

Abstract
Angiotensin-converting enzyme (ACE) regulates blood pressure by cleaving angiotensin I to produce angiotensin II. In the brain, ACE is especially abundant in striatal tissue, but the function of ACE in striatal circuits remains poorly understood. We find that ACE degrades an unconventional enkephalin heptapeptide, Met-enkephalin-Arg-Phe, in the nucleus accumbens of mice. ACE inhibition enhanced mu opioid receptor activation by Met-enkephalin-Arg-Phe, causing a cell type-specific long-term depression of glutamate release onto medium spiny projection neurons expressing the Drd1 dopamine receptor. Systemic ACE inhibition was not intrinsically rewarding, but decreased the conditioned place preference caused by fentanyl administration, and enhanced reciprocal social interaction. Our results raise the enticing prospect that central ACE inhibition can boost endogenous opioid signaling for clinical benefit, while mitigating risk of addiction.
One-Sentence Summary:
A novel pathway controls signaling via the brain’s own opioids and can be targeted to fine-tune neural circuit function.
As neural circuit dysfunction in brain disorders becomes increasingly well-defined, there is a growing need for interventions that specifically target dysfunctional circuit elements (1). Multiple brain disorders (2–4) involve imbalanced output of nucleus accumbens (NAc) medium spiny projection neurons expressing dopamine receptor Drd1 (D1-MSNs) or Drd2 (D2-MSNs). This imbalance has proven difficult to correct with standard interventions because these two MSN subtypes are physically intermingled, receive synaptic inputs from common sources, and have similar molecular profiles. A rare exception is ACE, which exhibits enriched expression by D1-MSNs in the dorsal striatum (5, 6) and the NAc (table S1; fig. S1). Inhibitors of ACE and other peptidases can be combined to regulate striatal excitatory synaptic transmission in an opioid-dependent fashion (7), suggesting that in addition to angiotensin conversion, ACE cleaves and degrades a peptide ligand for opioid receptors (Fig. 1A).
Fig. 1. ACE inhibition reduces excitatory input to D1-MSNs via endogenous opioid signaling.
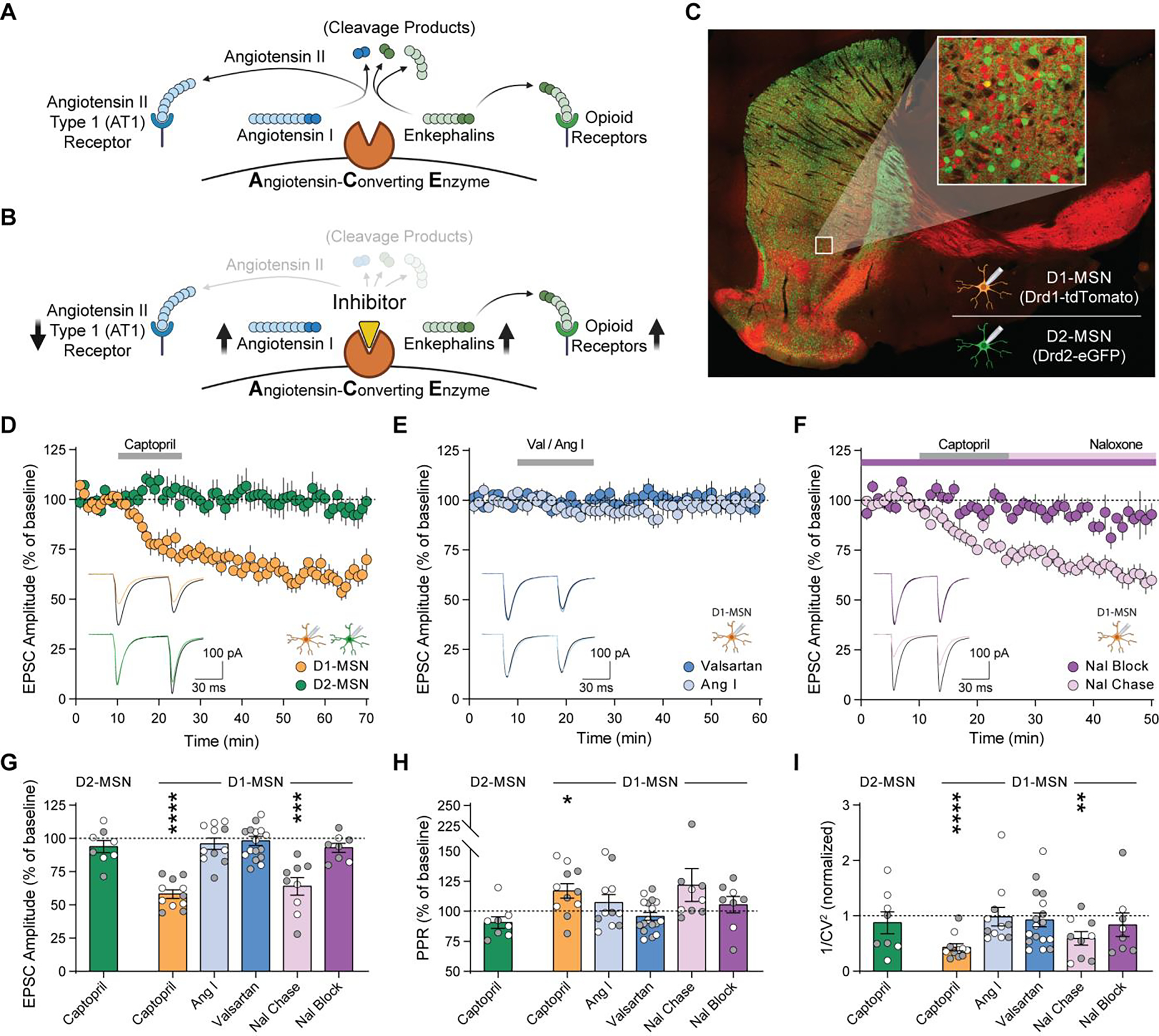
(A, B) Schematic of angiotensin and enkephalin regulation by ACE, in the absence (A) and presence (B) of ACE inhibition. (C) Drd1-tdTomato expression (red) in D1-MSNs, and Drd2-eGFP expression (green) in D2-MSNs. (D-F) EPSC amplitude before, during, and after 15 min bath perfusion (grey bar) of 10 μM captopril in D1-MSNs (orange, n=11) or D2-MSNs (green, n=8) (D); AT1R antagonist valsartan (dark blue, 2 μM n=8 and 20 μM n=9) or angiotensin I peptide (1 μM, light blue, n=11) in D1-MSNs (E); or captopril (10 μM) in continual presence of opioid receptor antagonist naloxone (10 μM, dark purple, n=8) or chased by naloxone (10 μM, light purple, n=9) in D1-MSNs (F). Bottom-left insets show traces before (black lines) and after (last 5 min of recording, colored lines). (G-I) EPSC parameters during the last 5 min of each recording, expressed as percentage of baseline prior to drug application: EPSC amplitude (G), paired-pulse ratio (H), and 1/CV2 (I). Data are mean ± s.e.m. for all panels; open and closed circles indicate recordings from female and male mice, respectively. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, ANOVA followed by one-sample t-test versus baseline; see Data S1 for complete statistics.
To separately measure how ACE inhibition affects excitatory synaptic transmission onto D1-MSNs and D2-MSNs, we performed whole-cell recordings in acute NAc brain slices from double-transgenic Drd1-tdTomato/Drd2-eGFP reporter mice (Fig. 1C, fig. S2). Brief exposure to captopril (10 μM), a prototypical ACE inhibitor (8), caused long-term depression (captopril-LTD) of excitatory synaptic transmission onto D1-MSNs (Fig. 1D). Captopril did not alter excitatory synaptic transmission onto D2-MSNs, which express ACE at a lower level than D1-MSNs (table S1; fig. S1). There was also no effect of captopril at excitatory synapses onto layer V pyramidal neurons in the anterior cingulate cortex (fig. S3), where ACE expression is low (5, 6).
Captopril and other ACE inhibitors canonically block conversion of angiotensin I to angiotensin II, preventing activation of the angiotensin II type 1 receptor (AT1R) and increasing levels of angiotensin I (Fig. 1B). However, LTD was not observed in D1-MSNs exposed to valsartan (2–20 μM), an AT1R antagonist, or exogenous angiotensin I peptide (1 μM) (Fig. 1E). In contrast, captopril-LTD in D1-MSNs was blocked in the continuous presence of naloxone (10 μM), an opioid receptor antagonist, but was not reversed by chasing captopril with naloxone (Fig. 1F). Captopril-LTD in D1-MSNs was associated with an increase in paired-pulse ratio and a decrease in 1/CV2 (Fig. 1, G to I), two changes that indicate decreased presynaptic probability of glutamate release, likely due to activation of presynaptic opioid receptors (7).
Local release of enkephalin peptides by D2-MSNs can regulate excitatory synaptic input to D1-MSNs (9). ACE can cleave enkephalin peptides, but is not principally responsible for degrading conventional Met-enkephalin or Leu-enkephalin in brain tissue (10). The proenkephalin gene (Penk) also encodes Met-enkephalin-Arg-Phe (MERF), a heptapeptide abundant in the NAc (11). MERF has high binding affinity for opioid receptors (12), is more potent than Met-enkephalin (13), and can be degraded by ACE (14). Using liquid chromatography-tandem mass spectrometry, we simultaneously quantified extracellular levels of enkephalins and other neuropeptides (15) released from mouse brain slices (Fig. 2A and fig. S4). After stimulation with KCl (50 mM), we observed increased extracellular levels of MERF as well as Met-enkephalin and Leu-enkephalin, along with dynorphins and substance P (Fig. 2B). Concentrations of MERF released from isolated NAc tissue punches were higher than dorsal striatum tissue punches (fig. S5, A to D). We could not detect appreciable release of angiotensin II (Fig. 2B) or bradykinin (fig. S5E). Enkephalin signals were absent in constitutive Penk knockout mice (fig. S5, F to K), indicating Penk is the primary source of enkephalin in this preparation.
Fig. 2. ACE selectively degrades MERF in the extracellular space.
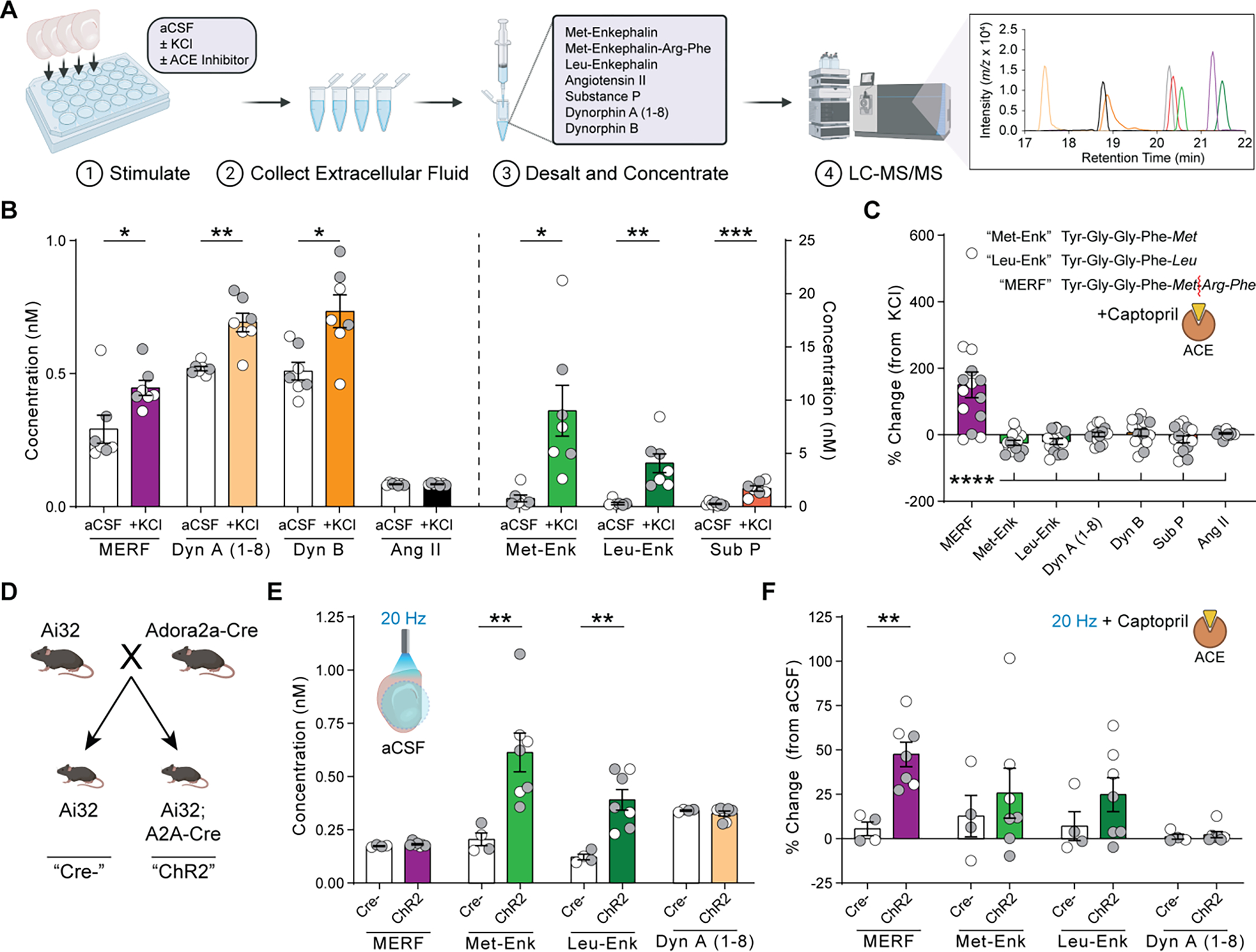
(A) Quantification of neuropeptide release from brain slices using LC-MS/MS. (B) Extracellular neuropeptide levels from slices submerged in normal aCSF or 50 mM KCl. (C) Percent change in extracellular neuropeptide levels after KCl stimulation in presence versus absence of captopril (10 μM). Inset shows enkephalin amino acid sequences and site of enzymatic cleavage of MERF by ACE (red line). (D) Breeding strategy to generate mice expressing channelrhodopsin-2 in D2-MSNs. (E) Extracellular neuropeptide levels from slices following optogenetic stimulation at 20 Hz. (F) Percent change in extracellular neuropeptide levels after optogenetic stimulation in presence versus absence of captopril (10 μM). Data are mean ± s.e.m. for all panels; open and closed circles indicate samples from female and male mice, respectively. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, ANOVA followed by simple effect test (B, E, F) or Fisher’s LSD post-hoc test (C); see Data S1 for complete statistics.
Inhibition of ACE with captopril (10 μM) robustly increased extracellular levels of MERF without affecting conventional enkephalins or other neuropeptides (Fig. 2C; table S2). We observed similar effects using trandolaprilat, a different ACE inhibitor that also caused LTD of excitatory synaptic input to D1-MSNs (fig. S6). In contrast, extracellular levels of MERF were not affected by pharmacological inhibition of aminopeptidase N and neprilysin, the enzymes responsible for degrading conventional enkephalins (10) (fig. S7A). A cocktail of inhibitors for all three enzymes blocked degradation of enkephalins as well as other neuropeptides (fig. S7B). D2-MSNs express high levels of Penk (6), making them a likely source of MERF. To evaluate this possibility, we bred mice with genetic expression of channelrhodopsin-2 in D2-MSNs (Fig. 2D and fig. S8). Optogenetic stimulation of acute brain slices from these mice increased extracellular levels of conventional enkephalins, but only MERF levels were elevated in the presence of captopril (Fig. 2, E to F; table S3).
To investigate how MERF regulates NAc synaptic transmission, we measured miniature excitatory postsynaptic currents (mEPSCs; fig. S9). Increasing concentrations of MERF caused a dose-dependent decrease in mEPSC frequency, without altering mEPSC amplitude (Fig. 3, A and B), consistent with a presynaptic reduction of glutamate release probability. Both MERF and Met-enkephalin (fig. S10, A to C) had similar effects on D1-MSNs and D2-MSNs, suggesting presynaptic terminals onto both cell types are equally sensitive to endogenous opioids. We used these data to construct dose-response curves, and found that MERF (IC50 = 438 nM; Fig. 3C) was more potent than Met-enkephalin (IC50 = 993 nM; fig. S10D), as previously reported (13).
Fig. 3. Captopril enhances MERF effects on presynaptic and postsynaptic opioid receptors.
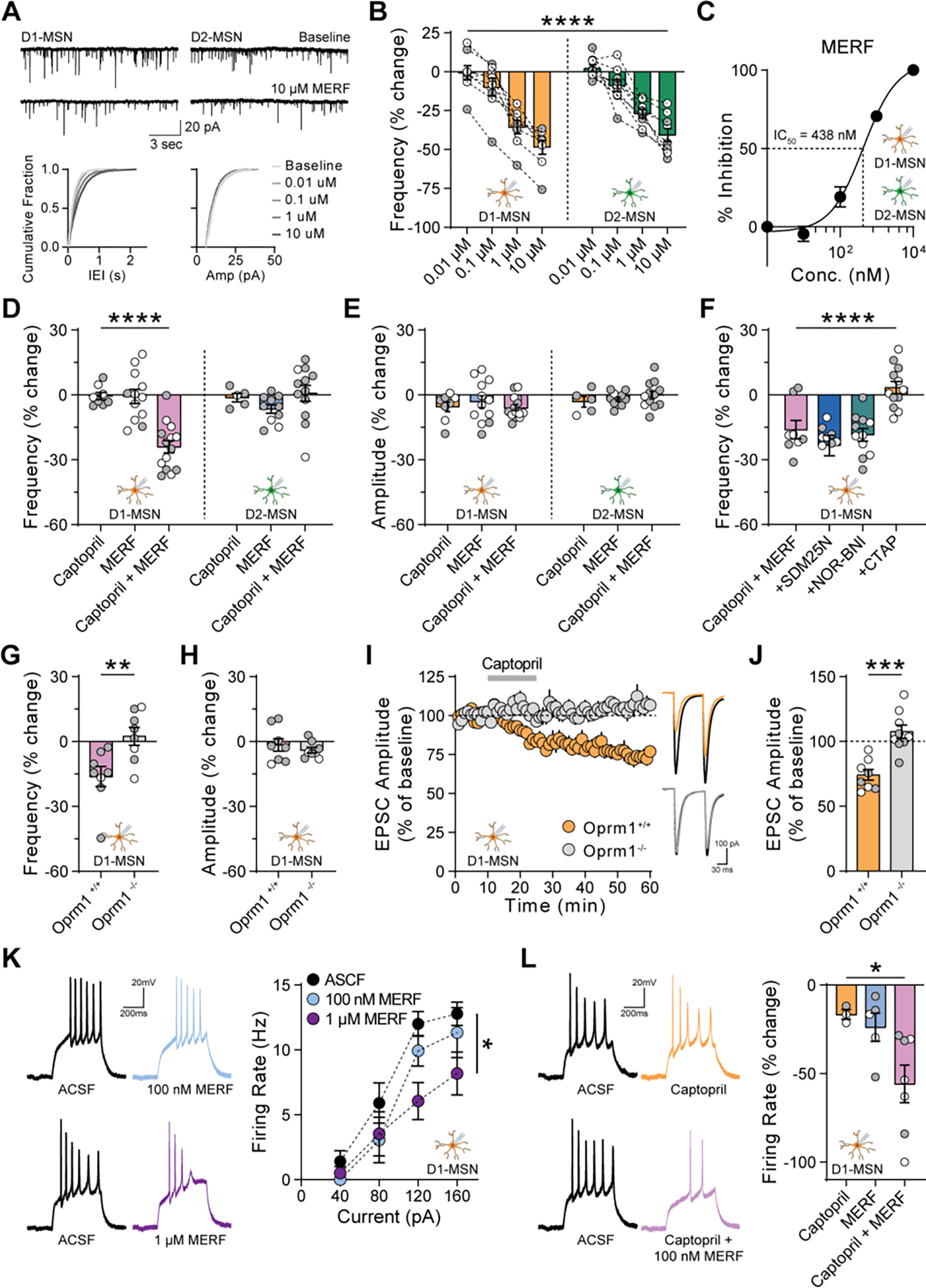
(A) Top, mEPSCs from D1-MSNs (left) and D2-MSNs (right) before and after bath perfusion of MERF (10 μM). Bottom, cumulative fraction plots of inter-event interval (left) and amplitude (right) of mEPSCs at increasing MERF concentrations (0.01–10 μM). (B) MERF caused a dose-dependent decrease in mEPSC frequency in D1-MSNs (left, orange, n=8) and D2-MSNs (right, green, n=9). (C) Sigmoidal interpolation of MERF dose-response normalized to maximal frequency change at 10 μM (IC50: 438 nM, 95% CI: 279–690 nM, n=17). (D, E) mEPSC frequency (D) and amplitude (E) after combined captopril (10 μM) and/or threshold MERF (100 nM) in D1-MSNs (left, n=14) and D2-MSNs (right, n=12). (F) Combined effect of captopril and threshold MERF in the presence of selective antagonists of delta (SDM25N, 500 nM, blue, n=9), kappa (NOR-BNI, 100 nM, green, n=11), or mu (CTAP, 1 μM, orange, n=12) opioid receptors. (G, H) Combined effect of captopril and threshold MERF on mEPSC frequency (G) and amplitude (H) in Oprm1−/− knockout mice (grey, n=8) and Oprm1+/+ littermates (purple, n=8). (I, J) EPSC amplitude time course (I) or average during last 5 min (J) of captopril-LTD in Oprm1+/+ (orange, n=8) and Oprm1−/− mice (grey, n=9). Inset shows traces before captopril (black lines) and during last 5 min (color lines). (K) Action potential firing rate of D1-MSNs (n=5–7) before and after exposure to MERF (0.1–1 μM). (L) Change in action potential firing rate of D1-MSNs (n=3–7) at 120 pA after combined captopril (10 μM) and/or threshold MERF (100 nM). Data are mean ± s.e.m. for all panels; open and closed circles indicate recordings from female and male mice, respectively. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, concentration main effect (B), treatment simple effect in D1-MSNs (D), genotype or treatment main effect (F, J, K, L), or two-sample t-test (G); see Data S1 for complete statistics.
These experiments identified a threshold MERF concentration (100 nM) that did not reliably affect synaptic transmission. Captopril alone (10 μM) also had no effect on frequency or amplitude of mEPSCs, which are measured in the absence of stimulation required to release endogenous opioids (7, 9). However, the combination of captopril and a threshold MERF concentration caused synergistic depression of mEPSC frequency in D1-MSNs, but not D2-MSNs (Fig. 3, D to E and fig. S11). This effect was absent following conditional genetic deletion of ACE from D1-MSNs (fig. S12). Inhibitors of aminopeptidase N and neprilysin did not enhance the effects of MERF, but did potentiate the effects of a threshold concentration of Met-enkephalin (100 nM) in both D1-MSNs and D2-MSNs (fig. S10, E to I).
To determine the opioid receptor subtype engaged by ACE inhibition, we recorded mEPSCs in D1-MSNs and applied captopril (10 μM) with threshold MERF (100 nM), in the presence of selective opioid receptor antagonists. Blocking delta opioid receptors with SDM25N (500 nM) or kappa opioid receptors with NOR-BNI (100 nM) did not prevent the decrease in mEPSC frequency (Fig. 3F and fig. S13). However, this effect was completely blocked by the mu opioid receptor (MOR) antagonist CTAP (1 μM), with no change in mEPSC amplitude (Fig. 3F and fig. S14, A to D). To confirm the role of MOR, we crossed Drd1-tdTomato reporter mice with constitutive MOR (Oprm1) knockout mice, generating offspring lacking functional MOR (Oprm1−/−) as well as littermate controls (Oprm1+/+) (fig. S14E). The synergistic effect of captopril (10 μM) and threshold MERF (100 nM) on mEPSC frequency in D1-MSNs was absent from Oprm1−/− mice (Fig. 3, G to H; fig. S14, F to I). Captopril-LTD of evoked EPSCs in D1-MSNs was also absent from Oprm1−/− mice (Fig. 3, I to J). MOR is also expressed postsynaptically by most D1-MSNs and fewer D2-MSNs (16, 17). In current-clamp recordings, MERF (1 μM) decreased action potential firing in D1-MSNs (Fig. 3K), but not D2-MSNs (fig. S15A). In combination with captopril (10 μM), a threshold MERF concentration (100 nM) also decreased action potential firing in D1-MSNs (Fig. 3L), but not D2-MSNs (fig. S15B).
Our experiments in brain slices have shown that ACE inhibition reduces excitatory synaptic input to D1-MSNs (Fig. 4A). To complement these analyses, we used fiber photometry in vivo, and found that systemic captopril administration observed reduced sensitivity of D1-MSNs to optogenetic stimulation of excitatory input arising from medial prefrontal cortex (Fig 4, B to D; fig. S16). Since the rewarding effects of addictive drugs are driven by D1-MSN activity and strengthening of excitatory synaptic input (18–22), we used an unbiased place conditioning assay to determine if systemic captopril administration can counteract the rewarding properties of fentanyl (Fig. 4E). Mice exhibited robust conditioned place preference (CPP) for a fentanyl-paired context (0.04 mg/kg, s.c.), but the magnitude of CPP was significantly attenuated when captopril (30 mg/kg, i.p.) was injected prior to fentanyl (Fig. 4, F and G). Trandolapril (the prodrug form of trandolaprilat) had a similar effect on fentanyl CPP (fig. S17). Captopril itself was not rewarding or aversive in the place conditioning assay (Fig. 4, H to J) and did not alter locomotion during conditioning (fig. S18). In a test of social interaction between two freely moving mice, captopril administration increased the amount of social interaction (Fig. 4, K to O), which is consistent with enhanced MOR signaling in the NAc (22) and rules out a general disruption of motivated behavior.
Fig. 4. Systemic captopril reduces excitatory input to D1-MSNs, counteracts fentanyl reward, and increases sociability.
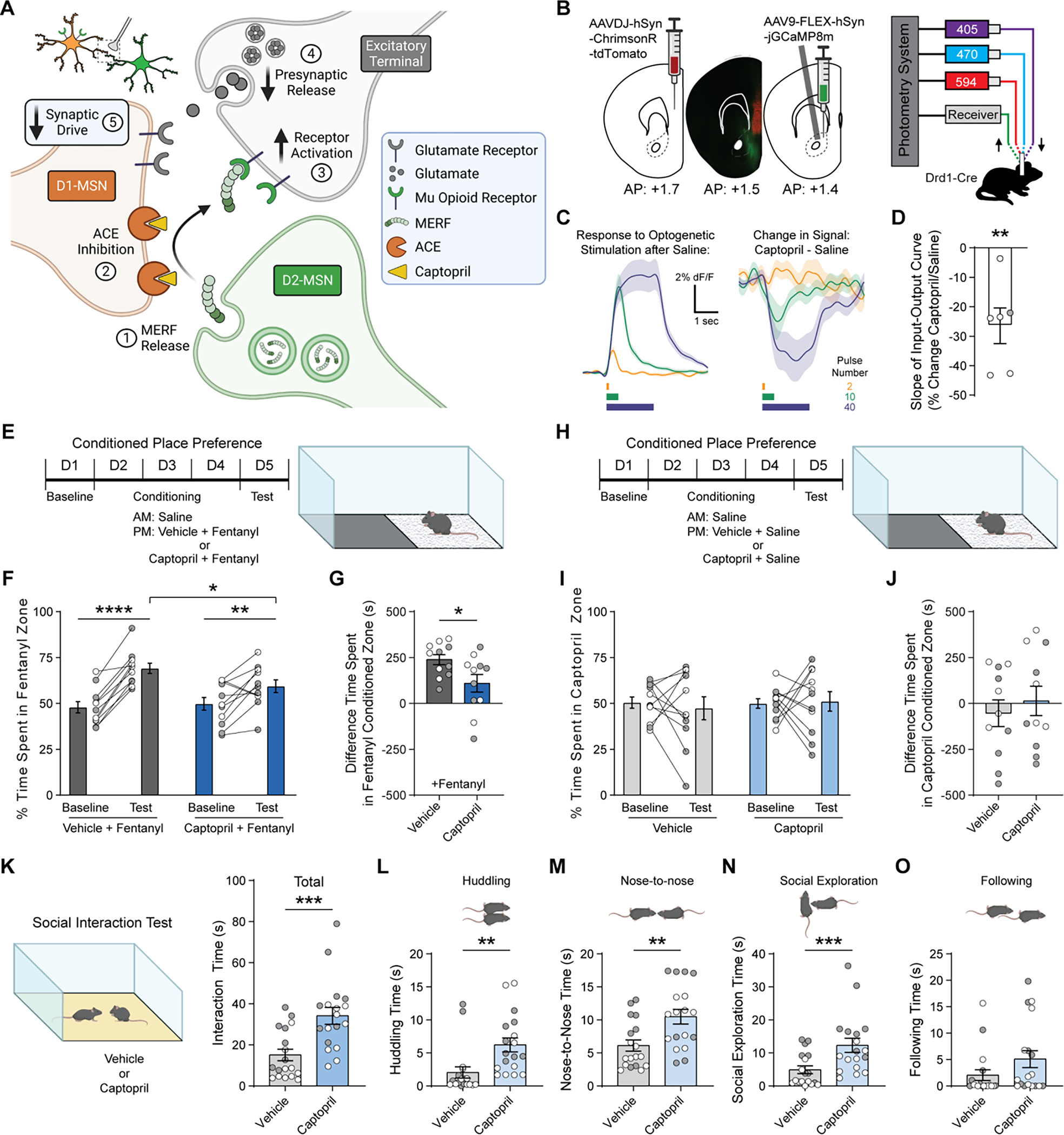
(A) Proposed mechanism by which captopril regulates glutamate release onto D1-MSNs via MERF. (B) Schematic showing viral injection of ChrimsonR-tdTomato in mPFC and Cre-dependent GCaMP8m in NAc, separated by fluorescent image showing viral expression (left), and setup for simultaneous optogenetic stimulation (594 nm) and fiber photometry recording (405/470 nm, right). (C) Traces showing average response to 2, 10, and 40 pulses of red light at 20 Hz after injection of saline (left), and average change in response following injection of captopril (30 mg/kg, i.p.; right). (D) Percent change in slope of the input-output curve following injection of captopril versus saline (n=6). (E-G) Schematic of unbiased place conditioning procedure (E), with percent time on fentanyl side (F) and CPP score (G) for groups receiving fentanyl (0.04 mg/kg, s.c.) preceded by vehicle (n=11, dark grey) or captopril (30 mg/kg, i.p.; n=11, dark blue). (H-J) Schematic of unbiased place conditioning procedure (H), with percent time on fentanyl side (I) and CPP score (J) for groups receiving saline preceded by vehicle (n=11, grey) or captopril (30 mg/kg, i.p.; n=11, blue). (K) Left, schematic of reciprocal social interaction test following injection of vehicle or captopril (30 mg/kg, i.p.). Right, total social interaction time after captopril (n=18, blue) or vehicle (n=18, grey). (L-O) Time spent huddling (L), interacting nose-to-nose (M), socially exploring (N), or following (O) the partner mouse throughout the assay. Data are mean ± s.e.m. for all panels; open and closed circles indicate female and male mice, respectively. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, one-sample t-test (D), simple effect of session/treatment (F), and treatment main effect (G, K-N); see Data S1 for complete statistics.
To translate an increasingly precise understanding of neural circuit function into therapeutic advances, one strategy is to target molecules with enriched expression in specific circuit elements (1). Our data show that enriched expression of ACE by D1-MSNs can be leveraged to induce synaptic plasticity in a brain circuit-specific fashion (fig. S19). Pharmacological inhibition of ACE prevents degradation of MERF, thereby enhancing endogenous MOR signaling in the NAc (Fig. 4A). This resembles the effects of selective reuptake inhibitors for other neurotransmitters, which have substantial therapeutic value for brain disorders. The circuit-specificity of these effects likely results from the high levels of MERF in the NAc, combined with enriched expression of ACE by D1-MSNs. ACE inhibition did not induce synaptic plasticity at excitatory synapses onto D2-MSNs or layer V pyramidal cells in the ACC, even though these synapses were sensitive to exogenous MERF application (Fig. 3B; fig. S3). By selectively and locally enhancing endogenous opioid signaling in the vicinity of D1-MSNs, central ACE inhibition may limit abuse liability by avoiding MOR activation in other brain circuits. In fact, systemic ACE inhibition significantly reduced the rewarding effects of fentanyl, and increased reciprocal social interaction. Conversely, rodents that exhibit reduced social interaction after chronic social stress have upregulated ACE expression in NAc tissue (23) and D1-MSNs (24). This behavioral phenotype is reversed by treatment with antidepressant drugs (25), and human patients taking centrally active ACE inhibitors can experience relief from depression (26–28), as well as improved quality of life (29, 30) and slower cognitive decline (31). Together, this evidence suggests that central ACE inhibition could have therapeutic potential for a variety of brain conditions. Our findings may thus herald a new era of repositioning and redesigning ACE inhibitors with central activity as a brain circuit-specific pharmacotherapy.
Supplementary Material
Acknowledgements:
We thank Drs. Hong Lu and Alan Daugherty (University of Kentucky) for generously providing the floxed ACE mouse line; Drs. Alfonso Araque and A. David Redish for helpful discussions; and the University of Minnesota Mouse Behavior Core for use of facilities to conduct behavioral tests. Some viral vectors used in this study were generated by the University of Minnesota Viral Vector and Cloning Core. Mass spectrometry was carried out in the Analytical Biochemistry Shared Resource of the Masonic Cancer Center, supported in part by the U.S. National Institutes of Health and National Cancer Institute (Cancer Center Support Grant CA-77598). Schematics were created with BioRender.com.
Funding:
This work was supported by the University of Minnesota’s MnDRIVE (Minnesota’s Discovery, Research, and Innovation Economy) initiative (B.H.T., E.M.L., P.E.R.), National Institutes of Health grant T32DA007234 (B.H.T., C.T., D.D.B.), National Institutes of Health grant F30DA049476 (B.H.T.), National Institutes of Health grant F31MH122094 (C.T.), National Institutes of Health grant F30DA052109 (D.D.B.), National Institutes of Health grant F30MH124404 (A.K.), National Institutes of Health grant T32MH115886 (R.M.D.), and National Institutes of Health grants R01DA048946 and R21DA050120 (P.E.R.).
Footnotes
Competing interests: Authors declare that they have no competing interests.
Supplementary materials:
Publisher's Disclaimer: This manuscript has been accepted for publication in Science. This version has not undergone final editing. Please refer to the complete version of record at http://www.sciencemag.org/. The manuscript may not be reproduced or used in any manner that does not fall within the fair use provisions of the Copyright Act without the prior, written permission of AAAS.
Data and materials availability:
All data are available in the main text or the supplementary material.
References and Notes
- 1.Gordon JA. Nat Neurosci 19, 1385–1386 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Rothwell PE et al. Cell 158, 198–212 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schwartz N et al. Science 345, 535–542 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Creed M, Pascoli VJ, Luscher C. Science 347, 659–664 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Strittmatter SM, Lo MM, Javitch JA, Snyder SH. Proc Natl Acad Sci U S A 81, 1599–1603 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saunders A et al. Cell 174, 1015–1030 e1016 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Atwood BK, Kupferschmidt DA, Lovinger DM. Nat Neurosci 17, 540–548 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ondetti MA, Rubin B, Cushman DW. Science 196, 441–444 (1977). [DOI] [PubMed] [Google Scholar]
- 9.Blomeley CP, Bracci E. J. Neurosci 31, 13346–13356 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roques BP, Fournie-Zaluski MC, Wurm M. Nat Rev Drug Discov 11, 292–310 (2012). [DOI] [PubMed] [Google Scholar]
- 11.Ploj K, Roman E, Gustavsson L, Nylander I. Brain Res Bull 53, 219–226 (2000). [DOI] [PubMed] [Google Scholar]
- 12.Mansour A, Hoversten MT, Taylor LP, Watson SJ, Akil H. Brain Research 700, 89–98 (1995). [DOI] [PubMed] [Google Scholar]
- 13.Inturrisi CE et al. Proc Natl Acad Sci U S A 77, 5512–5514 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benuck M, Berg MJ, Marks N. Neurochemistry International 4, 389–396 (1982). [DOI] [PubMed] [Google Scholar]
- 15.Al-Hasani R et al. , eLife 7, e36520 (2018).30175957 [Google Scholar]
- 16.Banghart MR, Neufeld SQ, Wong NC, Sabatini BL. Neuron 88, 1227–1239 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Charbogne P et al. Biol Psychiatry 81, 778–788 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lobo MK et al. Science 330, 385–390 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koo JW et al. Neuropsychopharmacology 39, 2646–2653 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pascoli V et al. Nature 509, 459–464 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Calipari ES et al. Proc Natl Acad Sci U S A 113, 2726–2731 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trezza V, Damsteegt R, Achterberg EJ, Vanderschuren LJ. J Neurosci 31, 6362–6370 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nam H et al. Neuropsychopharmacology 44, 1876–1885 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim HD et al. Mol Psychiatry, (2021). [Google Scholar]
- 25.Berton O et al. Science 311, 864–868 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Zubenko GS, Nixon RA. Am J Psychiatry 141, 110–111 (1984). [DOI] [PubMed] [Google Scholar]
- 27.Deicken RF. Biol Psychiatry 21, 1425–1428 (1986). [DOI] [PubMed] [Google Scholar]
- 28.Germain L, Chouinard G. Biol Psychiatry 23, 637–641 (1988). [DOI] [PubMed] [Google Scholar]
- 29.Croog SH et al. N Engl J Med 314, 1657–1664 (1986). [DOI] [PubMed] [Google Scholar]
- 30.Testa MA, Anderson RB, Nackley JF, Hollenberg NK. N Engl J Med 328, 907–913 (1993). [DOI] [PubMed] [Google Scholar]
- 31.Sink KM et al. Arch Intern Med 169, 1195–1202 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shansky RM. Science 364, 825–826 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Shuen JA, Chen M, Gloss B, Calakos N. J Neurosci 28, 2681–2685 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gong S et al. Nature 425, 917–925 (2003). [DOI] [PubMed] [Google Scholar]
- 35.Konig M et al. Nature 383, 535–538 (1996). [DOI] [PubMed] [Google Scholar]
- 36.Madisen L et al. Nat Neurosci 15, 793–802 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gerfen CR, Paletzki R, Heintz N. Neuron 80, 1368–1383 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen X et al. Arterioscler Thromb Vasc Biol 36, 1085–1089 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gong S et al. J Neurosci 27, 9817–9823 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matthes HW et al. Nature 383, 819–823 (1996). [DOI] [PubMed] [Google Scholar]
- 41.Shetty SS, DelGrande D. J Pharmacol Exp Ther 294, 179–186 (2000). [PubMed] [Google Scholar]
- 42.Matsuda H, Kurata Y, Imanishi S, Sato R, Shibamoto T. Pflugers Arch 448, 54–62 (2004). [DOI] [PubMed] [Google Scholar]
- 43.Wang J et al. J Clin Invest 117, 3393–3402 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sohn YI et al. Biochem Biophys Res Commun 439, 464–470 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Winters BL et al. Nat Commun 8, 14611 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ong EW, Xue L, Olmstead MC, Cahill CM. Br J Pharmacol 172, 615–629 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tejeda HA et al. Neuron 93, 147–163 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hjelmstad GO, Fields HL. J Neurophysiol 85, 1153–1158 (2001). [DOI] [PubMed] [Google Scholar]
- 49.Muñoz B, Fritz BM, Yin F, Atwood BK. Nat Commun 9, 1318 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chieng B, Connor M, Christie MJ. Mol Pharmacol 50, 650–655 (1996). [PubMed] [Google Scholar]
- 51.Bagley EE, Gerke MB, Vaughan CW, Hack SP, Christie MJ. Neuron 45, 433–445 (2005). [DOI] [PubMed] [Google Scholar]
- 52.Harrison JM, Allen RG, Pellegrino MJ, Williams JT, Manzoni OJ. J Neurophysiol 87, 2464–2470 (2002). [DOI] [PubMed] [Google Scholar]
- 53.Pennock RL, Hentges ST. J Physiol 592, 4247–4256 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Levitt ES, Abdala AP, Paton JF, Bissonnette JM, Williams JT. J Physiol 593, 4453–4469 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lau BK, Ambrose BP, Thomas CS, Qiao M, Borgland SL. J Neurosci 40, 5894–5907 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rappsilber J, Ishihama Y, Mann M. Anal. Chem 75, 663–670 (2003). [DOI] [PubMed] [Google Scholar]
- 57.Li D et al. Clin. Proteomics 15, 31 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lim BK, Huang KW, Grueter BA, Rothwell PE, Malenka RC. Nature 487, 183–189 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Toddes C, Lefevre EM, Brandner DD, Zugschwert L, Rothwell PE. J Neurosci, (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Terranova ML, Laviola G. Curr Protoc Toxicol Chapter 13, Unit13 10 (2005). [DOI] [PubMed] [Google Scholar]
- 61.Pisansky MT et al. Biol Psychiatry 86, 836–847 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lefevre EM et al. Neuropsychopharmacology 45, 1781–1792 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Savell KE et al. Sci Adv 6, eaba4221 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen R et al. Nat Neurosci 24, 1757–1771 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nieuwenhuis S, Forstmann BU, Wagenmakers EJ. Nat Neurosci 14, 1105–1107 (2011). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.