

Abstract
Innate lymphocytes are integral components of the cellular immune system that can coordinate host defense against a multitude of challenges and trigger immunopathology when dysregulated. Natural killer (NK) cells and innate lymphoid cells (ILCs) are innate immune effectors postulated to functionally mirror conventional cytotoxic T lymphocytes and helper T cells, respectively. Here, we showed that the cytolytic molecule granzyme C was expressed in cells with the phenotype of type 1 ILCs (ILC1s) in mouse liver and salivary gland. Cell fate-mapping and transfer studies revealed that granzyme C-expressing innate lymphocytes could be derived from ILC progenitors and did not interconvert with NK cells, ILC2s, or ILC3s. Granzyme C defined a maturation state of ILC1s. These granzyme C expressing ILC1s required the transcription factors T-bet and to a lesser extent Eomes and support from transforming growth factor-β (TGF-β) signaling for their maintenance in the salivary gland. In a transgenic mouse breast cancer model depleting ILC1s caused accelerated tumor growth. ILC1s gained granzyme C expression following interleukin-15 (IL-15) stimulation, which enabled perforin-mediated cytotoxicity. Constitutive activation of Stat5, a transcription factor regulated by IL-15, in granzyme C-expressing ILC1s triggered lethal perforin-dependent autoimmunity in neonatal mice. Thus, granzyme C marks a cytotoxic effector state of ILC1s, broadening their function beyond ‘helper-like’ lymphocytes.
One-Sentence Summary:
ILC1s express granzyme C, mediate perforin-dependent cytotoxicity, and can contribute to autoimmunity and cancer immunosurveillance.
INTRODUCTION
Conventional T cells, innate-like T cells, and innate lymphocytes constitute a system of multi-layered cellular immunity, differing in terms of spatial and temporal control but manifesting overlapping effector functions. Conventional CD4+ and CD8+ T cells recirculate and differentiate into helper and cytotoxic T cells in secondary lymphoid organs, while innate-like T cells are localized in peripheral tissues and are poised to exert helper and cytotoxic functions(1). Innate lymphocytes include three distinct lineages: lymphoid tissue inducer (LTi) cells, which hone to lymphoid organ anlagen and exert ‘helper-like’ functions to induce secondary lymphoid structure formation, natural killer (NK) cells, which recirculate through the vasculature and exhibit lytic granule-mediated cytotoxicity against target cells, and innate lymphoid cells (ILCs), which reside in peripheral tissues and produce an array of inflammatory cytokines(2). Although type 1 ILCs (ILC1s) are designated as the innate counterpart of T helper 1 cells and deemed non-cytotoxic, tissue-resident ILC1-phenotype cells with NK-like cytolytic activities have been reported(3–5). Thus, group 1 innate lymphocyte heterogeneity remains to be clarified(6).
In the following study, we broaden the effector spectrum of innate lymphocytes to include the cytotoxic ILC1 effector state, which was associated with expression of lytic granule-associated cytolytic molecules including specific expression of granzyme C. ILC1s with cytotoxic potential were found early in life, could differentiate from the ILC progenitor, and were distinct from S1pr5-expressing NK cells, inerleukin-5 (IL-5)-expressing ILC2s, and IL-17A- and IL-22-expressing ILC3s. Granzyme C-producing ILC1s expanded in mammary tumors and were dependent on transforming growth factor-β (TGF-β) signaling for their maintenance and cancer immunosurveillance function. Granzyme C-producing ILC1s also required IL-15 for their generation, and in the context of constitutive IL-15 signaling, triggered perforin-dependent lethal immunopathology. Together, these findings revealed that the lytic granule-mediated cytotoxicity program was not restricted to NK cells and could also occur in ILC1s.
RESULTS
S1pr5 and granzyme C mark subsets of circulating and tissue-resident group 1 innate lymphocytes, respectively
The chromatin landscape is a key determinant of gene expression programs and can mark cell differentiation states. To explore chromatin accessibility among group 1 innate lymphocytes, we isolated two populations with an NK cell phenotype (NK1.1+NKp46+CD49a−CD49b+) from the liver and spleen, and two populations with an ILC1 phenotype (NK1.1+NKp46+CD49a+) from the liver and salivary gland for ATAC-sequencing (ATACseq) (Fig. S1A). Pairwise comparisons between each of the NK cell populations with each of the ILC1 populations yielded 1,005 and 821 peaks with increased accessibility among NK cells and ILC1s, respectively (Fig. S1B and Table S1).
The differential peaks largely tracked with differential gene expression in NK cells and ILC1s (Fig. S1C)(7). Among the targets with positive association between locus accessibility and gene expression were those involved in cell trafficking and effector functions (Fig. 1A and Table S2). Of note, NK cell-enriched genes included Sell, S1pr1, and S1pr5 (Fig. 1A–B and Fig. S1D), which promote cell exit from bone marrow and migration through blood vasculature and lymph(8). ILC1-enriched genes included the integrin Itga1 and chemokine receptor Cxcr6 (Fig. S1D), which promote cell retention in peripheral tissues(1). Unexpectedly, the Gzmc locus, which encodes the lytic granule-associated cytotoxic molecule granzyme C(9) was highly accessible, and the Gzmc transcript was expressed in cells with an ILC1 phenotype (Fig. 1A–B). This raised the possibility that innate lymphocytes with cytotoxic potential resided among tissue-resident populations.
Figure 1. S1pr5 and granzyme C mark subsets of circulating and tissue-resident group 1 innate lymphocytes, respectively.

A. Log fold change (LFC) of gene expression from microarray of liver (Liv) ILC1s versus Liv NK cells (y-axis) versus mean accessibility LFC for Liv ILC1s versus Liv NK cells. Genes with significant differentially accessible peaks are included; genes with significant differential expression are shown in black, red (enriched in ILC1) or green (enriched in NK cell), and genes without significant differential expression are shown in gray. B. Gene accessibility tracks for S1pr5 and Gzmc, displaying average peaks for splenic (Spl) NK cell, Liv NK cell, Liv ILC1, and salivary gland (SG) ILC1, which had differential accessibility between overall ILC1 and NK cell as well as differential gene expression between Liv ILC1 and Liv NK cell. Differentially accessible peaks, as listed in Table S1, are highlighted in the red box. C. Representative histograms (left) and quantification (right) of CD49b, CD11b, KLRG1, CD49a, CD103, CXCR6, CD127, CD200R1, CD69, Eomes, Tbet, and IFN-γ expression in Spl, Liv, and SG S1pr5eGFP-positive or granzyme C (GzmC)-positive CD3−NK1.1+NKp46+ cells of S1pr5eGFP-iCre mice, or fluorescence minus one (FMO) staining. Each dot represents one mouse, 3–12 mice per group. All data are combined from three or more independent experiments and shown as mean +/− SEM (one-way ANOVA with Tukey’s multiple comparisons test, “ns” = not significant, * = p<0.05, ** = p<0.01, *** = p<0.001, **** = p<0.0001).
In order to identify and lineage trace NK cells specifically, we generated a knock-in mouse line. To this end, we selected S1pr5, given its specificity and importance in NK cell trafficking(10), whereas the genes S1pr1 and Sell were expressed more broadly in other lymphocyte populations. An expression cassette encoding enhanced green fluorescence protein (eGFP), T2A self-cleavage peptide, and improved Cre recombinase (iCre) was knocked into the 3’ untranslated region (UTR) of the S1pr5 locus (designated as S1pr5eGFP-T2A-iCre) (Fig. S2A–C). As expected, S1pr5eGFP and granzyme C protein were detected in NK and ILC1 phenotype cells, respectively (Fig. S2D). Neither marker was expressed by all cells within any of the populations analyzed for ATACseq, suggesting further heterogeneity or precursor states among group 1 innate lymphocytes (Fig. S2D).
When used in combination, S1pr5eGFP and granzyme C exhibited a more exclusive expression pattern than that of CD49a and CD49b among NK1.1+NKp46+ innate lymphocytes (Fig. 1C and Fig. S1A). Overall, S1pr5eGFP-positive cells had high expression of markers associated with mature NK cells, including CD49b, CD11b, and KLRG1 (Fig. 1C). S1pr5eGFP expression increased as splenic NK cells matured, resulting in high levels of expression in virtually all CD27−CD11b+ mature NK cells (Fig. S2E), in agreement with previous findings(10). Granzyme C-positive cells expressed low levels of CD49b, CD11b, and KLRG1, but high levels of ILC1 markers to varying extents, including CD49a, CD103, CXCR6, CD127, CD200R1, and CD69 (Fig. 1C). Furthermore, the mature NK cell transcription factor Eomes exhibited higher, but not exclusive, expression in S1pr5eGFP-positive cells compared to granzyme C-positive cells, while the transcription factor T-bet and the type 1 inflammatory cytokine IFN-γ were less differentially expressed (Fig. 1C). Thus, relative to S1pr5eGFP-positive cells, granzyme C-positive innate lymphocytes uniquely expressed markers of tissue residency, but exhibited overlapping expression of NK cell maturation markers.
Granzyme C-expressing innate lymphocytes differentiate from ILCps, but not from NK cells, ILC2s, or ILC3s
The shared properties of S1pr5-expressing NK cells and granzyme C-expressing ILC1-phenotype cells raised questions of their ontogenetic relationship. Previous adoptive cell transfer experiments show conflicting conclusions on the likelihood of NK cells giving rise to ILC1-like cells in tissues(11–13). To investigate whether granzyme C-positive innate lymphocytes might be converted from S1pr5-positive mature NK cells under physiological conditions (Fig. S3A), we crossed S1pr5eGFP-T2A-iCre to the Rosa26LSL-YFP allele, generating S1pr5 fate-mapper mice (S1pr5FM) in which cells with a history of S1pr5 expression and their progeny were permanently marked by YFP expression. Stochastic Cre activity during embryogenesis led to varying degrees of fate-mapping among hematopoietic stem cells (HSCs) (Fig. S3B). To bypass this non-specific fate-mapping, we transferred non-fate-mapped HSCs to irradiated recipient mice to measure history of S1pr5 expression during hematopoiesis (Fig. 2A). Analysis of the blood of chimeric mice revealed that S1pr5FM expression was restricted to cell populations that expressed S1pr5 at the time of analysis, namely circulating NK1.1+ innate lymphocytes and Ly6C− patrolling monocytes, with minimal fate-mapping in Ly6C+ monocytes and no fate mapping in T cells (Fig. S3C). Investigation of granzyme C and S1pr5FM expression among group 1 innate lymphocytes in the spleen, liver, and salivary gland revealed that the vast majority of granzyme C-expressing cells did not have a history of S1pr5 expression (Fig. 2A and Fig. S3D). The salivary gland ILC1-phenotype cells included subsets that expressed either CXCR6 or Eomes, both of which expressed granzyme C (Fig. 2B and Fig. S3E), thus explaining the heterogeneity of these markers in salivary gland granzyme C-expressing cells (Fig. 1C). Although Eomes was thought to mark all NK lineage cells, the vast majority of salivary gland Eomes+CD49a+ cells did not express S1pr5FM, as was true for the CXCR6+ ILC1 subsets in the liver and salivary gland, whereas almost all liver NK cells did (Fig. 2B). These findings demonstrated that S1pr5-positive mature NK cells largely did not give rise to granzyme C-positive innate lymphocytes, including both CXCR6+ and Eomes+ ILC1-phenotype cells.
Figure 2. Granzyme C-expressing innate lymphocytes differentiate from ILCps, but not from NK cells, ILC2s, or ILC3s.

A. Experimental design for S1pr5 fate-mapper (S1pr5FM) chimeric mice. Expression of S1pr5FM and granzyme C (GzmC) in splenic (Spl), liver (Liv), and salivary gland (SG) CD3−NK1.1+ cells. B. Expression of S1pr5FM in Liv and SG CXCR6+CD49a+, SG Eomes+CD49a+, and Liv CD49a−CD49b+ NK cells from S1pr5FM chimeric mice. C. Experimental design for ILCp (Lin−CD127+α4β7+Flt3−CD25−PD-1+) transfer. Expression of CD49a and GzmC in Liv ILCp-derived CD3−NK1.1+NKp46+ cells. D. Experimental design for IL-5 fate-mapper (Il5FM) mice. Expression of Il5FM and GzmC in Liv, SG, and small intestine lamina propria (SI LP) Lin−Thy1.2+ and/or NK1.1+ innate lymphocytes. E. Experimental design for IL-17A fate-mapper (Il17aFM) mice. Expression of Il17aFM and GzmC in Liv, SG, and SI LP Lin−Thy1.2+ and/or NK1.1+ innate lymphocytes. F. Experimental design for IL-22 fate-mapper (Il22FM) mice. Expression of Il22FM and GzmC in Liv, SG, and SI LP Lin−Thy1.2+ and/or NK1.1+ innate lymphocytes. Each dot represents one mouse, n = 3–6 mice per group. All data are combined from three or more independent experiments and shown as mean +/− SEM (one-way ANOVA with Tukey’s multiple comparisons test, “ns” = not significant, * = p<0.05, ** = p<0.01, *** = p<0.001, **** = p<0.0001).
To explore whether granzyme C-expressing innate lymphocytes were differentiated from the ILC lineage, we transferred previously defined ILC progenitors (ILCp)(14, 15) into Rag2−/−Il2rg−/− lymphopenic hosts (Fig. 2C and Fig. S3F). Indeed, ILCp gave rise to a sizable population of granzyme C-positive cells that expressed the tissue residency marker CD49a (Fig. 2C and Fig. S3G), while NK cells were not detected among the progenies of ILCp (data not shown), unlike findings using a phenotypically distinct progenitor(16). Studies have revealed plasticity among ILC compartments, including the potential for group 2 or 3 ILCs to convert to an ILC1 phenotype cell(17–19). To determine whether IL-5-producing ILC2s, IL-17A- or IL-22-producung ILC3s could give rise to granzyme C-expressing innate lymphocytes, we crossed Il5iCre, Il17aiCre, and Il22iCre each to Rosa26LSL-YFP to generate IL-5 fate-mapper (Il5FM), IL-17A fate-mapper (Il17aFM) and IL-22 fate-mapper (Il22FM) mice (Fig. 2D–F and Fig. S4A), where YFP expression denoted all cells and their progeny that had current or a history of cytokine expression. We examined total innate lymphocytes (Lin−Thy1+ and/or NK1.1+) across tissues (Fig. S4B). No co-expression of granzyme C with any cytokine fate-mapper was observed (Fig. 2D–F and Fig. S4C–E). These findings revealed that granzyme C-expressing innate lymphocytes could be differentiated from ILCp, but they were not converted from IL-5+ ILC2s, IL-17A+ or IL-22+ ILC3s at steady state.
Granzyme C-expressing cells are not precursors for NK cells, ILC2s, or ILC3s
Group 2 and 3 innate lymphocytes can seed tissues early in life(20, 21). As such, we investigated whether neonatal group 1 innate lymphocytes expressed granzyme C or S1pr5. There was a sizeable population of granzyme C-positive cells, which was associated with CD49a expression in livers of newborn mice and continuing through day seven (D7) and D14 of age (Fig. 3A). However, S1pr5 was not expressed through 14 days of age (Fig. 3A), supporting previous observations that ILC1-phenotype cells predominate in the liver of newborn mice while NK cells appear later in life(22).
Figure 3. Granzyme C-expressing cells are not precursors for NK cells, ILC2s, or ILC3s.

A. Representative flow cytometric analysis (left) and quantification (right) of granzyme C (GzmC) and S1pr5eGFP expression in NK1.1+NKp46+ innate lymphocytes in livers of 0.5 day-, 7 day-, 14 day-old and adult (8–12 weeks of age) mice. B. Representative plots of Gzmctd-Tomato and CD49a expression in splenic (Spl) and liver (Liv) NK phenotype and Liv and salivary gland (SG) ILC1 phenotype cells of GzmctdT-T2A-iCre mice. C. Experimental design of granzyme C fate-mapper (GzmcFM) mice. D. Quantification of GzmcFM expression in NK and ILC1-phenotype cells in the Spl, Liv and SG. E. Expression of GzmcFM and IL-5 in Liv, SG, and small intestine lamina propria (SI LP) Lin−Thy1.2+ and/or NK1.1+ innate lymphocytes after four-hour PMA/Ionomycin/Golgi stop treatment. F. Expression of GzmcFM and IL-17A in Liv, SG, and SI LP Lin−Thy1.2+ and/or NK1.1+ innate lymphocytes after four-hour PMA/Ionomycin/Golgi stop treatment. G. Expression of GzmcFM and IL-22 in Liv, SG, and SI LP Lin−Thy1.2+ and/or NK1.1+ innate lymphocytes after four-hour PMA/Ionomycin/Golgi stop treatment. Each dot represents one mouse, n = 3–10 mice per group. All data are combined from three or more independent experiments and shown as mean +/− SEM (one-way ANOVA with Tukey’s multiple comparisons test, “ns” = not significant, * = p<0.05, ** = p<0.01, *** = p<0.001, **** = p<0.0001).
To track and target granzyme C-expressing cells, we generated a mouse line with an expression cassette encoding tandem dimer Tomato (td-Tomato), T2A self-cleaving peptide, and iCre knocked into the 3’ UTR of the Gzmc locus (designated as GzmctdT-T2A-iCre) (Fig. S5A–C). Both granzyme C protein and td-Tomato reporter were associated with CD49a expression in the liver and salivary gland (Fig. 3B and Fig. S5D), and there was high correlation between Gzmctd-Tomato and granzyme C protein itself (Fig. S5E). Given the early tissue seeding by granzyme C-expressing cells and previous observations that fetal liver “T-bet+Eomes− immature NK cells” give rise to mature NK cells in cell transfer experiments(23, 24), we explored the possibility that granzyme C-expressing cells represented a precursor state for mature NK cells under physiological conditions (Fig. S5F). To this end, we generated a granzyme C fate-mapper mouse (GzmcFM) by crossing GzmctdT-T2A-iCre to the Rosa26LSL-YFP allele (Fig. 3C). Cells with a history of granzyme C expression and their progeny would be permanently marked by YFP expression. Analysis of the group 1 innate lymphocyte compartment revealed that a majority of ILC1-phenotype cells in the liver and salivary gland had a history of granzyme C expression (Fig. 3D and Fig. S5G). However, splenic and liver NK cells did not express YFP (GzmcFM) and thus had no history of granzyme C expression (Fig. 3D and Fig. S5G). To investigate whether granzyme C-expressing innate lymphocytes gave rise to other ILC populations, we measured production of IL-5, IL-17A, and IL-22 among innate lymphocytes in GzmcFM mice (Fig. S6A). Almost no cells that expressed both GzmcFM and any of these cytokines were detected across tissues (Fig. 3E–G and Fig. S6B–D). Taken together, these findings revealed that granzyme C expression defined a distinct tissue-resident ILC subset that did not differentiate from nor give rise to circulating NK cells, IL-5+ ILC2s, or IL-17A+or IL-22+ ILC3s at steady state.
Differential regulation of granzyme C-expressing ILC1s across tissues by T-bet, Eomes, and TGF-β
The transcription factors T-bet and Eomes control the differentiation of group 1 innate lymphocytes(11, 24). To investigate whether granzyme C-expressing ILC1s were dependent on these transcription factors for maintenance, we generated GzmcΔTbx21, GzmcΔEomes, and GzmcΔTbx21ΔEomes mice by crossing GzmctdT-T2A-iCre mice with mice carrying Tbx21fl/fl and Eomesfl/fl alleles. T-bet, but not Eomes, was required for the maintenance of all granzyme C-expressing ILC1s in the liver and thus a sizeable portion of the overall liver ILC1 population (Fig. 4A and Fig. S7A–B). In the salivary gland, where granzyme C-expressing ILC1s included CXCR6+ and Eomes+ subsets (Fig. 2B), T-bet promoted maintenance of both populations, while Eomes played an accessory role (Fig. 4A and Fig. S7B–C). In fact, Eomes itself was not required for granzyme C expression in most ILC1s, as GzmcΔEomes mice maintained sizeable granzyme C-expressing populations (Fig. 4A and Fig. S7B–C). Nonetheless, in the absence of T-bet, Eomes supported granzyme C-expressing ILC1s in the salivary gland, while GzmcΔTbx21ΔEomes mice exhibited the strongest loss of granzyme C-expressing cells (Fig. 4A and Fig. S7B–C).
Figure 4. Differential regulation of ILC1 populations across tissues by T-bet, Eomes and TGF-β.

A. Representative (left) flow cytometric analysis of granzyme C (GzmC) and CD49a expression among NK1.1+CD3− cells in the liver (upper) and salivary gland (lower) of wild-type (WT), GzmCiCreTbx21fl/fl (GzmcΔTbx21) (yellow), GzmCiCreEomesfl/fl (GzmcΔEomes) (green), and GzmCiCreTbx21fl/flEomesfl/fl (GzmcΔTbx21Δeomes) (blue) mice. (Right) abundance of GzmC+NK1.1+CD3− (GzmC+) and CD49a+NK1.1+CD3− (ILC1) cells out of total CD45+ cells in liver and salivary gland. B. Representative (left) and quantification (right) of flow cytometric analysis of group 1 innate lymphocytes in liver (upper) and salivary gland (lower) of WT (white bar) and GzmCiCreTgfbr2fl/fl (GzmcΔTgfbr2) (gray bar) mice, quantifying CD49a+ cells among NK1.1+CD3− cells and CD103+ and GzmC+ cells among CD49a+NK1.1+CD3− cells in each organ. Each dot represents one mouse (n = 4–10 mice per group). All data are combined from three or more independent experiments and shown as mean +/− SEM (unpaired student’s t-test for two groups, one-way ANOVA with Tukey’s multiple comparisons test with more than two groups, “ns” = not significant, * = p<0.05, ** = p<0.01, *** = p<0.001, **** = p<0.0001).
Salivary gland ILC1s require TGF-β signaling for their differentiation, whereas liver ILC1s do not(25). We observed co-expression of the TGF-β target CD103 with granzyme C in salivary gland ILC1s (Fig. S7D), prompting us to explore whether mature granzyme C-expressing ILC1s required continued TGF-β signaling. To this end, we generated GzmcΔTgfbr2 mice by crossing GzmctdT-T2A-iCre mice with mice carrying Tgfbr2fl/fl alleles. Indeed, loss of TGF-β signaling impaired homeostasis of granzyme C-expressing ILC1s in the salivary gland, but not in the liver, leading to decreased CD49a expression among group 1 innate lymphocytes and decreased CD103 and granzyme C expression among CD49a+NK1.1+CD3− cells (Fig. 4B). These observations demonstrated that TGF-β signaling was continually required for the maintenance of granzyme C-expressing ILC1s in the salivary gland.
Granzyme C-expressing ILC1s expand and can mediate cancer immunosurveillance in the PyMT model of breast cancer
Beyond tissues at steady-state, we next explored the identity and origin of group 1 innate lymphocytes in the context of cancer using the MMTV-PyMT spontaneous model of breast cancer(3). Relative to healthy mammary glands, we observed an increase in granzyme C-expressing innate lymphocytes in PyMT tumors (Fig. S8A). NK1.1+CD3− cells broadly included CD49a−CD49b+ NK cells and two subsets of CD49a+ cells differentiated by expression of CXCR6 and Eomes, all present at comparable frequencies (Fig. 5A). In the tumor, S1pr5eGFP was expressed in most NK cells, but minimally in Eomes+CD49a+ or CXCR6+CD49a+ cells (Fig. 5B). Granzyme C was highly expressed by both CXCR6+ and Eomes+ subsets of CD49a+ cells, with no expression in NK cells (Fig. 5C), similar to what was observed in steady state populations in the spleen, liver, and salivary gland (Fig. 1C). Conversion of NK cells to ILC1-like cells in tumors has been reported(13), and thus we generated S1pr5FM bone marrow chimera mice in PyMT recipients to track history of S1pr5 expression among tumor-infiltrating group 1 innate lymphocytes. Virtually all CXCR6+CD49a+ cells and about 75% of Eomes+CD49a+ cells did not express S1pr5FM and thus had no history of S1pr5 expression, whereas NK cells highly expressed S1pr5FM (Fig. 5D). Conversely, GzmcFMPyMT mice revealed that cells with a history of granzyme C expression were restricted to CXCR6- and Eomes-expressing CD49a+ cells, with no contribution to NK cells (Fig. 5E). Thus, most granzyme C-expressing tissue-resident phenotype innate lymphocytes appeared ontogenically distinct from circulating NK cells in PyMT tumors.
Figure 5. Granzyme C-expressing ILC1s expand and mediate cancer immunosurveillance in the PyMT model of breast cancer.
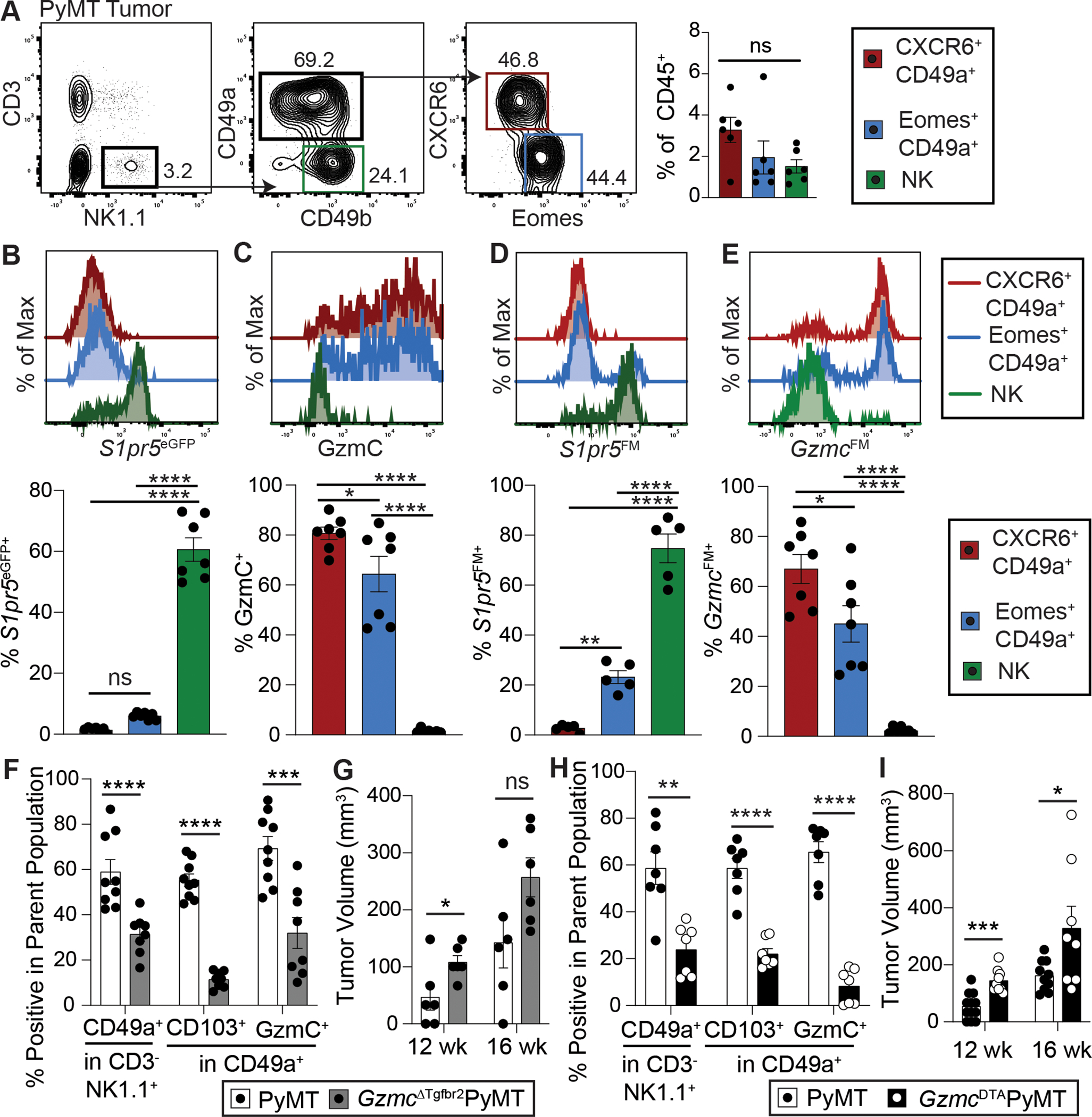
A. Abundance of CXCR6+CD49a+NK1.1+CD3− (CXCR6+CD49a+), Eomes+CD49a+NK1.1+CD3− (Eomes+CD49a+) and CD49a−CD49b+NK1.1+CD3− (NK) cells out of total CD45+ cells in PyMT tumors. Expression of S1pr5eGFP (B), granzyme C (GzmC) (C), S1pr5FM (D), and GzmcFM (E) in CXCR6+CD49a+ ILC1 (red), Eomes+CD49a+ ILC1 (blue), and CD49a−CD49b+ NK cell (green) populations in PyMT tumors of S1pr5eGFPPyMT (B), PyMT (C), S1pr5FMPyMT chimera (D), and GzmcFMPyMT (E) mice. F. Abundance of ILC1s in tumors of PyMT (white bar) and GzmCiCreTgfbr2fl/flPyMT (GzmcΔTgfbr2PyMT) (gray bar) mice, quantifying CD49a+ cells among NK1.1+CD3− cells and CD103+ and GzmC+ cells among CD49a+NK1.1+CD3− cells in the tumor. G. Tumor volume at 12 and 16 weeks of age from PyMT (white bar) and GzmcΔTgfbr2PyMT (gray bar) mice. H. Abundance of ILC1s in tumors of PyMT (white bar) and GzmCiCreRosa26LSL-DTA/+ PyMT (GzmcDTAPyMT) (black bar) mice, quantifying CD49a+ cells among NK1.1+CD3− cells and CD103+ and GzmC+ cells among CD49a+NK1.1+CD3− cells in the tumor. I. Tumor volume at 12 and 16 weeks of age from PyMT (white bar) and GzmcDTAPyMT (black bar) mice. Each dot represents one mouse (n = 5–9 mice per group). All data are combined from three or more independent experiments and shown as mean +/− SEM (unpaired student’s t-test for two groups, one-way ANOVA with Tukey’s multiple comparisons test with more than two groups, “ns” = not significant, * = p<0.05, ** = p<0.01, *** = p<0.001, **** = p<0.0001).
As in the salivary gland, ILC1-phenotype innate lymphocytes in mammary tumors can express CD103(3), suggesting that TGF-β signaling may control their differentiation and homeostasis. To this end, we generated GzmcΔTgfbr2PyMT mice, which had decreased CD49a expression among group 1 innate lymphocytes as well as decreased CD103 and granzyme C expression among CD49a+ cells (Fig. 5F and Fig. S8B), similar to what was observed in the salivary gland (Fig. 4B). Notably, these mice displayed accelerated tumor growth at early timepoints (Fig. 5G), revealing that TGF-β signaling is required for the maintenance of granzyme C-expressing innate lymphocytes and may contribute in part to their anti-tumor function in the PyMT breast cancer model.
To further explore the cancer surveillance function of granzyme C-expressing innate lymphocytes, we generated GzmcDTAPyMT mice by crossing GzmctdT-T2A-iCre mice with a conditional allele of Rosa26LSL-DTA encoding the lethal diphtheria toxin A preceded by a floxed translation-STOP cassette to deplete granzyme C-expressing cells (Fig. S8C). Indeed, these mice had large reductions in CD49a expression in NK1.1+CD3− cells as well as in CD103 and granzyme C expression among CD49a+ cells (Fig. 5H and Fig. S8D). There was also depletion of both CXCR6+ and Eomes+ subsets of CD49a+ cells while NK cell abundances were unchanged (Fig. S8E). Of note, these mice displayed tumor acceleration (Fig. 5I), demonstrating granzyme C-expressing innate lymphocytes can contribute to cancer immunosurveillance in the PyMT model of breast cancer, potentially through clearance of nascently transformed cells.
ILC1s broadly express cytotoxic molecules, with granzyme C expression marking a mature effector state
Given that ILC1-like cells from PyMT tumors can directly kill cancer cells in a perforin-dependent manner(3), and granzyme C can induce cell death in target cells(9), we revisited ILC1s in the liver and salivary gland for expression of other cytotoxic molecules. Indeed, we observed granzyme B expression in subsets of liver and salivary gland ILC1s and this expression was higher than that of NK cells in the spleen and liver (Fig. 6A). Additionally, all ILC1s and NK cells expressed perforin with liver NK cells expressing slightly higher levels (Fig. 6A). Using past and current granzyme C expression as a way to subset ILC1s in the liver and salivary gland, we were able to identify ILC1s with current granzyme C expression (GzmC+GzmcFM+, double-positive [DP]), past but not current granzyme C expression (GzmC−GzmcFM+, fate mapped single-positive [FMSP]), and no history of expression (GzmC−GzmcFM−, double-negative [DN]) that were present in varying proportions in these tissues (Fig. S9A). Granzyme B expression was highest in the DP and lowest in the DN populations in both liver and salivary gland, whereas perforin was high across ILC1 subsets (Fig. 6B).
Figure 6. ILC1s broadly express cytotoxic molecules and granzyme C expression marks a mature effector state.

A. Representative (upper) and quantification (lower) of granzyme B (GzmB) and perforin expression among splenic (Spl) and liver (Liv) NK cells as well as Liv and salivary gland (SG) ILC1s. B. Representative and quantification of GzmB and perforin protein expression among subsets of ILC1s in Liv (upper) and SG (lower) based on current and history of granzyme C expression (GzmC−GzmcFM−, double-negative [DN], blue; GzmC−GzmcFM+, fate-mapped single positive [FMSP], purple; GzmC+GzmcFM+, double positive [DP], red). C. The number of differentially expressed genes in each of six pairwise comparisons conducted between the four sequenced populations. The color indicates direction of higher expression. D. 62 of 74 genes significantly differentially expressed between DP and DN ILC1s with higher expression in DP, grouped by function and localization. 12 genes could not be grouped due to unknown cellular localization of the proteins they encode and are listed in Table S3. Each dot represents one mouse (n = 4–5 mice per group). Flow cytometry data are combined from three or more independent experiments and shown as mean +/− SEM (one-way ANOVA with Tukey’s multiple comparisons test, “ns” = not significant, * = p<0.05, ** = p<0.01, *** = p<0.001, **** = p<0.0001).
To further explore the heterogeneity among these ILC1 subsets, we performed RNA sequencing experiments on DP, FMSP, and DN ILC1s as well as S1pr5eGFP+ NK cells from the liver, carrying out six pairwise comparisons among the four populations (Fig. 6C, Fig. S9B and Table S3). The largest differences were observed between each of the ILC1 subsets and the NK cell population (Fig. 6C). We identified core NK cell and ILC1 genes based on differentially expressed genes shared across the three ILC1-NK cell comparisons (Fig. S9C and Table S3), including genes such as Sell, S1pr5, S1pr1, Gzmc, Itga1, and Cxcr6 identified in our analysis using ATACseq and microarray performed on bulk liver ILC1s (Fig. 1 and Fig. S1A–B). Another core ILC1 gene included Zfp683, encoding the protein Hobit, (Table S4), which controls ILC1 lineage commitment(4, 26, 27). Among comparisons within ILC1 subsets, gene expression was most different between DP and DN subsets, with 74 genes upregulated in DP and 245 genes upregulated in DN (Fig. 6D, S10, and Table S3). Genes enriched in DP population included those encoding secreted effector molecules such as Gzmb, Gzmc, Csf1, Ccl1, Cx3cl1, and Xcl1 (Fig. 6D). Most of the genes upregulated in DP were expressed in FMSP at intermediate levels, suggesting FMSP may be an intermediate state between DN and DP (Fig. 6D). There were many more genes upregulated in the DN population, many of which also showed intermediate levels of expression in the FMSP population (Fig. S10). Among DN-enriched genes were those encoding regulators of cell cycle progression such as Cdc45, Cdc6, and Cdca2 (Fig. S10), suggesting that DN cells may be more proliferative and represent more of a precursor population. Cd127, Cd160, and Gzma, which have been associated with different ILC1 subsets in the liver (4, 5, 27), were not differentially expressed among the three ILC1 populations we sequenced (Table S3). These findings suggested that the three ILC1 populations were relatively similar to one another, with granzyme C-expressing ILC1s in a mature effector state and granzyme C-nonproducing ILC1s including less differentiated cells.
IL-15 induces granzyme C expression in ILC1s that exhibit lytic granule-mediated cytotoxicity
Differentiation and function of group 1 innate lymphocytes are promoted by the common γ chain cytokine interleukin-15 (IL-15)(28). The sizeable FMSP population in liver CXCR6+ ILC1s suggested that granzyme C expression could be modulated in vivo. To investigate how IL-15 might regulate granzyme C expression in group 1 innate lymphocytes, we cultured DP, FMSP, and DN ILC1s and S1pr5+ NK cells from the liver with a IL-15/IL-15Rα complex (IL-15c). Notably, granzyme C protein expression was induced in both FMSP and DN ILC1s, but not NK cells (Fig. 7A), which was in agreement with the observation that Gzmc transcript was significantly higher in all three ILC1 subsets than in NK cells (Fig. S9C). These findings suggested that granzyme C expression may not have indicated a distinct subset of ILC1s but rather cells in a mature effector state, potentially induced by IL-15 stimulation. To further explore this possibility, we sorted GzmctdT+ and GzmctdT− CXCR6+ ILC1s from the liver of mice sufficient or deficient in the cytolytic pore-forming protein perforin, expanded them in vitro with IL-15c, and tested their ability to kill RMA-S target cells (Fig. S11A). After exposure to IL-15c, which can induce granzyme C expression, both subsets of ILC1s were able to robustly kill target cells in a perforin-dependent manner, with no death above background in either of the perforin-deficient effector conditions (Fig. 7B and Fig. S11B). Thus, CXCR6+ ILC1s had lytic granule-mediated cytotoxic capabilities.
Figure 7. CXCR6+ ILC1s from liver can mediate perforin-dependent cytotoxicity regardless of initial granzyme C expression.
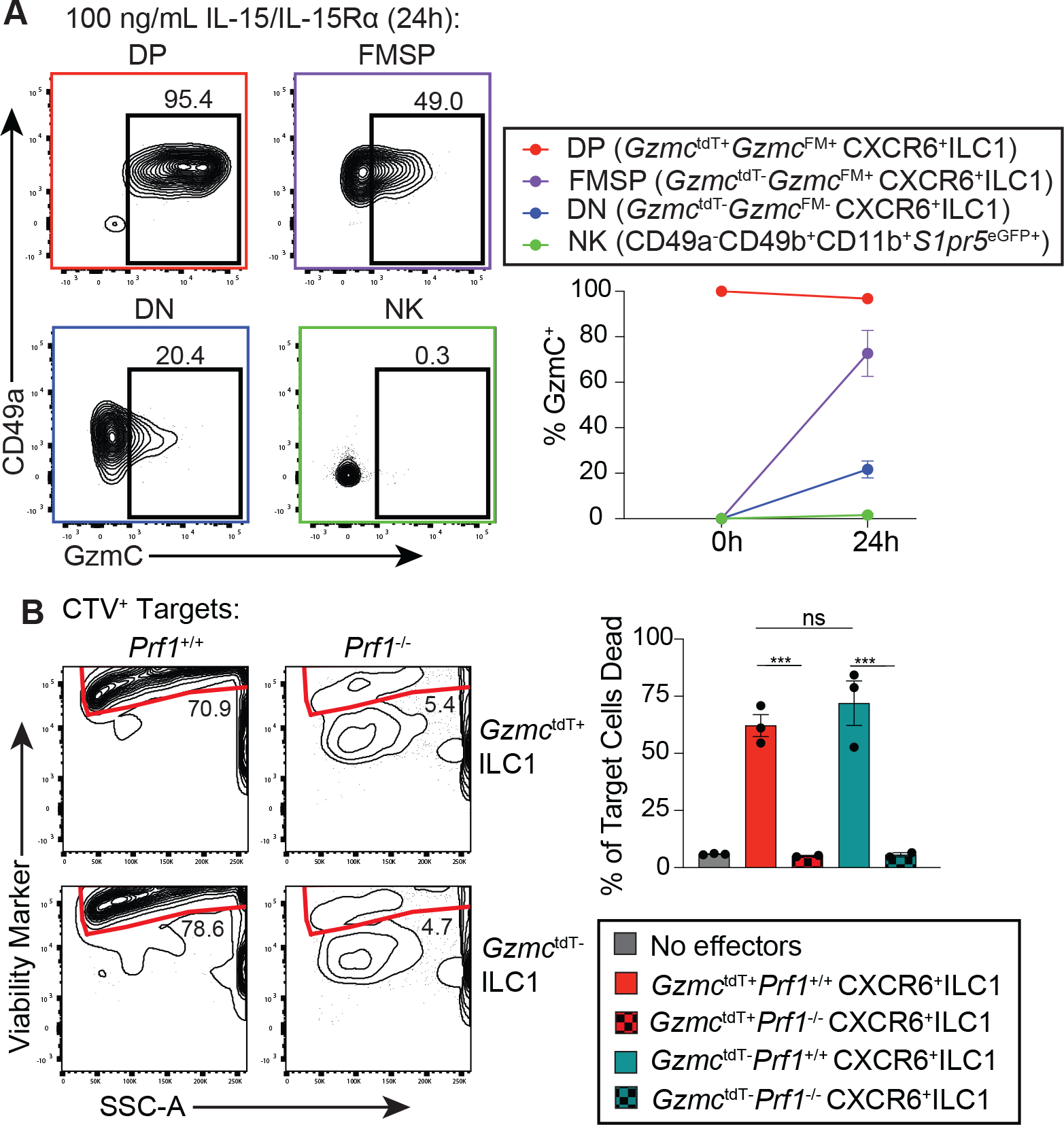
A. Expression of granzyme C (GzmC) in group 1 innate lymphocyte subsets sorted based on expression of GzmC protein and/or GzmcFM and cultured in 100 ng/mL IL-15/IL-15Rα for 24 hours (h). Sorted populations included DP (CD49a+CXCR6+GzmctdT+GzmcFM+) (red), FMSP (CD49a+CXCR6+GzmctdT−GzmcFM+) (purple), DN (CD49a+CXCR6+GzmctdT−GzmcFM−) (blue), and NK (CD49a−CD49b+CD11b+) (green) cell subsets. Data are combined from four independent experiments. B. Killing assay, displaying death rate of RMA-S target cells after coincubation with subsets of CXCR6+ ILC1s from the liver. Effector cells were either GzmctdT+ or GzmctdT− ILC1s (CXCR6+CD49a+NK1.1+CD3−) and were sorted from the livers of GzmctdT-T2A-iCre/+Prf1+/+ or GzmctdT-T2A-iCre/+Prf1−/− mice. Effectors were expanded in 100 ng/mL IL-15/IL-15Rα and cocultured with CTV-labeled RMA-S target cells for 16h at a 10:1 effector:target ratio in media supplemented with 100 ng/mL IL-15/IL-15Rα. Data are representative from one of three independent experiments. Each dot represents one mouse (n = 3–6 mice per group). All data are shown as mean +/− SEM (one-way ANOVA with Tukey’s multiple comparisons test for more than two groups, “ns” = not significant, * = p<0.05, ** = p<0.01, *** = p<0.001, **** = p<0.0001).
Constitutively active IL-15 signaling in granzyme C-expressing ILC1s causes perforin-dependent lethal autoimmunity
Given our findings in vitro, we next explored whether granzyme C expression required IL-15 in vivo. Indeed, there was a total loss of granzyme C-expressing ILC1s in the liver and salivary gland of Il15−/− mice (Fig. 8A and Fig. S12A). IL-15 signaling is predominantly transduced through Jak kinases that activate the Stat5a/b family of transcription factors(29). Of note, tissue-associated IL-15 may act as an alarmin to modulate the function of tissue-resident group 1 innate lymphocytes, and its availability may be restricted at steady state(30). To investigate whether enhanced IL-15 signaling in granzyme C-expressing ILC1s would alter their phenotype in vivo, we crossed GzmctdT-T2A-iCre mice with a conditional allele of Rosa26LSL-Stat5b-CA encoding a constitutively active (CA) form of Stat5b preceded by a floxed translation-STOP (LSL) cassette (Fig. 8B). Strikingly, all GzmcStat5b-CA/+ mice died at a median age of 16.5 days, and this lethality did not require Rag1-dependent adaptive lymphocytes (Fig. 8C). Granzyme C-expressing ILC1s were dramatically expanded in the liver of GzmcStat5b-CA mice (Fig. 8D and Fig. S12B). Multi-organ immunopathology was also detected, exemplified by multifocal to coalescing hepatitis with hepatocellular necrosis in association with enhanced immunoreactivity of NKp46 and cleaved caspase 3 (CC3), which marked innate lymphocytes and apoptotic cells, respectively (Fig. 8E). To determine whether lytic granule-mediated cytotoxicity contributed to the cell death phenotype, we crossed GzmcStat5b-CA mice onto a Prf1−/− background. Indeed, while GzmcStat5b-CA/+ and GzmcStat5b-CA/+Prf1−/− had comparable infiltration of NKp46+ innate lymphocytes, CC3-marked apoptosis and hepatocellular damage were attenuated in the absence of perforin (Fig. 8E). The neonatal lethal phenotype was substantially rescued, increasing the median survival time from 14 days in GzmcStat5b-CA/+ mice to 49 days in littermate GzmcStat5b-CA/+Prf1−/− mice (Fig. 8F). Thus, ILC1s could induce lethal autoimmunity in tissues including the liver via perforin-mediated cytotoxicity, and IL-15 may be an important regulator of this response.
Figure 8. Constitutively active IL-15 signaling in granzyme C-expressing ILC1s causes perforin-dependent lethal autoimmunity.

A. Granzyme C (GzmC) expression in liver (Liv) and salivary gland (SG) CD3−NK1.1+NKp46+ cells of wild-type (WT) and Il15−/− mice. B. Experimental design of GzmcStat5b-CA mice. C. Kaplan-Meier survival curves for GzmcStat5b-CA/+, GzmcStat5b-CA/+Rag1−/−, and littermate control mice. D. GzmC+NK1.1+NKp46+ cells per gram of Liv tissue from seven-day-old WT and GzmcStat5b-CA/+ mice. E. Representative images of hematoxylin and eosin staining (H&E, first row) and NKp46 (second row) and cleaved caspase 3 (CC3, third row) immunoreactivities (indicated by arrows) of Liv sections from 14-day-old WT, GzmcStat5b-CA/+, and GzmcStat5b-CA/+Prf1−/− mice. Images and inserts are 200X and 400X, respectively. NKp46 and CC3 immunoreactivity was also quantified on serial sections as percent of total tissue using digital pathology software (Halo). Scale bar indicates 100 μm. F. Kaplan-Meier survival curves for GzmcStat5b-CA/+ and GzmcStat5b-CA/+Prf1−/− littermate control mice (n = 15–18 per group). Each dot represents one mouse; A, D, E: n = 3–4 mice per group, C: n = 7–18 per group, F: n = 15–18 mice per group. All data are combined from three or more independent experiments and shown as mean +/− SEM (A, D, E: one-way ANOVA with Tukey’s multiple comparisons test, C, F: Log-rank [Mantel Cox] test; “ns” = not significant, * = p<0.05, ** = p<0.01, *** = p<0.001, **** = p<0.0001)
DISCUSSION
Heterogenous populations of group 1 innate lymphocytes are widely distributed in circulation and peripheral tissues and can be mobilized under immune challenging conditions. By generating two reporter and Cre mouse lines, this work allowed for specific genetic fate-mapping to clarify the ontogeny and plasticity of multiple innate lymphocyte effector populations at both neonatal and adult time points. S1pr5-expressing NK cells and granzyme C-expressing ILC1s were largely distinct and represented two discrete terminally differentiated populations at steady state. These two populations appeared at different points during development, with ILC1s present in peripheral organs at birth and S1pr5-expressing NK cells not detected until weeks later. Additionally, we did not observe interconversion between mature cytokine-producing ILC2s and ILC3s with granzyme C-expressing ILC1s, suggesting granzyme C-expressing ILC1s were terminally differentiated. However, interconversion of cells along the ILC1 and NKp46+ ILC3 lineage may remain possible in certain inflammatory conditions, as RORɣt appears to be a suppressor of this cytotoxic ILC1 state(17, 19).
The expression of Eomes has been proposed as a defining marker for NK cells. Our data uncovered more nuance in this definition. Given that S1pr5 was expressed by all CD27−CD11b+ mature NK cells in the spleen and given the lack of S1pr5-fate-mapping in a large majority of Eomes+ tissue-resident group 1 innate lymphocytes in the salivary gland or in the PyMT tumor, we concluded that a plurality of these Eomes+ populations did not differentiate from mature circulating NK cells. This is in agreement with the lack of input from wild-type circulating cells to the empty ILC niche in salivary glands of Ncr1ΔTgfbr2 parabionts(25). Given that Eomes is expressed before S1pr5, it remains possible the relevant precursor may be an NK cell-committed progenitor that retains the potential to gain tissue-residency, such as immature splenic NK (iNK)-phenotype cells. Splenic iNK had low S1pr5 expression, which may explain the partial fate-mapping of Eomes+ population in the tumor. However, iNK-phenotype cells may also be a mixture of ILC-lineage precursors and NK cell-lineage immature cells(22), and until NK cell and ILC1 precursor populations can be fully clarified(31, 32), whether all Eomes+ cells are NK lineage cells remains an open question.
Previous work demonstrated “ILC1-like” cells from PyMT tumors can directly kill cancer cells in a perforin-dependent manner(3). Here, we showed these “ILC1-like” cells in the PyMT tumor were a mixture of both CXCR6+ and Eomes+ tissue-resident cells. In the absence of a tumor, CXCR6+ ILC1s from the liver could also mediate perforin-dependent cytotoxicity, including both ILC1s that initially express granzyme C and those that did not. Although these ILC1s also express TRAIL, which can induce cell death in a perforin-independent manner (33), we observed that their killing capabilities against RMA-S cells required perforin. Other studies support our findings, as certain liver ILC1 subsets can kill YAC-1 target cells(4, 5). Given that these experiments did not include addition of IL-15c and had a shorter coculture period, this may explain the differences in killing rate observed compared to our findings. These subsets include CD127− mature ILC1, which are more enriched for granzyme B and C-expressing cells than the precursor CD127+ ILC1 population(4, 27). This maturation spectrum of ILC1s in the liver is in line with our RNAseq findings that DN ILC1s appear more proliferative and precursor-like, although we do not observe differential expression of Cd127 among the three subsets we sequenced. The fact that ILC1s can readily express cytolytic molecules and mediate cytotoxicity may help reconcile some observations in human, such as populations of ILC-like cells with cytotoxicity(34). This type of cytotoxic response may contribute to cancer immunosurveillance, as in both perforin and IL-15-deficient mice, tumor appearance and growth are accelerated(3). Here, we specifically disrupted granzyme C-expressing cells while leaving other IL-15-dependent and perforin-expressing immune cells such as NK cells intact and observed a similar acceleration in tumor growth. This confirms a that these cells can impact the growth of transformed mammary epithelium at early time points in the PyMT model and raises the possibility that they can contribute more broadly to cancer immunosurveillance in other models and tissues.
TGF-β signaling promoted maintenance and anti-tumor functions of granzyme C-expressing tissue-resident innate lymphocytes. TGF-β may represent a tissue niche-associated signal that helps maintain proper localization of these cells in epithelial tissues, for instance through direct lymphocyte-epithelial cell contact via CD103 and E-cadherin interactions, which may provide additional survival and differentiation cues. A study of human head and neck tumors revealed a CD49a+CD103+ ILC1-like population with potent cytolytic activities against cancer cells(34). These cells can be differentiated from peripheral NK-phenotype cells, especially the CD94+NKp80+CD16− immature subset, in a co-culture system with cancer cells and IL-15, and is dependent on TGF-β signaling(35). Nonetheless, TGF-β signaling-dependent conversion of NK-phenotype cells to ILC1-like cells appeared to suppress their cancer surveillance function in a murine fibrosarcoma model(13). Furthermore, excessive TGF-β signaling triggered by expression of a constitutively active form of TGF-β receptor in NK cells promotes fibrosarcoma development as a likely consequence of enhanced angiogenesis(13). These contrasting outcomes may be explained by the possibility that tumor types with epithelial cancers, but not mesenchymal fibrosarcoma, are subject to tissue-resident ILC1-mediated cancer immunosurveillance. In addition, inhibitory or stimulatory functions of TGF-β may be conditional on the strength and duration of tumor-associated signals such as IL-15.
IL-15 was a regulator of cytotoxic ILC1 differentiation and granzyme C expression. Our studies imply excessive cytotoxic ILC1 responses under conditions of enhanced Stat5 signaling can mediate immunopathology and autoimmunity. Of note, the phenotype of GzmcStat5b-CA/+ mice is reminiscent of human Aggressive NK-cell Leukemia (ANKL), which can be driven by JAK3 and STAT5A/B mutations and is characterized by hepatomegaly and necrotic tissue damage in organs such as the liver, with a median overall survival of less than two months(36). ANKL may represent a scenario of overactive STAT5 signaling in tissue-resident cytotoxic innate lymphocytes leading to immunopathology in humans, as ANKL cells can be sparse in the blood and bone marrow, expand in organs such as the liver, and exhibit a CD56highCD57− phenotype similar to purported human tissue-resident NK cells(37), which may be the human ortholog of mouse liver ILC1s.
ILC1s are not just cytokine-restricted helper populations but can also express high levels of cytotoxic molecules, including granzyme B, granzyme C, and perforin. They can mediate potent perforin-dependent cytotoxicity, contributing both to autoimmunity upon overactive IL-15 signaling and to cancer immunosurveillance of mammary epithelium in the PyMT model in mice. A true helper ILC1 population with no potential for cytotoxicity may exist, but will require further work to identify. Notably, both perforin and IFN-γ are required for NK1.1+ cells to suppress MCMV infection in the liver(39), and liver ILC1s have been shown to supply such a critical source of IFN-γ(40). Perhaps ILC1s maintain the ability to both produce cytokines and perform cytotoxicity depending on signals from their environment. Our exploration of the ontogeny and function of group 1 innate lymphocytes prompts a reframing of the ILC effector spectrum and may help pave the way for therapeutic targeting.
MATERIALS AND METHODS
Study design
The main goal of this study was to explore the heterogeneity of type 1 innate lymphocytes in mice using mouse genetics and molecular and cellular immunology approaches. The sample size for the flow cytometry and other experiments were based on previous studies and pilot experiments in the lab that allowed for conclusive data in measuring changes in cell populations. All experiments were performed at least two to three times independently, the number of individual mice or samples are always indicated in the figure legends, mice were age and sex-matched, and litter-mate or cage-mates were used where possible. The experiments were performed unblinded as mice of different genotypes were identified and kept track of, except for tumor measurements and scoring of histology slides.
Animal experimental models
Eomesfl, Il17aiCre, Il2rg−/−, Il22iCre, Il5tdT-iCre, Rag1−/−, Rag2−/−, Rosa26LSL-DTA, Rosa26FLPe, and Tbx21fl mice were purchased from Jackson Laboratories. Rosa26LSL-YFP, Il15−/−, Prf1−/−, MMTV-PyMT, Rosa26LSL-Stat5b-CA and Tgfbr2fl mouse strains were previously reported(3, 41–43), GzmctdTomato-T2A-iCre mice were generated by insertion of a targeting construct into the Gzmc locus by CRISPR/Cas9-mediated homologous recombination in embryonic stem cells (ESCs) on the albino C57BL/6 background. The targeting construct was generated by inserting 2 kb homology arms upstream and downstream of the 3’UTR of Gzmc derived from BAC RP23 (BACPAC Resources Center) into Zero blunt plasmid backbone, and then inserting IRES, tandem dimer (td) Tomato, T2A self-cleaving peptide, iCre recombinase, and frt site-flanked PGK-Neomycin resistance gene (NEO)-BGHpA cassette in between homology arms as previously described(44). Guide RNA targeting the 3’ UTR of Gzmc (5’-TCCTGGACTCAGCTATGGGG-3’) was cloned into the pX335 vector. The targeting construct and pX335 vector were co-injected into ESCs. Neomycin-resistant ESC clones were subject for Southern Blot analysis after EcoRI restriction enzyme treatment to confirm proper insertion of the construct, and for karyotyping to confirm chromosome integrity. Clones with successful insertions and stable chromosomes were injected into pseudo-pregnant female C57BL/6 mice mixed with non-albino C57BL/6 ESCs. Of resulting pups, the construct successfully went germline, as noted by white fur chimerism and genotyped offspring. Mice harboring the construct were then crossed with Rosa26FLPe mice to ensure removal of the NEO cassette. S1pr5eGFP-T2A-iCre mice were generated in the same manner as GzmctdT-T2A-iCre mice, except that the targeting vector contained eGFP in place of tdTomato and targeted the S1pr5 3’ UTR with associated homology arms, and BamHI was used in the Southern Blot analysis. All mice were bred and maintained in a specific pathogen-free facility at MSKCC and animal experimentation was conducted in accordance with institutional guidelines and under an approved IACUC animal use protocol.
Immune cell isolation
Immune cells were isolated from spleen, salivary gland, liver, small intestine lamina propria, and PyMT tumor as described previously(3, 45). Briefly, spleen single-cell suspension was obtained by tissue disruption with glass slides. For liver, tissue was manually dissociated using a razor blade and digested with 1 mg/ml Collagenase D (Sigma Aldrich) for 30 minutes at 37°C. For small intestine, tissue was dissected and washed in PBS. Intestines were cut in small pieces and incubated in 1 mM dithiothreitol and 1 mM EDTA in PBS with gentle agitation for 30 minutes. The supernatant containing intraepithelial lymphocytes was removed. Remaining tissue was digested in 1 mg/ml Collagenase Type III (Worthington) and 4 μg/ml DNase I (Sigma-Aldrich) in completed RPMI with gentle agitation for 20 minutes. For salivary gland and PyMT tumor, tissue was manually dissociated using a razor blade and digested for 1 hour in 1 mg/ml Collagenase Type III (Worthington) and 4 μg/ml DNase I (Sigma-Aldrich) in HBSS at 37°C with periodic vortex. After collagenase treatment, cells isolated from the liver, salivary gland, small intestine and tumor were filtered through a 70-μM cell strainer, layered in a 44% and 66% Percoll gradient (Sigma-Aldrich), and centrifuged at 1,900 g for 30 minutes without brake. Cells at the interface were collected and analyzed by flow cytometry. For bone marrow, bones were collected from limbs of mice, cleaned, sterilized, mashed with a mortar and pestle, and passed through 70 μm cell strainer. Lineage cells were depleted from bone marrow using MagniSort Lineage depletion kit (Invitrogen). For CLP and LSK, bones from one mouse were pooled per sample. For ILCp, bones from at least six mice were pooled per sample. Cells were then stained for sorting.
Flow cytometry
Cells were preincubated with 2.4G2 mAB (Bio X Cell) to block non-specific FcγR binding and Ghost Dye (Tonbo) to detect dead cells and were stained with panels of antibodies for 30 min on ice. Fluorochrome-conjugated or biotinylated antibodies against mouse CD103 (M290), CD11b (M1/70), CD27 (LG.3A10), CD4 (RM4-5), CD45 (30-F11), CD49a (Ha31/8), CD8β (H35-17.2), IL-17A (TC11-18H10.1), IL-5 (TRFK5), Ly6C (AL-21), and TCRγδ (GL3) were purchased from BD Biosciences. Antibodies against mouse αGalCer:CD1d (L363), CD127 (A7R34), CD19 (1D3), CD3ε (145-2C11), CD69 (H1.2F3), CD90.2/Thy1.2 (53–2.1), CXCR6 (SA051D1), granzyme C (SFC1D8), KLRG1 (2F1), NKp46 (29A1.4), and perforin (S16009A) were purchased from BioLegend. Antibodies against mouse CD200R1 (OX110), CD49b (Dx5), CD5 (53–7.3), Eomes (Dan11mag), IL-22 (1H8PWSR), NK1.1 (PK136), and T-bet (eBio4b10) and against Rabbit (anti-F(ab)2’) (polyclonal) were purchased from eBioscience, now Thermo Scientific. GFP (raised in rabbit) and human/mouse granzyme B (GB11) were purchased from Invitrogen. Mouse B220 (RA3-6B2), CD127 (A7R34), CD8α (53–6.7), F4/80 (BM8.1), IFN-γ (XMG1.2), Ly6G (1A8), and TCRβ (H57-597) were purchased from Tonbo Biosciences. CellTrace Violet was purchased from Thermo Fisher. For preservation of fluorescent proteins, cells were fixed in 4% PFA for 15 minutes on ice after surface staining. For intracellular antibodies, the Tonbo transcription factor kit was used to fix and permeabilize cells for 30 minutes on ice, followed by intracellular staining for 30 minutes on ice. All samples were acquired and analyzed with LSRII flow cytometer (Becton Dickson), FACSDiva (Becton Dickson) and FlowJo (TreeStar) software.
Cell sorting and transfer
After gating on morphology and singlets, CD45+Dead− cells were gated as follows: related to Fig. S1–S2 for ATACsequencing: lineage (CD19, CD3, CD5, Ly6G, TCRβ)-negative, NK1.1+NKp46+, for NK populations: CD49a-CD49b+ (spleen and liver), for ILC1 populations: CD49a+CD49b- (liver, salivary gland). Related to Fig. 3A and Fig. S3B, lineage (B220, CD11b, CD19, CD3e, Gr1, Ly6D, TER119)-, CD117+Sca1+, S1pr5FM− (non-FM, YFP−, LSKs). Cell sorting was conducted on Aria II (Becton Dickson). Related to Fig. S3E, Lineage (same as LSK)−NK1.1−CD127+α4β7+Flt3−PD-1+CD25− (ILCp). After sorting ILCp from pooled bone marrow of 6 mice (age 4–6 weeks), ILCp (around 4,000 cells) were transferred into one recipient Rag2−/−Il2rg−/− mouse. Recipient mice were analyzed around 4 weeks after transfer. Related to Fig. 6, S9 and S10 for RNAsequencing and Fig. 7A for IL-15 culture: NK1.1+CD3−CD49a+CXCR6+, then GzmctdT+GzmcFM+ (DP), GzmctdT−GzmcFM+ (FMSP), GzmctdT−GzmcFM− (DP) from livers of GzmcFM mice, and NK1.1+CD3−CD49a−CD49b+CD11b+S1pr5eGFP+ (NK) from livers of S1pr5eGFP mice. Related to Fig. 7B for killing assay: NK1.1+CD3−CD49a+CXCR6+, then GzmctdT+ or GzmctdT− from livers of either GzmctdT-T2A-iCre/+ or GzmctdT-T2A-iCre/+Prf1−/− mice.
ATAC sample preparation and sequencing
50,000 cells were sorted for each sample and frozen in FBS + 10% DMSO. Profiling of chromatin was performed by ATACseq as described previously(46). Briefly, 50,000 viably frozen cells were washed in cold PBS and lysed. The transposition reaction was incubated at 42°C for 45 minutes. The DNA was cleaned with the MinElute PCR Purification Kit (Qiagen catalog # 28004) and material was amplified for 5 cycles. After evaluation by real-time PCR, 8–10 additional PCR cycles were done. The final product was cleaned by aMPure XP beads (Beckman Coulter catalog # A63882) at a 1X ratio. Libraries were sequenced on a Hiseq 2500 in High Output mode in a PE50 run, using the TruSeq SBS Kit v4 (Illumina). An average of 57 million paired reads were generated per sample.
Bone marrow chimera
Recipient mice (6–8 weeks old, CD45.1.2+) were irradiated with 900 Gyz. Eighteen hours after irradiation, bone marrow was harvested from CD45.2+ S1pr5eGFP-iCre/+Rosa26LSL-YFP mice, lineage depleted, stained with flow antibodies, sorted for non-fate mapped LSK populations (detailed above), and transferred intravenously into recipient mice. Mice were maintained on sulfatrim antibiotic diet and aged to allow bone marrow to graft. Mice were analyzed 4–5 months after transfer.
Tumor measurement
At 12 and 16 weeks of age, tumors were measured with a caliper. Tumor volume was calculated using the equation [(L×W2) × (π/6)], in which L denotes length and W denotes width. Individual tumor volumes were added together to calculate total tumor burden per mouse. Researchers were blinded to genotypes of mice during measurements. Mice were euthanized if total tumor volume reached 3,000 mm3 or one tumor volume reached 2,000 mm3 in accordance with the IACUC-approved protocol.
RNA sample preparation and sequencing
All cells in a given population were sorted from one mouse per sample for DP, FMSP, DN and NK cells directly into 750 μL Trizol LS. After sort, total volume was brought up to 1 mL with sterile PBS, and samples were flash frozen. Four DP, four FMSP, three DN and four NK cell samples were processed and sequenced. For RNA extraction, phase separation in cells lysed in 1 mL TRIzol Reagent (ThermoFisher catalog # 15596018) was induced with 200 μL chloroform and RNA was extracted from the aqueous phase using the miRNeasy Micro Kit (Qiagen catalog # 217084) on the QIAcube Connect (Qiagen) according to the manufacturer’s protocol with 350 μL input. Samples were eluted in 15 μL RNase-free water. For transcriptome sequencing, after RiboGreen quantification and quality control by Agilent BioAnalyzer, 282–300 pg total RNA with RNA integrity numbers ranging from 7.8 to 9.4 underwent amplification using the SMART-Seq v4 Ultra Low Input RNA Kit (Clonetech catalog # 63488), with 12 cycles of amplification. Subsequently, 2.1–3 ng of amplified cDNA was used to prepare libraries with the KAPA Hyper Prep Kit (Kapa Biosystems KK8504) using 8 cycles of PCR. Samples were barcoded and run on a NovaSeq 6000 in a PE100 run, using the NovaSeq 6000 S4 Reagent Kit (200 Cycles) (Illumina). An average of 82 million paired reads were generated per sample and the percent of mRNA bases per sample ranged from 67% to 83%.
In vitro cell culture
Cells were first sorted as indicated above. Related to Fig. 7A, after sorting based on current and/or past expression of granzyme C, cells were cultured in T cell media (RPMI supplemented with 10% FCS, 1 mM sodium pyruvate, non-essential amino acids [Gibco], 10 mM Hepes, 55 μM 2-Mercaptoethanol, 100 U/mL Penicillin G and 0.1 mg/ml Streptomycin) supplemented with 100 ng/mL mouse IL-15/IL-15Rα (Invitrogen) for 24 hours at 37°C. Cells were then stained for flow cytometric analysis to measure expression of granzyme C.
Killing assay
The killing assay was performed as previously reported(3). Briefly, cells were sorted as described in the “cell sorting and transfer” subsection above, cultured in 100 ng/mL IL15/IL15Rα complex for one week, changing media every 3–4 days. Target RMA-S cells were maintained in T cell media. On the day of the experiment, RMA-S cells were stained with CellTrace Violet and mixed with sorted and expanded GzmctdT+ or GzmctdT− ILC1s at a ratio of 10 effector:1 target cell. Cells were incubated for 16 hours at 37°C in TCM supplemented with 100 ng/mL IL15/IL15Rα in 96-well U-bottom plate. After incubation, EDTA was added to separate cell conjugates. Cells were stained with Tonbo Ghost Dye to detect dead cells and were analyzed via flow cytometry for dead cells among CTV+ target cells. The experiment was repeated three times, each experiment including cells sorted from five mice.
Neonatal mouse necropsy phenotyping
GzmctdT-iCreRosa26LSL-STAT5b-CA, GzmctdT-iCreRosa26LSL-STAT5b-CAPerf−/−, or wild-type littermate mice were euthanized at 14 days of age with CO2. Following gross examination the liver was fixed in 10% neutral buffered formalin, followed by decalcification of bone in a formic acid solution (Surgipath Decalcifier I, Leica Biosystems). Tissues were then processed in ethanol and xylene and embedded in paraffin in a Leica ASP6025 tissue processor. Paraffin blocks were sectioned at 5 microns, stained with hematoxylin and eosin (H&E), and examined by a board-certified veterinary pathologist (AOM).
Immunohistochemistry
IHC was performed on a Leica Bond RX automated stainer using Bond reagents (Leica Biosystems, Buffalo Grove, IL), including a polymer detection system (DS9800, Novocastra Bond Polymer Refine Detection, Leica Biosystems). The chromogen was 3,3 diaminobenzidine tetrachloride (DAB), and sections were counterstained with hematoxylin. Staining for cleaved caspase 3 (CC3) used primary antibody (Cell signaling, Cat.9961) at 1:250 dilution and biotinylated anti-Rabbit IgG (H+L) as secondary antibody (Vector, Cat. BA1000) at 1:100 dilution. Staining for mouse NKp46 used primary antibody (R&D Systems, Cat. AF2225) at 1:1000 dilution and biotinylated anti-Goat IgG (H+L) (Vector, Cat. BA5000). Both markers used heat induced, pH 6.0 for epitope retrieval.
Quantification and statistical analysis
ATACseq analysis
For all replicates in all biological conditions, we first used seqtk to trim 10 bp from either end of raw paired-end ATAC-seq reads. The trimmed fastqs were then aligned to the mm10 reference genome using bowtie2(47). Duplicates were removed using samtools(48, 49), and peaks were called using MACS2(50) with parameters “shift 100 --extsize 200 -p 0.2 -B –SPMR”. To define an atlas of peaks reproducible across replicates within each biological condition, we used the irreproducible discovery rate framework (IDR) at FDR P < 0.1 for each pair of replicates within each condition, followed by merged peak calling across all replicates using MACS2. This defined a final atlas of 51412 peaks for downstream analysis. Peaks were annotated to the closest gene.
Differential accessibility analysis, CDF, differential gene expression
For differential accessibility analyses, we used DESeq2(51), and peaks were considered significantly differentially accessible at FDR P < 0.05. We first performed two rounds of differential accessibility analysis- liver ILC1 vs liver NK cell and splenic NK cell, and then salivary gland ILC1 vs liver NK cell and splenic NK cell. Peaks commonly differentially accessible across both comparisons were considered ILC1 lineage peaks, and we repeated this procedure with chromatin accessibility profiles from NK cells to define NK cell lineage peaks. Differentially expressed genes between liver ILC1 and liver NK cell genes were defined based on analysis of a publicly available microarray dataset (7). Genes were considered significantly differentially expressed at FDR P < 0.05. For CDF analyses, we overlapped all genes with significantly differentially accessible peaks from the comparison described above, and the Kolmogorov-Smirnov test was used to evaluate differences in log fold change distributions. Fold change versus fold change plot showing log fold change (LFC) of gene expression from microarray of liver ILC1 versus liver NK cell versus mean accessibility LFC for liver ILC1 versus liver NK cell was also generated.
RNA sequencing analysis
Paired-end reads in fastq format from 15 total samples (4 DP, 4 FMSP, 3 DN, and 4 NK) were quantified at the transcript level using the mm10 reference with Salmon (v1.6)(52) using default parameters, and then aggregated to gene-level counts using tximport (v1.2)(53). Differential expression analyses were conducted using DESeq2(51) separately for all six pairwise comparisons between the four cell types, and genes were included in the six lists of differentially expressed genes (Table S3) if they met all three of the following criteria: base mean expression > 50, false discovery rate (FDR) P < 0.05, and log2 fold change either > 1 or < −1. Genes were included in the ‘core’ list of NK cell and ILC1 genes (Table S4) if they were differentially expressed in the same direction in all three of the separate NK cell-ILC1 comparisons. Z-scores across groups of the log-regularized transformation values produced by DESeq2 were used to visualize the differentially expressed genes in heatmaps in Fig. 6D, S9C, and S10. Assignment of genes to the five classes in Fig. 6D and S10 (cell surface molecules, nuclear factors, metabolic enzymes, signaling proteins, and secreted molecules) was done manually.
Quantitative digital image analysis HALO
Expression of CC3 and NKp46 on IHC of mouse livers was evaluated quantitatively by automated image analysis. Whole-slide digital images were generated on a scanner (Pannoramic Flash 250, 3DHistotech, 20x/0.8NA objective, Budapest, Hungary), at a 0.243094 μm/pixel resolution. Image analysis was performed on HALO software (Indica Labs, Albuquerque, NM), employing the Area Quantification module v.2.1.2.0. Region of interest (ROI) was annotated manually, including liver tissue and excluding folded tissue. The area quantification module was used to detect area of CC3 and NKp46 based on the optical density (OD) of DAB immunoreactivity after determining the OD threshold. Tissues from three mice per genotype were analyzed.
Statistical analysis
All data are displayed as mean +/− SEM, each dot represents one mouse. For pair-wise comparisons between two samples, unpaired student t test, two-tailed, was used. For three or more samples, one-way ANOVA with Tukey’s multiple comparisons test was used. For survival analysis, Log-rank (Mantel Cox) test was performed. “ns” = not significant, * = p<0.05, ** = p<0.01, *** = p<0.001, **** = p<0.0001. All statistical analysis listed here was performed using GraphPad Prism software.
Supplementary Material
Table S2. Differentially accessible and differentially expressed genes in NK cells and ILC1s.
Table S1. Differentially accessible chromatin in NK cells and ILC1s.
Table S3. Pairwise comparisons among ILC1 subsets and NK cells from the liver.
Table S4. Core NK cell and ILC1 genes.
Table S5. Raw data and statistics for graphs in the manuscript.
Figure S1. Chromatin accessibility of circulating and resident group 1 innate lymphocytes reveals differentially accessible peaks.
Figure S2. S1pr5eGFP-T2A-iCre mice reveal single-cell resolution of S1pr5 expression that marks mature NK cells.
Figure S3. Granzyme C-expressing innate lymphocytes are not derived from S1pr5+ NK cells, but can be differentiated from ILCps.
Figure S4. Cytokine-producing effector ILC2s and ILC3s do not give rise to granzyme C-positive ILC1s.
Figure S5. GzmctdT-T2A-iCre/+ mice allow for lineage tracing of cells with a history of granzyme C expression.
Figure S6. Cells with a history of granzyme C expression do not give rise to IL-5-, IL-17A-, or IL-22-producing ILC2s or ILC3s.
Figure S7. Regulation of ILC1 by T-bet, Eomes, and TGF-β.
Figure S8. Granzyme C-expressing ILC1s in the tumor require TGF-β signaling and mediate cancer immunosurveillance.
Figure S9. Potential heterogeneity among ILC1 subsets with differential history of granzyme C expression.
Figure S10. Genes enriched in DN relative to DP ILC1s.
Figure S11. Liver ILC1s can mediate perforin-dependent cytotoxicity.
Figure S12. Granzyme C-expressing innate lymphocytes are lost in the absence of IL-15 and expanded upon gain-of-function in Stat5 signaling.
Acknowledgments:
We are grateful to members of the M.O.L Lab for helpful discussions. We acknowledge the use of the Integrated Genomics Operation Core, funded by the NCI Cancer Center Support Grant (CCSG, P30 CA08748), Cycle for Survival, and the Marie-Josée and Henry R. Kravis Center for Molecular Oncology.
Funding:
This work was funded by grants from the National Institutes of Health (F31CA210332 to BGN, R01CA243904-01A1 to MOL,U54 CA209975 to AYR, P30 CA008748 to AOM, AYR, CSL, and MOL, and 5T32CA009207-43 to AEC), by the Department of Defense (KC19008.e001 to MOL), by the Howard Hughes Medical Institute (Investigator to AYR and Faculty Scholar Award to MOL), by the Cancer Research Institute (CLIP grant to MOL), by the Ludwig Center for Cancer Immunotherapy (MOL), by the Functional Genomic Initiative (MOL), and by the Parker Institute for Cancer Immunotherapy (AYR).
Footnotes
Competing Interests:
MSKCC has filed a patent application with the U.S. Patent and Trademark Office directed toward targeting ILC1 IL-15 signaling for cancer immunotherapy. MOL is an SAB member of and holds equity or stock options in Amberstone Biosciences Inc. AYR is an SAB member of and holds equity or stock options in Sonoma Biotherapeutic, Surface Oncology, RAPT Therapeutics, and Vedanta Biosciences. He is an SAB member of BioInvent. A.Y.R. holds IP rights for therapeutic Treg cell depleting antibodies licensed to Takeda that is unrelated to the content of the present study.
Data Materials Availability:
The RNA-seq and ATAC-seq data are available at GEO under accession number GSE196716. All other data needed to evaluate the conclusions in the paper are present in the paper or Supplemental Materials.
References and Notes:
- 1.Fan X, Rudensky AY, Hallmarks of Tissue-Resident Lymphocytes. Cell 164, 1198–1211 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie ANJ, Mebius RE, Powrie F, Spits H, Innate Lymphoid Cells: 10 Years On. Cell 174, 1054–1066 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Dadi S, Chhangawala S, Whitlock BM, Franklin RA, Luo CT, Oh SA, Toure A, Pritykin Y, Huse M, Leslie CS, Li MO, Cancer Immunosurveillance by Tissue-Resident Innate Lymphoid Cells and Innate-like T Cells. Cell 164, 365–377 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Friedrich C, Taggenbrock R, Doucet-Ladeveze R, Golda G, Moenius R, Arampatzi P, Kragten NAM, Kreymborg K, Gomez de Aguero M, Kastenmuller W, Saliba AE, Grun D, van Gisbergen K, Gasteiger G, Effector differentiation downstream of lineage commitment in ILC1s is driven by Hobit across tissues. Nat Immunol 22, 1256–1267 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Di Censo C, Marotel M, Mattiola I, Muller L, Scarno G, Pietropaolo G, Peruzzi G, Laffranchi M, Mazej J, Hasim MS, Asif S, Russo E, Tomaipitinca L, Stabile H, Lee SH, Vian L, Gadina M, Gismondi A, Shih HY, Mikami Y, Capuano C, Bernardini G, Bonelli M, Sozzani S, Diefenbach A, Ardolino M, Santoni A, Sciume G, Granzyme A and CD160 expression delineates ILC1 with graded functions in the mouse liver. Eur J Immunol 51, 2568–2575 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spits H, Bernink JH, Lanier L, NK cells and type 1 innate lymphoid cells: partners in host defense. Nat Immunol 17, 758–764 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Robinette ML, Fuchs A, Cortez VS, Lee JS, Wang Y, Durum SK, Gilfillan S, Colonna M, Immunological Genome C, Transcriptional programs define molecular characteristics of innate lymphoid cell classes and subsets. Nat Immunol 16, 306–317 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gregoire C, Chasson L, Luci C, Tomasello E, Geissmann F, Vivier E, Walzer T, The trafficking of natural killer cells. Immunol Rev 220, 169–182 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson H, Scorrano L, Korsmeyer SJ, Ley TJ, Cell death induced by granzyme C. Blood 101, 3093–3101 (2003). [DOI] [PubMed] [Google Scholar]
- 10.Walzer T, Chiossone L, Chaix J, Calver A, Carozzo C, Garrigue-Antar L, Jacques Y, Baratin M, Tomasello E, Vivier E, Natural killer cell trafficking in vivo requires a dedicated sphingosine 1-phosphate receptor. Nat Immunol 8, 1337–1344 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Daussy C, Faure F, Mayol K, Viel S, Gasteiger G, Charrier E, Bienvenu J, Henry T, Debien E, Hasan UA, Marvel J, Yoh K, Takahashi S, Prinz I, de Bernard S, Buffat L, Walzer T, T-bet and Eomes instruct the development of two distinct natural killer cell lineages in the liver and in the bone marrow. J Exp Med 211, 563–577 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sojka DK, Plougastel-Douglas B, Yang L, Pak-Wittel MA, Artyomov MN, Ivanova Y, Zhong C, Chase JM, Rothman PB, Yu J, Riley JK, Zhu J, Tian Z, Yokoyama WM, Tissue-resident natural killer (NK) cells are cell lineages distinct from thymic and conventional splenic NK cells. Elife 3, e01659 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao Y, Souza-Fonseca-Guimaraes F, Bald T, Ng SS, Young A, Ngiow SF, Rautela J, Straube J, Waddell N, Blake SJ, Yan J, Bartholin L, Lee JS, Vivier E, Takeda K, Messaoudene M, Zitvogel L, Teng MWL, Belz GT, Engwerda CR, Huntington ND, Nakamura K, Holzel M, Smyth MJ, Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat Immunol 18, 1004–1015 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Constantinides MG, McDonald BD, Verhoef PA, Bendelac A, A committed precursor to innate lymphoid cells. Nature 508, 397–401 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu Y, Tsang JC, Wang C, Clare S, Wang J, Chen X, Brandt C, Kane L, Campos LS, Lu L, Belz GT, McKenzie AN, Teichmann SA, Dougan G, Liu P, Single-cell RNA-seq identifies a PD-1(hi) ILC progenitor and defines its development pathway. Nature 539, 102–106 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Xu W, Cherrier DE, Chea S, Vosshenrich C, Serafini N, Petit M, Liu P, Golub R, Di Santo JP, An Id2(RFP)-Reporter Mouse Redefines Innate Lymphoid Cell Precursor Potentials. Immunity 50, 1054–1068 e1053 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bernink JH, Krabbendam L, Germar K, de Jong E, Gronke K, Kofoed-Nielsen M, Munneke JM, Hazenberg MD, Villaudy J, Buskens CJ, Bemelman WA, Diefenbach A, Blom B, Spits H, Interleukin-12 and −23 Control Plasticity of CD127(+) Group 1 and Group 3 Innate Lymphoid Cells in the Intestinal Lamina Propria. Immunity 43, 146–160 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Silver JS, Kearley J, Copenhaver AM, Sanden C, Mori M, Yu L, Pritchard GH, Berlin AA, Hunter CA, Bowler R, Erjefalt JS, Kolbeck R, Humbles AA, Inflammatory triggers associated with exacerbations of COPD orchestrate plasticity of group 2 innate lymphoid cells in the lungs. Nat Immunol 17, 626–635 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fiancette R, Finlay CM, Willis C, Bevington SL, Soley J, Ng STH, Baker SM, Andrews S, Hepworth MR, Withers DR, Reciprocal transcription factor networks govern tissue-resident ILC3 subset function and identity. Nat Immunol 22, 1245–1255 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bando JK, Liang HE, Locksley RM, Identification and distribution of developing innate lymphoid cells in the fetal mouse intestine. Nat Immunol 16, 153–160 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schneider C, Lee J, Koga S, Ricardo-Gonzalez RR, Nussbaum JC, Smith LK, Villeda SA, Liang HE, Locksley RM, Tissue-Resident Group 2 Innate Lymphoid Cells Differentiate by Layered Ontogeny and In Situ Perinatal Priming. Immunity 50, 1425–1438 e1425 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Constantinides MG, Gudjonson H, McDonald BD, Ishizuka IE, Verhoef PA, Dinner AR, Bendelac A, PLZF expression maps the early stages of ILC1 lineage development. Proc Natl Acad Sci U S A 112, 5123–5128 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takeda K, Cretney E, Hayakawa Y, Ota T, Akiba H, Ogasawara K, Yagita H, Kinoshita K, Okumura K, Smyth MJ, TRAIL identifies immature natural killer cells in newborn mice and adult mouse liver. Blood 105, 2082–2089 (2005). [DOI] [PubMed] [Google Scholar]
- 24.Gordon SM, Chaix J, Rupp LJ, Wu J, Madera S, Sun JC, Lindsten T, Reiner SL, The transcription factors T-bet and Eomes control key checkpoints of natural killer cell maturation. Immunity 36, 55–67 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cortez VS, Cervantes-Barragan L, Robinette ML, Bando JK, Wang Y, Geiger TL, Gilfillan S, Fuchs A, Vivier E, Sun JC, Cella M, Colonna M, Transforming Growth Factor-beta Signaling Guides the Differentiation of Innate Lymphoid Cells in Salivary Glands. Immunity 44, 1127–1139 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mackay LK, Minnich M, Kragten NA, Liao Y, Nota B, Seillet C, Zaid A, Man K, Preston S, Freestone D, Braun A, Wynne-Jones E, Behr FM, Stark R, Pellicci DG, Godfrey DI, Belz GT, Pellegrini M, Gebhardt T, Busslinger M, Shi W, Carbone FR, van Lier RA, Kallies A, van Gisbergen KP, Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science 352, 459–463 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Yomogida K, Bigley TM, Trsan T, Gilfillan S, Cella M, Yokoyama WM, Egawa T, Colonna M, Hobit confers tissue-dependent programs to type 1 innate lymphoid cells. Proc Natl Acad Sci U S A 118, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kennedy MK, Glaccum M, Brown SN, Butz EA, Viney JL, Embers M, Matsuki N, Charrier K, Sedger L, Willis CR, Brasel K, Morrissey PJ, Stocking K, Schuh JC, Joyce S, Peschon JJ, Reversible defects in natural killer and memory CD8 T cell lineages in interleukin 15-deficient mice. J Exp Med 191, 771–780 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leonard WJ, Lin JX, O’Shea JJ, The gammac Family of Cytokines: Basic Biology to Therapeutic Ramifications. Immunity 50, 832–850 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Jabri B, Abadie V, IL-15 functions as a danger signal to regulate tissue-resident T cells and tissue destruction. Nat Rev Immunol 15, 771–783 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crinier A, Milpied P, Escaliere B, Piperoglou C, Galluso J, Balsamo A, Spinelli L, Cervera-Marzal I, Ebbo M, Girard-Madoux M, Jaeger S, Bollon E, Hamed S, Hardwigsen J, Ugolini S, Vely F, Narni-Mancinelli E, Vivier E, High-Dimensional Single-Cell Analysis Identifies Organ-Specific Signatures and Conserved NK Cell Subsets in Humans and Mice. Immunity 49, 971–986 e975 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McFarland AP, Yalin A, Wang SY, Cortez VS, Landsberger T, Sudan R, Peng V, Miller HL, Ricci B, David E, Faccio R, Amit I, Colonna M, Multi-tissue single-cell analysis deconstructs the complex programs of mouse natural killer and type 1 innate lymphoid cells in tissues and circulation. Immunity 54, 1320–1337 e1324 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.von Karstedt S, Montinaro A, Walczak H, Exploring the TRAILs less travelled: TRAIL in cancer biology and therapy. Nat. Rev. Cancer 17, 352–366 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Krabbendam L, Bernink JH, Spits H, Innate lymphoid cells: from helper to killer. Curr Opin Immunol 68, 28–33 (2021). [DOI] [PubMed] [Google Scholar]
- 35.Moreno-Nieves UY, Tay JK, Saumyaa S, Horowitz NB, Shin JH, Mohammad IA, Luca B, Mundy DC, Gulati GS, Bedi N, Chang S, Chen C, Kaplan MJ, Rosenthal EL, Holsinger FC, Divi V, Baik FM, Sirjani DB, Gentles AJ, Newman AM, Freud AG, Sunwoo JB, Landscape of innate lymphoid cells in human head and neck cancer reveals divergent NK cell states in the tumor microenvironment. Proc Natl Acad Sci U S A 118, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang YT, Wang D, Luo H, Xiao M, Zhou HS, Liu D, Ling SP, Wang N, Hu XL, Luo Y, Mao X, Ao QL, Huang J, Zhang W, Sheng LS, Zhu LJ, Shang Z, Gao LL, Zhang PL, Zhou M, Zhou KG, Qiu LG, Liu QF, Zhang HY, Li JY, Jin J, Fu L, Zhao WL, Chen JP, Du X, Huang G, Wang QF, Zhou JF, Huang L, Aggressive NK-cell leukemia: clinical subtypes, molecular features, and treatment outcomes. Blood Cancer J 7, 660 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Melsen JE, Lugthart G, Lankester AC, Schilham MW, Human Circulating and Tissue-Resident CD56(bright) Natural Killer Cell Populations. Front Immunol 7, 262 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McDonald BD, Jabri B, Bendelac A, Diverse developmental pathways of intestinal intraepithelial lymphocytes. Nat Rev Immunol 18, 514–525 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Loh J, Chu DT, O’Guin AK, Yokoyama WM, Virgin H. W. t., Natural killer cells utilize both perforin and gamma interferon to regulate murine cytomegalovirus infection in the spleen and liver. J Virol 79, 661–667 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weizman OE, Adams NM, Schuster IS, Krishna C, Pritykin Y, Lau C, Degli-Esposti MA, Leslie CS, Sun JC, O’Sullivan TE, ILC1 Confer Early Host Protection at Initial Sites of Viral Infection. Cell 171, 795–808 e712 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chinen T, Kannan AK, Levine AG, Fan X, Klein U, Zheng Y, Gasteiger G, Feng Y, Fontenot JD, Rudensky AY, An essential role for the IL-2 receptor in Treg cell function. Nat Immunol 17, 1322–1333 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, Pamer EG, Li MO, The cellular and molecular origin of tumor-associated macrophages. Science 344, 921–925 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ouyang W, Oh SA, Ma Q, Bivona MR, Zhu J, Li MO, TGF-beta cytokine signaling promotes CD8+ T cell development and low-affinity CD4+ T cell homeostasis by regulation of interleukin-7 receptor alpha expression. Immunity 39, 335–346 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Levine AG, Mendoza A, Hemmers S, Moltedo B, Niec RE, Schizas M, Hoyos BE, Putintseva EV, Chaudhry A, Dikiy S, Fujisawa S, Chudakov DM, Treuting PM, Rudensky AY, Stability and function of regulatory T cells expressing the transcription factor T-bet. Nature 546, 421–425 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Do MH, Wang X, Zhang X, Chou C, Nixon BG, Capistrano KJ, Peng M, Efeyan A, Sabatini DM, Li MO, Nutrient mTORC1 signaling underpins regulatory T cell control of immune tolerance. J Exp Med 217, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ, Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10, 1213–1218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Langmead B, Salzberg SL, Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome S Project Data Processing, The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li H, A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS, Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C, Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14, 417–419 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Soneson C, Love MI, Robinson MD, Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res 4, 1521 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2. Differentially accessible and differentially expressed genes in NK cells and ILC1s.
Table S1. Differentially accessible chromatin in NK cells and ILC1s.
Table S3. Pairwise comparisons among ILC1 subsets and NK cells from the liver.
Table S4. Core NK cell and ILC1 genes.
Table S5. Raw data and statistics for graphs in the manuscript.
Figure S1. Chromatin accessibility of circulating and resident group 1 innate lymphocytes reveals differentially accessible peaks.
Figure S2. S1pr5eGFP-T2A-iCre mice reveal single-cell resolution of S1pr5 expression that marks mature NK cells.
Figure S3. Granzyme C-expressing innate lymphocytes are not derived from S1pr5+ NK cells, but can be differentiated from ILCps.
Figure S4. Cytokine-producing effector ILC2s and ILC3s do not give rise to granzyme C-positive ILC1s.
Figure S5. GzmctdT-T2A-iCre/+ mice allow for lineage tracing of cells with a history of granzyme C expression.
Figure S6. Cells with a history of granzyme C expression do not give rise to IL-5-, IL-17A-, or IL-22-producing ILC2s or ILC3s.
Figure S7. Regulation of ILC1 by T-bet, Eomes, and TGF-β.
Figure S8. Granzyme C-expressing ILC1s in the tumor require TGF-β signaling and mediate cancer immunosurveillance.
Figure S9. Potential heterogeneity among ILC1 subsets with differential history of granzyme C expression.
Figure S10. Genes enriched in DN relative to DP ILC1s.
Figure S11. Liver ILC1s can mediate perforin-dependent cytotoxicity.
Figure S12. Granzyme C-expressing innate lymphocytes are lost in the absence of IL-15 and expanded upon gain-of-function in Stat5 signaling.
Data Availability Statement
The RNA-seq and ATAC-seq data are available at GEO under accession number GSE196716. All other data needed to evaluate the conclusions in the paper are present in the paper or Supplemental Materials.