

Abstract
Hepatitis B virus (HBV) is a major risk factor for the development and progression of hepatocellular carcinoma (HCC). It has been reported that viral infection can interfere with the expression of cellular microRNA (miRNA) to affect oncogenesis. In this study, we showed that miR-520c-3p was upregulated in liver tumor specimens, and we revealed that HBV infection enhanced the expression of miR-520c-3p through the interaction of viral protein HBV X protein (HBx) with transcription factor CREB1. We further showed that miR-520c-3p induced by HBV transfection/infection caused epithelial-mesenchymal transition (EMT). Using the miRNA target prediction database miRBase and luciferase reporter assays, we identified PTEN as a novel target gene of miR-520c-3p and miR-520c-3p directly targeted PTEN’s 3′-untranslated region. Moreover, we discovered that HBV promoted EMT via the miR-520c-3p-PTEN to activate AKT-NFκB signaling pathway, leading to increased HCC migration and invasion. Importantly, miR-520c-3p antagomir significantly represses invasiveness in HBx-induced hepatocellular xenograft models. Our findings indicate that miR-520c-3p is a novel regulator of HBV and plays an important role in HCC progression. It may serve as a new biomarker and molecular therapeutic target for HBV patients.
Keywords: MT: non-coding RNAs, hepatocellular carcinoma, miR-520c-3p, HBV/HBx, PTEN, migration and invasion, NF-κB
Graphical abstract
Introduction
Hepatitis B virus (HBV) is a major risk factor for the development and progression of hepatocellular carcinoma (HCC), which associates highly with poor prognosis and cancer metastasis. During HCC progression, hepatoma cells undergo epithelial-mesenchymal transition (EMT) to a mesenchymal phenotype with invasive capacities, thereby increasing the invasion and migratory capacity.1,2 But the molecular mechanisms of thus EMT are complex, with many players known to participate in EMT, including microRNAs (miRNAs).3
miRNAs are a class of conserved, small, noncoding RNA molecules that can negatively control gene expression at the transcriptional and/or post-transcriptional levels. miRNAs can function as either oncogenes or tumor suppressors through the suppression of key genes involved in cancer development and progression.4 Increasing evidence indicates that miRNAs are aberrantly expressed in HBV-HCC,5 and the dysfunction of miRNAs has a crucial role in tumor invasion and metastasis. Therefore, further understanding the involvement of miRNAs in HBV-related HCC metastasis and the associated molecular mechanisms is of great value for the development of new therapeutic strategies.
As an oncogenic virus, HBV can induce HCC (HBV-HCC) both directly and indirectly, where HBV X protein (HBx) is believed to play a key role. HBx, a 17-kDa protein, is not only essential for viral replication,6 but also contributes to HCC EMT. For example, HBx has been reported to induce EMT by repressing E-cadherin expression via the upregulation of transcriptional repressors E12/E477 or DNA methyltransferase 18 or insulin-like growth factor-II.9 Moreover, HBx has also been reported to regulate the transcription of some miRNAs to induce EMT, e.g., the repression of miR-148a to activate the AKT/mammalian target of rapamycin (mTOR) pathway to affect EMT.10 In contrast, Zhou et al.11 suggested that HBx-induced miR-3188 expression promotes tumor dissemination via the ZHX2-Notch1 signaling pathway. Although the pathological relevance and significance of HBx-regulated miRNAs in HBV-associated hepatocarcinogenesis have attracted much attention in recent years, the roles for other HBx-regulated miRNAs in hepatoma invasion and metastasis and the associated molecular mechanisms remain largely unknown.
miR-520c can function as an oncogene or a tumor suppressor, depending on the type of human cancer.12, 13, 14, 15, 16, 17 Interestingly, for HCC, there were two opposing findings. Miao et al.15 showed that miR-520c-3p acted as an antagonist of HCC, whereas Zheng et al.18 and Toffanin et al.19 showed the opposite results. To gain a better understanding of the roles of miR-520c-3p in HCC, we evaluated miR-520c-3p expression and function in HCC. We found that miR-520c-3p functions as an oncogene and is upregulated in HBV-infected primary human hepatocytes, hepatoma cells, and in HBV-associated HCC and adjacent tissues. We further demonstrated that PTEN, a major negative regulator of phosphoinositide 3-kinase (PI3K) activation, is a direct target of miR-520c-3p. In addition, miR-520c-3p promotes HCC cell migration and metastasis via PTEN-dependent activation of AKT/nuclear factor (NF)-κB pathway, leading to EMT. Thus, our findings identified a novel molecular mechanism for HBV enhancement of HCC invasion and metastasis and revealed a new potential therapeutic strategy for the adjuvant treatment of aggressive HCCs after surgical resection.
Results
miR-520c-3p expression is upregulated by HBV infection
To investigate if and how miR-520c-3p plays a role in HCC, especially for HBV-related HCC, a poor prognosis disease prevalent in Asia, we searched The Cancer Genome Atlas (TCGA) database and analyzed miR-520c-3p expression in HCC. We found that miR-520c-3p expression was elevated in HCC (Figure 1A) and that high miR-520c-3p expression correlated with decreased overall survival (Figure 1B). To independently confirm these interesting findings, we analyzed the expression of miR-520c-3p in a liver tumor tissue microarray with 30 primary HCC tissues as well as adjacent tissues by RNA fluorescence in situ hybridization (Figure 1C, left panel and Figure S1). We quantified the data by microarray scanning and found that miR-520c-3p expression was significantly higher in tumor tissues than in adjacent tissues and distal tissues (Figure 1C, right panel).
Figure 1.

HBV upregulates miR-520c-3p expression
(A) miR-520c-3p expression levels are elevated in HCC tissues based on analysis of the TCGA miRNA sequencing dataset. (B) TCGA data analyses show that HCC patients with high levels of miR-520c-3p expression had poorer survival than those with low levels of miR-520c-3p expression. (C) miRNA in situ hybridization (ISH) analyses show that miR-520c-3p level is higher in tumors than in adjacent non-tumor tissues or the distal normal tissue regions of 30 HCC patients’ microarray. (Left) A representative RNA fluorescence in situ hybridization result of miR-520c-3p in liver tissue microarray from tumors, adjacent non-tumors, and the distal regions of 30 patients with HCC, respectively. (Right) The microarray scanning data analysis of miR-520c-3p level in individual liver tissues. (D) miR-520c-3p expression is higher in HBV-positive HCC tissues. The 30 HCC patient tumors in (C) were separated into two groups based on the presence of HBV in the HCC. The miR-520c-3p ISH microarrays as shown in (C) for the 24 HBV-positive and 6 HBV-negative HCCs thus identified were scanned and presented individually. (E) qPCR shows that miR-520c-3p expression was much higher in HBV-infected HCC tissues compared with adjacent tissues. (F) HBV infection or transfection increases miR-520c-3p expression. (Left) PHHs were infected with HBV virus and miR-520c-3p expression was analyzed. (Right) Huh7 cells were transfected with pUC18 (mock) or pHBV1.3 plasmids and the miR-520c-3p level was measured by qPCR. (Three independent experiments were performed in [F], and representative data are shown.) ∗p < 0.05, ∗∗p < 0.01 in (C), (E), and (F).
HCC is known to be highly associated with a background of chronic and persistent HBV infection. Thus, to determine whether miR-520c-3p acts as a biomarker of HBV-induced HCC, we separated the HCC samples in the above microarray based on the presence or absence of HBV; the result showed that the expression of miR-520c-3p in HCC liver tumor tissues with HBV infection were much higher than that without HBV infection (Figure 1D). We also found that miR-520c-3p expression was much higher in HBV-infected HCC liver tissue samples than in adjacent tissues by quantitative PCR (qPCR) (Figure 1E). These results suggested that HBV infection may lead to increased miR-520c-3p expression. To test this, we infected primary human hepatocytes (PHHs) with HBV viruses (Figure 1F, left) and found that the expression of miR-520c-3p was upregulated by HBV. Similar results were obtained when we transfected Huh7 or HepG2 cells with pHBV1.3 plasmids (Figure 1F, right, Figure S2). Collectively, these results indicate that HBV infection upregulates the expression of miR-520c-3p.
HBV enhances cell migration and invasion by upregulating miR-520c-3p expression in hepatoma cells and in a nude mouse model
To investigate whether the upregulation of miR-520c-3p is involved in HBV-related HCC progression, we investigated whether miR-520c-3p expression affected the proliferation of liver cancer cells in the presence of HBV and found that neither overexpression nor knockdown of miR-520c-3p had any effect on proliferation in HepG2.2.15 cells (Figure S3). Next, we asked whether miR-520c-3p expression altered tumor cell migration and invasion, which are key for tumor metastasis. First, using the transwell Matrigel invasion assay, we showed that transfection of pHBV1.3 expression plasmid into Huh7 cells significantly enhanced cell invasion (Figure S4). Then, we transfected Huh7 cells with miR-520c-3p mimics, miR-520c-3p inhibitor, or the corresponding non-specific negative controls and found that cell migration, which was evaluated with the wound healing assay, and cell invasion, which was evaluated with the transwell cell invasion assay, were both enhanced with the miR-520c-3p mimic (Figure 2A) and reduced with the miR-520c-3p inhibitor (Figure 2B).
Figure 2.

HBV enhances hepatoma cell migration and invasion by upregulating miR-520c-3p
(A) miR-520c-3p promotes HCC migration and invasion. Huh7 cells were transfected with miR-520c-3p mimic or NC-mimic. The resulting cells were subjected to wound healing assay to measure the migration of the cells (left, top, representative photos of the assay; bottom, quantification of the assay) or Matrigel invasion assay to measure the invasiveness of the cells (right, top, representative photos of the assay; bottom, quantification of the assay). (B) Anti-miR-520c-3p inhibits HCC migration and invasion. Huh7 cells were transfected with miR-520c-3p inhibitor or NC-inhibitor. The cells were then assayed as in (A). (C) miR-520c-3p regulates the expression of proteins involved in EMT. NC-mimic or miR-520c-3p-mimic and NC-inhibitor or miR-520c-3p inhibitor (150 nM) were transfected into Huh7 cells, and the levels of Vimentin, E-cadherin, and MMP9 were examined by western blot. Note that MMP9 and Vimentin were positively regulated by miR-520c-3p while E-cadherin was negatively regulated by miR-520c-3p, suggesting that miR-520c-3p promotes EMT. (D) miR-520c-3p mediates HBV-induced cell migration and invasion. Huh7 cells were transfected with pUC18 or pHBV1.3 plasmids and NC-inhibitor or miR-520c-3p inhibitor before assaying for cell migration and invasion as in (A). (E) miR-520c-3p is required for enhanced EMT by HBV. Huh7 cells were transfected as in (D), and the levels of Vimentin, E-cadherin, and MMP9 were examined by western blot. (F) miR-520c-3p promotes metastasis in vivo. (Left) The levels of miR-520c-3p in LM3-miR-520c stably transfected cells or control cells were detected by qPCR. (Middle and Right) Huh7-miR-520c stably transfected cells were injected intravenously into male BALB/c nude. Eight weeks later, the animals were subjected to live imaging (middle) and the relative fluorescence intensity of the mice was calculated by using ImageJ. The scatter diagram was drawn by using GraphPad Prism software (right). (G) Representative HE images of mouse liver (left) and lung (right) tissues. (Three independent experiments were performed, and representative data are shown.) ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 in (A), (B), (D) and (F).
It is known that the increase in cell migration is typically associated with EMT. Thus, we examined three main protein markers of EMT, E-cadherin, vimentin and matrix metalloproteinase 9 (MMP9), by western blot analysis and found that miR-520c-3p overexpression in Huh7 cells increased the levels of vimentin and MMP9, but decreased the level of E-cadherin, which are indicative of induction of the EMT. The opposite trend was observed in Huh7 cells transfected with anti-miR-520c-3p (Figure 2C). These data suggest that miR-520c-3p plays a role in the EMT in HCC.
To determine whether HBV-induced metastasis is mediated by miR-520c-3p, we co-transfected Huh7 cells with miR-520c-3p-inhibitor and pHBV1.3 plasmid or their corresponding controls and found that ectopic expression of HBV increased cells migration and invasion and that this increase was blocked by miR-520c-3p-inhibitor (Figure 2D). Additionally, HBV transfection also increased the levels of vimentin and MMP9 level and reduced the E-cadherin level, indicative of increased EMT and all these changes were also blocked by miR-520c-3p-inhibitor (Figure 2E). Thus, HBV induces cell migration and invasion by upregulating miR-520c-3p.
To study the in vivo effects of altered miR-520c-3p expression, we studied an HCC-LM3-miR-520c stably transfected cell line with elevated levels of miR-520c-3p in mouse (Figure 2F, left). As expected, 8 weeks after the HCC-LM3-miR-520c stably transfected cells were injected intravenously into 4- to 5-week-old male BALB/c nude mice (six mice per group), live imaging of the mice showed that miR-520c-3p promoted metastasis in vivo (Figure 2F, middle and right). We performed a histologic analysis of liver and lung tissues from nude mice injected with HCC-LM3 cells stably expressing miR520c-3p by hematoxylin and eosin (HE) staining and found that the normal lung structure disappeared and lung tumors were visible (Figure 2G, lung panel) and that the liver was extensively occupied by HCC-like lesions consisting of lipid-rich and basophilic cells with high cytologic atypia (Figure 2G, liver panel). Thus, our results indicate that miR-520c-3p promotes cancer metastasis.
HBx of HBV upregulates miR-520c-3p expression by binding to CREB1 to activate miR-520c promoter
To investigate how HBV regulates miR-520c-3p expression, we examined the expression of miR-520c-3p in Huh7 cells transiently expressing individual HBV genes and found that miR-520c-3p expression was significantly higher in Huh7 cells transfected with HBx, but not with other HBV genes (Figure 3A, upper left). In addition, unlike the pHBV1.2 plasmid, the pHBV1.2-X– plasmid, in which the HBx gene was deleted, failed to enhance miR-520c-3p expression when transfected into cells (Figure 3A, lower left). Moreover, qPCR analysis showed that the HBx expression level was much higher in cancerous liver tissues than in adjacent liver tissues from HBV-infected HCC patients (Figure 3A, upper right). Further analysis showed that upregulation of miR-520c-3p correlated positively with HBx levels in HCC tissues (Figure 3A, lower right panel). Together, our findings indicate that upregulation of miR-520c-3p by HBV is dependent on the viral oncogene HBx.
Figure 3.

HBx interacts with CREB1 to upregulate miR-520c-3p expression
(A) HBx of HBV enhances miR-520c-3p expression. (Left top) The plasmid expressing viral protein HBp, HBc, HBs, or HBx, or the vector control was transfected into Huh7 cells, and the miR-520c-3p level was measured by qPCR. (Left bottom) Huh7 cells were transfected with pcDNA3.0, pcDNA3.0-HBV1.2, or pcDNA3.0-HBV1.2-X plasmids and the levels of miR-520c-3p was analyzed. (Right top) HBx expression was measured in cancer tissues or the adjacent tissues of HBV-related HCC by qPCR. (Right bottom) Correlation between relative expression of miR-520c -3p and that of HBx, as measured by qPCR. (B) Sequence of the promoter region showing the CREB1 binding sites likely required for CREB1 activation of the miR-520c promoter. Red underlined sequence: primer set 1; blue underlined sequence: primer set 2. The primer sets were used for ChIP analysis in D. Yellow color sequence: potential CREB1 binding sites. (C) CREB1 mediates the induction of miR-520c-3p by HBx. (Left) CREB1 upregulates miR-520c-3p expression. The plasmid expressing CREB1 or CREB2 or p53 or vector control were transfected into Huh7 cells, and the miR-520c-3p level was measured by qPCR. (Middle) The plasmid-expressing CREB1 was cotransfected with HBx plasmids or control plasmid into Huh7 cells, and the miR-520c-3p level was measured by qPCR. (Right) Knocking down CREB1 prevents the upregulation of miR-520c-3p by HBx. Huh7 cells were co-transfected with the plasmid expressing HBx and/or CREB1 siRNAs or siNC, and miR-520c-3p expression was analyzed. (D) HBx binds to CREB1 and is recruited to the miR-520c promoter by CREB1 to activate the promoter. (Left) HBx binds CREB1. Huh7 cells were transfected with the indicated plasmids and protein extracts were subjected to co-immunoprecipitation in Huh7 cells. (Right) A promoter region containing the CREB1 binding site is required for CREB1 activation of the miR-520c promoter. HEK293 cells were co-transfected with different miR-520c promoter constructs driving the expression of luciferase, pxj40-CREB1-Flag or empty vector and pRL-TK, and the luciferase activity of the transfected cells were measured. (E) HBx and CREB1 are associated with the promoter in vivo as shown by ChIP assay. Huh7 cells were transfected with Myc-CREB1 and/or Flag-HBx were subjected to ChIP assay with two sets of primers in the promoter region (top) and co-IP western blot analyses (bottom). (F) Inhibition of miR-520c-3p blocks HBx-induced cell migration and invasion. Huh7 cells were transfected with indicated plasmid and/or inhibitor and subjected to cell migration and invasion as in Figure 2D. (Three independent experiments were performed, and representative data are shown.) ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 in (A) and (B–F).
HBx, the most critical carcinogenic component of all HBV proteins, lacks double-strand DNA binding activity and functions as a trans-activator through interaction with transcription factors such as c-Myc,6 p53,10 and CREB,11,20 to activate several signal transduction cascades, leading to the regulation of a number of genes, including angiogenic factors, oncogenes, and those involved in metastasis. Interestingly, we discovered putative CREB-binding sites in the miR-520c-3p promoter region using ALGGE Promo (http://alggen.lsi.upc.edu/) and JASPAR (http://jaspar.genereg.net/) (Figure 3B, sequences labeled in yellow). To test whether CREB1 or CREB2 regulates the miR-520c-3p promoter, expression plasmids for CREB1, CREB2, or p53 were transfected into Huh7 cells, and only the CREB1 plasmid increased miR-520c-3p expression (Figure 3C, left). Furthermore, the co-expression of HBx with CREB1 further enhanced miR-520c-3p expression (Figure 3C, middle), whereas knockdown of CREB1 blocked the increased expression of miR-520c-3p by HBx (Figure 3C, right).
The above results suggested that HBx-mediated upregulation of miR-520c-3p by binding to CREB1. To further investigate this, we studied the interaction between HBx and CREB1. We co-transfected HBx and CREB1 into Huh7 cells and analyzed binding between CREB1 and HBx by co-immunoprecipitation assay (Figure 3D, left). In addition, when luciferase reporter constructs bearing the wild-type or deletion constructs of the miR-520c-3p promoter were analyzed, we found that the region containing the putative binding sites of CREB1 was required for transcriptional activation by CREB1 (Figure 3D, right). Finally, the chromatin immunoprecipitation (ChIP) assay showed that CREB1 was bound to the miR-520c-3p promoter region containing CREB1 binding sites in cells transfected the CREB 1 plasmid (Figure 3E, left). A two-step cross-linking ChIP assay of cells co-transfected with tagged HBx and CREB1 showed that HBx was also recruited to the miR-520c-3p promoter region (Figure 3E, right). Taken together, these data strongly indicate that HBx promotes miR-520c transcription through binding to the miR-520c promoter by associating with CREB1.
Earlier studies have showed that HBx promotes cell migration and invasion.21 We consistently found that HBx enhanced cell migration and invasion in Huh7 cells (Figure S5A). An X mutant in which the HBx gene was deleted lacked the ability to enhance cell migration and invasion (Figure S5B). To determine if HBx regulates cell migration and invasion by upregulating miR-520c-3p, we carried out co-transfection studies and found that the miR-520c-3p inhibitor blocked the enhancement of cell migration and invasion by HBx (Figure 3F).
miR-520c-3p downregulates PTEN expression via the PTEN 3′-untranslated region
To further investigate the pro-metastatic role of miR-520c-3p, we sought to identify the relevant endogenous targets of miR-520c-3p. We used two target prediction programs, namely, TargetScan and miRbase, to screen targets for miR-520c-3p, and PTEN, a well-known tumor suppressor that can regulate cancer development and progression,22, 23, 24, 25 was identified as a putative target of miR-520c-3p. To assess whether PTEN is regulated by miR-520c-3p, we generated a wild-type PTEN 3′-untranslated region (UTR) luciferase reporter plasmid (pMIR-PTEN 3′-UTR) and its mutant plasmid (pMIR-PTEN 3′-UTR-M), in which the miR-520c-3p-binding site was altered (Figure 4A, left), and analyzed the effect of miR-520c-3p on reporter activity. The data showed that miR-520c-3p decreased the luciferase activity of pMIR-PTEN 3′-UTR, but not of the pMIR-PTEN 3′-UTR-M mutant (Figure 4A, right). Furthermore, the miR-520c-3p mimics also dramatically decreased endogenous PTEN mRNA and protein levels, while the opposite results were obtained with the miR-520c-3p inhibitor (Figure 4B). These data indicate that PTEN is a direct target of miR-520c-3p. We also checked PTEN expression in HCC-LM3-miR-520c stably transfected cells and found that PTEN expression was also downregulated (Figure S6).
Figure 4.

miR-520c-3p targets PTEN to mediate HBV-induced cell migration and invasion
(A) Bioinformatics identification of the miR-520c-3p binding site in PTEN 3′-UTR and its validation with a reporter assay. PTEN 3′-UTR luciferase reporter plasmid (PTEN-wild type) or its mutant plasmid (PTEN-Mut) were transfected into Huh7 cells in the presence of miR-520c-3p mimic or scrambled control, and relative luciferase expression is shown as the mean ± standard deviation. (B) miR-520c-3p regulates the level of PTEN in cells. Huh7 cells were transfected with miR-520c-3p mimic/inhibitor or NC miRNA. At 48 h after transfection, the levels of PTEN were examined by qPCR (top) and western blot (bottom). (C) PTEN mRNA and protein levels are much lower in HBV-infected HCC tissues compared with their adjacent normal tissues and the reduction in PTEN level is accompanied by increased miR-520c-3p level in human patients. (Top) qPCR analysis of PTEN levels in HCC (T) or adjacent normal (N) tissues. (Middle) Western blot analysis of PTEN levels in HCC (T) or adjacent normal (N) tissues on randomly selected 18 out of patient tissues, the level of PTEN expression was significantly lower in HCC tumor tissues compared with their adjacent normal tissues by western blot. (Bottom) PTEN mRNA expression was negatively correlated with miR-520c-3p mRNA expression in HCC tumor tissues (R = −0.5000, p = 0.0016). (D) PTEN mRNA and protein levels were decreased by HBV in PHHs and Huh7 cells. (Left) HBV virus-infected PHHs; (right) HBV plasmids transfected into Huh7 cells. At 48 h after infection/transfection, PTEN mRNA (top) and protein (bottom) levels were determined. (E) Anti-miR-520c-3p prevented the downregulation of PTEN protein levels by HBV. Huh7 cells were co-transfected with HBV and/or miR-520c-3p-inhibitor or controls and subjected to western blot analysis of the level of PTEN. (F) PTEN is important for HBV-induced migration, invasion, and EMT via miR-520c-3p. Huh7 cells were transfected with pUC18 or pHBV1.3, NC-inhibitor, or miR-520c-3p inhibitor, and siNC or siPTEN and were then assayed for migration and invasion as in Figure 2A (left). The levels of the EMT-associated proteins Vimentin, E-cadherin, and MMP9 were determined by western blot analyses (right). Three independent experiments were performed, and representative data are shown. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 in (A) and (B) and (D–F).
To determine whether HBV downregulates PTEN by enhancing miR-520c-3p expression, we analyzed PTEN expression in hepatic tissues from a large number of HBV-infected patients with HCC. The results showed that PTEN mRNA and protein levels were lower in HBV-infected tumor tissues than in adjacent non-cancerous tissues (Figure 4C, top and middle). More important, PTEN expression correlated negatively with miR-520c-3p expression (Figure 4C, bottom). Next, total RNA and protein extracted from control or HBV virus-infected PHHs were subjected to qPCR and western blot analysis. The data showed that PTEN mRNA and protein levels were inhibited by HBV virus infection in PHHs (Figure 4D, left). Consistently, PTEN mRNA and protein levels were also lower in Huh7 and HepG2 cells transfected with pHBV1.3 than in cells transfected with the vector control (Figure 4D, right, and Figure S7).
To determine how pHBV1.3 regulates PTEN, we carried out co-transfection studies in Huh7 cells. As expected, we found that PTEN was downregulated by HBV and this downregulation was blocked by the miR-520c-3p inhibitor (Figure 4E). Importantly, HBx, but not the HBx mutant, also decreased the PTEN protein level (Figure S8A), and this decrease was reversed by an miR-520c-3p inhibitor (Figure S8B). Thus, HBV inhibits the PTEN level by increasing miR-520c-3p expression, mainly if not exclusively via HBx.
PTEN has phosphatase-dependent and phosphatase-independent (scaffold) activities in a wide variety of human cancers through regulating a variety of cellular processes, including cell migration, invasion, proliferation, survival, and energy metabolism.26 To investigate whether PTEN is involved in HBV-induced metastasis, we co-transfected cells with pHBV1.3 and/or miR-520c-3p inhibitor and/or small interfering RNA (siRNA) to knock down PTEN and found that miR-520c-3p inhibitor blocked the HBV-induced cell migration and invasion and this blockage was reversed by simultaneously knocking down of PTEN (Figure 4F, left and middle). Furthermore, these effects were accompanied by the corresponding changes in EMT as revealed by the expression of the three EMT marker genes (Figure 4F, right). Similar results were obtained by co-transfecting HBx with miR-520c-3p inhibitor and/or siPTEN (Figures S9A and S9B). These data strongly indicate that HBV/HBx represses PTEN expression by upregulating miR-520c-3p, thereby increasing the metastatic properties of hepatoma cells.
HBV regulates the miR-520c-3p-PTEN axis to induce the EMT in PHHs
To test whether HBV affects the EMT in PHHs, we infected PHHs with HBV virus before transfecting the miR-520c-3p antagomir. HBV infection upregulated miR-520c-3p and MMP9 expression and downregulated PTEN and E-cadherin expression, and incubation with the miR-520c-3p antagomir reversed the HBV-induced changes in EMT marker expression (Figures 5A and 5B). These data thus strongly suggest that the miR-520c-3p/PTEN axis mediates HBV-induced changes in EMT marker gene expression in PHHs. Although in the asymptomatic phase of chronic HBV-infection where liver damage does not occur, we speculate that these changes in the EMT marker genes by HBV infection of hepatocytes would be advantageous for the future development of HCC.
Figure 5.

HBV regulates the miR-520c-3p-PTEN axis to induce metastasis in PHHs
PHHs were infected with HBV viruses, followed by transfection with NC-antagomir or miR-520c-3p-antagomir. The levels of miR-520c-3p and PTEN were measured by qPCR (A) and the expression of EMT markers (B) were determined by western blot. ∗∗p < 0.01, ∗∗∗p < 0.001.
miR-520c-3p activates AKT via PTEN to mediate the effects of HBV/HBx
PTEN is known to negatively regulate PI3K/AKT signaling, which can affect EMT of HCC cells. Moreover, a loss-of-function mutant of PTEN enhanced human cancer aggressiveness by activating the Akt pathway.27 In addition, miRNAs can regulate the PI3K/AKT oncogenic pathway involved in human malignancies, such as liver cancer.28 It has also been reported that HBV induces AKT/GSK3β signaling to facilitate EMT and metastasis during HCC progression.29 Thus, to determine whether HBx activates AKT to induce EMT and hepatoma cells metastasis through miR-520c-3p-PTEN axis, Huh7 cells were transfected with an HBV- or an HBx-expressing plasmid. Western blot analysis showed a remarkable increase in tyrosine (T308) and serine (S473) phosphorylation of AKT, which indicates AKT activation, without altering the total protein level in cells transfected with HBV or HBx, but not in cells transfected with control vectors (Figure 6A left). Like that shown above, overexpression of HBV or HBx decreased E-cadherin and increased vimentin and MMP9 levels (Figure 6A left). Similar results were observed in HepG2 cells (Figure S10). Importantly, miR-520c-3p mimic also activated AKT, while miR-520c-3p inhibitor had the opposite effect (Figure 6A right). Again, similar results were observed in HepG2 cells (Figure S11A). These results indicate that miR-520c-3p activates AKT.
Figure 6.

HBx of HBV activates the PI3K/AKT pathway to promote cell migration and invasion via miR-520c-3p-mediated downregulation of PTEN
(A) Both miR-520c-3p and HBV/HBx activate AKT. Huh7 cells were transfected with HBV or HBx (left), miR-520c-3p mimics/inhibitors or corresponding NC miRNA (right). Huh7 cells were transfected with HBV/HBx or miR-520c-3p, after 48 h transfection, the cells were analyzed for the levels of AKT, p-AKT, Vimentin, E-cadherin, MMP9, and PTEN levels by western blot. (B) Downregulation of PTEN is required for the activation of AKT by miR-520c-3p. Huh7 cells were co-transfected with pEF-Flag (control) or PTEN plus miR-520c-3p mimic, and western blot analyses were performed for the indicated proteins. (C) Inhibiting miR-520c-3p prevents the activation of AKT by HBx, while knocking down PTEN reverses this effect. Huh7 cells were co-transfected with HBx, miR-520c-3p inhibitor, and siPTEN and western blot analyses were performed for the indicated proteins. (D) AKT activation is required for miR-520c-3p-induced cell migration and invasion. Huh7 cells were transfected with miR-520c-3p mimic or NC-mimic for 36 h, and treated with MK2206, a specific inhibitor of AKT phosphorylation, or dimethyl sulfoxide for 12 h. The cells were then subjected to western blot analyses for the indicated proteins (left) or cell migration and invasion assays as in Figure 2A (right). (E) AKT activation is required for HBx-induced cell migration and invasion. Huh7 cells at 36 h after transfection with pxj40-HBx-HA or empty vector were treated with MK2206 or dimethyl sulfoxide for 12 h. The cells were then subjected to western blot analyses for the indicated proteins (left) or cell migration and invasion assays as in Figure 2A (right). Three independent experiments were performed, and representative data are shown. ∗∗p < 0.01, ∗∗∗p < 0.001 in (D–E).
To determine whether this activation involves PTEN, we first confirmed that PTEN suppressed AKT activity in transfected HepG2 cells (Figure S11B). Next, we performed a rescue experiment. miR-520c-3p with pEF-flag-PTEN or pEF-flag were co-transfected into Huh7 cells. We found that PTEN expression prevented the activation of AKT or EMT by miR-520c-3p (Figure 6B). These findings raise an intriguing possibility that HBx-induced miR-520c-3p activates AKT pathway via inhibition of PTEN. Thus, we co-transfected HBx, miR-520c-3p-inhibitor, and/or siPTEN into Huh7 cells. As expected, anti-miR-520c-3p reversed the activation of AKT and EMT by HBx (Figure 6C), while simultaneous knockdown of PTEN rescued the effects of HBx (Figure 6C). Similar results were observed in HepG2 cells (Figure S12).
To further investigate the role of the AKT signaling pathway in HBx-induced metastasis via miR-520c-3p, we treated miR-520c-3p- or HBx-transfected Huh7 cells with MK2206, a specific inhibitor of AKT phosphorylation and found that phosphorylation of the AKT residues Thr308 and Ser473 were indeed repressed by MK2206 (Figures 6D and 6E, left). Importantly, wound healing and transwell assays with Matrigel (Figures 6D and 6E, right) revealed that the inhibition of AKT signaling with MK2206 resulted in a dramatic decrease in HBx/miR-520c-3p-induced cell migration and invasion, and this finding was also supported by western blot analysis of EMT markers (Figures 6D and 6E, left). Similar results were obtained with LY294002, another specific inhibitor of the PI3K/AKT pathway (Figure S13).
The HBx-miR-520c-3p-PTEN axis regulates AKT/NF-κB signaling to affect cell migration and invasion
Having demonstrated that HBx activates the AKT pathway to promote cell migration and invasion via the miR-520c-3p-PTEN pathway, we turned to events downstream of AKT. Previous studies have demonstrated that the effects of AKT activation involves the regulation of activated-IκB kinase, a kinase that induces the degradation of NF-κB inhibitor IκB30; glycogen synthase kinase-3β,29 a serine/threonine kinase involved in Wnt signaling, and the mTOR,31 a serine/threonine kinase that responds to nutrients to regulate cell cycle, survival, cell proliferation, and EMT. Accumulating evidence has demonstrated that NF-κB is a direct downstream target of the AKT pathway in endothelial injury30; thus, we hypothesized that the effects of HBx-induced miR-520c-3p expression on cell migration and invasion result from alteration of the NF-κB pathway. To test this, Huh7 cells were pre-treated with SC-514, a specific inhibitor of the native IKK complex and the IKK-α/IKK-β heterodimer, followed by transfection with HBx or miR-520c-3p. The results showed that the inhibition of NF-κB by SC-514 abolished the ability of miR-520c-3p or HBx to activate IκBα (Figures 7A and 7B left). Wound healing and Matrigel invasion assays showed that the cell migration and invasion effects of HBx were blocked by SC-514 (Figure 7B, right). Western blot analysis showed that the effect of HBx on EMT marker gene expression was also blocked by SC-514 (Figure 7B, left). The same conclusions were obtained with IMD-0354, another specific inhibitor of NF-κB (Figure S14). Furthermore, when a super-repressor6 that suppresses the canonical NF-κB pathway was used, we observed similar results (Figures 7C and 7D). To further determine whether NF-κB was specifically regulated by AKT or other events downstream of AKT also participated in HBx-miR-520c-3p axis-mediated cell migration, we used inhibitors that target downstream targets of AKT, such as inhibitor PD98058 that targets MAPK/ERK, inhibitor SP600125 that targets JNK, inhibitor XAV-939 that targets Wnt, inhibitor rapamycin for mTOR, and inhibitor AS1842856 for FOXO. We observed that none of these inhibitors affected miR-520c-3p-induced cell migration or invasion (Figure 7E). Taken together, these results revealed that the miR-520c-3p/PTEN/AKT/NF-κB pathway is essential for HBx-induced cell migration and invasion.
Figure 7.

PI3k/AKT-dependent activation of NF-κB by miR-520c-3p is important for HBx-induced migration and invasion
(A) miR-520c-3p activates NF-κB (p-IκBα). Huh7 cells were pretreated with 5 mM NF-κB inhibitor SC-514 or dimethyl sulfoxide for 4 h followed by transfection with NC or miR-520c-3p mimic. After 48 h, the levels of the indicated proteins were examined by western blot analyses. (B) HBx induces cell migration and invasion via activating NF-κB (p-IκBα). Huh7 cells were pretreated with SC-514 followed by transfection with pxj40-HA-HBx or empty vector. They were then subjected to western blot analyses for the indicated proteins (left) or cell migration and invasion assays as in Figure 2A (right). (C and D) Inhibition of the NF-κB pathway via inactivation of IκBα reverses the ability of miR-520c-3p or HBx to induce EMT and cell invasion. Huh7 were co-transfected with MIGR1-IκBα and miR-520c-3p or HBx for 48 h, and then the levels of the indicated proteins were examined by western blot analysis (C and D, left), and the cell migration and invasion assays were performed (D, right). (E) miR-520c-3p activates NF-κB (p-IκB). Huh7 cells were pretreated with signaling inhibitors (PD98058 for MAPK/ERK, SP600125 for JNK, XAV-939 for Wnt, rapamycin for mTOR, and AS1842856 for FOXO) for 4 h followed by transfection with NC or miR-520c-3p mimic and subjected to invasion assays. Three independent experiments were performed, and representative data are shown. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 in (B–D).
HBV regulates the miR-520c-3p-PTEN axis to promote tumor metastasis in vivo
To examine the effect of miR-520c-3p on HBV/HBx-induced tumor metastasis in vivo, we generated a stable HBx-overexpression HepG2 cell line (Figure 8A) and performed subcutaneous xenograft experiments to analyze the effect of miR-520c-3p on HBV/HBx-induced tumor progression. HepG2-GFP-HBX-Flag cells were injected into left dorsal subcutaneous tissues of nude mice. After 2 weeks, tumors of the same size were selected to be injected with negative control (NC) or miR-520c-3p antagomir twice a week (5 nM/mouse). After 4 weeks of treatment with the antagomir, no differences were observed in the growth or size of tumors treated with the miR-520c-3p antagomir and with the NC antagomir (Figure S15), which is consistent with our in vitro cell proliferation results. However, when mouse bioluminescence signals were measured to determine metastasis by using a live imaging system, the results showed that treatment with the miR-520c-3p antagomir inhibited HBx-induced metastasis compared with treatment with the NC antagomir (Figure 8B). As expected, treatment with the miR-520c-3p antagomir altered miR-520c-3p and PTEN expression in xenograft tumors (Figure 8C). We also measured PTEN, p-Akt, Akt, p-IκBα, and IκBα protein levels in xenograft tumors and found that treatment with the miR-520c-3p antagomir inhibited p-Akt and p-IκBα expression (Figure 8D), which is consistent with our in vitro results above. Furthermore, bioluminescence signals in the liver and lung also showed that the targeting of miR-520c-3p attenuated HBx-induced metastasis (Figure 8E).
Figure 8.

miR-520c-3p antagomir inhibition of HBX-induced tumor progression in vivo
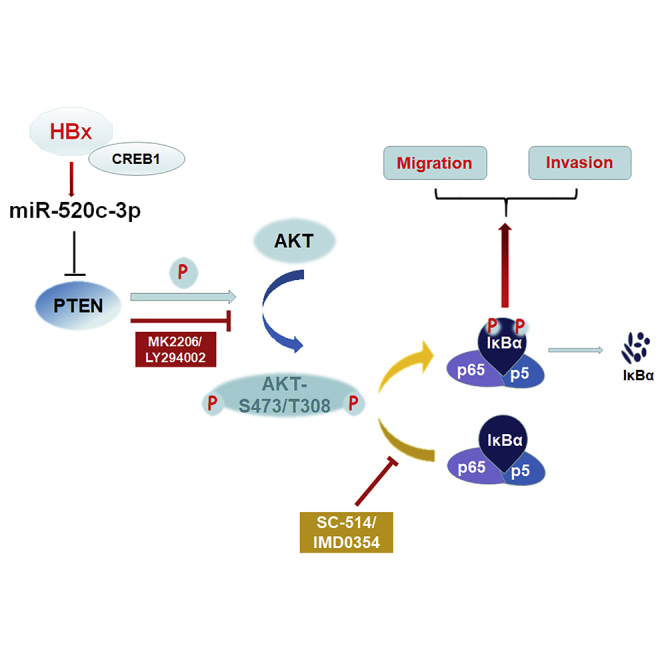
(A) qPCR and western blot assays showed a significantly higher expression of HBx in a stable HBx-overexpression HepG2 cell line. (B) Mice were imaged with live image equipment. The relative fluorescence intensity of the mice was calculated by using ImageJ, and the scatter diagram was drawn by using GraphPad Prism software. (C) Expression of PTEN and miR-520c-3p in tumors with NC/miR-520c-3p-antagomir-treatment were analyzed by qPCR. (D) Proteins in the AKT/NF-κB pathway were analyzed in tumor tissues by western blot. (E) Representative live images of mouse livers and lungs. (F) A model for HBV-induced cell migration and invasion. HBV, via HBx binding to CREB1, upregulates miR-520c-3p, which in turn downregulates PTEN, thereby leading to the activation of AKT/NF-κB pathway to enhance HCC EMT and thus cell migration and invasion. This study reveals that miR-520c-3p, upregulated by HBx of HBV, targets PTEN, an anti-oncogene, to promote hepatoma cell migration and metastasis through the activation of AKT/NF-κB pathway. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Discussion
The pathological relevance of HBx in HBV-associated hepatocarcinogenesis has attracted much attention in recent years. The enhanced migration and invasion capability of hepatoma cells elicited by HBx is closely related to HBV-induced initiation and progression of hepatocarcinogenesis.21,32 However, the mechanism remains poorly understood. Our study reveals a regulatory network initiated by the interaction between HBx and CREB1, which bind together to the promoter of miR-520c and increase the level of miR-520c-3p to activate AKT/NF-κB pathway by targeting PTEN. The final consequence is the promotion of metastasis of HCC derived from HBV infection (Figure 8F).
Over the past decade, miRNAs have been found to be involved in various biological processes, including tumorigenesis and metastasis.4 Chromosome 19 contains the largest miRNA cluster and encodes many miRNAs including miR-520a, b, and c-h, members of the miR-520 family. miR-520c-3p is encoded by the miR-520c gene located on chromosome 19q13.42, is a non-conserved miRNA, and its mature sequence contains 22 nt.33 miR-520c-3p functions as an oncogene12,13,16 or a tumor suppressor15, 16, 17,30 in different malignant tumors, even of the same cancer type, for example, breast cancer and HCC. Huang et al.12 suggested that miR-520c stimulates cancer cell migration and invasion by suppression of CD44 in breast cancer, whereas Tang et al.34 suggested that miR-520c-3p acts as a tumor suppressor in estrogen receptor-negative breast cancer. As in breast cancer, miR-520c-3p has paradoxical functions in liver cancer. Miao et al.15 used a liver cancer cell model to show that miR-520c-3p induced HCC cell apoptosis and inhibited cell growth and invasion. Similarly, Zhang et al.16 reported that miR-520c-3p was downregulated in HCC tissues compared with adjacent non-tumor tissues. However, Zheng et al.18 reported that miR-520c-3p was highly expressed in HCC tissues compared with the corresponding non-cancerous tissues. Toffanin et al.19 investigated 89 HCV-related HCC samples and also found higher miR-520c expression in the HCC tissues and further showed that miR-520c increased cell proliferation, migration, and invasion of Huh7 and SNU-449 cells. Although the reasons for these different findings are unclear, they may be due to the use of different cancer lines, genetic variabilities, various subclones of the same cell line, different types of tissue samples, and different experimental designs. To avoid single cell lines affecting the experiment results, in our study, we used multiple cell lines (Huh7, HepG2, HepG2.2.15, and HCC LM3 cells) to prove our conclusions. Given that the function of miR-520c-3p in HCC is still controversial, we analyzed the data in the TCGA database and found that the expression of miR-520c-3p in HCC tissues was significantly higher than that in normal tissues, which suggests that miR-520c-3p may be an oncogene in HCC. Because most Chinese patients with HCC are HBV positive and no studies have investigated the relationship between miR-520c-3p and HBV-related HCC progression, we focused on the role of miR-520c-3p in HBV-related HCC. We demonstrated that HBV infection significantly increases miR-520c-3p expression via HBx and that miR-520c-3p expression is higher in HBV-positive HCC tissues than in adjacent noncancerous tissues. Our findings may also provide a potential explanation for the previously observed discrepancy in the correlation between miR-520c-3p expression and HCC: HCC tissues with varying levels of HBV infection may have been used in the differing studies.
More important, we showed that HBV enhanced miR-520c-3p expression in HBV plasmid-transfected Huh7/HepG2 cells, HBV-expressing stable cell line HepG2.2.15, and HBV virus-infected PHHs. Mechanistically, we showed that this upregulation of miR-520c-3p was due to HBx binding to CREB1 to activate the miR-520c promoter in Huh7 cells. It is known that HBx can act as a coactivator to affect transcription through association with transcriptional factors.20,35,36 For example, HBx interacts with c-Myc to repress miR-192-3p expression.6 HBx also acts as a transcriptional coactivator by directly binding to CREB to increase the DNA binding affinity and transcriptional activity of CREB.11,20 However, the increase in the DNA binding affinity of CREB by HBx is necessary, but not sufficient, to explain the co-activation of CREB by HBx.36 Cougot et al.41 suggested that HBx might recruit CBP/p300 to enhance CREB activity. However, CREB includes CREB1 and CREB2, the previous study did not show which CREB bind to HBx. Our research showed that HBx binding to the miR-520c-5p promoter activates miR-520c-5p transcription via CREB1, not CREB2.
Functionally, we used Matrigel invasion and wound healing assays to evaluate cell invasion and cell migration, respectively, and analyzed the expression of EMT markers to evaluate EMT. We found that increased miR-520c-3p expression by HBV infection leads to increased metastasis via induction of the EMT in both Huh7 and HepG2 cells. We showed that the miR-520c-3p antagomir significantly attenuates HBx-induced liver and lung metastasis in vivo. Thus, we revealed a positive regulatory relationship between HBV upregulation of miR-520c-3p via HBx and advanced HCC progression, highlighting the potential of nucleic acid-based therapeutics for the treatment of HCC.
We discovered that PTEN is a direct target of miR-520c-3p and that the downregulation of PTEN by miR-520c-3p is critical for HBV-induced liver cancer progression. Consistent with a previous report by Tian et al.42, we further revealed that HBx played a major role in suppression of PTEN via miR-520c-3p in HBV expressing cells. PTEN is a potent inhibitor of the PI3K/Akt pathway. We consistently showed that miR-520c-3p induces phosphorylation of AKT, which activates the AKT pathway that inhibits PTEN expression in hepatoma cells.
In recent years, increasing attention has been given to the carcinogenic role of AKT via its activation of multiple signaling pathways, including ERK/P38, Wnt, mTOR, and NF-κB, that promote malignant transformation and growth of HBV-infected hepatomas. However, the NF-κB pathway has dual functions on inflammation and tumorigenesis.37 The activation of NF-κB has been observed in proliferating immune cells and in most tumor cells that progress to cancer, such as HCC.38 However, NF-κB can also function as a tumor suppressor. Wilson et al.39 reported that NF-κB1-mediated transcriptional repression of neutrophil-recruiting chemokines represses the development of HCC. By using pathway-specific inhibitors and a super-repressor to stably knockdown canonical NF-κB, we found that inhibition of the NF-κB signaling pathway markedly attenuated miR-520c-3p-induced hepatoma cell migration and invasion. Thus, HBV regulates the NF-κB pathway via the HBx/miR-520c-3p/PTEN/AKT axis to promote HCC EMT and metastasis.
Conclusion
In our system, we discovered that miR-520c-3p functions as a novel regulator of HBV-induced HCC metastasis. Our findings suggest a novel model in which HBx promotes HCC migration and invasion. HBx upregulates miR-520c-3p, which negatively regulates PTEN, resulting in the activation of the AKT/NF-κB pathway and induction of metastasis (Figure 7F). Our study has thus uncovered novel insights into a pivotal molecular mechanism underlying tumor invasion and metastasis in HBV-associated HCC and has revealed potentially novel therapeutic targets to treat metastatic HCC patients with chronic HBV infection.
Materials and methods
Cell cultures and transfection
Human hepatoma cells Huh7, HepG2, and HCCLM3 and HBV-expressing stable cell line HepG2.2.15 were cultured in DMEM. For HepG2.2.15 cells, G418 were also included in the culture medium. PHHs were purchased, cultured, transfected, and infected as described.6 siRNAs, the miR-520c-3p-mimic/inhibitor/antagomir, and the respective NCs were purchased from RiboBio. Plasmids (200 ng–2 μg) were introduced into hosts with Lipofectamine 2000 and miRNA/siRNA (50–100 nM) were introduced into hosts with Lipofectamine RNAiMAX (Invitrogen).
Plasmid construction
Plasmids pHBV1.3, pXJ40-HA-X, pXJ40-HA-P, pXJ40-HA-S, and pXJ40-HA-C were reported previously.7 Plasmid MIGR1-IκBα-DN was a generous gift from Prof. Zan Huang (College of Life Sciences, Wuhan University). Plasmids pXJ40-Flag-X and pXJ40-Flag-CREB1 were generated by using pXJ40-Flag vector. Plasmids MDH1-PGK-GFP_2.0-miR520c and MDH1-PGK-GFP_2.0-vector were generated by using retroviral vector of MDH1-PGK-GFP_2.0. The pMIR-PTEN 3′-UTR fragment including miR-520c-3p binding site (GCACTTA) was cloned into the pMIR-REPORT (Applied Biosystems). pMIR-PTEN-3′UTR-MUT, which contained the mutated site (GTGTAT) was made via site-directed mutagenesis. The 5′-flanking regions (nucleotides −3887 ∼ +31) of miR-520c was cloned into pGL3.0-basic vector to generate pGL3.0–3918. The regions (−3887 ∼ −1999, −3887 ∼ −3110, −3110 ∼ −1999, and −1999 ∼ +31) of miR-520c were PCR amplified from pGL3.0–3918 and were inserted into pGL3.0-basic to generate various luciferase reporter plasmids. All constructs were confirmed by DNA sequencing. Primers used in this study are shown in Table S1.
Chemicals, antibodies, and other reagents
MK2206 (T1952), SC-514 (T2118), IMD0354 (T6141), and Ly294002 (T2008) were purchased from Targetmol (MedChem Express), antibodies against PTEN (#9188S), Akt (#4685S), pAkt(T308) (#13038S), pAkt(S473) (#4060S), IKBα (#4812S) and p-IKBα (#9246S) were purchased from Cell Signaling Technology, antibodies against GAPDH, Flag-Tag (20543-1-AP), HA-Tag (51064-2-AP), Myc-Tag, Vimentin (10366-1-AP), MMP9 (10375-2-AP), and E-cadherin (20874-1-AP) were purchased from ProteinTech Group. Horseradish peroxidase (HRP)-conjugated secondary antibody (71045-M and 12–348) and Flag Beads (M8823) were purchased from Sigma (Aldrich).
HBV virus isolation
Hep2.2.15 cells were cultured in DMEM supplemented with 5% inactivated fetal bovine serum. After 4 days, HBV viruses in the medium culture was collected by ultracentrifugation at 40,000 rpm for 3 h at 30°C. Following centrifugation, the HBV viral particles appearing at the bottom or side of the tube as a faint white pellet were resuspended in 1/100 of original volume using DMEM.
Immunoblotting
Cellular proteins were extracted with RIPA (Beyotime) supplemented with PMSF (Beyotime) and PhosSTOP (Roche). After centrifugation, the supernatant was quantified by using bicinconinic acid protein kit (ThermoFisher Scientific). Total proteins (15–30 μg/lane) were separated by SDS-PAGE then transferred to a nitrocellulose membrane. The membranes were blocked in 5% milk for 1 h at room temperature (RT). After the membranes were incubated with primary antibodies overnight and followed by incubating with HRP-conjugated secondary antibodies (Sigma-Aldrich) at RT for 1 h. Undergoing washed by TBST, membranes were detected by an enhanced chemiluminescence system western Blotting Detection Kit (Bio-Rad).
RNA extraction, reverse transcription PCR, and quantitative real-time PCR
Total RNA was extracted from cells and tissues by using Trizol (Invitrogen) and was reversely transcribed into cDNA with random primers or miRNA with miRNA specific primers purchased from RiboBio by using the PrimeScript RT reagent Kit (TakaRa). The qPCR was performed by using gene primers or miRNA-specific qPCR primers purchased from RiboBio by using SYBR Select Master Mix (Life technologies). The PCR procedure was: 95°C for 3 minutes first, then 39 cycles at 95°C for 20 s, 60°C for 30 s, and 72°C for 30 s. The results were obtained with CFX96TM Real-time System 3.1 software (Applied Bio-Rad) and further analyzed with the 2−ΔΔct method. The expression level of an mRNA or miRNA was calculated relative to the level of control U6 or GAPDH, respectively. All primers are listed in Table S1.
Dual-luciferase reporter gene assay
Huh7 cells were seeded in 24-well plates for 12 h before transfection. To verify the target sites of miRNAs, cells were co-transfected with the firefly luciferase reporter constructs containing the wild-type or mutant’s 3′-UTR fragment of PTEN, Renilla vector (pRL-TK) and NC-mimic or miR-520c-3p mimic. To verify the binding between the miR-520c promoter region and CREB1, various constructs containing different lengths of the miR-520c 5′-flanking region or pGL3.0-basic, CREB1 and pRL-TK were introduced into Huh7 cells. The cell lysates were harvested for a Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer’s instructions. The firefly and Renilla luciferase activities were measured and the ratio of firefly/Renilla luciferase values was determined as a measure of the reporter activity.
ChIP assay
A ChIP assay was performed with Huh7 cells co-transfected with HBx and CREB1. Briefly, cells were fixed with 1% formaldehyde and then lysed. The chromatin was sheered with an ultrasonic cell disruptor. The protein-DNA complexes were precipitated with normal IgG and anti-FLAG M2 agarose beads (Sigma-Aldrich). After immunoprecipitation, the protein-DNA cross-links were reversed and the DNA was purified. The promoter region of miR-520c including the CREB1 binding sites were amplified from the immunoprecipitated DNA samples with specific primers.
Two-step cross-linking ChIP
After pouring out the medium from the cell culture dish, the cells were rinsed with PBS to remove the remaining medium. Cross-linking proteins to proteins was done by adding freshly made DSG solution (0.25 M disuccinimidyl glutarate [DSG] stock: dissolve 50 mg DSG in 100% dimethyl sulfoxide to a concentration of 0.25 M). This is followed by cross-linking proteins to DNA by adding 1% formaldehyde drop-wise directly to the media and rotate gently at RT for 10 min. The cells were then subjected to the rest of the ChIP assay as above.
Co-immunoprecipitation assay
We lysed 5 × 106 Huh7 cells in Western & IP Lysis Buffer (Beyotime) containing protease inhibitors. The supernatant was incubated with indicated antibodies, followed by the addition of protein A/G beads. The immunoprecipitated proteins were analyzed on western blot.
Transwell Matrigel invasion assay
The 24-well transwell chamber with 8.0 μm pore transparent PET membrane (BD Biosciences) was used to analyze the migratory and invasive ability of Huh7. At 24 h after indicated transfection, a total of 5 × 104 transfected cell numbers suspended in 100-μL serum-free medium were plated on the Matrigel-coated (BD Biosciences) membrane of the upper chamber and full serum medium was added at the bottom of the transwell. After incubation for 48 h, the cells remained in the upper chamber were removed with a cotton swab. Cells invaded through the pores to the opposite side of the membrane were fixed in 4% paraformaldehyde, and then stained with crystal violet for 20 min. Three randomized fields were captured for each transwell under the microscope and the cells in the field were counted.
Assays of wound healing
Transfected cells grown in six-well plates to 95% confluent monolayers were mechanically scratched by using a 10-μL pipette tip to create a wound. After the scratching, the cells were gently washed three times to remove detached cells. The culture dish was then placed in a cell culture incubator at 37°C and cell culture images were captured under a microscope at 0, 24, and 48 h with blue boxes outlining the starting wound area. The migration index (%) was determined from the wound area measured with ImageJ (National Institutes of Health) software. Three different batches of results were statistically analyzed using ImageJ.
CCK-8 assay
A CCK-8 kit (Dojindo) was used to measure the proliferation of HepG2.2.15 cells. A total of 3000 cells in a volume of 100 μL/well were cultured in six replicate wells in a 96-well plate in A medium containing 10% FBS. Then, 10 μL CCK-8 reagent were then added per well at 0, 24, 48, and 72 h and incubated for 1 h for cell quantification.
Tissue sample preparation
A total of 30 patients with HCC with chronic HBV infection for miR-520c-3p and PTEN mRNA and protein level analysis who underwent surgical resection and pathologically diagnosed with HCC in Hubei Cancer Hospital, Huazhong University of Science and Technology were enrolled in the study. All donors gave permission and written informed consent was obtained from all participants.
Generation of HCC-LM3 -miR-520c stably transfected cell line
To establish miR-520c-GFP overexpression cell lines, we used a vector-based retroviral technique. Recombinant retroviruses were produced by co-transfecting 293T cells with the MDH1-PGK-miR-520c-GFP retroviruses expression plasmid or MDH1-PGK-GFP_2.0-vector plasmid and packaging plasmids (ZV77) by using Lipofectamine 2000 (Invitrogen). Infectious retroviruses were collected 48 h after transfection. The supernatant was centrifuged to remove cell debris and filtered through 0.45-μm filters (Millipore). The viruses were concentrated by using Lenti-XTM Concentrator (Takara). HCC-LM3 cells were transduced with either empty vector or miR-520c-GFP retroviruses with 8μg/mL polybrene to obtain stable cell line. The miR-520c-3p expression efficiency was determined by qPCR.
Generation of the HepG2-GFP-HBX-flag stably transfected cell line
To establish the GFP-HBX stably overexpression cell lines, the full-length HBx cDNA was cloned into the empty lentivector lenti-CMV-GFP-Flag, and the recombinant plasmids were co-transfected with psPAX2 and pMD2.G into 293T cells by using Lipofectamine 2000 for lentivirus production. The methods for lentivirus concentration and transfection into the target cells were as described above. The HBx overexpression efficiency was determined by western blot with anti-flag antibodies.
Xenograft mouse model and the subcutaneous tumor model
Animal studies were approved by the Institutional Animal Care Committee of School of Basic Medical Sciences, Wuhan University. For the tail vein injection model, 2.0 × 106 LM3-miR-520c stable cells diluted into 100 μL PBS were injected intravenously into 4- to 5-week-old female BALB/c nude (six mice per group). Eight weeks post injection, mice were imaged live with live image equipment (PerkinElmer). The relative fluorescence intensity of the mice was calculated by using ImageJ, and the scatter diagram was drawn by using GraphPad Prism software. The lungs and livers of nude mice were used for HE staining analysis. For the subcutaneous tumor model, 5.0 × 106 HepG2-GFP-HBx-flag cells were diluted into 50 μL PBS, mixed with 50 μL Matrigel, and planted under the armpit of 3-week-old female mice by subcutaneous injection. When the tumor grows to 1.0 cm × 1.0 cm, 10 nM miR520c-3p antagomir and its corresponding control diluted into 100 μL PBS were injected into the tumor (six mice per group), twice a week for 2 weeks; 4 weeks after the first injection, the nude mice were analyzed for small animal live imaging and HE staining.
miRNA in situ hybridization
The miRNA-520 and scrambled oligonucleotides for in situ hybridization were purchased as digoxigenin-labeled locked nucleic acid (LNA) probes from Exiqon (Denmark). Liver cancer tissue microarrays (catalog HLivH090PG01) were purchased from Shanghai Outdo Biotech CO, LTD, and miR-ISH was performed by the same company, and slides were read as previously described. The intensity of the staining was scored as negative (–/0), weak (+/1), moderate (++/2), or strong (+++/3) as previously described.40
HE staining
The tissue samples were fixed with 10% formalin, and after 24 h, they were embedded to obtain paraffin blocks, then 4-μm-thick tissue sections were prepared with a Leica microtome. Subsequently, the tissue sections were deparaffinized, rehydrated, stained with hematoxylin for 3 to 5 min and stained with eosin for 5 min. Finally, the slices were dehydrated and covered with neutral gum. The sections were scanned with a panoramic scanner (3DHISTECH Case Viewer) to obtain high-resolution images.
Statistical analysis
All results are expressed as the mean ± standard deviation from at least three independent experiments. Unless otherwise noted, the differences between groups were analyzed by using the unpaired Student’s t test when only two groups were compared or by one-way ANOVA when more than two groups were compared. Differences with a p value of less than 0.05 were considered statistically significant. All analyses were performed using SPSS software, version 16.0 (SPSS, Inc).
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81572447, 31871427, 81572006, and 31370187). Y.B.S. was supported by the Intramural Research Program of NICHD, National Institutes of Health. The funders had no role in the design of the study, data collection, and interpretation, or in writing the manuscript.We thank Professor Yuchen Xia and the staffs at the Research Center for Medicine and Structural Biology of Wuhan University for technical assistance.
Author contributions
Study design: G.S. and M.G.; Data collection: Y.L., J.W., J.C., S.W., X.Z., Q.X., Y.G., J.S., F.S., J.X., S.Y., C.L., Y.H., M.W., M.G., and G.S.; Statistical analysis: G.S., J.W., Y.L., J.C., S.W., X.Z., Q.X., Y.G., and L.C.; Manuscript writing: G.S., J.W., M.G., and Y.L.; Critical revision of the manuscript: G.S., Y.B.S., D.G., J.W., and M.G. All authors read and approved the final manuscript.
Declarations of interests
The authors declare that they have no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2022.05.031.
Contributor Information
Mingxiong Guo, Email: guomx@whu.edu.cn.
Deyin Guo, Email: guodeyin@mail.sysu.edu.cn.
Guihong Sun, Email: ghsunlab@whu.edu.cn.
Supplemental information
References
- 1.Diepenbruck M., Christofori G. Epithelial-mesenchymal transition (EMT) and metastasis: yes, no, maybe? Curr. Opin. Cell Biol. 2016;43:7–13. doi: 10.1016/j.ceb.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Markopoulos G.S., Roupakia E., Tokamani M., Chavdoula E., Hatziapostolou M., Polytarchou C., Marcu K.B., Papavassiliou A.G., Sandaltzopoulos R., Kolettas E. A step-by-step microRNA guide to cancer development and metastasis. Cell. Oncol. 2017;40:303–339. doi: 10.1007/s13402-017-0341-9. [DOI] [PubMed] [Google Scholar]
- 4.Calin G.A., Croce C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 5.Sartorius K., Makarova J., Sartorius B., An P., Winkler C., Chuturgoon A., Kramvis A. The regulatory role of MicroRNA in hepatitis-B virus-associated hepatocellular carcinoma (HBV-HCC) pathogenesis. Cells. 2019;8:1504. doi: 10.3390/cells8121504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang J., Chen J., Liu Y., Zeng X., Wei M., Wu S., Xiong Q., Song F., Yuan X., Xiao Y., et al. Hepatitis B virus induces autophagy to promote its replication by the Axis of miR-192-3p-XIAP through NF kappa B signaling. Hepatology. 2019;69:974–992. doi: 10.1002/hep.30248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shin Kim S., Yeom S., Kwak J., Ahn H.J., Lib Jang K. Hepatitis B virus X protein induces epithelial-mesenchymal transition by repressing E-cadherin expression via upregulation of E12/E47. J. Gen. Virol. 2016;97:134–143. doi: 10.1099/jgv.0.000324. [DOI] [PubMed] [Google Scholar]
- 8.Xie Q., Chen L., Shan X., Shan X., Tang J., Zhou F., Chen Q., Quan H., Nie D., Zhang W., et al. Epigenetic silencing of SFRP1 and SFRP5 by hepatitis B virus X protein enhances hepatoma cell tumorigenicity through Wnt signaling pathway. Int. J. Cancer. 2014;135:635–646. doi: 10.1002/ijc.28697. [DOI] [PubMed] [Google Scholar]
- 9.Ha H.L., Kwon T., Bak I.S., Erikson R.L., Kim B.Y., Yu D.Y. IGF-II induced by hepatitis B virus X protein regulates EMT via SUMO mediated loss of E-cadherin in mice. Oncotarget. 2016;7:56944–56957. doi: 10.18632/oncotarget.10922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu X., Fan Z., Kang L., Han J., Jiang C., Zheng X., Zhu Z., Jiao H., Lin J., Jiang K., et al. Hepatitis B virus X protein represses miRNA-148a to enhance tumorigenesis. J. Clin. Invest. 2013;123:630–645. doi: 10.1172/jci64265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou S.J., Deng Y.L., Liang H.F., Jaoude J.C., Liu F.Y. Hepatitis B virus X protein promotes CREB-mediated activation of miR-3188 and Notch signaling in hepatocellular carcinoma. Cell Death Differ. 2017;24:1577–1587. doi: 10.1038/cdd.2017.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang Q., Gumireddy K., Schrier M., le Sage C., Nagel R., Nair S., Egan D.A., Li A., Huang G., Klein-Szanto A.J., et al. The microRNAs miR-373 and miR-520c promote tumour invasion and metastasis. Nat. Cell Biol. 2008;10:202–210. doi: 10.1038/ncb1681. [DOI] [PubMed] [Google Scholar]
- 13.Yang K., Handorean A.M., Iczkowski K.A. MicroRNAs 373 and 520c are downregulated in prostate cancer, suppress CD44 translation and enhance invasion of prostate cancer cells in vitro. Int. J. Clin. Exp. Pathol. 2009;2:361–369. [PMC free article] [PubMed] [Google Scholar]
- 14.Keklikoglou I., Koerner C., Schmidt C., Zhang J.D., Heckmann D., Shavinskaya A., Allgayer H., Guckel B., Fehm T., Schneeweiss A., et al. MicroRNA-520/373 family functions as a tumor suppressor in estrogen receptor negative breast cancer by targeting NF-κB and TGF-β signaling pathways. Oncogene. 2012;31:4150–4163. doi: 10.1038/onc.2011.571. [DOI] [PubMed] [Google Scholar]
- 15.Miao H.L., Lei C.J., Qiu Z.D., Liu Z.K., Li R., Bao S.T., Li M.Y. MicroRNA-520c-3p inhibits hepatocellular carcinoma cell proliferation and invasion through induction of cell apoptosis by targeting glypican-3. Hepatol. Res. 2014;44:338–348. doi: 10.1111/hepr.12121. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y., Xu J., Zhang S., An J., Zhang J., Huang J., Jin Y. HOXA-AS2 promotes proliferation and induces epithelial-mesenchymal transition via the miR-520c-3p/GPC3 Axis in hepatocellular carcinoma. Cell. Physiol. Biochem. 2018;50:2124–2138. doi: 10.1159/000495056. [DOI] [PubMed] [Google Scholar]
- 17.Li X., Fu Q., Li H., Zhu L., Chen W., Ruan T., Xu W., Yu X. MicroRNA-520c-3p functions as a novel tumor suppressor in lung adenocarcinoma. FEBS J. 2019;286:2737–2752. doi: 10.1111/febs.14835. [DOI] [PubMed] [Google Scholar]
- 18.Zheng J., Sadot E., Vigidal J.A., Klimstra D.S., Balachandran V.P., Kingham T.P., Allen P.J., D'Angelica M.I., DeMatteo R.P., Jarnagin W.R., Ventura A. Characterization of hepatocellular adenoma and carcinoma using microRNA profiling and targeted gene sequencing. PLoS One. 2018;13 doi: 10.1371/journal.pone.0200776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Toffanin S., Hoshida Y., Lachenmayer A., Villanueva A., Cabellos L., Minguez B., Savic R., Ward S.C., Thung S., Chiang D.Y., et al. MicroRNA-based classification of hepatocellular carcinoma and oncogenic role of miR-517a. Gastroenterology. 2011;140:1618–1628.e16. doi: 10.1053/j.gastro.2011.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang T., Zhang J., You X., Liu Q., Du Y., Gao Y., Shan C., Kong G., Wang Y., Yang X., et al. Hepatitis B virus X protein modulates oncogene Yes-associated protein by CREB to promote growth of hepatoma cells. Hepatology. 2012;56:2051–2059. doi: 10.1002/hep.25899. [DOI] [PubMed] [Google Scholar]
- 21.Liu H., Xu L., He H., Zhu Y., Liu J., Wang S., Chen L., Wu Q., Xu J., Gu J. Hepatitis B virus X protein promotes hepatoma cell invasion and metastasis by stabilizing Snail protein. Cancer Sci. 2012;103:2072–2081. doi: 10.1111/cas.12017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cao L.Q., Yang X.W., Chen Y.B., Zhang D.W., Jiang X.F., Xue P. Exosomal miR-21 regulates the TETs/PTENp1/PTEN pathway to promote hepatocellular carcinoma growth. Mol. Cancer. 2019;18:148. doi: 10.1186/s12943-019-1075-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang B., Feng X., Liu H., Tong R., Wu J., Li C., Yu H., Chen Y., Cheng Q., Chen J., et al. High-metastatic cancer cells derived exosomal miR92a-3p promotes epithelial-mesenchymal transition and metastasis of low-metastatic cancer cells by regulating PTEN/Akt pathway in hepatocellular carcinoma. Oncogene. 2020;39:6529–6543. doi: 10.1038/s41388-020-01450-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Song M.S., Salmena L., Pandolfi P.P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 2012;13:283–296. doi: 10.1038/nrm3330. [DOI] [PubMed] [Google Scholar]
- 25.Worby C.A., Dixon J.E. Pten. Annu. Rev. Biochem. 2014;83:641–669. doi: 10.1146/annurev-biochem-082411-113907. [DOI] [PubMed] [Google Scholar]
- 26.Lee Y.R., Chen M., Pandolfi P.P. The functions and regulation of the PTEN tumour suppressor: new modes and prospects. Nat. Rev. Mol. Cell Biol. 2018;19:547–562. doi: 10.1038/s41580-018-0015-0. [DOI] [PubMed] [Google Scholar]
- 27.Parsons R. Discovery of the PTEN tumor suppressor and its connection to the PI3K and AKT oncogenes. Cold Spring Harb. Perspect. Med. 2020;10:a036129. doi: 10.1101/cshperspect.a036129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rahmani F., Ziaeemehr A., Shahidsales S., Gharib M., Khazaei M., Ferns G.A., Ryzhikov M., Avan A., Hassanian S.M. Role of regulatory miRNAs of the PI3K/AKT/mTOR signaling in the pathogenesis of hepatocellular carcinoma. J. Cell. Physiol. 2020;235:4146–4152. doi: 10.1002/jcp.29333. [DOI] [PubMed] [Google Scholar]
- 29.Liu L., Dong Z., Liang J., Cao C., Sun J., Ding Y., Wu D. As an independent prognostic factor, FAT10 promotes hepatitis B virus-related hepatocellular carcinoma progression via Akt/GSK3β pathway. Oncogene. 2014;33:909–920. doi: 10.1038/onc.2013.236. [DOI] [PubMed] [Google Scholar]
- 30.Jiao Y., Zhao D., Gao F., Hu X., Hu X., Li M., Cui Y., Wei X., Xie C., Zhao Y., Gao Y. MicroRNA-520c-3p suppresses vascular endothelium dysfunction by targeting RELA and regulating the AKT and NF-κB signaling pathways. J. Physiol. Biochem. 2021;77:47–61. doi: 10.1007/s13105-020-00779-5. [DOI] [PubMed] [Google Scholar]
- 31.Garcia-Lezana T., Lopez-Canovas J.L., Villanueva A. Signaling pathways in hepatocellular carcinoma. Adv. Cancer Res. 2021;149:63–101. doi: 10.1016/bs.acr.2020.10.002. [DOI] [PubMed] [Google Scholar]
- 32.Sirma H., Giannini C., Poussin K., Paterlini P., Kremsdorf D., Brechot C. Hepatitis B virus X mutants, present in hepatocellular carcinoma tissue abrogate both the antiproliferative and transactivation effects of HBx. Oncogene. 1999;18:4848–4859. doi: 10.1038/sj.onc.1202867. [DOI] [PubMed] [Google Scholar]
- 33.Bentwich I., Avniel A., Karov Y., Aharonov R., Gilad S., Barad O., Barzilai A., Einat P., Einav U., Meiri E., et al. Identification of hundreds of conserved and nonconserved human microRNAs. Nat. Genet. 2005;37:766–770. doi: 10.1038/ng1590. [DOI] [PubMed] [Google Scholar]
- 34.Tang C.P., Zhou H.J., Qin J., Luo Y., Zhang T. MicroRNA-520c-3p negatively regulates EMT by targeting IL-8 to suppress the invasion and migration of breast cancer. Oncol. Rep. 2017;38:3144–3152. doi: 10.3892/or.2017.5968. [DOI] [PubMed] [Google Scholar]
- 35.Unger T., Shaul Y. The X protein of the hepatitis B virus acts as a transcription factor when targeted to its responsive element. EMBO J. 1990;9:1889–1895. doi: 10.1002/j.1460-2075.1990.tb08315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barnabas S., Andrisani O.M. Different regions of hepatitis B virus X protein are required for enhancement of bZip-mediated transactivation versus transrepression. J. Virol. 2000;74:83–90. doi: 10.1128/jvi.74.1.83-90.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Markopoulos G.S., Roupakia E., Tokamani M., Alabasi G., Sandaltzopoulos R., Marcu K.B., Kolettas E. Roles of NF-κB signaling in the regulation of miRNAs impacting on inflammation in cancer. Biomedicines. 2018;6:40. doi: 10.3390/biomedicines6020040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moles A., Butterworth J.A., Sanchez A., Hunter J.E., Leslie J., Sellier H., Tiniakos D., Cockell S.J., Mann D.A., Oakley F., Perkins N.D. A RelA(p65) Thr505 phospho-site mutation reveals an important mechanism regulating NF-κB-dependent liver regeneration and cancer. Oncogene. 2016;35:4623–4632. doi: 10.1038/onc.2015.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wilson C.L., Jurk D., Fullard N., Banks P., Page A., Luli S., Elsharkawy A.M., Gieling R.G., Bagchi Chakraborty J., Fox C., et al. Corrigendum: NFκB1 is a suppressor of neutrophil-driven hepatocellular carcinoma. Nat. Commun. 2015;6:8411. doi: 10.1038/ncomms9411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin Y., Zeng Y., Zhang F., Xue L., Huang Z., Li W., Guo M. Characterization of microRNA expression profiles and the discovery of novel microRNAs involved in cancer during human embryonic development. PLoS One. 2013;8:e69230. doi: 10.1371/journal.pone.0069230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cougot D., Wu Y., Cairo S., Caramel J., Renard C.A., Lévy L., et al. The hepatitis B virus X protein functionally interacts with CREB-binding protein/p300 in the regulation of CREB-mediated transcription. The Journal of biological chemistry. 2007;282:4277–4287. doi: 10.1074/jbc.M606774200. [DOI] [PubMed] [Google Scholar]
- 42.Tian Y., Xiao X., Gong X., Peng F., Xu Y., Jiang Y., et al. HBx promotes cell proliferation by disturbing the cross-talk between miR-181a and PTEN. Sci Rep. 2017;7:40089. doi: 10.1038/srep40089. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.