

Abstract
Malaria is a prevalent and lethal disease. The fast emergence and spread of resistance to current therapies is a major concern and the development of a novel line of therapy that could overcome, the problem of drug resistance, is imperative. Screening of a set of compounds with drug/natural product-based sub-structural motifs led to the identification of spirocyclic chroman-4-one 1 with promising antimalarial activity against the chloroquine-resistant Dd2 and chloroquine-sensitive 3D7 strains of the parasite. Extensive structure-activity and structure-property relationship studies were conducted to identify the essential features necessary for its activity and properties.
Keywords: Antimalarial, Drug resistance, Natural products, Spirocyclic chromane, Structure-activity relationship, Structure-property relationship
1. Introduction
Malaria remains a global and deadly infectious disease, with over 200 million annual cases and about half of the world’s population at risk.1–3 The heaviest burden of the disease is in sub-Sahara Africa, accounting for about 90% of all deaths predominantly children under five years.3 Malaria is caused by protozoa of the genus Plasmodium, which destroy infected red blood cells. Plasmodium falciparum and Plasmodium vivax are the most prevalent of the five Plasmodium species known to cause the human disease.4,5 The development of multi-drug resistant parasites has particularly made current available drugs less effective. Most drugs used for the treatment of malaria were either developed over 30 years ago or derived from older drugs, which makes the search for a novel line of therapy imperative.6–9
Natural products (NPs) and their derivatives or mimics have been a productive source of most of the active ingredients in drug development.10–12 Besides their diversity, they are known to occupy biologically important chemical space with interesting physiochemical properties.10,11 Interestingly, researchers have shown that the higher the fraction of saturated carbons, also known as the Fsp3 character, the more bioavailable it will be.13,14 Despite the advantages and successes of NPs, their use in drug discovery and development have declined due to the complexity of NP structures leading to synthetic challenges and concerns about intellectual property rights.10,11 With a view to identify novel antimalarial scaffolds that could overcome the problem of drug resistance, screening of a set of compounds with NP-based sub-structural motifs led to the discovery of a spirocyclic chromane 1 (Figure 1) with promising antimalarial activity against a chloroquine-resistant Dd2 strain and a chloroquine sensitive 3D7 strain of the parasite. Compound 1 exhibited excellent potency with a 50% effective concentration (EC50) of 350 nM against the chloroquine-resistant Dd2 strain displaying a selectivity of over 50 against human liver HepG2 cells. Analysis of the physiochemical properties of spirocyclic compound 1 showed close compliance with Lipinski’s parameters with an acceptable physiochemical profile. Biological evaluation indicated that spirocyclic chromane 1 is early-acting blocking parasite development at all erythrocytic stages including ring, trophozoite and schizont development as well as merozoite invasion. Most likely, spirocyclic chromane 1 also exhibits a cellular mechanism of action distinct from current antimalarials.15
Figure 1.

Structure of spirocyclic chromane 1
2. Results and Discussion
2.1. Chemistry
The synthesis of spirocyclic chromane 1 from α-naphthol was first reported by Roberts et al.12,13 Due to its promising antimalarial activity and physicochemical properties, detailed structure activity relationship (SAR) and structure-property relationship (SPR) studies were carried out. The initial SAR study focused on determining the importance of the spirocyclic chromane moiety to its activity while also exploring the essence of the two alcohol groups. This study was considered to deduce if the stereogenic carbons were integral for the potency of spirocyclic chromane 1. Various analogues of spirocyclic chromane 1 were synthesized from 4-oxo-3,4-dihydrospiro[benzo[h]chromene-2,4’-piperidine]-1’-carboxylate 2 as described below (Scheme 1).
Scheme 1.

Synthesis of analogues 2, 3, 5, 6 and 7
Reagents and conditions: (a) TFA, DCM; (b) i) NaBH4, EtOH, reflux ii) Pd/C, H2, EtOH; (c) for 2b-2i, 3a-3l, 5b-5h, 6b-6c and 7b-7c, RBr, K2CO3, DMF; (d) for 2j, 2k and 5i 2-(3,4-difluorophenyl)oxirane or 2-(3,4-dichlorophenyl)oxirane, EtOH reflux (e) for 2l and 5j 3,4-difluorobenzoic acid, EDCI, DMAP, DCM; (f) i) NaBH4, EtOH, reflux ii) PTSA(cat.), THF, reflux.
The deprotection of tert-butyl 4-oxo-3,4-dihydrospiro[benzo[h]chromene-2,4’piperidine]-1’-carboxylate 2 using trifluoroacetic acid (TFA) in dichloromethane (DCM) afforded amine 2a which was alkylated with alkyl halides to 2b-g and 2h-i in moderate to good yields. Sodium borohydride reduction of the corresponding alkylated carbonyl compounds yielded the hydroxy analogues 3a-g and 3j-k. For spirocyclic chromanes 2j-k, the corresponding epoxide, which was prepared from the sulfur ylide epoxidation of the aldehyde,16 was refluxed with amine 2a in ethanol and subsequently reduced to afford the hydroxy analogues 3h-i in good yields. The bis-chlorinated analogues 2i, 2k, 3i and 3k were inspired by the studies carried out by Medicine for Malaria Ventures and GlaxoSmithKline (GSK).17 Flexibility was also evaluated by altering the linker length between the spirocyclic moiety and the bis-fluorophenyl moiety. Commercially available benzylbromide, 2-phenethylbromide, and (3-bromopropyl)benzene were used to alkylate secondary amine 2a to tertiary amines 2c, d and f, which were then reduced to afford their 4-hydroxy analogues 3c, d and f. The rigidified amide analogues were also prepared in an EDCI amide coupling reaction with 2-(3,4-difluorophenyl)acetic acid and amine 2a to afford amide 2l, which was then reduced to its 4-hydroxy analogue 3l. All attempts to dehydroxylate alcohols 3a-3l using Pd/C, Pd/BaSO4 or Raney Nickel to afford the corresponding dehyroxylated products failed. An alternate approach consisting of an initial reduction of the carbonyl group of chromane 2 with sodium borohydride and subsequent hydrogenolysis of the 4-hydroxyl group using Pd/C in ethanol afforded the desired spirocyclic chromane 4 in good yield. However, a slightly less polar side product was also formed albeit in low yields. Interestingly, upon isolation and characterization, the side product was identified to be analogue 6, whose outermost aromatic ring was fully saturated. Compound 6 was considered an interesting analogue to study the effect of increased Fsp3 character by partial reduction of the naphthalene ring of the original compound 1. Boc-deprotection of tert-butyl carboxylate 4 and subsequent alkylation of the resultant amine 5a afforded the alkylated amines 5b-5h in good yields. Treatment of amine 5a with 2-(3,4-difluorophenyl)oxirane or 2-(3,4-difluorophenyl)acetic acid afforded alcohol 5i or amide 5j respectively. Deprotection of tert-butyl carboxylate 6 with TFA in DCM and subsequent alkylation afforded alkylated analogues 6a-6c. In order to study analogues with extended conjugation, 4-chromanone 2 was converted to chromene 7 via a carbonyl reduction followed by a PTSA-catalyzed dehydration. Boc-deprotection and N-alkylation afforded unsaturated analogues 7a-7c in good yields (Scheme 1).
The spirocyclic piperidine ring in analogue 1 was examined to determine whether the original angle of 180° between the benzo[h]4-chromanone plane and the trajectory of the N-substituent of the piperidine ring was optimal. Chiral spirocyclic chromanes were synthesized from easily available starting materials as outlined in scheme 2.
Scheme 2.

Synthesis of chiral spirocyclic chromanes
Reagents and conditions: (a) pyrrolidine, MeOH, reflux; (b) TFA, DCM; (c) (R)-2-acetoxy-2-phenylacetic acid, EDCI, DMAP, DCM; (d) for 10a and 10d, HCl, EtOH, reflux; then RBr, K2CO3, DMF (e) for 10b, 10c, 10e and 10f, NaBH4, EtOH, reflux.
Aldol condensation and cyclization of 1-(1-hydroxynaphthalen2-yl)ethan-1-one with Boc-protected piperidin-3-one in methanol using pyrrolidine afforded the racemic spirocyclic 4-chromanone 8 in moderate yield which was then deprotected. Resolution of the racemic spiro[benzo[h]chromene-2,3’-piperidin]-4(3H)-one (8a) was carried out by attaching the chiral auxiliary (R)-2-acetoxy-2-phenylacetic acid and separating the resulting two diastereomers (1R, 2R)-9a and (1R, 2S)-9b in good yield.18 The more polar diastereomer was crystallized using vapor diffusion with dimethyl sulfoxide (DMSO) as the solution solvent and hexanes as the diffusion solvent and its absolute configuration was determined through single crystal X-ray diffraction analysis to be S at C11 and R at C19 (Figure S1). Acid removal of the chiral auxiliary followed by chromatographic purification afforded the enantiomerically pure amines (2R)-10a and (2S)-10d that were then alkylated with selected alkyl bromides to corresponding amines (2R)-10b-10c and (2S)-10e-10f. The sodium borohydride reduction of the carbonyl group yielded 4-hydroxy analogues (2R)-11a-11b and (2S)-11c-11d in good yields (Scheme 2).
Lastly, the enantiopure isomers of the original spirocyclic compound 1 were synthesized (Scheme 3).
Scheme 3.

Synthesis of enantiopure analogues
Reagents and conditions: (a) CuBr2, CH3Cl/EtOAc, reflux, 5h; (b) (−)-DIP-Cl or (+)-DIP-Cl, THF, −41°C, 60 h; (c) 15% NaOH/EtOH, rt, 2 h (d) EtOH, reflux; (e) Noyori (R,R) or Noyori (S,S), FA/TEA(5:2), RT, 48 h.
The enantiopure bisfluoro epoxides (1R)-14a and (1S)-14b were prepared in a 3 step synthesis starting from 1-(3,4-difluorophenyl)ethan-1-one which was brominated using cupric bromide in a mixture of chloroform and ethyl acetate to afford halide 12 in good yield. Reduction with (−)DIP-chloride™ or (+)-DIP-chloride™ at −41 °C in THF for 60 hours afforded the enantiopure alcohol (1R)-13a and (1S)-13b respectively.19 The enantiopurity of (1R)-13a and (1S)-13b were determined by chiral SFC to be ≥ 98% (Figure S2). Ring opening reaction of both enantiopure epoxides (1R)-14a and (1S)-14b with amine 2a yielded the enantiopure spirocyclic chromanes (1’R)-2ma and (1’S)-2mb which retained the excellent enantiomeric excess despite the base-catalyzed epoxidation and the subsequent epoxide ring opening reaction (Figure S3). Initial attempts to stereoselectively reduce the carbonyl group on (1’R)-2ma and (1’S)-2mb using (−)DIP-chloride™ or (+)-DIP-chloride™ failed, nevertheless ruthenium (II) catalyzed asymmetric transfer hydrogenation using formic acid and trimethylamine azeotropic mixture was successful in converting the carbonyl group to the required hydroxyl group. To a solution of ketone (1’R)-2ma in formic acid-trimethylamine azeotrope was added (R)-RuCl[(1R,2R)-p-TsNCH(C6H5)CH(C6H5)NH2](ƞ6-mesitylene) and (S)-RuCl[(1S,2S)-p-TsNCH(C6H5)CH(C6H5)NH2](ƞ6-mesitylene), then stirred at room temperature for 48 h to afford the alcohol (4R,1’R)-3ma and (4S,1’R)-3mb in excellent diastereomeric excess. The diastereomers alcohol (4R,1’S)-3mc and (4S,1’S)-3md were prepared similarly from the ketone (1’S)-2mb. (Scheme 3 and Figure S4).20,21
2.2. Biological Evaluation
2.2.1. Structure-Activity Relationship (SAR) Studies
The initial SAR study focused on determining the importance of the spirocyclic chromane moiety to its activity while also exploring the essence of the benzylic alcohols. This study was considered to deduce if stereogenic centers were integral for the activity of the spirocyclic chromane. Furthermore, as Fsp3 benefits bioavailability, the optimization study focused on maintaining or increasing the Fsp3 while improving antimalarial potency. The first sub-series of analogues was designed to probe the importance of the N-substituent of the piperidine ring and to test for the need of the hydroxyl groups (Table 1). The spirocyclic chromanes 2a, 3a and 5a, whose piperidine nitrogen was unsubstituted, were 2-fold more potent than chromanes 2b, 3b and 5b in which the piperidine’s nitrogen was substituted with an ethyl chain. Replacing the hydroxyl group of the N-substituent by a carbonyl group or by a methylene in chromanes 3g and 3h reduced the potency by a factor of 10 and 2, respectively. Substituting the ortho-difluoro aryl group with an ortho-dichloro benzene moiety in compounds 3i and 3k did not display any significant improvements in potency. Similarly, replacement of the ortho-difluoro aryl group by a cyclohexyl ring 3e did not improve the potency as it displayed similar potency as reference compound 1. Finally, amide analogue 3l lacked antimalarial activity. This was consistent with other amides 2l and 5j. Next, the importance of the C-4 position was investigated. In this sub-series, the hydroxyl group in compounds 3e-3l appeared to play a more important role than close carbonyl analogues 2e-2l. In contrast, there was no clear potency pattern observed for compounds 5a-5i, in which the hydroxyl group in C-4 position was completely removed. However, it was evident that the hydroxyl group improved the selectivity as compound 3a and 1 displayed the highest selectivity of >53 against human liver HepG2 cells in this series of compounds tested. In summary, although no compounds of this first sub-series showed better potency than reference compound 1, cyclohexyl-substituted analogue 3e was equipotent to reference 1 with an EC50 value of 0.32 μM. The next sub-series of spirocyclic chromanes 2c-2f, 3c-3f and 5c-5f were synthesized to study the linker length between the piperidine nitrogen and the terminal aromatic residue. For example, for 4-hydroxy-substituted homologues 3c-3f, a potency trend was observed with compound 3d with 2 methylene units being more potent than analogue 3c containing 1 methylene unit, which was more potent than molecule 3f with 3 methylene units. Though such an activity pattern was not definite for compounds with a carbonyl or methylene group at the C-4 position, homologues 2c and 5c with a 1 methylene unit linker were the least potent in this sub-series. 4-Hydroxysubstituted analogue 3d with an EC50 of 0.32 μM was equipotent as reference compound 1 and it displays a slightly higher selectivity over HepG2 (Table 1).
Table 1.
Optimal oxidation state of C-4 and the N-substituent of the piperidine ring.
| Compound |
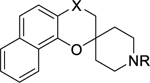
|
Dd2 EC50 (μM) | HepG2 EC50 (μM) | Selectivity | |
|---|---|---|---|---|---|
| X | R | ||||
| 2a | CO | H | 2.27 ± 0.1 | 7 | 3.2 |
| 2b | CO | CH2CH3 | >2.5 | 18 | 4.7 |
| 2c | CO | CH2Ph | >2.5 | 11 | 4.0 |
| 2d | CO | (CH2)2Ph | 0.53 ± 0.02 | 8 | 14.0 |
| 2e | CO | (CH2)2cyclohexyl | 0.71 ± 0.03 | 16 | 31.2 |
| 2f | CO | (CH2)3Ph | 0.48 ± 0.09 | >20 | >36.5 |
| 2g | CO | CH2CO(3,4-F)Ph | 1.30 ± 0.08 | 4 | 2.7 |
| 2h | CO | (CH2)2(3,4-F)Ph | >2.5 | >20 | 4.0 |
| 2i | CO | (CH2)2 (3,4-Cl)Ph | >2.5 | n.d. | n.d. |
| 2j | CO | CH2CHOH(3,4-F)Ph | 0.85 ± 0.02 | >20 | >6.1 |
| 2k | CO | CH2CHOH(3,4-Cl)Ph | >2.5 | n.d. | n.d. |
| 2l | CO | COCH2(3,4-F)Ph | >2.5 | 18 | <3.6 |
|
| |||||
| 3a | CHOH | H | 0.51 ± 0.04 | >20 | >54.5 |
| 3b | CHOH | CH2CH3 | >2.5 | 16 | 3.2 |
| 3c | CHOH | CH2Ph | 0.93 ± 0.07 | 6 | 5.7 |
| 3d | CHOH | (CH2)2Ph | 0.32 ± 0.09 | >20 | >68.9 |
| 3e | CHOH | (CH2)2cyclohexyl | 0.50 ± 0.09 | 14 | 35.1 |
| 3f | CHOH | (CH2)3Ph | 0.65 ± 0.03 | 1 | 2.0 |
| 3g | CHOH | CH2CO(3,4-F)Ph | >2.5 | 33 | <6.7 |
| 3h | CHOH | (CH2)2(3,4-F)Ph | 0.53 ± 0.04 | >20 | >32.5 |
| 3i | CHOH | (CH2)2 (3,4-Cl)Ph | 2.10 ± 0.09 | n.d. | n.d. |
| 1 | CHOH | CH2CHOH(3,4-F)Ph | 0.32 ± 0.05 | >20 | >53.3 |
| 3k | CHOH | CH2CHOH(3,4-Cl)Ph | 1.84 ± 0.07 | n.d. | n.d. |
| 3l | CHOH | COCH2(3,4-F)Ph | >2.5 | 5 | <1 |
|
| |||||
| 5a | CH2 | H | >2.5 | >20 | 4.0 |
| 5b | CH2 | CH2CH3 | 0.55 ± 0.02 | >20 | >52.1 |
| 5c | CH2 | CH2Ph | >2.5 | >20 | 4.0 |
| 5d | CH2 | (CH2)2Ph | 2.06 ± 0.15 | >20 | >9.6 |
| 5e | CH2 | (CH2)2cyclohexyl | 1.47 ± 0.19 | 2 | 1.1 |
| 5f | CH2 | (CH2)3Ph | 1.41 ± 0.24 | >20 | >45.6 |
| 5g | CH2 | CH2CO(3,4-F)Ph | >2.5 | >20 | >5.5 |
| 5h | CH2 | CH2CHOH(3,4-F)Ph | 1.91 ± 0.18 | >20 | 4.0 |
| 5i | CH2 | (CH2)2(3,4-F)Ph | 0.53 ± 0.04 | >20 | >10.4 |
| 5j | CH2 | COCH2(3,4-F)Ph | >2.5 | 5 | <1 |
n.d. = not determined
On reduction of the outermost ring of benzo[h]chromane yielded 7,8,9,10-tetrahydrobenzo[h] chromane 6a-6c, the antiplasmodial potency against the Dd2 strain was lost (Table 2). Next, a small set of benzo[h]chromenes 7a-7c (Table 2) were prepared and tested. Compound 7a unsubstituted at the piperidine’s nitrogen was the best in this series and slightly more potent than N-alkylated analogues 7b and 7c with EC50 values of 1.14 μM and 1.98 μM respectively. Compound 7a was also equipotent to reference compound 1 and also displayed the best selectivity in this entire sub-series.
Table 2.
SAR studies on the benzochromane moiety.
| Structure | Compound | R | Dd2 EC50 (μM) | HepG2 EC50 (μM) |
Selectivity |
|---|---|---|---|---|---|
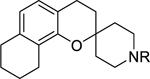
|
6a | H | >2.5 | >20 | 4.0 |
| 6b | CH2CH3 | >2.5 | 5 | <1 | |
| 6c | (CH2)2(3,4-F)Ph | >2.5 | >20 | 4.0 | |
|
| |||||
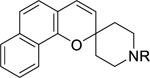
|
7a | H | 0.49 ± 0.08 | >20 | >45.6 |
| 7b | CH2CH3 | 1.14 ± 0.16 | 12 | 9.7 | |
| 7c | (CH2)2(3,4-F)Ph | 1.98 ± 0.08 | 13 | 7.2 | |
A set of compounds 10a-10f and 11a-11d (Table 3) differing in the 180° angle between the benzo[h]4-chromane plane and the N-substituent of the piperidine ring of reference compound 1. The S-enantiomer showed a clear pattern of at least a 3-fold higher potency than the corresponding R-enantiomer in all the sets of analogues made. Compound (S)-10c, the most potent compound in this sub-series with an EC50 value of 0.33 μM was over 5-fold more potent than (R)-10f with EC50 value of 1.84 μM. Besides potency, (S)-10c is also more selective and less toxic compared to (R)-10f. In contrast to previously observed activity trends, the analogues with a carbonyl at the C-4 position 10c and 10f were slightly more potent than those with a hydroxyl group in the C-4 position.
Table 3.
Optimal angle of the trajectory of the N-substituent of the piperidine ring.
| Structure | Compound | X | R | Dd2 EC50 (μM) | HepG2 EC50 (μM) | Selectivity |
|---|---|---|---|---|---|---|
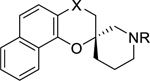
|
10a | CO | H | 0.63 ± 0.08 | 5 | 9.0 |
| 10b | CO | CH2CH3 | >2.5 | 8 | <1.6 | |
| 10c | CO | (CH2)2(3,4-F)Ph | 0.33 ± 0.04 | 14 | 34.2 | |
| 11a | CHOH | CH2CH3 | 0.63 ± 0.06 | >20 | >28.8 | |
| 11b | CHOH | (CH2)2(3,4-F)Ph | 0.61 ± 0.05 | >20 | >34.5 | |
|
| ||||||
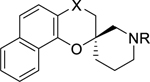
|
10d | CO | H | 1.20 ± 0.17 | 4 | 2.4 |
| 10e | CO | CH2CH3 | >2.5 | 5 | 1.3 | |
| 10f | CO | (CH2)2(3,4-F)Ph | 1.84 ± 0.15 | 10 | 5.8 | |
| 11c | CHOH | CH2CH3 | >2.5 | >20 | >7.6 | |
| 11d | CHOH | (CH2)2(3,4-F)Ph | >2.5 | 6 | 1.9 | |
Spirocyclic chromane 1 possess two stereogenic carbons and enantiopure analogues 3ma-3mb (Table 4) were prepared to assess whether one of the four possible stereoisomers is more potent than others. The cis analogue (4R,2”R)-3ma was the most potent isomer with an EC50 value of 1.06 μM. Strikingly, isomer (4R,2”R)-3ma is still over 2-fold less potent compared to the reference compound 1. In contrast, the enantiomer (4S,2”S)-3md was about 2-fold less potent and over 4-fold less potent than reference compound 1 comprising of four stereoisomers. This is an uncommon scenario and possibly implies a synergistic effect of the stereoisomers or the potential that the mixture of stereoisomers hit multiple targets.
Table 4.
Enantiopure analogues
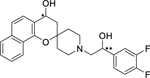
| |||||
|---|---|---|---|---|---|
| Compound | * | ** | Dd2 EC50 (μM) | HepG2 EC50 (μM) | Selectivity |
| 3ma | R | R | 1.06 ± 0.16 | 17.0 ± 0.94 | 16 |
| 3mb | R | S | >2.5 | 17.0 ± 0.88 | <7 |
| 3mc | S | R | 1.29 ± 0.23 | >20 | >15 |
| 3md | S | S | 1.79 ± 0.23 | >20 | 11 |
A selection of promising spirocyclic chromane analogues in addition to reference compound 1 were tested against the chloroquine sensitive D6 and the artemisinin resistant ARC08–022 strains of Plasmodium falciparum.22 The original compound 1 had an EC50 of 1.48 μM and 1.81 μM with D6 and ARC08–022 respectively. This represents at least a 3-fold loss in potency compared to the chloroquine-resistant Dd2 strain and the chloroquine sensitive 3D7 strain of the parasite. In comparison to spirocyclic chromane 1, analogues 2h and 5i with a hydroxyl group at C2”-position were more potent with 0.26 μM and 0.31 μM respectively against the D6 strain regardless of the substitution at the C-4 position (Table 5).
Table 5.
Potency against other Plasmodium strains
| Compound |
|
D6 EC50 (μM) | ARC08–022 EC50 (μM) | |
|---|---|---|---|---|
| X | R | |||
| 1 | CHOH | CH2CHOH(3,4-F)Ph | 1.48 | 1.81 |
| 2h | CO | CH2CHOH(3,4-F)Ph | 0.26 | 0.86 |
| 3e | CHOH | (CH2)2cyclohexyl | 0.50 | 0.63 |
| 3i | CHOH | CH2CHOH(3,4-Cl)Ph | 1.32 | 2.01 |
| 5i | CH2 | CH2CHOH(3,4-F)Ph | 0.31 | 0.97 |
2.2.2. Structure-Property Relationship (SPR) Studies
In parallel to the testing of spirocyclic chromanes for in vitro antimalarial activity, a structure-property relationship (SPR) study was conducted to identify potential physicochemical liabilities (Tables 6, 7, and 8). The log D3.0 and log D7.4, the distribution coefficient between octanol and water at pH 3.0 and pH 7.4, were experimentally determined via a previously described HPLC-based method.23 As previously disclosed, aqueous solubility at pH 7.4 was determined using a Biomek FX lab automation workstation with pION μSOL evolution software, whereas aqueous solubility at pH 2.0 was measured in an in-house implemented HPLC assay.24 In summary, spirocyclic chromanes’ aqueous solubility and distribution coefficient log D3.0 and log D7.4 displayed a pH dependence (Tables 6, 7, and 8).
Table 6.
Solubility and Log D
| Compound |
|
Log D | Solubilitya | |||
|---|---|---|---|---|---|---|
| X | R | pH 3.0 | pH 7.4 | pH 2.0 | pH 6.5 | |
| 2a | CO | H | 1.48 | 1.78 | ***** | ***** |
| 2b | CO | CH2CH3 | 1.65 | 2.19 | ***** | ***** |
| 2e | CO | (CH2)2cyclohexyl | 2.99 | 4.50 | *** | ** |
| 2g | CO | CH2CO(3,4-F)Ph | 1.52 | 1.82 | **** | **** |
| 2h | CO | CH2CHOH(3,4-F)Ph | 2.56 | 3.81 | ***** | ** |
| 2j | CO | (CH2)2(3,4-F)Ph | 2.79 | 4.51 | ***** | * |
| 2l | CO | COCH2(3,4-F)Ph | 4.00 | 4.03 | ND | * |
|
| ||||||
| 3a | CHOH | H | 2.04 | 2.46 | **** | * |
| 3b | CHOH | CH2CH3 | 1.51 | 1.88 | *** | *** |
| 3e | CHOH | (CH2)2cyclohexyl | 2.97 | 3.54 | **** | *** |
| 3g | CHOH | CH2CO(3,4-F)Ph | 1.53 | 0.89 | *** | *** |
| 3h | CHOH | CH2CHOH(3,4-F)Ph | 2.62 | 3.15 | ***** | ***** |
| 3j | CHOH | (CH2)2(3,4-F)Ph | 2.79 | 4.51 | ***** | * |
| 3l | CHOH | COCH2(3,4-F)Ph | 2.04 | 2.46 | ND | ND |
|
| ||||||
| 5a | CH2 | H | 1.97 | 2.33 | **** | *** |
| 5b | CH2 | CH2CH3 | 2.17 | 2.88 | * | * |
| 5e | CH2 | (CH2)2cyclohexyl | ND | 4.74 | **** | ND |
| 5g | CH2 | CH2CO(3,4-F)Ph | ND | 2.51 | *** | *** |
| 5h | CH2 | CH2CHOH(3,4-F)Ph | 3.29 | 4.51 | *** | ND |
| 5i | CH2 | (CH2)2(3,4-F)Ph | 3.05 | 4.00 | **** | * |
| 5j | CH2 | COCH2(3,4-F)Ph | 4.02 | 4.04 | ND | ND |
(*) For solubility ≤ 10 μM. (**) For 10 μM < solubility ≤ 20 μM. (***) For 20 μM < solubility ≤ 40 μM. (****) For 40 μM < solubility ≤ 80 μM. (*****) For solubility ≥ 80 μM.
Table 7.
Solubility and Log D
| Compound |
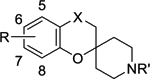
|
Log D | Solubilitya (μM) | ||||
|---|---|---|---|---|---|---|---|
| R | X | R’ | pH 3.0 | pH 7.4 | pH 2.0 | pH 6.5 | |
| 2aa | H | CO | H | 0.53 | 0.95 | ***** | ***** |
| 2ab | H | CO | CH2CH3 | 0.73 | 0.96 | ***** | **** |
| 2ac | H | CO | (CH2)2(3,4-F)Ph | 1.82 | 3.46 | ***** | *** |
| 3ac | H | CHOH | (CH2)2(3,4-F)Ph | 2.15 | 2.68 | *** | *** |
| 22a | H | CH2 | H | ND | 1.62 | ** | ND |
| 22b | H | CH2 | (CH2)2(3,4-F)Ph | ND | 3.79 | **** | *** |
|
| |||||||
| 5m | 7,8-cyclohexane | CH2 | H | 1.48 | 1.78 | * | ** |
| 5n | 7,8-cyclohexane | CH2 | CH2CH3 | 2.04 | 2.46 | *** | * |
| 5o | 7,8-cyclohexane | CH2 | (CH2)2(3,4-F)Ph | 1.97 | 2.33 | ** | * |
(*) For solubility ≤ 10 μM. (**) For 10 μM < solubility ≤ 20 μM. (***) For 20 μM < solubility ≤ 40 μM. (****) For 40 μM < solubility ≤ 80 μM. (*****) For solubility ≥ 80 μM.
Table 8.
Solubility and Log D
| Compound |
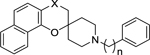
|
Log D | Solubilitya (μM) | |||
|---|---|---|---|---|---|---|
| X | n | pH 3.0 | pH 7.4 | pH 2.0 | pH 6.5 | |
| 2c | CO | 1 | 2.37 | 4.31 | ***** | **** |
| 2d | CO | 2 | 2.61 | 4.12 | ***** | **** |
| 2f | CO | 3 | 2.74 | 4.00 | **** | *** |
|
| ||||||
| 3c | CHOH | 1 | 2.27 | 2.97 | **** | **** |
| 3d | CHOH | 2 | 2.52 | 3.11 | **** | **** |
| 3f | CHOH | 3 | 2.71 | 3.19 | ***** | ***** |
|
| ||||||
| 5c | CH2 | 1 | ND | 4.49 | *** | ND |
(*) For solubility ≤ 10 μM. (**) For 10 μM < solubility ≤ 20 μM. (***) For 20 μM < solubility ≤ 40 μM. (****) For 40 μM < solubility ≤ 80 μM. (*****) For solubility ≥ 80 μM.
The majority of the compounds are highly soluble due to the piperidine moiety, whereas a very few analogues display a solubility below 20 μM. The majority of the compounds are slightly more soluble in more acidic environments. Similarly, the distribution coefficient log D is lower at low pH ranges. As the physicochemical properties of the potent compounds are well within the acceptable rages with solubility values of >20 μM and log D values of 1 – 4, the spirocyclic chromane compound series is considered to be well equipped for the development of orally bioavailable antimalarials.
During our optimization, we also calculated the fraction of saturated carbons Fsp3 to quantify the flatness of our molecule designs. According to the literature, for good bioavailability, the desired Fsp3 is over 0.45, and our compound optimization focused on maintaining or improving the Fsp3.13,14 Hit compound 1 displayed an Fsp3 of 0.36, whereas approximately 60% of the synthesized analogues displayed Fsp3 of 0.36 or higher (Figure 2). Molecule designs 3e and 5e, or 6a and 6b with significantly increased Fsp3 character were combined with a loss of potency.
Figure 2.

The fraction of saturated carbons Fsp3 of all synthesized compounds. Hit compound 1 possesses an Fsp3 of 0.36. Over 60% of the analogues possessed an Fsp3 of ≥0.36.
2.2.3. Discussion
Spirocyclic chromane 1 was identified when a compound collection containing NP-like structural motifs was screened to identify novel antimalarial compounds which interact with cellular targets different from those of current antimalarials. Its potency against the chloroquine resistant Dd2 strain underlines the possibility of its development to overcome the problem of drug-resistant malaria. Good physiochemical properties and compliance with Lipinski’s parameters makes spirocyclic chromane 1 a good candidate for hit-to-lead development of an orally bioavailable antimalarial. The SAR study revealed several trends namely that a larger N-substituent on the piperidine ring improved its potency. Importantly, the optimum linker length between the piperidine’s nitrogen and the terminating aryl moiety was 2 methylene units as antimalarial activity decreased with longer linker length. Furthermore, it was observed that the substitution of the C-4 position is essential for both activity and physiochemical properties. Although no clear deductions regarding activity was made from the effect of the hydroxyl group on the stereogenic carbons, these hydroxyls did have an effect in the solubility and log D. Flexibility was identified as essential for the activity of the spirocyclic chromanes. Antimalarial potency was completely lost in compounds 2l, 3l and 5j which were rigidified by an amidation of the piperidine’s nitrogen. Attempts to increase the Fsp3 character by either substituting the naphthalene moiety with a simple benzene ring or reducing one of the naphthalene rings did not show any improvements in potency. Changing the angle between the chromane plane and the N-substituent, basically the trajectory of the N-substituent in relation to the chromane, helped to identify the most potent enantiomer. However, these stereoisomers were at best equipotent to the original compound and hence these did not provide major improvements over the original compound 1. Substituting with benzo[f]chromane and benzo[g]chromane analogues thereby changing the geometry of the compounds revealed some interesting properties and activity patterns. Analogues 2ba–2bd and 3bb–3bd showed a decrease in potency compared to 2ca-2cd and 3cb-3cd. Surprisingly, the four enantiopure analogues of spirocyclic chromanes 1 were not as potent as their diastereomeric mixtures. This uncommon trend would require exploration of their mode of action to understand the reason for the synergy.
3. Conclusion
Spirocyclic chromane 1 was found to display antiplasmodial activity in both the chloroquine resistant Dd2 and chloroquine sensitive 3D7 strains. Inspired and encouraged by such an antimalarial activity and promising physiochemical properties, a library of 54 spirocyclic chromane analogues bearing a variety of N-substituents, varying angles of the trajectory of the N-substituent of the piperidine ring, different degree of oxidation of C-4 and different level of flexibility. Structural features essential for antimalarial activity were identified to be the spirocyclic chromane motif and flexibility. Compounds 3d and 10c were equipotent and 3d was also more selective and less toxic than the original spirocyclic chromane 1. Furthermore, the entire library of compounds was subjected to experimental profiling of physiochemical properties such as aqueous solubility and distribution coefficient log D in order to assess the potential for oral bioavailability. Most of the analogues possess an impressive solubility and log D with appreciable Fsp3 of approximately 0.4 indicating that spirocyclic chromanes have great potential as orally bioavailable antimalarial candidates.
4. Experimental Section
4.1. Compound Synthesis
General.
All reagents and solvents were obtained from Aldrich Chemical Co. and used without further purification. NMR spectra were recorded at ambient temperature on a 400 MHz or 500 MHz or 600 MHz Varian NMR spectrometer in the solvent indicated. All 1H NMR experiments are reported in δ units, parts per million (ppm) downfield of TMS, and were measured relative to the signals of chloroform (7.26 ppm) and dimethylsulfoxide (2.50 ppm) with 1H decoupled observation. Data for 1H NMR are reported as follows: chemicals shift (δ ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, sext = sextet, sept = septet, oct = octet, m = multiplet), integration and coupling constant (Hz) whereas 13C NMR analyses were reported in terms of chemical shift. NMR data was analyzed by using MestReNova Software version 6.0.2–5475. The purity of the final compounds was determined to be ≥95% by high-performance liquid chromatography (HPLC) using an Agilent 1100 LC/MSD-VL with electrospray ionization. Low resolution mass spectra were performed on an Agilent 1100 LC/MSD-VL with electrospray ionization. High-resolution mass spectra (HRMS) were performed on an Agilent LC/MSD TOF system G3250AA. Analytical thin layer chromatography (TLC) was performed on silica gel 60 F254 precoated plates (0.25 mm) from EMD Chemical Inc., and components were visualized by ultraviolet light (254 nm). EMD silica gel 230–400 (particle size 40–63 μm) mesh was used for all flash column chromatography.
General procedure A.
To a solution of the ethanone (1 equiv.) and carboxylate (1 equiv.) in absolute methanol (2.5 mL / mmol) was added pyrrolidine (2.6 equiv.) and heated to reflux for 8 h under N2. The methanol was removed under reduced pressure and the residue was purified by flash chromatography.
General procedure B.
A stirred solution of the protected carboxylate (1 equiv.) in DCM (1.0 M) at 0 °C was added TFA (1 equiv.) drop wise and the reaction mixture was allowed to warm up to room temperature. After 2 h, the reaction was diluted with water and extracted with DCM (2×). The aqueous layer was basified to pH 9.0 with 10% NaOH and then extracted with DCM (3×). The organic layer was combined, dried with sodium sulfate and concentrated under reduced pressure.
General procedure C.
To a solution of benzaldehyde (1 equiv.) and trimethylsulfonium iodide (Me3SI) (1.5 equiv.) in acetonitrile (2 mL) and water (5 μL) was added potassium hydroxide pellets (2 equiv.). The mixture was stirred at 80 °C for 4 h and then cooled; the acetonitrile was removed by rotatory evaporator. The residue was treated with a solution of water (5 mL) and sodium hypochlorite or bleach (1.5 mL) followed by extraction with diethyl ether (5 mL × 3). The organic layer was combined, dried with sodium sulfate and the solvent was removed under reduced pressure and then purified by flash chromatography.
General procedure D.
A solution of the amine (1 equiv.) and the epoxide (1 equiv.) in ethanol (0.1 M relative to the epoxide) was heated to reflux for 18 h. The ethanol was removed under reduced pressure and the residue was purified by flash chromatography.
General procedure E.
To a solution of the carbonyl (1 equiv.) in absolute ethanol (0.1 M relative to the carbonyl) was added sodium borohydride (3 equiv.). The reaction mixture was heated at reflux overnight and then cooled. The ethanol was removed under reduced pressure, the residue was diluted with water (1× the volume of ethanol) and extracted with ethyl acetate (5× the volume of ethanol). The combined organic layer was dried with sodium sulfate, the solvent was evaporated, and the residue was purified by flash chromatography.
General procedure F.
To a solution of the amine (1 equiv.) and halide (1 equiv.) in DMF (0.1 M relative to the halide) was added potassium carbonate (2 equiv.). The reaction mixture was stirred at room temperature until the starting halide was judged consumed by TLC analysis (8 h unless otherwise stated). The reaction was diluted with water (3× the volume of DMF) and extracted with ethyl acetate (5× the volume of DMF) thrice. The combine organic layer was washed with brine (5× the volume of DMF), dried with sodium sulfate. The solvent was removed under reduced pressure and the residue was purified by flash chromatography.
General procedure G.
To a solution of the chromane (1 equiv.) in ethanol (0.1 M relative to the SPC) was added Pd/C (0.5 equiv.) and stirred overnight under hydrogen balloon. The reaction mixture was filtered through a pad of Celite, rinsed with ethyl acetate (2× the volume of ethanol). The solvent was removed under reduced pressure and the residue was purified by flash chromatography.
General procedure H.
To a solution of the amine (1 equiv.) and the acid (1 equiv.) in DCM (0.1M relative to the amine) was added EDCI (2 equiv.) and DMAP (0.2 equiv.). The reaction mixture was stirred at room temperature until the starting material was judged consumed by TLC analysis (6 h unless otherwise stated). The reaction was quenched with water (2× the volume of DCM) and extracted with DCM thrice. The combine organic layer was dried with sodium sulfate, the solvent was removed under reduced pressure and the residue was purified by flash chromatography.
1’-Ethylspiro[benzo[h]chromene-2,4’-piperidin]-4(3H)-one (2b).
General procedure F was used to alkylate 2a (100 mg, 0.37 mmol) with bromoethane (40 mg, 0.37 mmol) using potassium carbonate (102 mg, 0.74 mmol) in DMF (4 mL). It was purified by flash chromatography (DCM:MeOH = 30:1) to afford 2b (83 mg, 75%) as white solid. Rf (DCM:MeOH = 10:1) = 0.45. 1H NMR (600 MHz, CDCl3) δ 8.35 (d, J = 8.3 Hz, 1H), 7.86 – 7.83 (m, 1H), 7.79 (t, J = 8.0 Hz, 1H), 7.64 – 7.60 (m, 1H), 7.57 – 7.52 (m, 1H), 7.40 – 7.37 (m, 1H), 2.83 – 2.74 (m, 4H), 2.54 – 2.47 (m, 4H), 2.26 – 2.19 (m, 2H), 1.87 (t, J = 10.7 Hz, 2H), 1.12 (t, 3H). 13C NMR (151 MHz, CDCl3) δ 191.69, 157.34, 137.90, 129.63, 128.09, 126.34, 125.56, 123.46, 121.64, 120.70, 115.24, 79.19, 52.53, 48.89, 47.79, 34.56, 12.31. HRMS (ESI) calcd for C19H24NO2 [M+H]+ : 296.1645, found: 296.1638.
1’-Benzylspiro[benzo[h]chromene-2,4’-piperidin]-4(3H)-one (2c).
General procedure F was used to alkylate 2a (300 mg, 1.12 mmol) with (bromomethyl)benzene (191 mg, 1.12 mmol) using potassium carbonate (310 mg, 2.24 mmol) in DMF (8 mL) for 1 h. It was purified by flash chromatography (Hexanes:EtOAc = 1:1) to afford 2c (375 mg, 94%) as yellow solid. Rf (EtOAc) = 0.35. 1H NMR (400 MHz, CDCl3) δ 8.31 (d, J = 8.3 Hz, 1H), 7.84 (d, J = 8.7 Hz, 1H), 7.78 (d, J = 8.1 Hz, 1H), 7.64 – 7.61 (m, 1H), 7.57 – 7.51 (m, 1H), 7.38 (s, 1H), 7.36 – 7.31 (m, 4H), 7.29 – 7.24 (m, 1H), 3.60 (s, 2H), 2.80 (s, 2H), 2.71 (d, J = 11.6 Hz, 2H), 2.61 – 2.53 (m, 2H), 2.18 (d, J = 12.3 Hz, 2H), 1.89 – 1.79 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 191.65, 157.31, 138.01, 137.83, 129.57, 129.30, 128.35, 128.02, 127.25, 126.30, 125.51, 123.46, 121.57, 120.63, 115.18, 79.12, 63.15, 49.00, 47.70, 34.51. HRMS (ESI) calcd for C24H24NO2 [M+H]+ : 358.1802, found: 358.1780.
1’-Phenethylspiro[benzo[h]chromene-2,4’-piperidin]-4(3H)-one (2d).
General procedure F was used to alkylate 2a (200 mg, 0.75 mmol) with (2-bromoethyl)benzene (138 mg, 0.75 mmol) using potassium carbonate (207 mg, 1.5 mmol) in DMF (5 mL). It was purified by flash chromatography (Hexanes:EtOAc = 1:1) to afford 2d (224 mg, 80%) as yellow solid. Rf (EtOAc) = 0.35. 1H NMR (400 MHz, CDCl3) δ 8.41 – 8.31 (m, 1H), 7.86 (d, J = 8.7 Hz, 1H), 7.83 – 7.76 (m, 1H), 7.67 – 7.60 (m, 1H), 7.59 – 7.52 (m, 1H), 7.39 (d, J = 8.7 Hz, 1H), 7.35 – 7.27 (m, 2H), 7.25 – 7.15 (m, 3H), 2.84 (d, J = 8.0 Hz, 6H), 2.77 – 2.67 (m, 2H), 2.67 – 2.54 (m, 2H), 2.29 – 2.15 (m, 2H), 1.96 – 1.76 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 191.66, 157.32, 140.37, 137.88, 129.62, 128.79, 128.53, 128.07, 126.34, 126.21, 125.54, 123.45, 121.62, 120.70, 115.23, 79.12, 60.70, 49.28, 47.79, 34.62, 33.98. HRMS (ESI) calcd for C25H26NO2 [M+H]+ : 372.1958, found: 372.1939.
1’-(2-Cyclohexylethyl)spiro[benzo[h]chromene-2,4’-piperidin]-4(3H)-one (2e).
General procedure F was used to alkylate 2a (300 mg, 1.12 mmol) with (2-bromoethyl)cyclohexane (214 mg, 1.12 mmol) using potassium carbonate (310 mg, 2.24 mmol) in DMF (8 mL). It was purified by flash chromatography (Hexanes:EtOAc = 2:1) to afford 2e (260 mg, 61%) as yellow solid. Rf (EtOAc) = 0.35. 1H NMR (400 MHz, CDCl3) δ 8.43 – 8.24 (m, 1H), 7.84 (d, J = 8.7 Hz, 1H), 7.78 (d, J = 8.1 Hz, 1H), 7.66 – 7.58 (m, 1H), 7.59 – 7.50 (m, 1H), 7.37 (d, J = 8.7 Hz, 1H), 2.80 (s, 2H), 2.77 – 2.63 (m, 2H), 2.56 – 2.37 (m, 4H), 2.26 – 2.13 (m, 2H), 1.91 – 1.78 (m, 2H), 1.75 – 1.56 (m, 5H), 1.47 – 1.35 (m, 2H), 1.33 – 1.10 (m, 4H), 1.01 – 0.83 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 191.77, 157.40, 137.88, 129.60, 128.06, 126.31, 125.57, 123.52, 121.63, 120.64, 115.23, 79.28, 56.83, 49.44, 47.80, 36.50, 34.95, 34.66, 33.63, 26.73, 26.45. HRMS (ESI) calcd for C25H32NO2 [M+H]+ : 378.2428, found: 378.2406.
1’-(3-Phenylpropyl)spiro[benzo[h]chromene-2,4’-piperidin]-4(3H)-one (2f).
General procedure F was used to alkylate 2a (200 mg, 0.75 mmol) with (3-bromopropyl)benzene (149 mg, 0.75 mmol) using potassium carbonate (207 mg, 1.5 mmol) in DMF (5 mL). It was purified by flash chromatography (Hexanes:EtOAc = 1:1) to afford 2f (159 mg, 55%) as yellow solid. Rf (EtOAc) = 0.24. 1H NMR (400 MHz, CDCl3) δ 8.38 – 8.31 (m, 1H), 7.85 (d, J = 8.7 Hz, 1H), 7.82 – 7.76 (m, 1H), 7.68 – 7.58 (m, 1H), 7.59 – 7.49 (m, 1H), 7.45 – 7.34 (m, 1H), 7.33 – 7.24 (m, 2H), 7.24 – 7.14 (m, 3H), 2.81 (s, 2H), 2.78 – 2.70 (m, 2H), 2.70 – 2.64 (m, 2H), 2.60 – 2.40 (m, 4H), 2.25 – 2.13 (m, 2H), 1.94 – 1.78 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 191.73, 157.37, 142.20, 137.87, 129.61, 128.50, 128.44, 128.06, 126.32, 125.90, 125.55, 123.50, 121.62, 120.65, 115.21, 79.23, 58.16, 49.30, 47.78, 34.64, 33.88, 28.97. HRMS (ESI) calcd for C25H32NO2 [M+H]+ : 386.2115, found: 386.2029.
1’-(2-(3,4-Difluorophenyl)-2-oxoethyl)spiro[benzo[h]chromene-2,4’-piperidin]-4(3H)-one (2g).
To a solution of 2a (200 mg, 0.74 mmol) and 2-bromo-1-(3,4-difluorophenyl)ethanone (176 mg, 0.74 mmol) in acetonitrile (6 mL) was added sodium bicarbonate (126 mg, 1.49 mmol) at 0 °C. The reaction mixture was allowed to stir to room temperature and held at room temperature for 4 hours. The acetonitrile was removed under reduced pressure, the residue was diluted with water and extracted with EtOAc (10 mL × 3). The combined organic layer was dried with sodium sulfate, the solvent was evaporated and the residue was purified by flash chromatography (DCM: MeOH = 75: 1) to afford 2g (180 mg, 57%) as yellow solid. Rf (DCM:MeOH = 10:1) = 0.71. 1H NMR (399 MHz, CDCl3) δ 8.40 – 8.28 (m, 1H), 7.94 – 7.85 (m, 1H), 7.85 – 7.72 (m, 3H), 7.66 – 7.58 (m, 1H), 7.58 – 7.50 (m, 1H), 7.37 (d, J = 8.6 Hz, 1H), 7.26 – 7.17 (m, 1H), 3.81 (s, 2H), 2.91 – 2.77 (m, 4H), 2.76 – 2.64 (m, 2H), 2.27 – 2.11 (m, 2H), 1.99 – 1.79 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 194.24, 191.50, 157.20, 153.64 (dd, 1JCF = 252.5, 2JCF = 14.5 Hz), 150.52 (dd, 1JCF = 252.5, 2JCF = 14.5 Hz), 137.90, 133.06 (dd, 3JCF = 7.5, 4JCF = 3.2 Hz), 129.70, 128.14, 126.43, 125.50 (dd, 3JCF = 7.6, 4JCF = 3.7 Hz), 123.40, 121.62, 120.84, 117.86 (d, 2JCF = 18.3 Hz), 117.63 (d, 2JCF = 17.8 Hz), 115.22, 78.56, 64.90, 49.56, 47.73, 34.44.
1’-(3,4-Difluorophenethyl)spiro[benzo[h]chromene-2,4’-piperidin]-4(3H)-one (2h).
General procedure F was used to alkylate 2a (110 mg, 0.4 mmol) with 4-(2-bromoethyl)-1,2-difluorobenzene (91 mg, 0.4 mmol) using potassium carbonate (110 mg, 0.8 mmol) in DMF (4 mL). It was purified by flash chromatography (Hexanes:EtOAc = 2:1) to afford 2j (117 mg, 72%) as yellow solid. Rf (EtOAc) = 0.26. 1H NMR (600 MHz, CDCl3) δ 8.53 – 8.26 (m, 1H), 7.84 (d, J = 8.6 Hz, 1H), 7.79 (d, J = 8.1 Hz, 1H), 7.70 – 7.58 (m, 1H), 7.60 – 7.49 (m, 1H), 7.38 (d, J = 8.6 Hz, 1H), 7.18 – 6.97 (m, 2H), 6.96 – 6.80 (m, 1H), 2.82 (s, 2H), 2.81 – 2.74 (m, 4H), 2.69 – 2.63 (m, 2H), 2.63 – 2.54 (m, 2H), 2.32 – 2.14 (m, 2H), 1.95 – 1.77 (m, 2H). HRMS (ESI) calcd for C25H24F2NO2 [M+H]+ : 408.1770, found: 408.1760.
1’-(3,4-Dichlorophenethyl)spiro[benzo[h]chromene-2,4’-piperidin]-4(3H)-one (2i).
General procedure F was used to alkylate 2a (260 mg, 0.97 mmol) with 4-(2-bromoethyl)-1,2dichlorobenzene (247 mg, 0.97 mmol) using potassium carbonate (270 mg, 1.9 mmol) in DMF (8 mL). It was purified by flash chromatography (Hexanes:EtOAc = 2:1) to afford 2k (316 mg, 74%) as yellow solid. Rf (EtOAc) = 0.26. 1H NMR (399 MHz, CDCl3) δ 8.35 (d, J = 8.2 Hz, 1H), 7.84 (d, J = 8.7 Hz, 1H), 7.79 (d, J = 8.1 Hz, 1H), 7.62 (t, J = 7.3 Hz, 1H), 7.55 (t, J = 7.6 Hz, 1H), 7.43 – 7.27 (m, 3H), 7.03 (dd, J = 8.2, 2.0 Hz, 1H), 2.81 (s, 2H), 2.80 – 2.72 (m, 4H), 2.71 – 2.53 (m, 4H), 2.31 – 2.13 (m, 2H), 1.94 – 1.75 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 191.61, 157.25, 140.65, 137.86, 132.30, 130.71, 130.38, 130.12, 129.65, 128.28, 128.09, 126.37, 125.48, 123.39, 121.60, 120.74, 115.19, 79.00, 59.97, 49.25, 47.76, 34.56, 33.04.
1’-(2-(3,4-Dichlorophenyl)-2-hydroxyethyl)spiro[benzo[h]chromene-2,4’-piperidin]-4(3H)-one (2k).
General procedure D was used with 2a (500 mg, 1.8 mmol) and 2-(3,4-difluorophenyl)oxirane11 (354 mg, 1.8 mmol) in ethanol (30 mL). It was purified by flash chromatography (hexanes:EtOAc = 2 :1) to afford 2i (628 mg, 74%) as a yellow solid. Rf (EtOAc) = 0.31. 1H NMR (399 MHz, CDCl3) δ 8.40 – 8.26 (m, 1H), 7.85 (d, J = 8.7 Hz, 1H), 7.80 (d, J = 8.1 Hz, 1H), 7.69 – 7.59 (m, 1H), 7.58 – 7.52 (m, 1H), 7.50 (d, J = 2.0 Hz, 1H), 7.45 – 7.37 (m, 2H), 7.25 – 7.16 (m, 1H), 4.71 (dd, J = 10.6, 3.4 Hz, 1H), 3.06 – 2.94 (m, 1H), 2.94 – 2.73 (m, 3H), 2.73 – 2.55 (m, 3H), 2.54 – 2.41 (m, 1H), 2.36 – 2.18 (m, 2H), 1.99 – 1.77 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 191.43, 157.14, 142.54, 137.89, 132.65, 131.41, 130.47, 129.73, 128.17, 127.93, 126.45, 125.43, 125.24, 123.31, 121.61, 120.91, 115.21, 78.74, 67.93, 66.02, 50.62, 47.77, 47.70, 34.73, 34.55.
1’-(2-(3,4-Difluorophenyl)acetyl)spiro[benzo[h]chromene-2,4’-piperidin]-4(3H)-one (2l).
General procedure H was used in the amidation reaction of 2a (600 mg, 2.24 mmol) and 2-(3,4-difluorophenyl)acetic acid (386 mg, 2.24 mmol) with EDCI (860 mg, 4.5 mmol) and DMAP (55 mg, 0.45 mmol) in DCM (15 mL). It was purified by flash chromatography (Hexanes:EtOAc = 2:1) to afford 2l (724 mg, 77%) as yellow solid. Rf (EtOAc) = 0.60. 1H NMR (600 MHz, CDCl3) δ 8.29 (d, J = 8.2 Hz, 1H), 7.85 – 7.78 (m, 2H), 7.66 – 7.60 (m, 1H), 7.57 – 7.52 (m, 1H), 7.41 (d, J = 8.6 Hz, 1H), 7.12 – 7.05 (m, 2H), 6.98 – 6.93 (m, 1H), 4.53 (d, J = 13.4 Hz, 1H), 3.75 – 3.64 (m, 3H), 3.56 (t, J = 11.9 Hz, 1H), 3.26 – 3.18 (m, 1H), 2.80 (q, J = 16.5 Hz, 2H), 2.28 – 2.17 (m, 2H), 1.73 – 1.64 (m, 1H), 1.54 – 1.46 (m, 1H). 13C NMR (151 MHz, CDCl3) δ 190.72, 168.69, 156.75, 150.47 (dd, 1JCF = 246.1, 2JCF =11.9 Hz), 149.59 (dd, 1JCF = 246.1, 2JCF = 11.9 Hz), 137.92, 131.92 (dd, 3JCF = 5.8, 4JCF = 3.9 Hz), 129.87, 128.24, 126.65, 125.23, 124.88 (dd, 3JCF = 5.9, 4JCF = 3.4 Hz), 123.01, 121.52, 121.26, 117.93 (d, 2JCF = 17.4 Hz), 117.54 (d, 2JCF = 17.4 Hz), 115.18, 78.85, 47.63, 42.04, 39.92, 37.81, 34.50, 34.33.
3,4-Dihydrospiro[benzo[h]chromene-2,4’-piperidin]-4-ol (3a).
General procedure B was used to deprotect 3 (1.48 g, 4.0 mmol) in DCM (40 mL) and TFA (13 mL) to afford 3a (1 g, 97%) as a brown solid. Rf (DCM:MeOH = 10:1) = 0.20. 1H NMR (600 MHz, CDCl3) δ 8.30 – 8.23 (m, 1H), 7.78 – 7.73 (m, 1H), 7.53 – 7.49 (m, 1H), 7.49 – 7.44 (m, 2H), 7.40 (d, J = 8.3 Hz, 1H), 4.96 – 4.88 (m, 1H), 3.22 – 3.13 (m, 1H), 3.13 – 3.03 (m, 1H), 2.94 – 2.56 (m, 4H), 2.22 – 2.13 (m, 1H), 2.08 – 2.01 (m, 1H), 1.97 – 1.82 (m, 2H), 1.75 – 1.53 (m, 2H). 13C NMR (151 MHz, CDCl3) δ 147.55, 134.34, 127.63, 126.58, 125.56, 125.54, 125.49, 122.24, 120.09, 120.05, 74.71, 63.12, 42.61, 42.17, 42.04, 36.74, 34.76.
1’-Ethyl-3,4-dihydrospiro[benzo[h]chromene-2,4’-piperidin]-4-ol (3b).
General procedure E was used to reduce 2a (150mg, 0.5mmol) in absolute ethanol (4 ml) using sodium borohydride (58mg, 1.5mmol). It was purified by flash chromatography (DCM:MeOH = 20:1) to afford 3b (134 mg, 89%) as white solid. Rf (DCM:MeOH = 10:1) = 0.22. 1H NMR (600 MHz, CDCl3) δ 8.29 – 8.14 (m, 1H), 7.89 – 7.65 (m, 1H), 7.56 – 7.41 (m, 3H), 7.43 – 7.30 (m, 1H), 4.99 – 4.75 (m, 1H), 2.82 – 2.63 (m, 2H), 2.57 – 2.43 (m, 4H), 2.44 – 2.35 (m, 1H), 2.23 – 2.02 (m, 2H), 1.97 – 1.86 (m, 2H), 1.86 – 1.77 (m, 1H), 1.77 – 1.66 (m, 1H), 1.21 – 1.00 (m, 3H). HRMS (ESI) calcd for C19H24NO2 [M+H]+ : 298.1802, found: 298.1788.
1’-Benzyl-3,4-dihydrospiro[benzo[h]chromene-2,4’-piperidin]-4-ol (3c).
General procedure E was used to reduce 2c (250 mg, 0.7 mmol) in absolute ethanol (5 mL) using sodium borohydride (79 mg, 2.1 mmol). It was purified by flash chromatography (DCM:MeOH = 30:1) to afford 3c (198 mg, 79%) as white solid. Rf (DCM:MeOH = 10:1) = 0.44. 1H NMR (400 MHz, CDCl3) δ 8.26 – 8.16 (m, 1H), 7.81 – 7.69 (m, 1H), 7.54 – 7.43 (m, 3H), 7.40 (d, J = 8.5 Hz, 1H), 7.38 – 7.31 (m, 4H), 7.31 – 7.23 (m, 1H), 5.02 – 4.85 (m, 1H), 3.62 (s, 2H), 2.80 – 2.72 (m, 1H), 2.72 – 2.64 (m, 1H), 2.64 – 2.58 (m, 1H), 2.58 – 2.48 (m, 1H), 2.20 (dd, J = 13.7, 6.1 Hz, 1H), 2.14 – 2.04 (m, 1H), 2.02 – 1.93 (m, 1H), 1.93 – 1.83 (m, 2H), 1.82 – 1.72 (m, 1H). 13C NMR (101 MHz, CDCl3) δ 147.63, 137.98, 134.37, 129.51, 128.36, 127.62, 127.27, 126.57, 125.55, 125.44, 122.29, 120.08, 118.07, 74.27, 63.46, 63.33, 49.32, 49.14, 42.18, 35.95, 34.11. HRMS (ESI) calcd for C24H26NO2 [M+H]+ : 360.1958, found: 360.1945.
1’-Phenethyl-3,4-dihydrospiro[benzo[h]chromene-2,4’-piperidin]-4-ol (3d).
General procedure E was used to reduce 2d (220 mg, 0.59 mmol) in absolute ethanol (5 mL) using sodium borohydride (67 mg, 1.78 mmol). It was purified by flash chromatography (DCM:MeOH = 30:1) to afford 3d (169 mg, 76%) as white solid. Rf (DCM:MeOH = 10:1) = 0.52. 1H NMR (400 MHz, CDCl3) δ 8.34 – 8.15 (m, 1H), 7.84 – 7.72 (m, 1H), 7.59 – 7.46 (m, 3H), 7.42 (d, J = 8.3 Hz, 1H), 7.31 (t, J = 7.3 Hz, 2H), 7.23 (d, J = 7.6 Hz, 3H), 4.97 (t, J = 6.7 Hz, 1H), 3.01 – 2.77 (m, 4H), 2.78 – 2.65 (m, 3H), 2.64 – 2.49 (m, 2H), 2.33 – 2.12 (m, 2H), 2.09 – 1.88 (m, 2H), 1.88 – 1.74 (m, 1H), 1.32 – 1.14 (m, 1H). 13C NMR (101 MHz, CDCl3) δ 147.58, 140.30, 134.38, 128.82, 128.55, 127.66, 126.59, 126.23, 125.57, 125.52, 122.22, 120.14, 118.12, 74.16, 63.38, 60.83, 49.48, 49.35, 42.23, 35.88, 34.15, 33.73. HRMS (ESI) calcd for C25H28NO2 [M+H]+ : 374.2115, found: 374.2102.
1’-(2-Cyclohexylethyl)-3,4-dihydrospiro[benzo[h]chromene-2,4’-piperidin]-4-ol (3e).
General procedure E was used to reduce 2e (200 mg, 0.53 mmol) in absolute ethanol (5 mL) using sodium borohydride (60 mg, 1.59 mmol). It was purified by flash chromatography (DCM:MeOH = 30:1) to afford 3e (143 mg, 71%) as white solid. Rf (DCM:MeOH = 10:1) = 0.60. 1H NMR (400 MHz, CDCl3) δ 8.33 – 8.18 (m, 1H), 7.84 – 7.72 (m, 1H), 7.56 – 7.45 (m, 3H), 7.40 (d, J = 8.5 Hz, 1H), 5.07 – 4.84 (m, 1H), 2.90 – 2.68 (m, 2H), 2.60 – 2.35 (m, 5H), 2.29 – 2.16 (m, 1H), 2.17 – 2.05 (m, 1H), 2.07 – 1.82 (m, 2H), 1.82 – 1.75 (m, 1H), 1.76 – 1.58 (m, 5H), 1.55 – 1.38 (m, 2H), 1.34 – 1.06 (m, 5H), 1.04 – 0.78 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 147.65, 134.38, 127.64, 126.55, 125.58, 125.52, 125.48, 122.28, 120.06, 118.15, 74.33, 63.40, 57.03, 49.62, 49.49, 42.30, 36.61, 36.00, 34.69, 34.12, 33.62, 26.73, 26.45. HRMS (ESI) calcd for C25H34NO2 [M+H]+ : 380.2584, found: 380.2576.
1’-(3-Phenylpropyl)-3,4-dihydrospiro[benzo[h]chromene-2,4’-piperidin]-4-ol (3f).
General procedure E was used to reduce 2f (200 mg, 0.57 mmol) in absolute ethanol (5 mL) using sodium borohydride (65 mg, 1.71 mmol). It was purified by flash chromatography (DCM:MeOH = 30:1) to afford 3f (161 mg, 73%) as white solid. Rf (DCM:MeOH = 10:1) = 0.53. 1H NMR (400 MHz, CDCl3) δ 8.35 – 8.17 (m, 1H), 7.82 – 7.70 (m, 1H), 7.58 – 7.46 (m, 3H), 7.40 (d, J = 8.5 Hz, 1H), 7.36 – 7.27 (m, 2H), 7.25 – 7.15 (m, 3H), 5.03 – 4.69 (m, 1H), 2.89 – 2.62 (m, 5H), 2.62 – 2.51 (m, 1H), 2.51 – 2.39 (m, 3H), 2.24 – 2.12 (m, 1H), 2.13 – 2.03 (m, 1H), 2.02 – 1.80 (m, 5H), 1.80 – 1.67 (m, 1H). 13C NMR (101 MHz, CDCl3) δ 147.60, 142.17, 134.33, 128.48, 128.43, 127.61, 126.52, 125.89, 125.54, 125.49, 122.26, 120.02, 118.15, 74.29, 63.27, 58.39, 49.46, 49.34, 42.24, 36.01, 34.01, 33.96, 28.78. HRMS (ESI) calcd for C26H30NO2 [M+H]+ : 388.2271, found: 388.2271.
1-(3,4-Difluorophenyl)-2-(4-hydroxy-3,4-dihydrospiro[benzo[h]chromene-2,4’-piperidin]-1’yl)ethanone (3g).
To a solution of 3a (500 mg, 1.85 mmol) and 2-bromo-1-(3,4-difluorophenyl)ethanone (436 mg, 1.85 mmol) in acetonitrile (15 mL) was added sodium bicarbonate (311 mg, 3.70 mmol) at 0 °C. The reaction mixture was allowed to stir to room temperature and held at room temperature for 4 hours. The acetonitrile was removed under reduced pressure, the residue was diluted with water and extracted with EtOAc (20 mL × 3). The combined organic layer was dried with sodium sulfate, the solvent was evaporated, and the residue was purified by flash chromatography (DCM: MeOH = 50:1) to afford 3g (400 mg, 51%) as yellow solid. Rf (DCM:MeOH = 10:1) = 0.45. 1H NMR (399 MHz, CDCl3) δ 8.47 – 8.08 (m, 1H), 7.94 – 7.87 (m, 1H), 7.85 – 7.72 (m, 3H), 7.66 – 7.58 (m, 1H), 7.58 – 7.50 (m, 1H), 7.37 (d, J = 8.6 Hz, 1H), 7.26 – 7.17 (m, 1H), 3.91 (s, 2H), 2.91 – 2.77 (m, 4H), 2.76 – 2.64 (m, 2H), 2.27 – 2.11 (m, 2H), 1.99 – 1.79 (m, 2H).
1’-(3,4-Difluorophenethyl)-3,4-dihydrospiro[benzo[h]chromene-2,4’-piperidin]-4-ol (3h).
General procedure E was used to reduce 2h (100 mg, 0.24 mmol) in absolute ethanol (3 mL) using sodium borohydride (28 mg, 0.73 mmol). It was purified by flash chromatography (DCM:MeOH = 30:1) to afford 3h (89 mg, 91%) as white solid. Rf (DCM:MeOH = 20:1) = 0.42. 1H NMR (600 MHz, CDCl3) δ 8.47 – 8.05 (m, 1H), 7.89 – 7.65 (m, 1H), 7.57 – 7.45 (m, 3H), 7.40 (d, J = 8.4 Hz, 1H), 7.24 – 6.96 (m, 2H), 6.97 – 6.80 (m, 1H), 5.09 – 4.76 (m, 1H), 3.03 – 2.69 (m, 4H), 2.68 – 2.58 (m, 3H), 2.59 – 2.48 (m, 2H), 2.29 – 2.15 (m, 1H), 2.15 – 2.03 (m, 1H), 2.01 – 1.90 (m, 2H), 1.90 – 1.81 (m, 1H), 1.83 – 1.66 (m, 1H). 13C NMR (151 MHz, CDCl3) δ 150.14 (dd, 1JCF = 246.4, 2JCF = 12.8 Hz), 148.89 (dd, 1JCF = 246.4, 2JCF = 12.8 Hz), 147.41, 137.23 (dd, 3JCF = 4.3, 4JCF=2.6 Hz), 134.24, 127.53, 126.47, 125.44, 125.40, 125.34, 124.46 (dd, 3JCF = 5.8, 4JCF = 3.3 Hz), 122.05, 120.03, 117.95, 117.37 (d, 2JCF = 17.0 Hz), 116.97 (d, 2JCF = 17.0 Hz), 73.98, 63.24, 60.19, 49.31, 49.20, 42.07, 35.76, 34.00, 32.76. HRMS (ESI) calcd for C25H26F2NO2 [M+H]+ : 410.1926, found: 410.1913.
1’-(3,4-Dichlorophenethyl)-3,4-dihydrospiro[benzo[h]chromene-2,4’-piperidin]-4-ol (3i).
General procedure E was used to reduce 2i (100 mg, 0.22 mmol) in absolute ethanol (3 mL) using sodium borohydride (28 mg, 0.68 mmol). It was purified by flash chromatography (DCM:MeOH = 30:1) to afford 3i (83 mg, 83%) as white solid. Rf (DCM:MeOH = 10:1) = 0.34. 1H NMR (500 MHz, CDCl3) δ 8.33 – 8.15 (m, 1H), 7.85 – 7.66 (m, 1H), 7.59 – 7.45 (m, 3H), 7.40 (d, J = 8.4 Hz, 1H), 7.38 – 7.29 (m, 2H), 7.11 – 6.92 (m, 1H), 5.06 – 4.83 (m, 1H), 2.85 – 2.69 (m, 4H), 2.68 – 2.55 (m, 3H), 2.57 – 2.46 (m, 1H), 2.28 – 2.15 (m, 1H), 2.15 – 2.06 (m, 1H), 2.02 – 1.89 (m, 2H), 1.90 – 1.80 (m, 1H), 1.79 – 1.64 (m, 1H). 13C NMR (100 MHz, CDCl3) δ 147.52, 140.66, 134.33, 132.27, 130.71, 130.36, 130.07, 128.28, 127.65, 126.60, 125.59, 125.50, 125.46, 122.18, 120.14, 118.08, 74.11, 63.31, 60.14, 49.39, 49.28, 42.21, 35.92, 34.00, 32.82.
1’-(2-(3,4-Dichlorophenyl)-2-hydroxyethyl)-3,4-dihydrospiro[benzo[h]chromene-2,4’-piperidin]-4-ol (3k).
General procedure E was used to reduce 2k(160 mg, 0.35 mmol) in absolute ethanol (3 mL) using sodium borohydride (240 mg, 1.0 mmol). It was purified by flash chromatography (DCM:MeOH = 30:1) to afford 3k (140 mg, 87%) as white solid. Rf (DCM:MeOH = 10:1) = 0.52. 1H NMR (500 MHz, CDCl3) δ 8.36 – 8.05 (m, 1H), 7.89 – 7.72 (m, 1H), 7.60 – 7.45 (m, 4H), 7.47 – 7.33 (m, 2H), 7.24 – 7.12 (m, 1H), 4.97 (t, J = 6.6 Hz, 1H), 4.81 – 4.59 (m, 1H), 3.11 – 2.70 (m, 2H), 2.73 – 2.48 (m, 3H), 2.42 (ddd, J = 12.5, 10.7, 8.4 Hz, 1H), 2.31 – 2.07 (m, 2H), 2.08 – 1.91 (m, 2H), 1.92 – 1.67 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 147.49, 142.72, 134.39, 132.61, 131.32, 130.43, 127.94, 127.71, 126.70, 125.66, 125.50, 125.44, 125.25, 122.14, 120.30, 117.90, 73.91, 67.85, 66.12, 66.10, 63.47, 63.46, 50.94, 50.79, 47.87, 47.72, 42.08, 36.12, 35.86, 34.63, 34.36.
2-(3,4-Difluorophenyl)-1-(4-hydroxy-3,4-dihydrospiro[benzo[h]chromene-2,4’-piperidin]-1’yl)ethanone (3l).
General procedure E was used to reduce 2l (340 mg, 0.81 mmol) in absolute ethanol (10 mL) using sodium borohydride (92 mg, 2.4 mmol). It was purified by flash chromatography (DCM:MeOH = 30:1) to afford 3l (250 mg, 73%) as white solid. Rf (DCM:MeOH = 20:1) = 0.69. 1H NMR (600 MHz, CDCl3) δ 8.31 – 8.04 (m, 1H), 7.84 – 7.68 (m, 1H), 7.56 – 7.44 (m, 3H), 7.44 – 7.37 (m, 1H), 7.08 (t, J = 9.1 Hz, 2H), 6.99 – 6.85 (m, 1H), 5.02 – 4.79 (m, 1H), 4.55 – 4.31 (m, 1H), 3.65 (s, 3H), 3.58 – 3.42 (m, 1H), 3.30 – 3.07 (m, 1H), 2.65 (s, 1H), 2.22 – 2.05 (m, 2H), 2.03 – 1.93 (m, 1H), 1.93 – 1.79 (m, 1H), 1.72 – 1.53 (m, 1H), 1.53 – 1.33 (m, 1H). HRMS (ESI) calcd for C25H24F2NO2 [M+H]+ : 424.1719, found: 424.1698.
tert-Butyl 3,4-dihydrospiro[benzo[h]chromene-2,4’-piperidine]-1’-carboxylate (4).
General procedure E followed by general procedure G was used to convert 2 to 4 over 2 steps in 47% yield. 1H NMR (500 MHz, CDCl3) δ 8.27 – 8.20 (m, 1H), 7.80 – 7.73 (m, 1H), 7.52 – 7.41 (m, 2H), 7.35 (d, J = 8.3 Hz, 1H), 7.17 (d, J = 8.3 Hz, 1H), 4.00 (s, 2H), 3.36 (d, J = 11.5 Hz, 2H), 2.90 (t, J = 6.8 Hz, 2H), 1.92 (q, J = 8.9, 6.8 Hz, 4H), 1.70 – 1.56 (m, 2H), 1.51 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 154.98, 147.67, 133.41, 127.71, 127.57, 125.76, 125.75, 125.33, 121.32, 119.56, 114.89, 79.53, 73.13, 39.88, 34.55, 32.25, 28.59, 21.82.
3,4-Dihydrospiro[benzo[h]chromene-2,4’-piperidine] (5a).
General procedure B was used to deprotect 4 (1.4 g, 4.0 mmol) in DCM (40 mL) and TFA (13 mL) to afford 5a (982 mg, 97%) as a brown solid. Rf (DCM:MeOH = 10:1) = 0.25. 1H NMR (400 MHz, CDCl3) δ 8.31 – 8.21 (m, 1H), 7.82 – 7.70 (m, 1H), 7.54 – 7.39 (m, 2H), 7.37 – 7.31 (m, 1H), 7.16 (d, J = 8.3 Hz, 1H), 3.22 (td, J = 12.2, 2.8 Hz, 2H), 3.14 (d, J = 4.3 Hz, 1H), 3.05 – 2.93 (m, 2H), 2.88 (t, J = 6.8 Hz, 2H), 2.00 – 1.83 (m, 4H), 1.73 – 1.61 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 147.85, 133.39, 127.75, 127.50, 125.84, 125.65, 125.18, 121.49, 119.29, 114.94, 73.36, 47.99, 42.22, 35.48, 34.82, 32.55, 21.74. HRMS (ESI) calcd for C17H20NO [M+H]+ : 254.1539, found: 254.1530.
1’-Ethyl-3,4-dihydrospiro[benzo[h]chromene-2,4’-piperidine] (5b).
General procedure F was used to alkylate 5a (60 mg, 0.23 mmol) with bromoethane (25 mg, 0.23 mmol) using potassium carbonate (64 mg, 0.46 mmol) in DMF (2.5 mL). It was purified by flash chromatography (DCM:MeOH = 30:1) to afford 5b (45 mg, 68 %) as brown oil.. Rf (DCM:MeOH = 10:1) = 0.28. 1H NMR (400 MHz, CDCl3) δ 8.35 – 8.14 (m, 1H), 7.96 – 7.64 (m, 1H), 7.58 – 7.39 (m, 2H), 7.33 (d, J = 8.3 Hz, 1H), 7.16 (d, J = 8.3 Hz, 1H), 2.89 (t, J = 6.8 Hz, 2H), 2.86 – 2.70 (m, 2H), 2.62 – 2.43 (m, 4H), 2.02 – 1.95 (m, 2H), 1.91 (t, J = 6.8 Hz, 2H), 1.82 – 1.72 (m, 2H), 1.15 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 147.94, 133.45, 127.84, 127.55, 125.92, 125.65, 125.20, 121.55, 119.29, 115.07, 73.06, 52.69, 49.17, 34.81, 32.33, 22.07, 12.37. HRMS (ESI) calcd for C19H24NO [M+H]+ : 282.1852, found: 282.1841.
1’-Benzyl-3,4-dihydrospiro[benzo[h]chromene-2,4’-piperidine] (5c).
General procedure F was used to alkylate 5a (50mg, 0.19mmol) with (bromomethyl)benzene (33mg, 0.19mmol) using potassium carbonate (55mg, 0.39mmol) in DMF (2 mL) for 1 h. It was purified by flash chromatography (Hexanes:EtOAc = 10:1) to afford 5c (55 mg, 81%) as white solid. Rf (EtOAc) = 0.54. 1H NMR (500 MHz, CDCl3) δ 8.24 (dd, J = 8.0, 1.7 Hz, 1H), 7.76 (dd, J = 7.4, 1.8 Hz, 1H), 7.56 – 7.42 (m, 2H), 7.41 – 7.31 (m, 5H), 7.31 – 7.27 (m, 1H), 7.16 (d, J = 8.3 Hz, 1H), 3.62 (s, 2H), 2.89 (t, J = 6.8 Hz, 2H), 2.83 – 2.68 (m, 2H), 2.67 – 2.51 (m, 2H), 2.09 – 1.83 (m, 4H), 1.84 – 1.71 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 147.97, 138.60, 133.44, 129.40, 128.31, 127.82, 127.53, 127.09, 125.93, 125.64, 125.20, 121.61, 119.25, 115.04, 73.04, 63.49, 49.46, 34.83, 32.25, 22.06. HRMS (ESI) calcd for C24H26NO [M+H]+ : 344.2009, found: 344.2003.
1’-Phenethyl-3,4-dihydrospiro[benzo[h]chromene-2,4’-piperidine] (5d).
General procedure F was used to alkylate 5a (50 mg, 0.19 mmol) with (2-bromoethyl)benzene (37 mg, 0.19 mmol) using potassium carbonate (55 mg, 0.39 mmol) in DMF (2 mL). It was purified by flash chromatography (Hexanes:EtOAc = 8:1) to afford 5d (57 mg, 81%) as white solid. Rf (EtOAc) = 0.51. 1H NMR (500 MHz, CDCl3) δ 8.32 – 8.19 (m, 1H), 7.81 – 7.72 (m, 1H), 7.55 – 7.42 (m, 2H), 7.39 – 7.29 (m, 3H), 7.29 – 7.21 (m, 3H), 7.18 (d, J = 8.3 Hz, 1H), 2.99 – 2.82 (m, 6H), 2.80 – 2.70 (m, 2H), 2.70 – 2.58 (m, 2H), 2.09 – 1.97 (m, 2H), 1.94 (t, J = 6.8 Hz, 2H), 1.87 – 1.72 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 147.93, 140.62, 133.45, 128.86, 128.53, 127.84, 127.57, 126.16, 125.92, 125.69, 125.25, 121.56, 119.33, 115.07, 72.99, 61.04, 49.58, 34.87, 34.06, 32.35, 22.08. HRMS (ESI) calcd for C25H28NO [M+H]+ : 358.2165, found: 358.2161.
1’-(2-Cyclohexylethyl)-3,4-dihydrospiro[benzo[h]chromene-2,4’-piperidine] (5e).
General procedure F was used to alkylate 5a (50 mg, 0.19 mmol) with (2-bromoethyl)cyclohexane (38 mg, 0.19 mmol) using potassium carbonate (55 mg, 0.39 mmol) in DMF (2 mL). It was purified by flash chromatography (Hexanes:EtOAc = 5:1;) to afford 5e (54 mg, 75%) as yellow solid. Rf (EtOAc) = 0.49. 1H NMR (500 MHz, CDCl3) δ 8.31 – 8.18 (m, 1H), 7.75 (dd, J = 7.7, 1.5 Hz, 1H), 7.51 – 7.39 (m, 2H), 7.33 (d, J = 8.3 Hz, 1H), 7.16 (d, J = 8.3 Hz, 1H), 2.88 (t, J = 6.8 Hz, 2H), 2.84 – 2.69 (m, 2H), 2.60 – 2.49 (m, 2H), 2.48 – 2.33 (m, 2H), 2.07 – 1.93 (m, 2H), 1.90 (t, J = 6.8 Hz, 2H), 1.84 – 1.58 (m, 7H), 1.53 – 1.37 (m, 2H), 1.35 – 1.10 (m, 5H), 1.05 – 0.78 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 147.99, 133.44, 127.84, 127.53, 125.94, 125.64, 125.18, 121.60, 119.24, 115.06, 73.11, 57.11, 49.74, 36.67, 35.01, 34.89, 33.66, 32.33, 26.77, 26.48, 22.08. HRMS (ESI) calcd for C25H34NO [M+H]+ : 364.2635, found: 364.2609.
1’-(3-Phenylpropyl)-3,4-dihydrospiro[benzo[h]chromene-2,4’-piperidine] (5f).
General procedure F was used to alkylate 5a (50 mg, 0.19 mmol) with (3-bromopropyl)benzene (39 mg, 0.19 mmol) using potassium carbonate (55 mg, 0.39 mmol) in DMF (2 mL). It was purified by flash chromatography (Hexanes:EtOAc = 5:1) to afford 5f (50 mg, 69%) as yellow solid. Rf (EtOAc) = 0.48. 1H NMR (500 MHz, CDCl3) δ 8.31 – 8.18 (m, 1H), 7.85 – 7.68 (m, 1H), 7.54 – 7.40 (m, 2H), 7.38 – 7.29 (m, 3H), 7.25 – 7.19 (m, 3H), 7.17 (d, J = 8.3 Hz, 1H), 2.90 (t, J = 6.8 Hz, 2H), 2.83 – 2.73 (m, 2H), 2.69 (t, J = 7.8 Hz, 2H), 2.61 – 2.43 (m, 4H), 2.02 – 1.95 (m, 2H), 1.95 – 1.86 (m, 4H), 1.83 – 1.71 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 147.95, 142.33, 133.42, 128.51, 128.43, 127.82, 127.53, 125.91, 125.86, 125.64, 125.19, 121.58, 119.25, 115.03, 73.06, 58.50, 49.60, 34.87, 34.03, 30.44, 29.09, 22.05. HRMS (ESI) calcd for C26H30NO [M+H]+: 372.2322, found: 372.2309.
1-(3,4-Difluorophenyl)-2-(1,4-dihydro-2H-spiro[phenanthrene-3,4’-piperidin]-1’-yl)ethan-1-one (5g)
To a solution of 5a (500 mg, 1.85 mmol) and 2-bromo-1-(3,4-difluorophenyl)ethanone (436 mg, 1.85 mmol) in acetonitrile (15 mL) was added sodium bicarbonate (311 mg, 3.70 mmol) at 0 °C. The reaction mixture was allowed to stir to room temperature and held at room temperature for 4 hours. The acetonitrile was removed under reduced pressure, the residue was diluted with water and extracted with EtOAc (20 mL × 3). The combined organic layer was dried with sodium sulfate, the solvent was evaporated and the residue was purified by flash chromatography (DCM: MeOH = 50:1) to afford 5g (400 mg, 49%) as yellow solid. Rf (DCM:MeOH = 10:1) = 0.55. 1H NMR (500 MHz, CDCl3) δ 8.35 – 8.14 (m, 1H), 7.96 – 7.64 (m, 1H), 7.58 – 7.39 (m, 2H), 7.33 (d, J = 8.3 Hz, 1H), 7.16 (d, J = 8.3 Hz, 1H), 2.89 (t, J = 6.8 Hz, 2H), 2.86 – 2.70 (m, 2H), 2.62 – 2.43 (m, 4H), 2.02 – 1.95 (m, 2H), 1.91 (t, J = 6.8 Hz, 2H), 1.89 – 1.80 (m, 2H), 1.66 – 1.51 (m, 1H).
1’-(3,4-Difluorophenethyl)-3,4-dihydrospiro[benzo[h]chromene-2,4’-piperidine] (5h).
General procedure F was used to alkylate 5a (130 mg, 0.51 mmol) with 4-(2-bromoethyl)-1,2-difluorobenzene (113 mg, 0.51 mmol) using potassium carbonate (142 mg, 1.0 mmol) in DMF (4 mL). It was purified by flash chromatography (DCM: MeOH = 30: 1) to afford 5h (134 mg, 67%) as yellow solid. Rf (DCM:MeOH = 20:1) = 0.46. 1H NMR (600 MHz, CDCl3) δ 8.33 – 8.16 (m, 1H), 7.83 – 7.66 (m, 1H), 7.54 – 7.39 (m, 2H), 7.34 (d, J = 8.3 Hz, 1H), 7.17 (d, J = 8.2 Hz, 1H), 7.12 – 7.01 (m, 2H), 6.98 – 6.82 (m, 1H), 2.90 (t, J = 6.8 Hz, 2H), 2.87 – 2.79 (m, 4H), 2.73 – 2.57 (m, 4H), 2.04 – 1.97 (m, 2H), 1.92 (t, J = 6.8 Hz, 2H), 1.83 – 1.69 (m, 2H). 13C NMR (151 MHz, CDCl3) δ 150.15 (dd, 1JCF = 246.73, 2JCF = 12.83 Hz), 148.87 (dd, 1JCF = 245.91, 2JCF = 12.28 Hz), 147.71, 137.39 (dd, 3JCF = 6.35, 4JCF = 4.23 Hz), 133.31, 127.68, 127.44, 125.75, 125.54, 125.10, 124.48 (dd, 3JCF = 6.21, 4JCF = 3.78 Hz), 117.39 (d, 2JCF = 15.96 Hz), 116.95 (d, 2JCF = 17.01 Hz), 114.90, 72.72, 60.26, 49.38, 34.61, 32.93, 32.15, 29.69, 21.89. HRMS (ESI) calcd for C25H26F2NO [M+H]+ : 394.1977, found: 394.1968.
1-(3,4-Difluorophenyl)-2-(3,4-dihydrospiro[benzo[h]chromene-2,4’-piperidin]-1’-yl)ethanol (5i).
General procedure D was used with 5a (200 mg, 0.79 mmol) and 2-(3,4-difluorophenyl)oxirane11 (123 mg, 0.79 mmol) in ethanol (12 mL). It was purified by flash chromatography (DCM: MeOH = 100:1) to afford 5i (194 mg, 60%) as white solid. Rf (DCM:MeOH = 10:1) = 0.52. 1H NMR (600 MHz, CDCl3) δ 8.23 (d, J = 8.0 Hz, 1H), 7.74 (d, J = 7.8 Hz, 1H), 7.43 (p, J = 6.9 Hz, 2H), 7.33 (d, J = 8.3 Hz, 1H), 7.24 (d, J = 8.2 Hz, 1H), 7.15 (d, J = 8.3 Hz, 1H), 7.10 (t, J = 8.8 Hz, 1H), 7.07 (d, J = 4.3 Hz, 1H), 4.79 – 4.58 (m, 1H), 3.05 – 2.84 (m, 4H), 2.75 – 2.54 (m, 3H), 2.43 (t, J = 11.6 Hz, 1H), 2.07 – 1.94 (m, 2H), 1.91 (t, J = 6.8 Hz, 2H), 1.82 – 1.68 (m, 2H). 13C NMR (151 MHz, CDCl3) δ 150.55 (dd, 1JCF = 247.78, 2JCF = 12.46 Hz), 149.72 (dd, 1JCF = 247.07, 2JCF = 12.46 Hz), 147.75, 139.63 (dd, 3JCF = 4.99, 4JCF =3.79 Hz), 133.46, 127.80, 127.61, 125.85, 125.71, 125.26, 121.74 (dd, 3JCF = 6.16, 4JCF = 3.34 Hz), 121.40, 119.48, 117.12 (d, 2JCF = 17.17 Hz), 115.00, 114.90 (d, 2JCF = 18.47 Hz), 72.69, 67.97, 66.28, 51.00, 47.90, 35.01, 34.76, 32.23, 21.98. HRMS (ESI) calcd for C25H26F2NO2 [M+H]+ : 410.1926, found: 410.1920.
2-(3,4-Difluorophenyl)-1-(3,4-dihydrospiro[benzo[h]chromene-2,4’-piperidin]-1’ yl)ethanone (5j).
General procedure H was used in the amidation reaction of 5a (100mg, 0.39mmol) and 2-(3,4-difluorophenyl)acetic acid (68 mg, 0.39 mmol) with EDCI (151 mg, 0.79 mmol) and DMAP (10 mg, 0.078 mmol) in DCM (15 mL). It was purified by flash chromatography (Hexanes:EtOAc = 2:1) to afford 5j (126 mg, 79%) as yellow solid. Rf (EtOAc) = 0.69. 1H NMR (400 MHz, CDCl3) δ 8.26 – 8.08 (m, 1H), 7.82 – 7.64 (m, 1H), 7.54 – 7.40 (m, 2H), 7.41 – 7.32 (m, 1H), 7.22 – 7.05 (m, 3H), 7.04 – 6.90 (m, 1H), 4.91 – 4.15 (m, 1H), 3.70 (s, 3H), 3.67 – 3.53 (m, 1H), 3.29 – 3.11 (m, 1H), 2.92 – 2.76 (m, 2H), 2.11 – 1.93 (m, 2H), 1.94 – 1.80 (m, 2H), 1.66 – 1.51 (m, 1H), 1.49 – 1.32 (m, 1H).
tert-Butyl-3,4,7,8,9,10-hexahydrospiro[benzo[h]chromene-2,4’-piperidine]-1’-carboxylate (6).
General procedure E followed by general procedure G was used to convert 2 to 6 over 2 steps in 17% yield. 1H NMR (500 MHz, CDCl3) δ 6.83 (d, J = 7.8 Hz, 1H), 6.61 (d, J = 7.7 Hz, 1H), 3.95 (s, 2H), 3.20 (s, 2H), 2.81 – 2.70 (m, 4H), 2.68 (t, J = 6.3 Hz, 2H), 1.88 – 1.69 (m, 8H), 1.54 (dd, J = 13.0, 5.0 Hz, 2H), 1.49 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 154.98, 150.51, 136.42, 126.18, 125.73, 120.61, 117.34, 79.46, 72.38, 39.54, 34.90, 32.35, 29.61, 28.58, 23.27, 23.04, 22.96, 21.40.
3,4,7,8,9,10-Hexahydrospiro[benzo[h]chromene-2,4’-piperidine] (6a).
General procedure B was used to deprotect 6 (200 mg, 0.56 mmol) in DCM (8 mL) and TFA (2.5 mL) to afford6a (117 mg, 81%) as a yellow solid. Rf (DCM:MeOH = 10:1) = 0.20. 1H NMR (500 MHz, CDCl3) δ 6.82 (d, J = 7.8 Hz, 1H), 6.61 (d, J = 7.8 Hz, 1H), 4.20 (s, 2H), 3.12 (t, J = 12.2 Hz, 2H), 2.98 (d, J = 11.8 Hz, 2H), 2.87 – 2.58 (m, 6H), 2.10 – 1.70 (m, 7H), 1.70 – 1.55 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 150.50, 136.33, 126.13, 125.67, 120.50, 117.34, 72.25, 41.85, 35.00, 32.52, 29.58, 23.28, 23.01, 22.93, 21.24. HRMS (ESI) calcd for C17H24NO [M+H]+ : 258.1852, found: 258.1848.
1’-Ethyl-3,4,7,8,9,10-hexahydrospiro[benzo[h]chromene-2,4’-piperidine] (6b).
General procedure F was used to alkylate 6a (30 mg, 0.11 mmol) with bromoethane (12 mg, 0.11 mmol) using potassium carbonate (32 mg, 0.233 mmol) in DMF (1 mL). It was purified by flash chromatography (Hexanes:EtOAc = 2:1) to afford 6b (19 mg, 57%) as white solid. Rf (EtOAc) = 0.44. 1H NMR (500 MHz, CDCl3) δ 6.81 (d, J = 7.7 Hz, 1H), 6.59 (d, J = 7.7 Hz, 1H), 3.35 (s, 1H), 2.90 – 2.77 (m, 2H), 2.77 – 2.68 (m, 4H), 2.66 (t, J = 6.3 Hz, 2H), 2.53 (q, J = 7.2 Hz, 2H), 2.43 (t, J = 11.8 Hz, 2H), 1.97 – 1.82 (m, 2H), 1.82 – 1.64 (m, 7H), 1.15 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 150.50, 136.33, 126.13, 125.67, 120.50, 117.34, 72.25, 41.85, 35.00, 32.52, 29.58, 23.28, 23.01, 22.93, 22.07, 21.24, 12.33. HRMS (ESI) calcd for C19H28NO [M+H]+ : 286.2165, found: 286.2154.
1’-(3,4-Difluorophenethyl)-3,4,7,8,9,10-hexahydrospiro[benzo[h]chromene-2,4’-piperidine] (6c).
General procedure F was used to alkylate 6a (50 mg, 0.19 mmol) with 4-(2-bromoethyl)1,2-difluorobenzene (43 mg, 0.19 mmol) using potassium carbonate (54 mg, 0.38 mmol) in DMF (2 mL). It was purified by flash chromatography (Hexanes:EtOAc = 10:1) to afford 6c (39 mg, 51%) as white solid. Rf (EtOAc) = 0.44. 1H NMR (500 MHz, CDCl3) δ 7.17 – 6.99 (m, 2H), 6.99 – 6.89 (m, 1H), 6.83 (d, J = 7.7 Hz, 1H), 6.60 (d, J = 7.7 Hz, 1H), 2.86 – 2.70 (m, 8H), 2.68 (t, J = 6.3 Hz, 2H), 2.66 – 2.60 (m, 2H), 2.54 – 2.40 (m, 2H), 1.95 – 1.83 (m, 2H), 1.83 – 1.72 (m, 6H), 1.72 – 1.63 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 150.72, 150.28 (dd, 1JCF = 246.4, 2JCF = 11.4 Hz), 149.00 (dd, 1JCF = 246.4, 2JCF = 11.4 Hz), 137.64 (dd, 3JCF = 5.7, 4JCF = 3.9 Hz), 136.34, 126.19, 125.75, 124.62 (dd, 3JCF = 6.0, 4JCF =3.4 Hz), 120.43, 117.54 (d, 2JCF = 16.6 Hz), 117.53, 117.07 (d, 2JCF = 17.0 Hz), 72.13, 60.54, 49.52, 34.93, 33.18, 32.38, 29.67, 23.44, 23.10, 23.06, 21.62. HRMS (ESI) calcd for C25H30F2NO [M+H]+ : 398.2290, found: 398.2279.
tert-Butyl spiro[benzo[h]chromene-2,4’-piperidine]-1’-carboxylate (7).
General procedure E was used to reduce 2 (1.4 g, 3.8 mmol) in absolute ethanol (30 mL) using sodium borohydride (432 mg, 11.4 mmol). To the above product was added PTSA·H2O (36 mg, 5 mol %) and refluxed for overnight. The reaction was cooled down, quenched with sodium bicarbonate and extracted with ethyl acetate. The organic layer was washed with brine, dried with sodium sulfate and the solvent was removed under reduced pressure. It was purified by flash chromatography (Hexanes:EA = 45:1) to afford 7 (800 mg, 61%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.32 – 8.14 (m, 1H), 7.82 – 7.70 (m, 1H), 7.55 – 7.41 (m, 2H), 7.37 (d, J = 8.4 Hz, 1H), 7.15 (d, J = 8.3 Hz, 1H), 6.51 (d, J = 9.6 Hz, 1H), 5.64 – 5.50 (m, 1H), 3.95 (s, 2H), 3.46 (t, J = 12.8 Hz, 2H), 2.38 – 2.03 (m, 2H), 1.80 – 1.59 (m, 2H), 1.51 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 154.87, 147.46, 134.54, 127.77, 127.76, 126.30, 125.59, 125.01, 124.57, 124.16, 121.68, 120.44, 115.99, 79.58, 75.51, 39.25, 34.90, 28.52.
Spiro[benzo[h]chromene-2,4’-piperidine](7a).
General procedure B was used to deprotect 7 (1.4 g, 4.0 mmol) in DCM (40 mL) and TFA (13 mL) to afford 7a (982 mg, 97%) as a brown solid. Rf (DCM:MeOH = 10:1) = 0.25. 1H NMR (400 MHz, CDCl3) δ 8.32 – 8.14 (m, 1H), 7.82 – 7.70 (m, 1H), 7.55 – 7.41 (m, 2H), 7.37 (d, J = 8.4 Hz, 1H), 7.15 (d, J = 8.3 Hz, 1H), 6.51 (d, J = 9.6 Hz, 1H), 5.64 – 5.50 (m, 1H), 3.95 (s, 2H), 3.46 (t, J = 12.8 Hz, 2H), 2.38 – 2.03 (m, 2H), 1.80 – 1.59 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 147.76, 134.54, 127.97, 127.76, 126.30, 125.59, 125.01, 124.57, 124.16, 121.68, 120.44, 115.99, 79.58, 74.51, 37.25.
1’-Ethylspiro[benzo[h]chromene-2,4’-piperidine] (7b).
General procedure F was used to alkylate 7a (60 mg, 0.23 mmol) with bromoethane (25 mg, 0.23 mmol) using potassium carbonate (64 mg, 0.46 mmol) in DMF (2.5 mL). It was purified by flash chromatography (DCM:MeOH = 30:1) to afford 7b (45 mg, 68%) as a brown oil. Rf (DCM:MeOH = 10:1) = 0.28. 1H NMR (400 MHz, CDCl3) δ 8.32 – 8.14 (m, 1H), 7.82 – 7.70 (m, 1H), 7.55 – 7.41 (m, 2H), 7.37 (d, J = 8.4 Hz, 1H), 7.15 (d, J = 8.3 Hz, 1H), 6.51 (d, J = 9.6 Hz, 1H), 5.64 – 5.50 (m, 1H), 3.95 (s, 2H), 3.46 (t, J = 12.8 Hz, 2H), 2.38 – 2.03 (m, 2H), 1.85 – 1.79 (m, 2H). 1.75 – 1.52 (m, 2H), 1.15 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 147.76, 134.54, 127.97, 127.76, 126.30, 125.59, 125.01, 124.57, 124.16, 121.68, 120.44, 115.99, 79.58, 74.51, 37.25, 22.07, 12.37.
1’-(3,4-Difluorophenethyl)spiro[benzo[h]chromene-2,4’-piperidine] (7c).
General procedure F was used to alkylate 7a (200 mg, 0.79 mmol) with 4-(2-bromoethyl)-1,2-difluorobenzene (176 mg, 0.79 mmol) using potassium carbonate (220 mg, 1.6 mmol) in DMF (5 mL). It was purified by flash chromatography (DCM: MeOH = 250:1) to afford 7c (157 mg, 51%) as a yellow solid. Rf (DCM:MeOH = 20:1) = 0.46. 1H NMR (500 MHz, CDCl3) δ 8.27 (t, J = 4.9 Hz, 1H), 7.76 (d, J = 8.1 Hz, 1H), 7.58 – 7.41 (m, 2H), 7.37 (d, J = 8.3 Hz, 1H), 7.16 (dd, J = 8.5, 2.2 Hz, 1H), 7.14 – 7.01 (m, 2H), 7.00 – 6.87 (m, 1H), 6.50 (dd, J = 9.8, 2.3 Hz, 1H), 5.61 (dd, J = 9.9, 2.2 Hz, 1H), 2.81 (t, J = 8.0 Hz, 2H), 2.79 – 2.65 (m, 6H), 2.31 – 2.19 (m, 2H), 1.95 – 1.75 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 150.28 (dd, 1JCF = 247.1, 2JCF = 12.5 Hz), 148.99 (dd, 1JCF = 245.8, 2JCF = 12.5 Hz), 147.71, 137.56 (dd, 3JCF = 8.5, 4JCF = 4.8 Hz), 134.61, 128.45, 127.83, 126.27, 125.52, 125.22, 124.70, 124.61 (dd, 3JCF = 5.7, 4JCF = 3.4 Hz), 124.03, 121.94, 120.31, 117.53 (d, 2JCF = 16.8 Hz), 117.07 (d, 2JCF = 16.8 Hz), 116.22, 75.43, 60.47, 48.81, 35.45, 33.14. HRMS (ESI) calcd for C25H24F2NO [M+H]+ : 392.1820, found: 392.1800.
(R)-Spiro[benzo[h]chromene-2,3’-piperidin]-4(3H)-one (10a).
A solution of 9a (300 mg, 0.6 mmol) in a 1:1 mixture of ethanol and hydrochloric acid (10 mL) was refluxed overnight. The organic solvent was removed under reduced pressure, neutralized with 10% NaOH and then extracted 3 times with DCM. The organic layer was combined, dried with sodium sulfate and concentrated under reduced pressure. It was purified by flash chromatography (DCM:MeOH= 30:1) to afford 10a (160 mg, 89%) as a yellow solid. Rf (DCM:MeOH = 10:1) = 0.6. 1H NMR (500 MHz, DMSO-d6) δ 8.74 – 8.61 (m, 1H), 7.95 – 7.85 (m, 1H), 7.77 – 7.64 (m, 2H), 7.64 – 7.55 (m, 1H), 7.54 – 7.44 (m, 1H), 3.32 – 3.23 (m, 1H), 3.09 – 2.94 (m, 3H), 2.84 – 2.73 (m, 1H), 2.74 – 2.64 (m, 1H), 2.12 – 1.98 (m, 1H), 1.80 – 1.64 (m, 2H), 1.56 – 1.45 (m, 1H). 13C NMR (126 MHz, DMSO-d6) δ 190.56, 156.52, 137.17, 129.82, 127.78, 126.50, 124.80, 124.05, 120.86, 120.61, 114.53, 78.28, 51.13, 44.24, 43.84, 30.38, 20.60. (2R)-10a [α]D20 −0.07 (c 1.0, CH3Cl). HRMS (ESI) calcd for C17H17NO2 [M+H]+ : 268.1332, found: 268.1317.
(R)-1’-Ethylspiro[benzo[h]chromene-2,3’-piperidin]-4(3H)-one (10b).
General procedure F was used to alkylate 10a (135 mg, 0.51 mmol) with bromoethane (55 mg, 0.51 mmol) using potassium carbonate (140 mg, 1.0 mmol) in DMF (4 mL). It was purified by flash chromatography (DCM:MeOH = 100:1) to afford 10b (113 mg, 75%) as a yellow solid. Rf (DCM:MeOH = 10:1) = 0.6. 1H NMR (400 MHz, CDCl3) δ 8.44 – 8.28 (m, 1H), 7.83 (d, J = 8.7 Hz, 1H), 7.79 – 7.71 (m, 1H), 7.66 – 7.55 (m, 1H), 7.56 – 7.45 (m, 1H), 7.44 – 7.31 (m, 1H), 3.05 (d, J = 16.8 Hz, 1H), 2.86 (d, J = 16.7 Hz, 1H), 2.77 – 2.61 (m, 2H), 2.58 – 2.49 (m, 1H), 2.48 – 2.33 (m, 3H), 2.09 – 1.91 (m, 1H), 1.91 – 1.77 (m, 2H), 1.73 – 1.58 (m, 1H), 1.00 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 191.76, 157.85, 137.79, 129.53, 127.85, 126.12, 125.48, 123.88, 121.58, 120.45, 114.98, 80.04, 60.18, 53.17, 52.16, 44.94, 33.91, 22.12, 12.14. (2R)-10b [α]D20 −0.06 (c 1.0, CH2Cl2). HRMS (ESI) calcd for C19H22NO2 [M+H]+ : 296.1645, found: 296.1624.
(R)-1’-(3,4-Difluorophenethyl)spiro[benzo[h]chromene-2,3’-piperidin]-4(3H)-one (10c).
General procedure F was used to alkylate 10a (150 mg, 0.6 mmol) with 4-(2-bromoethyl)-1,2difluorobenzene (124 mg, 0.6 mmol) using potassium carbonate (166 mg, 1.2 mmol) in DMF (5 mL). It was purified by flash chromatography (DCM: MeOH = 200: 1) to afford 10c (169 mg, 69%) as yellow solid. Rf (DCM:MeOH = 20:1) = 0.70. 1H NMR (500 MHz, CDCl3) δ 8.44 – 8.22 (m, 1H), 7.83 (d, J = 8.7 Hz, 1H), 7.80 – 7.71 (m, 1H), 7.67 – 7.58 (m, 1H), 7.57 – 7.50 (m, 1H), 7.42 – 7.33 (m, 1H), 7.07 – 6.94 (m, 1H), 6.95 – 6.86 (m, 1H), 6.84 – 6.72 (m, 1H), 3.00 – 2.93 (m, 1H), 2.83 (t, J = 15.3 Hz, 2H), 2.71 – 2.53 (m, 6H), 2.51 – 2.33 (m, 1H), 2.14 – 1.99 (m, 1H), 2.00 – 1.85 (m, 1H), 1.83 – 1.67 (m, 1H), 1.71 – 1.51 (m, 1H). 13C NMR (126 MHz, CDCl3) δ 191.44, 157.64, 150.03 (dd, 1JCF = 247.6, 2JCF = 12.6 Hz), 148.82 (dd, 1JCF = 246.3, 2JCF = 14.1 Hz), 137.72, 137.3 (dd, 3JCF = 8.5, 4JCF = 5.2 Hz), 129.58, 127.78, 126.17, 125.32, 124.52 (dd, 3JCF = 6.1, 4JCF = 3.7 Hz), 123.73, 121.39, 120.50, 117.33 (d, 2JCF = 17.2 Hz), 116.74 (d, 2JCF = 17.2 Hz), 114.79, 79.63, 60.01, 59.14, 53.52, 45.11, 33.59, 32.37, 21.80. (2R)-10c [α]D20 −0.52 (c 1.0, CH2Cl2). HRMS (ESI) calcd for C25H24F2NO2 [M+H]+ : 408.1770, found: 408.1755.
(S)-Spiro[benzo[h]chromene-2,3’-piperidin]-4(3H)-one (10d).
A solution of 9b (300 mg, 0.6 mmol) in a 1:1 mixture of ethanol and hydrochloric acid (10 mL) was refluxed overnight. The organic solvent was removed under reduced pressure, neutralized with 10% NaOH and then extracted 3 times with DCM. The organic layer was combined, dried with sodium sulfate and concentrated under reduced pressure. It was purified by flash chromatography (DCM:MeOH= 30:1) to afford 10d (160 mg, 89%) as a yellow solid. Rf (DCM:MeOH = 10:1) = 0.6. 1H NMR (500 MHz, DMSO-d6) δ 8.74 – 8.61 (m, 1H), 7.95 – 7.85 (m, 1H), 7.77 – 7.64 (m, 2H), 7.64 – 7.55 (m, 1H), 7.54 – 7.44 (m, 1H), 3.32 – 3.23 (m, 1H), 3.09 – 2.94 (m, 3H), 2.84 – 2.73 (m, 1H), 2.74 – 2.64 (m, 1H), 2.12 – 1.98 (m, 1H), 1.80 – 1.64 (m, 2H), 1.56 – 1.45 (m, 1H). 13C NMR (126 MHz, DMSO-d6) δ 190.13, 156.25, 137.18, 129.89, 127.74, 126.50, 124.72, 124.35, 120.87, 120.80, 114.54, 77.92, 49.87, 44.09, 42.98, 29.17, 19.29. (2S)-10d [α]D20 +0.04 (c 1.0, CH3Cl). HRMS (ESI) calcd for C17H17NO2 [M+H]+ : 268.1332, found: 268.1320.
(S)-1’-Ethylspiro[benzo[h]chromene-2,3’-piperidin]-4(3H)-one (10e).
General procedure F was used to alkylate 10d (180 mg, 0.67 mmol) with bromoethane (73 mg, 0.67 mmol) using potassium carbonate (186 mg, 1.3 mmol) in DMF (4 mL). It was purified by flash chromatography (DCM:MeOH = 100:1) to afford 10e (146 mg, 73%) as yellow solid. Rf (DCM:MeOH = 10:1) = 0.6. 1H NMR (400 MHz CDCl3) δ 8.39 – 8.28 (m, 1H), 7.83 (d, J = 8.7 Hz, 1H), 7.78 – 7.71 (m, 1H), 7.65 – 7.56 (m, 1H), 7.55 – 7.46 (m, 1H), 7.39 – 7.28 (m, 1H), 3.05 (d, J = 16.8 Hz, 1H), 2.86 (d, J = 16.7 Hz, 1H), 2.76 – 2.61 (m, 2H), 2.57 – 2.46 (m, 1H), 2.46 – 2.27 (m, 3H), 2.02 – 1.91 (m, 1H), 1.90 – 1.76 (m, 2H), 1.74 – 1.56 (m, 1H), 0.99 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 191.74, 157.84, 137.78, 129.52, 127.84, 126.12, 125.47, 123.87, 121.57, 120.44, 114.97, 80.03, 60.17, 53.16, 52.15, 44.93, 33.91, 22.12, 12.14. (2S)-10e [α]D20 +0.15 (c 1.0, CH2Cl2). HRMS (ESI) calcd for C19H22NO2 [M+H]+ : 296.1645, found: 296.1634.
(S)-1’-(3,4-Difluorophenethyl)spiro[benzo[h]chromene-2,3’-piperidin]-4(3H)-one (10f).
General procedure F was used to alkylate 10d (150 mg, 0.6 mmol) with 4-(2-bromoethyl)-1,2-difluorobenzene (124 mg, 0.6 mmol) using potassium carbonate (166 mg, 1.2 mmol) in DMF (5 mL). It was purified by flash chromatography (DCM: MeOH = 200: 1) to afford 10f (169 mg, 69%) as a yellow solid. Rf (DCM:MeOH = 20:1) = 0.70. 1H NMR (500 MHz, CDCl3) δ 8.32 (s, 1H), 7.83 (d, J = 8.6 Hz, 1H), 7.78 (d, J = 8.1 Hz, 1H), 7.68 – 7.59 (m, 1H), 7.57 – 7.48 (m, 1H), 7.37 (d, J = 8.6 Hz, 1H), 7.02 – 6.95 (m, 1H), 6.95 – 6.86 (m, 1H), 6.84 – 6.71 (m, 1H), 3.02 – 2.93 (m, 1H), 2.92 – 2.76 (m, 2H), 2.69 – 2.53 (m, 6H), 2.52 – 2.39 (m, 1H), 2.17 – 1.99 (m, 1H), 2.00 – 1.88 (m, 1H), 1.83 – 1.72 (m, 1H), 1.71 – 1.58 (m, 1H). 13C NMR (126 MHz, CDCl3) δ 191.59, 157.76, 150.02 (dd, 1JCF = 246.8, 2JCF = 13.3 Hz), 148.76 (dd, 1JCF = 246.8, 2JCF = 13.3 Hz), 137.81, 137.38 (dd, 3JCF = 5.7, 4JCF = 3.8 Hz), 129.66, 127.88, 126.23, 125.43, 124.51 (dd, 3JCF = 6.1, 4JCF = 3.1 Hz), 123.81, 121.50, 120.56, 117.33 (d, 2JCF = 16.9 Hz), 116.72 (d, 2JCF = 16.8 Hz), 114.89, 79.76, 60.18, 59.25, 53.64, 45.21, 33.73, 32.52, 21.97. (2S)-10f [α]D20 +0.57 (c 1.0, CH2Cl2). HRMS (ESI) calcd for C25H24F2NO2 [M+H]+ : 408.1770, found: 408.1746.
(2R)-1’-Ethyl-3,4-dihydrospiro[benzo[h]chromene-2,3’-piperidin]-4-ol (11a).
General procedure E was used to reduce 10b (90 mg, 0.31 mmol) in absolute ethanol (3 mL) using sodium borohydride (35 mg, 0.91 mmol). It was purified by flash chromatography (DCM:MeOH = 20:1) to afford 11a (63 mg, 69%) as a white solid. Rf (DCM:MeOH = 10:1) = 0.39. 1H NMR (400 MHz, CDCl3) δ 8.36 – 8.17 (m, 1H), 7.90 – 7.66 (m, 1H), 7.55 – 7.49 (m, 1H), 7.50 – 7.43 (m, 2H), 7.43 – 7.38 (m, 1H), 5.41 – 4.63 (m, 1H), 2.73 (d, J = 11.3 Hz, 1H), 2.70 – 2.57 (m, 1H), 2.58 – 2.17 (m, 5H), 2.03 – 1.66 (m, 4H), 1.26 (s, 3H), 1.17 – 0.96 (m, 3H). 13C NMR (101 MHz, CDCl3) δ 148.16, 148.02, 134.32, 134.25, 127.86, 127.47, 127.46, 126.46, 126.41, 125.93, 125.43, 125.34, 125.32, 125.25, 122.49, 119.95, 119.83, 118.13, 117.82, 114.02, 75.96, 75.31, 63.27, 62.97, 60.87, 59.69, 53.88, 53.60, 52.45, 52.35, 38.35, 35.69, 34.26, 29.81, 22.41, 21.97, 12.03, 11.72. HRMS (ESI) calcd for C19H24NO2 [M+H]+ : 298.1802, found: 298.1792.
(2R)-1’-(3,4-Difluorophenethyl)-3,4-dihydrospiro[benzo[h]chromene-2,3’-piperidin]-4-ol (11b).
General procedure E was used to reduce 10c (98 mg, 0.24 mmol) in absolute ethanol (3 mL) using sodium borohydride (27 mg, 0.72 mmol). It was purified by flash chromatography (DCM:MeOH = 75:1) to afford 11b (69 mg, 70%) as a white solid. Rf (DCM:MeOH = 10:1) = 0.58. Rf (DCM:MeOH = 10:1) = 0.58. 1H NMR (500 MHz, CDCl3) δ 8.38 – 8.15 (m, 1H), 7.87 – 7.68 (m, 1H), 7.59 – 7.44 (m, 3H), 7.40 (d, J = 8.6 Hz, 1H), 7.12 – 6.87 (m, 2H), 6.88 – 6.71 (m, 1H), 5.03 – 4.67 (m, 1H), 2.84 – 2.61 (m, 3H), 2.62 – 2.36 (m, 6H), 2.31 – 2.09 (m, 1H), 2.00 – 1.86 (m, 2H), 1.87 – 1.74 (m, 1H), 1.74 – 1.49 (m, 1H). HRMS (ESI) calcd for C25H26F2NO2 [M+H]+ : 410.1926, found: 410.1916.
(2S)-1’-Ethyl-3,4-dihydrospiro[benzo[h]chromene-2,3’-piperidin]-4-ol (11c).
General procedure E was used to reduce 10e (90 mg, 0.31 mmol) in absolute ethanol (3 mL) using sodium borohydride (35 mg, 0.91 mmol). It was purified by flash chromatography (DCM:MeOH = 20:1) to afford 11c (63 mg, 69 %) as a white solid. Rf (DCM:MeOH = 10:1) = 0.39. 1H NMR (400 MHz, CDCl3) δ 8.36 – 8.17 (m, 1H), 7.90 – 7.66 (m, 1H), 7.55 – 7.49 (m, 1H), 7.50 – 7.43 (m, 2H), 7.43 – 7.38 (m, 1H), 5.41 – 4.63 (m, 1H), 2.73 (d, J = 11.3 Hz, 1H), 2.70 – 2.57 (m, 1H), 2.58 – 2.17 (m, 5H), 2.03 – 1.66 (m, 4H), 1.26 (s, 3H), 1.17 – 0.96 (m, 3H). 13C NMR (101 MHz, CDCl3) δ 148.16, 148.02, 134.32, 134.25, 127.86, 127.47, 127.46, 126.46, 126.41, 125.93, 125.43, 125.34, 125.32, 125.25, 122.49, 119.95, 119.83, 118.13, 117.82, 114.02, 75.96, 75.31, 63.27, 62.97, 60.87, 59.69, 53.88, 53.60, 52.45, 52.35, 38.35, 35.69, 34.26, 29.81, 22.41, 21.97, 12.03, 11.72. HRMS (ESI) calcd for C19H24NO2 [M+H]+ : 298.1802, found: 298.1787.
(2S)-1’-(3,4-Difluorophenethyl)-3,4-dihydrospiro[benzo[h]chromene-2,3’-piperidin]-4-ol (11d).
General procedure E was used to reduce 10f (98 mg, 0.24 mmol) in absolute ethanol (3 mL) using sodium borohydride (27 mg, 0.72 mmol). It was purified by flash chromatography (DCM:MeOH = 75:1) to afford 11d (69 mg, 70%) as a white solid. Rf (DCM:MeOH = 10:1) = 0.58. 1H NMR (500 MHz, CDCl3) δ 8.38 – 8.15 (m, 1H), 7.87 – 7.68 (m, 1H), 7.59 – 7.44 (m, 3H), 7.40 (d, J = 8.6 Hz, 1H), 7.12 – 6.87 (m, 2H), 6.88 – 6.71 (m, 1H), 5.03 – 4.67 (m, 1H), 2.84 – 2.61 (m, 3H), 2.62 – 2.36 (m, 6H), 2.31 – 2.09 (m, 1H), 2.00 – 1.86 (m, 2H), 1.87 – 1.74 (m, 1H), 1.74 – 1.49 (m, 1H). HRMS (ESI) calcd for C25H26F2NO2 [M+H]+ : 410.1926, found: 410.1910.
(S)-2-Bromo-1-(3,4-difluorophenyl)ethan-1-ol (13b).
A solution of 12 (1 g, 4.2 mmol) in anhydrous tetrahydrofuran (15 mL) was added to a solution of (−)-DIP-chloride™ (1.5 g, 4.7 mmol) in anhydrous tetrahydrofuran (9 mL) at −25 °C under nitrogen. The resulting solution was stirred at −25 °C for 60 h, and then warmed to 0 °C, and diethanolamine (894 mg g, 8.5 mmol) was added dropwise. The mixture was warmed to room temperature and stirred for further 2 h, whereupon the boranes precipitated as a complex which was filtered and washed with pentane. The combined solvents were removed under pressure, and the residue was purified by flash chromatography (hexanes:EtOAc = 4 :1) to afford 13 (755 mg, 75%) as a colorless oil. 1H NMR (399 MHz, CDCl3) δ 7.30 – 7.20 (m, 1H), 7.20 – 7.12 (m, 1H), 7.12 – 7.06 (m, 1H), 5.24 – 4.61 (m, 1H), 3.89 – 3.52 (m, 1H), 3.55 – 3.29 (m, 1H), 2.85 – 2.45 (m, 1H).
(S)-2-(3,4-Difluorophenyl)oxirane (14b).
A 15% solution of NaOH in water (12 mL) was added to a solution of 13 (500 mg, 2.1 mmol) in ethanol (12 mL). The mixture was stirred at room temperature for 2 h. The reaction mixture was concentrated, treated with water, and extracted with ethyl acetate. The organic layer was washed with brine and water, and dried. The filtrate was evaporated under reduced pressure and the residue was purified by flash chromatography (hexanes:EtOAc = 10:1) to afford 14 (160 mg, 49 %) as a colorless oil. 1H NMR (399 MHz, CDCl3) δ 7.19 – 7.11 (m, 1H), 7.11 – 7.05 (m, 1H), 7.05 – 7.00 (m, 1H), 4.22 – 3.59 (m, 1H), 3.34 – 2.96 (m, 1H), 2.86 – 2.32 (m, 1H). 13C NMR (100 MHz, CDCl3) δ 150.80 (dd, 1JCF = 248.5, 2JCF = 13.0 Hz), 150.39 (dd, 1JCF = 248.8, 2JCF = 13.0 Hz), 134.96 (dd, 3JCF = 5.5, 4JCF = 3.5 Hz), 121.83 (dd, 3JCF = 6.0, 4JCF = 3.6 Hz), 117.54 (d, 2JCF = 16.9 Hz), 114.41 (d, 2JCF = 18.0 Hz), 51.51 (d, 4JCF = 1.8 Hz), 51.42.
(R)-1’-(2-(3,4-Difluorophenyl)-2-hydroxyethyl)spiro[benzo[h]chromene-2,4’-piperidin]-4(3H)-one (2ma).
General procedure D was used to synthesize 2ma in 63 % yield. 1H NMR (399 MHz, CDCl3) δ 8.34 (d, J = 8.2 Hz, 1H), 7.85 (d, J = 8.7 Hz, 1H), 7.80 (d, J = 8.1 Hz, 1H), 7.69 – 7.59 (m, 1H), 7.59 – 7.44 (m, 1H), 7.40 (d, J = 8.7 Hz, 1H), 7.29 – 7.18 (m, 1H), 7.17 – 6.94 (m, 2H), 4.75 – 4.58 (m, 1H), 3.06 – 2.93 (m, 1H), 2.94 – 2.79 (m, 3H), 2.73 – 2.57 (m, 3H), 2.53 – 2.39 (m, 1H), 2.34 – 2.17 (m, 2H), 1.98 – 1.76 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 191.38, 157.12, 150.54 (dd, 1JCF = 248.2, 2JCF = 13.0 Hz), 149.76 (dd, 1JCF = 247.9, 2JCF =13.0 Hz), 139.31 (dd, 3JCF = 4.7, 4JCF =3.9 Hz), 137.87, 129.69, 128.13, 126.40, 125.42, 123.28, 121.75 (dd, 3JCF = = 6.3, 4JCF = 3.8 Hz), 121.57, 120.86, 117.16 (d, 2JCF = 19.1 Hz), 115.19, 114.90 (d, 2JCF = 19.1 Hz), 78.74, 68.03, 66.14, 50.57, 47.80, 47.65, 34.70, 34.51.
(R)-1’-((R)-2-(3,4-Difluorophenyl)-2-hydroxyethyl)-3,4-dihydrospiro[benzo[h]chromene-2,4’-piperidin]-4-ol (3ma).
To a stirred solution of 2ma in trimethylamine (120mg, 1.1mmol) at 0 °C was added formic acid (54 mg, 1.1mmol) slowly under argon. After the white smoke subsided, RuCl[(R,R)-TsDPEN](mesitylene) (0.4 mg, 0.6 μmol) was added. The resulting mixture was stirred at room temperature for 24 h. The reaction was quenched by adding water and extracted with EtOAc. The combined organic layer was washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The crude residue was purified by flash chromatography (DCM:MeOH = 20:1) to afford 3ma (33mg, 66 %) as a white solid. 1H NMR (399 MHz, CDCl3) δ 8.27 – 8.19 (m, 1H), 7.86 – 7.67 (m, 1H), 7.60 – 7.45 (m, 3H), 7.42 (d, J = 8.5 Hz, 1H), 7.30 – 7.18 (m, 1H), 7.17 – 7.08 (m, 1H), 7.08 – 7.01 (m, 1H), 4.96 (t, J = 6.6 Hz, 1H), 4.82 – 4.56 (m, 1H), 3.16 – 2.77 (m, 2H), 2.71 – 2.58 (m, 2H), 2.58 – 2.52 (m, 1H), 2.49 – 2.37 (m, 1H), 2.28 – 2.16 (m, 1H), 2.16 – 2.07 (m, 1H), 2.07 – 1.92 (m, 2H), 1.92 – 1.80 (m, 1H), 1.78 – 1.66 (m, 1H). 13C NMR (100 MHz, CDCl3) δ 150.55 (dd, 1JCF = 247.7, 2JCF = 13.4 Hz), 149.75 (dd, 1JCF = 247.1, 2JCF = 13.4 Hz), 147.50, 139.45 (dd, 3JCF = 5.3, 4JCF = 3.5 Hz), 134.38, 127.70, 126.69, 125.64, 125.50, 125.44, 122.14, 121.76 (dd, 3 JCF = 6.0, 4JCF =3.4 Hz), 120.27, 117.93, 117.15 (d, 2JCF = 17.3 Hz), 114.91 (d, 2JCF = 17.8 Hz), 73.95, 67.94, 66.26, 63.46, 50.94, 47.71, 42.06, 36.17, 34.32. (4R,2”R)-3ma [α]D20 −0.59 (c 1.0, CH2Cl2).
(S)-1’-((R)-2-(3,4-Difluorophenyl)-2-hydroxyethyl)-3,4-dihydrospiro[benzo[h]chromene-2,4’-piperidin]-4-ol (3mc).
1H NMR (399 MHz, CDCl3) δ 8.48 – 8.10 (m, 1H), 7.99 – 7.67 (m, 1H), 7.56 – 7.45 (m, 3H), 7.43 (d, J = 8.5 Hz, 1H), 7.30 – 7.19 (m, 1H), 7.18 – 7.02 (m, 2H), 4.99 (t, J = 6.5 Hz, 1H), 4.85 – 4.58 (m, 1H), 2.98 (d, J = 11.0 Hz, 1H), 2.92 – 2.78 (m, 1H), 2.74 – 2.56 (m, 3H), 2.50 – 2.32 (m, 1H), 2.32 – 2.13 (m, 2H), 2.08 – 1.90 (m, 2H), 1.91 – 1.76 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 150.58 (dd, 1JCF = 248.5, 2JCF = 13.9 Hz), 149.78 (dd, 1JCF = 247.7, 2JCF = 12.9 Hz), 147.51, 139.45 (dd, 3JCF = 5.2, 4JCF = 3.9 Hz), 134.42, 127.72, 126.70, 125.66, 125.51, 125.45, 122.14, 121.78 (dd, 3JCF = 6.3, 4JCF = 3.5 Hz), 120.33, 117.92, 117.18 (d, 2JCF = 17.4 Hz), 114.94 (d, 2JCF = 18.2 Hz), 73.92, 67.96, 66.30, 63.50, 50.83, 47.94, 42.10, 35.85, 34.66. (4R,2”S)-3mc [α]D20 +0.42 (c 1.0, CH2Cl2).
(S)-1’-((S)-2-(3,4-Difluorophenyl)-2-hydroxyethyl)-3,4-dihydrospiro[benzo[h]chromene-2,4’-piperidin]-4-ol (3md).
1H NMR (399 MHz, CDCl3) δ 8.34 – 8.12 (m, 1H), 7.85 – 7.63 (m, 1H), 7.55 – 7.45 (m, 3H), 7.43 (d, J = 8.5 Hz, 1H), 7.28 – 7.19 (m, 1H), 7.17 – 7.10 (m, 1H), 7.09 – 7.03 (m, 1H), 4.99 (t, J = 6.6 Hz, 1H), 4.81 – 4.59 (m, 1H), 3.01 – 2.83 (m, 2H), 2.75 – 2.52 (m, 3H), 2.51 – 2.38 (m, 1H), 2.33 – 2.19 (m, 1H), 2.20 – 2.11 (m, 1H), 2.08 – 1.95 (m, 2H), 1.95 – 1.84 (m, 1H), 1.82 – 1.69 (m, 1H). 13C NMR (100 MHz, CDCl3) δ 150.57 (dd, 1JCF = 247.4, 2JCF =12.7 Hz), 149.27 (dd, 1JCF = 247.4, 2JCF =12.7 Hz), 147.52, 139.46 (dd, 3JCF = 5.1, 4JCF =3.7 Hz), 134.41, 127.71, 126.70, 125.66, 125.52, 125.43, 122.15, 121.77 (dd, 3JCF = 6.4, 4JCF =3.7 Hz), 120.30, 117.92, 117.16 (d, 2JCF = 17.4 Hz), 114.92 (d, 2JCF = 17.8 Hz), 73.96, 67.95, 66.27, 63.51, 50.96, 47.74, 42.09, 36.17, 34.40. (4S,2”S)-3md [α]D20 +0.49 (c 1.0, CH2Cl2).
4.2. Antiplasmodial Activities of the Compounds
All synthesized compounds were tested, as previously reported, against a chloroquine-resistant Dd2 strain and a chloroquine sensitive 3D7 strain of the parasite.15 Additionally, selected compounds were tested, as previously reported, against D6 and artemisinin-resistant strain ARC08–022.22
4.3. Cytotoxicity
An important quality of antimalarial drug compounds is their selectivity to inhibit parasite growth over mammalian cells. Therefore, the majority of the spirocyclic chromanes were evaluated for cytotoxicity using HepG2 human hepatoma cells in a clear bottom 384 well-plate (SantaCruz Biotechnology) which was seeded with 2,500 cells/well and incubated for 24 h. Serial dilutions (0–40 μM concentration range) of the compound were added to the plate followed by incubation for an additional 48 h. Viability of cells were assessed by MTS [(3-(4,5dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) cell proliferation assay.25 The effective concentration EC50 was calculated (n = 3) from a dose response curve. Furthermore, the selectivity index was calculated as the ratio of the effective concentrations of Dd2 and HepG2 (SI = EC50(HepG2)/EC50(Dd2). Generally, the majority of the spirocyclic chromanes exhibited EC50 of 20 μM or higher against HepG2 (Tables 1–4). Spirocyclic chromanes with a methylene group at the C-4 position were generally less toxic than those with a C-4 hydroxyl groups. Especially compounds 2e, 3d, 5p, and 10c displaying potent antimalarial activity had appreciable selectivity index values of >30 indicating that they are selective and nontoxic compounds.
4.4. X-ray Crystallography
The X-ray diffraction data for compound 8b were measured on a Bruker D8 Venture PHOTON 100 CMOS system equipped with a Cu Kα INCOATEC ImuS micro-focus source (λ = 1.54178 Å). Indexing was performed using APEX2 (Difference Vectors method).26 Data integration and reduction were performed using SaintPlus 6.01.27 Absorption correction was performed by multiscan method implemented in SADABS.28 Space group was determined using XPREP implemented in APEX2.26 Structure was solved using SHELXS-97 (direct methods) and refined using SHELXL-201529−31(full-matrix least-squares on F2) through OLEX2 interface program.32 All non-hydrogen atoms were refined anisotropically. Hydrogen atoms of –CH, –CH2, -CH3 groups were placed in geometrically calculated positions and were included in the refinement process using riding model with isotropic thermal parameters: Uiso(H) = 1.2[1.5]Ueq(-CH,-CH2,[-CH3]). Crystal data and refinement conditions are shown in Table S1. Absolute configuration was established based on Flack parameter, Bijvoet-Pair Analysis and Bayesian Statistics (Tables S1 and S2). P2 is the probability that the current model is correct assuming two possibilities – one out of two enantiomers present. The asymmetric unit of 8a is shown in Fig S1.
Supplementary Material
Acknowledgements
We thank National Institutes of Health (RO1AI144464 and R03AI117298) for supporting the herein presented studies financially.
References
- 1.Murray CJL, Rosenfeld LC, Lim SS, Andrews KG, Foreman KJ, Haring D, Fullman N, Naghavi M, Lozano R, Lopez AD, Global malaria mortality between 1980 and 2010: a systematic analysis, The Lancet. 379 (2012) 413–431. [DOI] [PubMed] [Google Scholar]
- 2.Malaria Fact Sheet. http://www.who.int/medicacentre/factsheets/fs094/en/
- 3.World Malaria Report 2018; World Health Organization, 2018. [Google Scholar]
- 4.Moody A, Rapid diagnostic tests for malaria parasites, Clin. Microbiol. Rev. 15 (2002) 66–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rafael ME, Taylor T, Magill A, Lim Y, Girosi F, Allan R, Reducing the burden of childhood malaria in Africa: the role of improved, Nature 444 (2006) 39–48. [DOI] [PubMed] [Google Scholar]
- 6.Sáenz FE, LaCrue AN, Cross RM, Maignan JR, Udenze KO, Manetsch R, Kyle DE, 4-(1H)-Quinolones and 1,2,3,4-Tetrahydroacridin-9(10H)-Ones Prevent the Transmission of Plasmodium falciparum to Anopheles freeborni, Antimicrob. Agents and Chemother. 57 (2013) 6178–6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Monastyrskyi A, Kyle DE, Manetsch R, 4(1H)-pyridone and 4(1H)-quinolone derivatives as antimalarials with erythrocytic, exoerythrocytic, and transmission blocking activities. Curr. Top. Med. Chem. 14 (2014) 1693–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maignan JR, Lichorowic CL, Giarrusso J, Blake LD, Casandra D, Mutka TS, LaCrue AN, Burrows JN, Willis PA, Kyle DE, Manetsch R, ICI 56,780 Optimization: Structure–Activity Relationship Studies of 7-(2-Phenoxyethoxy)-4(1H)-quinolones with Antimalarial Activity, J. Med. Chem. 59 (2016) 6943–6960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neelarapu R, Maignan JR, Lichorowic CL, Monastyrskyi A, Mutka TS, LaCrue AN, Blake LD, Casandra D, Mashkouri S, Burrows JN, Willis PA, Kyle DE, Manetsch R, Design and Synthesis of Orally Bioavailable Piperazine Substituted 4(1H)-Quinolones with Potent Antimalarial Activity: Structure–Activity and Structure–Property Relationship Studies. J. Med. Chem. 61 (2018) 1450–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Camp D, Davis RA, Campitelli M, Ebdon J, Quinn RJ, Drug-like properties: Guiding principles for the design of natural product libraries, J. Nat. Prod. 75 (2012) 72–81. [DOI] [PubMed] [Google Scholar]
- 11.Harvey AL, Natural products in drug discovery, Drug Discov. Today 13 (2008) 894–901. [DOI] [PubMed] [Google Scholar]
- 12.Newman DJ, Cragg GM, Snader KM, Natural products as sources of new drugs over the period 1981–2002, J. Nat. Prod. 66 (2003) 1022–1037. [DOI] [PubMed] [Google Scholar]
- 13.Lovering F, Bikker J, Humblet C, Escape from flatland: increasing saturation as an approach to improving clinical success, J. Med. Chem. 52 (2009) 6752–6756. [DOI] [PubMed] [Google Scholar]
- 14.Walters WP, Green J, Weiss JR, Murcko MA, What do medicinal chemists actually make? A 50-year retrospective, J. Med. Chem. 54 (2011) 6405–6416. [DOI] [PubMed] [Google Scholar]
- 15.Roberts BF, Iyamu ID, Lee S, Lee E, Ayong L, Kyle DE, Yuan Y, Manetsch R, Chakrabarti D, Spirocyclic chromanes exhibits antiplamodial activities and inhibit all intraerythrocytic life cycle stages, Int. J. Parasitol. Drugs Drug Resist. 6 (2016) 85–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bentley TM, Jones RVH, Larder AH, Lock SJ, Solvent as phase transfer catalysts. Reaction of trimethylsulfonium iodide and solid potassium hydroxide in acetonitrile leading to an epoxide of benzophenone, J. Chem. Soc., Perkin Trans. 2 (1998) 1407–1412. [Google Scholar]
- 17.Chaparro MJ, Vidal J, Angulo-Barturen I, Bueno JM, Burrows J, Cammack N, Castaneda P, Colmenarejo G, Coteron JM, Heras L, Fernandez E, Ferrer S, Gabarro R, Gamo FJ, Garcia M, Jimenez-Diaz MB, Lafuente MJ, Leon ML, Martinez MS, Minick D, Prats S, Puente M, Rueda L, Sandoval E, Santos-Villarejo A, Witty M, Claderon F, Case study of small molecules as antimalarials: 2-amino-1-phenylethanol (APE) derivatives, ACS Med. Chem. Lett. 5 (2014) 657–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang L, Morriello G, Prendergast K, Cheng K, Jacks T, Chan WW, Schleim KD, Smith RG, Patchett AA, Potent 3-spiropiperidine growth hormone secretagogues, Bioorg. & Med. Chem. Lett. 8 (1998) 107–112. [DOI] [PubMed] [Google Scholar]
- 19.Cao G, Hu A, Zou K, Xu L, Chen J, Tan W, Highly enantioselective synthesis, crystal structure, and circular dichroism spectroscopy of (R)-bambuterol hydrochloride, Chirality 20 (2008) 856–862. [DOI] [PubMed] [Google Scholar]
- 20.Fujii A, Hashiguchi S, Uematsu N, Ikariya T, Noyori R, Ruthenium (II)-catalyzed asymmetric transfer hydrogenation of ketones using a formic acid-triethylamine mixture. J. Am. Chem. Soc. 118 (1996) 2521–2522. [Google Scholar]
- 21.Bajaj SO, Farnsworth JR, O’Doherty G, Enantioselective synthesis of α- and β-bocprotected, 6-hydroxy-pyranones: Carbohydrate building blocks, Org. Synth. 91 (2014) 338–355. [Google Scholar]
- 22.Hott A, Casandra D, Sparks KN, Morton LC, Castanares G, Rutter A, Kyle DE, Artemisinin-resistant Plasmodium falciparum parasites exhibit altered patterns of development in infected erythrocytes, Antimicrob. Agents and Chemother. 59 (2015) 3156–3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Donovan SF, Pescatore MC, Method for measuring the logarithm of the octanol–water partition coefficient by using short octadecyl-poly(vinyl alcohol) high-performance liquid chromatography columns, J. Chromatogr. A. 952 (2002) 47–61. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y, A Clark J, Connelly MC, Zhu F, Min J, Guiguemde WA, Pradhan A, Iyer L, Furimsky A, Gow J, Parman T, El MF, Phillips MA, Kyle DE, Mirsalis J, Guy RK, Lead optimization of 3-carboxyl-4(1H)-quinolones to deliver orally bioavailable antimalarials, J. Med. Chem. 55 (2012) 4205–4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cory AH, Owen TC, Barltrop JA, Cory JG, Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture, Cancer Commun. 3 (1991) 207–212. [DOI] [PubMed] [Google Scholar]
- 26.Bruker (2013). APEX2 Bruker AXS Inc., 2013 Madison, Wisconsin, USA. [Google Scholar]
- 27.Bruker (2013) SAINT Data Reduction Software. [Google Scholar]
- 28.Sheldrick GM, (1996). SADABS. Program for Empirical Absorption Correction. University of Gottingen, Germany. [Google Scholar]
- 29.Sheldrick GM, (1997) SHELXL-97. Program for the Refinement of Crystal [Google Scholar]
- 30.Sheldrick GM, Acta Cryst. A 46 (1990) 467–473. [Google Scholar]
- 31.Sheldrick GM, Acta Cryst. A 64 (2008) 112–122. [DOI] [PubMed] [Google Scholar]
- 32.Dolomanov OV, Bourhis LJ, Gildea RJ, Howard JAK, Puschmann H, OLEX2: A complete structure solution, refinement and analysis program, J. Appl. Crystallogr. 42 (2009) 339–341. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.