

Abstract
Ultrasound imaging presents many positive attributes, including safety, real-time imaging, universal accessibility, and cost. However, inherent difficulties in discrimination between soft tissues and tumors prompted development of stabilized microbubble contrast agents. This presents the opportunity to develop agents in which drug is entrapped in the microbubble shell. We describe preparation and characterization of theranostic poly(lactide) (PLA) and pegylated PLA (PEG-PLA) shelled microbubbles that entrap gemcitabine, a commonly used drug for pancreatic cancer (PDAC). Entrapping 6 wt% gemcitabine did not significantly affect drug activity, microbubble morphology, or ultrasound contrast activity compared with unmodified microbubbles. In vitro microbubble concentrations yielding ≥ 500nM entrapped gemcitabine were needed for complete cell death in MIA PaCa-2 PDAC drug sensitivity assays, compared with 62.5 nM free gemcitabine. In vivo administration of gemcitabine-loaded microbubbles to xenograft MIA PaCa-2 PDAC tumors in athymic mice was well tolerated and provided substantial tumoral image enhancement before and after destructive ultrasound pulses. However, no significant differences in tumor growth were observed among treatment groups, in keeping with the in vitro observation that much higher doses of gemcitabine are required to mirror free gemcitabine activity.
Keywords: Polymer microbubbles, Ultrasound, Targeted drug delivery, Pancreatic adenocarcinoma, Gemcitabine
1. Introduction
According to the American Cancer Society, pancreatic cancer remains one of the deadliest cancers, with predictions of close to 57,600 people (30,400 men and 27,200 women) being diagnosed in 2020, and about 45,050 deaths in the United States alone, representing roughly 3% of all cancers in the United States and about 7% of all cancer deaths [1]. Pancreatic cancer is predicted to be the leading cause of cancer death by 2030, the most common type being pancreatic ductal adenocarcinoma (PDAC), which is characterized by a thick stroma surrounding the tumor, arising from pancreatic stellate cells that are activated to proliferate and produce collagens, laminin, and fibronectin [2,3]. This dense stroma and hostile tumor microenvironment is implicated in tumor growth and metastasis, and plays a role in inhibiting access of drugs and imaging agents to tumor tissue [4,5]. The resulting increased interstitial pressure leads to constriction of the blood vessels, further restricting access of agents [6]. Even after surgical resection, the best therapeutic option, 5-year survival is only 5–35% [7,8]. In addition, nearly 80% of all patients are diagnosed with unresectable disease, leading to systemic chemotherapy as the only currently clinically-available therapeutic option for the vast majority of PDAC patients.
One of the most commonly used PDAC chemotherapies involves gemcitabine (GEM) - (2′,2′-difluorodeoxycytidine, Gemzar) either alone or in combination with other chemotherapeutics [9]. Relative to other chemotherapeutics, GEM is well tolerated, and shows modest survival (5–7 months) as a monotherapy, [10,11] which is slightly improved when combined with other adjuvant therapies such as cisplatin and nab-paclitaxel [9,11,12]. Despite its frequent use, studies have shown that GEM is associated with a marginal survival extension of approximately one month [13]. This low response rate is likely due to factors such as a short plasma half-life (8–11 minutes) due to rapid deamination by cytidine deaminase, the inability of the drug to penetrate pancreatic stroma and/or the development of resistance, and the advanced stage at which PDAC is generally diagnosed [14,15]. There have also been many attempts to overcome barriers encountered with the tumor microenvironment through anti-stromal therapies [16]. However, the initial optimism has not been realized in clinical trials, not least due to the increasingly evident, highly complex relationship between tumor cells and the tumor microenvironment [17–19]. Other approaches to targeted delivery of GEM include conjugating the drug with targeting agents [20,21] and RNA aptamers, [22,23] and use of prodrugs such as 4-(N)-stearoyl GEM [24]. Many of these approaches still involve systemic administration of toxic chemotherapeutics, which remain susceptible to degradation in the plasma, whereas encapsulation of the drug in a particulate carrier shields both the drug from degradation and the healthy tissues from exposure [25–30].
Targeted drug delivery can help administer therapeutic concentrations of drug to the intended tumor cells, while reducing systemic exposure, limiting uptake by healthy cells, and improving the overall therapeutic index [31–34]. In ultrasound-based approaches, drugs can be infused simultaneously with empty microbubbles (MBs) or contrast agents, in an effort to take advantage of numerous mechanisms whereby MB-ultrasound interaction at a membrane causes enhanced drug entry into the cell, particularly when targeting the blood brain barrier [35–41]. The safety and toxicity of this approach have been investigated in clinical trials [42].
The delivery platform discussed here is a biodegradable polymeric MB that acts as an ultrasound contrast agent by virtue of the gas core. Previously, we have demonstrated enhanced doxorubicin delivery to a hepatocellular carcinoma model using MBs to enhance drug delivery when exposed to ultrasound focused at the tumor site [43–47]. We propose that if GEM remains viable through this approach, an added advantage would be protection from cytidine deaminase degradation before being delivered directly to the tumor at the focus of an ultrasound beam (Fig. 1).
Fig. 1.
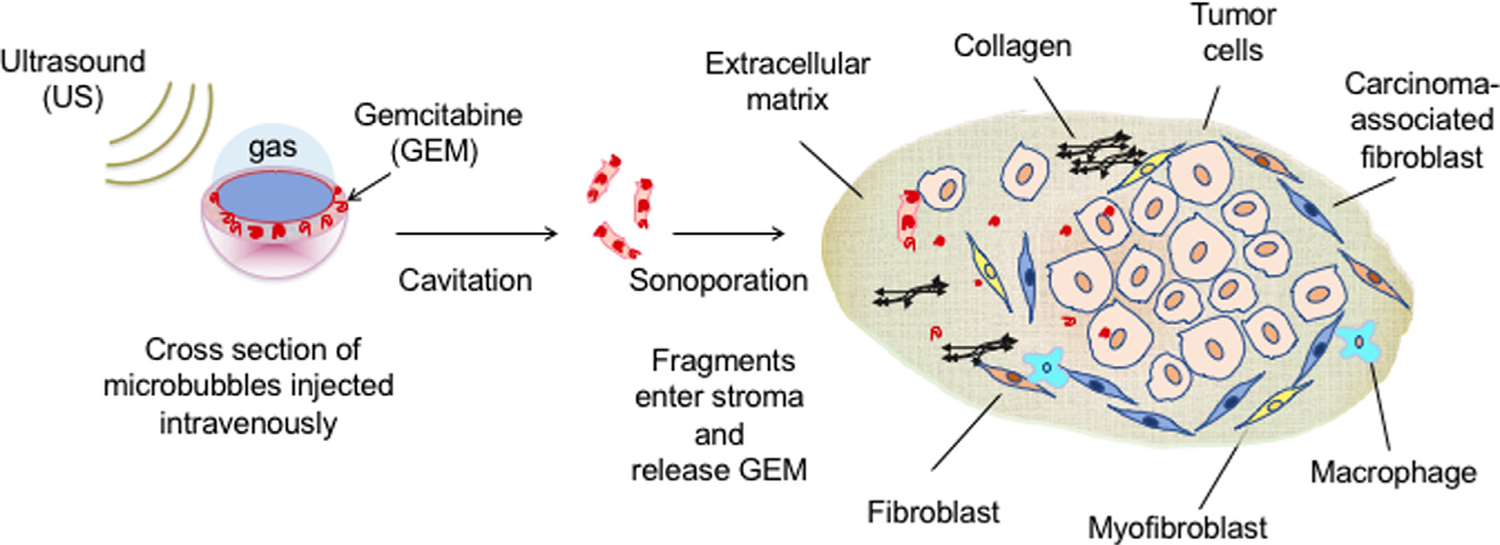
Representation of ultrasound-triggered delivery of gemcitabine to pancreatic ductal adenocarcinoma stroma via drug-loaded ultrasound contrast microbubbles (not to scale).
Controlled release of GEM encapsulated in a biodegradable polymer-shelled MB would also be an advantage for this cell cycle-dependent drug [48]. Our previous studies show that the ultrasound-generated, drug-carrying MB fragments (nano shards or nSh) can be forced through the leaky pores found in angiogenic tumor vessels (approximately 300–700 nm pore diameter) [44]. This is especially relevant when the pores in the vasculature are also enlarged by the interplay between the incident ultrasound beam and the MBs [49,50]. Here we investigate the feasibility of entrapping GEM in polylactic acid (PLA)-stabilized ultrasound contrast agents, and test the retention of drug activity compared with free GEM. We further assess the effects of inclusion of polyethylene glycol-polylactic acid (PEG-PLA) within the shell, to facilitate intravenous injection and reduce the potential for immunogenicity [47]. Finally, we investigate the ability of the MBs to be visualized penetrating the stroma-dense environment of PDAC tumors in mice and compare the results with the in vitro cell studies.
2. Materials & methods
2.1. Preparation of drug-loaded microbubbles
2.1.1. Microbubble fabrication
GEM-loaded MBs were prepared by modifying the water/oil/water (w/o/w) double emulsion process that is well-established in our lab, using camphor and ammonium carbamate as porogens [45–47]. Briefly, for PLA MBs (no PEG), 0.5 g of PLA (100 DL 7E, 118 kDa, Evonik Biomaterials, Essen, Germany) shown to be the correct choice for combined drug encapsulation and acoustic properties, [43] and 0.05 g camphor (Sigma Aldrich, St. Louis, MO) were dissolved in 10mL methylene chloride (Sigma), with 3, 6, and 10 weight percent (wt%) gemcitabine hydrochloride (GEM, Abcam, Cambridge, MA) added during the organic phase [45,47]. This counter intuitive addition of a water soluble drug to the organic phase was found to produce better results than addition to the water phase, and recapitulates previous findings using doxorubicin [45]. Then, 1 mL of 0.4 M ammonium carbamate solution was added to the organic phase and sonicated to form the first emulsion, which was then added to 50 mL of 5% w/v polyvinyl alcohol (PVA, PolySciences, Warrington, PA) solution and homogenized to create the second w/o/w emulsion. Resulting MBs were then washed with 2% v/v isopropanol (Sigma) and stirred to evaporate organic material. The MB solution was then centrifuged, washed with hexane, washed with DI water, and then flash frozen in liquid nitrogen and lyophilized (10–30 μbar) for 48 hours (Virtis, Gardiner, NY), then exposed to room air which forms the gas core of the MBs [46]. For the inclusion of PEG into the MBs, the same procedure was followed except that the 0.5 g polymer used in the first emulsion was proportionally comprised of 5 wt% PEG-PLA (100 DL mPEG 5000 6CE, 67 mol% PLA, 33 mol% PEG, 69 kDa, Evonik) and 95 wt% PLA (100 DL 7E). Of the 0.5 g total polymer mass, the proportion of PEG to PLA is 8.25 mg (1.65%) PEG to 491.75 mg (98.35%) PLA [47].
2.1.2. Encapsulation efficiency
GEM loading was determined by dissolving loaded MBs in dimethyl sulfoxide (DMSO, Sigma) and though ultraviolet-visible spectrophotometry (UV-Vis) at 296nm (Tecan Life Sciences, Mannedorf, Switzerland) and comparing with a standard curve prepared in DMSO. Encapsulation efficiency (EE %) of GEM-MBs was defined as:
2.2. Physical characterization
Surface morphology was determined by using a Philips FEI XL30 environmental scanning electron microscopy (SEM), with acceleration voltage 5 kV and spot size 3, at 5000 × magnification. Microbubble diameter, polydispersity index (PDI), and zeta potential (ζ) were recorded using a Malvern Nano ZetaSizer (Worcestershire, United Kingdom), which uses dynamic light scattering (DLS). For size and PDI, 1 mg of MBs was suspended in 1 mL of phosphate buffered saline (PBS) at a pH of 7.4 and measured at room temperature in triplicate. Zeta potential was measured in triplicate at room temperature by suspending 1 mg of MB in 1 mL of deionized (DI) water and loading into a Malvern zeta capillary cuvette. Salts retained on the dried capsules supplied the minimum required level of conductivity so that an electric field could be applied.
2.3. C3 complement activation evaluation
In order to fabricate MBs which would elicit minimum immunological response when injected intravenously, PEG-PLA was added to the MB shell and C3 complement activation was determined ex vivo under sterile conditions for the GEM-MB by methods described previously for unloaded MB [47]. Briefly, MBs were sterilized under ultraviolet (UV) irradiation for 30 minutes before a 3 mL MB suspension containing approximately 107 MBs/mL sterile PBS was mixed with 2 mL human complement-preserved serum (pooled from several human donors, Valley Biomedical, Winchester, VA) and shaken at room temperature for 30 minutes. The solution was then centrifuged at 300 G for 3 min to separate the MBs, and the supernatant was collected and diluted 1:3 with 20 mM EDTA. The amount of activated C3a for MBs containing 3 wt% and 6 wt% GEM was assayed using an ELISA kit purchased from BD Biosciences (Franklin Lakes, NJ, catalog #550499) following the manufacturer’s instructions.
2.4. Acoustic characterization
In order to gain an understanding of the effects that the various MB shell manipulations had on acoustic behavior, in vitro acoustic characterization tests were performed. Testing was performed in a custom-built acoustic setup equipped with a 5 MHz, 12.7 mm, single element ultrasound transducer (Panametrics, Waltham, MA), with 6 dB bandwidth of 91% and focal length of 50.8 mm, held at 37° C in a gas-free water bath and focused through the acoustic window of a 50 mL sample chamber [45]. The 50 mL sample chamber is a custom-made, cylindrical, 1.5-inch inner diameter, acrylic tube with a 1-inch square portion of the tube wall removed and covered with clear polypropylene tape to create an acoustic window for measuring the performance of the microbubbles. Continuous stirring via a magnetic stirrer kept the sample suspended, and blank runs in the absence of MBs confirmed that no extraneous gas was entrained due to stirring. A pulser/receiver (5072 PR, Panametrics) connected to the transducer was used to generate an acoustic pulse with pulse repetition frequency (PRF) of 100 Hz, resulting in a peak positive pressure (PPP) amplitude of 0.69 MPa and a peak negative pressure (PNP) amplitude of 0.45 MPa at the focus (equivalent to a mechanical index of 0.20), determined using a 0.5 mm polyvinylidene fluoride needle hydrophone (Precision Acoustics, Dorset, United Kingdom). Reflected signals were measured using the transducer and amplified 40 dB before being read by an oscilloscope (Lecroy 9350, Chestnut Ridge, NY). Data acquisition and processing were done using LabView 7 Express (National Instruments, Austin, TX). These tests allow for rapid and straightforward comparison of acoustic performance as the MBs are developed, in a general representation of in vivo conditions (i.e., pH 7.4, PBS buffer, 37° C, and constant movement through the acoustic beam). The choice of 5 MHz was dictated by previous studies, which determined this to be the resonance frequency (optimal for MB destruction) under these conditions [51].
Dose response curves were generated by plotting backscattering enhancement (in dB) as a function of MB concentration. Microbubbles were suspended in a mixture of 3 mg of MBs in 800 μL of PBS (pH 7.4, 37° C) and vortexed briefly. A baseline was recorded to establish the background signal of the PBS solution before acoustic testing. A cumulative dose response was constructed by adding ten 20 μL increments of MB suspension (one every 30 seconds) and recording the reflected signal after each addition. After each addition, MBs were mixed for 10 seconds to ensure a homogenous media before measurement. Enhancement in relationship to a baseline reading was plotted for each dosage ranging from 0 to 15 μg/mL in 1.5 μg/mL (20 μL aliquots) increments.
Comparison of microbubble stability in the ultrasound beam was measured by adding 4 mg/mL of MBs to the sample holder and continuously insonating with stirring, using the setup described above. Enhancement was measured over the course of 15 min and normalized with respect to the initial value to allow for inter-sample comparison.
In order to generate the ultrasound-triggered MB fragments (nSh) for in vitro cytotoxicity studies, GEM-loaded MBs were exposed to ultrasound, but using deionized water in place of PBS to avoid problems with excess salt in subsequent administration to the PDAC cells. This solution was frozen at −20° C overnight and then lyophilized for 48 hours using a Virtis Benchtop freeze drier (as described previously) to collect the nSh.
2.5. Cell studies
2.5.1. Drug sensitivity assays
In vitro cell studies were conducted using the cell lines MIA PaCa-2 (ATCC® CRL-1420™) and Panc1 (ATCC® CRL-1469™), using UV-sterilized PLA MBs. This was a classic sensitivity study in which the cells were exposed to media that contained the intact MBs or nSh, and none of the cells were directly insonated. Roughly 1000 cells/well in 96-well plates (Corning Costar, Sigma) were plated and allowed to adhere overnight. A dose escalating amount of the samples (free GEM, unloaded MBs, ultrasound-treated unloaded MBs, GEM-loaded MBs, and ultrasound-treated GEM-loaded MBs) were added to the plates (via media containing the relevant treatment), which were then incubated at 37° C for seven days, after which a PicoGreen assay (Quant-iT PicoGreen dsDNA Assay Kit, Thermo Fisher Scientific, Waltham, MA) was conducted. Concentrations between 0 and 1 μM GEM were prepared by serial dilution for all plates that were treated with free GEM.
2.5.2. Cell viability studies
To compare drug loaded MBs and free GEM cytotoxicity daily, UV-sterilized PEG-PLA MBs were used. Cells (MIA PaCa-2) were grown in a Corning™ 96-well black clear bottom plate (Thermo Fischer Scientific, Waltham, MA) in DMEM with 10% fetal bovine serum (FBS) at 37° C and 5% CO2. Seeding was performed with 1000 cells in 100 μL of media per well and allowed to adhere for 24 hours before being treated with 200 μL media or supplemented medium (unloaded MBs, ultrasound-treated unloaded MBs, GEM-loaded MBs, and ultrasound-treated GEM-loaded MBs). MBs of equal weight to the drug loading conditions were used (1.2 mg). Wells treated with drug were brought to a final GEM concentration of 750 nM, based on the drug sensitivity assay. Negative controls consisted of media alone or supplemented with UV-sterilized unloaded MBs or nSh. The effect of both empty MBs and nSh in the presence of free GEM was also investigated. For 7 consecutive days, cell growth was analyzed using a Quant-iT PicoGreen dsDNA Assay Kit as described above, according to the manufacturer’s protocol and measured using a Tecan fluorimeter. Data was reported as % cell viability normalized to the initial reading on Day 1.
2.6. In vivo evaluation in xenograft mouse model
Tolerance of intravenously injected MBs, and the ability to track penetration into the tumor using ultrasound, were tested in a xenograft MIA PaCa-2 PDAC murine model. All animal work was conducted in compliance with the IACUC and Animal Care Policies of Thomas Jefferson University. Tumor models were generated by injection of 106 MIA PaCa-2 cells subcutaneously into the right hind limb of aythmic 42–49 day old mice (Charles River, Horsham, PA) split evenly by gender. Tumor growth was monitored until reaching volumes of 50–70 mm3 (approximately 3 weeks post injection), before being randomized to imaging arms.
UV-sterilized, GEM-loaded PEG-PLA MBs, free GEM, unloaded (blank) PEG-PLA MBs, and sterile saline were administered to 60 nude, athymic mice (12 mice per treatment group split evenly by gender). Flash/replenishment sequences were performed with an S3000 Helx Evolution scanner using a 9L4 probe (Siemens Healthineers, Mountain View, CA). Ultrasound imaging was performed at 4 MHz in cadence pulse sequencing mode at a frame rate of 33 Hz and 100% acoustic output power at a focal depth of 1 cm during retro-orbital bolus injection of 0.1 mL of the appropriate treatment solution. For drug-loaded samples, the 0.1 mL injection consisted of 0.12 mg MBs resulting in 2.2 μg GEM per injection. Quantities were based on reasonable tolerated dose of MBs/mouse.
Flash/replenishment sequences consisting of a series of 4 second destructive pulses at a mechanical index of 1.35 were generated to cavitate and rupture the MBs within the visualized tumor region, followed by 10 seconds of nonlinear imaging at lower intensity (mechanical index = 0.06) to allow for MB reperfusion. The imaging plane was maintained at the tumor midline for the flash/replenishment sequences over a period of up to 10 minutes until contrast was no longer visible, to maximize MB activation and delivery potential. Treatments were repeated 4 times over 2 weeks, with attempts to image different tumor planes with each treatment to treat the largest volume of tumor tissue possible. Tumor size was measured twice per week with calipers until sacrifice. Animals were monitored for disease progression and sacrificed as required by our protocol (i.e., tumor size exceeded allowable limit (> 20 mm in largest dimension), signs of distress, etc.).
2.7. Statistical analysis
Statistical analysis was performed with GraphPad Prism 7 (GraphPad Software, La Jolla, CA), using a one-way ANOVA analysis to determine significance across multiple groups (for α < 0.05), while Student’s t tests with Bonferroni correction for multiple comparisons were used for individual comparisons within groups. For the acoustic and physical characterizations, immunological assay, and cytotoxicity assays, data are presented as mean ± standard error about the mean (SEAM).
3. Results and discussion
Conventional systemic treatments for PDAC using GEM have proven ineffective in penetrating the excessive desmoplastic connective tissue surrounding the tumor to reach the target tumor, and in protecting drugs from enzymatic degradation [4,52]. We investigated ultrasound insonation combined with drug-loaded polymer MBs as a potential means to circumvent these issues through ultrasound-triggered delivery of GEM, while also protecting the GEM from degradation in the plasma. Once at the site, the MB shells are shattered by the ultrasound and the fragments (nSh) hydrolyze to lactic acid over time, creating a sustained localized release of GEM to tumor tissue. The length of time needed for this hydrolysis depends on the size and thickness of the MB shell but can be tuned to provide sustained release over time as opposed to bolus release and systemic exposure. This preliminary study explores the feasibility of encapsulating GEM within the shell of polymer MBs without affecting the morphology and the efficacy in vitro in MIA PaCa-2 cell cultures. Since the end application involves intravenous injection, the possibility of adding PEG to the shell to decrease immune response is also investigated, together with the effects these modifications have on acoustic and drug release properties. Finally, we study the ability to visualize these MBs penetrating the tumor microenvironment in a xenograft pancreatic cancer mouse model.
3.1. Drug loading
3.1.1. Morphology
Scanning electron microscopy (SEM) was used to establish the effect of incorporating GEM via the w/o/w emulsion method used to produce the MBs on morphology. Representative SEMs of unloaded MBs, and 3 wt%, 6 wt% and 10 wt% GEM-loaded PLA MBs are shown in Fig. 2. At 10 wt% GEM (Fig. 2D), the images show many broken and misshapen MBs. However, GEM encapsulation up to 6 wt% does not significantly affect MB morphology.
Fig. 2.

Scanning electron micrographs showing effect of increasing GEM loading on PLA MBs. A) Unloaded MBs, b) 3 wt % loading C) 6 wt % loading D) 10 wt % loading. (Size bar = 5 μm).
3.1.2. Drug-loading encapsulation efficiency
Based on our experience with various chemotherapeutics, including doxorubicin and paclitaxel, we anticipated encapsulation efficiency between 14 – 80%, depending on the starting concentration and individual drug properties [43,45]. We also expect the GEM to be encapsulated within the polymeric shell, given our experience with doxorubicin and its visualization within the shell via confocal microscopy [45]. Gemcitabine-loaded PLA MBs were used to investigate loading by the w/o/w emulsion method (Table 1). Encapsulation efficiency ranged from between 8–15%, representing 4.3 – 11.7 μg GEM/mg MB. As observed previously, encapsulation efficiency drops off considerably at higher initial loadings, with little advantage in the final loading amount between 6 and 10 wt% loading of drug [45]. Based on these results, 6 wt% loading was considered to represent the best compromise between loading and encapsulation efficiency, and 10 wt% loading was not considered further.
Table 1.
GEM encapsulation based on initial loading (Average ± standard error).
| Initial Loading | GEM Encapsulation (μg GEM/mg MB) | Encapsulation Efficiency (%) |
|---|---|---|
| 3 wt % | 4.3 ± 0.3 | 10.3 |
| 6 wt % | 12.4 ± 0.2 | 14.8 |
| 10 wt% | 11.7 ± 0.1 | 8.4 |
3.1.3. Drug sensitivity studies
Having determined the loading at which incorporation of GEM gave the optimal encapsulation efficiency without compromising MB integrity (6 wt%), a drug sensitivity assay was performed to verify that microencapsulation by the w/o/w method did not inactivate the GEM, to determine an optimal MB concentration compared to free GEM, and to investigate the cytotoxicity of products of GEM-MB insonation to MIA PaCa-2 cells. As expected, due to the need for PLA hydrolysis for GEM release from the MBs, the profiles for free GEM and PLA-encapsulated GEM differ in the concentration required to achieve zero cell survival at the completion of the 7 day assay (62.5 nM vs. 500 nM, Fig. 3). This is further exacerbated by the presence of cytidine deaminase, both in the culture medium and intracellularly, [53] which will start to deactivate the GEM by deamination. We will investigate this possible deactivation mechanism in future studies with this platform. Unloaded MBs had no significant effect on 7 day survival, including when added at concentration greater than the equivalent weight of GEM-MBs that resulted in 0% cell survival (500 nM).
Fig. 3.
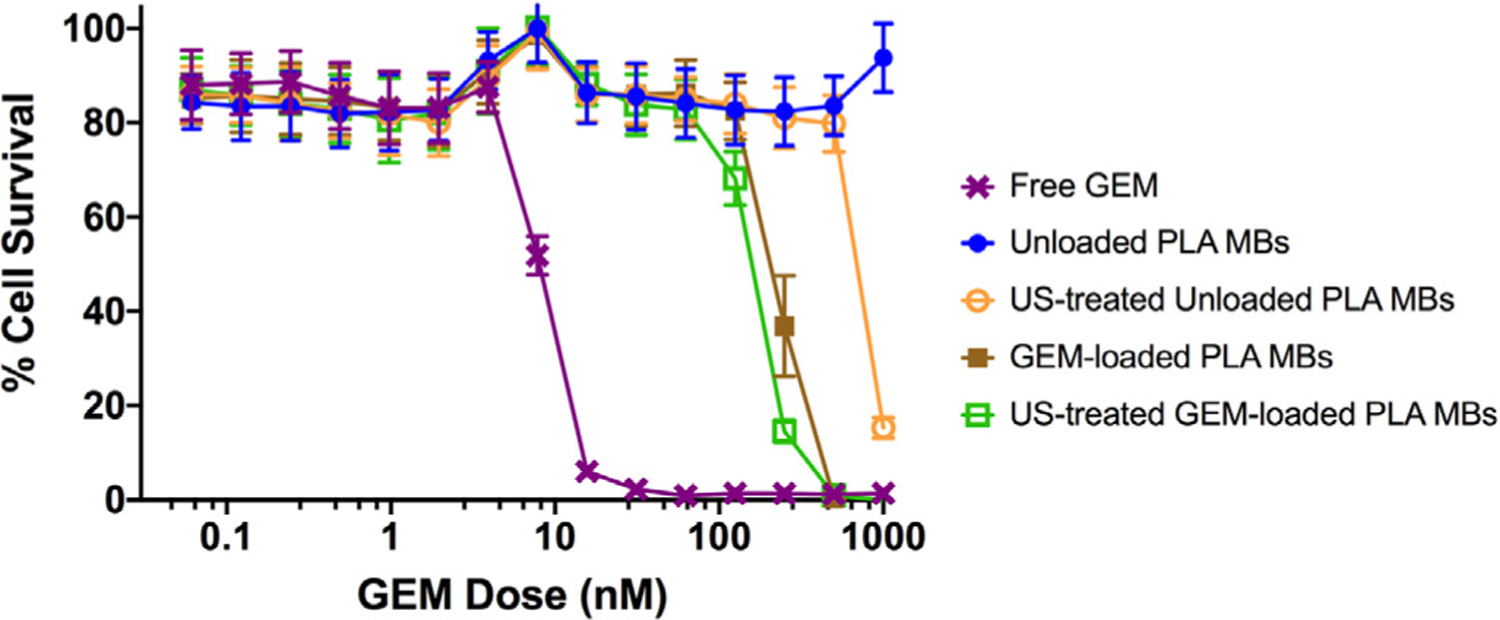
Effect of method of presentation of GEM to MIA PaCa-2 cells on cell viability. -X- Free GEM, -●- Unloaded PLA MBs, -○- US-treated unloaded PLA MBs, -■- GEM-loaded PLA MBs, -□- US-treated GEM-loaded PLA MBs (n = 6).
Both intact GEM-MBs and GEM-MBs previously subjected to ultrasound (nSh) cause 100% cell death at a GEM concentration 8 fold higher than free GEM (500 nM compared with 62.5 nM, Fig. 3). This raises several possibilities. Firstly, the process of embedding GEM into the polymers could have compromised GEM function in some way. However, if that is the case, the GEM is not completely compromised by the encapsulation process in either the intact MBs or US-GEM-MBs, both of which still induce cell death, albeit at a higher concentration. The higher dose required to achieve 0% cell survival is more likely due to the fact that the GEM-MBs and ultrasound-treated GEM-MBs are delivering GEM in a controlled fashion, due to the slow process of polymer hydrolysis, and hence a higher dose is required to eventually achieve the lethal dose in the culture medium. We might have expected the ultrasound-treated MBs to hydrolyze more rapidly, but this was not evident. An alternative explanation could also be that the amount of GEM in the free GEM (direct weighing of GEM) and the MB samples (derived by MB dissolution and spectroscopic analysis) are not identical. As mentioned above, there may also be some deactivation of the GEM being released from the polymer due to the presence of cytidine deaminase in the cell culture medium. An unexpected result came from the observation that unloaded ultrasound-treated MBs (Fig. 3, hollow -○-) track along with the profile of empty MBs up to equivalent polymer weight of the “500 nM” samples, then cause a massive decline in cell survival at increasingly higher doses, approaching 1000 nM equivalent weight. The robustness of this result was reinforced by the fact that identical results were obtained when repeating the study with Panc1 cells (Fig. S1).
At these very high particle concentrations, the empty MB fragments may be undergoing endocytosis by the cells at concentrations that cause cell death. One possible scenario, suggested by others in the literature, is that endocytosed polymeric nanoparticles escape rapidly from the endo-lysosomal compartment by selective reversal of the surface charge from anionic to cationic in the acidic environment, causing the nanoparticles to escape into the cytosol and become cytotoxic [54]. We have previously observed that doxorubicin-loaded, ultrasound-generated MB fragments are endocytosed [43]. These toxic effects seen in the nSh, while intriguing and warranting further investigation, do not appear until very high concentrations of the polymeric fragments are reached. These concentrations are considerably higher than would be achievable in a preclinical or clinical setting. It is also important to note that the cells were not directly insonated in these cytotoxicity tests, but that the MBs were previously treated with ultrasound and the resulting lyophilized fragments were then exposed to the cells under static conditions.
3.2. Incorporation of PEG into the polymeric shell
3.2.1. Effect of PEG-PLA on MB physical characteristics
Quantified physical characteristics of the PEGylated MBs are summarized in Table 2, comparing unloaded and loaded PLA MBs and the effect of incorporating PEG-PLA into the shell. All MB formulations satisfied the criteria for parenteral injection. The unloaded and GEM-loaded MBs had an average diameter between 1–3 μm, all well below the 8 μm diameter accepted as the upper limit to allow free passage throughout the vasculature, as well as polydispersity indices (PDI) below 0.4 (between 0.3 and 0.2), indicating a relatively uniform size distribution. For the PLA MBs, both 3 wt% (1.3 ± 0.1 μm) and 6 wt% GEM MBs (1.4 ± 0.1 μm) were significantly smaller than unloaded MBs (2.2 ± 0.1 μm, p < 0.034) as well as from the 10 wt% GEM MBs (2.5 ± 0.3 μm, p< 0.001, not shown). These larger 10% loaded MBs were not significantly different from the unloaded MBs (p > 0.99). We believe that this lack of size change in the 10 wt% GEM MBs compared to the unloaded MBs is due to the gross morphological changes that were shown in the SEM images (cf., Fig. 2), which is why these MBs were not pursued further in this study.
Table 2.
Effect of PEG incorporation on physical characteristics of microbubbles (Average ± standard error).
| Unloaded PLA | PLA with 3 wt% GEM | PLA with 6 wt% GEM | Unloaded 5% PEG-PLA | 5% PEG-PLA with 3 wt% GEM | 5% PEG-PLA with 6 wt% GEM | |
|---|---|---|---|---|---|---|
| Average Diameter (μm) and Polydispersity | 2.2 ± 0.1 | 1.3 ± 0.1 | 1.4 ± 0.1 | 1.9 ± 0.1 | 2.2 ± 0.1 | 2.5 ± 0.1 |
| PDI | PDI | PDI | PDI | PDI | PDI | |
| 0.2 ± 0.0 | 0.3 ± 0.0 | 0.2 ± 0.0 | 0.3 ± 0.0 | 0.3 ± 0.0 | 0.1 ± 0.0 | |
| Average Zeta Potential (mV) | −23.2 ± 1.1 | −34.0 ± 0.8 | −34.1 ± 1.2 | −32.7 ± 0.3 | −22.0 ± 0.9 | −17.7 ± 1.1 |
There were no significant differences in PDI for any level of GEM loading (range: 0.2 ± 0.0 to 0.3 ± 0.0, p > 0.57). Incorporating PEG-PLA into the MBs resulted in 6 wt% GEM loading becoming significantly larger in diameter (2.5 ± 0.1 μm) than 3 wt% GEM loading (2.2 ± 0.1 μm, p = 0.008) and unloaded MBs (1.9 ± 0.1 μm, p < 0.0001). There was no difference in size between unloaded PEG-PLA MBs and 3 wt% GEM PEG-PLA MBs (p = 0.07). The unloaded PEG-PLA MBs had a less uniform distribution than unloaded PLA MBs (p = 0.012), but there was no difference in PDI between the two formulations for 3 wt% and 6 wt% GEM loading (p > 0.09).
All MBs, irrespective of shell, maintained a negative zeta potential of between −34.1 ± 1.2 and −17.7 ± 1.1 mV, which will inhibit aggregation upon suspension (Table 2). However, comparison of data for unloaded MBs indicates that incorporation of PEG into the shell had significant influence on the zeta potential, moving it from −23.2 ± 1.1 mV to −32.7 ± 0.3 mV (p < 0.0001). The PLA MBs demonstrated more negative zeta potential upon loading with 3 wt% (−34.0 ± 0.8 mV) and 6 wt% GEM (−34.1 ± 1.2 mV) compared to unloaded MBs (p < 0.002), but there was no statistical difference between unloaded MBs and 10 wt% GEM MBs (−30.8 ± 0.6 mV, p > 0.99, not shown). The PEG-PLA MBs loaded with 3 wt% GEM exhibited a less negative zeta potential than unloaded MBs (−22.0 ± 0.9 mV vs. −32.7 ± 0.3 mV, p < 0.0001). Furthermore, the MBs loaded with 6 wt% GEM (−17.7 ± 1.1 mV) had a less negative zeta potential than those loaded with 3 wt% GEM (p = 0.004). However, when compared with their first generation counterparts, both 3 wt% and 6 wt% GEM resulted in a less negative zeta potential (p < 0.0001). It has been reported that PLA/PEG co-polymers possess greater hydrophilicity and lower glass transition temperature than PLA homopolymer, [55] and this in turn also affects the resulting shell properties of our microbubbles. This undoubtedly also affects the interactions between any incorporated drug and the polymer, which in turn will affect the zeta potential.
3.2.2. Acoustic characteristics
In vitro tank testing of the MBs is a convenient tool to rapidly assess the effect that varying parameters, such as shell composition and drug loading, have on the acoustic performance. While not all in vivo conditions are reproduced in this setup, we previously showed that in vitro dose response curves closely mirror those obtained with the same contrast agent in vivo [56]. Additionally, we have shown that acoustic backscatter measured in vitro down to 15 dB can give a readily detectable contrast-enhanced image in vivo [44].
As with the physical characteristics, addition of PEG to the shell had an effect on acoustic response, but all values were within our acceptable limits (Table 3). Adding PEG to the MB shell reduced the enhancement measured for the initial dose, indicating that fewer echogenic MBs were present in the sample.
Table 3.
Summary of acoustic characteristics for PLA and PEG-PLA MBs (Average ± standard error)
| Unloaded PLA | PLA with 3 wt% GEM | PLA with 6 wt% GEM | Unloaded 5% PEG-PLA | 5% PEG-PLA with 3 wt% GEM | 5% PEG-PLA with 6 wt% GEM | |
|---|---|---|---|---|---|---|
| Initial Enhancement (dB) | 15.6 ± 0.7 | 14.4 ± 0.9 | 10.5 ± 0.9 | 13.2 ± 0.7 | 11.2 ± 0.7 | 13.7 ± 0.7 |
| Maximum Enhancement (dB) | 19.4 ± 0.3 | 16.6 ± 0.3 | 14.5 ± 0.7 | 19.4 ± 0.5 | 17.6 ± 0.7 | 15.7 ± 0.5 |
| In vitro Acoustic Half-life (Min) | >15 | >15 | >15 | >15 | >15 | >15 |
Loading of GEM also influenced echogenicity, being reduced but acceptable compared to unloaded controls (p=0.0004; Fig. 4) [44]. The average echogenicity measured at the first MB dose decreased from 13.2 ± 0.7 dB for unloaded PEG-PLA MBs to 11.2 ± 0.7 for 3 wt% GEM-loaded PEG-PLA MBs, but then rebounded to 13.7 ± 0.7 dB with the 6 wt% PEG-PLA sample. The maximum achievable response decreased from unloaded through 3 wt% to 6 wt%, being 19.4 ± 0.5 dB at a dose of 15 μg/mL unloaded MBs, to 17.6 ± 0.7 dB at a dose of 13.5 μg/mL for 3 wt% loading, to 15.7 ± 0.5 dB at the 7.5 μg/mL dose for 6 wt% loaded samples.
Fig. 4.
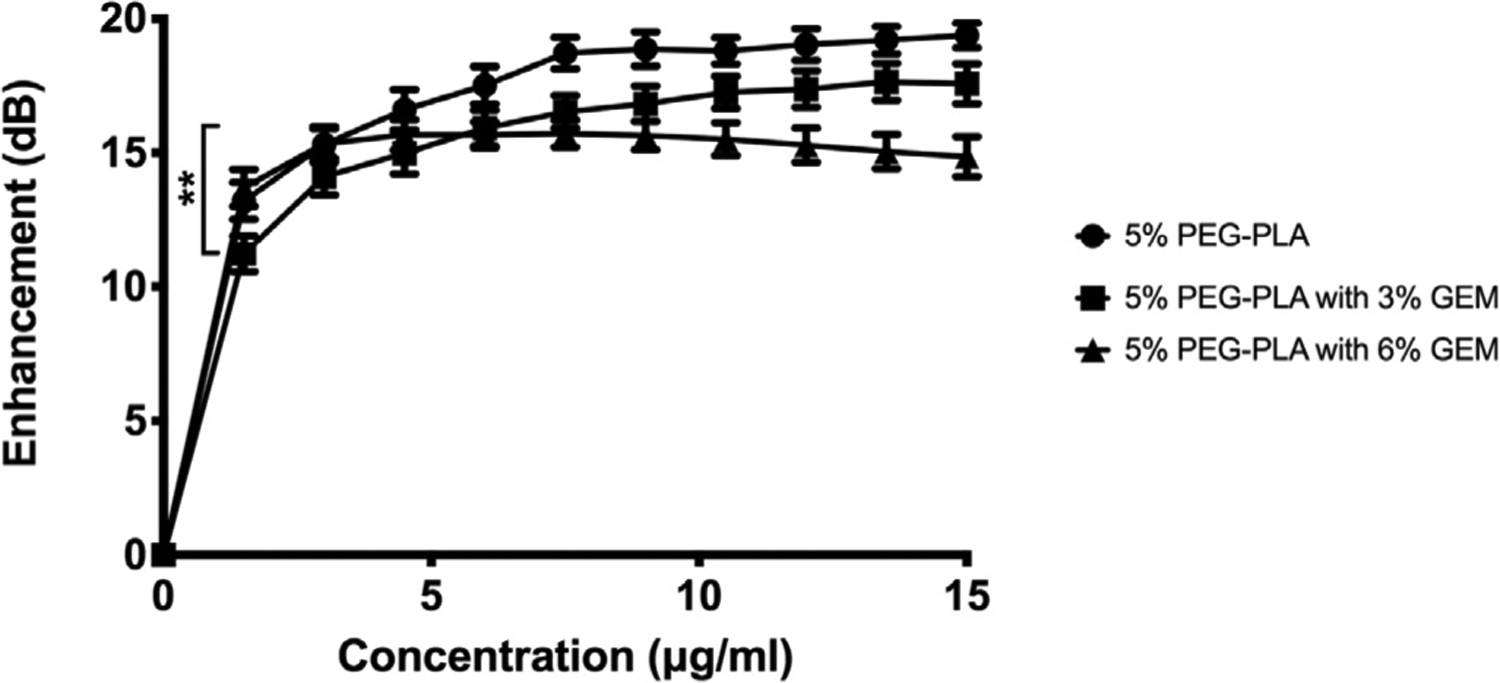
Effect of drug loading on in vitro acoustic enhancement with cumulative increasing dose of PEG-PLA MBs. -●- Unloaded MBs, -■- 3% GEM, -▲- 6% GEM. (**p = 0.004).
The individual profiles reflect both intrinsic echogenicity and microbubble populations. Adding 6 wt% GEM to the shell appears to increase the echogenicity achieved at the first dose, and shadowing begins to be detectable at ~7.5 μg/mL. Both observations suggest a greater population of echogenic bubbles, possibly due to production of a larger proportion of intact bubbles.
In terms of long-term stability within an ultrasound beam (Fig. S2), slight loss of echogenicity was seen at low PNP (approximately 0.45 MPa, mechanical index = 0.15) for all MBs, giving an in vitro acoustic half-life (defined as time at which normalized echogenicity decreases to 0.5) of greater than 15 minutes (Table 3). The stability trends were similar between PLA and PEG-PLA MBs (data not shown). It is interesting to note that incorporation of GEM was the major driver in reducing echogenicity for both PLA and PEG-PLA MBs. However, incorporation of PEG into the MB shell has little effect on the stability in the ultrasound beam.
3.3. Immunological assay
An assay of C3a activation was conducted to measure the extent to which the immunological impact of GEM-loaded MBs could be reduced by adding a PEGylated surface. These results are summarized in Fig. 5 and are consistent with our prior findings in unloaded MBs [47].
Fig. 5.
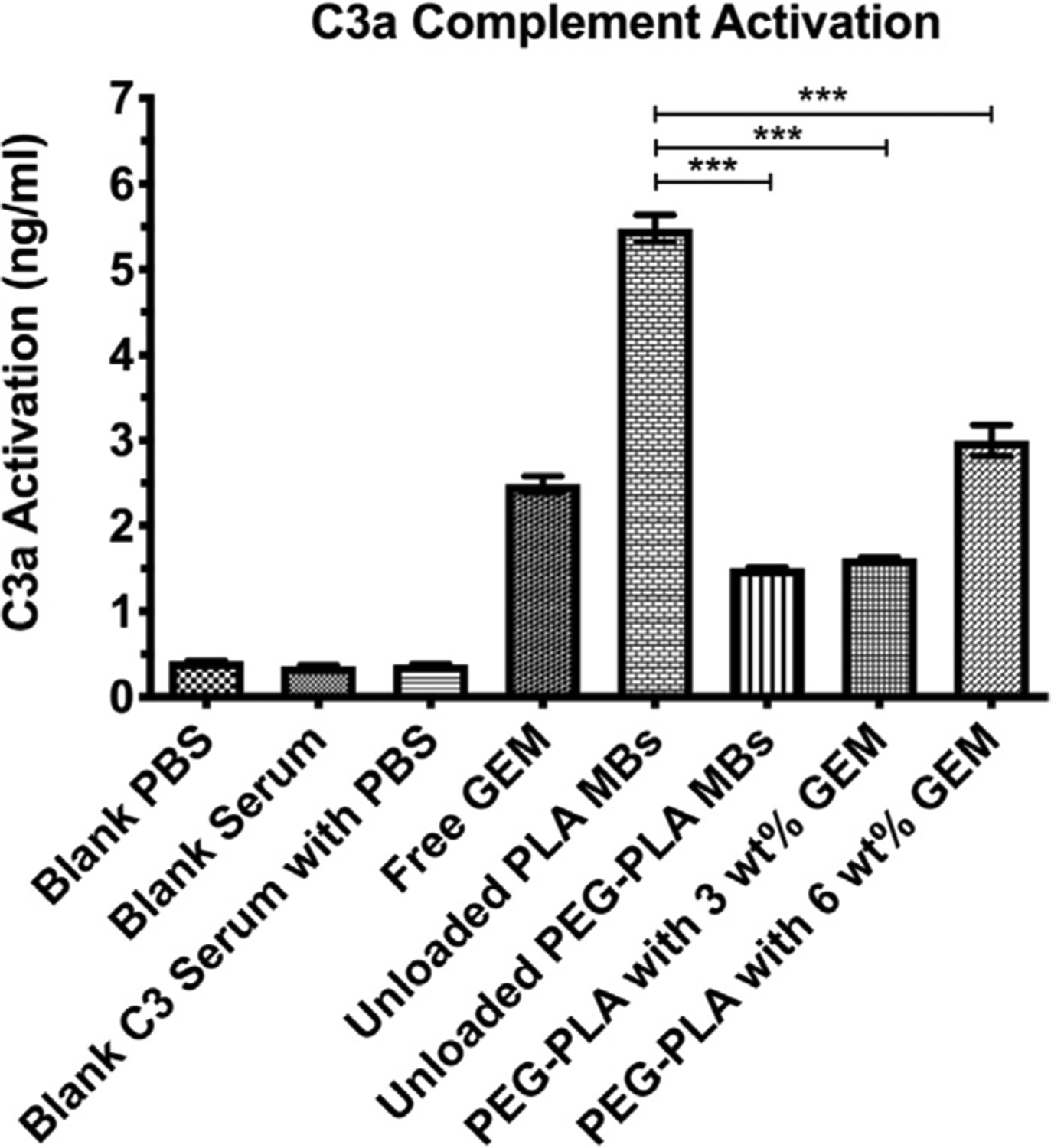
C3a compliment activation assay (n = 3, ***p < 0.0001).
The negative controls of PBS, serum, and a mixture of the two registered less than 0.5 ng/mL activation. Unmodified, unloaded PLA MBs registered the highest C3a activation at 5.5 ng/mL, but this was reduced to 1.6 ng/mL by addition of 5 wt% PEG into the shell. When GEM was encapsulated within PEG-PLA MBs at 3 wt%, the activation was not statistically different, but increasing the GEM in the preparation to 6 wt% produced a value of 3.0 ng/mL (p < 0.0001). Free GEM, on the other hand, generated around 2.5 ng/mL activation, greater than the 3 wt% GEM loading but less than the 6 wt% (p = 0.003).
While very low activation is the goal, [57] even the high loading MBs initiated around 50% less activation than empty, unmodified PLA MBs. Most PLA micro- and nano-capsules are subject to these restrictions, and it is thought that an interaction between the PLA carboxylic acid end groups on the MB surface and the unstable thioester bond in the C3 protein facilitates binding of the C3 protein to the MB surface, activating the immune response, which may be proportional to the number of exposed reactive groups [57–59].
3.4. Viability studies with PEG-PLA MBs
Monitoring the daily effect of GEM and GEM-MBs on MIA PaCa-2 cells in vitro (Fig. 6), we found that distinguishing effects on cell survival began between day 2 and day 3. Blank MB have no detrimental effect on cell growth, as expected, where cells continue to proliferate at a rate equal to or greater than the control (p > 0.99). Blank nSh delayed cell growth for 6 days (p = 0.0002 compared to control), and then cell growth rebounded. The GEM-nSh and blank nSh with accompanying free GEM had somewhat reduced rates of growth compared to controls (p > 0.36), but still did not reduce or halt tumor cell growth. Importantly, by day 3, free GEM, free GEM plus blank MB, and GEM-loaded MB had all dropped to an average of about 40% survival, dropping to total loss of viability by day 5 (p < 0.0198). These results suggest that GEM-loaded PEG-PLA MBs are a viable drug delivery vehicle. Furthermore, this method of assessing in vitro drug release profiles has advantages over the traditional static release into PBS, since it simultaneously shows both release and active drug effect.
Fig. 6.
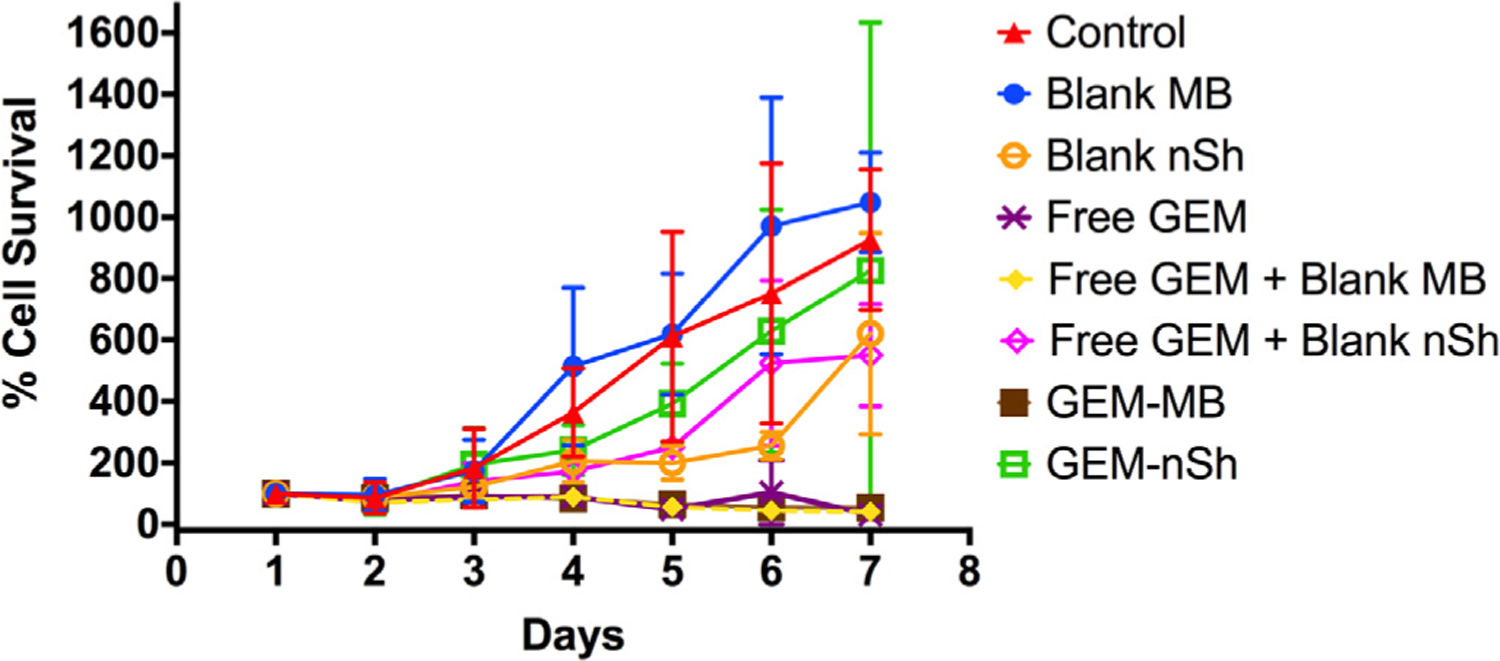
Effect of method of presentation of GEM to MIA PaCa-2 cells on cell viability. -△- control (no intervention), -●- Unloaded PLA MBs, -○- US-treated unloaded PLA nSh, -X- Free GEM, -♦- Free GEM + Blank MBs, -◊- Free GEM + Blank nSh, -■- GEM-loaded PLA MBs, -□- US-treated GEM-loaded PLA MBs (n = 6).
Interestingly, the GEM-MBs treated with ultrasound (nSh) prior to addition to the culture display a somewhat different effect compared to the GEM-loaded PEG-PLA MBs in the sensitivity assay. This may be due to the pre-treated MBs losing encapsulated GEM during the insonation and lyophilization process, used to create fragments in vitro. The ultrasound-treated unloaded nSh again display unexpected toxicity, as was observed in the drug sensitivity assays using PLA MBs, but as with the GEM-loaded ultrasound-treated MBs, the effect was less, and shorter lived, than with the PLA MBs. The difference in the degree of effect could be related to the higher (less negative) zeta potential displayed by the PEG-MBs used here compared with the PLA MBs used in the drug sensitivity assays, or the presence of PEG itself, both of which have been shown to reduce the rate of endocytosis and cytotoxicity [60].
It is also important to note that in this assay, the free GEM is exposed directly to the MIA PaCa-2 cells, without having to penetrate a stroma surrounding a tumor nor being subject to systemic uptake or enzymatic breakdown. Additionally, we were surprised to find that the free GEM was not as effective in the presence of the blank nSh. We hypothesize that this is due to the increased surface area of PEG-PLA compared to the intact MBs, which could lead to increased GEM adsorption into the polymer and away from the cells or formation of a protective polymeric barrier between the free GEM in the medium and the cells. More research is needed to elucidate the mechanism behind these results. However, this assay does demonstrate that GEM-loaded MBs are capable of sustained GEM release and cytotoxicity to MIA PaCa-2 cells in an in vitro environment.
3.5. In vivo evaluation
In vivo, administration of PEGylated GEM-MBs was well tolerated and provided substantial tumoral contrast enhancement (Fig. 7). This is clearly visible in Fig. 7B, where the tumor is delineated and differentiated from the surrounding tissue when infused with GEM-MBs, compared to prior to injection (Fig. 7A). Destruction of MBs within the tumor was confirmed by evaluating differences in enhancement before (B) and after (C) destructive pulses, and MB reperfusion was observed within tumor tissue for repeated destruction/reperfusion sequences up to approximately 10 minutes following injection, up to the point where contrast was no longer visible to maximize MB activation and delivery potential. This process causes the burst of the MBs to allow for extravascular escape of the nSh and their subsequent deposition within the surrounding tumor tissue to allow for sustained, targeted GEM delivery well beyond the imaging session. Any nSh not directed into the tumor will continue to circulate and are available for regular nanoparticle uptake mechanisms such as the enhanced permeability and retention (EPR) effect.
Fig. 7.
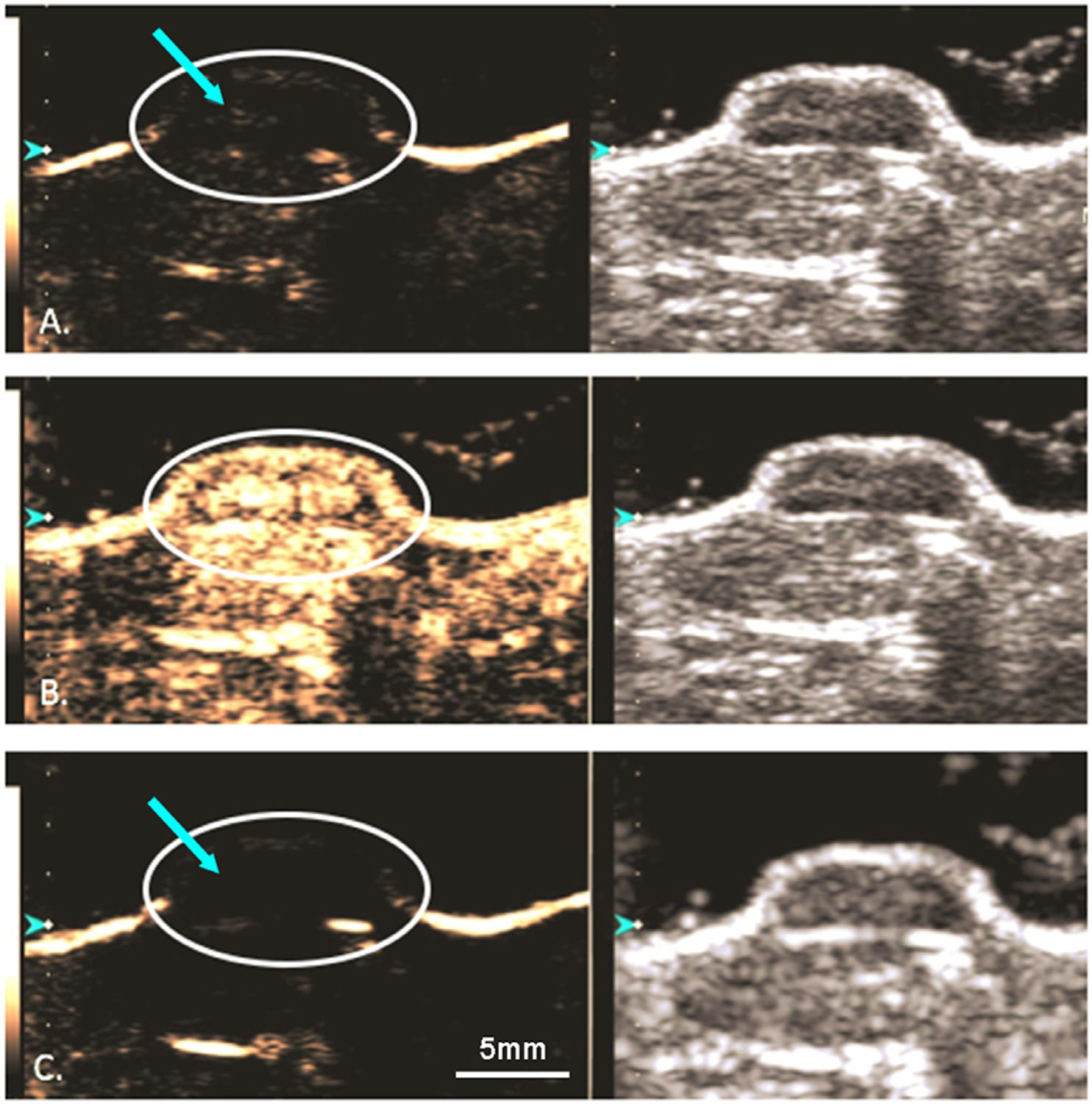
Dual ultrasound of a contrast enhanced (left) and B-mode (right) human PDAC xenograft in a mouse model, MIA PaCa-2. A) GEM-MB are visualized within the tumor (arrow). B) A 4 second, 1.35 mechanical index destructive pulse was delivered to the tumor. C) Post destructive pulse displaying an absence of microbubbles within the tumor. Scale bar: 5mm.
Monitoring of tumor growth indicated no observable reduction in tumor growth across any of the treatment groups (Fig. 8), indicating insufficient amounts of GEM were able to reach the tumor tissue.
Fig. 8.
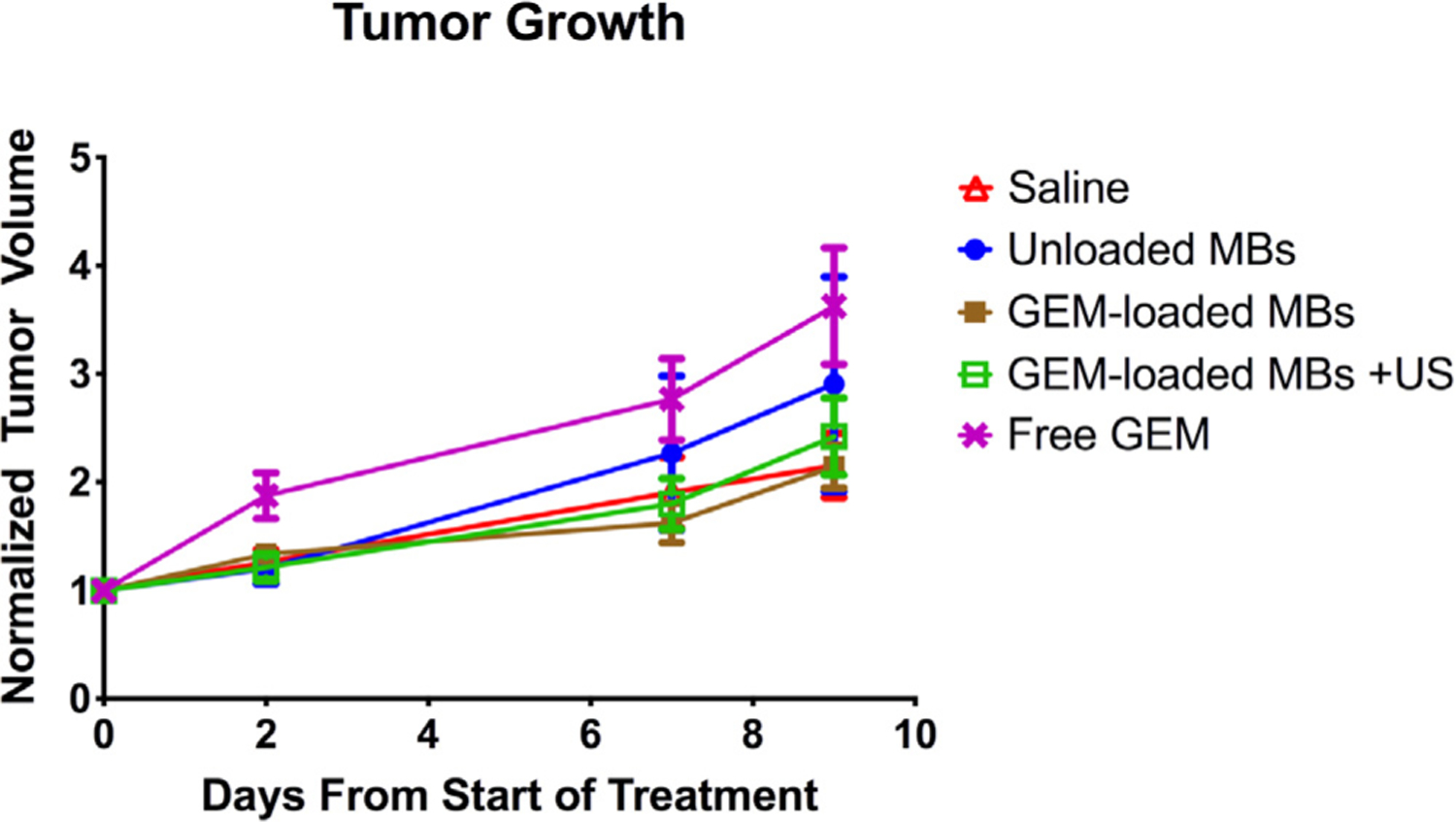
Normalized tumor volume over time in xenograft MIA PaCa-2 mouse model. -△- Control saline, -●- Unloaded PEG PLA MBs, -■- GEM-loaded PEG PLA MBs, -□- US-treated GEM-loaded PEG PLA MBs, -X- Free GEM (n = 12 mice per group).
This is expected for the free GEM treatment, given the difficulty in penetrating the surrounding stroma with free drug. In the case of the GEM MBs, the encapsulated payload was also not sufficiently high to result in any significant reduction in tumor volume during the observation period. While tolerability and the ability to locally deliver GEM are encouraging, these findings point to the need for improved drug loading to achieve in vivo tumor control. We are currently exploring increasing the drug load by methods other than direct incorporation into the shell, since we show here that 10 wt% GEM loading results in unacceptable microbubble formation (cf., Fig. 2). To date, we have shown that nanoparticles, such as gold and iron, can be incorporated into the polymeric shell, and the next step is to pre-coat these nanoparticles with drug [61].
4. Conclusion
Encapsulation of GEM within the shell of polymer MBs resulted in a promising, new design and methodology involving ultrasound triggering of a drug delivery platform for PDAC, with the potential to become a viable method of neoadjuvant chemotherapy to treat the population with pancreatic cancer deemed ineligible for surgical resection. The study showed that these agents retained acceptable echogenic/acoustic, morphological, and physical properties, when compared to unloaded MBs, and displayed inertial cavitation disruption upon exposure to clinical ultrasound, while the encapsulated GEM retained its cytotoxicity to MIA PaCa-2 cells in culture. Evidence from drug sensitivity curves highlighted the requirement for polymer hydrolysis to release the encapsulated GEM resulting in the need for a high GEM concentration when encapsulated. Ongoing efforts are in progress to increase GEM loading within the MBs by co-encapsulation of drug-loaded nanoparticles or therapeutic gases for improved treatment efficacy at the tumor site.
Overall, these functionalized agents represent a first step to-wards an innovative approach to overcoming the multiple challenges associated with pancreatic cancer treatment, including the ability to shield healthy tissues from systemic administration, to effectively deliver therapy to the tumor tissue using an innovative, dual-functioning platform to create a truly “theranostic” modality.
Supplementary Material
Statement of significance.
The preliminary results shown here are encouraging and support further investigation into increased gemcitabine loading. Encapsulation of gemcitabine within polylactic acid (PLA) microbubbles does not damage its activity towards pancreatic cancer (pancreatic ductal adenocarcinoma, PDAC) cells. Excellent imaging and evidence of penetration into the highly desmoplastic PDAC tumors is demonstrated. Microbubble destruction was confirmed in vivo, showing that elevated mechanical index shatters the microbubbles for enhanced delivery. The potential to slow PDAC growth in vivo is shown, but higher gemcitabine concentrations are required. Current efforts are directed at increasing drug loading by inclusion of drug-carrying nanoparticles for effective in vivo treatment.
Acknowledgements
The authors wish to thank Averie Palovcak, Stephen Zachariah, Timothy Hoang, Rebecca Sheridan, and Jaap Patel from Drexel University for their contributions to this work. Invaluable assistance with cell culture at Drexel University was provided by Dolores Conover. This study was funded in part by the Coulter-Drexel Translational Research Partnership, W.W. Smith Charitable Foundation, and NIH grants R01CA212600, R01EB026881, R01CA199646, S10OD010408 and U01CA224012. Lauren Delaney is supported by F32 AR072491. Masaya Jimbo was supported by NIH 5 T32 AR 52273. This work was also supported by a SKCC (TJU) Cancer Center Support Grant 5P30CA056036-17 (JRB). Flemming Forsberg acknowledges equipment support from Siemens Healthineers.
Declaration of Competing Interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: Flemming Forsberg acknowledges equipment support from Siemens Healthineers. John Eisenbrey acknowledges equipment support from Siemens, grant funding and equipment support from GE Healthcare, and drug support and speaker fees from Lantheus Medical Imaging. Lauren Delaney, David Brown, Jonathan Brody, Masaya Jimbo, Brian Oeffinger, Maria Stanczak, Ji-Bin Liu, and Margaret Wheatley have no competing interests to report.
Footnotes
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi: 10.1016/j.actbio.2021.05.046.
References
- [1].American Cancer Society, Key statistics for pancreatic cancer, 2019. http://www.cancer.org/cancer/pancreaticcancer/detailedguide/pancreatic-cancer-key-statistics. Accessed July 2019).
- [2].Apfel RE, Holland CK, Gauging the likelihood of cavitation from short-pulse, low-duty cycle diagnostic ultrasound, Ultrasound Med. Biol 17 (2) (1991) 179–185. [DOI] [PubMed] [Google Scholar]
- [3].Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM, Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States, Cancer Res. 74 (11) (2014) 2913–2921. [DOI] [PubMed] [Google Scholar]
- [4].Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA, The pancreas cancer microenvironment, Clin. Cancer Res 18 (16) (2012) 4266–4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR, Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma, Cancer Cell 21 (3) (2012) 418–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, Feig C, Nakagawa T, Caldwell ME, Zecchini HI, Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer, Gut 62 (1) (2013) 112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bilimoria KY, Bentrem DJ, Ko CY, Stewart AK, Winchester DP, Talamonti MS, National failure to operate on early stage pancreatic cancer, Ann. Surg 246 (2) (2007) 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kleeff J, Michalski C, Friess H, Büchler M, Surgical treatment of pancreatic cancer: the role of adjuvant and multimodal therapies, Eur. J. Surg. Oncol 33 (7) (2007) 817–823. [DOI] [PubMed] [Google Scholar]
- [9].Oettle H, Post S, Neuhaus P, Gellert K, Langrehr J, Ridwelski K, Schramm H, Fahlke J, Zuelke C, Burkart C, Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer, J. Am. Med. Assoc 297 (3) (2007) 267–277. [DOI] [PubMed] [Google Scholar]
- [10].Carmichael J, Fink U, Russell R, Spittle M, Harris A, Spiessi G, Blatter J, Phase II study of gemcitabine in patients with advanced pancreatic cancer, Br. J. Cancer 73 (1) (1996) 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ergun Y, Ozdemir NY, Guner EK, Esin E, Sendur MA, Koksoy EB, Demirci NS, Eren T, Dede I, Sezer A, Comparison of Gemcitabine monotherapy with Gemcitabine and Cisplatin combination in metastatic pancreatic cancer: a retrospective analysis, J. Balkan Union Oncol 23 (7) (2018) 116–121. [PubMed] [Google Scholar]
- [12].Javed MA, Beyer G, Le N, Vinci A, Wong H, Palmer D, Morgan RD, Lamarca A, Hubner RA, Valle JW, Impact of intensified chemotherapy in metastatic pancreatic ductal adenocarcinoma (PDAC) in clinical routine in Europe, Pancreatology 19 (1) (2019) 97–104. [DOI] [PubMed] [Google Scholar]
- [13].Burris HA, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P, Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial, J. Clin. Oncol 15 (6) (1997) 2403–2413. [DOI] [PubMed] [Google Scholar]
- [14].Garrido-Laguna I, Hidalgo M, Pancreatic cancer: from state-of-the-art treatments to promising novel therapies, Nat. Rev. Clin. Oncol 12 (6) (2015) 319. [DOI] [PubMed] [Google Scholar]
- [15].Neesse A, Algül H, Tuveson DA, Gress TM, Stromal biology and therapy in pancreatic cancer: a changing paradigm, Gut 64 (9) (2015) 1476–1484. [DOI] [PubMed] [Google Scholar]
- [16].Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer, Science 324 (5933) (2009) 1457–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gore J, Korc M, Pancreatic cancer stroma: friend or foe? Cancer Cell 25 (6) (2014) 711–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu C-C, Simpson TR, Laklai H, Sugimoto H, Kahlert C, Novitskiy SV, Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival, Cancer Cell 25 (6) (2014) 719–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, Dekleva EN, Saunders T, Becerra CP, Tattersall IW, Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma, Cancer Cell 25 (6) (2014) 735–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Patra CR, Bhattacharya R, Wang E, Katarya A, Lau JS, Dutta S, Muders M, Wang S, Buhrow SA, Safgren SL, Targeted delivery of gemcitabine to pancreatic adenocarcinoma using cetuximab as a targeting agent, Cancer Res. 68 (6) (2008) 1970–1978. [DOI] [PubMed] [Google Scholar]
- [21].Quinn BA, Wang S, Barile E, Das SK, Emdad L, Sarkar D, De SK, Kharagh SM, Stebbins JL, Pandol SJ, Therapy of pancreatic cancer via an EphA2 receptor-targeted delivery of gemcitabine, Oncotarget 7 (13) (2016) 17103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ray P, Cheek MA, Sharaf ML, Li N, Ellington AD, Sullenger BA, Shaw BR, White RR, Aptamer-mediated delivery of chemotherapy to pancreatic cancer cells, Nucl. Acid Ther 22 (5) (2012) 295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Yoon S, Huang K-W, Reebye V, Mintz P, Tien Y-W, Lai H-S, Sætrom P, Reccia I, Swiderski P, Armstrong B, Targeted delivery of C/EBP α-saRNA by pancreatic ductal adenocarcinoma-specific RNA aptamers inhibits tumor growth in vivo, Mol. Ther 24 (6) (2016) 1106–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Daman Z, Ostad S, Amini M, Gilani K, Preparation, optimization and in vitro characterization of stearoyl-gemcitabine polymeric micelles: A comparison with its self-assembled nanoparticles, Int. J. Pharm 468 (1) (2014) 142–151. [DOI] [PubMed] [Google Scholar]
- [25].Chung W-G, Sandoval MA, Sloat BR, Lansakara-P DS, Cui Z, Stearoyl gemcitabine nanoparticles overcome resistance related to the over-expression of ribonucleotide reductase subunit M1, J. Control. Release 157 (1) (2012) 132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Immordino ML, Brusa P, Rocco F, Arpicco S, Ceruti M, Cattel L, Preparation, characterization, cytotoxicity and pharmacokinetics of liposomes containing lipophilic gemcitabine prodrugs, J. Control. Release 100 (3) (2004) 331–346. [DOI] [PubMed] [Google Scholar]
- [27].Li J.-m., Chen W, Wang H, Jin C, Yu X.-j., Lu W.-y., Cui L, Fu D.-l., Ni Q.-x., Hou H.-m., Preparation of albumin nanospheres loaded with gemcitabine and their cytotoxicity against BXPC-3 cells in vitro, Acta Pharmacol. Sin 30 (9) (2009) 1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Maksimenko A, Caron J, Mougin J, Desmaële D, Couvreur P, Gemcitabine-based therapy for pancreatic cancer using the squalenoyl nucleoside monophosphate nanoassemblies, Int. J. Pharm 482 (1–2) (2015) 38–46. [DOI] [PubMed] [Google Scholar]
- [29].Stella B, Arpicco S, Rocco F, Marsaud V, Renoir J-M, Cattel L, Couvreur P, Encapsulation of gemcitabine lipophilic derivatives into polycyanoacry-late nanospheres and nanocapsules, Int. J. Pharm 344 (1–2) (2007) 71–77. [DOI] [PubMed] [Google Scholar]
- [30].Yu X, Di Y, Xie C, Song Y, He H, Li H, Pu X, Lu W, Fu D, Jin C, An in vitro and in vivo study of gemcitabine-loaded albumin nanoparticles in a pancreatic cancer cell line, Int. J. Nanomed 10 (2015) 6825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Arya G, Vandana M, Acharya S, Sahoo SK, Enhanced antiproliferative activity of Herceptin (HER2)-conjugated gemcitabine-loaded chitosan nanoparticle in pancreatic cancer therapy, Nanomedicine 7 (6) (2011) 859–870. [DOI] [PubMed] [Google Scholar]
- [32].Bilalis P, Skoulas D, Karatzas A, Marakis J, Stamogiannos A, Tsimblouli C, Sereti E, Stratikos E, Dimas K, Vlassopoulos D, Self-healing pH-and enzyme stimuli-responsive hydrogels for targeted delivery of gemcitabine to treat pancreatic cancer, Biomacromolecules 19 (9) (2018) 3840–3852. [DOI] [PubMed] [Google Scholar]
- [33].Mura S, Nicolas J, Couvreur P, Stimuli-responsive nanocarriers for drug delivery, Nature materials 12 (11) (2013) 991–1003. [DOI] [PubMed] [Google Scholar]
- [34].Yu X, Zhang Y, Chen C, Yao Q, Li M, Targeted drug delivery in pancreatic cancer, Biochimica et Biophysica Acta (BBA)-Reviews on Cancer 1805 (1) (2010) 97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Bressand D, Novell A, Girault A, Raoul W, Fromont-Hankard G, Escoffre J-M, Lecomte T, Bouakaz A, Enhancing Nab-paclitaxel delivery using microbubble-assisted ultrasound in a pancreatic cancer model, Mol. Pharm 16 (9) (2019) 3814–3822. [DOI] [PubMed] [Google Scholar]
- [36].Eisenbrey JR, Shraim R, Liu J-B, Li J, Stanczak M, Oeffinger B, Leeper DB, Keith SW, Jablonowski LJ, Forsberg F, Sensitization of hypoxic tumors to radiation therapy using ultrasound-sensitive oxygen microbubbles, Int. J. Radiat. Oncol. * Biol. * Phys 101 (1) (2018) 88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Geers B, De Wever O, Demeester J, Bracke M, De Smedt SC, Lentacker I, Targeted liposome-loaded microbubbles for cell-specific ultrasound-triggered drug delivery, Small 9 (23) (2013) 4027–4035. [DOI] [PubMed] [Google Scholar]
- [38].Luo T, Sun J, Zhu S, He J, Hao L, Xiao L, Zhu Y, Wang Q, Pan X, Wang Z, Ultrasound-mediated destruction of oxygen and paclitaxel loaded dual-targeting microbubbles for intraperitoneal treatment of ovarian cancer xenografts, Cancer Lett. 391 (2017) 1–11. [DOI] [PubMed] [Google Scholar]
- [39].Sirsi SR, Borden MA, State-of-the-art materials for ultrasound-triggered drug delivery, Adv. Drug Del. Rev 72 (2014) 3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Snipstad S, Berg S, Mørch Ý, Bjørkøy A, Sulheim E, Hansen R, Grimstad I, van Wamel A, Maaland AF, Torp SH, Ultrasound improves the delivery and therapeutic effect of nanoparticle-stabilized microbubbles in breast cancer xenografts, Ultrasound Med. Biol 43 (11) (2017) 2651–2669. [DOI] [PubMed] [Google Scholar]
- [41].Zhang Y, Chang S, Sun J, Zhu S, Pu C, Li Y, Zhu Y, Wang Z, Xu RX, Targeted microbubbles for ultrasound mediated short hairpin RNA plasmid transfection to inhibit survivin gene expression and induce apoptosis of ovarian cancer A2780/DDP cells, Mol. Pharm 12 (9) (2015) 3137–3145. [DOI] [PubMed] [Google Scholar]
- [42].Dimcevski G, Kotopoulis S, Bjånes T, Hoem D, Schjøtt J, Gjertsen BT, Biermann M, Molven A, Sorbye H, McCormack E, A human clinical trial using ultrasound and microbubbles to enhance gemcitabine treatment of inoperable pancreatic cancer, J. Control. Release 243 (2016) 172–181. [DOI] [PubMed] [Google Scholar]
- [43].Cochran MC, Eisenbrey J, Ouma RO, Soulen M, Wheatley MA, Doxorubicin and paclitaxel loaded microbubbles for ultrasound triggered drug delivery, Int. J. Pharm 414 (1–2) (2011) 161–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Cochran MC, Eisenbrey JR, Soulen MC, Schultz SM, Ouma RO, White SB, Furth EE, Wheatley MA, Disposition of Ultrasound Sensitive Polymeric Drug Carrier in a Rat Hepatocellular Carcinoma Model, Acad. Radiol 18 (11) (2011) 1341–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Eisenbrey J, Burstein OM, Kambhampati R, Forsberg F, Liu J-B, Wheatley M, Development and optimization of a doxorubicin loaded poly (lactic acid) contrast agent for ultrasound directed drug delivery, J. Control. Release 143 (1) (2010) 38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Eisenbrey J, Huang P, Hsu J, Wheatley M, Ultrasound triggered cell death in vitro with doxorubicin loaded poly lactic-acid contrast agents, Ultrasonics 49 (8) (2009) 628–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Jablonowski LJ, Alfego D, Andorko JI, Eisenbrey JR, Teraphongphom N, Wheatley MA, Balancing stealth and echogenic properties in an ultrasound contrast agent with drug delivery potential, Biomaterials 103 (2016) 197–206. [DOI] [PubMed] [Google Scholar]
- [48].Johnston MJ, Semple SC, Klimuk SK, Edwards K, Eisenhardt ML, Leng EC, Karlsson G, Yanko D, Cullis PR, Therapeutically optimized rates of drug release can be achieved by varying the drug-to-lipid ratio in liposomal vincristine formulations, Biochim. Biophys. Acta 1758 (1) (2006) 55–64. [DOI] [PubMed] [Google Scholar]
- [49].Arvanitis CD, Bazan-Peregrino M, Rifai B, Seymour LW, Coussios CC, Cavitation-enhanced extravasation for drug delivery, Ultrasound Med. Biol 37 (11) (2011) 1838–1852. [DOI] [PubMed] [Google Scholar]
- [50].Geers B, Dewitte H, De Smedt SC, Lentacker I, Crucial factors and emerging concepts in ultrasound-triggered drug delivery, J. Control. Release 164 (3) (2012) 248–255. [DOI] [PubMed] [Google Scholar]
- [51].El-Sherif DM, Wheatley MA, Development of a novel method for synthesis of a polymeric ultrasound contrast agent, J. Biomed. Mater. Res. Part A 66 (2) (2003) 347–355. [DOI] [PubMed] [Google Scholar]
- [52].Mitragotri S, Burke PA, Langer R, Overcoming the challenges in administering biopharmaceuticals: formulation and delivery strategies, Nat. Rev. Drug Discov 13 (9) (2014) 655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Bjånes TK, Jordheim LP, Schjøtt J, Kamceva T, Cros-Perrial E, Langer A, Ruiz de Garibay G, Kotopoulis S, McCormack E, Riedel B, Intracellular Cytidine Deaminase Regulates Gemcitabine Metabolism in Pancreatic Cancer Cell Lines, Drug Metab. Disposition 48 (3) (2020) 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Panyam J, Zhou W-Z, Prabha S, Sahoo SK, Labhasetwar V, Rapid endo-lyso-somal escape of poly (DL-lactide-co-glycolide) nanoparticles: implications for drug and gene delivery, FASEB J 16 (10) (2002) 1217–1226. [DOI] [PubMed] [Google Scholar]
- [55].Huang Y-Y, Chung T-W, Tzeng T.-w., Drug release from PLA/PEG microparticulates, Int. J. Pharm 156 (1) (1997) 9–15. [Google Scholar]
- [56].Wheatley MA, Forsberg F, Oum K, Ro R, El-Sherif D, Comparison of in vitro and in vivo acoustic response of a novel 50: 50 PLGA contrast agent, Ultrasonics 44 (4) (2006) 360–367. [DOI] [PubMed] [Google Scholar]
- [57].Janeway CA, Travers P, Walport M, Shlomchik M, Immunobiology: the immune system in health and disease, Curr. Biol. Lond (1996). [Google Scholar]
- [58].Chen CC, Borden MA, The role of poly (ethylene glycol) brush architecture in complement activation on targeted microbubble surfaces, Biomaterials 32 (27) (2011) 6579–6587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Janssen BJ, Huizinga EG, Raaijmakers HC, Roos A, Daha MR, Nilsson-Ekdahl K, Nilsson B, Gros P, Structures of complement component C3 provide insights into the function and evolution of immunity, Nature 437 (7058) (2005) 505. [DOI] [PubMed] [Google Scholar]
- [60].Wang W, Zhou S, Guo L, Zhi W, Li X, Weng J, Investigation of endocytosis and cytotoxicity of poly-d, l-lactide-poly (ethylene glycol) micro/nano-particles in osteoblast cells, Int. J. Nanomed 5 (2010) 557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Teraphongphom N, Chhour P, Eisenbrey JR, Naha PC, Witschey WRT, Opasanont B, Jablonowski L, Cormode DP, Wheatley MA, Nanoparticle Loaded Polymeric Microbubbles as Contrast Agents for Multimodal Imaging, Langmuir 31 (43) (2015) 11858–11867. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.