

Abstract
Asobara japonica is an endoparasitic wasp that parasitizes Drosophila flies. It synthesizes various toxic components in the venom gland and injects them into host larvae during oviposition. To identify and characterize these toxic components for enabling parasitism, we performed the whole-genome sequencing (WGS) and devised a protocol for RNA interference (RNAi) with A. japonica. Because it has a parthenogenetic lineage due to Wolbachia infection, we generated a clonal strain from a single wasp to obtain highly homogenous genomic DNA. The WGS analysis revealed that the estimated genome size was 322 Mb with a heterozygosity of 0.132%. We also performed RNA-seq analyses for gene annotation. Based on the qualified WGS platform, we cloned ebony-Aj, which encodes the enzyme N-β-alanyl dopamine synthetase, which is involved in melanin production. The microinjection of double-stranded RNA (dsRNA) targeting ebony-Aj led to body colour changes in adult wasps, phenocopying ebony-Dm mutants. Furthermore, we identified putative venom genes as a target of RNAi, confirming that dsRNA injection-based RNAi specifically suppressed the expression of the target gene in wasp adults. Taken together, our results provide a powerful genetic toolkit for studying the molecular mechanisms of parasitism.
Keywords: endoparasitoid wasp, Asobara japonica, whole-genome sequencing, RNA interference, parasitism
1. Introduction
Parasitism is an ecological lifestyle whereby the parasite organism exploits resources from the host organism to sustain life. In insects, parasitoid wasps belong to the order Hymenoptera, exploiting other insect species and arthropods as hosts. It is estimated that a group of parasitoid wasps accounts for more than 20% of all insect species.1 A large number of species have highly diversified strategies for successful parasitism on earth.2 For example, parasitoid wasps lay eggs at a certain developmental stage of their host arthropods, including the embryo, larva, pupa, and adult stages. Even in close relatives, some wasp species are adapted to many host species (generalist), whereas others only parasitize a limited number of host species (specialist). Many studies have reported ecological relationships and coevolution mechanisms between parasitoid wasps and their hosts.3 Previous studies have also revealed a wide variety of bioactive molecules, venom proteins, and virus-like particles produced in parasitoid wasps.4 Conversely, host arthropods acquire several repellant behaviours and immune defense systems against parasites.5,6 However, the underlying molecular mechanism of wasp parasitism is still poorly understood compared with the immune defense system of their hosts.
To study the molecular mechanisms of parasitism, it is crucial to obtain whole-genome information for both parasitoid wasps and their host arthropods. In the fruit fly Drosophila melanogaster, an excellent model organism, ∼50 drosophilid parasitoids have been reported.7,8 Among them, the genus Leptopilina (Hymenoptera: Figitidae) has been the best studied. These wasps lay their eggs in fruit fly larvae and the developing wasp larvae eventually consume and kill their fly hosts. Leptopilinaheterotoma is a generalist, whereas Leptopilinaboulardi is a specialist. Recent studies have revealed that these wasps have developed different strategies to defeat the immune defense of Drosophila.9 Whole-genome sequencing (WGS) analysis has been performed, and distinct sets of venom proteins have been reported.10
In this study, we focussed on Asobara japonica Belokobylskij (Hymenoptera: Braconidae). Asobarajaponica is a typical generalist parasitoid wasp that lays eggs in the larvae of various Drosophila species.11,12 Similar to the genus Leptopilina, an adult female A. japonica wasp oviposits an egg into the host larva, enabling a wasp larva to hatch inside the host. The parasitoid wasp larva grows with the host fly larva until pupariation. Inside the host pupa, the wasp kills the host after pupariation and eventually ecloses from the host pupal case. It takes 15–17 days to become an adult wasp after oviposition. Notably, A. japonica has a high parasitic success rate (>90%), implying that the host immune defense system hardly affects wasp development. This feature enables us to consistently study the developmental process of parasitoid wasps, in contrast to studying the ‘tug-of-war’ relationship between host immune defense response and parasitic strategy. Moreover, it has been reported that the venom of A. japonica has a severe deleterious effect on the host larvae13,14: Interruption of their oviposition behaviour before egg laying causes high larval mortality in host Drosophila species. This suggests that the presence of toxic venom components as well as their neutralizer components underlie the unique parasitic strategy in A. japonica, which is distinct from the venom components of other parasitoid wasps.
Based on host specificity and the death of host animals, many parasitic species are used as biological control agents for the management of pest species.15 Recent studies have evaluated the parasitism of A. japonica, because it parasitizes Drosophila suzukii (Matsumura), commonly known as spotted wing Drosophila, which is a serious economic threat to the production of fruits worldwide.16,17 Considering the deleterious effect of A. japonica on the hosts,13,14 it is crucial to understand the molecular mechanisms of parasitism in order to develop novel insecticide seed strategies.
Here, we performed WGS analysis of A. japonica and devised a protocol for RNA interference (RNAi) with A. japonica. To obtain a homogenous genome sequence, we generated the clonal strain ‘Genome #3’ from a single female wasp and successfully constructed the highly qualified WGS platform for gene annotation, compared with the previous study.18 The well-organized WGS platform allows us to conduct genetic approaches from both hosts and parasites in a bidirectional manner.
2. Materials and methods
2.1 Biological materials
A parthenogenetic strain of the endoparasitoid wasp A.japonica ‘Tokyo (TK)’ was originally collected near Tokyo by Dr. Masato Kimura and kindly provided by Dr. Kazuo Takahashi at Okayama University.19 The clonal strain ‘Genome #3’ was established from a single female in the Tokyo strain. In D.melanogaster, the Oregon R (OR) strain was used as the wild-type and host in this study. The e1 mutant (DGRC #106436) was obtained from KYOTO Stock Center (DGRC) at Kyoto Institute of Technology. A.japonica and D. melanogaster were reared on standard agar-cornmeal medium at 25°C under a 12:12-h light/dark cycle.
2.2 Genome preparation
In the clonal strain ‘Genome #3’, 200 Wolbachia-free wasps were collected from host fly larvae reared on a standard cornmeal diet supplemented with tetracycline (4 mg/2 g food). Wasps were flash-frozen in liquid nitrogen and stored at −80°C. Genomic DNA was extracted using the genomic DNA buffer set and Genomic-tip 100/G (QIAGEN). After the genomic DNA samples were resuspended in 1× Tris-EDTA buffer (pH 8.0), the concentration of DNA was measured using a Nanodrop and Qubit dsDNA BR Assay kit (Thermo Fisher Scientific). The quality of the samples was examined by DNA electrophoresis. A total of 38 µg of 200 kb DNA was used for WGS.
2.3 Genome sequencing and assembly
Genome sequencing was performed using Illumina HiSeq 2500, for paired-end short reads, and PacBio Sequel II platforms. Genome size and heterozygosity were estimated using the GenomeScope 2.0.20,21 The primary assembly was performed using FALCON-UNZIP22 with default settings, and the assembled contigs were polished using Arrow23 and Pilon.24 Completeness of the assembly was assessed using BUSCO v5.2.2 with the ‘insecta_odb10’ and ‘hymenoptera_odb10’ datasets.25 For comparison, we performed BUSCO analysis with other Hymenopteran species: Apis mellifera, Orussus abietinus, Trichogramma pretiosum, Copidosoma floridanum, Nasonia vitripennis, Ceratosolen solmsi, Diachasma alloeum, Fopius arisanus, Microplitis demolitor, Macrocentrus cingulum, Cotesia typhae, Cotesia glomerata, Chelonus insularis, and Aphidius gifuensis. The references of the genomes of all species are listed in Supplementary Fig. S1.26–38
2.4 RNA sequencing
Total RNA was extracted from whole bodies of 200 female wasps, venom glands of 300 female wasps at 1 day after eclosion, and venom glands of 300 female wasps at 10 days after eclosion. Samples were dissected and ground with pestles in RNAiso Plus reagent (TaKaRa). After DNase treatment, the concentration of RNA was measured using a Nanodrop and Qubit double-stranded RNA (dsRNA) BR Assay kit (Thermo Fisher Scientific). Sequencing libraries were prepared by means of polyA selection step with TruSeq Stranded mRNA Library Prep kit (Illumina). Then, libraries were sequenced on an Illumina HiSeq 2500 (101 cycles) for paired-end short reads with a length of 100 bp. After filtering the low-quality and adapter sequences, 82,282,218, 84,554,634, and 85,120,902 RNA-Seq reads were obtained, respectively.
2.5 Gene annotation
Protein-coding gene prediction was conducted using an in-house annotation pipeline based on a combination of RNA-seq-based prediction, homology-based prediction, and ab initio prediction methods. To map the first RNA-seq-based prediction, RNA-seq reads were mapped to assembled genomes using HiSat2 (version 2.1.0),39 and gene structure was predicted using StringTie (version 1.3.4d).40 For de novo first RNA-seq-based prediction, Trinity41 and Oases42 were used. Assembled RNA contigs were spliced and mapped by GMAP43 and the open reading frame (ORF) was predicted using ORFfinder. For homology-based predictions, protein sequences from four species (D. alloeum, F. arisanus, M. demolitor, and M. cingulum) were aligned to the assembled genomes using Spaln.44 For ab initio gene prediction, Augustus45 and SNAP46 were trained with RNA-seq-based predicted genes and then used. Moreover, functional annotation was performed using BLASTp for homology signatures against NCBI databases and InterProScan for Pfam, and the gene ontology terms associated with the proteins were retrieved using the InterPro ID. The subcellular localization was predicted using SignalP.47,48
2.6 Phylogenetic analysis
To compare orthologues, we used protein sequences of 14 hymenopteran species available from three repositories. Protein sequences of O. abietinus, D. alloeum, F. arisanus, T. pretiosum, M. cingulum, M. demolitor, C. floridanum, N. vitripennis, and C. solmsi were downloaded from WaspBase (http://www.insect-genome.com/waspbase/); A. mellifera, C. typhae, C. insularis, and A. gifuensis from NCBI (https://www.ncbi.nlm.nih.gov); and C. glomerata from Figshare (https://figshare.com). Subsequently, the identification of orthologous groups of proteins was performed. First, protein sequences from all species were grouped into gene families and 2,381 orthologue groups with a one-to-one relationship across all species were extracted using Proteinortho.49 For each group, multiple alignments were performed with MAFFT50 and sites containing gaps (‘–’) or ambiguous characters (‘X’) were excluded. All alignments were concatenated, and 1,110,464 amino acid sites were used for the phylogenetic analysis. A phylogenetic tree was constructed using RAxML (version 8.2.12).51 Here, we applied the JTT substitution matrix with a gamma model of rate heterogeneity (-m PROTGAMMAJTT).
2.7 Data availability
The raw sequence data generated on the Illumina and PacBio platforms were deposited in the DNA Databank of Japan (DDBJ, BioProject ID: PRJDB12783). The accession numbers of genome assembled scaffold data are BQMI01000001–BQMI01000601. Gene annotation data are available upon request.
2.8 Observation of the developmental progression of A. japonica
Wild-type fly eggs were laid for 2 h on a grape plate with yeast paste to synchronize host development. After 24 h, the first instar (L1) larvae were transferred into vials containing standard cornmeal food. At 96 h after egg-laying, the third instar (L3) larvae and adult wasps were transferred into the bulk infection arena (Fig. 3A). Wasps were allowed to freely lay eggs in L3 host larvae for 1 h and were then removed from the arena to synchronize the timing of oviposition. At a certain time point after infection, a wasp larva was carefully removed from the host pupal case with forceps under a dissection microscope and placed on a 2% agar plate. When the wasp larvae were removed from the host pupal case at 4 days post-infection (dpi), development proceeded on the agar plate and wasp adults eclosed normally.
Figure 3.

Schematic representation of the RNAi protocol during A. japonica development. (A) A standard setup of a bulk infection arena for wasp parasitism. (B) Representative images of developing wasps on an agar plate. dpi, days post-infection. Scale bar, 1 mm.
2.9 Identification of the A. japonica ebony gene and putative venom genes
We identified the A. japonica ebony gene (gene003270, ebony-Aj) as an orthologue of the D. melanogaster ebony (ebony-Dm) gene. Ebony encodes the enzyme N-β-alanyl dopamine (NBAD) synthetase, which converts dopamine to NBAD.52 Loss of this enzymatic function increases dark melanin and decreases tan pigment, resulting in a dark coloured body phenotype. Thus, we chose ebony-Aj to be the target of RNAi for the visible loss-of-function phenotype in the first trial. The D. melanogaster amino acid sequence of the ebony gene was obtained from FlyBase (http://flybase.org/). The A. japonica orthologue of ebony (ebony-Aj) was identified by tBLASTn analysis using the amino acid sequence of Ebony-Dm as a query.
To identify putative venom genes, the fragments per kilobase of transcript per million mapped reads (FPKM) values were calculated with RNA-seq data of the venom gland sample (1 day after eclosion). Adapter sequences were trimmed from raw RNA-seq reads using Trimmomatic (version 0.39).53 RNA-seq reads were mapped to assembled genomes using HiSat2 (version 2.1.0).39 Mapped read data were sorted, merged and counted using SAMtools (version 1.9)54 and StringTie (version 2.0.6).40 FPKM values were calculated with R (version 3.6.1) and Ballgown (version 2.18.0).55
2.10 Quantitative reverse transcription PCR
To confirm if these putative venom genes are strongly expressed in the venom gland, we performed quantitative reverse transcription PCR (qRT-PCR) analysis of the top five genes with high FPKM values (Table 7). The venom glands and the other carcass from 20 wasp adults were dissected and total RNA was extracted using RNAiso Plus reagent (TaKaRa). cDNA was prepared with ReverTra Ace qPCR RT Master Mix with gDNA Remover (TOYOBO). Quantitative PCR was conducted using the Universal SYBR Select Master Mix (Applied Biosystems) with a Thermal Cycler Dice TP800 system (TaKaRa). For the relative quantification of expression, the A. japonica orthologue of Ribosomal protein L32 (RpL32-Aj) was identified by tBLASTn analysis using amino acid sequence of D. melanogaster RpL32. The target gene expression level was normalized to RpL32 and then relative fold changes were calculated by the delta-delta Ct method. The mean values were from three independent experiments. The primers used for qRT-PCR are described in Table 1.
Table 7.
Top five genes with high FPKM values in the venom gland sample
| Gene ID | FPKM value | Relative expression level (venom gland/other carcase) | |
|---|---|---|---|
| Mean value (n = 3) | |||
| 1 | gene006533 | 403151.38 | 110,082.6 |
| 2 | gene010987 | 395514.72 | N.A. |
| 3 | gene010975 | 113373.65 | 1,401,477.8 |
| 4 | gene003054 | 112626.62 | 1,185,641.8 |
| 5 | gene000789 | 34218.91 | 258,595.8 |
N.A. (not available): The coding sequence of gene010987 was an exact match with that of gene006533 and the gene locus of gene010987 was just next to gene006533. qRT-PCR was not performed for gene010987.
Table 1.
Primer sequences used in this study
| Primer name | Sequence (5′> 3′) | Purpose |
|---|---|---|
| Ajebony_ex8_Fwd | TATTTTAACCGAAAGTTTCTACTTGAAAGCTG | dsRNA template |
| Ajebony_ex8_Rev | CTGTGTAAGAGGACTCGTATTGGTG | dsRNA template |
| AjRpL32_qPCR_Fwd | CCCGTCACATGCTTCCTACT | qRT-PCR |
| AjRpL32_qPCR_Rev | GAATTTGCGATTCTGCATCA | qRT-PCR |
| Aj_gene003054_ex1_Fwd | ATGTACTCCCACTGTAGGTTCCAAG | CDS cloning |
| Aj_gene003054_ex3_Rev | TTACTTCCTCCCCGTAAGCGCTC | CDS cloning, dsRNA template |
| Aj_gene003054_RNAi_Fwd | CAAGAAAGCAAACGGCAAACCTTGG | dsRNA template |
| T7_Aj_gene003054_RNAi_Fwd | GGATCCTAATACGACTCACTATAGCAAGAAAGCAAACGGCAAACCTTGG | dsRNA template |
| T7_Aj_gene003054_ex3_Rev | GGATCCTAATACGACTCACTATAGGTTACTTCCTCCCCGTAAGCGCTC | dsRNA template |
| qPCR_gene006533_Fwd | CATCGGAACTACAGGGCATT | qRT-PCR |
| qPCR_gene006533_Rev | TGCCAATGTCTTCACACTCC | qRT-PCR |
| qPCR_gene003054_Fwd | CTGATTGTCGTGCTCGGTTA | qRT-PCR |
| qPCR_gene003054_Rev | CCACCCTGAGGATGTGTTTC | qRT-PCR |
| qPCR_gene010975_Fwd | GTATCTTCGGGATGCTCTGC | qRT-PCR |
| qPCR_gene010975_Rev | CCCTCCGCTAACTCACACAT | qRT-PCR |
| qPCR_gene010975_Fwd2 | CCTGATAATCGTCGGTATCTTC | qRT-PCR |
| qPCR_gene010975_Rev2 | TGCCCACTGTTCCTGACATC | qRT-PCR |
| qPCR_gene000789_Fwd | TTCTGAGACAGAGCCCGAAT | qRT-PCR |
| qPCR_gene000789_Rev | GGAATATGCAGTGGGTCGTC | qRT-PCR |
Underline indicates the T7 promoter sequence. dsRNA, double-strand RNA; qRT-PCR, quantitative reverse transcription PCR; CDS, coding sequence.
2.11 Primer design for RNAi target regions
To conduct ebony-Aj RNAi experiments, we chose the exon 8 sequence as an RNAi target region because it is the longest single exon among the ebony-Aj (gene003270) gene region. For the putative venom gene gene003054, the 151 bp region of the coding sequence was chosen as the RNAi target region. RNAi target regions were distinct from the qPCR target regions. For qPCR, each primer was designed using Primer3Plus.56 The primer sequences are listed in Table 1.
2.12 dsRNA synthesis and injection
For dsRNA synthesis, a gene-specific sequence was used as a template for transcription. Template cDNAs were obtained from total RNA extracted from the whole body of A. japonica using RNAiso Plus reagent (TaKaRa) and PrimeScript™ Reverse Transcriptase (TaKaRa). PCR was performed using KOD Plus Neo (TOYOBO) and the amplified specific DNA fragments were inserted into the SmaI site of the pBluescript SK (-) plasmid with Ligation high Ver. 2 (TOYOBO). These plasmids were digested with NotI and EcoRI restriction enzymes, and specific DNA fragments were ligated into NotI and EcoRI cut pBluescript KS (+) in the opposite direction to the T7 promoter sequence. Finally, these two complementary DNA fragments were digested with NotI and EcoRI to generate linearized DNA templates in the T7 RiboMAX™ Express RNAi system (Promega) following the manufacturer’s instructions. The Green fluorescent protein (GFP)-coding sequence containing the p-GEM easy plasmid was a gift from Dr. Manabu Kamimura (National Agriculture and Food Research Organization, NARO) and was used for the synthesis of a control GFP dsRNA.
dsRNA microinjection was performed with a glass needle made from a glass capillary with filament (GD-1, NARISHIGE) using a puller (PC-10, NARISHIGE) and polished with a micro grinder (EG-401, NARISHIGE). The dsRNA solution was injected into the abdomen of A. japonica larvae, after the host pupal case was carefully removed and then placed on a 2% agar plate. For ebony-Aj RNAi, dsRNA was concentrated to 10 μg/μl by ethanol precipitation for microinjection. In the case of the putative venom gene-RNAi, dsRNA solution was diluted to 1,000, 500, and 250 ng/μl with ultrapure water. Although we could not control the exact amount of synthesized dsRNA for microinjection because of limitations imposed by our injection apparatus, we could estimate the required amount of 100–200 nl.
2.13 Evaluation of body colour
The dorsal side of the wasp bodies was imaged using a digital camera (Leica MC120 HD) attached to a dissection microscope under constant lighting conditions. A square area of the wasp thorax was extracted to quantify the body colour of individual wasps. To minimize the effects of highlights and shadows in the images, we selected an area devoid of pixels with overexposure brightness and underexposure darkness. Then, 8-bit integer RGB values (0 ≤ R, G, B ≤ 255) were measured and averaged using Fiji software (National Institutes of Health), and the redness index was calculated by Σ(R—mean[R, G, B])/n (n = the number of pixels).57
3. Results and discussion
3.1 Generation of a clonal strain derived from a single female wasp
To obtain a homogenous genome sequence with high quality, it is essential to collect enough amount of uniform genome DNA from individual samples. In A. japonica, the Wolbachia-infected strain ‘Tokyo (TK)’ is available, which produces only female wasps from unfertilized eggs by Wolbachia-induced parthenogenesis. Therefore, we generated a clonal strain ‘Genome #3’ from a single female wasp in TK strain (Fig. 1). After Genome #3 was established, Wolbachia was removed from these wasp clones to avoid contamination with Wolbachia DNA of the wasp genomic DNA samples. When host fly larvae were reared on a standard diet supplemented with tetracycline, parasitoid wasp eggs and larvae were exposed to tetracycline through the haemolymph of the hosts and Wolbachia was eliminated from wasp bodies.58,59 Under these conditions, eclosed female wasps produced not only female but also male offspring in the next generation. We confirmed the removal of Wolbachia at 4 mg tetracycline in 2 g fly food, and collected 200 female wasps with an identical genetic background for genomic DNA extraction (Supplementary Fig. S2A).
Figure 1.
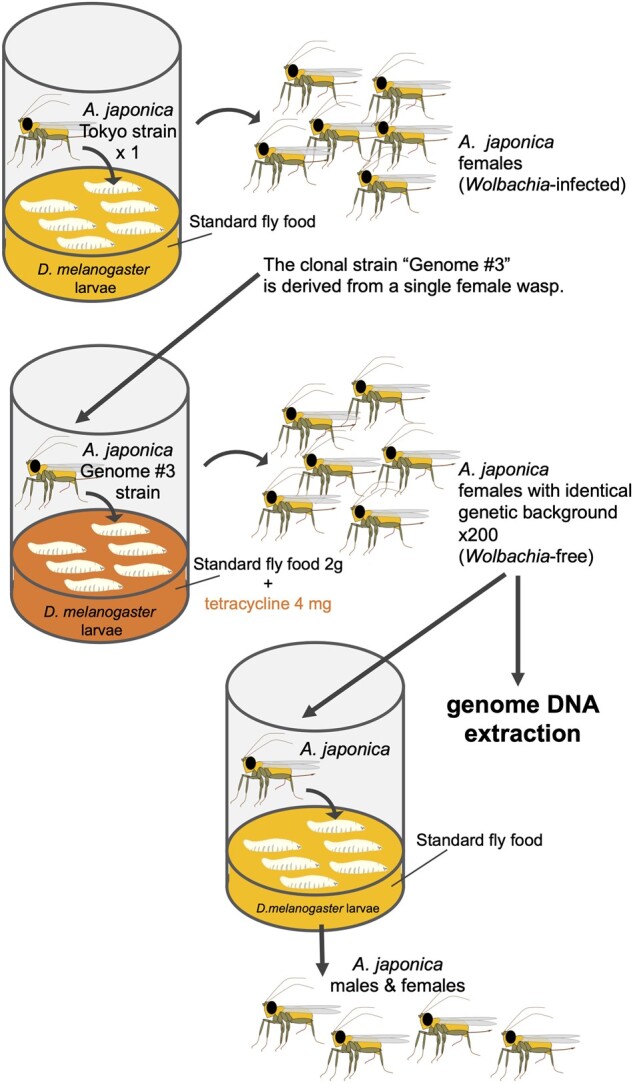
Generation of a clonal strain ‘Genome #3’ from a single female wasp. To obtain the high-quality homogenous genome sequence, we generated a clonal strain from a single female wasp derived from the Wolbachia-infected parthenogenetic strain ‘Tokyo’. After the clonal strain ‘Genome #3’ was established, we removed Wolbachia from these wasp clones by rearing host fly larvae on the standard food supplemented with tetracycline. 200 Wolbachia-free wasps were collected for genome DNA extraction. At the same time, we confirmed the removal of Wolbachia by the appearance of male wasp offspring in the next generation.
3.2 Genome assembly and assessment of genome completeness
We sequenced DNA extracted from adult female A. japonica wasps with an identical genetic background. PacBio Sequel of 27.7 Gb on 1,627,155 reads (average read length: 16 kb) and Illumina Hiseq2500 reads of 64.3 Gb on 257,165,308 read-pairs (read length: 250 bp, average insert size: 631 bp) were obtained (Table 2). From the short reads, the genome size was estimated to be 322 Mb.20,21 The heterozygosity was estimated to be 0.132%, indicating that a homogenous genome sequence was successfully obtained (Supplementary Fig. S2B). This value reflects the clonal population of Genome #3. Assembly using FALCON-UNZIP resulted in 601 primary contigs with a total length of 322.2 Mb and an N50 of 2.64 Mb (Table 2). This assembled size is highly consistent with the estimated genome size and comparable to that of other strains of A. japonica, which was recently reported to be 281–304 Mb (Kagoshima, asexual strain) and 280–284 Mb (Amami-Oshima, sexual strain).18 The total length of our WGS was 322.2 Mb, which is longer than that of previous study (270.9 Mb), indicating that our WGS covers broader, unspecified genomic regions than the previous study (Table 3). Moreover, the genome size is comparable to that of other Hymenopteran species (Table 3).
Table 2.
Determination of the WGS of A. japonica
| (1) PacBio Sequel | ||||
|---|---|---|---|---|
| Total number of bases (bp) | Total reads | Max read length (bp) | N50 (bp) | Average read length (bp) |
| 27,657,055,104 | 1,627,155 | 213,716 | 29,875 | 16,997 |
| (2) Illumina Hiseq2500 | |||
|---|---|---|---|
| Total number of bases (bp) | Total reads | Read length (bp) | Average insert size (bp) |
| 64,291,327,000 | 257,165,308 | 250 | 631 |
| (3) Summary statistics of the assembled genome | |||
|---|---|---|---|
| Type of assembled sequence | Primary | Alternative | mtDNA |
| Total number of bases (bp) | 322,176,672 | 71,031,528 | 19,152 |
| Number of scaffolds | 601 | 1,004 | 1 |
| Smallest scaffold (bp) | 8,253 | 4,233 | 19,152 |
| Longest scaffold (bp) | 7,850,940 | 790,158 | 19,152 |
| N50 (bp) | 2,638,563 | 76,239 | 19,152 |
| Average length (bp) | 536,067 | 70,748 | 19,152 |
N50 is the shortest contig length that needs to be included for covering 50% of the genome.
Assembly: Falcon (v0.7) + FalconUnzip (v0.4.0) error correction: Arrow (SMRT Link v6.0.0) + Pilon (v1.22).
Table 3.
Genome assembly values among Hymenopteran species
| (1) Comparison of statistics of genome | |||||||
|---|---|---|---|---|---|---|---|
| Total length (Mb) | Number of scaffolds | Average of scaffold (kb) | Longest scaffold (Mb) | N50 (Mb) | L50 | GAP (%) | |
| A. japonica (this study) | 322.2 | 601 | 536.1 | 7.9 | 2.6 | 38 | 0 |
| A. japonica 18 | 270.9 | 774 | 350 | 5.8 | 1.7 | 47 | 0 |
| A. mellifera a | 225.3 | 177 | 1,272.6 | 27.8 | 13.6 | 7 | 0.6 |
| O. abietinus | 201.2 | 936 | 215 | 6.44 | 2.4 | 28 | 7.3 |
| T. pretiosum | 195.1 | 357 | 546.5 | 8.8 | 3.7 | 19 | 7.7 |
| C. floridanum | 555.0 | 5,445 | 101.9 | 8.1 | 1.0 | 153 | 18.0 |
| N. vitripennis | 295.8 | 6,098 | 48.5 | 33.6 | 0.9 | 21 | 19.3 |
| C. solmsi | 277.1 | 2,457 | 112.8 | 27.4 | 9.6 | 10 | 0.5 |
| (2) Assessment of genome completeness (insecta_odb10; number of BUSCOs 1,353) | |||||
|---|---|---|---|---|---|
| Complete_all (%) | Complete_Single (%) | Complete_Double (%) | Fragment (%) | Missing (%) | |
| A. japonica (this study) | 98.8 | 98.0 | 0.8 | 0.2 | 1.0 |
| A. japonica 18 | 97.4 | 96.6 | 0.8 | 0.5 | 2.1 |
| A. mellifera a | 99.4 | 99.3 | 0.1 | 0 | 0.6 |
| O. abietinus | 98.6 | 98.2 | 0.4 | 0.4 | 1.0 |
| T. pretiosum | 98.0 | 94.7 | 3.3 | 0.2 | 1.8 |
| C. floridanum | 96.3 | 95.0 | 1.3 | 1.8 | 2.0 |
| N. vitripennis | 96.0 | 95.0 | 1.0 | 2.2 | 1.8 |
| C. solmsi | 98.6 | 98.2 | 0.4 | 0.2 | 1.2 |
| (3) Assessment of genome completeness (hymenoptera_odb10; number of BUSCOs 5,991) | |||||
|---|---|---|---|---|---|
| Complete_all (%) | Complete_Single (%) | Complete_Double (%) | Fragment (%) | Missing (%) | |
| A. japonica (this study) | 93.8 | 93.2 | 0.6 | 1.5 | 4.8 |
| A. japonica 18 | 92.5 | 91.9 | 0.7 | 1.8 | 5.7 |
| A. mellifera a | 97.7 | 97.6 | 0.1 | 0.3 | 2.0 |
| O. abietinus | 94.2 | 94.0 | 0.2 | 1.5 | 4.3 |
| T. pretiosum | 90.7 | 89.1 | 1.6 | 1.3 | 8.0 |
| C. floridanum | 85.7 | 84.7 | 1.0 | 4.4 | 9.9 |
| N. vitripennis | 89.7 | 89.0 | 0.7 | 3.2 | 7.1 |
| C. solmsi | 92.1 | 91.7 | 0.4 | 1.7 | 6.2 |
The Bold values were discriminated from the other values that have been already reported in other references.
Refseq is used for analyses. Refseq is a curated genome data by NCBI using genome data which is uploaded by users.
The completeness of the genomic assembly data was evaluated by detecting a set of unique single-copy genes in the primary sequence with Benchmarking Universal Single-Copy Orthologues (BUSCO, v5.2.2). Of the 1,353 BUSCOs in the insect dataset ‘insecta_odb10’, 1,337 (98.8%) complete BUSCOs were detected in the assembly (Table 3). The number of complete and single-copy BUSCOs was 1,326 (98.0%). In addition, we also performed BUSCO analysis using the Hymenoptera (‘hymenoptera_odb10’; 5,991 BUSCOs) dataset and identified 93.8% complete BUSCOs in our assembly (Table 3). This rate is sufficiently high and comparable to that of the previous study18 (92.5%) and other species, including A. mellifera (97.7%) and N. vitripennis (89.7%). In the family of Braconidae, complete BUSCOs were calculated in D. alloeum (95.3%), F. arisanus (94.5%), M. demolitor (94.4%), M. cingulum (93.5%), C. typhae (93.2%), C. glomerata (93.6%), C. insularis (93.5%), and A. gifuensis (91.3%) (Table 4). Taken together, these results indicate that the generated assembly was of high quality for gene annotation in A. japonica.
Table 4.
Genome assembly values among Blaconidae species
| (1) Statistics of genome | |||||||
|---|---|---|---|---|---|---|---|
| Total length (Mb) | Number of scaffolds | Average of scaffold (kb) | Longest scaffold (Mb) | N50 (Mb) | L50 | GAP content (%) | |
| A. japonica (this study) | 322.2 | 601 | 536.1 | 7.9 | 2.6 | 38 | 0 |
| A. japonica 18 | 270.9 | 774 | 350 | 5.8 | 1.7 | 47 | 0 |
| D. alloeum | 388.8 | 3,968 | 98.0 | 6.6 | 0.6 | 128 | 6.2 |
| F. arisanus | 153.6 | 1,042 | 147.4 | 5.5 | 1.0 | 49 | 8.2 |
| M. demolitor | 241.2 | 1,794 | 134.4 | 7.1 | 1.1 | 50 | 14.7 |
| M. cingulum | 132.4 | 5,696 | 23.2 | 1.4 | 0.2 | 179 | 3.0 |
| C. typhae | 186.7 | 72 | 2,592.5 | 20.0 | 6.8 | 8.0 | 0 |
| C. glomerata | 288.8 | 3,354 | 86.1 | 37.5 | 27.8 | 5.0 | 0 |
| C. insularis a | 135.7 | 455 | 298.3 | 4.7 | 1.2 | 33.0 | 0 |
| A. gifuensis | 156.9 | 24 | 6,539.4 | 32.1 | 27.5 | 3.0 | 0 |
| (2) Assessment of genome completeness (insecta_odb10; number of BUSCOs 1,353) | |||||
|---|---|---|---|---|---|
| Complete_all (%) | Complete_Single (%) | Complete_Double (%) | Fragment (%) | Missing (%) | |
| A. japonica (this study) | 98.8 | 98.0 | 0.8 | 0.2 | 1.0 |
| A. japonica 18 | 97.4 | 96.6 | 0.8 | 0.5 | 2.1 |
| D. alloeum | 99.0 | 98.6 | 0.4 | 0.6 | 0.4 |
| F. arisanus | 97.4 | 97.1 | 0.2 | 1.3 | 1.3 |
| M. demolitor | 99.3 | 98.3 | 1.0 | 0.3 | 0.4 |
| M. cingulum | 98.4 | 96.9 | 1.5 | 0.6 | 1.0 |
| C. typhae | 98.8 | 97.6 | 1.2 | 0.3 | 0.9 |
| C. glomerata | 99.5 | 97.7 | 1.8 | 0.1 | 0.4 |
| C. insularis a | 99 | 98.6 | 0.4 | 0.1 | 0.8 |
| A. gifuensis | 98.8 | 94.9 | 3.9 | 0.1 | 1.1 |
| (3) Assessment of genome completeness (hymenoptera_odb10; number of BUSCOs 5,991) | |||||
|---|---|---|---|---|---|
| Complete_all (%) | Complete_Single (%) | Complete_Double (%) | Fragment (%) | Missing (%) | |
| A. japonica (this study) | 93.8 | 93.2 | 0.6 | 1.5 | 4.8 |
| A. japonica 18 | 92.5 | 91.9 | 0.7 | 1.8 | 5.7 |
| D. alloeum | 95.3 | 94.8 | 0.5 | 1.8 | 2.9 |
| F. arisanus | 94.5 | 94.1 | 0.4 | 2.0 | 3.5 |
| M. demolitor | 94.4 | 93.7 | 0.7 | 1.6 | 4.1 |
| M. cingulum | 93.5 | 92.5 | 0.9 | 1.8 | 4.8 |
| C. typhae | 93.2 | 92.6 | 0.6 | 1.4 | 5.4 |
| C. glomerata | 93.6 | 92.9 | 0.7 | 1.4 | 5.0 |
| C. insularis a | 93.5 | 92.9 | 0.6 | 1.5 | 5.0 |
| A. gifuensis | 91.3 | 88.5 | 2.8 | 1.4 | 7.3 |
Refseq is used for analyses. Refseq is a curated genome data by NCBI using genome data which is uploaded by users.
3.3 Gene prediction, annotation, and phylogenetic tree analysis with hymenopteran species
Protein-coding gene prediction was conducted using an in-house annotation pipeline based on a combination of RNA-seq-based prediction, homology-based prediction, and ab initio prediction methods. The final gene model set consisted of 12,508 genes with a mean coding sequence (CDS) length of 1,594 bp (Table 5). The statistics of gene annotation data were comparable to other Hymenopteran species. Moreover, the completeness of the gene annotation data was evaluated by BUSCO in the insect dataset and the Hymenoptera dataset: 12,283 (98.2%) and 11,933 (95.4%) complete BUSCOs were detected in the annotated genes, respectively. We also compared these rates with other Braconidae species (Table 6). Overall, these results strongly support that our gene annotation data is reliable for the identification of genes in A. japonica.
Table 5.
Comparison of gene annotations between A. japonica and other Hymenopteran species
| (1) Statistics of gene annotation | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Number of genes | Number of exons per gene | Number of single exon gene | Total exon length (Mbp) | Mean exon length (bp) | Mean CDS length (bp) | Total intron length (Mbp) | Mean intron length (bp) | GT-AG splicing site (%) |
|
| A. japonica | 12,508 | 5.7 | 608 | 19.9 | 282.2 | 1,594.1 | 103.2 | 1,774.5 | 99.4 |
| A. mellifera a | 9,922 | 7.1 | 503 | 18.1 | 257.4 | 1,824.1 | 102.6 | 1,699.7 | 98.6 |
| O. abietinus | 9,996 | 7.0 | 533 | 18.1 | 260.0 | 1,810.2 | 77.0 | 1,290.9 | 98.6 |
| T. pretiosum | 12,422 | 5.8 | 1,238 | 21.0 | 290.5 | 1,689.5 | 67.9 | 1,134.6 | 98.3 |
| C. floridanum | 11,907 | 6.2 | 989 | 19.2 | 259.2 | 1,612.8 | 195.7 | 3,147.1 | 98.0 |
| N. vitripennis | 13,185 | 6.1 | 1,226 | 21.0 | 259.2 | 1,590.6 | 86.9 | 1,283.4 | 98.0 |
| C. solmsi | 9,823 | 6.9 | 601 | 16.5 | 244.9 | 1,683.8 | 106.0 | 1,837.2 | 96.5 |
| (2) Assessment of gene annotation completeness (insecta_odb10; number of BUSCOs 1,353) | |||||
|---|---|---|---|---|---|
| Complete_all (%) | Complete_Single (%) | Complete_Double (%) | Fragment (%) | Missing (%) | |
| A. japonica | 98.2 | 97.4 | 0.9 | 0.5 | 1.2 |
| A. mellifera a | 99.0 | 99.0 | 0.1 | 0.2 | 0.7 |
| O. abietinus | 98.5 | 98.0 | 0.5 | 0.3 | 1.2 |
| T. pretiosum | 98.6 | 95.5 | 3.1 | 0.1 | 1.3 |
| C. floridanum | 96.8 | 95.0 | 1.8 | 1.3 | 1.9 |
| N. vitripennis | 93.9 | 92.8 | 1.2 | 2.9 | 3.1 |
| C. solmsi | 98.0 | 97.6 | 0.4 | 0.7 | 1.3 |
| (3) Assessment of gene annotation completeness (hymenoptera_odb10; number of BUSCOs 5,991) | |||||
|---|---|---|---|---|---|
| Complete_all (%) | Complete_Single (%) | Complete_Double (%) | Fragment (%) | Missing (%) | |
| A. japonica | 95.4 | 94.6 | 0.8 | 1.0 | 3.6 |
| A. mellifera a | 98.8 | 98.6 | 0.2 | 0.3 | 0.9 |
| O. abietinus | 96.5 | 96.3 | 0.2 | 0.7 | 2.7 |
| T. pretiosum | 92.5 | 90.7 | 1.8 | 0.6 | 6.9 |
| C. floridanum | 89.5 | 88.0 | 1.5 | 3.6 | 6.9 |
| N. vitripennis | 88.9 | 88.0 | 0.9 | 3.5 | 7.6 |
| C. solmsi | 92.7 | 92.3 | 0.4 | 1.9 | 5.4 |
Refseq is used for analyses. Refseq is a curated genome data by NCBI using genome data which is uploaded by users.
Table 6.
Comparison of gene annotations between A. japonica and other Blaconidae species
| (1) Statistics of Gene annotation | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Number of genes | Number of exons per gene | Number of single exon gene | Total exon length (Mbp) | Mean exon length (bp) | Mean CDS length (bp) | Total intron length (Mbp) | Mean intron length (bp) | GT-AG splicing site (%) | |
| A. japonica | 12,508 | 5.7 | 608 | 19.9 | 282.2 | 1,594.1 | 103.2 | 1774.5 | 99.4 |
| D. alloeum | 12,837 | 4.8 | 1,759 | 21.1 | 338.7 | 1,640.8 | 124.4 | 2,520.0 | 98.8 |
| F. arisanus | 10,991 | 5.0 | 819 | 18.5 | 334.9 | 1,686.1 | 53.4 | 1,204.5 | 98.4 |
| M. demolitor | 12,144 | 5.3 | 1,339 | 20.4 | 318.9 | 1,676.5 | 79.1 | 1,529.7 | 98.2 |
| M. cingulum | 11,993 | 5.1 | 1,218 | 18.5 | 300.5 | 1,541.5 | 19.0 | 384.1 | 100.0 |
| C. typhae | 8,591 | 5.0 | 483 | 12.8 | 299.9 | 1,487.4 | 20.4 | 598.6 | 99.4 |
| C. glomerata | 19,218 | 4.2 | 820 | 22.8 | 284.4 | 1,187.8 | 45.6 | 746.7 | 99.7 |
| C. insularis a | 10,548 | 5.6 | 826 | 18.7 | 315.7 | 1,773.4 | 45.2 | 928.6 | 98.8 |
| A. gifuensis | 11,504 | 5.4 | 60 | 19.5 | 316.3 | 1,695.3 | 48.6 | 969.6 | 99.3 |
| (2) Assessment of gene annotation completeness (insecta_odb10; number of BUSCOs 1,353) | |||||
|---|---|---|---|---|---|
| Complete_all (%) | Complete_Single (%) | Complete_Double (%) | Fragment (%) | Missing (%) | |
| A. japonica | 98.2 | 97.4 | 0.9 | 0.5 | 1.2 |
| D. alloeum | 99.0 | 98.6 | 0.4 | 0.3 | 0.7 |
| F. arisanus | 97.5 | 97.4 | 0.1 | 1.0 | 1.5 |
| M. demolitor | 99.3 | 98.0 | 1.4 | 0.2 | 0.4 |
| M. cingulum | 88.5 | 86.8 | 1.7 | 1.2 | 10.2 |
| C. typhae | 70.7 | 69.7 | 1.0 | 6.2 | 23.0 |
| C. glomerata | 95.2 | 93.5 | 1.7 | 1.2 | 3.7 |
| C. insularis a | 99.1 | 98.5 | 0.7 | 0.1 | 0.7 |
| A. gifuensis | 94.6 | 90.4 | 4.2 | 0.4 | 5.0 |
| (3) Assessment of gene annotation completeness (hymenoptera_odb10; number of BUSCOs 5,991) | |||||
|---|---|---|---|---|---|
| Complete_all (%) | Complete_Single (%) | Complete_Double (%) | Fragment (%) | Missing (%) | |
| A. japonica | 95.4 | 94.6 | 0.8 | 1.0 | 3.6 |
| D. alloeum | 97.7 | 97.0 | 0.7 | 0.9 | 1.4 |
| F. arisanus | 95.9 | 95.5 | 0.4 | 1.8 | 2.4 |
| M. demolitor | 96.3 | 95.2 | 1.1 | 0.9 | 2.8 |
| M. cingulum | 84.8 | 84.1 | 0.8 | 2.8 | 12.3 |
| C. typhae | 59.2 | 58.8 | 0.4 | 6.7 | 34.1 |
| C. glomerata | 86.4 | 85.5 | 0.8 | 4.6 | 9.0 |
| C. insularis a | 96.1 | 95.2 | 0.9 | 0.3 | 3.6 |
| A. gifuensis | 88.1 | 85.0 | 3.1 | 0.7 | 11.3 |
Refseq is used for analyses. Refseq is a curated genome data by NCBI using genome data which is uploaded by users.
Single-copy orthologous genes were determined from 10 species (A. japonica, D. alloeum, F. arisanus, M. demolitor, M. cinglum, C. typhae, C. glomerata, C. insularis, A. gifuensis, and A. mellifera) and 3,263 groups were extracted with a one-to-one relationship across all species. For each group, multiple alignments were performed using the MAFFT.50 A phylogenetic tree was constructed using RAxML (version 8.2.12).51 The orthologues of A. mellifera were used as the outer group. A total of 1,000 replicates were used for bootstrap analysis (Fig. 2). Our results are consistent with the phylogeny based on mitochondrial DNA, as previously reported.60,61
Figure 2.

Phylogenetic analysis of A. japonica and other Hymenopteran species. (A) The phylogenetic tree was generated with 2,381 pairs of single-copy orthologues between A. japonica and 14 related Hymenoptera species. (B) The phylogenetic tree was generated with 3,263 pairs of single-copy orthologues between A. japonica and 8 related species in the family Braconidae. Apis mellifera was used as an out group. All nodes have 100% bootstrap support after 1,000 replications.
3.4 RNAi technique protocol with A. japonica
To the best of our knowledge, there have been no previous studies on genetic manipulation in A. japonica. To perform functional analyses of A. japonica genes, we devised a protocol for dsRNA microinjection-based RNAi. This procedure is a post-transcriptional gene silencing mechanism that applies exogenous dsRNA to wasp bodies. dsRNAs are cleaved into small interfering RNA fragments that bind to complementary endogenous mRNAs. Target mRNAs result in degradation such that the corresponding gene products are decreased. The effects of RNAi have already been reported in various Hymenoptera species, including the ectoparasitoid wasp N.vitripennis,62,63 the endoparasitoid wasp Microplitis,64,65 and L. boulardi.66 Therefore, we reasoned that these methods could be applied to experiments with A. japonica.
As in L. boulardi, A. japonica larvae undergo metamorphosis inside the host puparium, which makes it difficult to conduct dsRNA microinjection experiments. To examine whether the development of A. japonica progresses outside the host puparium, we set up a bulk infection arena to synchronize the timing of oviposition (Fig. 3A). In addition, third-instar (L3) larvae (at 96 h after egg-laying) were used as hosts. At a certain time point after infection, we carefully removed the host pupal case with forceps and observed wasp development on the 2% agar plate (Fig. 3B). When the wasp larvae were removed from the host pupal case at 4 dpi, development proceeded on the agar plate and wasp adults eclosed normally. During this time, the wasp larva completely consumed the fly host body. We recommend that the appropriate timing for dsRNA injection is after 4 dpi.
For the RNAi experiment, we chose the ebony gene to be the target of RNAi for the first trial. Ebony encodes the enzyme NBAD synthetase, which converts dopamine to NBAD.52 Loss of this enzymatic function increases dark melanin and decreases tan pigment, resulting in a dark body colour phenotype. We identified gene003270 (ebony-Aj) as an orthologue of the D. melanogaster ebony (ebony-Dm) gene from the annotated A. japonica gene set using BLAST.
It appeared that eye and body colour pigmentation started from 7 to 9 dpi (Fig. 3B). We removed the pupal case and injected dsRNA into A. japonica at 6 dpi, prior to body colour pigmentation. Therefore, the body colour of ebony-Aj dsRNA-injected wasps became darker than that of the control GFP dsRNA-injected wasp (Fig.4A and B and Supplementary Fig. S3). To quantitatively evaluate the body colour, we calculated the redness index57 from the captured images (Fig. 4C). In our analysis, the redness index was significantly decreased in ebony-Aj dsRNA-injected wasps, which is similar to the body colour difference between Drosophila wild-type and ebony mutant animals (Supplementary Fig. S4). From this result, we concluded that the function of Ebony was successfully silenced in A. japonica using RNAi.
Figure 4.

RNAi effects of RNAi wasps were evaluated by phenotype analysis and qPCR. (A) Representative images of wasp adults being injected with GFP dsRNA as a control. A square indicates the region of interest (ROI) for RGB value measurements. (B) Representative images of wasp adults being injected with ebony-Aj dsRNA as ebony RNAi. (C) Quantitative evaluation of wasp body colour. The Y-axis indicates the redness of the ROI. ***P < 0.005 from Student’s t-test. n = 18 (GFP dsRNA), 21 (ebony-Aj dsRNA). (D, E) The relative expression levels of gene003054 (D) and gene010975 (E) in GFP-RNAi or gene003054-RNAi wasps were quantified using the delta-delta Ct method. The expression of RpL32-Aj was used to normalize the values. All values represent the means ± SD with all data points (n = 3). *P < 0.05, **P < 0.01, and ***P < 0.005 from Student’s t-test. n.s., non-significant (P > 0.05).
3.5 Knock down of the putative venom gene in A. japonica
After confirming the RNAi effect by ebony-Aj-dsRNA microinjection, we applied this method to knock down the putative venom gene in A. japonica. Our RNA-seq analysis indicates that a group of genes are strongly expressed in the venom gland, which is likely to be involved in venom production. We listed these putative venom genes in descending order of FPKM values and examined the expression of the top five genes (gene006533; gene010987; gene010975; gene003054; gene000789) by qRT-PCR (Table 7). Consistent with RNA-seq data, these genes were exclusively expressed in the venom gland compared with other tissues. These gene products showed little homology signatures against NCBI database and few functional domains against Pfam and SMART, suggesting that they are specific to A. japonica. Among them, gene003054 encodes a putative transmembrane protein, and we chose it as a target of RNAi.
dsRNA microinjection was performed with wasp larvae at 6 dpi. GFP-dsRNA was used as the negative control. To determine the appropriate concentration of dsRNA, we used 1,000, 500, and 250 ng/μl dsRNA-containing ultrapure water for microinjection. When dsRNA-injected wasps were eclosed, total RNA was extracted from these wasps, and the relative expression level of each gene was measured by qRT-PCR. In sharp contrast to GFP-dsRNA injection, the expression levels of the target gene003054 were drastically reduced in all three concentrations of dsRNA-injected wasps (Fig. 4D). To test the specificity of RNAi, we also examined the expression of gene010975 (Fig. 4E). The expression did not decrease in 1,000, 500, and 250 ng/µl dsRNA injection in gene003054–RNAi wasps. These results suggest that dsRNA injection-based RNAi specifically suppressed the expression of the target gene (Fig. 4D and E). In addition, 250 ng/μl of dsRNA was sufficient for effective RNAi against putative venom genes. After eclosion, the dsRNA-injected wasps were viable but less tolerant to the behavioural assay. It is possible that microinjection or rearing conditions may affect wasps with a weak constitution. Taken together, we confirmed that the RNAi method provides an efficient way to silence putative venom genes in A. japonica.
4. Conclusions
Here, we performed WGS analysis of A. japonica, annotating a set of 12,508 genes. These genes include highly conserved sets of genes in Hymenoptera, indicating the high reliability of genome data information. The qualified WGS platform provides us with the references needed to identify venom components for parasitizing Drosophilidae and the diversity of species during coevolution. Considering the virulence effects of venoms and the specificity of host insects, including D. suzukii, venom genes and their active products are crucial for developing insecticides in agricultural sciences. Moreover, we succeeded in devising a protocol for RNAi techniques using A. japonica. Given the genetic tools available in the host fly D. melanogaster, the development of genetic tools in the parasitoid wasp A. japonica enables a bidirectional approach to examine host-parasite interactions. Based on the WGS and functional analyses, we confirmed that it is possible to explore these molecular mechanisms in non-model organisms.
Supplementary data
Supplementary data are available at DNARES online.
Author contributions
T.Ka., M.K., H.M., T.Ku., A.K., and Y.S.-N. conducted the experiments. A.T., H.T., T.I., and T.Ka. performed WGS and RNA-seq analyses. T.Ka., Y.S.-N, H.T., and R.N. wrote the article with feedback from all the co-authors.
Supplementary Material
Acknowledgements
We thank Kazuo Takahashi, Shunsuke Furihata, Masato Kimura, Manabu Kamimura, Takaaki Daimon, Ryo Futahashi, and Genta Okude for providing reagents and critical comments. We also thank Hitomi Takemata, Reiko Kise, Mitsuki Fujii, Kazuki Seike, and Masako Iida for their technical assistance. We are grateful to all members of the Niwa laboratory for their insightful discussions and comments on the article. For WGS analysis, sequencing data were obtained with the support of the Platform for Advanced Genome Science. T.Ka. is a recipient of a fellowship from the Japan Society for the Promotion of Science.
Funding
This work was supported by JSPS KAKENHI Grant Number 16H06279 (PAGS), 16K20945 (Y.S.-N.), 18K05670 (Y.S.-N), 21J10894 (T.Ka.), and the Japan Science and Technology Agency (JST)/PRESTO .
Accession numbers
The raw sequence data generated on the Illumina and PacBio platforms were deposited in the DNA Databank of Japan (DDBJ, BioProject ID: PRJDB12783). The accession numbers are SAMD00434239-00434241, SAMD00436468, DRA013279. The accession numbers of genome assembled scaffold data are BQMI01000001-BQMI01000601.
Conflict of inteest
None declared.
Contributor Information
Takumi Kamiyama, Graduate School of Life and Environmental Sciences, University of Tsukuba, Tsukuba 305-8577, Japan; Life Science Center for Survival Dynamics, Tsukuba Advanced Research Alliance (TARA), University of Tsukuba, Tsukuba 305-8577, Japan.
Yuko Shimada-Niwa, Life Science Center for Survival Dynamics, Tsukuba Advanced Research Alliance (TARA), University of Tsukuba, Tsukuba 305-8577, Japan; Precursory Research for Embryonic Science and Technology (PREST), Japan Science and Technology Agency (JST), Tokyo 102-0076, Japan.
Hiroyuki Tanaka, Department of Biological Information, Tokyo Institute of Technology, Meguro, Tokyo 152-8550, Japan.
Minami Katayama, Graduate School of Life and Environmental Sciences, University of Tsukuba, Tsukuba 305-8577, Japan.
Takayoshi Kuwabara, College of Biological Sciences, University of Tsukuba, Tsukuba 305-8577, Japan.
Hitoha Mori, College of Biological Sciences, University of Tsukuba, Tsukuba 305-8577, Japan.
Akari Kunihisa, College of Biological Sciences, University of Tsukuba, Tsukuba 305-8577, Japan.
Takehiko Itoh, Department of Biological Information, Tokyo Institute of Technology, Meguro, Tokyo 152-8550, Japan.
Atsushi Toyoda, Comparative Genomics Laboratory, National Institute of Genetics, Mishima, Shizuoka 411-8540, Japan.
Ryusuke Niwa, Life Science Center for Survival Dynamics, Tsukuba Advanced Research Alliance (TARA), University of Tsukuba, Tsukuba 305-8577, Japan.
References
- 1. Pennacchio F., Strand M.R.. 2006, Evolution of developmental strategies in parasitic hymenoptera, Annu. Rev. Entomol., 51, 233–58. [DOI] [PubMed] [Google Scholar]
- 2. Quicke D.L. (ed.) 1997, Parasitic Wasps. Springer, Germany, 221–55. [Google Scholar]
- 3. Quicke D.L. 2015, The Braconid and Ichneumonid Parasitoid Wasps. Wiley-Blackwell, USA. [Google Scholar]
- 4. Moreau S., Asgari S.. 2015, Venom proteins from parasitoid wasps and their biological functions, Toxins (Basel), 7, 2385–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yang L., Qiu L.M., Fang Q., Stanley D.W., Ye G.Y.. 2021, Cellular and humoral immune interactions between Drosophila and its parasitoids, Insect Sci., 28, 1208–27. [DOI] [PubMed] [Google Scholar]
- 6. Leitão A.B., Arunkumar R., Day J.P., et al. 2020, Constitutive activation of cellular immunity underlies the evolution of resistance to infection in Drosophila, Elife, 9, e59095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Basden E. 1972, The Hymenopterous Parasites of the Drosophilidae. Drosoph. Inf. Serv., 48, 70–72.
- 8. Carton Y., Bouletreau M.V., Lenteren J.V., Alphen J.. 1986, The Drosophila parasitic wasps. In: Ashburner M., Carson H., Thompson J., eds. The Genetics and Biology of Drosophila, Vol. 3, pp. 347–394. New York: Academic Press. [Google Scholar]
- 9. Schlenke T. A., Morales J., Govind S., Clark A.G.. 2007, Contrasting infection strategies in generalist and specialist wasp parasitoids of Drosophila melanogaster. PLoS Pathog., 3, 1486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Huang J., Chen J., Fang G., et al. 2021, Two novel venom proteins underlie divergent parasitic strategies between a generalist and a specialist parasite, Nat. Commun., 12, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mitsui H., Achterberg K., Van Nordlander G., Kimura M.T.. 2007, Geographical distributions and host associations of larval parasitoids of frugivorous Drosophilidae in Japan, J. Nat. Hist., 41, 1731–8. [Google Scholar]
- 12. Ideo S., Watada M., Mitsui H., Kimura M.T.. 2008, Host range of Asobara japonica (Hymenoptera: Braconidae), a larval parasitoid of Drosophilid flies, Entomol. Sci., 11, 1–6. [Google Scholar]
- 13. Furihata S.X., Kimura M.T.. 2009, Effects of Asobara japonica venom on larval survival of host and nonhost Drosophila species, Physiol. Entomol., 34, 292–5. [Google Scholar]
- 14. Mabiala-Moundoungou A.D.N., Doury G., Eslin P., Cherqui A., Prévost G.. 2010, Deadly venom of Asobara japonica parasitoid needs ovarian antidote to regulate host physiology, J. Insect Physiol., 56, 35–41. [DOI] [PubMed] [Google Scholar]
- 15. LaSalle J., Gauld I.D.. 1993, Hymenoptera: their biodiversity, and their impact on the diversity of other organisms. In: LaSalle, J. and Gauld, I.D., eds. Hymenoptera and Biodiversity, pp. 1–26. CAB International, Wallingford (UK). [Google Scholar]
- 16. Wang X., Biondi A., Daane K.M.. 2020, Functional responses of three candidate Asian larval parasitoids evaluated for classical biological control of Drosophila suzukii (Diptera: Drosophilidae), J. Econ. Entomol., 113, 73–80. [DOI] [PubMed] [Google Scholar]
- 17. Girod P., Lierhmann O., Urvois T., Turlings T.C.J., Kenis M., Haye T.. 2018, Host specificity of Asian parasitoids for potential classical biological control of Drosophila suzukii, J. Pest Sci., 91, 1241–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ma W.‐J., Pannebakker B.A., Li X., et al. 2021, A single QTL with large effect is associated with female functional virginity in an asexual parasitoid wasp, Mol. Ecol., 30, 1979–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Amano S., Akada T., Takahashi K.H.. 2019, In vivo and in vitro developmental profiling of Asobara japonica, a larval endoparasitoid of drosophilid flies, Entomol. Exp. Appl., 167, 442–56. [Google Scholar]
- 20. Vurture G.W., Sedlazeck F.J., Nattestad M., et al. 2017, GenomeScope: fast reference-free genome profiling from short reads, Bioinformatics, 33, 2202–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ranallo-Benavidez T.R., Jaron K.S., Schatz M.C.. 2020, GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes, Nat. Commun., 11, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chin C.S., Peluso P., Sedlazeck F.J., et al. 2016, Phased diploid genome assembly with single-molecule real-time sequencing, Nat. Methods, 13, 1050–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chin C.S., Alexander D.H., Marks P., et al. 2013, Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data, Nat. Methods, 10, 563–9. [DOI] [PubMed] [Google Scholar]
- 24. Walker B.J., Abeel T., Shea T., et al. 2014, Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement, PLoS One, 9, e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Manni M., Berkeley M.R., Seppey M., Simão F.A., Zdobnov E.M.. 2021, BUSCO Update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes, Mol. Biol. Evol., 38, 4647–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wallberg A., Bunikis I., Pettersson O.V., et al. 2019, A hybrid de novo genome assembly of the honeybee, Apis mellifera, with chromosome-length scaffolds, BMC Genomics, 20, 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Misof B., Liu S., Meusemann K., et al. 2014, Phylogenomics resolves the timing and pattern of insect evolution, Science, 346, 763–7. [DOI] [PubMed] [Google Scholar]
- 28. Lindsey A.R.I., Kelkar Y.D., Wu X., et al. 2018, Comparative genomics of the miniature wasp and pest control agent Trichogramma pretiosum, BMC Biol., 16, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bonasio R., Zhang G., Ye C., et al. 2010, Genomic comparison of the ants Camponotus floridanus and Harpegnathos saltator, Science, 329, 1068–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Werren J.H., Richards S., Desjardins C.A., Nasonia Genome Working Group., et al. 2010, Functional and evolutionary insights from the genomes of three parasitoid Nasonia species, Science, 327, 343–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xiao J.H., Yue Z., Jia L.Y., et al. 2013, Obligate mutualism within a host drives the extreme specialization of a fig wasp genome, Genome Biol., 14, R141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tvedte E.S., Walden K.K.O., McElroy K.E., et al. 2019, Genome of the parasitoid wasp Diachasma alloeum, an emerging model for ecological speciation and transitions to asexual reproduction, Genome Biol. Evol., 11, 2767–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Geib S.M., Liang G.H., Murphy T.D., Sim S.B.. 2017, Whole genome sequencing of the braconid parasitoid wasp Fopius arisanus, an important biocontrol agent of pest Tepritid fruit flies, G3 Genes|Genomes|Genetics, 7, 2407–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Burke G.R., Walden K.K.O., Whitfield J.B., Robertson H.M., Strand M.R.. 2014, Widespread genome reorganization of an obligate virus mutualist, PLoS Genet., 10, e1004660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yin C., Li M., Hu J., et al. 2018, The genomic features of parasitism, Polyembryony and immune evasion in the endoparasitic wasp Macrocentrus cingulum, BMC Genomics, 19, 420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Muller H., Chebbi M.A., Bouzar C., et al. 2021, Genome-wide patterns of bracovirus chromosomal integration into multiple host tissues during parasitism, J. Virol., 95, e0068421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pinto B.J., Weis J.J., Gamble T., Ode P.J., Paul R., Zaspel J.M.. 2021, A chromosome-level genome assembly of the parasitoid wasp, Cotesia glomerata (Hymenoptera: Braconidae), J. Hered., 112, 558–64. [DOI] [PubMed] [Google Scholar]
- 38. Feng Z., Wu Y., Yang C., et al. 2020, Evolution of tRNA gene rearrangement in the mitochondrial genome of ichneumonoid wasps (Hymenoptera: Ichneumonoidea), Int. J. Biol. Macromol., 164, 540–7. [DOI] [PubMed] [Google Scholar]
- 39. Kim D., Langmead B., Salzberg S.L.. 2015, HISAT: a fast spliced aligner with low memory requirements, Nat. Methods, 12, 357–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pertea M., Pertea G.M., Antonescu C.M., Chang T.C., Mendell J.T., Salzberg S.L.. 2015, StringTie enables improved reconstruction of a transcriptome from RNA-seq reads, Nat. Biotechnol., 33, 290–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Grabherr M.G., Haas B.J., Yassour M., et al. 2011, Full-length transcriptome assembly from RNA-Seq data without a reference genome, Nat. Biotechnol., 29, 644–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schulz M.H., Zerbino D.R., Vingron M., Birney E.. 2012, Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels, Bioinformatics, 28, 1086–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wu T.D., Watanabe C.K.. 2005, GMAP: a genomic mapping and alignment program for mRNA and EST sequences, Bioinformatics, 21, 1859–75. [DOI] [PubMed] [Google Scholar]
- 44. Iwata H., Gotoh O.. 2012, Benchmarking spliced alignment programs including Spaln2, an extended version of Spaln that incorporates additional species-specific features, Nucleic Acids Res., 40, e161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Stanke M., Keller O., Gunduz I., Hayes A., Waack S., Morgenstern B.. 2006, AUGUSTUS: ab initio prediction of alternative transcripts, Nucleic Acids Res., 34, W435–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Korf I. 2004, Gene finding in novel genomes, BMC Bioinformatics, 5, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nielsen C.H.H., Engelbrecht J., Brunak S., von Heijne G.. 1997, A neural network method for identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites, Int. J. Neural Syst., 8, 581–99. [DOI] [PubMed] [Google Scholar]
- 48. Almagro Armenteros J.J., Tsirigos K.D., Sønderby C.K., et al. 2019, SignalP 5.0 improves signal peptide predictions using deep neural networks, Nat. Biotechnol., 37, 420–3. [DOI] [PubMed] [Google Scholar]
- 49. Lechner M., Findeiß S., Steiner L., Marz M., Stadler P.F., Prohaska S.J.. 2011, Proteinortho: detection of (co-)orthologs in large-scale analysis, BMC Bioinformatics, 12, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Katoh K., Misawa K., Kuma K.I., Miyata T.. 2002, MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform, Nucleic Acids Res., 30, 3059–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Stamatakis A. 2014, RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies, Bioinformatics, 30, 1312–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hovemann B.T., Ryseck R.P., Walldorf U., Störtkuhl K.F., Dietzel I.D., Dessen E.. 1998, The Drosophila ebony gene is closely related to microbial peptide synthetases and shows specific cuticle and nervous system expression, Gene, 221, 1–9. [DOI] [PubMed] [Google Scholar]
- 53. Bolger A.M., Lohse M., Usadel B.. 2014, Trimmomatic: a flexible trimmer for Illumina sequence data, Bioinformatics, 30, 2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li H., Handsaker B., Wysoker A.. et al. ; 1000 Genome Project Data Processing Subgroup. 2009, The Sequence Alignment/Map format and SAMtools, Bioinformatics, 25, 2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pertea M., Kim D., Pertea G.M., Leek J.T., Salzberg S.L.. 2016, Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown, Nat. Protoc., 11, 1650–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Untergasser A., Nijveen H., Rao X., Bisseling T., Geurts R., Leunissen J.A.M.. 2007, Primer3Plus, an enhanced web interface to Primer3, Nucleic Acids Res., 35, W71–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Anbutsu H., Moriyama M., Nikoh N., et al. 2017, Small genome symbiont underlies cuticle hardness in beetles, Proc. Natl. Acad. Sci. U. S. A., 114, E8382–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dedeine F., Vavre F., Fleury F., Loppin B., Hochberg M.E., Boulétreau M.. 2001, Removing symbiotic Wolbachia bacteria specifically inhibits oogenesis in a parasitic wasp, Proc. Natl. Acad. Sci. U. S. A., 98, 6247–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Furihata S., Hirata M., Matsumoto H., Hayakawa Y.. 2015, Bacteria endosymbiont, wolbachia, promotes parasitism of parasitoid wasp Asobara japonica, PLoS One, 10, e0140914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Li Q., Wei S.J., Tang P., et al. 2016, Multiple lines of evidence from mitochondrial genomes resolve phylogenetic relationships of parasitic wasps in Braconidae, Genome Biol. Evol., 8, 2651–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wei S., jun Shi M., Sharkey M.J., van Achterberg C., Chen X. x.. 2010, Comparative mitogenomics of Braconidae (Insecta: Hymenoptera) and the phylogenetic utility of mitochondrial genomes with special reference to Holometabolous insects, BMC Genomics, 11, 371–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lynch J.A., Desplan C.. 2006, A method for parental RNA interference in the wasp Nasonia vitripennis, Nat. Protoc., 1, 486–94. [DOI] [PubMed] [Google Scholar]
- 63. Werren J.H., Loehlin D.W.. 2009, Larval RNAi in Nasonia (Parasitoid Wasp), Cold Spring Harb. Protoc., 10, pdb.prot5311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Li K.-M., Ren L.-Y., Zhang Y.-J., Wu K.-M., Guo Y.-Y.. 2012, Knockdown of microplitis mediator odorant receptor involved in the sensitive detection of two chemicals, J. Chem. Ecol., 383, 287–94. [DOI] [PubMed] [Google Scholar]
- 65. Burke G.R., Thomas S.A., Eum J.H., Strand M.R.. 2013, Mutualistic polydnaviruses share essential replication gene functions with pathogenic ancestors, PLoS Pathog., 9, e1003348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Colinet D., Kremmer L., Gatti J.-L., et al. 2014, Development of RNAi in a Drosophila endoparasitoid wasp and demonstration of its efficiency in impairing venom protein production, J. Insect Physiol., 63, 56–61. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.