

Abstract
New antibiotics with either a novel mode-of-action (MoA) or novel mode-of-inhibition (MoI) are urgently needed to overcome the threat of drug-resistant tuberculosis (TB). The present study profiles new spiropyrimidinetriones (SPTs), DNA gyrase inhibitors having activity against drug resistant Mycobacterium tuberculosis (Mtb), the causative agent of TB. While the clinical candidate zoliflodacin has progressed to Phase 3 trials for the treatment of gonorrhea, compounds herein demonstrated higher inhibitory potency against Mtb DNA gyrase (e.g., Compound 42 with an IC50 = 2.0) and lower Mtb MICs (0.49 μM for 42). Notably, 42 and analogues showed selective Mtb activity relative to representative Gram-positive and Gram-negative bacteria. DNA gyrase inhibition was shown to involve stabilization of double-cleaved DNA while on-target activity was supported by hypersensitivity against a gyrA hypomorph. Finally, a docking model for SPTs with Mtb DNA gyrase was developed and a structural hypothesis was built for SAR expansion.
Keywords: Tuberculosis, Mycobacterium tuberculosis, DNA gyrase, drug discovery, spiropyrimidinetriones
Graphical Abstract
INTRODUCTION
The increasing prevalence of Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis (TB), has been largely due to resistance to first- and second-line TB antibiotics and is exacerbating the global TB epidemic.1 Multidrug resistant (MDR) Mtb strains resistant to both rifampicin (RIF) and isoniazid (INH), the most effective first-line drugs. Extensively drug resistant (XDR) Mtb strains are defined as those with MDR plus resistance to any fluoroquinolone (FQ) and one of the second line injectable drugs. As per the most recent World Health Organization report, in 2020 a global total of about 3 million people were diagnosed with pulmonary TB, of which 2.1 million showed rifampicin resistance and 202 000 showed MDR- or rifampicin-resistant TB (RR-TB).1 Hence, there is an urgent need to replenish the TB drug discovery and development pipeline with new drug candidates having a novel mode-of-action (MoA) or novel mode-of-inhibition (MoI) of a known clinically validated target to combat resistance and to add to the arsenal of existing TB drugs.2 Among the various approaches to overcome TB drug resistance, revisiting clinically validated targets by designing next generation compounds that overcome resistance or by identifying novel scaffolds that inhibit the validated targets differently remain worthwhile approaches to explore.3–4 Herein, we present work around the spiropyrimidinetrione (SPT) class of compounds acting through the inhibition of DNA gyrase (hereafter referred to as gyrase), a clinically validated target for the treatment of TB.
DNA type II topoisomerases, comprised of the homologous enzyme gyrases and Topoisomerase IV, have drugs in the clinic representing four different classes of antibacterial agents.5–7 Notably, these four classes that have all shown efficacy in humans avoid cross-resistance with one another. Gyrase is formed by two subunits each designated GyrA and GyrB operating as an A2B2 complex, while Topoisomerase IV similarly is formed by two subunits each designated ParC and ParE in Gram-negative bacteria and GrlA and GrlB in Gram-positive bacteria. The primary role of gyrase is to adjust DNA topology by introducing negative supercoils necessary for DNA replication. Topoisomerase IV functions primarily to decatenate replicating DNA, also necessary for replication. Notably, Mtb differs from most other human bacterial pathogens, including both Gram-negative and Gram-positive bacteria, in that it contains only one type II topoisomerase, namely gyrase, that carries out both supercoiling and decatenation.8–9
FQs comprise the most widely prescribed class of topoisomerase inhibitor (Figure 1) and are used for the treatment of a variety of bacterial infections.5, 7, 10 Three FQs – moxifloxacin, levofloxacin and gatifloxacin – are currently used as second-line TB treatments. These compounds bind to a complex wherein both strands of the DNA are cleaved, covalently bonded to the topoisomerases.11–12 As a second class of type II topoisomerase inhibitors, aminocoumarins are notable as competitive binders to the ATP-binding site situated well away from the topoisomerase DNA binding domain, a site that has received extensive investigation for inhibitor and drug design. Novobiocin, the leading example of an aminocoumarin, was on the market for Gram-positive infections associated with skin and respiratory tract infections for over 40 years, but its use was limited due to there being more potent drugs with better safety profiles and it was withdrawn in 2007.13 Several other ATPase inhibitors have demonstrated potent Mtb activity,14–16 though no clinical candidate has been revealed. Gepotidacin, representing a third class and having a novel triazaacenaphthylene moiety, inhibits type II topoisomerases at a distinct site from both FQs and aminocoumarins interacting with both DNA and the enzyme.17 The compound shows broad spectrum activity against a variety of Gram-positive and Gram-negative pathogens and has progressed to Phase III clinical trials for treatment of uncomplicated urinary tract infection and urogenital gonorrhea.18 The quite diverse class of compounds represented by gepotidacin has been referred to as “novel bacterial topoisomerase inhibitors” (NBTIs) with several members having been identified with Mtb activity.19–22 The SPT zoliflodacin, the leading compound of a fourth class of type II topoisomerase inhibitor, is currently in Phase 3 clinical trials as a single dose cure for uncomplicated gonorrhoea.23 We have recently published an exploration of SPTs for the treatment of TB wherein activity was seen for a variety of analogues including zoliflodacin and the methyloxadiazole analogue 5 of Figure 1. Compound 5, which showed higher Mtb activity than zoliflodacin, demonstrated cidality against the pathogen, activity against FQ-resistant Mtb strains and activity in a mouse infection model of TB.24
Figure 1.
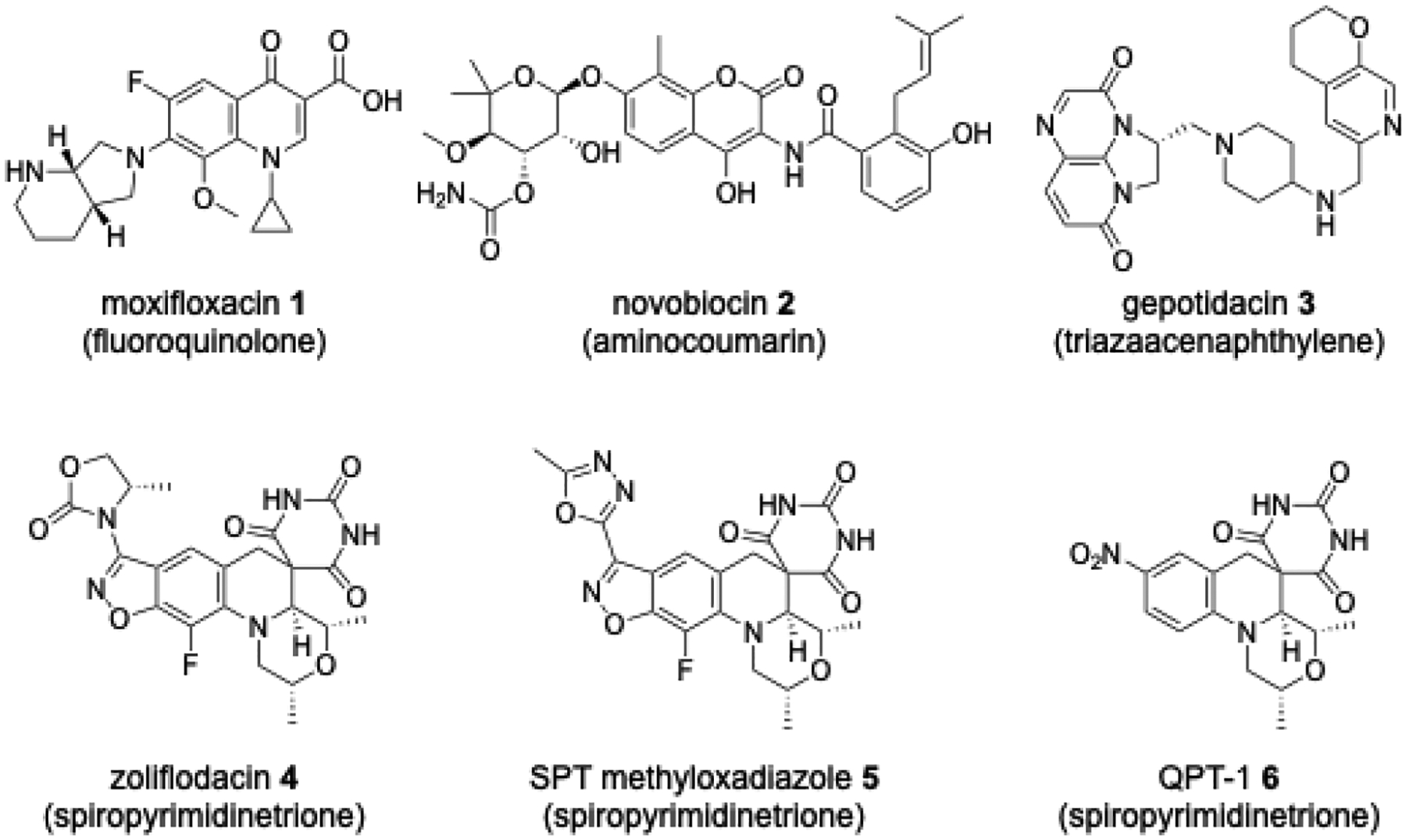
Known inhibitors of gyrase and Topoisomerase IV - compound class in parentheses.
Here, we present the synthesis, structure-activity relationships (SAR) and biological assessments of an expanded series of SPTs with the goal of identifying a novel treatment for TB. One issue that is seen with FQs, ATPase inhibitors, quinolines and SPTs is that they have significant activity beyond Mtb against a broader spectrum of bacterial pathogens. It would be preferable to have a Mtb-selective agent to minimize the hardships stemming from the killing of commensal bacteria.25 Furthermore, optimized dosing for treating TB may promote resistance in co-infections if such dosing were suboptimal against other pathogens. Therefore, activity of the new SPTs was determined against a Gram-positive and Gram-negative pathogen alongside Mtb towards understanding trends that would foster selectivity.
Results and Discussions
Model for SPTs within the Mtb gyrase binding site.
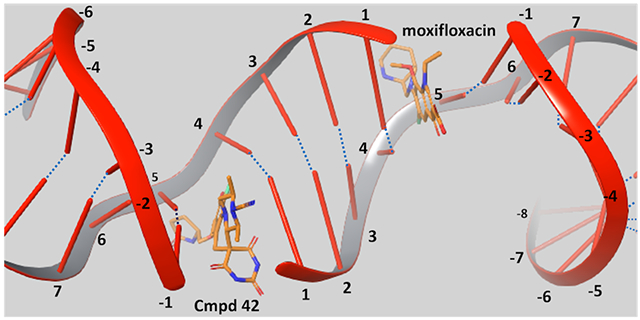
Despite the lack of cross resistance between FQs and SPTs across a variety of pathogens, moxifloxacin and a SPT analogue, QPT-1 (Figure 1), have been shown to share binding sites as determined via X-ray crystallography with the Staphylococcus aureus gyrase.26–27 Gyrase creates a double stranded break with covalent phosphate bonds to Y123 (Y129 in Mtb) of GyrA, four bases apart on complementary DNA strands. Subsequently, a transported (T-segment) of duplex DNA is passed through, and the break is re-ligated. Moxifloxacin and other FQs bind at both the points of the double-stranded DNA breaks in both S. aureus and Mtb gyrase, inserting themselves between the base involved in a scissile bond and the −1 base and intercalating DNA bases (Figure 2A). Several GyrA residues lying beyond the π-stacking bases that sandwich the scaffold are associated with the moxifloxacin carboxylate and a Mg2+ and form part of the quinolone resistance-determining region (QRDR) including G88, A90, S91 and D94 (for Mtb, see Figure S1A and PDB 5BS8)28–29 Like the FQs, QPT-1 forms π-stacking interactions with the complement of the −1 base as seen in its S. aureus co-crystal structure (Figure S1B). The pyrimidinetrione moiety extends into a region that is unoccupied by FQs and lies orthogonal to the scissile nucleotide and the Y123 phosphate acceptor. The two pyrimidinetrione NH groups each form a hydrogen bond with waters, one that bridges to the carboxylate of D437 and the other that bridges to the released ribose 3′-hydroxyl of the cleavage site DNA cytosine. The QPT-1 morpholino group is oriented towards the same solvated pocket as the chelated Mg2+ seen in the FQ binding mode. Residues abutting the morpholine ring include S84, S85, E88 and R92 of the GyrA QRDR (transformed to A90, S91, D94 and R98 in Mtb and to S89, A90, D93 and R97 in E. coli, see Supporting Information for GyrA and GyrB sequence alignments). The QPT-1 nitro group resides in a large open region lined by a tetrad of polar residues (R458, N475, N476 and E477). This region is occupied by the much larger tertiary amine of FQs at the protein interface with the −1 base complement. Sequence alignment showed that these residues are transformed to R482, N499, T500 and E501 in Mtb and K447, S464, Q465 and E466 in E. coli. Hence, the only difference between Mtb and S. aureus in the region is that of threonine to asparagine. Overall, functionality of similar polarity is retained across the three isozymes.
Figure 2.
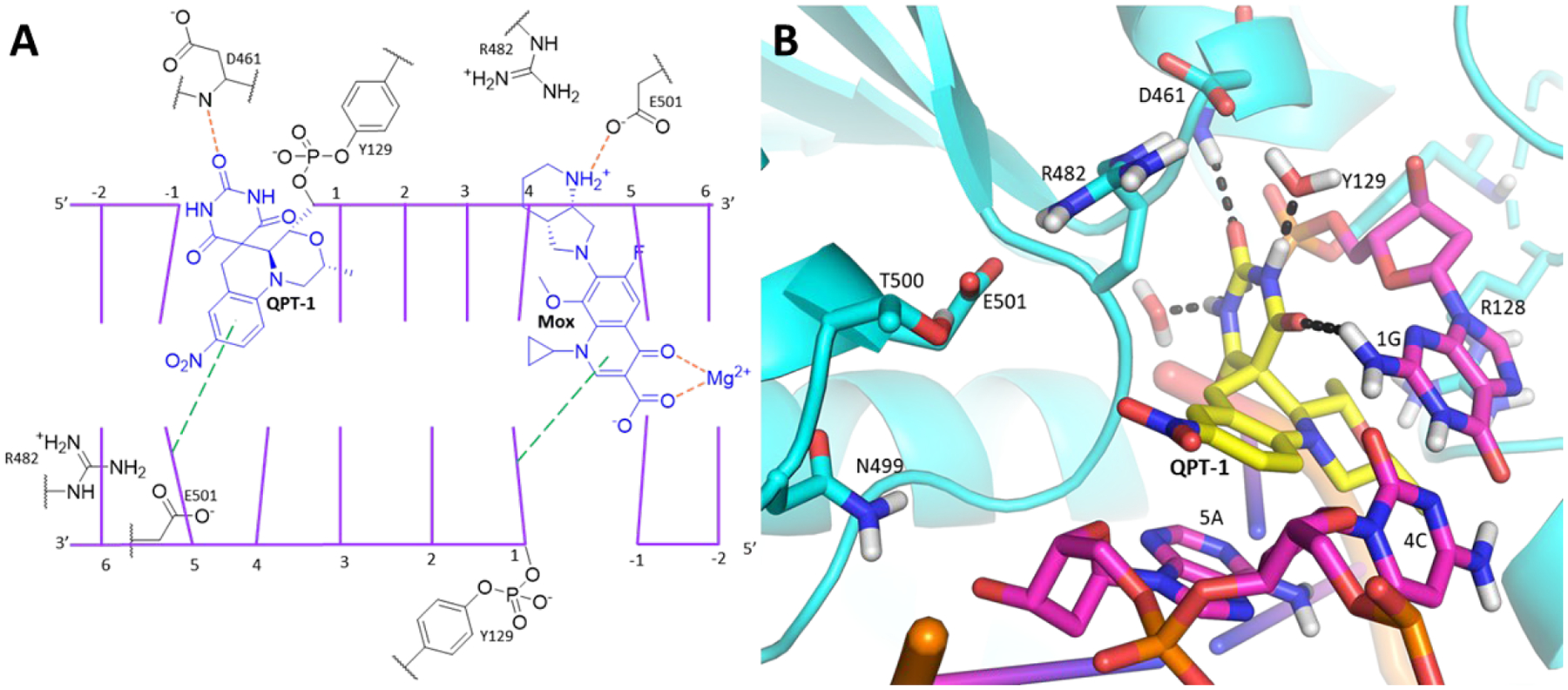
(A) A hybrid illustration of the double-stranded DNA break on complementary strands four bases apart with each break creating a FQ/SPT binding site, one showing QPT-1 and the other moxifloxacin. (B) Energy minimized structure of QPT-1 (yellow carbons) translocated into the Mtb gyrase binding site.
We generated a model for SPT binding to Mtb gyrase using the available Mtb gyrase structure with bound moxifloxacin (PDB 5BS8) and doubly cleaved DNA (Figure 2B). FQs and SPTs may stack with different bases at the site of DNA scission as suggested by optimal base sequences selected for co-crystallization work.30 Several manipulations had to be made to the Mtb gyrase structure before SPT docking was possible. The Mtb structure was aligned with the QPT-1 S. aureus gyrase co-crystal structure (PDB 5CDM). Then the moxifloxacin ligand and its chelated Mg2+ ion were removed from one of the inhibitor binding sites, into which the QPT-1 ligand and the two bound waters associated with the pyrimidinetrione were transposed. An extended minimization of enzyme residues and DNA bases within a 7.5 Å radius of the transposed ligand in the Mtb structure was carried out. Following the minimization, QPT-1 was found to form a similar hydrogen bonding network around the pyrimidinetrione as was seen in its S. aureus co-crystal structure, that is with the D461 backbone NH H-bonding to the distal carbonyl and the two bound water molecules H-bonding with each of the pyrimidinetrione NH groups. Additionally, as in the S. aureus co-crystal structure with QPT-1, one face of the SPT scaffold stacked on the adenosine-thymidine base pair (DNA −1 and DNA +5 relative to the scission sites) and the other face stacked on the guanosine DNA +1 base (relative to the scission site) while forming a tilted edge-to-π interaction with a cytidine base in the +4 position of complementary DNA strand. A limitation of our model could be that nucleotide recognition for cleavage of DNA duplex may vary by inhibitor and gyrase isozyme,30 as this transposition using a 24-mer DNA recognized by moxifloxacin may not be best recognized by QPT-1. Nonetheless, it is gratifying that the main contacts seen in this Mtb model mirror those seen in the S. aureus QPT-1 co-crystal structure offering a measure of validation. Notably, the pyrimidinetrione binding region might not have been predicted from the Mtb gyrase-moxifloxacin co-crystal structure alone.
After the SPT translocation and minimization, a Glide grid was generated from the ligand transposed structure31–32 and validated by docking QPT-1 to afford a closely realigned pose. By extension, this pose aligned closely with that in the co-crystal of QPT-1 bound to the S. aureus gyrase with a negligible RMSD of 0.64 Å (Figure S2). Compound 5 was then docked into this validated grid (Figure 1) creating a pose in line with the QPT-1 binding mode wherein the pyrimidinetrione and morpholine moieties maintained their interactions at the binding site (Figure 3) and the fluorobenzisoxazole scaffold maintained the π-stacking interactions. The benzisoxazole oxygen atom abutted the tilted π-face of the cytosine. The methyloxadiazole extended well beyond the space surrounding the QPT-1 nitro group occupying a larger region bounded by the sidechains of the tetrad of GyrB polar residues (R482, N499, T500 and E501) as well as two consecutive DNA ribose rings and their phosphate bridge. The E501 carboxylate formed a salt bridge with the R482 guanidine and a H-bond with the T500 hydroxyl. The ring nitrogen atoms of the methyloxadiazole were positioned to accept a hydrogen bond from the guanidine of R482. Compound 5 also showed activity against S. aureus and Escherichia coli (Table 1) in line with similar binding interactions in the region surrounding the methyloxadiazole across species. As the region otherwise opens towards solvent along with apparent flexibility in the resident amino acid sidechains, there is considerable leeway in the diversity of substituents that can be tolerated on the benzisoxazole ring. At one level, the dockings of 5 and other compounds offer a measure of self-validation in that the benzisoxazole is tolerated for activity as is the variety of benzisoxazole substituents.
Figure 3.
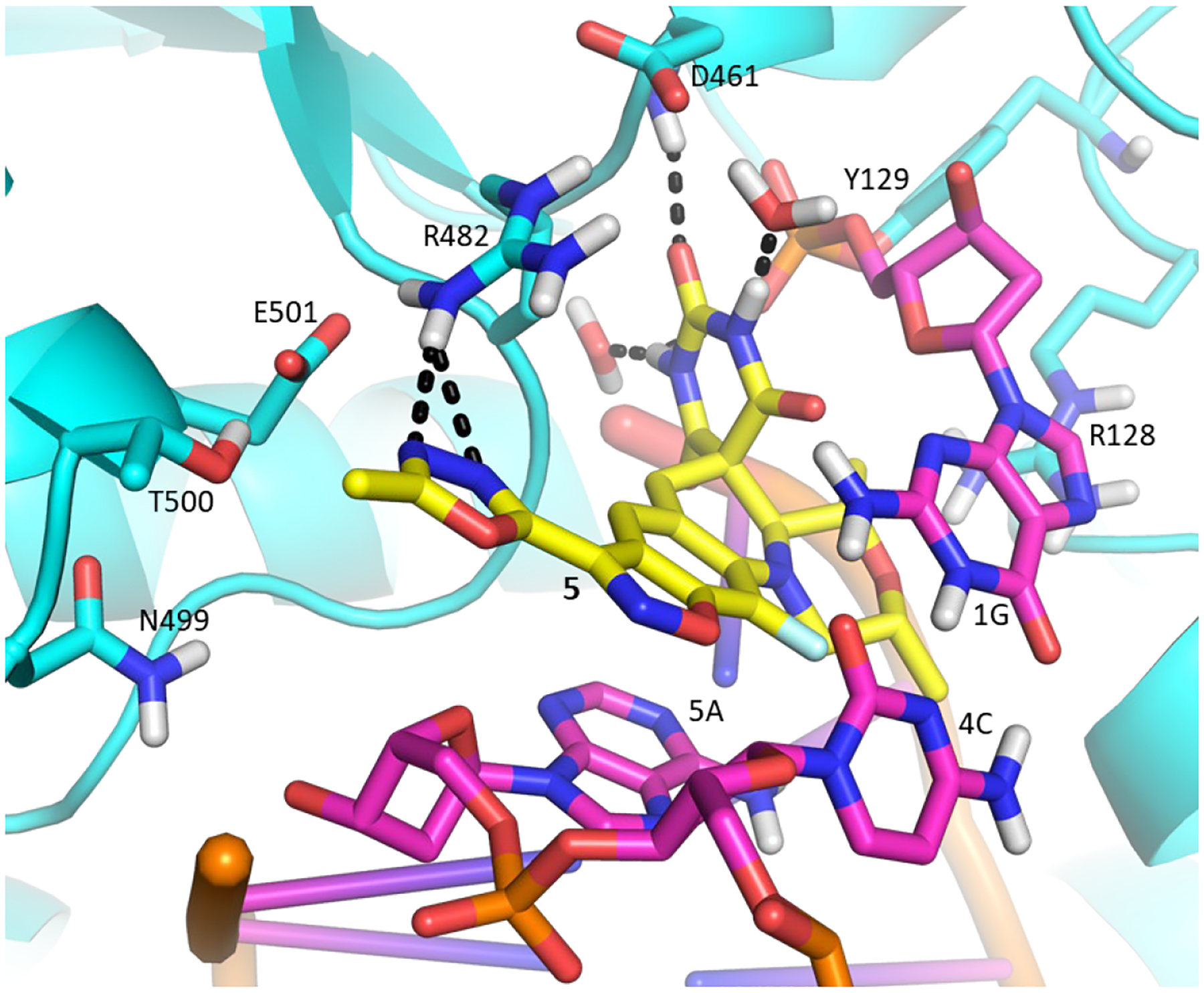
Model of 5 (yellow carbons) docked into Mtb gyrase making the same contacts as the modelled QPT-1 and showing an additional H-bond contact between the methyloxadiazole and R482.
Table 1.
Data for reference compounds
| Compound | R | Solubility (μM) | logD | Mtb gyrase IC50 (μM) | Sau gyrase IC50 (μM) | Mtba MIC (μM) | Saub MIC (μM) | Ecolc MIC (μM) | HepG2 IC50 (μM) |
|---|---|---|---|---|---|---|---|---|---|
| Zoliflodacin 4 |
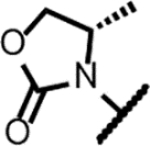
|
160 | 2.0 | 32 | 4.3 | 7.0 | 0.35 | 1.4 | >50 |
| Methyl-oxadiazole 5 |
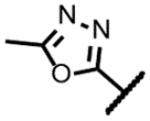
|
140 | 2.3 | 2 | ND | 1.7 | 0.45 | 31 | >300 |
| Triazole 7 |
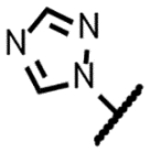
|
115 | 1.6 | 18 | 1.7 | 2.0 | 0.24 | 2.0 | 26 |
| Moxifloxacin | >20045 | 0.345 | 10 | 6.8 | 0.17 | 0.14 | 0.14 | >50 |
M. tuberculosis H37Rv;
S. aureus ATTC 25923
E. coli ATTC 25922
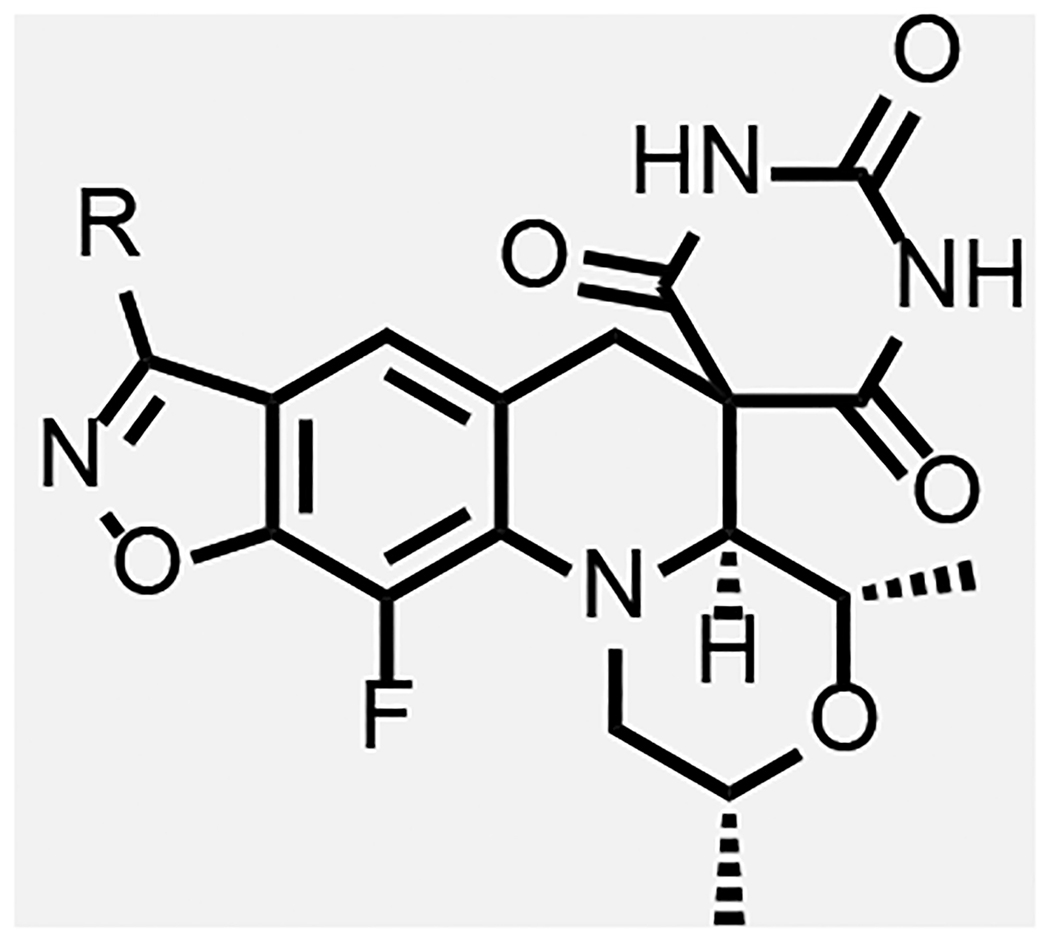
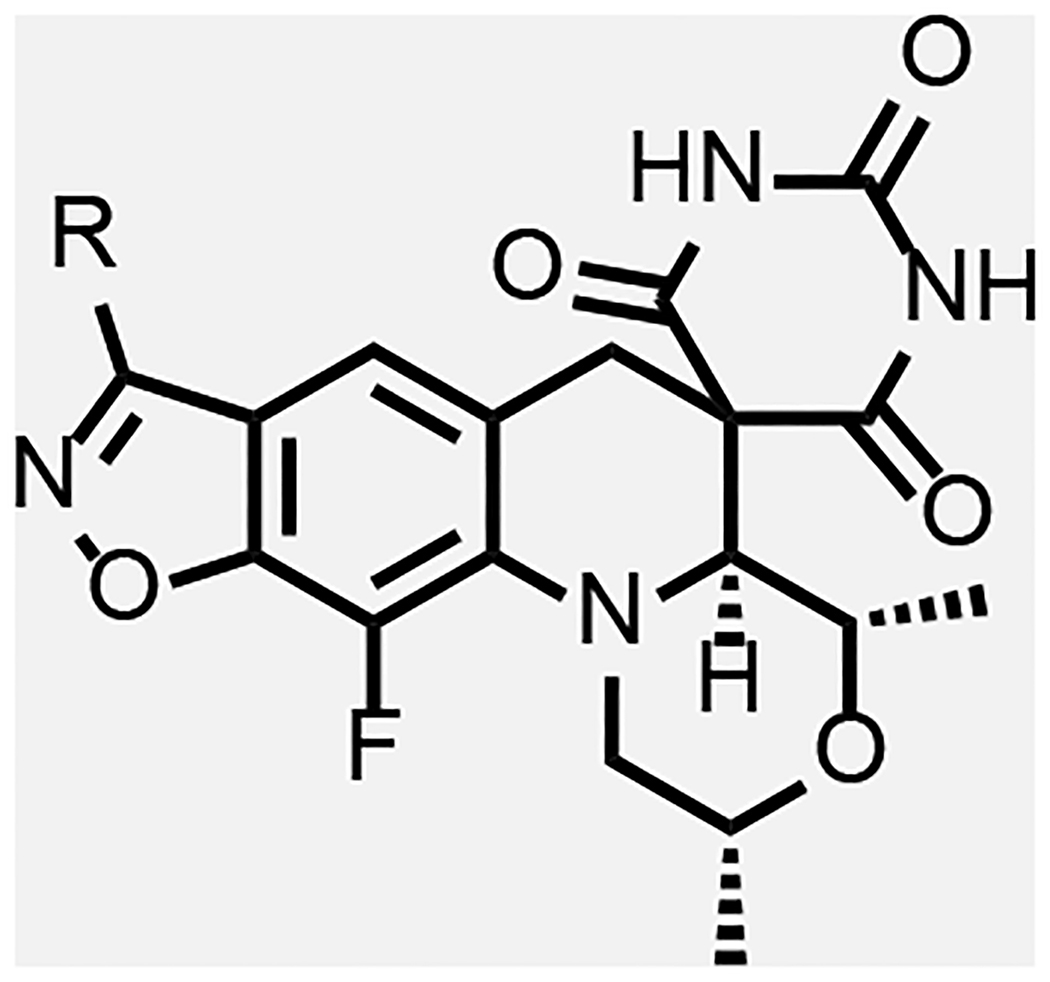
Overall, the modelling work pointed to two regions of the enzyme adjacent to the SPT molecule where it was deemed that further detailed SAR explorations would be fruitful as summarized in Figure 4. As already implied, the region surrounding the benzisoxazole R-substituent offers ample opportunity for variation, which aligns with extensive SAR work already published. Various compounds with N- or O-linked R-substituents having polar functionality (compounds 8–33, Table 2 and 3) were designed to probe the tetrad of polar residues (R482, N499, T500 and E501) previously mentioned. The second binding region deemed exploitable surrounds the morpholine ring; this region is part of the QRDR and is considerably smaller and seemingly less amenable to incorporation of a broad array of substituents. By the model, the morpholine ring of SPTs adopted a chair conformation placing the two methyl groups in equatorial orientations, while alternate conformations and orientations create a clash with stacking DNA bases. The surrounding region is where the FQ’s chelate Mg2+ ion and is located between the scissile base, the −1 base and the enzyme. The immediate environment around the morpholine oxygen suggested a small accessible space for functionality in equatorial (or pseudo-equatorial) orientations. Hence compounds were made replacing the morpholine with substituted piperidines and piperazines (34–45, Table 4). The accessible space is bounded by the guanidine of the conserved R128 (R122 and R121 in S. aureus and E. coli, respectively) that displayed two occupancies for the sidechain guanidine in the moxifloxacin co-crystal structure with Mtb gyrase: one oriented towards a backbone DNA phosphate and the other to the carboxylate of the inhibitor, with the latter being chosen for the modelling. The pyrimidinetrione functionality was kept constant in the analogues herein, as it is perceived to be critical for potency based on the multiple hydrogen bond interactions and the snug fit in its binding pocket. To date, there are few published examples where the pyrimidinetrione was modified, and none where significant activity was observed.33
Figure 4.
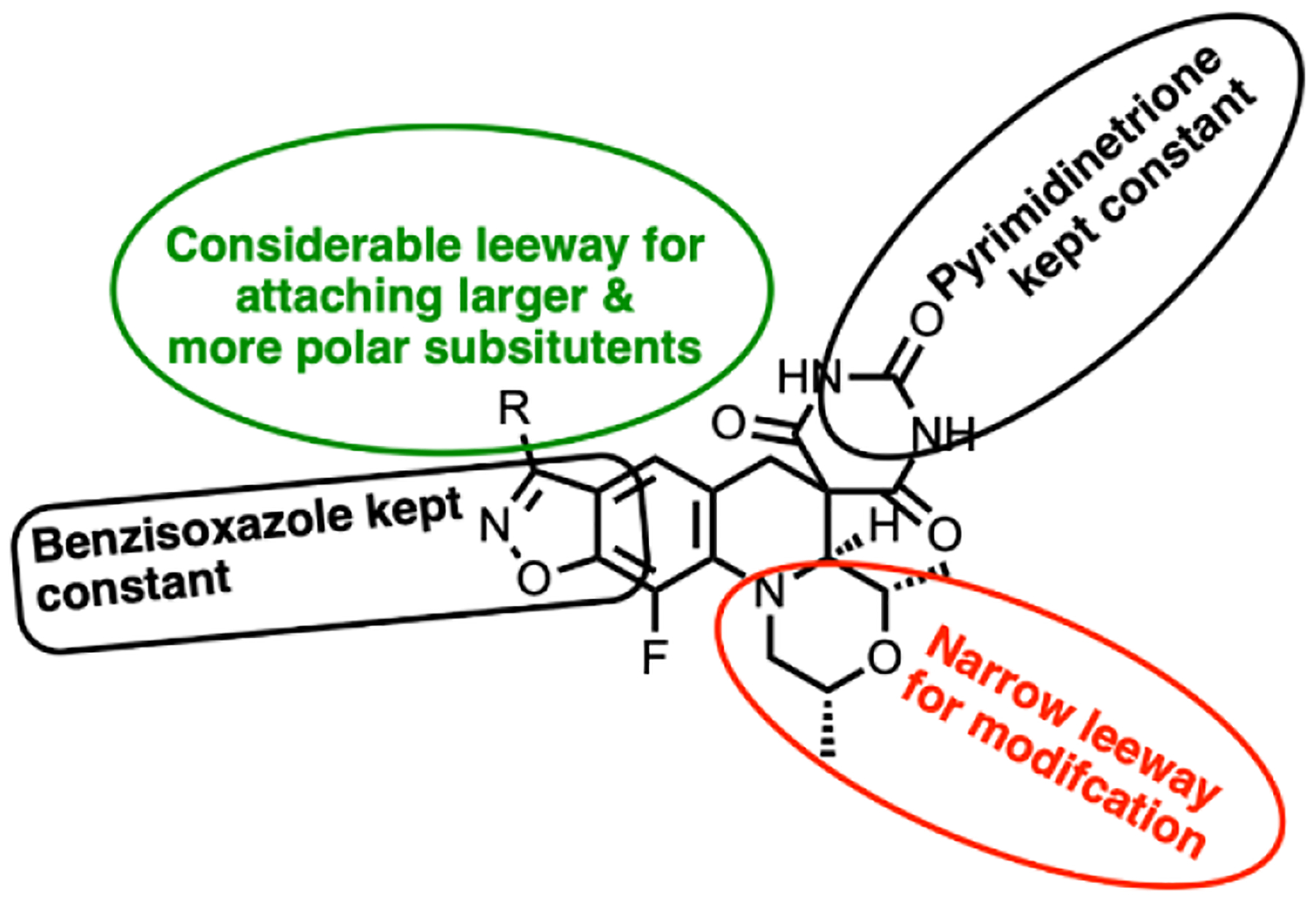
SAR explorations.
Table 2.
Data for SPTs with pyrrolidine and piperidine benzisoxazole substituents
| Compound | R | Solubility (μM) | logD | Mtb gyrase IC50 (μM) | Sau gyrase IC50 (μM) | Mtba MIC (μM) | Saub MIC (μM) | Ecolc MIC (μM) | HepG2 IC50 (μM) |
|---|---|---|---|---|---|---|---|---|---|
| 8 |
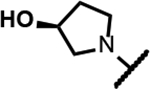
|
190 | 1.4 | 20 | 6.9 | 5.9 | 64 | >125 | >50 |
| 9 |
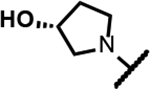
|
>200 | 1.4 | - | - | 3.9 | 64 | >125 | >50 |
| 10 |
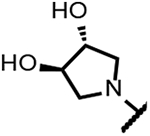
|
>200 | 1.2 | 16 | 6.6 | 5.9 | >125 | >125 | >50 |
| 11 |
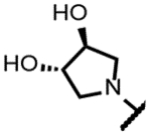
|
190 | 0.89 | - | - | 31 | >125 | >125 | - |
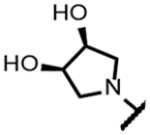
| 12 |
|
>200 | 0.76 | - | - | 16 | >125 | >125 | >300 |
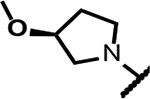
| 13 |
|
140 | 2.0 | 77 | - | 31 | >125 | >125 | >300 |
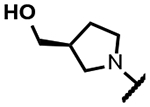
| 14 |
|
170 | 1.7 | 18 | - | 16 | >125 | >125 | >300 |
| 15 |
|
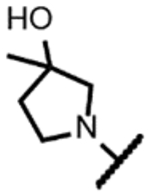
175 | 1.6 | - | - | >250 | >250 | >250 | >50 |
| 16 |
|
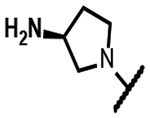
>200 | 0.16 | 34 | - | 63 | 125 | 125 | >50 |
| 17 |
|
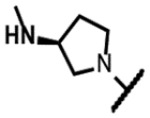
185 | 0.27 | - | - | 63 | 125 | 125 | >300 |
| 18 |
|
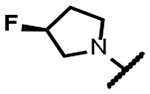
5 | 2.2 | - | - | 4.0 | 2.0 | 31 | >300 |
| 19 |
|
>200 | 1.7 | 10 | - | 4.0 | >125 | 31 | ND |
| 20 |
|
150 | - | - | - | >125 | >125 | >125 | ND |
| 21 |
|
>200 | 1.9 | - | - | 16 | 94 | >125 | >50 |
| 22 |
|
>200 | 1.8 | - | - | 10 | 94 | >125 | >50 |
M. tuberculosis H37Rv;
S. aureus ATTC 25923
E. coli ATTC 25922
Table 3.
Data for SPTs with ether-linked benzisoxazole substituents
| Compound | R | Solubility (μM) | logD | Mtb gyrase IC50 (μM) | Mtba MIC (μM) | Saub MIC (μM) | Ecolc MIC (μM) | HepG2 IC50 (μM) |
|---|---|---|---|---|---|---|---|---|
| 23 |
|
185 | 1.4 | 2.6 | 1.0 | >125 | >125 | >100 |
| 24 |
|
< 5 | 1.4 | - | >125 | >125 | >125 | >300 |
| 25 |
|
190 | 1.6 | - | 3.9 | 63 | 63 | >300 |
| 26 |
|
170 | 1.6 | - | 2.0 | 3.9 | >125 | >300 |
| 27 |
|
155 | 1.5 | - | 3.9 | >250 | >250 | >300 |
| 28 |
|
180 | 1.3 | - | 2.0 | 63 | >250 | >300 |
| 29 |
|
190 | 1.3 | - | 2.0 | 63 | >250 | >300 |
| 30 |
|
190 | 1.3 | - | 2.0 | >125 | >250 | >300 |
| 31 |
|
200 | 1.4 | - | 2.0 | 31 | 63 | >300 |
| 32 |
|
190 | 1.7 | - | 3.9 | 31 | >125 | >300 |
| 33 |
|
190 | 1.6 | - | 63 | >125 | >125 | >300 |
M. tuberculosis H37Rv;
S. aureus ATTC 25923
E. coli ATTC 25922
Table 4.
Data for SPTs with piperidine and piperazine replacements for morpholine
| Compound | R | Morpholine Variation | Solubility (μM) | logD | Mtb gyrase IC50 (μM) | Sau gyrase IC50 (μM) | Mtba MIC (μM) | Saub MIC (μM) | E. colic MIC (μM) | HepG2 IC50 (μM) |
|---|---|---|---|---|---|---|---|---|---|---|
| 34 |
|
|
50 | 0.30 | - | - | >125 | 63 | 31 | >50 |
| 35 |
|
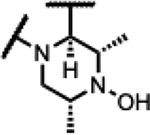
|
>200 | 0.58 | 207 | - | >125 | >125 | >125 | - |
| 36 |
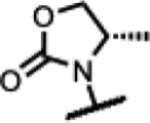
|
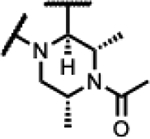
|
>200 | 1.4 | >1000 | - | >125 | >125 | >125 | >300 |
| 37 |
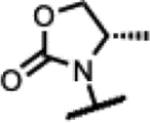
|
|
95 | - | - | - | >125 | >125 | >125 | >50 |
| 38 |
|
|
<5 | 1.8 | - | - | 3.9 | 1.0 | 3.9 | >50 |
| 39 |
|
|
50 | 1.7 | - | - | 0.98 | 16 | 63 | >300 |
| 40 |
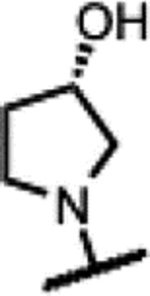
|
|
155 | 1.3 | 19 | 3.1 | 7.8 | >125 | >125 | >50 |
| 41 |
|
|
145 | 1.2 | - | - | 7.8 | >125 | >125 | >50 |
| 42 |
|
|
130 | 1.3 | 2.6 | - | 0.49 | >125 | >125 | >300 |
| 43 |
|
|
40 | 3.9 | - | - | 16 | 7.8 | 63 | >300 |
| 44 |
|
|
45 | 1.9 | - | - | 1.5 | >125 | >125 | >300 |
| 45 |
|
|
15 | 3.2 | - | - | 16 | >250 | >250 | >50 |
M. tuberculosis H37Rv;
S. aureus ATTC 25923
E. coli ATTC 25922
Synthesis.
The literature synthesis of zoliflodacin 1 ends with the tertiary amino effect reaction (T-reaction) wherein chiral intermediate 46 and barbituric acid are heated at 110 °C to afford a thermodynamic 9:1 diastereomeric mixture; purification can be achieved by chromatography or crystallization (Scheme 1).34–36 As the synthesis of 46 uses a rather expensive chiral (2R,6R)-2,6-dimethylmorpholine as a reactant,37–38 we incorporated the inexpensive meso-2,6-dimethylmorpholine to set up the T-reaction in racemic form as depicted for the conversion of the transient intermediate aldehyde in Scheme 2. Hence, 47 with the meso-morpholine was prepared on a multi-gram scale as set out in the literature.37 The chlorine atom of 47 was displaced with a variety of cyclic amines by refluxing in acetonitrile in the presence of DBU to give 48a-o and with a variety of alcohols using NaH in DMF to give O-linked 48p-z. Subsequent reaction of intermediates 48 with barbituric acid in ethanol containing HCl at 80 °C effected ketal hydrolysis to the transient aldehyde and in situ T-reaction to afford compounds 8–33 (Tables 2 and 3). As the T-reaction temperature was kept to the lower temperature of 80 °C, there was no formation of the other morpholine diastereomer as seen in Scheme 1.39–40 The racemic mixtures (or diastereomeric mixtures when the R-substituent is chiral) were generally separated by chiral HPLC to give the enantiopure compounds. The stereochemistry of the isolated isomers could easily be assigned as the desired (2R,4S,4aS) isomers invariably have a negative optical rotation and showed activity against Mtb. In the case of 20, the corresponding ethyl ester (S1) was maintained through the T-reaction conditions and separated by chiral chromatography before being hydrolyzed with lithium hydroxide to the acid.
Scheme 1.
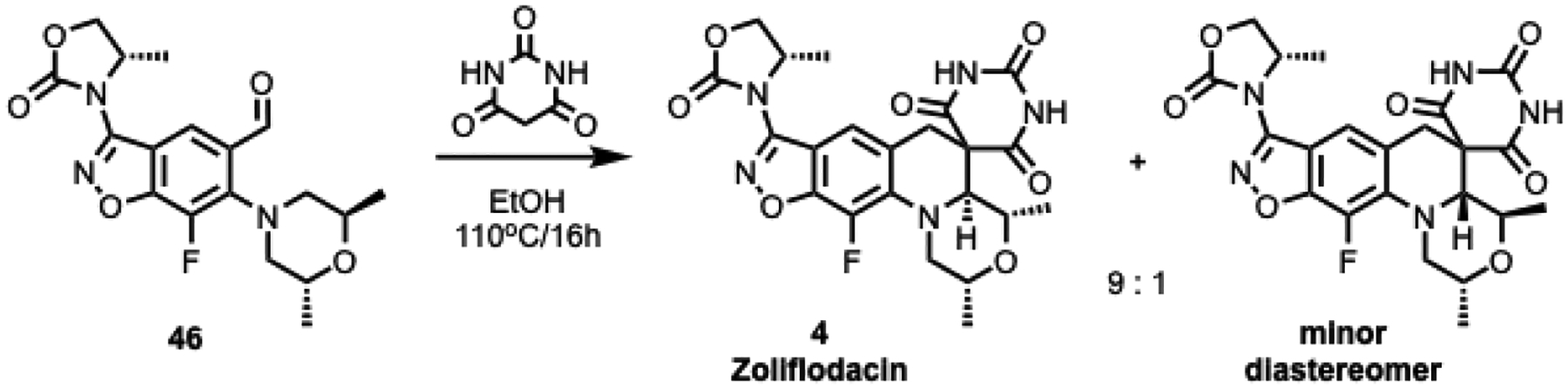
Synthesis of 1 via tertiary amino effect reaction (T-reaction) − chiral enablement
Scheme 2.
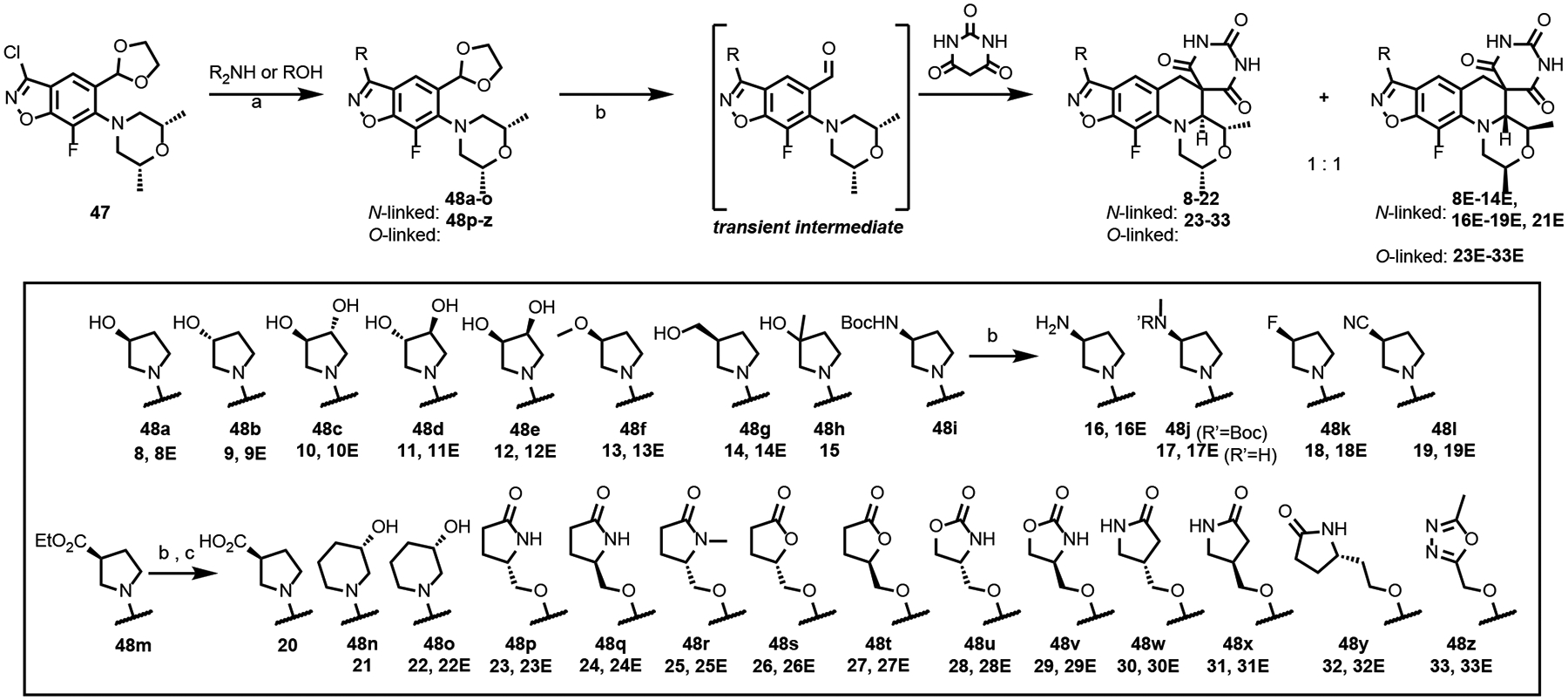
Synthesis of Compounds 8–33.
Reagents and conditions: (a) For cyclic amines: DBU, CH3CN, 90 °C, 16 h, 30–95%; For alcohols: NaH, DMF, 0–30°C, 1 h, 24–95%; (b) EtOH/2M HCl (10:1), 80 °C, 16 h, 3–65%, (c) LiOH, 1,4-dioxane: H2O (3:1), 30°C, 1 h, 36%.
Scheme 3 exemplifies the sequence that was used to synthesize compounds whereby the dimethylmorpholine was changed to substituted piperazines. As mentioned, having an equatorially orientated substituent on the X-atom in Scheme 3 was predicted to interact with the conserved GyrA Arg128 of Mtb. Towards this, intermediate 4937 was assembled with the chiral oxazolidinone substituent on the benzisoxazole, set up for displacement of the fluorine atom ortho to the carboxaldehyde by various amines (syntheses of new amines shown in Scheme S1–S2) to afford 50a-d. Intermediates 50c and 50d were derived from 50a by reacting with acetic anhydride and bromoacetonitrile, respectively. Compounds 34–37 were prepared from 50a-d via the T-reaction with barbituric acid and were purified via chiral chromatography. The N-cyano analogue 38 was prepared by cyanation of racemic mixture of 34 with cyanogen bromide, followed by chromatographic separation on a chiral column.
Scheme 3.
Synthesis of Compounds 34–38.
Reagents and conditions: (a) amine [meso-2,6-dimethylpiperazine (for 50a) or meso-2,6-dimethylpiperazin-1-ol hydrochloride S3 (for 50b)], K2CO3, CH3CN, 80°C (MW), 1–2 h or Et3N, DMSO, 90–110°C, 2 h, 41–86%; (b) (Ac)2O, pyridine, CH2Cl2, 25°C, 24 h, 80% (for 50c) or BrCH2CN, K2CO3, acetone, 25°C, 16 h, 68% (for 50d) (c) barbituric acid, AcOH:H2O (4:1) or EtOH, 80°C, 2 h, 2–23%; (d) Br-CN, K2CO3, acetone, 25°C, 16 h, 14%.
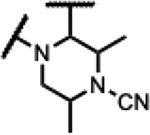
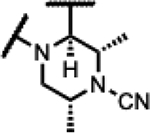
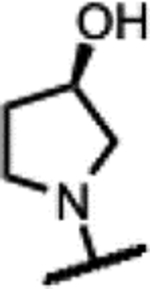
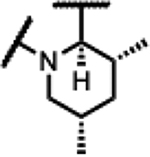
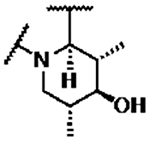
Compounds 39–45 were made wherein the dimethylmorpholine ring was replaced with either a dimethylpiperidine or N-cyanodimethylpiperazine combining with more favorable benzisoxazole R-substituents (Scheme 4). For 39–43, the syntheses involved the incorporation of amines (meso-1-cyano-2,6-dimethylpiperazine (S5) or meso-3,5-dimethylpiperidine) by SNAr displacement of the fluorine ortho to the carboxaldehyde of 5136 to afford 52a-b. Subsequently, the aldehydes were protected as the ethylene acetals affording 53a-b. The R-substituents were then introduced by SNAr displacement on the chlorobenzisoxazole using 1,2,4-triazol, (S)-3-hydroxypyrrolidine, (R)-3-hydroxypyrrolidine or (S)-5-(hydroxymethyl)pyrrolidin-2-one to afford 54a-e. Finally, 54a-e were transformed to 39–43 along with their diastereomeric or enantiomeric pairs by in situ acetal hydrolysis and T-reaction with barbituric acid. Compound 39 was only isolated as the racemic mixture as it did not succumb to separation of enantiomers, whereas 40-43 were separated from the corresponding isomers 40E-43E via chiral chromatography. In Scheme 5, the known intermediate 5541 was treated with chiral (R,R)-2,6-dimethylpiperazine followed by cyanation with cyanogen bromide to afford 57 that is set up for the higher temperature T-reaction to afford 44. For 45 (Scheme 5), 55 was treated with (meso-3S,4r,5R)-3,5-dimethylpiperidin-4-ol hydrochloride (supporting information intermediate S11, preparation outlined in Scheme S3) and reacted with barbituric acid in the T-reaction for subsequent purification by chiral chromatography. Importantly, the stereochemistry of S11 was established to have the 4-hydroxyl trans-to the cis-dimethyl groups (see Figure S2). The stereochemistry was maintained on conversion to 45 as confirmed by 1H-NMR spectroscopy, wherein the carbinol CH appears as a quartet with 9 Hz coupling consistent with a trans-diaxial orientation relative to the two hydrogens beneath each of the piperidine methyl groups. In the 1H-NMR spectrum, there was a coincident 9 Hz coupling to the OH. Having the hydroxyl in the equatorial orientation was predicted to have a more favorable interaction in the enzyme binding site relative to the axial orientation wherein the OH would clash with the stacking guanosine base.
Scheme 4.
Synthesis of compounds 39–43.
Reagents and conditions: (a) meso-2,6-dimethylpiperazine-1-carbonitrile hydrochloride (S5) for 52a or meso-3,5-dimethylpiperidine for 52b, K2CO3, CH3CN, 80–110 °C, 1–16 h, 26–81%; (b) ethylene glycol, p-TSA, toluene, 130 °C, 16 h, 57–87%; (c) 1H-1,2,4-triazol for 54a, [(S)-3-hydroxypyrrolidine for 54b, (R)-3-hydroxypyrrolidine for 54c, [(S)-5-(hydroxymethyl)pyrrolidin-2-one for 54d and 54e, DBU in CH3CN (for N-linked analogues) or NaH in DMF (for O-linked analogues), 30–110 °C, 1–16 h, 69–95%; (d) barbituric acid, EtOH:2M HCl (10:1), 80 °C, 16 h, 10–27%.
Scheme 5:
Synthesis of compounds 44 and 45.
Reagents and conditions: (a) (2R,6R)-2,6-dimethylpiperazine dihydrochloride, K2CO3, CH3CN:water (20:1), 90 °C, 13 h, 92%; (b) Br-CN, K2CO3, acetone, 27 °C, 9 h, 77%; (c) barbituric acid, EtOH, 110 °C, 20 h, 4–10%; (d) (meso-3S,4r,5R)-3,5-dimethylpiperidin-4-ol hydrochloride, K2CO3, CH3CN:water (10:1), 90 °C, 1 h, 89%; (e) barbituric acid, EtOH, 80 °C, 16 h, 10–18%.
Structure-activity relationships.
Analogues were profiled for the minimum inhibitor concentration (MIC) against Mtb as well as a Gram-positive (S. aureus) and Gram-negative (E. coli) pathogen, the latter two assays towards assessing selectivity for Mtb (Tables 1, 2, 3 and 4). Physical properties (solubility and logD) and cytotoxicity against HepG2 cells42ref were also determined; gratifyingly, solubilities for most of the compounds were high (>100 μM) and cytotoxicities (>50 μM) were low. Selected compounds were assessed for inhibition in gel based Mtb and S. aureus gyrase assays for correlation with whole cell activity. The SPTs of Table 1 showed considerably lower MIC values for S. aureus than those seen for Mtb, while the E. coli MICs were more variable. However, the SAR for Mtb activity diverges from that seen for S. aureus and E. coli as previously described.24 The reasons for pathogen-to-pathogen divergence in SAR could be differences in the inhibitor binding domain as the surrounding residues do vary despite being quite similar. Additionally, the optimal DNA cleavage sites and stacking bases that abut the inhibitors may differ depending on the pathogen. However, the data supports a larger influence of a second factor for the variable pathogen-to-pathogen SAR, namely the disparate nature of the outer cell envelope and efflux pumps among Mtb, Gram-positive and Gram-negative pathogens differentially affecting the compound permeability into the bacterial cytoplasm where gyrase is localized.43–44 Indeed, the large influence of efflux was demonstrated for SPTs relative to E. coli activity, wherein considerably lower MICs (generally 200-fold or more) were seen for a tolC− efflux pump debilitated mutant relative to wild-type.36 The key role of bacterial permeability is supported by comparing 5 with moxifloxacin (Table 1) wherein the former is 5-fold more potent as an inhibitor of Mtb gyrase inhibitor but 10-fold less active against the pathogen suggesting greatly enhanced permeability for the latter. It is worth noting that the gel-based DNA supercoiling assay requires a much higher amount of supercoiled DNA substrate to monitor conversion to relaxed DNA relative to concentrations encountered inside the cell. Hence, inhibitory potencies can often be considerably higher than MICs recorded in culture.
SAR of benzisoxazole substituent.
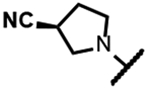
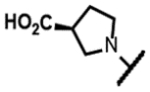
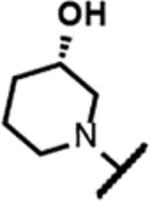
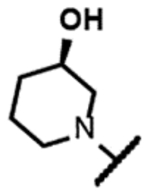
The series of SPTs in Table 2 with pyrrolidine benzisoxazole substituents showed MICs as low as 4 μM against Mtb and markedly higher MICs for both S. aureus and E. coli. For example, 8 with the S-hydroxypyrrolidine showed about equal activity relative to zoliflodacin against Mtb but was upwards of 200-fold less active against both S. aureus and E. coli. However, 8 (logD = 1.4) and zoliflodacin (logD = 2.0) showed comparable inhibitory potencies (IC50s = 6.9 and 4.3 μM, respectively) against gyrase from S. aureus. Compound 7 (logD = 1.6) also showed a comparable S. aureus gyrase inhibitory potency (IC50 = 1.7 μM) to zoliflodacin and was similarly active against the pathogen. The data are in line with correlations made previously for SPTs wherein higher lipophilicity was associated with higher bacterial cell permeability and lower MICs against E. coli.37 Hence, it is apparent that permeability for S. aureus and E. coli, whether it is due to passive outer cell envelop diffusion or active efflux, is much more sensitive to compound polarity than is Mtb. Compound 9 with the (R)-hydroxypyrrolidine maintained the Mtb activity compared to 8. A binding model supports that both the R- and S-hydroxypyrrolidine configurations can form a favorable H-bond with the sidechain hydroxyl of T500 or the guanidine of R482. Incorporation of a second hydroxyl onto the pyrrolidine forming the dihydroxypyrrolidine diastereomers 10, 11 and 12 were well-tolerated. A binding model for 10 (Figure 5A), the most active of the three, suggests H-bond interactions with both T500 and R482. The IC50s for gyrase from both Mtb and S. aureus (16 and 6.6 μM, respectively) were similar to the values seen for 8, as were the MICs against Mtb, S. aureus and E. coli, providing further confirmation that bacterial permeability is driving selectivity for Mtb. The methoxypyrrolidine 13 (IC50 = 77 μM against gyrase, MIC = 31 μM) showed diminished Mtb activity relative to 8 suggesting that the hydroxyl group better donates a H-bond versus being on the receiving end of a H-bond and, indeed, the T500 hydroxyl looks to be tied up with a H-bond to E501. The Mtb gyrase IC50’s for 8, 10, 14 and 19 were equal to or only around 2-fold higher than the value for moxifloxacin (10 μM), yet Mtb MICs were considerably worse, again attributable to inferior bacterial permeability for SPTs. Compound 15 with 3-methyl-3-hydroxypyrrolidine (obtained as a mixture of inseparable diastereomers) showed a significant loss of Mtb activity (MIC >125 μM); both configurations for the compound place the methyl substituent in a clash with T500 that is thought to receive a H-bond from the hydroxyl. Compounds 16 and 17 with the basic 3-aminopyrrolidine and 3-methylaminopyrrolidine showed 9 to16-fold higher MICs compared to 8. As 16 was only about 2-fold less potent against Mtb gyrase relative to moxifloxacin, it is likely that the positively charged basic amine diminishes bacterial permeability accounting for the lower antibacterial activity. Interestingly, 18 and 19 with 3-F and 3-CN-pyrrolidines, respectively, retained Mtb activities similar to 8, suggesting that both functionalities can interact productively to the surrounding enzyme functionality. Notably, 18 being more lipophilic than 8, showed improved activity against S. aureus and E. coli. Compound 20 with the 3-pyrrolidine carboxylate substituent showed significantly lower Mtb activity (MIC >125 μM) indicating intolerance for the charged carboxylate group for binding affinity and/or diminished bacterial permeability. The two 3-hydroxypiperidine diastereomers 21 and 22 had moderate MICs indicating a slightly less favorable positioning of the hydroxyl group and a limitation of the 6-membered ring relative to the pyrrolidine.
Figure 5.
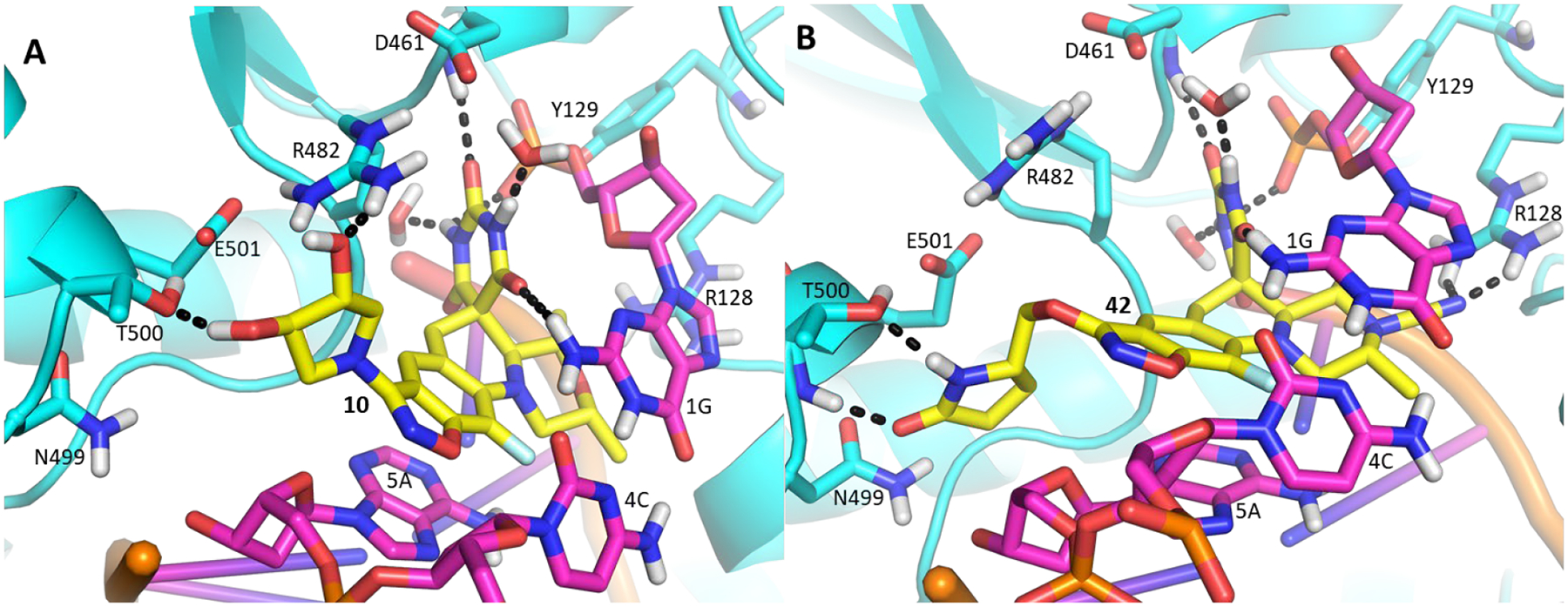
(A) Model of 10 (yellow) with the dihydroxypyrrolidine forming H-bond interactions with the sidechain functionality of T500 and R482; (B) Model of 42 (yellow) with the CN forming H-bond with the guanidine of R128 and the pyrrolidinone forming H-bonds with the backbone NH and hydroxyl of T500.
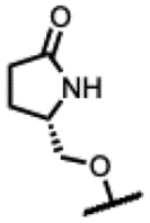
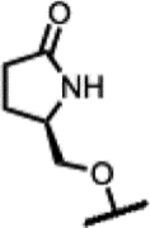
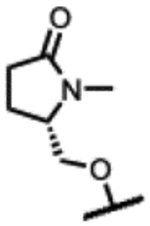
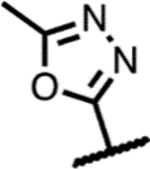
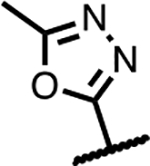
Another series of Mtb active SPTs were identified having heterocycles in a two-atom bridge linked to the benzisoxazole through an ether oxygen (Table 3). Compound 23 with the pyrrolidinone heterocycle showed a high Mtb gyrase inhibitory potency (IC50 = 2.6 μM) and a low Mtb MIC (1.0 μM). Compound 24 with the epimeric configuration at the lactam ring was found to be inactive (MIC >125 μM); however, the compound was extremely insoluble in aqueous media, which might compromise the expression of activity. The reason for extremely low solubility for 24 is not understood and may involve extraordinary tight packing in a crystal lattice. Capping of the lactam NH of 23 with methyl affording 25 was detrimental to Mtb activity by 4-fold. Replacing the lactam with a lactone as in 26 and 27 reduced the activity 2- to 4-fold compared to 23. Oxazolidinones 28 and 29 maintained Mtb MICs of 2 μM. Changing the attachment of the 2-atom linker to the lactam ring from the 5- to 4-position (compounds 30 and 31) was also well-tolerated for Mtb activity with MICs = 2 μM. As well, 32 with 3-atom linker was ~4-fold less active than 23. Finally in Table 3, 33 with a 2-atom linker to the methyloxadiazole was considerably less active than the compounds otherwise. Overall, these observations indicate considerable leeway in the region surrounding the benzisoxazole, though there is a measure of specific binding.
SAR of morpholine modifications.
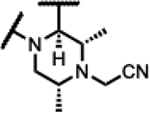
In general, modifications of the morpholine ring of SPTs showed little leeway for variation (Table 4), in line with the docking model. Compound 43 with dimethylpiperidine was 16-fold less active than the corresponding morpholine matched pair 23. The binding model has the axial hydrogen for the CH2 for O replacement approaching a stacking DNA base, which may contribute to the lower activity. However, relative to 8 and 23, 43 was more active against S. aureus and E. coli, in line with improved permeability due to the piperidine increasing the logD about one unit over the morpholine. For S. aureus, it is hypothesized that the optimal DNA stacking for the inhibition complex is different such that binding with the piperidine would be favored, though structural work with a variety of DNA duplex motifs would need to be carried out for confirmation. Replacing the morpholine with a piperidine substituted at the 4-position by an equatorial hydroxyl (compound 45) led to a 10-fold reduction in Mtb activity relative to the matched pair 5 of Table 1.
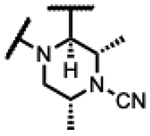
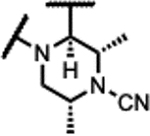
Piperazine derivatives were made eliminating the potential negative impact of the axial hydrogen atom of the piperidine. Compound 34 with the weakly basic 2,6-dimethylpiperazine and various N-substituted analogues thereof such as N-hydroxy 35, N-acetyl 36 and N-cyanomethyl 37 showed considerably lower Mtb activity (>125 μM). It is likely that N-acetyl derivative 36 alters the conformation of piperidine ring clashing with the adjacent methyl groups and is too large to fit into the adjacent space. The N-cyanomethyl group is also likely too large to fit into the space. The parent piperazine and N-hydroxypiperazine may not make favorable interactions especially for the former if it is likely protonated at physiological pH. However, the N-cyano analogue 38, was nearly 2-fold more active against Mtb than its matched pair, zoliflodacin; the compound also showed significant activity (though lower than zoliflodacin) against S. aureus and E. coli. Hence, the series of compounds 39-42 maintaining the N-cyanopiperazine in place of the morpholine but varying the R-substituent were made, consistently showing about equal activity to the corresponding matched pair. Compound 39 (MIC = 0.9 μM) with the triazole benzisoxazole substituent was 2-fold more active relative to its matched pair 7 (MIC = 2.0 μM); since 39 could only be isolated as the racemate, the active enantiomer could have a 4-fold lower MIC. Notably, 39 showed lower HepG2 cytotoxicity (>300 versus 26 μM) compared to 7 of Table 1, which might be associated with incompatibility of the N-cyanopiperazine with the human orthologs, topoisomerase IIa and topoisomerase IIb.37 Compound 42 showed the highest activity against Mtb (MIC = 0.49 μM) of the N-cyanopiperazines as well as potent inhibition (IC50 = 2.6 μM) of Mtb gyrase while retaining many of the same attributes as its morpholine matched pair 23, including selectivity relative to S. aureus and E. coli activity, high solubility and clean cytotoxicity at the highest concentration tested (300 μM). Figure 5B shows a proposed model of the N-cyano group within the surrounding region of the gyrase QRDR wherein the piperazine maintains a conformation to display the two methyl and cyano groups in pseudo-equatorial orientations. The cyano extends out towards the guanidine of the conserved GyrA R128, the residue assuming an orientation similar to that seen in the moxifloxacin co-crystal structure wherein it bridged to the inhibitor carboxylate. The pyrrolidinone tethered off the benzisoxazole reaches over to T500. Though the equivalent of T500 in S. aureus and E. coli are N476 and Q465, it is possible that these residues contribute to variations in pathogen MICs. However, there is a fair statistical correlation (R2 = 0.8) between Mtb inhibitory potency and MIC for the ten compounds where the first value was determined (Figure 6). The ten compounds displayed broad range of lipophilicity (logD’s 0.16– 2.3); a poor correlation was seen for SPTs with a similar logD range against E. coli.30 Again the data indicate that E. coli and S. aureus SPT permeability are more sensitive to compound lipophilicity than is Mtb, in line with what has already been noted for 10. Explicitly, more polar compounds are apparently better able to transit into the Mtb cytoplasm relative to Gram-positive and Gram-negative bacteria.
Figure 6.
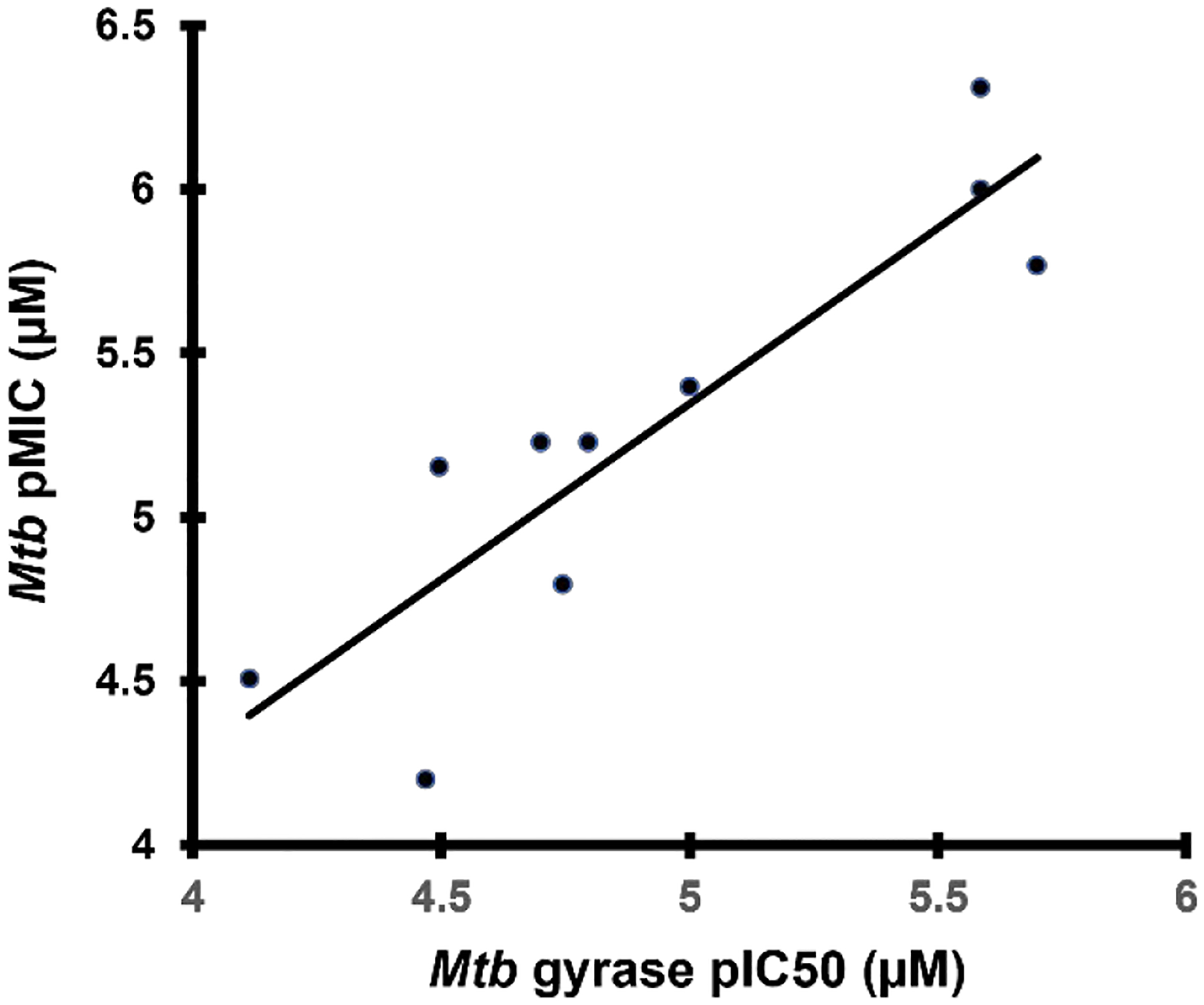
Compound MIC versus gyrase inhibitory potency.
Mode-of-action characterization.
To validate that Mtb gyrase is the primary target of SPTs and to confirm the on-target activity, we evaluated whether conditional depletion of GyrA in Mtb would sensitize the growth inhibitory effects of SPTs. Previously, 5 was shown to be devoid of cross-resistance to two strains of Mtb having point mutations (A90V and G88C) in GyrA that impart resistance to FQs.24 Herein, an anhydrotetracycline (ATc)-regulated conditional knockdown mutant (hypomorph) in the Tet-ON configuration underexpressing gyrA46 was assessed for inhibitory activity by SPTs. Due to the sensitivity of gyrA hypomorph to ATc and the nature of the assay, we compared both MIC50 (for 50% growth inhibition) and MIC (for 99% growth inhibition) and calculated the fold-hypersensitivity in the form of the ratio (WT/hypomorph MICs, Table 5) thereby getting a reading of variations in the concentration response curves. The gyrA hypomorph consistently displayed hypersusceptibility to SPTs versus wildtype H37Rv control, similar to and/or greater than what is seen for moxifloxacin confirming that the SPTs inhibit Mtb growth via enzymatic inhibition of gyrase (Table 5). Rifampicin as a negative control validated that the MIC differences are not artefacts due to fitness of the hypomorph construct. These data support the conclusion that SPTs operate by inhibition of gyrase Mtb thereby thwarting DNA replication. However, it is well understood that varying enzyme levels could be complicated by the formation of DNA strand breaks, which also contributes to cell death via the SOS response (see below).24, 48 A much more involved investigation would be needed to assess the influence of increasing and decreasing levels of either gyrA and gyrB (or both simultaneously) to understand the relative role to cell cidality around blocking DNA replication versus generating DNA strand breaks.
Table 5.
Modulation of MICs against gyrA hypomorph
| WT H37Rv | gyrA hypomorph | Ratio (WT/hypomorph) | ||||
|---|---|---|---|---|---|---|
| Compound | MIC50 (μM) | MIC (μM) | MIC50 (μM) | MIC (μM) | MIC50 | MIC |
| Rifampicin | 0.24 | 0.041 | 0.021 | 0.037 | 1.3 | 1.1 |
| Moxifloxacin | 0.12 | 0.19 | 0.041 | 0.07 | 2.9 | 2.5 |
| Zoliflodacin | 20 | 44 | 4.6 | 12 | 4.3 | 3.6 |
| 7 | 1.8 | 3.8 | 0.26 | 0.65 | 7.1 | 5.8 |
| 8 | 3.3 | 5.9 | 0.92 | 2.4 | 3.6 | 2.5 |
| 10 | 3.65 | 6.75 | 1.24 | 3.2 | 2.9 | 2.1 |
| 40 | 5.35 | 10.3 | 1.75 | 5.05 | 3.1 | 2.0 |
WT: wild-type; gyrA mutant: gyrA-FDAS-TetON-1; MIC50: The concentration of compound that inhibits bacterial growth to 50% of maximal (DMSO control) growth; MIC: the lowest concentration of the given compound that completely inhibits bacterial growth.
We also set out to determine whether the inhibitory mechanism of SPTs correlates with stabilization of the doubly cleaved complex of DNA with gyrase as had been demonstrated for other pathogens.5, 7, 10 Assays were carried out via incubation of Mtb gyrase with negative supercoiled plasmid in the presence of drug, and, after denaturing, DNA was analyzed by gel electrophoresis (Figure 7).47 Both moxifloxacin and 23 displayed similar accumulations of double-stranded cleaved DNA (linear DNA), supporting that they operate by binding to DNA doubly cleaved to Mtb gyrase and supporting the binding model that has been used herein. It is likely that there is a contribution to cidality associated with production of the cleaved DNA due to induction of the cellular hypersensitive SOS response.24, 48 Taken together with the hypomorph data, the MoIs for SPTs and FQs are similar in that they show bactericidal activity by inhibition of gyrase. That SPTs are not cross-resistant to gyrase mutations that impart decreased FQ sensitivity likely invokes that SPTs are unaffected by the association of the QRDR with FQ-Mg2+ complexes.49
Figure 7.
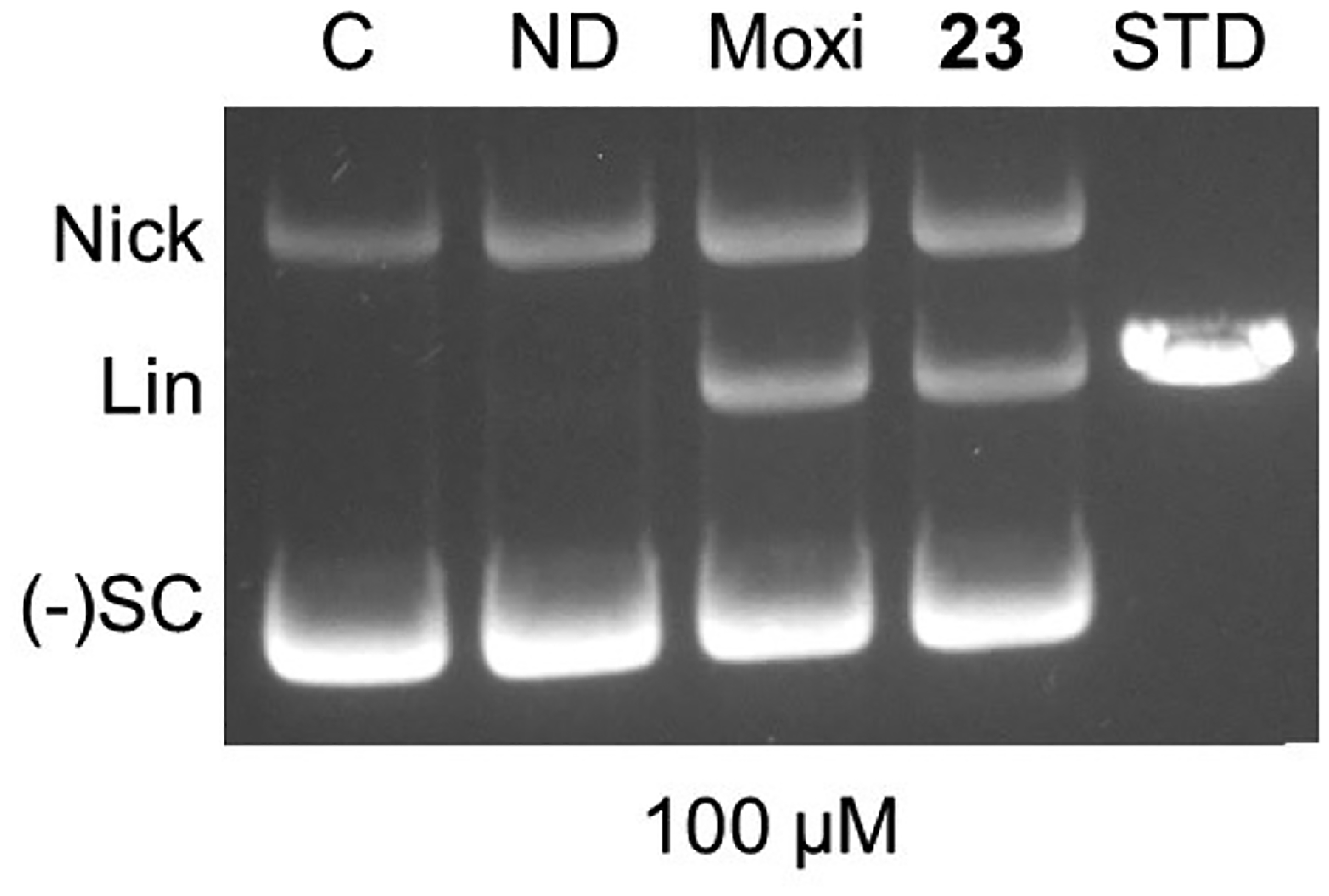
Stabilization of doubly cleaved DNA complex for gyrase by 23 and moxifloxacin each at 100 μM. Lanes from left to right are 1) negatively supercoiled DNA control, 2) Mtb gyrase + DNA with no drug, 3) Mtb gyrase + DNA + moxifloxacin, 4) Mtb gyrase + DNA + 23, 5) Linear DNA standard. Nick = nicked DNA (single-stranded DNA break), Lin = linear DNA (double-stranded DNA break), (−)SC = negatively supercoiled pBR2322 DNA.
Conclusion
Compounds were identified with improved Mtb activity over the two previously characterized compounds, zoliflodacin and 5. Importantly, a docking model for SPTs was developed using a Mtb gyrase crystal structure to build a structural hypothesis for the observed SAR. The selectivity seen for Mtb over other Gram-positive and Gram-negative bacteria (as exemplified by S. aureus and E. coli, respectively) was attributed to compound lipophilicity and differential bacterial permeability. Hence, SPTs are less sensitive to changes in lipophilicity relative to permeability in mycobacteria, and selectivity was not due to drastic differences in target inhibition. The benefit for such selectivity would be ease of dosing in the clinic by minimizing the upset due to disruption of beneficial gastrointestinal microbiota and by removing the pressure for resistance development to the class when co-infection occurs. Previous publications around SPTs disclose compounds that displayed both S. aureus and E. coli activities. The compounds of Table 1, including moxifloxacin, show activity against Mtb and appreciable cross-over to the other pathogens. Two parts of the SPT molecule were identified for analogue optimization leading to expansion of SAR over what had previously been established. First, the scaffold benzisoxazole substituent was modified to afford novel substituents with N-linked hydroxypyrrolidines and with heterocycles linked via an O-linked two-atom bridge. Second, replacing the morpholine with an N-cyanopiperazine, hypothesized to interact with the conserved Arg128, was shown to be operable for inhibition of gyrase representing perhaps the only SPT morpholine modification disclosed to date that maintains target potency and antibacterial activity. Overall, 42 having the pyrrolidinone heterocycle linked to the benzisoxazole and the N-cyanopiperazine replacement for morpholine provided the highest Mtb activity herein, while maintaining favorable solubility and low cytotoxicity. Notably, the mode-of-inhibition for a representative SPT (compound 23) correlated with binding to the DNA cleaved complex of Mtb gyrase in line with observations seen for other pathogens. The significance is that gyrase functions differently in Mtb due to its enhanced role for decatenation in addition to supercoiling, yet its interaction with the SPT is similar. Previously it was shown that SPTs are not cross-resistant with FQs despite the shared mode-of-action; herein we showed that the Mtb activity is due to gyrase inhibition as demonstrated by the upward shift in MICs against the down-regulated gyrA hypomorph. These studies thus lay the foundation for further compound optimization of activity towards achieving in vivo efficacy and identifying a valuable treatment for TB.
Experimental section
MIC assays.
The minimum inhibitory concentration (MIC) that inhibits 90% of growth of the bacterial population was determined using the broth micro-dilution method against the M. tuberculosis H37RvMa strain (ATCC 27294), as described earlier.50 MIC values represent the average of at least two biological replicates. Briefly, a 10 mL culture of the M. tuberculosis H37RvMa strain was grown to an optical density (OD600) of 0.6 – 0.7. Duplicate two-fold serial dilutions of the test compounds were prepared across 10 wells in a 96-well microtiter plate, in a volume of 50 μL, after which, 50 μL of the diluted M. tuberculosis culture (1:500) was added to each well in the plate. The final volume per well was 100 μL. A positive growth (DMSO =< 2.5%), a negative growth (Rifampicin at 2xMIC), and a Rifampicin dose response (range 0.15 – 0.0002 μM) controls were used to measure the contamination and/or plate-to-plate variations. The microtiter plate was sealed in a secondary container and incubated at 37°C with humidification. The AlamarBlue (Bio-Rad) reagent was added to each well of the assay plate at day 7 and incubated for 24 h. The measurement of MIC values was done at day 8, either visually (the lowest concentration of compound displaying no visible growth was scored as the MIC, blue colour – no growth, pink/purple colour - growth) or by measuring relative fluorescence (excitation 540 nm; emission 590 nm) using a SpectraMax i3x Plate reader. The raw fluorescent data were used to calculate % inhibition using a 4-parameter curve fit protocol (Softmax® Pro 6 Version 6.5.1).
MICs against gyrA hypomorph.
Wild-type H37Rv and gyrA-FDAS-TetON-1 were grown in 7H9 broth (supplemented with 5 g/L bovine serum albumin, 2 g/L dextrose, 0.85 g/L NaCl, 0.2% glycerol, and 0.05% Tyloxapol, and +50 μg/mL Hygromycin, +25 μg/mL Streptomycin, +500 ng/mL Anhydrotetracycline for the gyrA hypomorph) in a humidified incubator at 37 °C and 5% CO2 until logarithmic growth was reached. The cultures were washed with fresh drug-free 7H9 and suspended to an OD580 of 0.01. The diluted suspensions were added to 384-well black, clear-bottom plates at 50 μL/well, containing 16-point 2-fold drug dilutions in DMSO in triplicate, for a final DMSO concentration of 1%. Plates were wrapped with aluminum foil in stacks of no more than six and incubated at 37 °C and 5% CO2 in a humidified incubator. The OD580 of the plates was read after incubating 11 days. Growth data was normalized to inhibitor-free wells, and MIC values were determined by fitting the normalized data to a Gompertz model in GraphPad Prism. MIC values represent the average of two replicates.
DNA cleavage.
DNA cleavage reactions were based on the procedure of Aldred et al.47 Negatively supercoiled pBR322 plasmid DNA was prepared from E. coli using a Plasmid Mega Kit (Qiagen) as described by the manufacturer. Reactions were carried out in the presence or absence of 23 or moxifloxacin (LKT) and contained 100 nM wild-type (1.5:1 GyrA:GyrB ratio) and 10 nM negatively supercoiled pBR322 in a total volume of 20 μL of 10 mM Tris-HCl (pH 7.5), 40 mM KCl, 6 mM MgCl2, 0.1 mg/mL bovine serum albumin, and 10% glycerol. 20 mM stocks of 23 and moxifloxacin were made with 100% DMSO and stored at −20 °C. Dilutions (200 μM) of each compound was made in 40% DMSO. Reactions were incubated at 37 °C for 10 min. Covalent gyrase-cleaved DNA complexes were trapped by adding 2 μL of 4% sodium dodecyl sulfate followed by 1 μL of 375 mM Na2EDTA and 2 μL of 0.8 mg/mL Proteinase K (Affymetrix). Reaction mixtures were incubated at 45 °C for 30 min to digest the enzyme. Samples were mixed with 2 μL of 60% sucrose, 10 mM Tris-HCl (pH 7.9), 0.5% bromophenol blue; and 0.5% xylene cyanol FF and incubated at 45 °C for 2 min before loading onto 1% agarose gels. Reaction products were subjected to electrophoresis in 40 mM Tris-acetate (pH 8.3) and 2 mM EDTA containing 0.5 μg/mL ethidium bromide. DNA bands were visualized with medium-range ultraviolet light and quantified them using an Alpha Innotech digital imaging system. DNA single- or double-stranded cleavage was monitored by the conversion of supercoiled plasmid to nicked or linear molecules.
Docking model generation.
The Mtb gyrase structure was prepared from the crystal structure (PDB ID: 5BS8)27 co-crystallised in complex with moxifloxacin using the Maestro Protein Preparation wizard. The dsDNA portion of the structure was solved for two strands with alternate sequences on the same coordinates. With two DNA strands (chains D and E; F and G), the F and G chains were deleted while saving their two Mg2+ ions to coordinate with the two moxifloxacin molecules from the D and E chains. With the overlapping DNA chains removed, the missing covalent bonds between the catalytic Y129 residues and the scissile bases were added. The charges around the moxifloxacin binding site were then corrected to give zero order bonds between the ligand and its coordinating Mg2+ ions. The Maestro protein preparation wizard51 preprocess was then run,52 followed by the H-bond optimizer, water removal and restrained minimization. Once minimized, the S. aureus gyrase co-crystal structure with QPT-1 (PDB ID: 5CDM)26 was aligned with the prepared Mtb gyrase structure and prepared using the Protein preparation wizard. Once minimized, the QPT-1 and the two water molecules mediating a hydrogen bond between the two pyrimidinetrione NH groups and the receptor were transposed into the Mtb gyrase structure followed by a 7.5 Å radius minimization around the transposed ligand using Prime.53 The minimized structure was then inspected and the aromatic portion of the transposed QPT-1 ligand was found to stack with the adenine base of the −1 DNA cleavage site, forming a hydrogen bond between the distal pyrimidinetrione carbonyl and the backbone amide of D461 as seen analogously in the S. aureus crystal structure.
The minimized structure was then validated by creating a docking grid around the new ligand binding site using the Glide grid generation tool.31–32 The grid was centered around the transposed ligand using a hydrogen bond constraint with the backbone amide of D461. Docking QPT-1 into this grid using Glide SP precision reproduced this pose exactly. Tests also suggested rotating the α-β torsion of T500 by 180° would help break the hydrogen bond with its neighboring E501 freeing up these residues for possible polar and H-bond interactions with various substituents on the benzisoxazole. Two docking grids were therefore created to sample both possible T500 orientations. All ligands were then docked into both grids using Glide SP docking enforcing the hydrogen bond constraint. Docked ligand poses were then visually inspected before Prime MM-GBSA calculations with 5 Å radius of minimization with the R482 residue fixed.
Chemistry.
All reagents and solvents were obtained from commercial sources and used as received. Compound 1 (moxifloxacin) was purchased from Sigma-Aldrich. The following compounds37 4,38 5,34 7,41 47,37 49,37 51,36 and 5541 were prepared according to literature procedures. Reactions were monitored by thin layer chromatography (TLC) using Merck TLC silica gel 60 F254 aluminium-backed precoated plates and were visualized by ultraviolet light at 254 nm. Purification of compounds was carried out by either column chromatography on silica gel 60 (Fluka), particle size 0.063–0.2 mm (70–230 mesh ASTM), as the stationary phase, or by preparative HPLC using a Waters’ HPLC fitted with a X-bridge C18 5μm OBD column (4.6 × 150 mm); organic phase: 10 mM ammonium acetate (pH 3.7) in HPLC grade methanol, aqueous phase: 10 mM ammonium acetate (pH 3.7) in HPLC grade water; flow rate = 15.00 mL/min; detector: photodiode array (PDA). Isomers were separated by chiral HPLC using a Waters’ HPLC fitted with a Chiralpak IA (5 μm, 20mm diameter × 250mm) or Chiralpak IC (5 μm, 20mm diameter × 250mm) column with HPLC grade solvents (see individual compounds for specific details); flow rate = 15.00 mL/min; detector: photodiode array (PDA). Optical rotations were measured with an Autopol I-S3 (single wavelength) using a 1.5 mL glass cell with a 10 cm path length. All target compounds and intermediates were characterized by 1H NMR, 13C NMR, 19F NMR and/or MS. NMR spectra were recorded on either a Varian Mercury-300 (1H 300.1 MHz, 13C 75.5 MHz) or Bruker-400 (1H 400.2 MHz, 13C 100.6 MHz, 19F 377 MHz) instrument using CDCl3, MeOH-d4 or DMSO-d6 as solvents. The 1H NMR data are reported as follows: chemical shift in parts per million (δ) downfield of tetramethylsilane (TMS), multiplicity (s = singlet, bs = broad singlet, d = doublet, t = triplet, q = quartet, dd =doublets of doublets, dt = triplets of doublets and m = multiplet), coupling constant (Hz), and integrated value. The 13C and 19F NMR spectra were measured with complete proton decoupling. LC–MS analysis was performed using an Agilent® 1260 Infinity Binary Pump, Agilent® 1260 Infinity Diode Array Detector (DAD), Agilent® 1290 Infinity Column Compartment, Agilent® 1260 Infinity Standard Autosampler, and a Agilent® 6120 Quadrupole (Single) MS, with APCI/ESI multimode ionisation source. The purities were determined by Agilent® LCMS/MS (Method 1) or Waters’ HPLC (Method 2) using X-bridge C18 5μm column (4.6 × 150 mm); organic phase: 10 mM Ammonium acetate (pH 3.7) in HPLC grade methanol, aqueous phase: 10 mM Ammonium acetate (pH 3.7) in HPLC grade water; flow rate = 1.20 mL/min; detector: photodiode array (PDA). The purities of all compounds were found to be >95% or otherwise stated.
(2R,4S,4aS)-11-fluoro-8-((S)-3-hydroxypyrrolidin-1-yl)-2,4-dimethyl-1,2,4,4a-tetrahydro-2’H,6H-spiro[isoxazolo[4,5-g][1,4]oxazino[4,3-a]quinoline-5,5’-pyrimidine]-2’,4’,6’(1’H,3’H)-trione (8).
A mixture of 48a (293mg, 1.21 mmol) and pyrimidine-2,4,6(1H, 3H, 5H)-trione (170mg, 1.33mmol) in 10mL of EtOH and 1mL of 2M HCl was heated to 80°C for 16H. The solvents were removed, and the reaction mixture was precleaned by flash chromatography on silica gel, using a 4g column (over 16min) and a linear gradient 0–10% (DCM:0.05M NH3 in MeOH). The major band was isolated and contained a mixture of isomers. The mixture was separated using chiral chromatography (hexane:EtOH, 50:50, isocratic, 15mL/min, Daicel IA column). Compound 8 was isolated as a white solid (150 mg, 26%). 1H NMR (300 MHz, MeOD) δ 7.06 (s, 1H), 4.60 − 4.39 (m, 1H), 4.12 − 4.02 (m, 1H), 3.98 − 3.88 (m, 1H), 3.86 − 3.73 (m, 1H), 3.72 − 3.47 (m, 4H), 3.43 − 3.33 (m, 1H), 3.22 − 3.18 (m, 1H), 3.08 − 2.88 (m, 2H), 2.14 − 1.87 (m, 2H), 1.11 (d, J = 6.2 Hz, 3H), 0.90 (d, J = 6.3 Hz, 3H). 13C NMR (101 MHz, MeOD) δ 172.75, 169.43, 160.00, 154.73 (d, J = 12.8 Hz), 151.13, 136.40 (d, J = 239.5 Hz), 136.18, 122.21, 117.30, 110.15, 74.29, 73.87, 71.77, 66.50, 58.14 (d, J = 9.8 Hz), 57.90, 55.48, 47.84, 41.22, 35.03, 18.97, 18.64. RP-HPLC tR = 2.939 min (method 1, purity 95%); LC-MS APCI, m/z 472.1 [M-H]− (anal. calcd. for C22H24FN5O6, m/z = 473.5). [α]D20 = −128.2° (c 0.26, MeOH).
(2R,4S,4aS)-11-fluoro-8-((R)-3-hydroxypyrrolidin-1-yl)-2,4-dimethyl-1,2,4,4a-tetrahydro-2’H,6H-spiro[isoxazolo[4,5-g][1,4]oxazino[4,3-a]quinoline-5,5’-pyrimidine]-2’,4’,6’(1’H,3’H)-trione (9).
Prepared following the preparation of 8 using 48b (229 mg, 0.56 mmol) and pyrimidine-2,4,6(1H, 3H, 5H)-trione (79 mg, 0.62 mmol). Chiral column chromatography using hexane:EtOH (65:35, isocratic, 15mL/min, Daicel IA column). White solid (44 mg, 17%). 1H NMR (300 MHz, MeOD) δ 7.18 (s, 1H), 4.62 − 4.49 (m, 1H), 4.28 − 4.14 (m, 1H), 4.09 − 4.01 (m, 1H), 3.97 − 3.59 (m, 5H), 3.59 − 3.41 (m, 1H), 3.35 (d, J = 4.6 Hz, 1H), 3.23 − 3.00 (m, 2H), 2.25 − 1.97 (m, 2H), 1.22 (d, J = 6.2 Hz, 3H), 1.01 (d, J = 6.3 Hz, 3H). 13C NMR (101 MHz, MeOD) δ 171.17, 167.85, 158.37, 153.13 (d, J = 12.8 Hz), 149.57, 134.68 (d, J = 119.8 Hz), 134.52, 120.68, 115.66, 108.49, 72.68, 72.24, 70.11, 64.91, 56.51 (d, J = 9.7 Hz), 56.21, 53.89, 46.25, 39.60, 33.40, 17.37, 17.05. RP-HPLC tR = 2.929 min (method 1, purity 99%); LC-MS APCI, m/z 472.1 [M-H]− (anal. calcd. for C22H24FN5O6, m/z = 473.5). [α]D20 = −165.2° (c 0.27, MeOH).
(2R,4S,4aS)-8-((3R,4R)-3,4-dihydroxypyrrolidin-1-yl)-11-fluoro-2,4-dimethyl-1,2,4,4a-tetrahydro-2’H,6H-spiro[isoxazolo[4,5-g][1,4]oxazino[4,3-a]quinoline-5,5’-pyrimidine]-2’,4’,6’(1’H,3’H)-trione (10).
Prepared following the preparation of 8 using 48c (481 mg, 1.14 mmol) and pyrimidine-2,4,6(1H, 3H, 5H)-trione (160 mg, 1.25 mmol). Chiral column chromatography using hexane:EtOH (70:30, isocratic, 15mL/min, Daicel IA column). White solid (119 mg, 21%). 1H NMR (300 MHz, MeOD) δ 7.09 (d, J = 3.3 Hz, 1H), 4.17 − 4.01 (m, 3H), 4.00 − 3.89 (m, 1H), 3.86 − 3.63 (m, 4H), 3.58 − 3.45 (m, 1H), 3.31 − 3.15 (m, 2H), 3.10 − 2.92 (m, 2H), 1.09 (d, 3H), 0.90 (d, J = 6.4, 3.3 Hz, 3H). 13C NMR (101 MHz, MeOD) δ 172.76, 169.44, 160.14, 154.74 (d, J = 12.9 Hz), 151.14, 136.41 (d, J = 240.0 Hz), 136.23, 122.25, 117.25, 110.09, 76.61 (2C), 74.32, 73.89, 66.47, 58.55, 58.15 (d, J = 9.5 Hz), 55.58 (2C), 41.22, 18.98, 18.65. RP-HPLC tR = 2.831 min (method 1, purity 98%); LC-MS APCI, m/z 488.1 [M-H]− (anal. calcd. for C22H24FN5O7, m/z = 489.5). [α]D20 = −133.2° (c 0.26, MeOH).
(2R,4S,4aS)-8-((3S,4S)-3,4-dihydroxypyrrolidin-1-yl)-11-fluoro-2,4-dimethyl-1,2,4,4a-tetrahydro-2’H,6H-spiro[isoxazolo[4,5-g][1,4]oxazino[4,3-a]quinoline-5,5’-pyrimidine]-2’,4’,6’(1’H,3’H)-trione (11).
Prepared following the preparation of 8 using 48d (142 mg, 0.34 mmol) and pyrimidine-2,4,6(1H, 3H, 5H)-trione (47 mg, 0.37 mmol). Chiral column chromatography using hexane:EtOH (65:35, isocratic, 15mL/min, Daicel IA column). Off-white solid (10 mg, 6%). 1H NMR (300 MHz, MeOD) δ 7.25 (s, 1H), 4.23 (d, J = 3.6 Hz, 3H), 4.17 (d, J = 2.2 Hz, 1H), 4.06 (d, J = 8.8 Hz, 1H), 3.98 − 3.79 (m, 4H), 3.55 (d, J = 10.8 Hz, 2H), 3.16 − 3.05 (m, 2H), 1.22 (d, J = 6.2 Hz, 3H), 1.01 (d, J = 6.3 Hz, 3H). 13C NMR (151 MHz, MeOD) δ 171.14, 167.81, 158.52, 153.21, 149.54, 135.57, 134.60, 133.98, 120.82, 115.61, 108.44, 74.94, 72.66, 72.24, 64.91, 56.51, 54.47, 53.94, 42.40, 39.57, 17.36, 17.03. RP-HPLC tR = 2.856 min (method 1, purity 99%); LC-MS APCI, m/z 488.1 [M-H]− (anal. calcd. for C22H24FN5O7, m/z = 489.5). [a]d20 = −137.8° (c 0.26, MeOH).
(2R,4S,4aS)-8-((3R,4S)-3,4-dihydroxypyrrolidin-1-yl)-11-fluoro-2,4-dimethyl-1,2,4,4a-tetrahydro-2’H,6H-spiro[isoxazolo[4,5-g][1,4]oxazino[4,3-a]quinoline-5,5’-pyrimidine]-2’,4’,6’(1’H,3’H)-trione (12).
Prepared following the preparation of 8 using 48e (589 mg, 1.39 mmol) and pyrimidine-2,4,6(1H, 3H, 5H)-trione (196 mg, 1.53 mmol). Chiral column chromatography using hexane:EtOH (70:30, isocratic, 15mL/min, Daicel IA column). White solid (42 mg, 6%).1H NMR (300 MHz, MeOD) δ 7.19 (s, 1H), 4.37 − 4.27 (m, 2H), 4.24 − 4.13 (m, 1H), 3.85 − 3.70 (m, 3H), 3.59 − 3.47 (m, 2H), 3.41 − 3.35 (m, 1H), 3.20 − 3.04 (m, 2H), 1.86 − 1.72 (m, 2H), 1.22 (d, J = 6.2 Hz, 3H), 1.01 (d, J = 6.4 Hz, 3H). 13C NMR (151 MHz, MeOD) δ 171.14, 167.84, 158.33, 153.15 (d, J = 12.8 Hz), 149.54, 134.75 (d, J = 239.7 Hz), 134.61, 120.78, 115.57, 108.27, 72.67, 72.24, 70.83, 64.89, 56.51 (d, J = 9.7 Hz), 53.84, 52.76, 52.50, 39.54, 24.23, 17.36, 17.04. RP-HPLC tR = 2.826 min (method 1, purity 99%); LC-MS APCI, m/z 488.1 [M-H]− (anal. calcd. for C22H24FN5O7, m/z = 489.5). [α]D20 = −116.0° (c 0.25, MeOH).
(2R,4S,4aS)-11-fluoro-8-((S)-3-methoxypyrrolidin-1-yl)-2,4-dimethyl-1,2,4,4a-tetrahydro-2’H,6H-spiro[isoxazolo[4,5-g][1,4]oxazino[4,3-a]quinoline-5,5’-pyrimidine]-2’,4’,6’(1’H,3’H)-trione (13).
Prepared following the preparation of 8 using 48f (255 mg, 0.61 mmol) and pyrimidine-2,4,6(1H, 3H, 5H)-trione (85 mg, 0.67 mmol). Chiral column chromatography using hexane:EtOH:DCM (70:20:10, isocratic, 15mL/min, Daicel IA column). Off-white solid (32 mg, 11%). 1H NMR (300 MHz, MeOD) δ 7.16 (s, 1H), 4.23 − 4.11 (m, 2H), 4.09 − 3.99 (m, 1H), 3.97 − 3.73 (m, 2H), 3.73 − 3.44 (m, 5H), 3.38 (s, 3H), 3.15 − 3.05 (m, 2H), 2.27 − 2.02 (m, 2H), 1.22 (d, J = 6.2 Hz, 3H), 1.01 (d, J = 6.4 Hz, 3H). 13C NMR (101 MHz, MeOD) δ 171.15, 167.79, 158.64, 153.30 (m), 149.46, 134.82 (d, J = 218.1 Hz), 134.60, 120.73, 115.57, 108.69, 79.82, 72.69, 72.22, 65.06, 56.58 (m), 55.39, 54.03, 53.38, 46.35, 39.74, 30.28, 17.33, 16.98. RP-HPLC tR = 3.046 min (method 1, purity 99%); LC-MS APCI, m/z 486.1 [M-H]− (anal. calcd. for C23H26FN5O6, m/z = 487.5). [α]D20 = −149.4° (c 0.24, MeOH).
(2R,4S,4aS)-11-fluoro-8-((S)-3-(hydroxymethyl)pyrrolidin-1-yl)-2,4-dimethyl-1,2,4,4a-tetrahydro-2’H,6H-spiro[isoxazolo[4,5-g][1,4]oxazino[4,3-a]quinoline-5,5’-pyrimidine]-2’,4’,6’(1’H,3’H)-trione (14).
Prepared following the preparation of 8 using 48g (279 mg, 0.66 mmol) and pyrimidine-2,4,6(1H, 3H, 5H)-trione (93 mg, 0.73 mmol). Chiral column chromatography using hexane:EtOH:DCM (80:15:5, isocratic, 15mL/min, Daicel IA column). Off-white solid (73 mg, 23%). 1H NMR (300 MHz, MeOD) δ 7.17 (s, 1H), 4.24 − 4.10 (m, 1H), 4.12 − 3.99 (m, 1H), 3.98 − 3.74 (m, 2H), 3.74 − 3.46 (m, 6H), 3.40 − 3.35 (m, 1H), 3.19 − 3.01 (m, 2H), 2.66 − 2.45 (m, 1H), 2.25 − 2.00 (m, 1H), 1.94 − 1.76 (m, 1H), 1.22 (d, J = 6.2 Hz, 3H), 1.01 (d, J = 6.3 Hz, 3H). 13C NMR (151 MHz, MeOD) δ 171.12, 167.83, 158.29, 153.09 (d, J = 12.7 Hz), 149.53, 134.74 (d, J = 239.5 Hz), 134.51, 120.61, 115.76, 108.51, 72.66, 72.24, 64.87, 63.22, 56.49 (d, J = 9.8 Hz), 53.88, 50.90, 48.15, 41.04, 39.58, 27.44, 17.37, 17.05. RP-HPLC tR = 3.027 min (method 1, purity 99%); LC-MS APCI, m/z 486.1 [M-H]− (anal. calcd. for C23H26FN5O6, m/z = 487.5). [α]D 20 = −137.3° (c 0.25, MeOH).
11-fluoro-8-(3-hydroxy-3-methylpyrrolidin-1-yl)-2,4-dimethyl-1,2,4,4a-tetrahydro-2’H,6H-spiro[isoxazolo[4,5-g][1,4]oxazino[4,3-a]quinoline-5,5’-pyrimidine]-2’,4’,6’(1’H,3’H)-trione (15).
Prepared following the preparation of 8 using 48h (22 mg, 0.050 mmol) and pyrimidine-2,4,6(1H, 3H, 5H)-trione (7 mg, 0.060 mmol) (Note: compound isolated as a mixture of isomers). Pale yellow solid (9 mg, 36%). 1H NMR (300 MHz, DMSO-d6) δ 11.46 (br s, 2H), 7.36 (s, 1H), 4.07 (d, J= 13.6 Hz, 1H), 3.93 (d, J= 8.7 Hz, 1H), 3.88 − 3.38 (m, 5H), 3.16 − 3.00 (m, 1H), 2.95 (d, J= 14.2 Hz, 1H), 2.47 − 2.19 (m, 4H), 1.14 (d, J= 6.1 Hz, 3H), 0.89 (d, J= 6.3 Hz, 3H). RP-LC tR = 1.060 min (method 2, purity 100%); LC-MS ESI, m/z 486.2 [M-H]− (anal. calcd. for C23H26FN5O6, m/z = 487.5).
(2R,4S,4aS)-8-((S)-3-aminopyrrolidin-1-yl)-11-fluoro-2,4-dimethyl-1,2,4,4a-tetrahydro-2’H,6H-spiro[isoxazolo[4,5-g][1,4]oxazino[4,3-a]quinoline-5,5’-pyrimidine]-2’,4’,6’(1’H,3’H)-trione (16).
Prepared following the preparation of 8 using 48i (192 mg, 0.38 mmol) and pyrimidine-2,4,6(1H, 3H, 5H)-trione (53 mg, 0.42 mmol). Chiral column chromatography using hexane:0.1% Et2NH in EtOH (65:35, isocratic, 15mL/min, Daicel IA column). Beige solid (10 mg, 6%). 1H NMR (300 MHz, MeOD) δ 7.20 (s, 1H), 4.23 − 4.14 (m, 1H), 4.08 − 4.02 (m, 1H), 3.96 − 3.86 (m, 1H), 3.85 − 3.70 (m, 3H), 3.69 − 3.56 (m, 1H), 3.42 − 3.34 (m, 1H), 3.17 − 3.01 (m, 3H), 2.39 − 2.19 (m, 1H), 2.02 − 1.92 (m, 2H), 1.22 (d, J = 6.2 Hz, 3H), 1.01 (d, J = 6.3 Hz, 3H). 13C NMR (101 MHz, MeOD) δ 171.65, 168.27, 158.34, 153.21 (d, J= 13.0 Hz), 150.17, 134.77 (d, J= 239.6 Hz), 134.64, 120.87, 115.61, 108.42, 72.72, 72.24, 64.95, 56.57, 55.12, 53.80, 50.66, 46.55, 42.14, 39.65, 17.35, 17.04. RP-UPLC tR = 0.677 min (method 2, purity 99%); LC-MS ESI, m/z 471.2 [M-H]− (anal. calcd. for C22H25FN6O5, m/z = 472.5). [α]D20 = −94.° (c 0.22, MeOH).
(2R,4S,4aS)-11-fluoro-2,4-dimethyl-8-((S)-3-(methylamino)pyrrolidin-1-yl)-1,2,4,4a-tetrahydro-2’H-spiro[isoxazolo[5’,4’:4,5]benzo[1,2-b][1,4]oxazino[4,3-d][1,4]oxazine-5,5’-pyrimidine]-2’,4’,6’(1’H,3’H)-trione (17).
Prepared following the preparation of 8 using 48j (365 mg, 0.70 mmol) and pyrimidine-2,4,6(1H, 3H, 5H)-trione (99 mg, 0.77 mmol). Chiral column chromatography using hexane:0.1% Et2NH in EtOH (50:50, isocratic, 15mL/min, Daicel IA column). Off-white solid (41 mg, 11%). 1H NMR (300 MHz, MeOD) δ 7.23 (s, 1H), 4.22 − 4.12 (m, 1H), 4.08 − 4.01 (m, 1H), 3.95 − 3.83 (m, 1H), 3.82 − 3.68 (m, 2H), 3.68 − 3.54 (m, 1H), 3.51 − 3.38 (m, 2H), 3.18 − 2.96 (m, 3H), 2.48 (s, 3H), 2.36 − 2.20 (m, 1H), 2.05 − 1.92 (m, 2H), 1.22 (d, J = 6.2 Hz, 3H), 1.01 (d, J = 6.3 Hz, 3H). 13C NMR (151 MHz, MeOD) δ 171.43, 168.06, 158.33, 153.20 (d, J= 12.7 Hz), 149.91, 134.76 (d, J= 239.3 Hz), 134.63, 120.86, 115.63, 108.39, 72.69, 72.24, 64.91, 58.95, 56.49 (d, J= 9.5 Hz), 53.81, 52.82, 46.65, 39.59, 32.86, 30.11, 17.35, 17.03. RP-HPLC tR = 2.569 min (method 1, purity 95%); LC-MS APCI, m/z 485.1 [M-H]− (anal. calcd. for C23H27FN6O5, m/z = 486.5). [α]D 20 = −88.6° (c 0.28, MeOH).
(2R,4S,4aS)-11-fluoro-8-((S)-3-fluoropyrrolidin-1-yl)-2,4-dimethyl-1,2,4,4a-tetrahydro-2’H,6H-spiro[isoxazolo[4,5-g][1,4]oxazino[4,3-a]quinoline-5,5’-pyrimidine]-2’,4’,6’(1’H,3’H)-trione (18).
Prepared following the preparation of 8 using 49k (247 mg, 0.60 mmol) and pyrimidine-2,4,6(1H, 3H, 5H)-trione (85 mg, 0.66 mmol). Chiral column chromatography using hexane:EtOH:EtOAc (70:5:25, isocratic, 15mL/min, Daicel IA column). Off-white solid (52 mg, 18%). 1H NMR (300 MHz, MeOD) δ 7.17 (s, 1H), 5.53 − 5.26 (m, 1H), 4.25 − 4.13 (m, 1H), 4.10 − 3.99 (m, 1H), 3.94 − 3.86 (m, 1H), 3.84 − 3.61 (m, 4H), 3.39 − 3.26 (m, 2H), 3.22 − 2.98 (m, 2H), 2.45 − 2.07 (m, 2H), 1.22 (d, J = 6.2 Hz, 3H), 1.01 (d, J = 6.3 Hz, 3H). 13C NMR (151 MHz, MeOD) δ 171.10, 167.86, 158.17, 153.19 (d, J= 12.9 Hz), 149.52, 134.74 (d, J= 239.9 Hz), 134.65, 120.80, 115.49 (d,J= 3.4 Hz), 108.27, 93.27, 92.11, 72.67, 72.25, 64.85, 56.52 (d, J= 9.6 Hz), 54.77 (d, J= 23.0 Hz), 46.04, 39.61, 31.75 (d, J= 21.7 Hz), 17.37, 17.04. RP-UPLC tR = 0.959 min (method 2, purity 99%); LC-MS ESI, m/z 474.1 [M-H]− (anal. calcd. for C22H23F2N5O5, m/z = 475.5). [α]D20 = −144.6° (c 0.27, MeOH).
(S)-1-((2R,4S,4aS)-11-fluoro-2,4-dimethyl-2’,4’,6’-trioxo-1,1’,2,3’,4,4a,4’,6’-octahydro-2’H,6H-spiro[isoxazolo[4,5-g][1,4]oxazino[4,3-a]quinoline-5,5’-pyrimidin]-8-yl)pyrrolidine-3-carbonitrile (19).
Prepared following the preparation of 8 using 48l (109 mg, 0.26 mmol) and pyrimidine-2,4,6(1H, 3H, 5H)-trione (37 mg, 0.29 mmol). Chiral column chromatography using hexane:0.1% Et2NH in EtOH (55:45, isocratic, 15mL/min, Daicel IA column). Off-white solid (6 mg, 5%). 1H NMR (300 MHz, MeOD) δ 7.23 (s, 1H), 4.23 − 4.15 (m, 1H), 4.08 − 4.03 (m, 1H), 3.97 − 3.84 (m, 2H), 3.84 − 3.75 (m, 2H), 3.75 − 3.63 (m, 1H), 3.56 − 3.44 (m, 1H), 3.17 − 3.06 (m, 2H), 3.04 − 2.93 (m, 2H), 2.53 − 2.29 (m, 2H), 1.22 (d, J = 6.2 Hz, 3H), 1.01 (d, J = 6.3 Hz, 3H). 13C NMR (151 MHz, MeOD) δ 171.73, 168.35, 158.02, 153.34 (d, J= 12.5 Hz), 150.30, 134.78, 134.71 (d, J= 240.2 Hz), 121.28, 120.24, 115.32, 108.01, 72.71, 72.23, 64.96, 56.49 (d, J= 9.4 Hz), 51.23, 42.20, 39.58, 29.38, 27.87, 17.34, 17.03, 10.53. RP-HPLC tR = 2.764 min (method 1, purity 96%); LC-MS APCI, m/z 481.2 [M-H]− (anal. calcd. for C23H23FN6O5, m/z = 482.5). [α]D20 = −89.6° (c 0.12, MeOH).
(S)-1-((2R,4S,4aS)-11-fluoro-2,4-dimethyl-2’,4’,6’-trioxo-1,1’,2,3’,4,4a,4’,6’-octahydro-2’H,6H-spiro[isoxazolo[4,5-g][1,4]oxazino[4,3-a]quinoline-5,5’-pyrimidin]-8-yl)pyrrolidine-3-carboxylic acid (20).
To a solution of ethyl (S)-1-((2R,4S,4aS)-11-fluoro-2,4-dimethyl-2’,4’,6’-trioxo-1,1’,2,3’,4,4a,4’,6’-octahydro-2’H,6H-spiro[isoxazolo[4,5-g][1,4]oxazino[4,3-a]quinoline-5,5’-pyrimidin]-8-yl)pyrrolidine-3-carboxylate (S1) (20 mg, 0.038mmol) in 1mL (1,4-dioxane:H2O, 3:1), lithium hydroxide monohydrate (4.8mg, 0.110mmol) was added. The reaction mixture was heated at 30°C for 1 h. The mixture was cooled and neutralized with 2M HCl to pH 6. Organic solvent was removed under reduced pressure and the remaining water removed on the freeze dryer overnight. To remove any salts present, the remaining residue was taken up in a minimum amount of MeOH and passed through a 2g Salicycle (SPE-R5123003–064) propylsulfonic acid cartridge. The cartridge was pre-washed with MeOH. Pressure was used to move the solvent through the cartridge. The solvent was removed, and material dried to afford 20 as a white solid (8 mg, 36%). 1H NMR (300 MHz, MeOD) δ 7.19 (s, 1H), 4.19 (d, J= 12.6 Hz, 2H), 4.04 (d, J= 8.8 Hz, 1H), 3.97 − 3.85 (m, 1H), 3.83 − 3.65 (m, 6H), 3.19 − 3.03 (m, 2H), 2.40 − 2.23 (m, 2H), 1.21 (d, J= 6.1 Hz, 3H), 1.00 (d, J= 6.2 Hz, 3H). 13C NMR (151 MHz, MeOD) δ 175.24, 171.11, 167.83, 158.15, 153.17 (d, J= 13.1 Hz), 149.51, 134.68 (d, J= 240.0 Hz), 134.60, 120.82, 115.62, 108.36, 72.66, 72.23, 64.88, 56.49 (d, J= 9.6 Hz), 53.86, 50.39, 50.28, 42.72, 39.53, 28.35, 17.35, 17.03. RP-UPLC tR = 0.896 min (method 2, purity 99%); LC-MS ESI, m/z 500.1 [M-H]− (anal. calcd. for C23H24FN5O7, m/z = 501.5). [α]D20 = −120.3° (c 0.08, MeOH).
(2R,4S,4aS)-11-fluoro-8-((S)-3-hydroxypiperidin-1-yl)-2,4-dimethyl-1,2,4,4a-tetrahydro-2’H,6H-spiro[isoxazolo[4,5-g][1,4]oxazino[4,3-a]quinoline-5,5’-pyrimidine]-2’,4’,6’(1’H,3’H)-trione (21).
Prepared following the preparation of 8 using 48n (109 mg, 0.26 mmol) and pyrimidine-2,4,6(1H, 3H, 5H)-trione (36 mg, 0.28 mmol). Chiral column chromatography using hexane:EtOH (65:35, isocratic, 15mL/min, Daicel IA column). Off-white solid (64 mg, 51%). 1H NMR (300 MHz, MeOD) δ 7.16 (s, 1H), 4.24 − 4.12 (m, 1H), 4.09 − 4.00 (m, 1H), 3.98 − 3.74 (m, 4H), 3.70 − 3.57 (m, 2H), 3.18 − 3.05 (m, 3H), 3.06 − 2.94 (m, 1H), 2.07 − 1.97 (m, 1H), 1.97 − 1.85 (m, 1H), 1.75 − 1.47 (m, 2H), 1.22 (d, J = 6.2 Hz, 3H), 1.01 (d, J = 6.3 Hz, 3H). 13C NMR (101 MHz, MeOD) δ 171.91, 168.62, 161.81, 154.41 (d, J = 13.3 Hz), 150.31, 135.09, 135.56 (d, J = 239.9 Hz), 121.80, 116.29, 116.26, 109.17, 73.46, 73.03, 66.17, 65.68, 57.30 (d, J = 9.8 Hz), 55.44, 54.68, 40.43, 32.96, 22.93, 18.17, 17.85. RP-HPLC tR = 3.018 min (method 1, purity 99%); LC-MS APCI, m/z 486.1 [M-H]− (anal. calcd. for C23H26FN5O6, m/z = 487.5). [α]D 20 = −124.3° (c 0.27, MeOH).
(2R,4S,4aS)-11-fluoro-8-((R)-3-hydroxypiperidin-1-yl)-2,4-dimethyl-1,2,4,4a-tetrahydro-2’H,6H-spiro[isoxazolo[4,5-g][1,4]oxazino[4,3-a]quinoline-5,5’-pyrimidine]-2’,4’,6’(1’H,3’H)-trione (22).
Prepared following the preparation of 8 using 48o (251 mg, 0.60 mmol) and barbituric acid (84 mg, 0.66 mmol). Chiral column chromatography using hexane:EtOH (65:35, isocratic, 15mL/min, Daicel IA column). Off-white solid (86 mg, 30%). 1H NMR (300 MHz, MeOD) δ 7.20 (s, 1H), 4.19 (dd, J = 14.2, 2.2 Hz, 1H), 4.05 (d, J = 8.8 Hz, 1H), 3.99 − 3.74 (m, 5H), 3.73 − 3.59 (m, 1H), 3.40 − 3.28 (m, 1H), 3.20 − 2.93 (m, 4H), 2.10 − 1.86 (m, 1H), 1.84 − 1.62 (m, 1H), 1.62 − 1.45 (m, 1H), 1.22 (d, J = 6.2 Hz, 3H), 1.01 (d, J = 6.4 Hz, 3H). 13C NMR (101 MHz, MeOD) δ 171.91, 168.62, 161.88, 154.45 (d, J = 12.9 Hz), 150.31, 135.58 (d, J = 240.3 Hz), 135.14, 121.88, 116.28, 109.19, 73.46, 73.03, 66.22, 65.69, 57.29 (d, J = 9.5 Hz), 55.48, 54.72, 48.70, 40.45, 32.96, 22.96, 18.16, 17.83. RP-HPLC tR = 3.002 min (method 1, purity 99%); LC-MS APCI, m/z 486.1 [M-H]− (anal. calcd. for C23H26FN5O6, m/z = 487.5). [a]d20 = −163.2° (c 0.29, MeOH).
(2R,4S,4aS)-11-fluoro-2,4-dimethyl-8-(((S)-5-oxopyrrolidin-2-yl)methoxy)-1,2,4,4a-tetrahydro-2’H,6H-spiro[isoxazolo[4,5-g][1,4]oxazino[4,3-a]quinoline-5,5’-pyrimidine]-2’,4’,6’(1’H,3’H)-trione (23).
Prepared following the preparation of 8 using 48p (265 mg, 0.61 mmol) and pyrimidine-2,4,6(1H, 3H, 5H)-trione (86 mg, 0.67 mmol). Chiral column chromatography using hexane:EtOH (50:50, isocratic, 15mL/min, Daicel IA column). Off-white solid (42 mg, 14%). 1H NMR (300 MHz, DMSO-d6) δ 7.87 (s, 1H), 7.09 (s, 1H), 4.35 − 4.19 (m, 2H), 4.11 − 4.02 (m, 1H), 4.02 − 3.91 (m, 2H), 3.84 − 3.63 (m, 2H), 3.57 − 3.46 (m, 1H), 3.15 − 3.03 (m, 1H), 2.94 − 2.83 (m, 1H), 2.82 − 2.68 (m, 1H), 2.25 − 2.13 (m, 2H), 1.95 − 1.88 (m, 1H), 1.14 (d, J = 6.2 Hz, 3H), 0.88 (d, J = 6.3 Hz, 3H). 13C NMR (101 MHz, MeOD) δ 179.75, 172.65, 169.14, 166.07, 153.65 (d, JCF= 31.2 Hz), 135.43 (d, JCF= 25.1 Hz), 133.29, 122.81, 113.34, 105.81, 72.76, 72.48, 72.21, 65.18, 56.52 (d,JCF= 9.7 Hz), 53.65, 53.43, 42.24, 39.53, 29.51, 22.58, 17.31, 17.04. RP-UPLC tR = 0.901 min (method 2, purity 100%); LC-MS ESI, m/z 500.1 [M-H]− (anal. calcd. for C23H24FN5O7, m/z = 501.5). [α]D 20 = −135.8° (c 0.27, MeOH).
(2R,4S,4aS)-11-fluoro-2,4-dimethyl-8-(((R)-5-oxopyrrolidin-2-yl)methoxy)-1,2,4,4a-tetrahydro-2’H,6H-spiro[isoxazolo[4,5-g][1,4]oxazino[4,3-a]quinoline-5,5’-pyrimidine]-2’,4’,6’(1’H,3’H)-trione (24).
Prepared following the preparation of 8 using 48q (346 mg, 0.79 mmol) and pyrimidine-2,4,6(1H, 3H, 5H)-trione (112 mg, 0.87 mmol). Chiral column chromatography using hexane:EtOH:DCM (60:20:20, isocratic, 15mL/min, Daicel IA column). Beige solid (42 mg, 14%). 1H NMR (300 MHz, DMSO-d6) δ 11.56 (s, 2H), 7.89 (s, 1H), 7.12 (s, 1H), 4.40 − 4.18 (m, 2H), 4.13 − 4.03 (m, 1H), 3.97 (dd, J = 13.1, 6.5 Hz, 2H), 3.85 − 3.74 (m, 1H), 3.74 − 3.62 (m, 1H), 3.62 − 3.50 (m, 1H), 3.18 − 3.03 (m, 1H), 2.97 − 2.86 (m, 1H), 2.35 − 2.06 (m, 3H), 1.99 − 1.82 (m, 1H), 1.14 (d, J = 6.2 Hz, 3H), 0.89 (d, J = 6.3 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 177.35, 171.37, 168.10, 166.29, 153.17 (d, J = 12.4 Hz), 149.95, 135.69, 134.24 (d, J = 240.7 Hz), 123.16, 114.50, 105.76, 73.26, 72.52, 72.15, 64.88, 56.82 (d, J = 7.4 Hz), 53.57, 52.55, 39.07, 30.05, 23.14, 18.67, 18.59. RP-UPLC tR = 0.910 min (method 2, purity 100%); LC-MS ESI, m/z 500.1 [M-H]− (anal. calcd. for C23H24FN5O7, m/z = 501.5). [α]D20 = −185.1° (c 0.35, MeOH).
(2R,4S,4aS)-11-fluoro-2,4-dimethyl-8-(((S)-1-methyl-5-oxopyrrolidin-2-yl)methoxy)-1,2,4,4a-tetrahydro-2’H,6H-spiro[isoxazolo[4,5-g][1,4]oxazino[4,3-a]quinoline-5,5’-pyrimidine]-2’,4’,6’(1’H,3’H)-trione (25).
Prepared following the preparation of 8 using 48r (208 mg, 0.46 mmol) and pyrimidine-2,4,6(1H, 3H, 5H)-trione (65 mg, 0.51 mmol). Chiral column chromatography using hexane:EtOH:2-propanol (65:35:5, isocratic, 15mL/min, Daicel IA column). Beige solid (66 mg, 28%). 1H NMR (300 MHz, DMSO-d6) δ 7.10 (s, 1H), 4.62 − 4.33 (m, 2H), 4.13 − 4.02 (m, 1H), 4.00 − 3.89 (m, 2H), 3.83 − 3.72 (m, 1H), 3.72 − 3.63 (m, 1H), 3.63 3.51 (m, 1H), 3.15 − 3.03 (m, 1H), 2.94 − 2.83 (m, 1H), 2.78 (s, 3H), 2.47 − 2.32 (m, 1H), 2.28 2.11 (m, 2H), 2.01 − 1.84 (m, 1H), 1.16 − 1.12 (m, 3H), 0.88 (d, J = 6.3 Hz, 3H). 13C NMR (151 MHz, DMSO) δ 175.24, 171.86, 168.49, 166.19, 153.13 (d, JCF= 12.8 Hz), 150.49, 135.76, 134.17 (d, JCF= 240.1 Hz), 123.44, 114.18, 105.51, 72.58, 72.23, 70.74, 64.89, 58.49, 56.73 (d, JCF= 9.3 Hz), 53.42, 38.95, 30.12, 28.13, 21.07, 18.53, 18.48. RP-UPLC tR = 0.941 min (method 2, purity 100%); LC-MS ESI, m/z 514.2 [M-H]− (anal. calcd. for C24H26FN5O7, m/z = 515.5). [α]D 20 = −149.0° (c 0.24, MeOH).
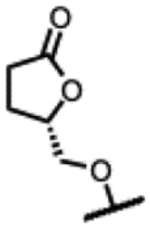
(2R,4S,4aS)-11-fluoro-2,4-dimethyl-8-(((S)-5-oxotetrahydrofuran-2-yl)methoxy)-1,2,4,4a-tetrahydro-2’H,6H-spiro[isoxazolo[4,5-g][1,4]oxazino[4,3-a]quinoline-5,5’-pyrimidine]-2’,4’,6’(1’H,3’H)-trione (26).
Prepared following the preparation of 8 using 48s (247 mg, 0.57 mmol) and pyrimidine-2,4,6(1H, 3H, 5H)-trione (80 mg, 0.62 mmol). Chiral column chromatography using hexane:EtOH:MTBE:DCM (45:5:30:20, isocratic, 15mL/min, Daicel IC column). Off-white solid (28 mg, 10%). 1H NMR (300 MHz, DMSO-d6) δ 11.82 (br s, 1H), 11.46 (br s, 1H), 7.15 (s, 1H), 4.99 − 4.93 (m, 1H), 4.61 − 4.52 (m, 1H), 4.53 − 4.44 (m, 1H), 4.12 − 4.03 (m, 1H), 3.98 − 3.91 (m, 1H), 3.83 − 3.74 (m, 1H), 3.72 − 3.64 (m, 1H), 3.62 − 3.56 (m, 1H), 3.17 − 3.03 (m, 1H), 2.97 − 2.86 (m, 1H), 2.62 − 2.54 (m, 2H), 2.42 − 2.26 (m, 1H), 2.16 − 2.05 (m, 1H), 1.14 (d, J = 6.1 Hz, 3H), 0.89 (d, J = 6.3 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 177.38, 171.32, 168.09, 166.05, 153.28 (d, J = 12.9 Hz), 149.89, 135.81, 131.58 (d, J = 303.7 Hz), 123.41, 114.35, 105.57, 77.60, 72.51, 72.16, 72.01, 64.90, 56.82 (d, J = 9.0 Hz), 53.67, 39.03, 28.32, 23.45, 18.67, 18.59. RP-UPLC tR = 0.954 min (method 2, purity 98%); LC-MS ESI, m/z 501.1 [M-H]− (anal. calcd. for C23H23FN4O8, m/z = 502.5). [α]D20 = −118.2° (c 0.26, MeOH).
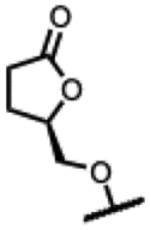
(2R,4S,4aS)-11-fluoro-2,4-dimethyl-8-(((R)-5-oxotetrahydrofuran-2-yl)methoxy)-1,2,4,4a-tetrahydro-2’H,6H-spiro[isoxazolo[4,5-g][1,4]oxazino[4,3-a]quinoline-5,5’-pyrimidine]-2’,4’,6’(1’H,3’H)-trione (27).
Prepared following the preparation of 8 using 48t (191 mg, 0.44 mmol) and pyrimidine-2,4,6(1H, 3H, 5H)-trione (62 mg, 0.48 mmol). Chiral column chromatography using hexane:EtOH:MTBE:DCM (45:5:30:20, isocratic, 15mL/min, Daicel IC column). Off-white solid (24 mg, 10%). 1H NMR (300 MHz, DMSO-d6) δ 7.15 (s, 1H), 5.01 − 4.89 (m, 1H), 4.60 − 4.51 (m, 1H), 4.51 − 4.41 (m, 1H), 4.13 − 4.02 (m, 1H), 3.98 − 3.90 (m, 1H), 3.85 − 3.73 (m, 1H), 3.71 − 3.63 (m, 1H), 3.61 − 3.53 (m, 1H), 3.16 − 3.04 (m, 1H), 2.92 (d, J = 14.3 Hz, 1H), 2.62 − 2.53 (m, 2H), 2.42 − 2.25 (m, 1H), 2.17 − 2.04 (m, 1H), 1.14 (d, J = 6.1 Hz, 3H), 0.89 (d, J = 6.4 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 177.37, 171.32, 168.07, 166.07, 153.27 (d, J = 13.1 Hz), 149.88, 135.79, 134.28 (d, J = 240.4 Hz), 123.37, 114.35, 105.57, 77.64, 72.52, 72.16, 72.06, 64.88, 56.83 (d, J = 9.3 Hz), 53.63, 39.05, 28.31, 23.44, 18.67, 18.59. RP-UPLC tR = 0.954 min (method 2, purity 95%); LC-MS ESI, m/z 501.1 [M-H]− (anal. calcd. for C23H23FN4O8, m/z = 502.5). [α]D20 = −152.8° (c 0.23, MeOH).
(2R,4S,4aS)-11-fluoro-2,4-dimethyl-8-(((R)-2-oxooxazolidin-4-yl)methoxy)-1,2,4,4a-tetrahydro-2’H,6H-spiro[isoxazolo[4,5-g][1,4]oxazino[4,3-a]quinoline-5,5’-pyrimidine]-2’,4’,6’(1’H,3’H)-trione (28).
Prepared following the preparation of 8 using 48u (276 mg, 0.63 mmol) and pyrimidine-2,4,6(1H, 3H, 5H)-trione (89 mg, 0.69 mmol). Chiral column chromatography using hexane:EtOAc (55:45, isocratic, 15mL/min, Daicel IA column). Off-white solid (34 mg, 11%). 1H NMR (300 MHz, DMSO-d6) δ 7.93 (s, 1H), 7.07 (s, 1H), 4.51 − 4.41 (m, 1H), 4.35 (dd, J = 6.2, 3.9 Hz, 2H), 4.29 − 4.19 (m, 2H), 4.12 − 4.03 (m, 1H), 3.98 − 3.90 (m, 1H), 3.85 − 3.72 (m, 1H), 3.71 − 3.60 (m, 2H), 3.17 − 3.05 (m, 1H), 2.95 − 2.84 (m, 1H), 1.14 (d, J = 6.1 Hz, 3H), 0.89 (d, J = 6.3 Hz, 3H). 19F NMR (377 MHz, DMSO) δ −157.61 (s, 1H). RP-UPLC tR = 0.888 min (method 2, purity 100%); LC-MS ESI, m/z 502.1 [M-H]− (anal. calcd. for C22H22FN5O8, m/z = 503.4). [α]D20 = −118.5° (c 0.24, MeOH).
(2R,4S,4aS)-11-fluoro-2,4-dimethyl-8-(((S)-2-oxooxazolidin-4-yl)methoxy)-1,2,4,4a-tetrahydro-2’H,6H-spiro[isoxazolo[4,5-g][1,4]oxazino[4,3-a]quinoline-5,5’-pyrimidine]-2’,4’,6’(1’H,3’H)-trione (29).
Prepared following the preparation of 8 using 48v (281 mg, 0.64 mmol) and pyrimidine-2,4,6(1H, 3H, 5H)-trione (91 mg, 0.71 mmol). Chiral column chromatography using hexane:EtOAc (55:45, isocratic, 15mL/min, Daicel IA column). Off-white solid (32 mg, 10%). 1H NMR (300 MHz, DMSO-d6) δ 7.93 (s, 1H), 7.07 (s, 1H), 4.50 − 4.42 (m, 1H), 4.41 − 4.29 (m, 2H), 4.29 − 4.18 (m, 2H), 4.12 − 4.02 (m, 1H), 3.98 − 3.90 (m, 1H), 3.85 − 3.72 (m, 1H), 3.71 − 3.59 (m, 2H), 3.17 − 3.04 (m, 1H), 2.95 − 2.85 (m, 1H), 1.14 (d, J = 6.1 Hz, 3H), 0.89 (d, J = 6.2 Hz, 3H). RP-UPLC tR = 0.887 min (method 2, purity 100%); LC-MS ESI, m/z 502.1 [M-H]− (anal. calcd. for C22H22FN5O8, m/z = 503.4). [α]D20 = −113.3° (c 0.23, MeOH).
(2R,4S,4aS)-11-fluoro-2,4-dimethyl-8-(((S)-5-oxopyrrolidin-3-yl)methoxy)-1,2,4,4a-tetrahydro-2’H,6H-spiro[isoxazolo[4,5-g][1,4]oxazino[4,3-a]quinoline-5,5’-pyrimidine]-2’,4’,6’(1’H,3’H)-trione (30).
Prepared following the preparation of 8 using 48w (282 mg, 0.65 mmol) and pyrimidine-2,4,6(1H, 3H, 5H)-trione (91 mg, 0.71 mmol). Chiral column chromatography using hexane:EtOH:DCM (60:30:10, isocratic, 15mL/min, Daicel IA column). Off-white solid (70 mg, 22%). 1H NMR (300 MHz, DMSO-d6) δ 7.00 (s, 1H), 4.29 (d, J = 6.3 Hz, 2H), 4.07 − 3.96 (m, 1H), 3.94 − 3.83 (m, 1H), 3.82 − 3.66 (m, 1H), 3.66 − 3.54 (m, 1H), 3.52 − 3.39 (m, 1H), 3.39 − 3.28 (m, 1H), 3.19 − 3.10 (m, 1H), 3.09 − 2.82 (m, 3H), 2.47 − 2.34 (m, 1H), 2.17 − 2.05 (m, 1H), 1.10 (d, J = 6.4 Hz, 3H), 0.85 (d, J = 6.3 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 177.94, 171.94, 168.41, 166.24, 153.01 (d, J = 13.0 Hz), 150.52, 135.63, 134.12 (d, J = 241.3 Hz), 122.98, 114.24, 105.59, 72.71, 72.48, 72.23, 64.86, 56.53, 53.32, 44.53, 41.91, 33.33, 33.15, 18.28, 18.17. RP-UPLC tR = 0.894 min (method 2, purity 100%); LC-MS ESI, m/z 500.1 [M-H]− (anal. calcd. for C23H24FN5O7, m/z = 501.5). [α]D20 = −152.2° (c 0.23, MeOH).
(2R,4S,4aS)-11-fluoro-2,4-dimethyl-8-(((R)-5-oxopyrrolidin-3-yl)methoxy)-1,2,4,4a-tetrahydro-2’H,6H-spiro[isoxazolo[4,5-g][1,4]oxazino[4,3-a]quinoline-5,5’-pyrimidine]-2’,4’,6’(1’H,3’H)-trione (31).
Prepared following the preparation of 8 using 48x (224 mg, 0.51 mmol) and pyrimidine-2,4,6(1H, 3H, 5H)-trione (72 mg, 0.57 mmol). Chiral column chromatography using hexane:EtOH:MTBE:DCM (40:10:30:20, isocratic, 15mL/min, Daicel IA column). Off-white solid (34 mg, 13%).1H NMR (600 MHz, DMSO-d6) δ 7.55 (s, 1H), 7.06 (d, J = 1.2 Hz, 1H), 4.36 − 4.28 (m, 2H), 4.08 − 4.00 (m, 1H), 3.94 − 3.88 (m, 1H), 3.79 − 3.71 (m, 1H), 3.68 − 3.61 (m, 1H), 3.51 − 3.46 (m, 1H), 3.43 − 3.37 (m, 1H), 3.13 − 3.03 (m, 2H), 2.95 − 2.84 (m, 2H), 2.34 − 2.27 (m, 1H), 2.10 − 2.03 (m, 1H), 1.11 (d, J = 6.2 Hz, 3H), 0.85 (d, J = 6.3 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 176.12, 171.84, 168.53, 166.26, 153.13 (d, J = 12.5 Hz), 150.55, 135.71, 134.24 (d, J = 239.9 Hz), 123.38, 114.28, 105.67, 72.57, 72.45, 72.13, 64.92, 56.81 (d, J = 9.2 Hz), 53.46, 44.29, 39.07, 33.71, 33.41, 18.64, 18.60. RP-UPLC tR = 0.889 min (method 2, purity 100%); LC-MS ESI, m/z 500.2 [M-H]− (anal. calcd. for C23H24FN5O7, m/z = 501.5). [α]D 20 = −170.8° (c 0.21, MeOH).
(2R,4S,4aS)-11-fluoro-2,4-dimethyl-8-(2-((S)-5-oxopyrrolidin-2-yl)ethoxy)-1,2,4,4a-tetrahydro-2’H,6H-spiro[isoxazolo[4,5-g][1,4]oxazino[4,3-a]quinoline-5,5’-pyrimidine]-2’,4’,6’(1’H,3’H)-trione (32).
Prepared following the preparation of 8 using 48y (359 mg, 0.80 mmol) and pyrimidine-2,4,6(1H, 3H, 5H)-trione (113 mg, 0.88 mmol). Chiral column chromatography using hexane:EtOH:DCM (55:35:15, isocratic, 15mL/min, Daicel IA column). Beige solid (38 mg, 9%). 1H NMR (300 MHz, DMSO-d6) δ 11.61 (s, 2H), 7.80 (s, 1H), 7.10 (s, 1H), 4.48 − 4.34 (m, 2H), 4.12 − 4.00 (m, 1H), 3.98 − 3.89 (m, 1H), 3.85 − 3.60 (m, 3H), 3.60 − 3.50 (m, 1H), 3.17 − 3.02 (m, 1H), 2.97 − 2.87 (m, 1H), 2.24 − 2.09 (m, 3H), 2.03 − 1.87 (m, 2H), 1.79 − 1.64 (m, 1H), 1.14 (d, J = 6.1 Hz, 3H), 0.89 (d, J = 6.3 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 177.03, 171.42, 168.11, 166.19, 153.12 (d, J = 12.5 Hz), 149.98, 135.64, 134.31 (d, J = 240.5 Hz), 123.16, 114.34, 105.95, 72.54, 72.15, 68.18, 64.93, 56.85 (d, J = 10.5 Hz), 53.58, 51.12, 39.06, 35.99, 30.29, 27.25, 18.67, 18.60. RP-UPLC tR = 0.954 min (method 2, purity 100%); LC-MS ESI, m/z 514.1 [M-H]− (anal. calcd. for C24H26FN5O7, m/z = 515.5). [α]D20 = −165.3° (c 0.27, MeOH).
(2R,4S,4aS)-11-fluoro-2,4-dimethyl-8-((5-methyl-1,3,4-oxadiazol-2-yl)methoxy)-1,2,4,4a-tetrahydro-2’H,6H-spiro[isoxazolo[4,5-g][1,4]oxazino[4,3-a]quinoline-5,5’-pyrimidine]-2’,4’,6’(1’H,3’H)-trione (33).
Prepared following the preparation of 8 using 48z (77 mg, 0.177 mmol) and pyrimidine-2,4,6(1H, 3H, 5H)-trione (25 mg, 0.194 mmol). Chiral column chromatography using hexane:EtOH:DCM (56:40:4, isocratic, 15mL/min, Daicel IC column). Off-white solid (65 mg, 73%). 1H NMR (400 MHz, DMSO-d6) δ 11.62 (br s, 2H), 7.13 (s, 1H), 5.66 (s, 2H), 4.14 − 4.04 (m, 1H), 3.99 − 3.89 (m, 1H), 3.85 − 3.74 (m, 1H), 3.73 − 3.61 (m, 1H), 3.60 − 3.52 (m, 1H), 3.16 − 3.06 (m, 1H), 2.97 − 2.89 (m, 1H), 2.54 (s, 3H), 1.14 (d, J = 6.2 Hz, 3H), 0.90 (d, J = 6.3 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 171.39, 168.13, 165.40, 165.33, 161.88, 153.51 (d, J = 13.2 Hz), 149.98, 135.97, 134.20 (d, J= 240.0 Hz), 123.70, 114.23, 105.10, 72.55, 72.16, 64.95, 61.68, 56.83 (d, J = 8.9 Hz), 53.50, 39.01, 18.66, 18.58, 10.93. 19F NMR (377 MHz, DMSO-d6) δ −157.50. RP-UPLC tR = 0.983 min (method 2, purity 99%); LC-MS ESI, m/z 499.1 [M-H]− (anal. calcd. for C22H21FN6O7, m/z = 500.4). [α]D20 = −203° (c 0.29, MeOH).
(2R,4S,4aS)-11-fluoro-2,4-dimethyl-8-((S)-4-methyl-2-oxooxazolidin-3-yl)-2,3,4,4a-tetrahydro-1H,2’H,6H-spiro[isoxazolo[4,5-g]pyrazino[1,2-a]quinoline-5,5’-pyrimidine]-2’,4’,6’(1’H,3’H)-trione (34).
A mixture of 50a (120 mg, 0.319 mmol) and pyrimidine-2,4,6(1H, 3H, 5H)-trione (41 mg, 0.319 mmol) in AcOH (2 mL) and water (0.5 mL) was stirred for 4 h at 80 °C. The solvents were removed, and the reaction mixture was precleaned by flash chromatography on silica gel, using a 4g column (over 16min) and a linear gradient 0–10% (DCM:MeOH). The major band was isolated and contained a mixture of isomers. The mixture of isomers was separated using reverse phase preparative HPLC [(10 mM ammonium acetate buffer containing 0.4% acetic acid in water):(10 mM ammonium acetate buffer containing 0.4% acetic acid in methanol), gradient, 20–60%, 15mL/min, C18 column). Compound 34 was isolated as a white solid (15 mg, 3.0%). 1H NMR (300 MHz, DMSO-d6) δ 7.55 (s, 1H), 4.65 (m, 2H), 4.19 (dd, J = 7.6, 4.2 Hz, 1H), 4.01 (d, J = 12.0 Hz, 1H), 3.83 (d, J = 9.1 Hz, 1H), 3.56 (d, J = 14.5 Hz, 1H), 3.05 − 2.80 (m, 4H), 1.41 (d, J = 5.7 Hz, 3H), 1.02 (d, J = 5.5 Hz, 3H), 0.78 (d, J = 6.4 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 171.79, 168.31, 154.46, 153.93 (d, J = 12.8 Hz), 152.58, 150.06, 135.90, 133.71 (d, J = 235.6 Hz), 123.24, 118.24, 106.99, 70.70, 66.34, 53.65, 53.30, 52.00, 51.02, 40.91, 39.06, 21.50, 19.00, 18.01. RP-UPLC tR = 2.61 min (method 1, purity 100%); LC-MS ESI, m/z 487.2 [M+H]+ (anal. calcd. for C22H23FN6O6, m/z = 486.5). [α]D20 = −55° (c 0.5, MeOH).
(2R,4S,4aS)-11-fluoro-3-hydroxy-2,4-dimethyl-8-((S)-4-methyl-2-oxooxazolidin-3-yl)-2,3,4,4a-tetrahydro-1H,2’H,6H-spiro[isoxazolo[4,5-g]pyrazino[1,2-a]quinoline-5,5’-pyrimidine]-2’,4’,6’(1’H,3’H)-trione (35).
A mixture of 50b (400 mg, 1.02 mmol) and pyrimidine-2,4,6(1H, 3H, 5H)-trione (131 mg, 1.02 mmol) in ethanol (3 mL) was stirred for 2 h at 80 °C. The solvents were removed, and the reaction mixture was precleaned by flash chromatography on silica gel, using a 4g column (over 16min) and a linear gradient 0–10% (DCM:MeOH). The major band was isolated and contained a mixture of isomers. The mixture was separated using reverse phase preparative HPLC [(10 mM ammonium acetate buffer containing 0.4% acetic acid in water):(10 mM ammonium acetate buffer containing 0.4% acetic acid in methanol), gradient, 20–60%, 15mL/min, C18 column). Compound 35 was isolated as a white solid (30 mg, 6.0 % yield). 1H NMR (300 MHz, DMSO-d6) δ 11.76 (s, 1H), 11.42 (s, 1H), 8.04 (s, 1H), 7.58 (s, 1H), 4.73–4.62 (m, 2H), 4.23 − 4.15 (m, 1H), 4.03–4.00 (m, 2H), 3.59 (d, J = 16.0 Hz, 1H), 3.15 (d, J = 17.3 Hz, 1H), 2.91 (d, J = 14.5 Hz, 1H), 2.74–2.70 (m, 2H), 1.42 (d, J = 5.7 Hz, 3H), 1.12 (d, J = 6.0 Hz, 3H), 0.88 (d, J = 6.2 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 171.51, 168.34, 154.52, 153.88 (d, J = 12.9 Hz), 152.56, 149.93, 134.86 (d, J = 239.5 Hz), 129.81, 123.16, 118.34, 107.29, 70.64, 64.07, 62.83, 61.56, 54.91, 54.04, 53.00, 39.05, 17.76, 17.18, 16.17. RP-UPLC tR = 3.403 min (method 1, purity 100%); LC-MS ESI, m/z 503.1 [M+H]+ (anal. calcd. for C22H23FN6O7, m/z = 502.5). [α]D20 = −63° (c 1.0, MeOH).
(2R,4S,4aS)-3-acetyl-11-fluoro-2,4-dimethyl-8-((S)-4-methyl-2-oxooxazolidin-3-yl)-2,3,4,4a-tetrahydro-1H,2’H,6H-spiro[isoxazolo[4,5-g]pyrazino[1,2-a]quinoline-5,5’-pyrimidine]-2’,4’,6’(1’H,3’H)-trione (36).
Prepared as described for 35 using 50c (220 mg, 0.526 mmol) and pyrimidine-2,4,6(1H, 3H, 5H)-trione (67 mg, 0.526 mmol). Flash column chromatography using CH2Cl2:MeOH (90:10). The mixture of isomers were separated using reverse phase preparative HPLC [(10 mM ammonium acetate buffer containing 0.4% acetic acid in water):(10 mM ammonium acetate buffer containing 0.4% acetic acid in methanol), gradient, 20–60%, 15mL/min, C18 column). Compound 36 was isolated as a white solid (40 mg, 14%). 1H NMR (300 MHz, MeOD+CDCl3) δ 7.73 (s, 1H), 5.04 (d, J = 7.5 Hz, 1H), 4.79 − 4.66 (m, 2H), 4.19 (q, J = 3.1 Hz, 1H), 4.11 − 4.07 (m, 1H), 3.98 − 3.90 (m, 2H), 3.74 (t, J = 11.6 Hz, 1H), 3.51 (d, J = 16.6 Hz, 1H), 3.24 (d, J = 16.7 Hz, 1H), 2.06 (s, 3H), 1.53 (d, J = 6.0 Hz, 3H), 1.48 (d, J = 7.2 Hz, 3H), 1.31 (d, J = 6.1 Hz, 3H). 13C NMR (101 MHz, MeOD+CDCl3) δ 171.80 (2C), 154.79, 153.02 (d, J = 12.9 Hz), 152.20, 149.34, 136.13, 135.64, 133.72, 120.65, 118.80, 109.23, 70.53 (2C), 65.82, 53.15 (2C), 50.20 (2C), 37.22 (2C), 20.60, 17.55 (2C). RP-UPLC tR = 0.762 min (method 2, purity 100%); LC-MS ESI, m/z 529.2 [M+H]+ (anal. calcd. for C24H25FN6O7, m/z = 528.4). [α]D20 = −63° (c 1.0, MeOH).
2-((2R,4S,4aS)-11-fluoro-2,4-dimethyl-8-((S)-4-methyl-2-oxooxazolidin-3-yl)-2’,4’,6’-trioxo-1,1’,2,3’,4,4a,4’,6’-octahydro-2’H,3H,6H-spiro[isoxazolo[4,5-g]pyrazino[1,2-a]quinoline-5,5’-pyrimidin]-3-yl)acetonitrile (37).
Prepared as described for 35 using 50d (350 mg, 0.843 mmol) and pyrimidine-2,4,6(1H, 3H, 5H)-trione (108 mg, 0.843 mmol). Flash column chromatography using CH2Cl2:MeOH (90:10). The mixture of isomers were separated using reverse phase preparative HPLC [(10 mM ammonium acetate buffer containing 0.4% acetic acid in water):(10 mM ammonium acetate buffer containing 0.4% acetic acid in methanol), gradient, 20–60%, 15mL/min, C18 column). Compound 37 was isolated as a major component (Note: minor diastereomer was observed but was not isolated). White solid (23 mg, 5.0%). 1H NMR (300 MHz, DMSO-d6) δ 11.81 (s, 1H), 11.48 (s, 1H), 7.61 (s, 1H), 4.74 − 4.62 (m, 2H), 4.22 − 4.16 (m, 1H), 4.13 (d, J = 9.1 Hz, 1H), 4.04 − 3.97 (m, 3H), 3.63 (d, J = 14.3 Hz, 1H), 3.18 (t, J = 12.2 Hz, 1H), 2.93 (d, J = 14.3 Hz, 1H), 2.73–2.68 (m, 2H), 1.42 (d, J = 5.7 Hz, 3H), 1.12 (d, J = 6.1 Hz, 3H), 0.84 (d, J = 6.4 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 171.41, 168.36, 154.53, 153.84 (d, J = 12.5 Hz), 152.59, 149.85, 135.21, 133.92 (d, J = 239.7 Hz), 123.22, 118.39, 115.57, 107.64, 70.65, 64.80, 56.65 (d, J = 8.0 Hz), 56.11, 54.80 (2C), 53.01, 38.99, 37.63, 17.73, 17.28, 16.44. RP-UPLC tR = 0.913 min (method 2, purity 96%); LC-MS ESI, m/z 526.0 [M+H]+ (anal. calcd. for C24H24FN7O6, m/z = 525.5). [α]D20 = −62° (c 1.0, DMSO).
(2R,4S,4aS)-11-fluoro-2,4-dimethyl-8-((S)-4-methyl-2-oxooxazolidin-3-yl)-2’,4’,6’-trioxo-1,1’,2,3’,4,4a,4’,6’-octahydro-2’H,3H,6H-spiro[isoxazolo[4,5-g]pyrazino[1,2-a]quinoline-5,5’-pyrimidine]-3-carbonitrile (38).
Cyanic bromide (65 mg, 0.617 mmol) and K2CO3 (85 mg, 0.617 mmol) were added to a non-separated mixture of 34+34E (200 mg, 0.411 mmol) in acetone (20 mL) and resulting reaction mixture was stirred at 25 °C for 16 h. Then the solvent was removed under reduced pressure and residue was taken up in EtOAc (20 mL) and washed with water (2×10 mL). The organic phase was isolated, dried over Na2SO4, filtered and solvent removed under reduced pressure. The mixture of isomers were separated using reverse phase preparative HPLC [(10 mM ammonium acetate buffer containing 0.4% acetic acid in water):(10 mM ammonium acetate buffer containing 0.4% acetic acid in methanol), gradient, 20–60%, 15mL/min, C18 column). Compound 38 was isolated as a major component (Note: minor diastereomer was observed but was not isolated). White solid (30 mg, 14%). 1H NMR (300 MHz, DMSO-d6) δ 11.83 (s, 1H), 11.55 (s, 1H), 7.64 (s, 1H), 4.75 − 4.61 (m, 2H), 4.22 − 4.16 (m, 1H), 4.12 − 4.06 (m, 2H), 3.67 (d, J = 14.3 Hz, 1H), 3.49 (t, J = 9.0 Hz, 1H), 3.41 (dd, J = 9.3, 6.8 Hz, 1H), 3.22 (d, J = 13.1 Hz, 1H), 3.00 (d, J = 14.3 Hz, 1H), 1.42 (d, J = 5.9 Hz, 3H), 1.30 (d, J = 6.5 Hz, 3H), 1.04 (d, J = 6.7 Hz, 3H). RP-UPLC tR = 4.02 min (method 1, purity 99%); LC-MS ESI, m/z 512.2 [M+H]+ (anal. calcd. for C23H22FN7O6, m/z = 511.2). [α]D20 = −75° (c 0.4, MeOH).
11-fluoro-2,4-dimethyl-2’,4’,6’-trioxo-8-(1H-1,2,4-triazol-1-yl)-1,1’,2,3’,4,4a,4’,6’-octahydro-2’H,3H,6H-spiro[isoxazolo[4,5-g]pyrazino[1,2-a]quinoline-5,5’-pyrimidine]-3-carbonitrile (39).
To a solution of 54a (190 mg, 0.470 mmol) in 2mL (EtOH:2M HCl, 10:1), pyrimidine-2,4,6(1H, 3H, 5H)-trione (66 mg, 0.510 mmol) was added. The solution instantly turned an orange colour. The reaction mixture was heated at 80°C for 16 h. The reaction was cooled, and solvent removed to afford an orange-yellow residue. The crude residue was pre-cleaned using normal phase chromatography, by dry loading onto flash silica and purification was performed by flash chromatography using 0–10% (MeOH in DCM). The major fraction was isolated, solvent removed to afford the product as a mixture of isomers Attempts to dissolve the mixture proved difficult and thus HPLC chiral separation was not performed. The racemic mix was found to be soluble in DMF and DMSO, two solvents which would have detrimental effects to chiral chromatography separation. The title compound was isolated as a mixture of isomers (125 mg, 53%) as a pale brown solid. 1H NMR (300 MHz, DMSO-d6) δ 11.91 (s, 1H), 11.56 (s, 1H), 9.44 (s, 1H), 8.52 (s, 1H), 7.66 (s, 1H), 4.19 − 4.06 (m, 2H), 3.82 (d, J= 14.2 Hz, 1H), 3.60 − 3.39 (m, 2H), 3.01 (d,J= 14.3 Hz, 1H), 1.32 (d, J= 6.4 Hz, 3H), 1.06 (d, J= 5.6 Hz, 3H). 1 proton masked under water peak. 19F NMR (377 MHz, DMSO-d6) δ −156.30. RP-UPLC tR = 1.029 min (method 2, purity 94%); LC-MS ESI, m/z 378.1 [M-H]− (anal. calcd. for C21H18FN9O4, m/z = 379.1).
(2R,4S,4aS)-11-fluoro-8-((S)-3-hydroxypyrrolidin-1-yl)-2,4-dimethyl-2’,4’,6’-trioxo-1,1’,2,3’,4,4a,4’,6’-octahydro-2’H,3H,6H-spiro[isoxazolo[4,5-g]pyrazino[1,2-a]quinoline-5,5’-pyrimidine]-3-carbonitrile (40).
Prepared following the preparation of 8 using 54b (433 mg, 1.00 mmol) and pyrimidine-2,4,6(1H, 3H, 5H)-trione (141 mg, 1.01 mmol). Chiral column chromatography using hexane:EtOH:DCM (60:30:10, isocratic, 15mL/min, Daicel IA column). Off-white solid (132 mg, 27%). 1H NMR (300 MHz, DMSO-d6) δ 11.88 (s, 1H), 11.51 (s, 1H), 7.37 (s, 1H), 4.41 (d, J = 4.7 Hz, 1H), 4.06 (dd, J = 17.0, 11.7 Hz, 2H), 3.61 − 3.31 (m, 7H), 3.30 − 3.07 (m, 1H), 2.98 (d, J = 14.1 Hz, 1H), 2.16 − 1.79 (m, 2H), 1.29 (d, J = 6.4 Hz, 3H), 1.04 (d, J = 6.7 Hz, 3H). 13C NMR (101 MHz, MeOD) δ 172.21, 169.40, 160.01, 150.97, 137.96, 135.88, 135.57, 122.44, 117.33, 114.94, 111.12, 71.74, 65.06, 57.93, 56.51, 55.97, 47.86, 41.15, 35.04, 30.86, 24.41, 16.77, 16.44. RP-UPLC tR = 2.763 min (method 1, purity 71%); LC-MS ESI, m/z 496.1 [M-H]− (anal. calcd. for C23H24FN7O5, m/z = 497.5). [α]D 20 = −115° (c 0.26, MeOH).
(2R,4S,4aS)-11-fluoro-8-((R)-3-hydroxypyrrolidin-1-yl)-2,4-dimethyl-2’,4’,6’-trioxo-1,1’,2,3’,4,4a,4’,6’-octahydro-2’H,3H,6H-spiro[isoxazolo[4,5-g]pyrazino[1,2-a]quinoline-5,5’-pyrimidine]-3-carbonitrile (41).
Prepared following the preparation of 8 using 54c (236 mg, 0.549 mmol) and pyrimidine-2,4,6(1H, 3H, 5H)-trione (77 mg, 0.604 mmol). Isomers were separated using reverse phase preparative HPLC [(10 mM ammonium acetate buffer containing 0.4% acetic acid in water):(10 mM ammonium acetate buffer containing 0.4% acetic acid in methanol), gradient, 20–60%, 15mL/min, C18 column). Compound 41 was isolated as a major component. (Note: minor diastereomer was observed but was not isolated). White solid (141 mg, 52%). 1H NMR (300 MHz, DMSO-d6) δ 11.88 (d, J = 1.6 Hz, 1H), 11.51 (t, J = 1.7 Hz, 1H), 7.43 − 7.32 (m, 1H), 4.54 − 4.29 (m, 1H), 4.15 − 3.96 (m, 2H), 3.69 − 3.30 (m, 7H), 3.25 − 3.13 (m, 1H), 2.98 (d, J = 14.1 Hz, 1H), 2.11 − 1.83 (m, 2H), 1.29 (d, J = 6.4 Hz, 3H), 1.04 (d, J = 6.7 Hz, 3H). RP-UPLC tR = 2.394 min (method 1, purity 100%); LC-MS ESI, m/z 495.8 [M-H]− (anal. calcd. for C23H24FN7O5, m/z = 497.5). [α]D20 = −45° (c 0.24, MeOH).
(2R,4S,4aS)-11-fluoro-2,4-dimethyl-2’,4’,6’-trioxo-8-(((S)-5-oxopyrrolidin-2-yl)methoxy)-1,1’,2,3’,4,4a,4’,6’-octahydro-2’H,3H,6H-spiro[isoxazolo[4,5-g]pyrazino[1,2-a]quinoline-5,5’-pyrimidine]-3-carbonitrile (42).
Prepared following the preparation of 8 using 54d (312 mg, 0.679 mmol) and pyrimidine-2,4,6(1H, 3H, 5H)-trione (96 mg, 0.747 mmol). Chiral column chromatography using hexane:EtOH:IPA (45:35:20, isocratic, 15mL/min, Daicel IA column). Off-white solid (35 mg, 9.3%). 1H NMR (600 MHz, DMSO-d6) δ 7.85 (s, 1H), 7.12 (d, J = 1.2 Hz, 1H), 4.26 (ddd, J = 42.2, 10.3, 4.7 Hz, 2H), 4.09 (d, J = 9.3 Hz, 1H), 4.03 − 3.98 (m, 1H), 3.95 (dd, J= 8.3, 4.4 Hz, 1H), 3.52 (d, J = 14.2 Hz, 1H), 3.48 − 3.41 (m, 1H), 3.41 − 3.36 (m, 1H), 3.22 3.16 (m, 1H), 2.90 (dd, J = 14.2, 1.5 Hz, 1H), 2.29 − 2.21 (m, 1H), 2.21 − 2.09 (m, 2H), 1.91 − 1.85 (m, 1H), 1.26 (d, J = 6.5 Hz, 3H), 1.00 (d, J = 6.7 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 177.38, 171.88, 169.04, 166.32, 152.96 (d, J = 12.3 Hz), 151.13, 135.63, 134.55 (d, J = 240.8 Hz), 123.81, 114.38, 113.69, 106.47, 73.22, 63.51, 55.88 (d, J = 7.2 Hz), 54.98, 54.36, 54.20, 52.56, 39.06, 30.07, 23.13, 16.28, 16.23. RP-UPLC tR = 0.888 min (method 2, purity 100%); LC-MS ESI, m/z 524.2 [M-H]− (anal. calcd. for C24H24FN7O6, m/z = 525.5). [α]D20 = −122° (c 0.23, MeOH).
(2S,4R,4aR)-11-fluoro-2,4-dimethyl-8-(((S)-5-oxopyrrolidin-2-yl)methoxy)-2,3,4,4a-tetrahydro-1H,2’H,6H-spiro[isoxazolo[4,5-g]pyrido[1,2-a]quinoline-5,5’-pyrimidine]-2’,4’,6’(1’H,3’H)-trione (43).
Prepared following the preparation of 8 using 54e (371 mg, 0.855 mmol) and pyrimidine-2,4,6(1H, 3H, 5H)-trione (120 mg, 0.941 mmol). Chiral column chromatography using hexane:EtOH:EtOAc (65:10:25, isocratic, 15mL/min, Daicel IC column). White solid (52 mg, 12%). 1H NMR (300 MHz, DMSO-d6) δ 7.87 (s, 1H), 7.07 (s, 1H), 4.28 (qd, J = 10.4, 4.7 Hz, 2H), 4.06 − 3.88 (m, 2H), 3.78 (d, J = 9.9 Hz, 1H), 3.54 (d, J = 13.7 Hz, 1H), 2.95 − 2.82 (m, 2H), 2.35 2.09 (m, 3H), 1.99 − 1.70 (m, 4H), 0.90 (d, J = 6.4 Hz, 3H), 0.64 (d, J = 6.4 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 178.24, 172.76, 169.21, 167.20, 154.11 (d, J = 12.7 Hz), 150.98, 137.69, 135.23 (d, J = 240.0 Hz), 124.64, 114.99, 106.15, 74.02, 67.49, 59.43 (d, J = 8.9 Hz), 55.49, 53.47, 44.32, 39.81, 33.75, 32.71, 30.96, 24.05, 20.02, 19.74. RP-UPLC tR = 1.028 min (method 2, purity 100%); LC-MS ESI, m/z 498.2 [M-H]− (anal. calcd. for C24H26FN6O6, m/z = 499.5). [α]D20 = −154° (c 0.24, MeOH).
(2R,4S,4aS)-11-fluoro-2,4-dimethyl-8-(5-methyl-1,3,4-oxadiazol-2-yl)-2’,4’,6’-trioxo-1,1’,2,3’,4,4a,4’,6’-octahydro-2’H,3H,6H-spiro[isoxazolo[4,5-g]pyrazino[1,2-a]quinoline-5,5’-pyrimidine]-3-carbonitrile (44).
A mixture of 57 (350 mg, 0.963 mmol) and pyrimidine-2,4,6(1H, 3H, 5H)-trione (123 mg, 0.963 mmol) in ethanol (4 mL) was stirred for 20 h at 110 °C. The solvents were removed, and the reaction mixture was precleaned by flash chromatography on silica gel, using a 4g column (over 16min) and a linear gradient 0–10% (DCM:MeOH). The major band was isolated and contained a mixture of isomers. The mixture was separated using chiral chromatography hexane:MeOH (20:80), isocratic, 15mL/min, Daicel IC column. Compound 44 was isolated as a white solid (64 mg, 10%). 1H NMR (300 MHz, DMSO-d6) δ 11.72 (s, 2H), 7.70 (s, 1H), 4.21 − 4.07 (m, 2H), 3.86 (d, J = 14.3 Hz, 1H), 3.60 − 3.49 (m, 1H), 3.49 − 3.39 (m, 1H), 3.25 (s, 1H), 3.01 (d, J = 14.2 Hz, 1H), 2.68 (s, 3H), 1.32 (d, J = 6.4 Hz, 3H), 1.10 − 1.01 (m, 3H). 13C NMR (101 MHz, DMSO-d6) δ 171.09, 168.41, 165.81, 156.67, 153.32 (d, J = 12.8 Hz), 150.11, 144.98, 136.14, 134.19 (d, J = 241.8 Hz), 126.32, 116.11, 113.58, 111.65, 63.59, 55.92 (d, J = 8.5 Hz), 54.98, 54.36, 54.18, 38.74, 16.34, 16.25, 11.08. 19F NMR (377 MHz, DMSO-d6) δ −155.40. RP-UPLC tR = 0.981 min (method 2, purity 100%); LC-MS ESI, m/z 493.1 [M-H]− (anal. calcd. for C22H19FN8O5, m/z = 494.4). [α]D20 = −148° (c 0.11, MeOH).
(2R,3S,4S,4aR)-11-fluoro-3-hydroxy-2,4-dimethyl-8-(5-methyl-1,3,4-oxadiazol-2-yl)-2,3,4,4a-tetrahydro-1H,2’H,6H-spiro[isoxazolo[4,5-g]pyrido[1,2-a]quinoline-5,5’-pyrimidine]-2’,4’,6’(1’H,3’H)-trione (45).
Prepared following the preparation of 8 using 58 (200 mg, 0.534 mmol) and pyrimidine-2,4,6(1H, 3H, 5H)-trione (68 mg, 0.534 mmol). Chiral column chromatography using hexane:EtOH:EtOAc (70:14:16, isocratic, 15mL/min, Daicel IC column). Light yellow solid (27 mg, 10%). 1H NMR (300 MHz, DMSO-d6) δ 11.62 (brs, 2H), 7.60 (s, 1H), 4.73 (d, J = 7.6 Hz, 1H), 3.98 − 3.93 (m, 2H), 3.74 (d, J = 14.3 Hz, 1H), 3.01 (t, J = 13.1 Hz, 1H), 2.91 (d, J = 14.2 Hz, 1H), 2.75 (q, J = 9.24 Hz, 1H), 2.68 (s, 3H), 1.72 − 1.63 (m, 2H), 1.01 (d, J= 6.4 Hz, 3H), 0.76 (d, J = 6.6 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 171.82, 168.46, 165.72, 156.77, 153.54 (d, J = 12.6 Hz), 150.13, 144.78, 137.02, 133.89 (d, J = 241.63 Hz), 126.64, 115.72, 110.49, 78.92, 64.95, 55.86 (d, J = 8.8 Hz), 54.54, 40.90, 38.77, 15.58, 14.78, 11.07. Note: one of the peak is for two carbons. 19F NMR (377 MHz, DMSO-d6) δ −157.66. RP-UPLC tR = 0.939 min (method 2, purity 99%); LC-MS ESI, m/z 485.1 [M+H]+ (anal. calcd. for C22H21FN6O6, m/z = 484.4). [α]D20 = −190° (c 0.19, THF).
Supplementary Material
ACKNOWLEDGMENT
The Mtb DNA gyrase assay was carried out by Inspiralis, Ltd. of Norfolk, United Kingdom with the experimental method described in the Supplementary Information. The authors wish to acknowledge Tando Ntsabo of H3D for the routine MIC screening; Dr Dale Taylor, Mr Virgil Verhoog and Ms Sumaya Salie for cytotoxicity data; Dr Mathew Njoroge, Dr Nina Lawrence, Ms Nesia Barnes. Mr Warren Oliphant, and Mr Ronald Olkers for logD and solubility determinations; Marianna de Kock and Claudia Spies, SAMRC and Stellenbosch University, South Africa for excellent technical assistance in generating MICs against clinical TB isolates. Lina Castro, Aaron Korkegian and Anu Kumar from Infectious Disease Research Institute for technical assistance and support.
Funding Sources
The project was funded through Global Health Grants (Number OPP1066878 and OPP1024038) received from the Bill and Melinda Gates Foundation, the Division of Intramural Research of the NIAID/NIH and the Strategic Health Innovation Partnerships (SHIP) unit of the South African Medical Research Council (SAMRC). Additional support from the University of Cape Town and the South African Research Chairs Initiative of the Department of Science and Technology, administered through the South African National Research Foundation, are gratefully acknowledged (K.C.). N.O. was funded was supported by the U.S. Veterans Administration Merit Review award I01Bx002198 and the National Institutes of Health (R01 GM126363)
ABBREVIATIONS
- ADC
albumin dextrose catalase
- ATc
anhydrotetracycline
- DAD
diode array detector
- FQ
fluoroquinolone
- INH
isoniazid
- MDR
multidrug resistant
- MGIT
mycobacteria growth indicator tube
- MIC
minimum inhibitory concentration
- MoA
mode-of-action
- MoI
mode-of-inhibition
- Mtb
Mycobacterium tuberculosis
- MW
molecular weight
- NBTIs
novel bacterial topoisomerase inhibitors
- NMR
nuclear magnetic resonance
- PDA
photodiode array
- QRDR
quinoline resistance-determining region
- RIF
rifampicin
- RR-TB
rifampicin-resistant-TB
- SAR
structure-activity relationship
- RMSD
root mean square deviation
- SPT
spiropyrimidinetrione
- T-reaction
tertiary amino effect reaction
- TB
tuberculosis
- TLC
thin-layer chromatography
- TMS
tetramethylsilane
- WT
wild-type
- XDR
extensively drug resistant
Footnotes
The authors declare no competing financial interest.
Supporting information includes Figure S1 (illustrations of moxifloxacin and QPT-1 crystal structures), Figure S2 (illustration of QPT-1 docking validation in Mtb DNA gyrase), the synthesis and characterization of inter- mediates, characteriazation of inactive isomers, the experimental method for the DNA gyrase supercoiling assay, the experimental methods for in vitro ADMET assays, sequence alignments of GyrA and GyrB (M. tuberculosis, S. aureus and E. coli), and QC data (NMR and UPLC-MS) for selected compounds (PDF)Coordinate files for Mtb gyrase binding models of compounds 5, 6, 10 and 42 (PDB).
Coordinate files for Mtb gyrase binding models of compound 5 (PDB)
Coordinate files for Mtb gyrase binding models of compound 6 (PDB)
Coordinate files for Mtb gyrase binding models of compound 10 (PDB)
Coordinate files for Mtb gyrase binding models of compound 42 (PDB)
Molecular formula strings and some data (CSV).
REFERENCES
- (1).Tuberculosis: Key facts. https://www.who.int/news-room/fact-sheets/detail/tuberculosis (accessed February 14, 2022).
- (2).New TB drugs. https://tbfacts.org/new-tb-drugs/ (accessed February 14, 2022).
- (3).Bandodkar B; Shandil RK; Bhat J; Balganesh TS, Two decades of TB drug discovery efforts—what have we learned? Applied Sciences 2020, 10, 5704. [Google Scholar]
- (4).Singh V; Chibale K, Strategies to combat multi-drug resistance in tuberculosis. Acc. Chem. Res 2021, 54, 2361–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Aldred KJ; Kerns RJ; Osheroff N, Mechanism of quinolone action and resistance. Biochemistry 2014, 53, 1565–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Gibson EG; Ashley RE; Kerns RJ; Osheroff N, Fluoroquinolone interactions with bacterial type II topoisomerases and target-mediated drug resistance. In Antimicrobial Resistance in the Twenty-first Century, Fong B; Shlaes D; Drlica K, Eds. Springer: New York, 2018; pp 507–529. [Google Scholar]
- (7).Bax BD; Murshudov G; Maxwell A; Germe T, DNA topoisomerase inhibitors: trapping a DNA-cleaving machine in motion. J. Mol. Biol 2019, 431, 3427–3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Aubry A; Fisher LM; Jarlier V; Cambau E, First functional characterization of a singly expressed bacterial type II topoisomerase: the enzyme from Mycobacterium tuberculosis. Biochem. Biophys. Res. Commun 2006, 348, 158–165. [DOI] [PubMed] [Google Scholar]
- (9).Cole ST; Brosch R; Parkhill J; Garnier T; Churcher C; Harris D; Gordon SV; Eiglmeier K; Gas S; Barry CE; Tekaia F; Badcock K; Basham D; Brown D; Chillingworth T; Connor R; Davies R; Devlin K; Feltwell T; Gentles S; Hamlin N; Holroyd S; Hornsby T; Jagels K; Krogh A; McLean J; Moule S; Murphy L; Oliver K; Osborne J; Quail MA; Rajandream MA; Rogers J; Rutter S; Seeger K; Skelton J; Squares R; Squares S; Sulston JE; Taylor K; Whitehead S; Barrell BG, Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. [DOI] [PubMed] [Google Scholar]
- (10).Basarab GS, Four Ways to Skin a Cat: Inhibition of Bacterial Topoisomerases Leading to the Clinic. In Antibacterials: Volume I, Fisher JF; Mobashery S; Miller MJ, Eds. Springer International Publishing: Cham: 2018; pp 165–188. [Google Scholar]
- (11).Moadebi S; Harder CK; Fitzgerald MJ; Elwood KR; Marra F, Fluoroquinolones for the treatment of pulmonary tuberculosis. Drugs 2007, 67, 2077–2099. [DOI] [PubMed] [Google Scholar]
- (12).Dawson R; Diacon AH; Everitt D; van Niekerk C; Donald PR; Burger DA; Schall R; Spigelman M; Conradie A; Eisenach K; Venter A; Ive P; Page-Shipp L; Variava E; Reither K; Ntinginya NE; Pym A; von Groote-Bidlingmaier F; Mendel CM, Efficiency and safety of the combination of moxifloxacin, pretomanid (PA-824), and pyrazinamide during the first 8 weeks of antituberculosis treatment: a phase 2b, open-label, partly randomised trial in patients with drug-susceptible or drug-resistant pulmonary tuberculosis. Lancet 2015, 385, 1738–1747. [DOI] [PubMed] [Google Scholar]
- (13).Bisacchi GS; Manchester JI, A new-class antibacterial—Almost. Lessons in drug discovery and development: A critical analysis of more than 50 years of effort toward ATPase inhibitors of DNA gyrase and topoisomerase IV. ACS Infectious Diseases 2015, 1, 4–41. [DOI] [PubMed] [Google Scholar]
- (14).Kale RR; Kale MG; Waterson D; Raichurkar A; Hameed SP; Manjunatha MR; Kishore Reddy BK; Malolanarasimhan K; Shinde V; Koushik K; Jena LK; Menasinakai S; Humnabadkar V; Madhavapeddi P; Basavarajappa H; Sharma S; Nandishaiah R; Mahesh Kumar KN; Ganguly S; Ahuja V; Gaonkar S; Naveen Kumar CN; Ogg D; Boriack-Sjodin PA; Sambandamurthy VK; de Sousa SM; Ghorpade SR, Thiazolopyridone ureas as DNA gyrase B inhibitors: optimization of antitubercular activity and efficacy. Bioorg. Med. Chem. Lett 2014, 24, 870–879. [DOI] [PubMed] [Google Scholar]
- (15).Kale MG; Raichurkar A; Hameed PS; Waterson D; McKinney D; Manjunatha MR; Kranthi U; Koushik K; Jena LK; Shinde V; Rudrapatna S; Barde S; Humnabadkar V; Madhavapeddi P; Basavarajappa H; Ghosh A; Ramya VK; Guptha S; Sharma S; Vachaspati P; Kumar KNM; Giridhar J; Reddy J; Panduga V; Ganguly S; Ahuja V; Gaonkar S; Kumar CNN; Ogg D; Tucker JA; Boriack-Sjodin PA; de Sousa SM; Sambandamurthy VK; Ghorpade SR, Thiazolopyridine ureas as novel antitubercular agents acting through inhibition of DNA gyrase B. J. Med. Chem 2013, 56, 8834–8848. [DOI] [PubMed] [Google Scholar]
- (16).Locher CP; Jones SM; Hanzelka BL; Perola E; Shoen CM; Cynamon MH; Ngwane AH; Wiid IJ; van Helden PD; Betoudji F; Nuermberger EL; Thomson JA, A novel inhibitor of gyrase B is a potent drug candidate for treatment of tuberculosis and nontuberculosis mycobacterial infections. Antimicrob. Agents Chemother 2015, 59, 1455–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Gibson EG; Bax B; Chan PF; Osheroff N, Mechanistic and structural basis for the actions of the antibacterial gepotidacin against Staphylococcus aureus gyrase. ACS Infect. Dis 2019, 5, 570–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).O’Riordan W; Tiffany C; Scangarella-Oman N; Perry C; Hossain M; Ashton T; Dumont E, Efficacy, safety, and tolerability of gepotidacin (GSK2140944) in the treatment of patients with suspected or confirmed gram-positive acute bacterial skin and skin structure infections. Antimicrob. Agents Chemother 2017, 61, e02095–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Hameed PS; Patil V; Solapure S; Sharma U; Madhavapeddi P; Raichurkar A; Chinnapattu M; Manjrekar P; Shanbhag G; Puttur J; Shinde V; Menasinakai S; Rudrapatana S; Achar V; Awasthy D; Nandishaiah R; Humnabadkar V; Ghosh A; Narayan C; Ramya VK; Kaur P; Sharma S; Werngren J; Hoffner S; Panduga V; Kumar CNN; Reddy J; Kumar Kn M; Ganguly S; Bharath S; Bheemarao U; Mukherjee K; Arora U; Gaonkar S; Coulson M; Waterson D; Sambandamurthy VK; de Sousa SM, Novel N-linked aminopiperidine-based gyrase inhibitors with improved hERG and in vivo efficacy against Mycobacterium tuberculosis. J. Med. Chem 2014, 57, 4889–4905. [DOI] [PubMed] [Google Scholar]
- (20).Hameed PS; Manjrekar P; Raichurkar A; Shinde V; Puttur J; Shanbhag G; Chinnapattu M; Patil V; Rudrapatana S; Sharma S; Kumar CNN; Nandishaiah R; Madhavapeddi P; Sriram D; Solapure S; Sambandamurthy VK, Left-hand side exploration of novel bacterial topoisomerase inhibitors to improve selectivity against hERG binding. ACS Med. Chem. Lett 2015, 6, 741–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Blanco D; Perez-Herran E; Cacho M; Ballell L; Castro J; González del Río R; Lavandera JL; Remuiñán MJ; Richards C; Rullas J; Vázquez-Muñiz MJ; Woldu E; Zapatero-González MC; Angulo-Barturen I; Mendoza A; Barros D, Mycobacterium tuberculosis gyrase inhibitors as a new class of antitubercular drugs. Antimicrob. Agents Chemother 2015, 59, 1868–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Gibson EG; Blower TR; Cacho M; Bax B; Berger JM; Osheroff N, Mechanism of action of Mycobacterium tuberculosis gyrase Inhibitors: a novel class of gyrase poisons. ACS Infect. Dis 2018, 4, 1211–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Bradford PA; Miller AA; O’Donnell J; Mueller JP, Zoliflodacin: An oral spiropyrimidinetrione antibiotic for the treatment of Neisseria gonorrheae, including multi-drug-resistant isolates. ACS Infect. Dis 2020, 6, 1332–1345. [DOI] [PubMed] [Google Scholar]
- (24).Basarab GS; Ghorpade S; Gibhardt L; Muller R; Njoroge M; Peton N; Govender P; Massoudi LM; Robertson GT; Lenaerts AJ; Boshoff HIM; Joerss D; Parish T; Durand-Reville TF; Perros M; Singh V; Chibale K, Spiropyrimidinetriones: a class of DNA gyrase inhibitors with activity against Mycobacterium tuberculosis and without cross-resistance to fluoroquinolones. Antimicrob. Agents Chemother 2021, 66, e02192–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Eribo OA; du Plessis N; Ozturk M; Guler R; Walzl G; Chegou NN, The gut microbiome in tuberculosis susceptibility and treatment response: guilty or not guilty? Cell. Mol 2020, 77, 1497–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Chan PF; Srikannathasan V; Huang J; Cui H; Fosberry AP; Gu M; Hann MM; Hibbs M; Homes P; Ingraham K; Pizzollo J; Shen C; Shillings AJ; Spitzfaden CE; Tanner R; Theobald AJ; Stavenger RA; Bax BD; Gwynn MN, Structural basis of DNA gyrase inhibition by antibacterial QPT-1, anticancer drug etoposide and moxifloxacin. Nat. Commun 2015, 6, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Blower TR; Williamson BH; Kerns RJ; Berger JM, Crystal structure and stability of gyrase–fluoroquinolone cleaved complexes from mycobacterium tuberculosis. Proc. Natl. Acad. Sci 2016, 113, 1706–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Hooper DC; Jacoby GA, Mechanisms of drug resistance: quinolone resistance. Ann. N. Y. Acad. Sci 2015, 1354, 12–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Avalos E; Catanzaro D; Catanzaro A; Ganiats T; Brodine S; Alcaraz J; Rodwell T, Frequency and geographic distribution of gyrA and gyrB mutations associated with fluoroquinolone resistance in clinical Mycobacterium tuberculosis isolates: a systematic review. PLoS One 2015, 10, e0120470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Srikannathasan V; Wohlkonig A; Shillings A; Singh O; Chan PF; Huang J; Gwynn MN; Fosberry AP; Homes P; Hibbs M; Theobald AJ; Spitzfaden C; Bax BD, Crystallization and initial crystallographic analysis of covalent DNA-cleavage complexes of Staphyloccocus aureus DNA gyrase with QPT-1, moxifloxacin and etoposide. Acta Cryst. 2015, F71, 1242–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Friesner RA; Banks JL; Murphy RB; Halgren TA; Klicic JJ; Mainz DT; Repasky MP; Knoll EH; Shelley M; Perry JK; Shaw DE; Francis P; Shenkin PS, Glide: a new approach for rapid, accurate docking and scoring. 1. method and assessment of docking accuracy. J. Med. Chem 2004, 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- (32).Halgren TA; Murphy RB; Friesner RA; Beard HS; Frye LL; Pollard WT; Banks JL, Glide: a new approach for rapid, accurate docking and scoring. 2. enrichment factors in database screening. J. Med. Chem 2004, 47, 1750–1759. [DOI] [PubMed] [Google Scholar]
- (33).Briones JF; Basarab GS, Expedient synthesis of tetrahydroquinoline-3-spirohydantoin derivatives via the lewis acid-catalyzed tert-amino effect reaction. Chem. Comm 2016, 52, 8541–8544. [DOI] [PubMed] [Google Scholar]
- (34).Bravian K; Basarab GS; Gowravaram MR; Hauck SI; Zhou F Fused, spirocyclic heteroaromatic componds for the treatment of bacterial infections. WO 2010/043893 A1, 2010.
- (35).Bridges AJ; Lee A; Maduakor EC; Schwartz CE, Fluorine as an ortho-directing group in aromatic metalation: Generality of the reaction and the high position of fluorine in the Dir-Met potency scale. Tetrahedron Lett. 1992, 33, 7495–7498. [Google Scholar]
- (36).Basarab GS; Brassil P; Doig P; Galullo V; Haimes HB; Kern G; Kutschke A; McNulty J; Schuck VJA; Stone G; Gowravaram MR, Novel DNA gyrase inhibiting spiropyrimidinetriones with a benzisoxazole scaffold: SAR and in vivo characterization. J. Med. Chem 2014, 57, 9078–9095. [DOI] [PubMed] [Google Scholar]
- (37).Basarab GS; Doig P; Galullo V; Kern G; Kimzey A; Kutschke A; Newman JP; Morningstar M; Mueller J; Otterson L; Vishwanathan K; Zhou F; Gowravaram M, Discovery of novel DNA gyrase inhibiting spiropyrimidinetriones: benzisoxazole fusion with N-linked oxazolidinone substituents leading to a clinical candidate (ETX0914). J. Med. Chem 2015, 58, 6264–6282. [DOI] [PubMed] [Google Scholar]
- (38).Basarab GS; Gowravaram MR; Hauck SI; Zhou F Preparation of spiro[oxazino[4,3-a]isoxazolo[4,5-g]quinoline- pyrimidine]trione. Compounds and methods for treating bacterial infections. US 20140206677, 2014.
- (39).Ruble JC; Hurd AR; Johnson TA; Sherry DA; Barbachyn MR; Toogood PL; Bundy GL; Graber DR; Kamilar GM, Synthesis of (−)-PNU-286607 by asymmetric cyclization of alkylidene barbiturates. J. Am. Chem. Soc 2009, 131, 3991–3997. [DOI] [PubMed] [Google Scholar]
- (40).Basarab GS; Galullo V; DeGrace N; Hauck S; Joubran C; Wesolowski SS, Synthesis of a tetrahydronaphthyridine spiropyrimidinetrione DNA gyrase inhibiting antibacterial agent - differential substitution at all five carbon atoms of pyridine. Org. Lett 2014, 16, 6456–6459. [DOI] [PubMed] [Google Scholar]
- (41).Basarab GS; Kern GH; McNulty J; Mueller JP; Lawrence K; Vishwanathan K; Alm RA; Barvian K; Doig P; Galullo V; Gardner H; Gowravaram M; Huband M; Kimzey A; Morningstar M; Kutschke A; Lahiri SD; Perros M; Singh R; Schuck VJA; Tommasi R; Walkup G; Newman JV, Responding to the challenge of untreatable gonorrhea: ETX0914, a first-in-class agent with a distinct mechanism-of-action against bacterial type II topoisomerases. Sci. Rep 2015, 5, 11827–11840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Le Manach C; Dam J; Woodland JG; Kaur G; Khonde LP; Brunschwig C; Njoroge M; Wicht KJ; Horatscheck A; Paquet T; Boyle GA; Gibhard L; Taylor D; Lawrence N; Yeo T; Mok S; Eastman RT; Dorjsuren D; Talley DC; Guo H; Simeonov A; Reader J; van der Watt M; Erlank E; Venter N; Zawada JW; Aswat A; Nardini L; Coetzer TL; Lauterbach SB; Bezuidenhout BC; Theron A; Mancama D; Koekemoer LL; Birkholtz L-M; Wittlin S; Delves M; Ottilie S; Winzeler EA; von Geldern TW; Smith D; Fidock DA; Street LJ; Basarab GS; Duffy J; Chibale K, Identification and profiling of a novel diazaspiro[3.4]octane chemical series active against multiple stages of the human malaria parasite Plasmodium falciparum and optimization efforts. J. Med. Chem 2021, 64, 2291–2309. [DOI] [PubMed] [Google Scholar]
- (43).Zgurskaya HI; Rybenkov VV, Permeability barriers of gram-negative pathogens. Ann. N. Y. Acad. Sci 2019, 1459, 5–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Schindler BD; Kaatz GW, Multidrug efflux pumps of gram-positive bacteria. Drug Resist. Updat 2016, 27, 1–13. [DOI] [PubMed] [Google Scholar]
- (45).Sharapova A; Ol’khovich M; Blokhina S; Zhirova E; Perlovich G, Solubility and partition behavior of moxifloxacin: experimental results and thermodynamics properties. J. Mol. Liq 2021, 339, 116814–116826. [Google Scholar]
- (46).Johnson EO; LaVerriere E; Office E; Stanley M; Meyer E; Kawate T; Gomez JE; Audette RE; Bandyopadhyay N; Betancourt N; Delano K; Da Silva I; Davis J; Gallo C; Gardner M; Golas AJ; Guinn KM; Kennedy S; Korn R; McConnell JA; Moss CE; Murphy KC; Nietupski RM; Papavinasasundaram KG; Pinkham JT; Pino PA; Proulx MK; Ruecker N; Song N; Thompson M; Trujillo C; Wakabayashi S; Wallach JB; Watson C; Ioerger TR; Lander ES; Hubbard BK; Serrano-Wu MH; Ehrt S; Fitzgerald M; Rubin EJ; Sassetti CM; Schnappinger D; Hung DT, Large-scale chemical–genetics yields new M. tuberculosis inhibitor classes. Nature 2019, 571, 72–78. [DOI] [PubMed] [Google Scholar]
- (47).Aldred KJ; McPherson SA; Wang P; Kerns RJ; Graves DE; Turnbough CL Jr.; Osheroff N, Drug interactions with Bacillus anthracis topoisomerase IV: biochemical basis for quinolone action and resistance. Biochemistry 2012, 51, 370–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Iacobino A; Piccaro G; Pardini M; Fattorini L; Giannoni F, Moxifloxacin activates the SOS response in Mycobacterium tuberculosis in a dose- and time-dependent manner. Microorganisms 2021, 9, 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Aldred KJ; Blower TR; Kerns RJ; Berger JM; Osheroff N, Fluoroquinolone interactions with mycobacterium tuberculosis gyrase: enhancing drug activity against wild-type and resistant gyrase. Proc. Natl. Acad. Sci 2016, 113, E839–E846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Singh V; Donini S; Pacitto A; Sala C; Hartkoorn RC; Dhar N; Keri G; Ascher DB; Mondésert G; Vocat A; Lupien A; Sommer R; Vermet H; Lagrange S; Buechler J; Warner DF; McKinney JD; Pato J; Cole ST; Blundell TL; Rizzi M; Mizrahi V, The inosine monophosphate dehydrogenase, GuaB2, is a vulnerable new bactericidal drug target for tuberculosis. ACS Infect. Dis 2017, 3, 5–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Schrödinger Release2021–1: Maestro, Schrödinger, LLC: New York, NY, 2021. [Google Scholar]
- (52).Sastry GM; Adzhigirey M; Day T; Annabhimoju R; Sherman W, Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided. Mol. Des 2013, 27, 221–34. [DOI] [PubMed] [Google Scholar]
- (53).Jacobson MP; Friesner RA; Xiang Z; Honig B, On the role of the crystal environment in determining protein side-chain conformations. J. Mol. Biol 2002, 320, 597–608. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.