensions were sonicated for up to 3.5 min with high energy level and centrifuged at 12,000 rpm for 20 min. The supernatants were harvested as the nuclear fraction.
For total protein extraction from cultured hippocampal neurons, cells were lysed by using SDS-sample buffer (188 mm Tris, pH 6.8, 3% SDS, 3% glycerol, and 0.01% bromophenol blue) with addition of 15% 2-Mercaptoethanol before use. The lysates were incubated at 55°C for 10 min and resolved by Western blot analysis.
For immunoblotting analysis of mouse brains, tissues were homogenized by tight douncing in buffered sucrose (4 mm HEPES, 320 mm sucrose, 2 mm DTT, 2 mm MgCl2, 1 mm EDTA, protease, and phosphatase inhibitors), lysed with addition of 10% SDS to a final concentration of 1% SDS. Then, the lysates were sonicated and centrifuged at 13,000 rpm for 10 min; the supernatant was collected as total protein lysates for immunoblotting.
Obtaining the subcellular fractionation of mouse brains was previously described (Wang et al., 2017); tissues were homogenized by loose douncing in buffered sucrose and centrifuged at 800 × g for 10 min. The supernatants were collected as the cytoplasmic fractions and pellets were re-suspended by using buffered sucrose containing 1% SDS, followed by sonication and collection for supernatants as the nuclear fractions.
For Western blot analyses, signals were developed by use of Super Signal West Pico Chemiluminescent Substrate (Thermo). The intensity of bands was captured by x-ray film and measured by using ImageQuant TL 1D 7.0 (GE Healthcare Life Sciences) and ImageJ (NIH).
RNA preparation, RT-PCR, and splicing analysis
Total RNA was extracted by using TRIzol reagent (Invitrogen) and used for cDNA synthesis followed by PCR as described (Wang et al., 2007). Primers used for Mbnl2 and Gapdh amplification were Mbnl2-F, 5′-CTCTGCAGCAACAACTCCTGC-3′; Mbnl2-R, 5′-TAGCAGAACTAGCCTTAGGG-3′; Gapdh-F, 5′-TGCACCACCAACTGCTTA-3′; Gapdh-R, 5′-TAGCAGAA CTAGCCTTAGGG-3′. For RT-PCR splicing analysis, the primers used for detecting alternative splicing of Cacna1d exon 12a, Mapt exons 3 and 8 and alternative polyadenylation of Sptb were previously described (Goodwin et al., 2015). PCR products were analyzed with 5% nondenaturing polyacrylamide gels. Quantification of the percentage of exon inclusion was as described (Kalsotra et al., 2008).
For detecting the mRNA level of exon 12a-containing Cacna1d isoform (Cacna1d-Ex12a+ mRNA) in the cortical tissues of ICR mice and CA1-enriched tissues of THY-Tau22 mice, total extracted RNA was divided into two parts and subjected to cDNA synthesis with the oligo(dT)12–18 primer or Cacna1d-specific primer CACCAGGACAATCACCAGCCAGTAAA. Primers used for detecting Cacna1d-Ex12a+ mRNA were Cacna1d-ex12a-F, 5′-CATGCCCACCAGCGAGACTGA-3′; Cacna1d-ex12a-R, 5′-CACCCAGAGGGCCCAGTCTTG-3′ (located within exon 12a).
For the detection of exon 3-containing Mapt isoform (Mapt-Ex3+ mRNA) in the cortical tissues of ICR mice and CA1-enriched tissues of THY-Tau22 mice, cDNA was synthesized with the oligo(dT)12–18 primer. Primers used for detecting Mapt-Ex3+ mRNA were Mapt-ex3-F, 5′-GCTGACCCTCGCCAGGAGTTTG-3′; Mapt-ex3-R, 5′-ACCCTGGTTCCTCCGCTCCATC-3′ (located within exon 3).
Immunofluorescence staining, immunohistochemistry, quantification, and imaging
For detecting endogenous protein expression in neurons, immunofluorescence staining was performed as described (Wang et al., 2018). Briefly, cells were fixed with 4% paraformaldehyde (PFA) and 4% sucrose in PBS for 15 min at room temperature. Fixed cells underwent permeabilization and blocking in 3% normal goat serum (NGS) in tris-buffered saline (TBS) containing 0.2% Triton X-100 for 1 h, followed by incubation with primary antibodies overnight in incubation solution (1% NGS and 0.2% Triton X-100 in TBS), then with secondary antibodies conjugated with Alexa Fluor (Invitrogen) and with DAPI for nuclear labeling for 2 h. After a washing, cells were mounted with Fluoro-gel (EMS) for imaging.
Immunohistochemistry and immunofluorescence staining of mouse brain sections was as described (Wang et al., 2017). Briefly, mice were anesthetized and transcardially perfused with PBS and 4% PFA sequentially. Brain tissues were postfixed in 4% PFA for 16 h at 4°C and sectioned by vibratome sectioning. To detect endogenous MBNL2 expression by using immunohistochemistry, brain sections first underwent bleaching with 3% H2O2 in TBS for 10 min and permeabilization in TBS containing 0.5% Triton X-100 for 10 min. After incubation with blocking solution (3% NGS and 0.2% Triton X-100 in TBS) for 1 h, sections were incubated with anti-MBNL2 antibody in incubation solution (1% NGS and 0.2% Triton X-100 in TBS) overnight at 4°C. After washing, sections were incubated with biotinylated secondary antibody (Vector Laboratories) for 2 h, then with ABC reagent (Vector Laboratories) for 2 h, followed by signal development by using the SG substrate kit (Vector Laboratories). Sections were counterstained with Nuclear Fast Red to label the nucleus and coverslipped by using permount mounting medium (Vector Laboratories). To detect endogenous MBNL2 and CAPN2 in the brain sections by using immunofluorescence staining, similar staining procedures were performed except for those after the primary antibody incubation. For the immunofluorescence staining, sections were incubated with secondary antibody conjugated with Alexa Fluor (Invitrogen) and DAPI after the primary antibody incubation, followed by washing with PBS and mounting with Fluoro-Gel (EMS).
Quantification of the total MBNL2 immunoreactivity and ratio of nuclear to cytoplasmic fraction of CAPN2 immunoreactivity intensity in cultured neurons and THY-Tau22 and APP/PS1 brains was as described (Wang et al., 2017). The CA1 cells in the dorsal hippocampal regions in THY-Tau22 brains and the cortical Layer V neurons in APP/PS1 brains were used. For quantification of MBNL2 immunoreactivity intensity, images of MBNL2 staining were measured by using MetaMorph 7.7.5.0 (Molecular Devices). CAPN2 intensity in the nucleus and cytoplasm was measured by using ImageJ (NIH).
Quantification of the total dendrite length and number of primary dendrites was as described (Wang et al., 2018). Briefly, the thin, slender processes with constant diameter along the process was identified as axons (Bartlett and Banker, 1984; Dotti et al., 1988). Processes directly emerging from the soma that were longer than 20 µm were identified as primary dendrites. The total dendrite length included the length of primary dendrites and their branches.
Fluorescent images were acquired under a fluorescent microscope (AxioImager M2, Carl Zeiss) equipped with a 40×/0.75 objective lens (Plan-Apochromat, Carl Zeiss) or a confocal laser scanning microscope (LSM700, Carl Zeiss) equipped with a 40×/1.4/oil objective lens (Plan-Apochromat, Carl Zeiss). Images for immunohistochemistry were acquired under an upright microscope (BX51, Olympus) equipped with a 10×/0.4 or 20×/0.75 objective lenses (UPlanSApo, Olympus).
For all experiments with cultured hippocampal neurons, the image acquisition involved the same settings for experimental groups, with transfections performed at the same time and used for comparison.
Antibodies
Antibodies used were anti-neurofilament-160 (NF160; Biosensis, M-1394, 1:10,000 for immunoblotting), anti-MBNL2 (Santa Cruz Biotechnology, sc-136167, 1:2500 for immunoblotting; 1:400 for immunostaining), anti-calpain-1 (CAPN1; Abcam, ab39170, 1:5000 for immunoblotting), anti-CAPN2 (Santa Cruz Biotechnology, sc-373966, 1:10,000 for immunoblotting; 1:250 for immunostaining in cells; 1:10 for immunostaining in brain sections), anti-NeuN (Abcam, ab177487, 1:1000 for immunostaining), anti-β-tubulin (Novus, NB600-936, 1:20,000 for immunoblotting), anti-GAPDH (Millipore, MAB374, 1:40,000 for immunoblotting), anti-Histone H3 (Santa Cruz Biotechnology, sc-8654, 1:5000 for immunoblotting), and anti-Myc (Abcam, ab9106, 1:1000 for immunostaining).
Plasmids
The target-specific shRNA clones TRCN 0000072224 (shLacZ, CGCGATCGTAATCACC CGAGT), TRCN0000178919 (shMbnl2-1, CAACACCGTAACCGTTTGTAT), TRCN0000102459 (shMbnl2-2, CAAAGAGACAAGCACTTGAAA), and TRCN0000030665 (shCapn1, GCCGTGGACTTTGACAACTTT) were purchased from an RNAi core facility (Academia Sinica). The Capn2-specific shRNA clone (shCapn2, GGAGTGCCCTTCTGGAGAA) and the construct of rat CAPN2 sequence in pcDNA3.1 (Wang and Huang, 2012) was kindly provided by Yi-Shuian Huang (Academia Sinica). The rat CAPN2 sequence was subcloned by using the forward primer AATTGGTACCTCATGGCGGGCATCGCGATG and reverse primer AATGTCGACTCAGAGTACTGAAAAACTC into GW1-Myc backbone (Myc-CAPN2). The nuclear localization signal (NLS) of SV40 large-T antigen was inserted by subcloning the Myc-CAPN2 by using the sense primer AATTGGTACCTAATGGCGGGCATCGCGATG and antisense primer carrying NLS AATGTCGACTCAAACCTTGCGCTTCTTCTTAGGGAGTACTGAAAAACTC at the C terminus of Myc tagged-CAPN2 (generation of Myc-CAPN2-NLS construct). The second NLS was then inserted into Myc-CAPN2-NLS by using the sense primer GCTTGCCACCATGCCTAAGAAGAAGCGCAAGGTTGAGCAAAAGCTG and antisense primer CAGCTTTTGCTCAACCTTGCGCTTCTTCTTAGGCATGGTGGCAAGC at the N terminus of CAPN2 (generation of NLS-Myc-CAPN2-NLS construct).
Experimental design and statistical analysis
For the experiments of Western blot analysis, drug treatments using hippocampal neurons were repeated at least three times as indicated in each figure legend with different batches of neuronal preparations from embryos derived from different pregnant rats. For the experiments of immunostaining in drug treated neuronal cells (Figs. 3D,H, 5B, 7F), cells for quantification were obtained from 1 batch of neuronal preparation. For the experiments of neuronal transfection (Figs. 1E, 5F), cells for quantification were collected from three batches of neuronal preparation. Total number of cells and cultures used were described in the figures and figure legends.
Figure 3.
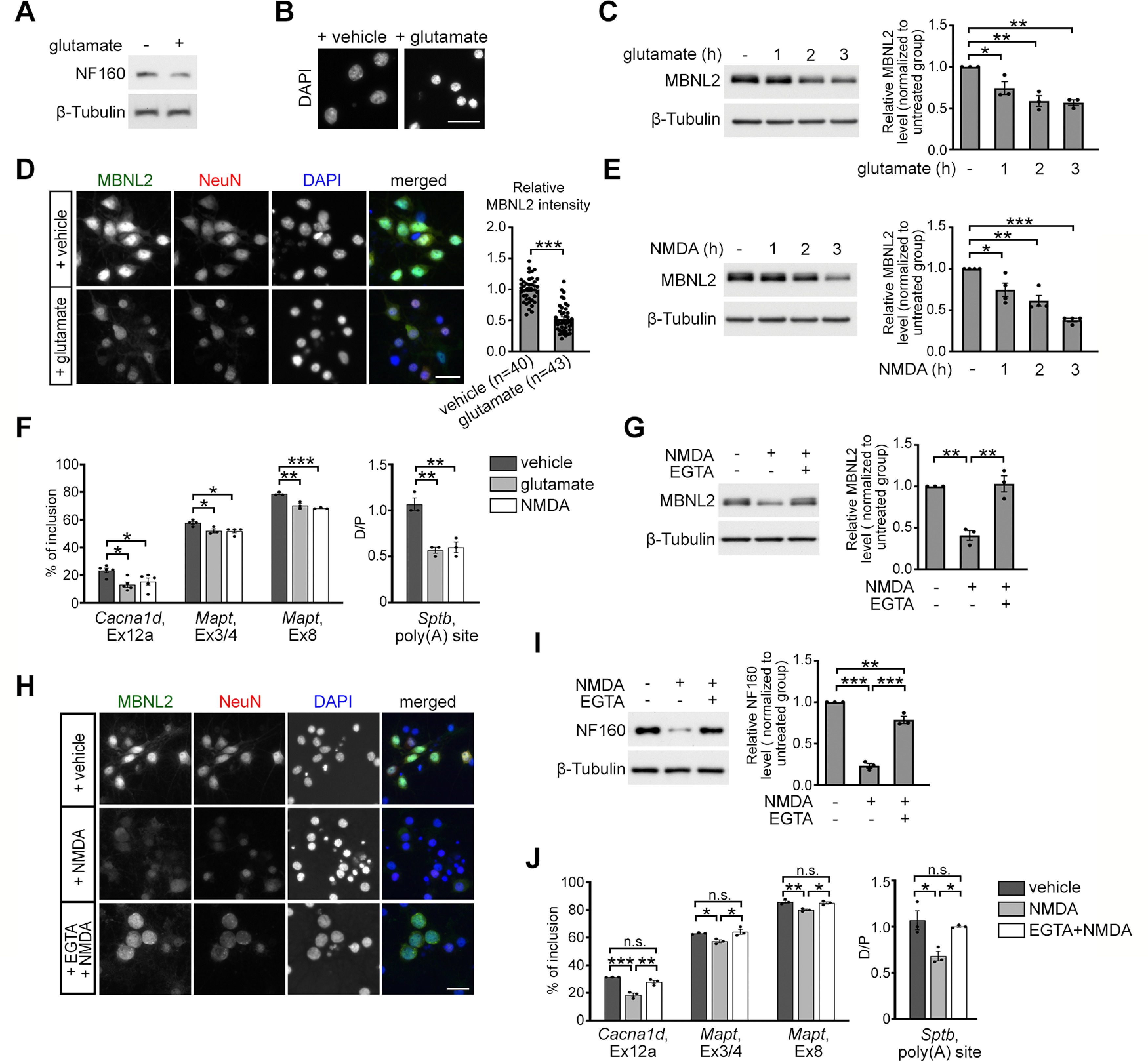
Glutamate-induced excitotoxicity reduces MBNL2 expression. A, B, Glutamate treatment for 3 h in neurons caused degeneration features including reduced level of NF160 (A) and condensed nuclei (B). C, MBNL2 level was reduced in mature cultured hippocampal neurons treated with glutamate for the indicated times. β-Tubulin was a loading control. Relative amount of MBNL2 for treatments was compared by normalization with β-tubulin (1 h: p = 0.0371; 2 h: p = 0.0026; 3 h: p = 0.0019). D, Representative images of MBNL2 immunoreactivity in neurons treated with vehicle or glutamate. NeuN staining was a neuronal marker. Quantification of overall MBNL2 intensity is shown at the right (p < 0.0001). Number of neurons (n, from 1 cultured neuron preparation) used for quantification is indicated. E, MBNL2 level was reduced in neurons treated with NMDA for the indicated times (1 h: p = 0.0220; 2 h: p = 0.0011; 3 h: p < 0.0001). F, Aberrant MBNL2-regulated RNA processing events in glutamate-treated or NMDA-treated neurons (for Cacna1d, Ex12a, glutamate: p = 0.0119; NMDA: p = 0.0455, for Mapt, Ex3/4, glutamate: p = 0.0323; NMDA: p = 0.0181, for Mapt, Ex8, glutamate: p = 0.0019; NMDA: p = 0.0006, for Sptb, poly(A) site, glutamate: p = 0.0015; NMDA: p = 0.0022). G, Pretreatment with calcium chelator EGTA preserved MBNL2 level in NMDA-treated neurons (NMDA: p = 0.0018; NMDA+EGTA: p = 0.0014). H, Immunofluorescent staining of MBNL2 expression and DAPI used for examining the nuclear morphology in the NeuN+ neurons. I, Effect of EGTA pretreatment on NF160 expression in the NMDA-treated neurons. Relative amount of NF160 was compared by normalization with β-tubulin (untreated vs NMDA: p < 0.0001; NMDA vs NMDA+EGTA: p < 0.0001; untreated vs NMDA+EGTA: p = 0.0052). J, Effect of EGTA pretreatment before NMDA stimulation on the splicing pattern of MBNL2-regulated RNA processing events (for Cacna1d, Ex12a, vehicle vs NMDA: p = 0.0004; NMDA vs NMDA+EGTA: p = 0.0020; vehicle vs NMDA+EGTA: p = 0.1301, for Mapt, Ex3/4, vehicle vs NMDA: p = 0.0433; NMDA vs NMDA+EGTA: p = 0.0204; vehicle vs NMDA+EGTA: p = 0.8062, for Mapt, Ex8, vehicle vs NMDA: p = 0.0055; NMDA vs NMDA+EGTA: p = 0.0109; vehicle vs NMDA+EGTA: p = 0.7879, for Sptb, poly(A) site, vehicle vs NMDA: p = 0.0132; NMDA vs NMDA+EGTA: p = 0.0302; vehicle vs NMDA+EGTA: p = 0.7621). Three to four independent experiments were used for quantification. Data are mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001, by one-way ANOVA with Tuckey's test (C, E–G, I, J) or Student t test (D). n.s., not significant. Scale bar: 20 µm (B), 50 µm (D), and 25 µm (H).
Figure 5.
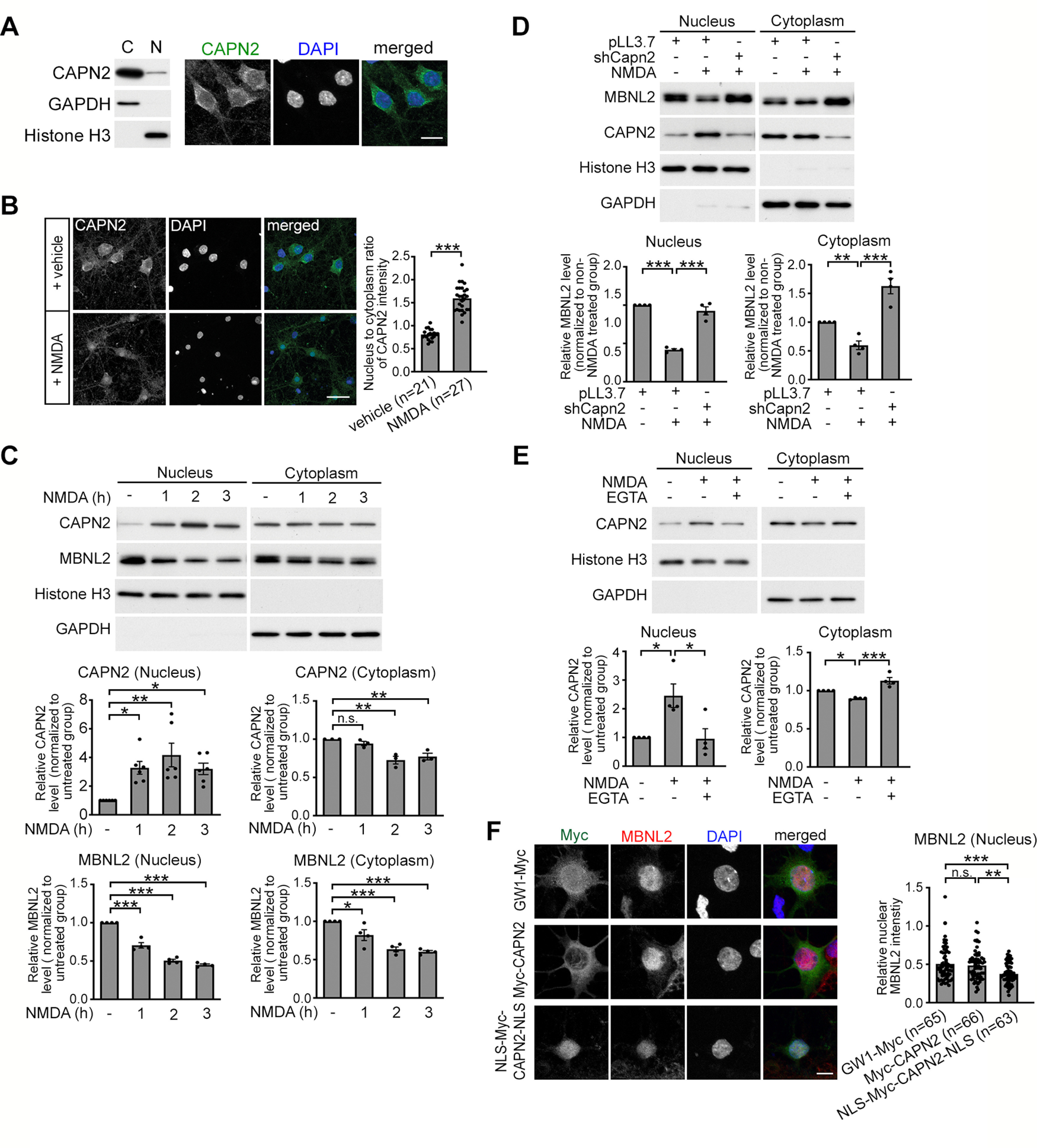
Nuclear translocation of calpain-2 is calcium-dependent and required for MBNL2 degradation. A, Subcellular distribution of endogenous CAPN2 in mature hippocampal neurons by biochemical fractionation (left) and immunofluorescence staining (right). B, NMDA treatment increased the nuclear fraction of CAPN2. Quantification of nucleus-to-cytoplasm ratio of CAPN2 immunoreactivity at right (p < 0.0001). Number of neurons (n, from 1 cultured neuron preparation) used for quantification is indicated. C, Nuclear and cytoplasmic distribution of CAPN2 and MBNL2 in neurons treated with NMDA for the indicated times (for CAPN2 nucleus, 1 h: p = 0.0233; 2 h: p = 0.0014; 3 h: p = 0.0288, for CAPN2 cytoplasm, 1 h: p = 0.6990; 2 h: p = 0.0032; 3 h: p = 0.0098, for MBNL2 nucleus, p < 0.0001 for all, for MBNL2 cytoplasm, 1 h: p = 0.0311; 2 h: p = 0.0001; 3 h: p < 0.0001). D, CAPN2 knock-down preserved MBNL2 expression in both nuclear and cytoplasmic fractions of NMDA-treated neurons (for nucleus, pLL3.7 vs pLL3.7+NMDA: p < 0.0001; pLL3.7+NMDA vs shCapn2+NMDA: p = 0.0001, for cytoplasm, pLL3.7 vs pLL3.7+NMDA: p = 0.0013; pLL3.7+NMDA vs shCapn2+NMDA: p < 0.0001). E, Pretreatment with EGTA inhibits the nuclear translocation of CAPN2 in NMDA-treated neurons (for nucleus, untreated vs NMDA: p = 0.0225; NMDA vs NMDA+EGTA: p = 0.0192, for cytoplasm, untreated vs NMDA: p = 0.0414; NMDA vs NMDA+EGTA: p = 0.0003). F, Representative images of Myc and MBNL2 immunoreactivity in neurons transfected with GW1-myc (top), Myc-CAPN2 (middle), or NLS-Myc-CAPN2-NLS (bottom). Quantification of intensity of nuclear MBNL2 immunoreactivity in neurons transfected with the indicated constructs was shown at right (GW1-Myc vs Myc-CAPN2: p = 0.8074; Myc-CAPN2 vs NLS-Myc-CAPN2-NLS: p = 0.0027; GW1-Myc vs NLS-Myc-CAPN2-NLS: p = 0.0003). Number of neurons (n, from 3 independent cultured neuron preparations and transfections) used for quantification is indicated. Histone H3 and GAPDH were loading controls for nuclear and cytoplasmic fractions, respectively (A, C–E). Quantification of cytoplasmic and nuclear fractions of CAPN2 and MBNL2 relative to loading controls is shown (C–E). Three to five independent experiments were used for quantification. Data are mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001, by one-way ANOVA with Tuckey's test (C–F) or Student t test (B). n.s., not significant. Scale bar: 25 µm (A), 20 µm (B), and 5 µm (F).
Figure 7.
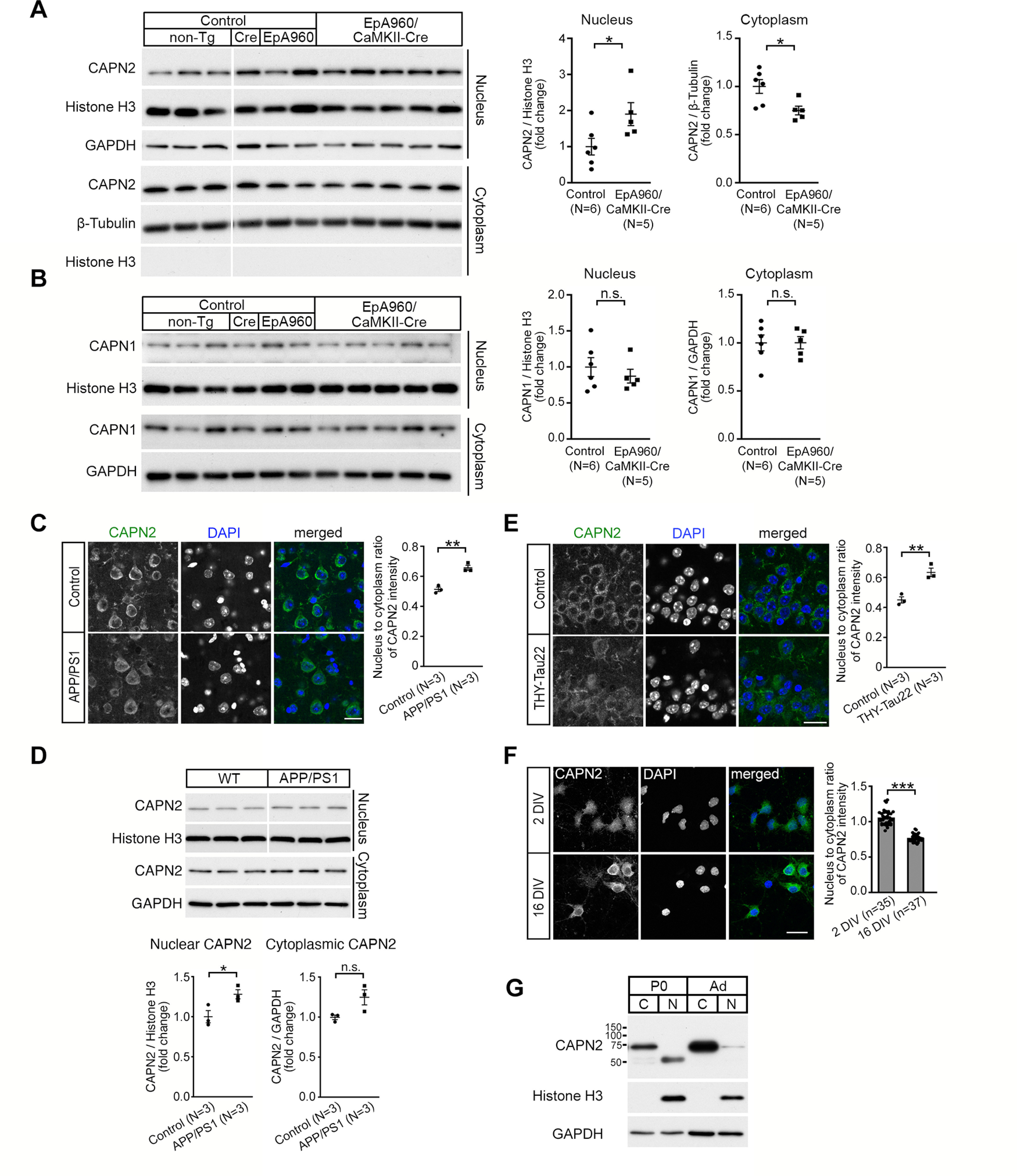
Increased level of nuclear CAPN2 is observed in the DM1 and AD mouse brains. A, B, CAPN2 (A, nucleus: p = 0.0303; cytoplasm: p = 0.0173) and CAPN1 (B, nucleus: p = 0.4716; cytoplasm: p = 0.9857) content in nuclear and cytoplasmic fractions of lysates from the cortical regions of control and EpA960/CaMKII-Cre brains at age 12–16 months. Brains from animals with different genotypes including non-Tg, CaMKII-Cre (Cre) and EpA960 brains were controls. Same lysates were used for detecting CAPN2 (A) and CAPN1 (B). GAPDH and histone H3 were used as loading controls for the cytoplasmic and nuclear fraction, respectively. C, Representative images of CAPN2 immunostaining in the cortical Layer V of brain sections from non-Tg control and APP/PS1 mice at age 12 months (p = 0.0029). D, The level of nuclear and cytoplasmic CAPN2 in the cortex of non-Tg control and APP/PS1 mice at age 10 months. Quantification of relative CAPN2 expression in the nuclear (p = 0.0424) and cytoplasmic (p = 0.0634) fractions, normalized with Histone H3 and GAPDH, respectively. Same lysates were used for detecting MBNL2 (Fig. 2B) and CAPN2. E, Representative images of CAPN2 immunostaining in the CA1 region of sections from non-Tg control and THY-Tau22 mice at age 15 months (p = 0.0065). F, Representative images of CAPN2 immunoreactivity in neurons at 2 and 16 DIV (p < 0.0001). G, CAPN2 expression in the cytoplasmic (C) and nuclear (N) fraction from the cortex of neonatal (P0) and adult (Ad) wild-type mice. Nucleus-to-cytoplasm ratio of CAPN2 immunoreactivity is quantified and shown at right (C, E, F). Number of animals (N) or neurons (n, from 1 cultured neuron preparation) used for quantification is indicated. Data are mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001, by Student t test. n.s., not significant. Scale bar: 20 µm (C, E, F).
Figure 1.
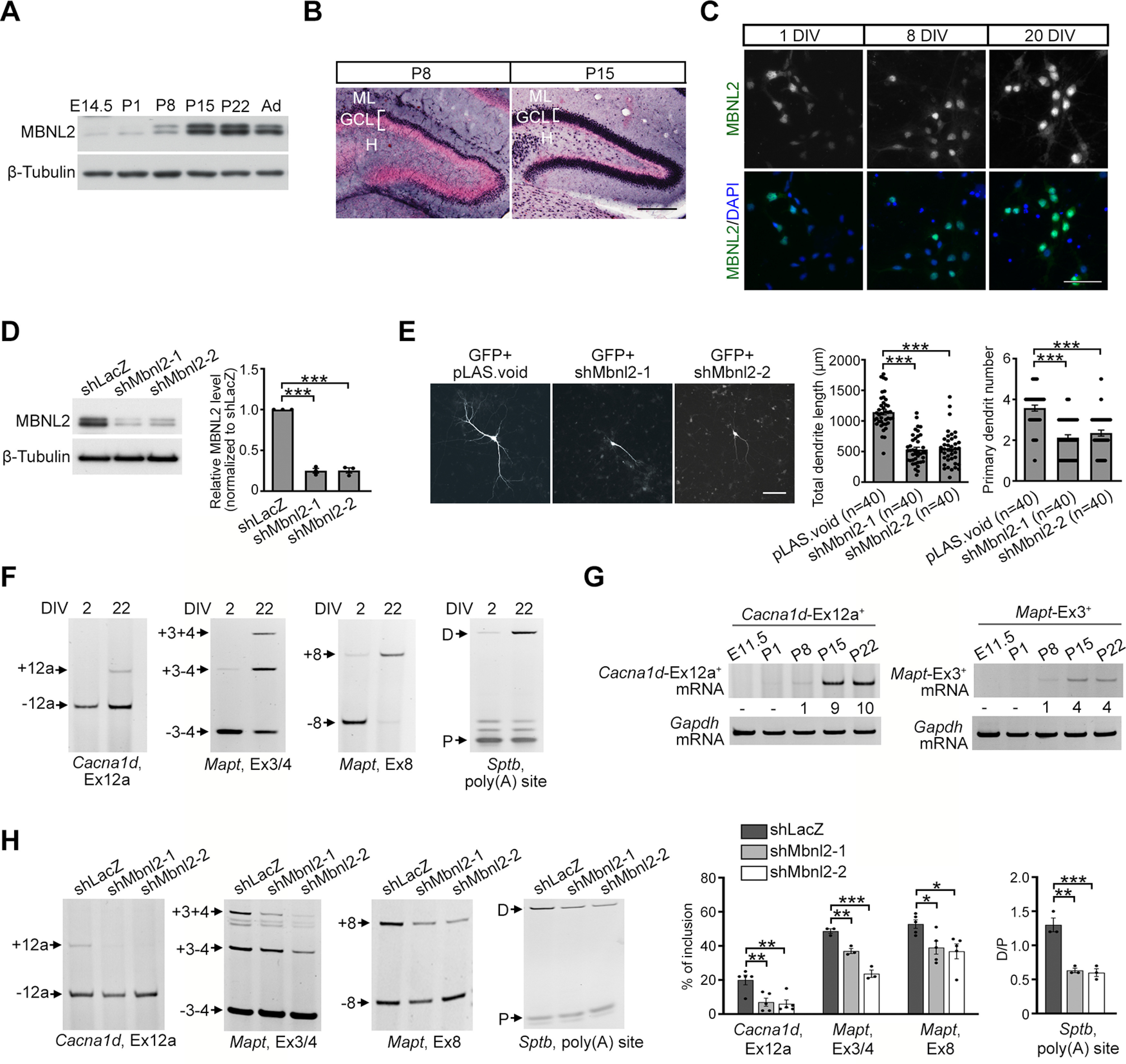
MBNL2 is required for regulating neuronal morphogenesis and the developmental transition of RNA processing. A, Temporal expression of MBNL2 in the mouse forebrain. Protein lysates were prepared from mice at different ages including E14.5, P1, P8, P15, and P22 and two months (Ad). B, MBNL2 immunoreactivity with nuclear fast red used for nuclei labeling in the dentate gyrus of the P8 and P15 hippocampus. C, MBNL2 immunoreactivity in the cultured hippocampal neurons at 1, 8, 20 DIV. DAPI was used for nuclear staining. D, Knock-down of MBNL2 by specific shRNAs in the neurons. Cultured hippocampal neurons were infected with lentivirus carrying plasmids expressing MBNL2 shRNA (shMbnl2-1 or shMbnl2-2) or control shRNA (shLacZ). Quantification of the knock-down efficiency of MBNL2 (p < 0.0001 for both). E, MBNL2 knock-down impairs dendrite development. Neurons were transfected with plasmids expressing MBNL2 shRNA (shMbnl2-1 or shMbnl2-2) or control shRNA (pLAS.void). Plasmid expressing GFP was co-transfected to label neuronal morphology. Quantification of total dendrite length and primary dendrite number (p < 0.0001 for all). Number of neurons (n, from 3 independent cultured neuron preparations and transfections) used for quantification is indicated. F, Pattern of MBNL2-regulated RNA processing events including the Cacna1d exon 12a, Mapt exon 3/4 and 8 and distal (D) to proximal (P) poly(A) utilization of Sptb in neurons at 2 and 22 DIV. G, Temporal expression of Cacna1d-Ex12a+ (left) and Mapt-Ex3+ (right) mRNAs during development in the mouse forebrains. Total RNA was prepared from mice at different ages including E11.5, P1, P8, P15, and P22. Numbers shown represent relative quantification of Cacna1d-Ex12a+ and Mapt-Ex3+ mRNA levels to that in P8 after normalization to Gapdh level. H, Aberrant MBNL2-regulated RNA processing in Mbnl2-knock-down neurons. Representative gel images and the quantification of the inclusion of Cacna1d exon 12a (shMbnl2-1: p = 0.0082; shMbnl2-2: p = 0.0049), Mapt exons 3/4 (shMbnl2-1: p = 0.0072; shMbnl2-2: p = 0.0001) and 8 (shMbnl2-1: p = 0.0495; shMbnl2-2: p = 0.0232), and distal (D) to proximal (P) poly(A) utilization of Sptb (shMbnl2-1: p = 0.0012; shMbnl2-2: p = 0.0009). Three to five independent experiments were used for quantification. Data are mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001, by one-way ANOVA with Tuckey's test (D, E, H). ML, molecular layer; GCL, granule cell layer; H, hilus. Scale bar: 200 µm (B), 50 µm (C), and 100 µm (E).
For experiments using animals, as indicated in Figures 2 and 7, three animals per group were used for APP/PS1 and THY-Tau22 mice and their littermate wild-type controls; five to six animals per group were used for EpA960/CaMKII-Cre mice and the age-matched control (includes different control genotypes as indicated in the figure and figure legend). For the quantification of the total MBNL2 immunoreactivity in the sections of APP/PS1 and THY-Tau22 mice, 300–400 cells per animal were used. Total number of cells collected for quantifying the nucleus-to-cytoplasm ratio of CAPN2 immunoreactivity was as follows: 100 cells from the Thy1-Tau dorsal CA1 region; 135 cells from littermate controls; 128 cells from the APP/PS1 Layer V and 143 cells from littermate controls. On average, 35–60 cells from each animal were used for measurement.
Figure 2.
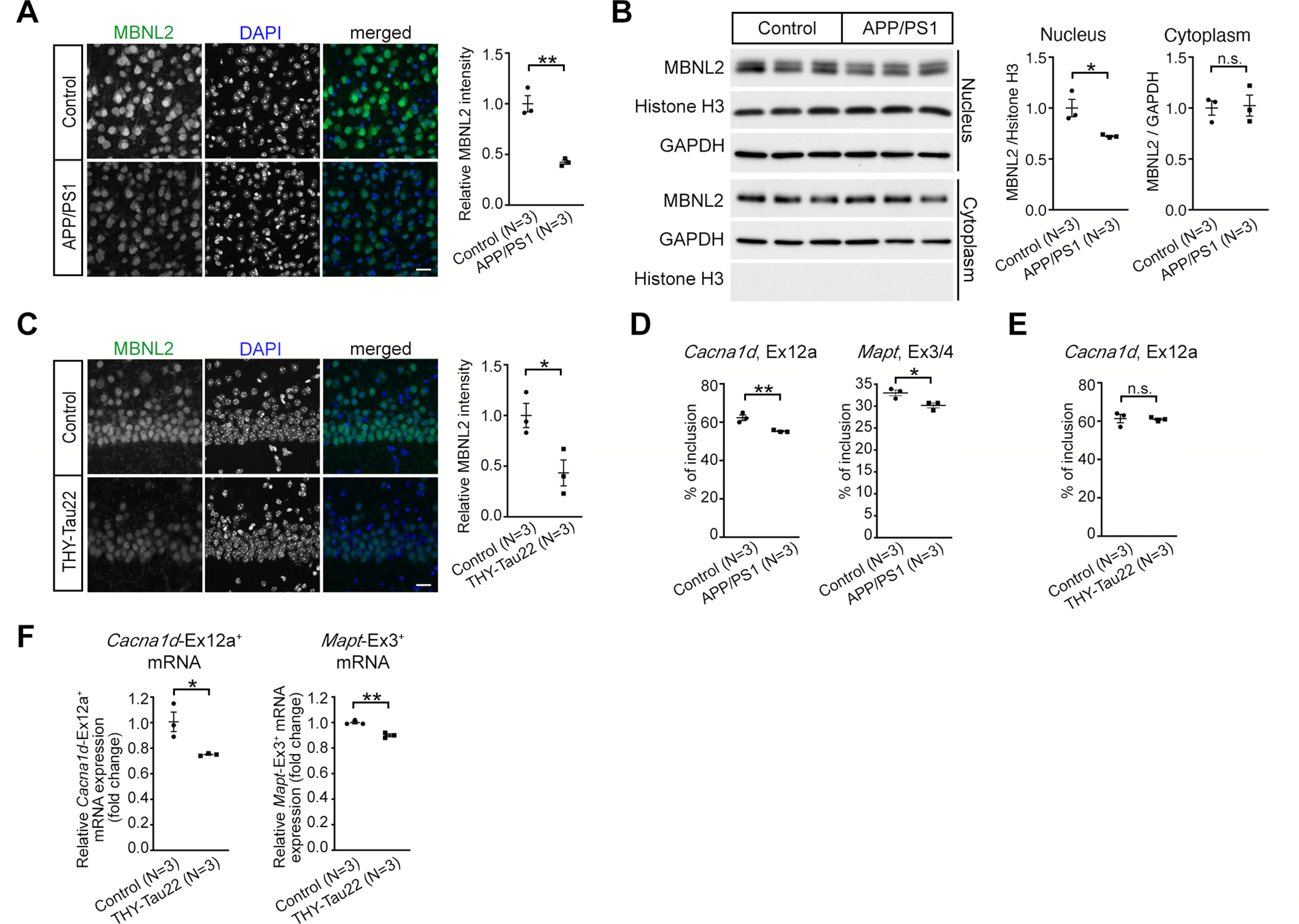
Neurodegeneration-reduced MBNL2 expression and aberrant MBNL2-reuglated splicing are detected in the mouse brains for AD. A, The intensity of MBNL2 immunoreactivity was reduced in cortical Layer V neurons of female APP/PS1 mice at age 12 months (p = 0.0022). Non-Tg animals at the same age were used as controls. B, Western blot analysis of MBNL2 expression in the nuclear and cytoplasmic fraction from the cortical region of the control (non-Tg) and APP/PS1 mice at age 10 months. Relative MBNL2 level in the nuclear and cytoplasmic fraction, normalized with Histone H3 and GAPDH, respectively, was quantified (Nucleus: p = 0.0322; Cytoplasm: p = 0.8609). C, Immunofluorescent staining of MBNL2 expression in the caudal CA1 neurons of female non-Tg (Control) and THY-Tau22 mice at age 15 months (p = 0.0318). D, The percentage of inclusion of Cacna1d exon 12a (p = 0.0093) and Mapt exons 3/4 (p = 0.0295) in the cortical tissues of APP/PS1 mice. E, The percentage of inclusion of Cacna1d exon 12a using RNA collected from the entire hippocampal tissues of THY-Tau22 mice (p = 0.8899). F, Quantification of the levels of exon 12a-containing Cacna1d (Cacna1d-Ex12a+) and exon 3-containing Mapt (Mapt-Ex3+) mRNA by RT-qPCR after normalization to Gapdh level using RNA collected from the CA1-enriched tissues of THY-Tau22 mice (Cacna1d-Ex12a+: p = 0.0284; Mapt-Ex3+: p = 0.0028). Number of animals (N) in each group is indicated. Data are mean ± SEM; *p < 0.05, **p < 0.01, by Student t test. n.s., not significant. Scale bar: 20 µm (A and C).
All data were expressed as mean ± SEM. Statistical analysis was performed using GraphPad Prism version 9.2.0 for macOS (GraphPad Software). Unpaired two-tailed Student's t test was used for comparing two groups and one-way ANOVA followed by Tukey's multiple comparisons test for multiple groups. The statistical values and the statistical tests used for all quantitative results were described in each figure legend. p < 0.05 was considered statistically significant.
Results
MBNL2 is required for neuronal maturation and regulating the developmental transition of RNA processing
MBNL2 mediates the developmental transition of alternative splicing and polyadenylation during brain development (Charizanis et al., 2012; Batra et al., 2014), and reduced MBNL2 expression occurred after axon and dendrite degeneration in DM1 mouse brain (Wang et al., 2017). Hence, we investigated the involvement of MBNL2 in neuronal maturation. We first examined MBNL2 expression during mouse brain development. MBNL2 was barely detected in early developing brains, including at E14.5 and postnatal day (P)1 (Fig. 1A) but became detectable by P8 and increased afterward during postnatal maturation (Fig. 1A, P15). Development of granular neurons of dentate gyrus in the hippocampus follows an “ectal-to-endal” gradient pattern (Bayer, 1980; Wang et al., 2002). We detected MBNL2 in the outermost granule cell layer of the external dentate limb (Fig. 1B, P8). The number of MBNL2-expressing cells increased in a gradient as a function of time, from external to internal layers of the dentate limb (Fig. 1B, P15), which indicates consistency with the pattern of maturation. Consistently, in the cultured hippocampal neurons derived from the E18.5 rat embryos, the intensity of MBNL2 immunoreactivity was low in the neurons at 1 DIV, and gradually increased with neuronal differentiation from 8 to 20 DIV (Fig. 1C).
To determine whether MBNL2 was required for neuronal morphogenesis, we used two different Mbnl2-specific shRNA constructs to deplete MBNL2 in cultured hippocampal neurons (Fig. 1D). A GFP expression construct for showing the neuronal morphology was co-transfected with Mbnl2-shRNA constructs on 11 DIV and dendrite development was analyzed at 14 DIV. MBNL2 knocked-down neurons exhibited shorter total dendrite length and reduced primary dendrite number as compared with control neurons, which suggests that loss of MBNL2 function impairs dendrite maturation (Fig. 1E).
We next determined the association of MBNL2-regulated alternative splicing and polyadenylation with neuronal maturation. During hippocampal neuron maturation, the inclusion of the Cacna1d exon 12a and Mapt exons 3/4 and 8 (exons 2/3 and 10 in human) was increased, and utilization of Sptb alternative poly(A) sites was toward the distal site (Fig. 1F; Charizanis et al., 2012; Goodwin et al., 2015). Similarly, during brain development, the expression levels of Cacna1d exon 12a-containing and Mapt exon 3-containing transcripts were also increased (Fig. 1G). However, MBNL2 depletion in cultured hippocampal neurons impaired the transition of MBNL2-regulated RNA processing events toward mature patterns; inclusion of the Cacna1d exon 12a and Mapt exons 3/4 and 8 was decreased, and utilization of Sptb alternative poly(A) sites was not shifted toward the distal site (Fig. 1H). Our results suggest that MBNL2 is maturation-associated and required for dendrite development and regulation of RNA processing transition toward the adult stage and that pathogenesis-reduced MBNL2 expression in DM1 mouse brain resembles its embryonic expression pattern.
Reduced MBNL2 expression associated with aberrant MBNL2-regulated alternative splicing in mouse models of AD
Next, we determined whether MBNL2 reduction may also occur in other neurodegenerative diseases. We examined MBNL2 expression in two mouse models of AD, the most prevalent neurodegenerative disorder. The models APP/PS1 and THY-Tau22 exhibit two pathologic hallmarks of AD, amyloid plaque deposition and tau aggregation, respectively (Jankowsky et al., 2004; Schindowski et al., 2006). Apparent amyloid plaque with reduced number of neuron was shown in the cortex of the APP/PS1 mice, while abundant expression of hyperphosphorylated tau with neuronal loss was detected in the hippocampal CA1 region of the THY-Tau22 mice (Radde et al., 2006; Schindowski et al., 2006; Kuhla et al., 2017). Therefore, we examined MBNL2 level in the cortical region of the APP/PS1 brain and the CA1 region of the THY-Tau22 brain. The total level of MBNL2 immunoreactivity was reduced in cortical Layer V neurons of 12-month-old APP/PS1 brains as compared with wild-type controls (Fig. 2A). Using biochemical fractionation to separate cytoplasmic and nuclear fractions, we found that MBNL2 expression in the nuclear fraction was reduced (Fig. 2B). Similarly, the total level of MBNL2 immunoreactivity was reduced in the caudal hippocampal CA1 neurons of 15-month-old THY-Tau22 brains (Fig. 2C).
We next examined the alternative splicing of MBNL2-regulated targets and found decreased inclusion of Cacna1d exon 12a and Mapt exons 3/4 in the APP/PS1 cortical region (Fig. 2D). However, examination of inclusion of Cacna1d exon 12a showed no change in the THY-Tau22 hippocampal region (Fig. 2E). We wondered whether the unchanged Cacna1d exon 12a splicing was likely because of a weakening effect with use of the entire hippocampal region for detection. Therefore, we used isoform-specific primers with RNA collected from the hippocampal CA1 region to enrich Cacna1d exon 12a-containing and Mapt exon 3-containing transcripts and performed RT-qPCR to examine the expression level of these two transcripts. The levels of both Cacna1d exon 12a-containing and Mapt exon 3-containing transcripts were reduced in the THY-Tau22 hippocampal region as compared with wild-type controls (Fig. 2F), thus indicating decreased inclusion of Cacna1d exon 12a and Mapt exon 3 and showing a reversal of the developmental transition pattern (Fig. 1G). Hence, reduced MBNL2-associated reversal of MBNL2-regulated alternative splicing to embryonic pattern also occurred in AD models.
Glutamate-induced excitotoxicity reduces MBNL2 expression
The reduced MBNL2 expression in both DM1 and AD mouse brains suggested a neurodegeneration-induced regulatory mechanism. Glutamate-induced excitotoxicity is implicated in neurodegeneration (Lau and Tymianski, 2010). Therefore, we used cultured hippocampal neurons to study the effect of glutamate-induced excitotoxicity on MBNL2 expression. Glutamate-induced excitotoxicity and degeneration was shown by reduced level of NF160 and condensed nuclei on DAPI staining (Fig. 3A,B). In mature neurons cultured at 20–24 DIV and treated with 20 µm glutamate, MBNL2 protein level was decreased at 1 h with glutamate stimulation (Fig. 3C). With increased glutamate treatment time, MBNL2 expression continued to decrease. On immunofluorescence staining under the same exposure time, the intensity of MBNL2 immunoreactivity was reduced in neurons stained with anti-NeuN antibody (Fig. 3D). Glutamate-elicited excitotoxicity is mediated by calcium influx via the activation of NMDA receptor (NMDAR; Lau and Tymianski, 2010). Hippocampal neurons treated with 50 µm NMDA showed a similar pattern of MBNL2 downregulation as glutamate-treated neurons (Fig. 3E). We next determined whether glutamate-reduced MBNL2 expression affected the developmental transition of RNA processing by analyzing the alternative splicing and polyadenylation of MBNL2 targets in glutamate-treated and NMDA-treated neurons. The pattern of alternative splicing and polyadenylation in neurons treated with glutamate or NMDA was dysregulated and similar to that in MBNL2-depleted neurons (Figs. 1H, 3F).
Next, we determined whether NMDAR-mediated calcium influx involves in the MBNL2 downregulation. Before NMDA stimulation, blockade of extracellular calcium influx by EGTA prevented NMDA-reduced MBNL2 expression (Fig. 3G,H), as well as the DAPI condensation and NF160 downregulation (Fig. 3H,I). Before NMDA stimulation, pretreatment of EGTA also preserved MBNL2 function in regulating RNA processing. The pattern of dysregulated RNA processing events in the NMDA-treated neurons was preserved by the EGTA pretreatment as compared with control neurons (Fig. 3J). The results suggest that NMDAR-mediated excitotoxicity reduced MBNL2 expression and induced a developmental pattern of MBNL2-regulated RNA processing.
NMDAR-mediated calpain-2 activation causes MBNL2 degradation
We next investigated how NMDAR-mediated excitotoxicity reduces MBNL2 expression. In cultured hippocampal neurons treated with glutamate or NMDA, Mbnl2 mRNA level remained unchanged as compared with that in unstimulated neurons (Fig. 4A), so NMDA-reduced MBNL2 expression was not because of downregulation at the mRNA level. We next determined whether NMDA-reduced MBNL2 expression was because of proteasome degradation by pretreating neurons with two proteasome inhibitors, MG132 or lactacystin, before NMDA treatment. Pretreatment with MG132 but not lactacystin prevented the reduced MBNL2 expression (Fig. 4B, lanes 1–4). The lack of response to lactacystin pretreatment suggested that NMDA-reduced MBNL2 expression was not because of proteasome degradation. In addition to proteasome inhibition, MG132 also inhibits activities of other cysteine proteases including cathepsins and calpains (Lee and Goldberg, 1998). Calpains belong to a family of calcium-dependent cysteine proteases, and their activity can be induced by NMDAR-induced calcium influx (Goll et al., 2003; Vosler et al., 2008). We next assessed whether calpain was involved in regulating MBNL2 downregulation. We pretreated neurons with N-acetyl-L-leucyl-L-leucyl-L-norleucinal (calpain inhibitor I) or carbobenzoxy-valinyl-phenylalaninal (calpain inhibitor III) before NMDA and found that both inhibitors prevented NMDA-reduced MBNL2 expression (Fig. 4B, lanes 5–6). Thus, NMDAR-mediated MBNL2 downregulation was likely via calpain activation.
Figure 4.

Calpain-2 is required for NMDAR-mediated MBNL2 degradation. A, RT-PCR detection of Mbnl2 mRNA in neurons treated with glutamate or NMDA. Gapdh was a loading control. Relative Mbnl2 mRNA level after normalization to Gapdh was shown at right (p = 0.4428, one-way ANOVA). B, MBNL2 level detected by immunoblotting in neurons treated with combinations of drugs as indicated (NMDA– vs NMDA+: p < 0.0001; NMDA+ vs MG132: p = 0.0001; NMDA+ vs lactacystin: p > 0.9999; NMDA+ vs calpain inh I: p = 0.0017; NMDA+ vs calpain inh III: p = 0.0004). C, Change in CAPN1 and CAPN2 protein levels in NMDA-treated neurons. Quantitative results of relative CAPN1 or CAPN2 level normalized to β-Tubulin were shown at right (CAPN1: p = 0.0037, N = 7; CAPN2: p < 0.0001, N = 5). D, The efficiency and specificity of CAPN1 and CAPN2 knock-down in cultured hippocampal neurons. Lentivirus expressing specific shRNAs to knock down Capn1 (shCapn1) or Capn2 (shCapn2) was introduced into cultured hippocampal neurons. Virus that expressed shLacZ or pLL3.7 was used as a control. β-Tubulin was used as a loading control. Relative CAPN1 and CAPN2 level normalized to β-tubulin is shown as indicated numbers. E, CAPN1 knock-down failed to prevent the reduction of MBNL2 level in NMDA-treated neurons (shLacZ vs shLacZ+NMDA: p < 0.0001; shLacZ+NMDA vs shCapn1+NMDA: p = 0.1259). F, CAPN2 knock-down preserved levels of MBNL2 and NF160 in NMDA-treated neurons (for MBNL2, pLL3.7 vs pLL3.7+NMDA: p = 0.0005; pLL3.7+NMDA vs shCapn2+NMDA: p < 0.0001, for NF160, p < 0.0001 for both). G, Knock-down of CAPN2 preserved the developmental RNA processing transition in NMDA-treated neurons (Cacna1d, Ex12a: vehicle vs shCapn2: p = 0.0249; vehicle vs NMDA: p = 0.0051; NMDA vs shCapn2+NMDA: p = 0.0444; vehicle vs shCapn2+NMDA: p = 0.0973; Mapt, Ex3/4, vehicle vs shCapn2: p = 0.0448; vehicle vs NMDA: p = 0.0448; NMDA vs shCapn2+NMDA: p = 0.0023; vehicle vs shCapn2+NMDA: p = 0.1749; Mapt, Ex8: vehicle vs shCapn2: p = 0.7697; vehicle vs NMDA: p = 0.0001; NMDA vs shCapn2+NMDA: p = 0.0217; vehicle vs shCapn2+NMDA: p = 0.0056; Sptb, poly(A) site, vehicle vs shCapn2: p = 0.0017; vehicle vs NMDA: p = 0.0110; NMDA vs shCapn2+NMDA: p = 0.0017; vehicle vs shCapn2+NMDA: p = 0.4494). Three to four independent experiments were used for quantification. Data are mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001, by one-way ANOVA with Tuckey's test (B, E–G) or student t test (C). n.s., not significant.
To further determine whether NMDA-induced MBNL2 degradation was regulated by calpain, we tested whether calpain knock-down could prevent MBNL2 degradation under NMDA treatment. The major calpains expressed in neurons are calpain-1 and calpain-2 (Vosler et al., 2008). Both contain a distinct catalytic subunit, CAPN1 or CAPN2, respectively, and a common regulatory subunit, CAPNS1 (Goll et al., 2003). We first determined the levels of calpain-1 and calpain-2 by examining those of CAPN1 and CAPN2 in response to NMDA treatment in cultured hippocampal neurons. In neurons treated with NMDA for 3 h, total CAPN1 level was increased, whereas CAPN2 level was slightly reduced as compared with that in unstimulated neurons (Fig. 4C). We next determined which calpain regulates MBNL2 degradation by depleting endogenous CAPN1 and CAPN2 by infection with a lentivirus expressing Capn1-specific or Capn2-specific shRNA, respectively (Fig. 4D). In NMDA-treated neurons, CAPN1 knock-down failed to prevent MBNL2 degradation (Fig. 4E), but CAPN2 knock-down prevented MBNL2 degradation after NMDA stimulation (Fig. 4F). In addition, CAPN2 knock-down preserved the level of NF160 in NMDA-treated neurons (Fig. 4F). We next determined whether depletion of CAPN2 could prevent NMDA-induced aberrant RNA processing regulated by MBNL2. We first determined the effect of CAPN2 knock-down on MBNL2-regulated RNA processing events in vehicle-treated neurons. In CAPN2-depleted neurons, MBNL2 expression was slightly increased (Fig. 4D) as well as increased inclusion of Cacna1d exon 12a, Mapt exons 3/4 and the distal selection of Sptb alternative polyadenylation sites compared with pLL3.7-infected neurons (Fig. 4G). In CAPN2-depleted neurons treated with NMDA, the inclusion of Cacna1d exon 12a or Mapt exons 3/4 and 8 was preserved and the selection of Sptb alternative polyadenylation sites was toward the distal site (Fig. 4G), which indicated inhibition of aberrant RNA processing with CAPN2 depletion in NMDA-treated neurons. These results suggest that calpain-2 regulated MBNL2 degradation and the developmental transition of RNA processing.
Calcium-dependent nuclear translocation of calpain-2 causes MBNL2 degradation
Immunofluorescence staining and biochemical fractionation examination of CAPN2 localization in cultured hippocampal neurons revealed a predominantly cytoplasmic distribution (Fig. 5A). However, MBNL2 distribution was relatively abundant in the nucleus of cultured hippocampal neurons (Fig. 3D, top). To determine how cytoplasmic-localized calpain-2 may mediate MBNL2 degradation, we wondered whether it was because of NMDA-induced nuclear translocation of calpain-2. The nucleus-to-cytoplasm ratio of CAPN2 immunoreactivity was higher in NMDA-treated than vehicle-treated control neurons (Fig. 5B). Using biochemical fractionation, we found that NMDA treatment increased CAPN2 level in the nuclear fraction but decreased it in the cytoplasmic fraction (Fig. 5C), which suggests a possible involvement of nuclear translocation. Despite reduced MBNL2 expression detected in both cytoplasmic and nuclear fractions, the extent of reduction was greater in the nuclear than cytoplasmic fraction (remainder: nuclear, 45%; cytoplasmic, 60%; Fig. 5C), that was similar to the patterns seen in EpA960/CaMKII-Cre mouse brain (Wang et al., 2017). Knock-down of CAPN2 in NMDA-treated neurons preserved MBNL2 levels in both nuclear and cytoplasmic fractions (Fig. 5D), which suggests that calpain-2 undergoing nuclear translocation mediates MBNL2 degradation in both the nucleus and cytoplasm. Calpain-2 activation requires calcium binding, so we next determined whether the nuclear translocation of CAPN2 was also calcium-dependent. Pretreatment with EGTA preserved the subcellular distribution of CAPN2 in NMDA-treated neurons (Fig. 5E), which suggests that the nuclear translocation of CAPN2 requires calcium.
We further determined whether nuclear localization of CAPN2 affected MBNL2 degradation. We generated a Myc-tagged CAPN2 mutant construct by adding nuclear localization signals (NLS) at both the N and C termini of Myc-CAPN2 (NLS-Myc-CAPN2-NLS) to enhance the nuclear translocation of CAPN2. The wild-type Myc-tagged CAPN2 (Myc-CAPN2) was mainly distributed in the cytoplasm (Fig. 5F). The expression of Myc-CAPN2 in neurons did not affect endogenous MBNL2 level as compared with GW1-Myc-transfected control neurons (Fig. 5F). In contrast, the expression of NLS-Myc-CAPN2-NLS in neurons reduced endogenous MBNL2 level as compared with GW1-Myc-transfected and Myc-CAPN2-transfected neurons (Fig. 5F), which suggests that nuclear localization of CAPN2 is involved in nuclear MBNL2 degradation. Our results suggest that calcium-dependent CAPN2 nuclear translocation is associated with MBNL2 degradation.
Dysregulated calcium homeostasis reduces MBNL2 expression
In addition to NMDAR-mediated excitotoxicity, dysregulation of calcium homeostasis is implicated in the pathogenesis of neurodegeneration (Bezprozvanny, 2009). To test whether aberrant calcium homeostasis may also induce CAPN2 nuclear translocation and MBNL2 reduction, we used a calcium ionophore, A23187, to treat neurons for mimicking the dysregulated calcium homeostasis. Consistent with the findings in NMDA-treated neurons, in cultured neurons treated with A23187, MBNL2 expression was reduced in both the cytoplasmic and nuclear fractions, associated with an increase in CAPN2 level in the nuclear fraction and a decrease in CAPN2 level in the cytoplasmic fraction (Fig. 6A, lanes 1, 2, 4, 5). We further determined whether preventing CAPN2 nuclear translocation would prevent MBNL2 degradation. Pretreatment with ivermectin, an inhibitor of importin-mediated nuclear import (Wagstaff et al., 2012), prevented A23187-reduced MBNL2 expression and CAPN2 nuclear translocation (Fig. 6A, lanes 3, 6). In addition, knock-down of CAPN2 in neurons treated with A23187 prevented MBNL2 degradation (Fig. 6B) and increased inclusion of Cacna1d exon 12a and Mapt exon 3/4 (Fig. 6C). We noticed that in neurons treated with A23187, the inclusion of Cacna1d exon 12a was decreased whereas the alternative splicing of Mapt exon 3/4 and 8 were unaffected. Our results suggest that dysregulated calcium homeostasis caused CAPN2 nuclear translocation associated with reduced MBNL2 expression and re-induction of a developmental RNA processing program.
Figure 6.

Dysregulated calcium homeostasis causes calpain-2 nuclear translocation and reduces MBNL2 expression. A, Detection of MBNL2 and CAPN2 levels in the nuclear and cytoplasmic fractions of A23187-treated neurons with or without pretreatment of ivermectin. For nuclear and cytoplasmic fractions, Histone H3 and GAPDH were used as loading controls, respectively. Quantification of cytoplasmic and nuclear fractions of CAPN2 and MBNL2 relative to loading controls is shown (for MBNL2 nucleus, untreated vs A23187: p = 0.0047; A23187 vs A23187+Ivermectin: p = 0.0035, for MBNL2 cytoplasm, untreated vs A23187: p = 0.0417; A23187 vs A23187+Ivermectin: p = 0.0452, for CAPN2 nucleus, untreated vs A23187: p = 0.0079; A23187 vs A23187+Ivermectin: p = 0.0118, for CAPN2 cytoplasm, untreated vs A23187: p = 0.0051; A23187 vs A23187+Ivermectin: p = 0.0069). B, CAPN2 knock-down prevented MBNL2 reduction in A23187-treated neurons (pLL3.7 vs pLL3.7+NMDA: p = 0.0001; pLL3.7+NMDA vs shCapn2+NMDA: p = 0.0018; pLL3.7 vs shCapn2+NMDA: p = 0.0154). C, Effect of CAPN2 knock-down on MBNL2-regulated splicing events in the A23187-treated neurons. Quantification of relative percentage of inclusion (Cacna1d, Ex12a: vehicle vs A23187: p = 0.0009; A23187 vs shCapn2+A23187: p ≤ 0.0001; vehicle vs shCapn2+A23187: p ≤ 0.0001; Mapt, Ex3/4, vehicle vs A23187: p = 0.9988; A23187 vs shCapn2+A23187: p = 0.0065; vehicle vs shCapn2+A23187: p = 0.0068; Mapt, Ex8: p = 0.7251). Three independent experiments were used for quantification. Data are mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001, by one-way ANOVA with Tuckey' test (A–C). n.s., not significant.
Nuclear translocation of CAPN2 in the degenerative brains of DM1 and AD mouse models recapitulates its developmental pattern
We next determined the distribution of CAPN2 in DM1 and AD mouse brains. The nuclear fraction of CAPN2 was increased and the cytoplasmic fraction of CAPN2 was decreased in the EpA960/CaMKII-Cre brain as compared with other control brains (Fig. 7A), but the subcellular distribution of CAPN1 was unchanged among all animals (Fig. 7B). Similarly, the nucleus-to-cytoplasm ratio of CAPN2 immunoreactivity was increased in cortical Layer V neurons of the APP/PS1 brain (Fig. 7C), as well as the nuclear expression of CAPN2 examined by the biochemical fractionation was increased in the cortical regions of APP/PS1 brains (Fig. 7D). In addition, the nucleus-to-cytoplasm ratio of CAPN2 expression detected in hippocampal CA1 neurons of the THY-Tau22 brain was also increased as compared with the control brains (Fig. 7E). These results suggest that the increased nuclear calpain-2 level accompanied by reduced MBNL2 level and re-induction of the developmental RNA processing program is likely a common feature across models of neurodegenerative diseases.
The reduced MBNL2 level associated with reversal of developmental RNA processing suggested re-induction of a developmental program under neurodegenerative conditions. We wondered whether calpain-2 nuclear translocation under neurodegeneration also represents a reversal of the developmental transition associated with MBNL2 expression. To address this question, we examined the cellular distribution of CAPN2 in cultured hippocampal neurons at early and mature stages. CAPN2 was present in both the cytoplasm and nucleus of cultured neurons at 2 DIV but mainly distributed in the cytoplasm at 16 DIV (Fig. 7F). As compared with CAPN2 distribution in neurons at 2 DIV, in neurons at 16 DIV, the ratio of nucleus-to-cytoplasm fraction of CAPN2 immunoreactivity was reduced (Fig. 7F). Examination of the distribution of CAPN2 in the cortical regions of neonatal (P0) and adult mouse brains revealed a similar pattern. In P0 brain, CAPN2 was distributed in both the cytoplasmic and nuclear fractions but with a lower molecular weight shown in the nuclear fraction (Fig. 7G), whereas CAPN2 was predominantly distributed in the cytoplasmic fraction in the adult brain, which suggests a developmental transition from nucleus-to-cytoplasm distribution during neuronal maturation. The neurodegenerative condition may have induced a developmental transition for CAPN2 from the cytoplasm to nucleus, thus reducing MBNL2 level accompanied by reversal of the developmental RNA processing transition.
Discussion
Reversal of transcriptomic profiles to an embryonic pattern is often induced on injury or stress stimulation for adapting the pathologic condition and produces a protective or adverse effect (Mayr and Bartel, 2009; Bangru et al., 2018; Poplawski et al., 2020). In the present study, we demonstrated that neurodegeneration also re-induced a developmental program of enhanced calpain-2 nuclear translocation and caused MBNL2 degradation-associated reversal of MBNL2-regulated alternative splicing and polyadenylation toward the embryonic pattern.
Neurodegeneration-induced MBNL2 loss of function seen in the DM1 mouse model EpA960/CaMKII-Cre suggests an additional mechanism regulating reduced MBNL2 level besides nuclear sequestration (Goodwin et al., 2015). We showed that neurodegenerative conditions induced calpain-2 nuclear translocation resulting in MBNL2 degradation and reversal of MBNL2-mediated RNA processing to an embryonic pattern. Calpain activation has been found in several neurodegenerative disorders (Vosler et al., 2008; Ono et al., 2016). We showed that NMDAR-mediated excitotoxicity altered the levels of calpain-1 and calpain-2. However, genetic knock-down of calpain-2 but not calpain-1 preserved MBNL2 level and MBNL2-mediated function, which supports the notion that differences of calpain-1 and calpain-2 in the structures and requirement of calcium concentration for activation may contribute to distinct roles during the progression of neurodegenerative disorders (Goll et al., 2003; Baudry and Bi, 2016). Increased nuclear level of calpain-2 and reduced MBNL2 level occurred in different models of neurodegenerative diseases including DM1 and AD, which suggests that re-induction of a developmental RNA processing program mediated by MBNL2 via nuclear calpain activity is a shared mechanism of neurodegenerative diseases.
The major molecular feature of DM1 is reversal of alternative splicing and polyadenylation to the embryonic pattern (Charizanis et al., 2012; Batra et al., 2014; Goodwin et al., 2015). Indeed, re-induction of the fetal/neonatal gene program has been reported under several pathologic conditions. After myocardial infarction, CUGBP Elav-like family member 1 (CELF1) is upregulated, which is normally downregulated in the adult heart, resulting in mRNA degradation of genes involved in cardiac conduction and contractility (Chang et al., 2017). Upon toxin-induced injury, adult hepatocyte regeneration is associated with reactivation of neonatal translation and splicing programs (Bangru et al., 2018). Similarly, after spinal cord injury, the transcriptional profile of the corticospinal tract motor neurons temporarily exhibits an embryonic pattern (Poplawski et al., 2020). Grafting of spinal cord-derived neural progenitor cells sustains the embryonic transcriptional profile in corticospinal tract motor neurons, thus enabling the regeneration of corticospinal axons (Poplawski et al., 2020). In the present study, we showed that nuclear localization of calpain-2 and low level of MBNL2 seen in neurodegenerative conditions recapitulates the developmental expression pattern. Therefore, re-induction of a fetal gene program is likely a common feature induced by the pathologic conditions. How re-induction of a fetal gene program contributing a beneficial or adverse effect is determined is worthy of investigation.
MBNL2 has been found involved in regulating splicing switches during postnatal neuronal maturation (Weyn-Vanhentenryck et al., 2018). Cacna1d exon 12a is the most responsive MBNL2 target (Charizanis et al., 2012). Cacna1d gene encoding a low voltage-activated L-type Ca channel Cav1.3 plays a key role in regulating neuronal excitability (Hofer et al., 2021). Inclusion of exon 12a results in a negative shift in the voltage-dependence of activation in Cav1.3 (Hofer et al., 2021). Shift to more negative potentials has been implicated to enhance synaptic transmission (van den Maagdenberg et al., 2010), which suggests an association of exon 12a inclusion in Cav1.3 with neuronal maturation. Therefore, reduced Cacna1d exon 12a-containing transcripts may be associated with reduced neurotransmission in degenerative brains and serve as a marker for neurodegeneration.
Footnotes
This work was supported by the Institute of Biomedical Sciences, Academia Sinica (Y.C. and G.-S.W.). We thank Dr. Yi-Shuian Huang and Dr. Chia-Fang Wang (Institute of Biomedical Sciences, Academia Sinica) for providing Capn2-specific shRNA clone and the construct of rat CAPN2 sequence in pcDNA3.1 (detailed information is described in the plasmid section of Materials and Methods).
The authors declare no competing financial interests.
References
- Bangru S, Arif W, Seimetz J, Bhate A, Chen J, Rashan EH, Carstens RP, Anakk S, Kalsotra A (2018) Alternative splicing rewires Hippo signaling pathway in hepatocytes to promote liver regeneration. Nat Struct Mol Biol 25:928–939. 10.1038/s41594-018-0129-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett WP, Banker GA (1984) An electron microscopic study of the development of axons and dendrites by hippocampal neurons in culture. I. Cells which develop without intercellular contacts. J Neurosci 4:1944–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batra R, Charizanis K, Manchanda M, Mohan A, Li M, Finn DJ, Goodwin M, Zhang C, Sobczak K, Thornton CA, Swanson MS (2014) Loss of MBNL leads to disruption of developmentally regulated alternative polyadenylation in RNA-mediated disease. Mol Cell 56:311–322. 10.1016/j.molcel.2014.08.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudry M, Bi X (2016) Calpain-1 and calpain-2: the yin and yang of synaptic plasticity and neurodegeneration. Trends Neurosci 39:235–245. 10.1016/j.tins.2016.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer SA (1980) Development of the hippocampal region in the rat. I. Neurogenesis examined with 3H-thymidine autoradiography. J Comp Neurol 190:87–114. 10.1002/cne.901900107 [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I (2009) Calcium signaling and neurodegenerative diseases. Trends Mol Med 15:89–100. 10.1016/j.molmed.2009.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KT, Cheng CF, King PC, Liu SY, Wang GS (2017) CELF1 mediates connexin 43 mRNA degradation in dilated cardiomyopathy. Circ Res 121:1140–1152. 10.1161/CIRCRESAHA.117.311281 [DOI] [PubMed] [Google Scholar]
- Charizanis K, et al. (2012) Muscleblind-like 2-mediated alternative splicing in the developing brain and dysregulation in myotonic dystrophy. Neuron 75:437–450. 10.1016/j.neuron.2012.05.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotti CG, Sullivan CA, Banker GA (1988) The establishment of polarity by hippocampal neurons in culture. J Neurosci 8:1454–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallais B, Gagnon C, Mathieu J, Richer L (2017) Cognitive decline over time in adults with myotonic dystrophy type 1: a 9-year longitudinal study. Neuromuscul Disord 27:61–72. 10.1016/j.nmd.2016.10.003 [DOI] [PubMed] [Google Scholar]
- Goll DE, Thompson VF, Li H, Wei W, Cong J (2003) The calpain system. Physiol Rev 83:731–801. 10.1152/physrev.00029.2002 [DOI] [PubMed] [Google Scholar]
- Goodwin M, Mohan A, Batra R, Lee KY, Charizanis K, Fernández Gómez FJ, Eddarkaoui S, Sergeant N, Buée L, Kimura T, Clark HB, Dalton J, Takamura K, Weyn-Vanhentenryck SM, Zhang C, Reid T, Ranum LP, Day JW, Swanson MS (2015) MBNL sequestration by toxic RNAs and RNA misprocessing in the myotonic dystrophy brain. Cell Rep 12:1159–1168. 10.1016/j.celrep.2015.07.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofer NT, Pinggera A, Nikonishyna YV, Tuluc P, Fritz EM, Obermair GJ, Striessnig J (2021) Stabilization of negative activation voltages of Cav1.3 L-Type Ca(2+)-channels by alternative splicing. Channels (Austin) 15:38–52. 10.1080/19336950.2020.1859260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, Copeland NG, Lee MK, Younkin LH, Wagner SL, Younkin SG, Borchelt DR (2004) Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet 13:159–170. 10.1093/hmg/ddh019 [DOI] [PubMed] [Google Scholar]
- Jimenez-Marin A, Diez I, Labayru G, Sistiaga A, Caballero MC, Andres-Benito P, Sepulcre J, Ferrer I, Lopez de Munain A, Cortes JM (2021) Transcriptional signatures of synaptic vesicle genes define myotonic dystrophy type I neurodegeneration. Neuropathol Appl Neurobiol 47:1092–1108. 10.1111/nan.12725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalsotra A, Xiao X, Ward AJ, Castle JC, Johnson JM, Burge CB, Cooper TA (2008) A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proc Natl Acad Sci U S A 105:20333–20338. 10.1073/pnas.0809045105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhla A, Rühlmann C, Lindner T, Polei S, Hadlich S, Krause BJ, Vollmar B, Teipel SJ (2017) APPswe/PS1dE9 mice with cortical amyloid pathology show a reduced NAA/Cr ratio without apparent brain atrophy: a MRS and MRI study. Neuroimage Clin 15:581–586. 10.1016/j.nicl.2017.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau A, Tymianski M (2010) Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch 460:525–542. 10.1007/s00424-010-0809-1 [DOI] [PubMed] [Google Scholar]
- Lee DH, Goldberg AL (1998) Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol 8:397–403. 10.1016/s0962-8924(98)01346-4 [DOI] [PubMed] [Google Scholar]
- Lester E, Ooi FK, Bakkar N, Ayers J, Woerman AL, Wheeler J, Bowser R, Carlson GA, Prusiner SB, Parker R (2021) Tau aggregates are RNA-protein assemblies that mislocalize multiple nuclear speckle components. Neuron 109:1675–1691.e9. 10.1016/j.neuron.2021.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Pattamatta A, Zu T, Reid T, Bardhi O, Borchelt DR, Yachnis AT, Ranum LP (2016) C9orf72 BAC mouse model with motor deficits and neurodegenerative features of ALS/FTD. Neuron 90:521–534. 10.1016/j.neuron.2016.04.005 [DOI] [PubMed] [Google Scholar]
- Maurage CA, Udd B, Ruchoux MM, Vermersch P, Kalimo H, Krahe R, Delacourte A, Sergeant N (2005) Similar brain tau pathology in DM2/PROMM and DM1/Steinert disease. Neurology 65:1636–1638. 10.1212/01.wnl.0000184585.93864.4e [DOI] [PubMed] [Google Scholar]
- Mayr C, Bartel DP (2009) Widespread shortening of 3'UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 138:673–684. 10.1016/j.cell.2009.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM (2003) Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39:409–421. 10.1016/s0896-6273(03)00434-3 [DOI] [PubMed] [Google Scholar]
- Ono Y, Saido TC, Sorimachi H (2016) Calpain research for drug discovery: challenges and potential. Nat Rev Drug Discov 15:854–876. 10.1038/nrd.2016.212 [DOI] [PubMed] [Google Scholar]
- Poplawski GHD, Kawaguchi R, Van Niekerk E, Lu P, Mehta N, Canete P, Lie R, Dragatsis I, Meves JM, Zheng B, Coppola G, Tuszynski MH (2020) Injured adult neurons regress to an embryonic transcriptional growth state. Nature 581:77–82. 10.1038/s41586-020-2200-5 [DOI] [PubMed] [Google Scholar]
- Radde R, Bolmont T, Kaeser SA, Coomaraswamy J, Lindau D, Stoltze L, Calhoun ME, Jäggi F, Wolburg H, Gengler S, Haass C, Ghetti B, Czech C, Hölscher C, Mathews PM, Jucker M (2006) Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep 7:940–946. 10.1038/sj.embor.7400784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rau F, Freyermuth F, Fugier C, Villemin JP, Fischer MC, Jost B, Dembele D, Gourdon G, Nicole A, Duboc D, Wahbi K, Day JW, Fujimura H, Takahashi MP, Auboeuf D, Dreumont N, Furling D, Charlet-Berguerand N (2011) Misregulation of miR-1 processing is associated with heart defects in myotonic dystrophy. Nat Struct Mol Biol 18:840–845. 10.1038/nsmb.2067 [DOI] [PubMed] [Google Scholar]
- Schindowski K, Bretteville A, Leroy K, Bégard S, Brion JP, Hamdane M, Buée L (2006) Alzheimer's disease-like tau neuropathology leads to memory deficits and loss of functional synapses in a novel mutated tau transgenic mouse without any motor deficits. Am J Pathol 169:599–616. 10.2353/ajpath.2006.060002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahidullah M, Le Marchand SJ, Fei H, Zhang J, Pandey UB, Dalva MB, Pasinelli P, Levitan IB (2013) Defects in synapse structure and function precede motor neuron degeneration in Drosophila models of FUS-related ALS. J Neurosci 33:19590–19598. 10.1523/JNEUROSCI.3396-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton CA (2014) Myotonic dystrophy. Neurol Clin 32:705–719. 10.1016/j.ncl.2014.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Maagdenberg AM, Pizzorusso T, Kaja S, Terpolilli N, Shapovalova M, Hoebeek FE, Barrett CF, Gherardini L, van de Ven RC, Todorov B, Broos LA, Tottene A, Gao Z, Fodor M, De Zeeuw CI, Frants RR, Plesnila N, Plomp JJ, Pietrobon D, Ferrari MD (2010) High cortical spreading depression susceptibility and migraine-associated symptoms in Ca(v)2.1 S218L mice. Ann Neurol 67:85–98. 10.1002/ana.21815 [DOI] [PubMed] [Google Scholar]
- Vosler PS, Brennan CS, Chen J (2008) Calpain-mediated signaling mechanisms in neuronal injury and neurodegeneration. Mol Neurobiol 38:78–100. 10.1007/s12035-008-8036-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagstaff KM, Sivakumaran H, Heaton SM, Harrich D, Jans DA (2012) Ivermectin is a specific inhibitor of importin α/β-mediated nuclear import able to inhibit replication of HIV-1 and dengue virus. Biochem J 443:851–856. 10.1042/BJ20120150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CF, Huang YS (2012) Calpain 2 activated through N-methyl-D-aspartic acid receptor signaling cleaves CPEB3 and abrogates CPEB3-repressed translation in neurons. Mol Cell Biol 32:3321–3332. 10.1128/MCB.00296-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, Cody NA, Jog S, Biancolella M, Wang TT, Treacy DJ, Luo S, Schroth GP, Housman DE, Reddy S, Lécuyer E, Burge CB (2012) Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell 150:710–724. 10.1016/j.cell.2012.06.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GS, Chang NC, Wu SC, Chang AC (2002) Regulated expression of alpha2B adrenoceptor during development. Dev Dyn 225:142–152. 10.1002/dvdy.10141 [DOI] [PubMed] [Google Scholar]
- Wang GS, Kearney DL, De Biasi M, Taffet G, Cooper TA (2007) Elevation of RNA-binding protein CUGBP1 is an early event in an inducible heart-specific mouse model of myotonic dystrophy. J Clin Invest 117:2802–2811. 10.1172/JCI32308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang PY, Lin YM, Wang LH, Kuo TY, Cheng SJ, Wang GS (2017) Reduced cytoplasmic MBNL1 is an early event in a brain-specific mouse model of myotonic dystrophy. Hum Mol Genet 26:2247–2257. 10.1093/hmg/ddx115 [DOI] [PubMed] [Google Scholar]
- Wang PY, Chang KT, Lin YM, Kuo TY, Wang GS (2018) Ubiquitination of MBNL1 is required for its cytoplasmic localization and function in promoting neurite outgrowth. Cell Rep 22:2294–2306. 10.1016/j.celrep.2018.02.025 [DOI] [PubMed] [Google Scholar]
- Weyn-Vanhentenryck SM, Feng H, Ustianenko D, Duffié R, Yan Q, Jacko M, Martinez JC, Goodwin M, Zhang X, Hengst U, Lomvardas S, Swanson MS, Zhang C (2018) Precise temporal regulation of alternative splicing during neural development. Nat Commun 9:2189. 10.1038/s41467-018-04559-0 [DOI] [PMC free article] [PubMed] [Google Scholar]