

Abstract
Nonalcoholic fatty liver disease (NAFLD) is the most common type of chronic liver disease in children. The mechanisms that drive NAFLD disease progression in this specific patient population remain poorly defined. In this study, we obtained liver biopsy samples from a multiethnic cohort of pediatric patients with NAFLD (n = 52, mean age = 13.6 years) and healthy liver controls (n = 5). We analyzed transcriptomic changes associated with NAFLD stages using high‐throughput RNA sequencing. Unsupervised clustering as well as pairwise transcriptome comparison distinguished NAFLD from healthy livers. We identified perturbations in pathways including calcium and insulin/glucose signaling occurring early in NAFLD disease, before the presence of histopathologic evidence of advanced disease. Transcriptomic comparisons identified a 25‐gene signature associated with the degree of liver fibrosis. We also identified expression of the insulin‐like growth factor binding protein (IGFBP) gene family (1/2/3/7) as correlating with disease stages, and it has the potential to be used as a peripheral biomarker in NAFLD. Comparing our data set with publicly available adult and adolescent transcriptomic data, we identified similarities and differences in pathway enrichment and gene‐expression profiles between adult and pediatric patients with NAFLD. Regulation of genes including interleukin‐32, IGFBP1, IGFBP2, and IGFBP7 was consistently found in both NAFLD populations, whereas IGFBP3 was specific to pediatric NAFLD. Conclusion: This paper expands our knowledge on the molecular mechanisms underlying pediatric NAFLD. It identifies potential biomarkers and directs us toward new therapies in this population.
The mechanisms that drive NAFLD disease progression in pediatrics remains poorly defined. Using RNA sequencing in liver biopsies of pediatric patients with NAFLD, we identified genes and pathways that are common and distinct to pediatric and adult disease. This work identifies potential biomarkers and mechanisms in the pediatric population.

INTRODUCTION
Over the past few decades, there has been an epidemic of worldwide obesity.[ 1 ] Paralleled with this trend is the growing prevalence of nonalcoholic fatty liver diseases (NAFLD) across all age groups, countries, and ethnicities.[ 2 ] NAFLD consists of a spectrum of pathology hallmarked by the hepatic accumulation of triglycerides, which can progress to nonalcoholic steatohepatitis (NASH).[ 3 , 4 ] Development of NAFLD/NASH is strongly associated with cardiometabolic disease, liver fibrosis/cirrhosis, and hepatocellular carcinoma (HCC).[ 5 , 6 , 7 ] An estimated 25% of the worldwide population is affected, and the rate is on the rise.[ 2 ] Meanwhile, the more alarming trend is the growing prevalence of NAFLD in children and adolescents, which portends an increasing burden on health care systems in the future.[ 3 , 8 ] In the United States, NAFLD is estimated to affect 7 million children and has become the most common cause of pediatric chronic liver disease.[ 4 , 9 ]
Transcriptomic sequencing (RNA sequencing [RNA‐seq]) has proven to be a comprehensive method to examine the changes in gene expression during the development of disease and offers valuable information on disease biology, risk factors, and biomarkers. Two factors should be considered when choosing the cohort to map gene signatures. First, NAFLD is a spectrum of disease covering progressive stages of steatosis, inflammation, and fibrosis; likely distinct transcriptional profiles are associated with disease progression from normal liver to NAFLD to NASH. Second, the pathogenesis of NAFLD in children and adolescents is likely multifactorial, with genetic predisposition playing a significant role. Multiethnic studies of pediatric NAFLD in the United States clearly indicate that Hispanics have the highest prevalence of NAFLD, and African Americans have the lowest.[ 10 , 11 ] Previous studies focused primarily on adults,[ 12 , 13 , 14 ] and therefore may not accurately represent the gene signatures of pediatric patients with NAFLD. In addition, these studies used patient cohorts composed primarily of Western Europeans and may not fully describe differences in other ethnic groups.
In this study, our goal was to use RNA‐seq data to define molecular pathways and gene signatures in pediatric patients associated with different features of NAFLD assessed by liver biopsy. We compared our data set to adolescent and adult NAFLD studies to discover similarities and differences in the mechanisms of NAFLD progression. Importantly, we also aimed to discover genes unique to our data set that may point to distinct mechanistic differences in this age group and are potential targets for future investigations.
MATERIALS AND METHODS
Study participants/approvals/liver biopsy
Fifty‐two pediatric patients with an elevated alanine aminotransferase (ALT) level (> 40 U/l) agreed to participate in this study. Subjects reported no alcohol consumption or medical treatment before biopsy. The nature and potential risks of the study were explained to all subjects/guardians before obtaining their written consent. The study was approved by the ethics committee of Yale University.
Clinically indicated ultrasound‐guided liver biopsies were performed using 15G TruCut liver biopsy needles with a portion of the liver biopsy archived. Biopsies were immediately immersed in RNAlater (Thermo Fisher Scientific) according to the manufacturer’s instructions. Deidentified control liver tissues/RNA from healthy subjects were obtained from University of Pittsburgh Medical Center (n = 3), Mount Sinai Medical Center (n = 1), and Thermo Fisher Scientific (AM7960, Lot No. 2280511).
Histopathological analyses
The histopathology was evaluated by experienced pediatric hepato‐pathologists. Composite histologic activity was assessed using the validated NAFLD activity score (NAS) according to the NASH Clinical Research Network (CRN) scoring system.[ 15 ] NAS is the subtotal of three subscores: hepatocyte ballooning, steatosis, and lobular inflammation. A subscore > 0 indicates presence of these histologic features. NAS was used to categorize the cohort into control, steatosis (NAS ≤ 3), borderline (NAS = 4 or NAS ≥ 5 with one or more subscores = 0), and NASH (NAS ≥ 5, subscores > 0), as described by Govaere et al.[ 12 ] for application of the transcriptional statistical analysis. Fibrosis was evaluated separately from the NAS following established histologic criteria.[ 15 ] In addition, biopsies were categorized on the base of the pattern of injury and presence and degree of specific lesions in diagnostic categories as “NAFL,” “borderline NASH,” or “definite NASH.”[ 16 ]
RNA extraction, RNA‐seq library preparation, and sequencing
RNA was isolated using Total RNeasy Mini Kit (QIAGEN Inc.). RNA‐seq libraries were prepared using NEBNext Poly(A) messenger RNA (mRNA) Magnetic Isolation Module (NEB #E7490) and NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (NEB #E7760). RNA‐seq libraries were validated by MedGenome (San Francisco, CA) before being sequenced on a Nova‐Seq 6000 (Illumina).
RNA‐seq preprocessing
An average of 47.2 million reads were recorded for each sample. For each sample, a pipeline including FastQC,[ 17 ] adapter, and low‐quality reads filtering by Trimmomatic[ 18 ] and alignment to the human reference genome (hg38) with STAR aligner[ 19 ] was performed. Gene read counts were quantified by STAR quantMode GeneCount setting. All tools were run by default parameter setting.
Statistical analysis on transcriptomic and clinical data
To detect the correlation between gene expression and Glu120, Pearson correlation, and linear regression model were applied. Partial least‐squares discriminant analysis (PLS‐DA) was performed by R package ropls [ 20 ] to classify all patients into NASH and fibrosis categories. Euclidean distances for all the pairwise samples were calculated, and hierarchical clustering was performed on all samples grouping them into two major clusters.
Differential expression and Ingenuity Pathway Analysis
Differential expression analysis was performed on raw read count data, comparing steatosis versus control, NASH versus control, NASH versus steatosis, S0 versus S1, S0 versus S2, and S0 versus S3. For each comparison, R package DESeq2 [ 21 ] was used to perform the differential test, and the differentially expressed genes (DEGs) were defined by false discovery rate (FDR) < 5% and absolute fold change ≥ 1.5. For the comparison with control samples, stronger signals were detected in general. Stringent gene‐filtering criteria were applied with FDR < 0.0001 and fold change ≥ 3. Y chromosome genes were filtered to eliminate sex bias. Selected DEGs were applied into Ingenuity Pathway Analysis (IPA; Qiagen) to detect enriched pathways. Significant pathways were defined by FDR < 5%, with positive Z‐scores indicating activated pathways and negative Z‐scores for inhibited pathways. Based on gene‐expression profile, gene‐set variation analysis was performed by R package GSVA [ 22 ] to evaluate the enrichment score of the Kyoto Encyclopedia of Genes and Genomes[ 23 ] metabolic pathways for each sample.
Public data mining
Two adult studies (GSE135251 [ 12 ] and GSE89632 [ 14 ]) and one adolescent study (GSE66676)[ 24 ] were compared with the current pediatric study to check for similarities and differences among pediatric, adolescent, and adult cohorts. A minimum FDR < 0.05% and fold change of 1.5 were used as cutoffs. Differential expression and pathway analyses were applied to all three public studies independently. Detailed data preprocessing steps as well as information on each study cohort are described in the Supporting Methods.
Statistical analysis
All the statistical analysis was performed by R programming and the available R/Bioconductor packages. Data were visualized by R packages ComplexHeatmap [ 25 ] and ggplot2 (https://ggplot2.tidyverse.org).
Oral glucose tolerance test
After 12‐h overnight fasting, 17 participants underwent a 3‐h oral glucose tolerance test (OGTT; 1.75 g glucose/kg body weight, up to 75 g) at the Yale Center for Clinical Investigation, as previously described.[ 26 ] Plasma glucose levels were measured using the Yellow Springs Instruments 2700 STAT Analyzer.
RESULTS
We examined a multiethnic cohort of 52 pediatric patients who received a clinically indicated liver biopsy. The mean age of this cohort was 13.6 ± 2.9 years with 61.5% of the cohort being male (Table 1). The body mass index (BMI) mean was 32.0 ± 5.0 and the mean ALT of this cohort was 206.9 ± 135.5 (U/dl) (Table 1). The patient cohort was also evaluated by its histopathologic diagnostic categories (Table S1)[ 16 ] and according to the NASH CRN guidelines.[ 15 ] There was no statistical difference between Hispanic (69.2%) and non‐Hispanic (28.8%) members of the cohort with respect to several demographic and biochemical measures, except for total cholesterol (Table 1; p = 0.03; 171.4 vs. 203.8, respectively). We incorporated livers from 5 healthy adults (mean age: 47.4 ± 13.8 years; BMI: 28.5 ± 11.5; ALT: 21.3 ± 16.3) to compare their liver transcriptional profiles to the pediatric patients in this study (Table S2).
TABLE 1.
Pathological features of NASH patient samples by ethnicity
| Feature | Total | Hispanic | Non‐Hispanic | p‐value (Hispanic vs. non‐Hispanic) |
|---|---|---|---|---|
| n | 52 | 36 | 15 | |
| Age (years) | 13.6 ± 2.9 | 13.2 ± 2.9 | 14.7 ± 2.8 | 0.10 |
| ♂/♀ | 32/20 | 25/12 | 7/7 | |
| BMI (kg/m2) | 32.0 ± 5.0 | 31.2 ± 5.2 | 33.8 ± 5.4 | 0.13 |
| Z‐score (weight for age) | 2.31 ± 0.52 | 2.28 ± 0.55 | 2.39 ± 0.44 | 0.5 |
| ALT (U/L) | 206.9 ± 135.5 | 208.5 ± 146.5 | 201.5 ± 110.8 | 0.86 |
| AST (U/L) | 108.5 ± 76.3 | 111.14 ± 78.0 | 103.8 ± 77.0 | 0.77 |
| ALP | 215.6 ± 126.1 | 234.6 ± 122.3 | 169.8 ± 131.5 | 0.14 |
| GGT | 74.4 ± 49.1 | 69.4 ± 49.4 | 89.9 ± 48.2 | 0.21 |
| Total cholesterol | 180.4 ± 36.6 | 171.4 ± 33.0 | 203.8 ± 36.7 | 0.03 |
| Triglycerides | 186.4 ± 89.6 | 182.1 ± 93.5 | 197.4 ± 81.9 | 0.63 |
| HbA1c (%) | 5.6 ± 0.5 | 5.6 ± 0.3 | 5.7 ± 0.8 | 0.47 |
| Type 2 diabetes mellitus | 0 | 0 | 0 | |
| Steatosis | 59.8 ± 24.5 | 59.7 ± 23.7 | 58.5 ± 27.9 | 0.89 |
| Steatosis grade | ||||
| 0 | 3 | 1 | 2 | 0.30 |
| 1 | 4 | 4 | 0 | |
| 2 | 16 | 12 | 4 | |
| 3 | 29 | 20 | 8 | |
| Fibrosis stage | ||||
| 0 | 6 | 6 | 0 | 0.42 |
| 1 | 37 | 25 | 11 | |
| 2 | 4 | 3 | 1 | |
| 3 | 5 | 3 | 2 | |
| Lobular inflammation | ||||
| 0 | 7 | 6 | 1 | 0.81 |
| 1 | 37 | 25 | 11 | |
| 2 | 3 | 3 | 0 | |
| 3 | 4 | 3 | 1 | |
| Portal inflammation | ||||
| 0 | 16 | 13 | 3 | 0.39 |
| 1 | 29 | 21 | 7 | |
| 2 | 6 | 3 | 3 | |
| NAS | ||||
| ≥ 5 | 18 | 14 | 3 | 0.33 |
| < 5 | 34 | 23 | 11 |
p‐value calculated by Fisher's exact test. Non‐Hispanic: Caucasian (n = 8), Asian (n = 4), African American (n = 1), non‐Hispanic with unknown ethnicities (n = 2), and unknown (n = 1).
Abbreviations: ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; GGT: gamma‐glutamyltransferase; HbA1C, hemoglobin A1c.
Unsupervised clustering and PLS‐DA divides cohort into two groups
Bulk RNA‐seq analysis was performed using biopsy samples from our pediatric NAFLD cohort and healthy adult controls. Unsupervised clustering divided the cohort into two distinct groups (Figure 1). Group 1 consisted of all five of the control samples as well as two NAFLD cases. PLS‐DA strongly discriminated the transcriptomic profiles between the controls and pediatric NAFLD samples (Figure S1A). No clear distinction was present between the NAFLD samples, indicating highly similar transcriptomic profiles between these groups (Figure 2A).
FIGURE 1.
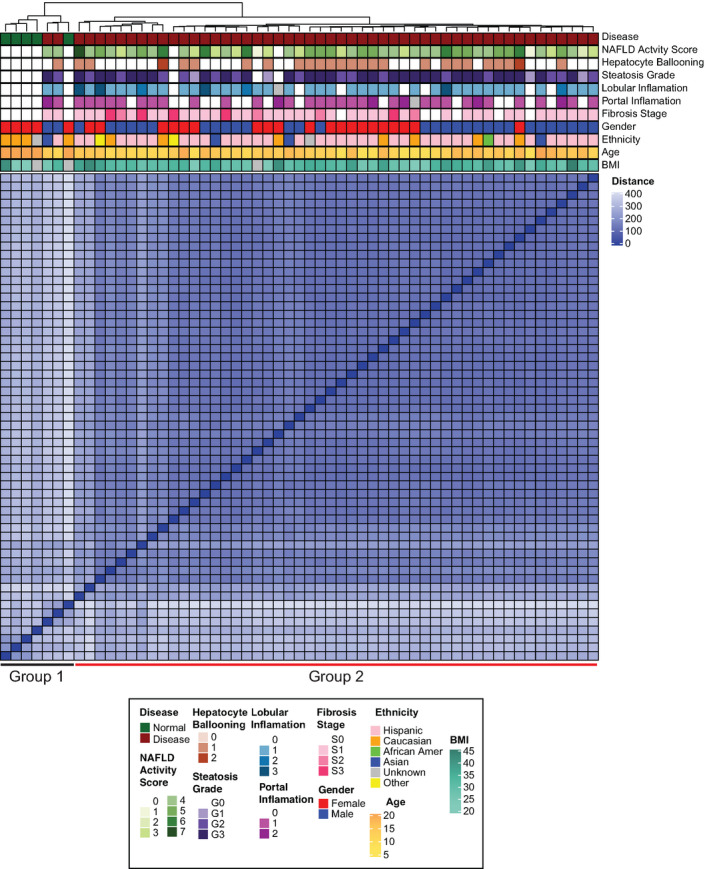
Unsupervised clustering of pediatric patients with nonalcoholic fatty liver disease (NAFLD) (n = 52) and healthy liver controls (n = 5) using RNA sequencing. The heatmap shows the pairwise distance of the samples, and the top annotation bar indicates clinical features of the individual subjects. BMI, body mass index
FIGURE 2.
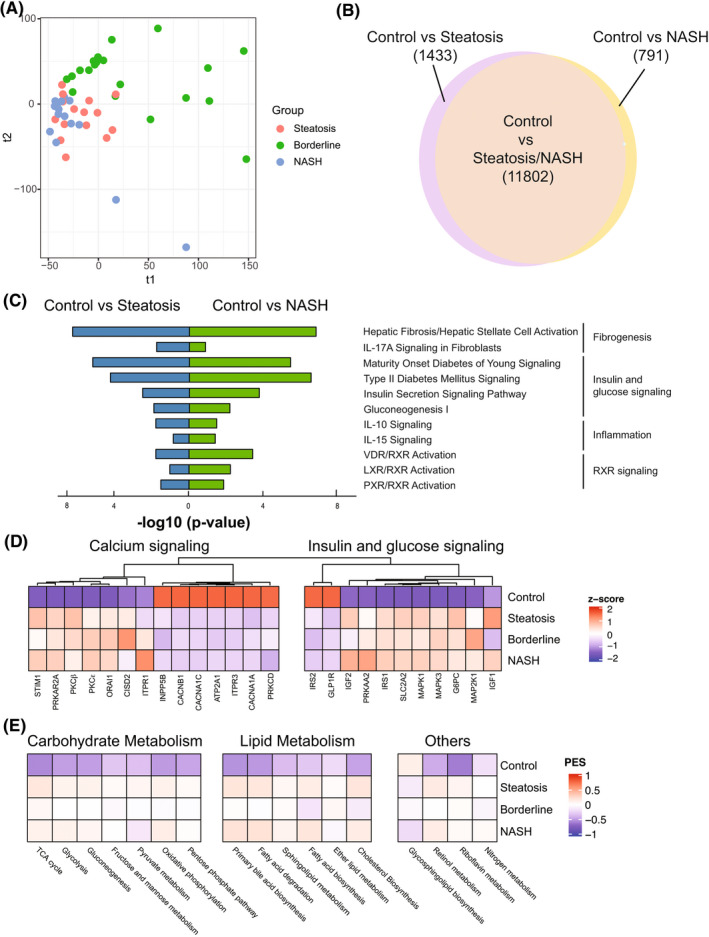
Transcriptomic differences between different stages of control and pediatric NAFLD. (A) Patients with NAFLD visualized by the top two components of partial least‐squares discriminant analysis (PLS‐DA). (B) Venn diagram showing the number of differentially expressed genes (DEGs) in control versus indicated group. A total of 13,235 differentially regulated genes were identified between steatosis and control; 12,593 differentially regulated genes were identified between NASH and control. (C) Selected top enriched pathways grouped by biologic function. (D) Heatmap showing the normalized gene expression (represented by z‐score) of candidate NAFLD genes involved in “calcium signaling” and “insulin & glucose signaling.” (E) Pathway Enrichment Score (PES) of selected Kyoto Encyclopedia of Genes and Genomes (KEGG) metabolic pathways in normal liver and indicated NAFLD grade. IL, interleukin; LXR, liver X receptor; NASH, nonalcoholic steatohepatitis; PXR, pregnane X receptor; RXR, retinoid X receptor; VDR, vitamin D receptor
Performing DEG and pathway analysis between groups 1 and 2, pathways such as “type 2 diabetes mellitus signaling,” “insulin secretion pathway,” “agranulocyte adhesion and diapedesis,” and “hepatic fibrosis/stellate cell activation” were enriched, consistent with the development of steatosis, inflammation, and fibrosis in these subjects. A full list of enriched pathways between group 1 and group 2 is presented in Table S3. The two group‐1 individuals displaying similar transcriptomic profiles with controls, but histologically defined as NAFLD, were excluded from further downstream analysis.
Defining liver transcriptome changes associated with different stages of NAFLD
To understand the mechanisms of NAFLD progression in children, we divided our cohort (Table S4) into control, steatosis (NAS ≤ 3), borderline NASH (NAS = 4 or NAS ≥ 5 with one or more subscores = 0), and NASH (NAS ≥ 5, subscores ≥ 1), as described in the “Materials and Methods” section. Pairwise comparisons among the control, steatosis, and NASH groups defined 13,235 DEGs in control versus steatosis, and 12,593 DEGs in control versus NASH (Figure 2B). Pathways significantly activated in fibrosis, inflammation, insulin/glucose signaling, and retinoid X receptor (RXR) signaling were present when examining control versus steatosis as well as in control versus NASH (Figure 2C; see Tables S5 and S6 for a full list of significant pathways).
“Calcium signaling” was the top differentially regulated pathway in both the steatosis and NASH comparisons to controls. Calcium signaling plays an important role in hepatic metabolism. Liver steatosis can disrupt calcium homeostasis in hepatocytes and cause endoplasmic reticulum stress, leading to insulin resistance.[ 27 , 28 ] Therefore, we examined gene expression associated with calcium signaling in hepatocytes and insulin resistance.[ 29 ] Compared with controls, pediatric patients with NAFLD have an overall decrease in calcium ion transport (CANA1A, CANA1C, CACNB1), increased protein kinase C expression (PKCβ, PKCε), increased gluconeogenesis and glucose transport (G6PC, SLC2A2), as well as activation of the mitogen‐activated protein kinase kinase/extracellular signal–regulated kinase pathway (Figure 2D and Figure S1B).
We also examined a list of candidate genes previously reported by adult and mouse NAFLD studies.[ 30 , 31 ] Compared with healthy controls, pediatric NAFLD samples showed an overall up‐regulation of farnesoid X receptor signaling/bile acid synthesis, immune system activation, and Hippo pathway activation (Figure S1C). However, there was no clear trend in apoptosis and stellate cell activation (Figure S1D), which may explain a paucity of advanced disease in this cohort. Expression changes of these candidate genes occurred early after the onset of NAFLD and persisted through steatohepatitis.
Additionally, we plotted the enrichment of selected metabolic pathways to determine their differences in NAFLD stages (Figure 2E). There was an overall up‐regulation of metabolic gene expression in NAFLD compared with normal liver controls, including carbohydrate, lipid, and retinol metabolism. The only down‐regulated metabolic pathway we identified was “glycosphingolipid biosynthesis.” There was a lack of differential gene expression as NAFLD progresses from steatosis to NASH in this cohort, except a downward trend in oxidative phosphorylation.
Identifying DEGs and pathways with progression of fibrosis in pediatric NAFLD
To identify molecular mechanisms associated with fibrotic progression in pediatric NAFLD, we first performed PLS‐DA on the transcriptome profiles of different fibrosis stages (S0–S3) within NAFLD (Figure S2A). There is a clear separation between no (S0) and advanced fibrosis (S3) (Figure 3A). Comparing S0 against S3, we identified 500 differentially up‐regulated genes and 439 down‐regulated genes (Figure 3B).
FIGURE 3.
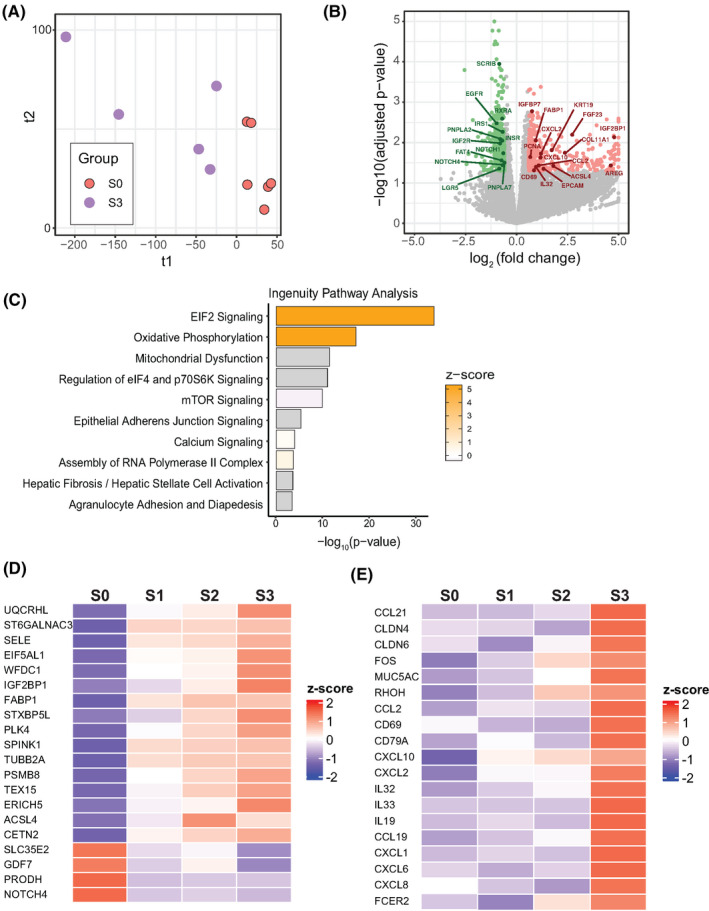
Gene‐set analysis of fibrotic progression in pediatric patients with NAFLD. (A) PLS‐DA of S0 versus S3 patients in this study. (B) 939 DEGs were differentially regulated in S0 versus S3. A total of 500 were significantly up‐regulated; 439 were significantly down‐regulated. Genes of interest are marked. (C) Top 10 enriched biological pathways in NAFLD S0 versus S3 by Ingenuity Pathway Analysis (IPA), ranked by p‐value. (D) Normalized expression of a 20‐protein coding gene set in different stages of fibrosis within NAFLD. All 20 genes are significantly regulated (p < 0.05) compared with NAFLD S0. (E) Normalized expression of selected genes related to inflammation and immune cell activation. Genes are significantly up‐regulated (p < 0.05) between S0 and S3. Z‐score represents normalized expression. EIF2, eukaryotic initiation factor 2; mTOR, mammalian target of rapamycin
IPA identified the top enriched biological pathways in our S0 versus S3 subjects (Figure 3C). The top 5 regulated pathways can be subdivided into two categories: translation regulation (i.e., “EIF2 signaling,” “mTOR signaling”), which is driven by the up‐regulation of several ribosomal protein subunit genes, and mitochondrial stress (“mitochondria dysfunction,” “oxidative phosphorylation”), which is driven by up‐regulation genes in the electron transport chain, including cytochrome c oxidase subunits, complex I subunits, complex III subunits, and adenosine triphosphate synthase subunits. This suggests a general up‐regulation of protein synthesis and mitochondrial activity at advanced stages of fibrosis. This is consistent with the up‐regulation of several genes encoding enzymes in the glycolysis pathway (Figure S2B).
Next, we sought to find common genes that are differentially expressed throughout progression of fibrosis in pediatric NAFLD. Using NAFLD S0 as a baseline, we performed pairwise analysis of DEG and pathway enrichment in NAFLD S1–S3. We found the intersection of DEGs between the three pairwise comparisons that contain 25 genes, 20 of which are protein‐coding (Figure S2C). Among the 20 protein‐coding genes, 16 of them are consistently up‐regulated throughout fibrosis progression, while four are consistently down‐regulated (Figure 3D).
In our cohort, subjects with S3 fibrosis did not consistently have high NAS scores (> 4) and none showed signs of hepatic ballooning. S3 patients showed significantly higher expression of various pro‐inflammatory cytokines and receptors including chemokine (C‐C motif) ligand 21, CD69, chemokine (C‐X‐C motif) ligand 10 (CXCL10), interleukin (IL)–32 (Figure 3E). In addition, IL11 and IL17A, two cytokines associated with liver fibrogenesis or NAFLD progression in mice and humans,[ 32 , 33 ] were up‐regulated in our S3 subjects, although not significantly (p = 0.07 and 0.09, respectively). This cohort of advanced fibrotic pediatric patients with NAFLD is hallmarked by dysregulation of pro‐inflammatory cytokines and immune activation markers.
Insulin‐like growth factor binding protein and insulin‐like growth factor binding protein 2 family proteins in NASH and fibrosis regulation
One purpose of this study was to identify potential noninvasive markers for the progression of NAFLD and fibrosis that are detectable in peripheral blood. The insulin‐like growth factor binding protein (IGFBP) family may satisfy these requirements, as it is associated with adult NALFD[ 34 ] and detectable in serum.[ 35 ]
RNA‐seq data showed that hepatic IGFBP1 and IGFBP2 expression significantly decreased between steatosis to NASH livers and is significantly lower compared with healthy livers (Figure 4A). Their expression also decreased with increasing fibrotic stage, although it was not statistically significant (Figure 4B). In contrast, IGFBP3 significantly increased in pediatric NAFLD compared with control, but it does not distinguish NASH and simple steatosis. There was an upward trend with increasing fibrosis (Figure 4A,B). IGFBP7 expression was significantly increased in S3 fibrosis (Figure 4B; p = 3.92E−6).
FIGURE 4.
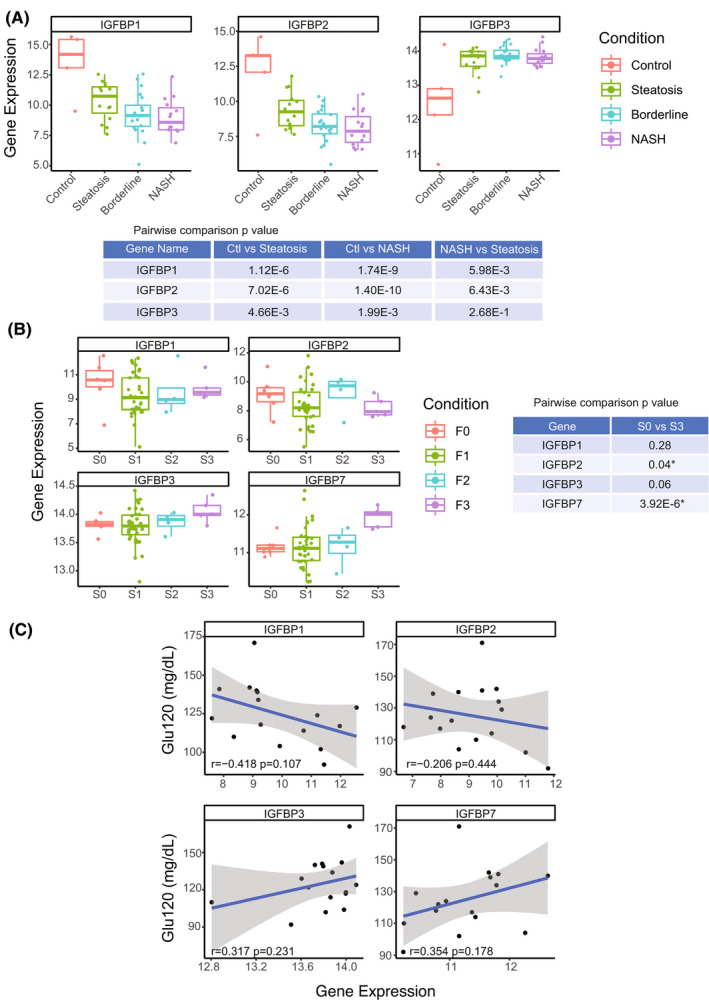
Hepatic expression of insulin‐like growth factor binding protein (IGFBP) family correlates with pediatric NAFLD/fibrosis severity. (A) Hepatic IGFBP1, IGFBP2, and IGFBP3 expression in different stages of NAFLD: normal (n = 5), steatosis (NAFLD activity score [NAS] ≤ 3; n = 15), borderline NASH (NAS = 4 or NAS ≥ 5 with one or more subscores = 0; n = 19), and NASH (NAS > 5, subscores ≥ 1; n = 15). (B) Hepatic expression of IGFBP1, IGFBP2, IGFBP3, and IGFBP7 in different stages of fibrosis in pediatric patients with NAFLD: S0 (n = 6), S1 (n = 35), S2 (n = 4), and S3 (n = 5). *p‐value < 0.05. (C) Pearson correlation between 2‐h glucose (Glu120) value and hepatic expression of IGFBP1, IGFBP2, IGFBP3, and IGFBP7
NAFLD progression is directly associated with type 2 diabetes development.[ 36 , 37 ] In this cohort, we investigated whether 2‐h glucose values from OGTT are related to IGFBP expression. IGFBP1 and IGFBP2 was negatively correlated with 2‐h glucose (r = −0.42 and −0.21, respectively), whereas IGFBP3 and IGFBP7 showed a positive correlation (r = 0.32 and 0.35, respectively) (Figure 4C). These correlations were not statistically significant, partially due to the lack of OGTT data, but the trends were consistent with our gene‐expression/histopathologic comparisons (Figure 4A,B).
Meta‐analysis comparing adolescent/adult NASH transcriptomic data reveals common and unique gene‐expression patterns
We then examined whether differentially expressed genes/pathways in this study are shared in previously published studies of patients with NAFLD/NASH. First, we compared our data with the transcriptomic data set of adolescent liver biopsies undergoing bariatric surgery for extreme obesity.[ 24 ] We identified seven common up‐regulated genes between the two studies (Table S7). Several of the genes are involved in lipid metabolism or have been implicated in NAFLD/NASH.
We then evaluated whether our cohort recapitulates aspects of adult NAFLD by exploring two transcriptomic adult data sets. These data sets included healthy controls and had well‐annotated NAS and fibrosis scores for their samples.[ 12 , 14 ] By combining the histological scores of the two adult cohorts, we found that, in adults, NAS strongly correlates with increasing fibrosis (Figure S3A; p = 0.0005). There was no similar clear correlation in our pediatric cohort (Figure S3B; p = 0.6), suggesting potentially different mechanisms driving fibrosis progression.
We then performed DEG and pathway enrichment analyses with the two adult studies alongside our pediatric cohort. This included pairwise comparison of controls versus NASH and no fibrosis (S0) with advanced fibrosis (S3/S4). Some pathways were similarly regulated in both pediatric and adult NAFLD patients (Figure 5A,B) such as hepatic fibrosis and type 2 diabetes mellitus signaling. Other pathways were only enriched in pediatric patients, such as “calcium signaling pathway” in NASH and “oxidative phosphorylation” in fibrosis.
FIGURE 5.
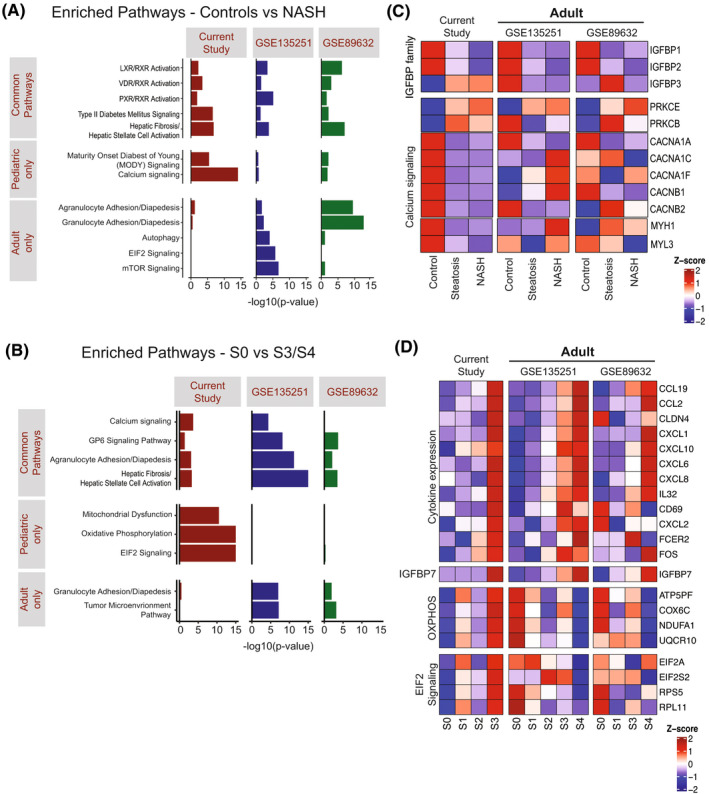
Similarities and differences comparing pediatric with adult NAFLD studies. (A) Selected enriched pathways in pediatric and two adult NAFLD studies using all DEGs identified comparing controls versus NASH. (B) Selected enriched pathways in pediatric and two adult NAFLD studies using DEGs comparing S0 versus S3/S4 fibrosis. (C) Heatmap showing hepatic expression of selected genes related to IGFBP and calcium signaling pathway in different NAFLD stages. (D) Heatmap showing hepatic expression of selected genes/pathways with progression of fibrosis in NAFLD. OXPHOS, Oxidative phosphorylation. GSE89632: S2 patients were excluded due to small number (n = 2). (E) Diagram showing the common DEGs (false discovery rate [FDR] < 0.05, fold change > 1.5) from NASH versus control in different age groups. All of the identified DEGs here are up‐regulated in NASH
We examined a recently proposed 25‐gene expression panel for fibrosis/NASH[ 12 ] in our data set (Figure S3C). Only 4 of 25 genes (CFAP221 [cilia and flagella associated protein 221], HECW1 [HECT, C2 and WW domain containing E3 ubiquitin protein ligase 1], regulator of G protein signaling 4 [RGS4], and IL‐32) were significantly associated with progression in our pediatric cohort. Then, we examined genes across all three studies that we identified as associated with increasing NAS. There was a reduction of hepatic IGFBP1 and IGFBP2 expression across all of the studies in the progression from normal to steatosis to NASH (Figure 5C; IGFBP family). In contrast, IGFBP3 increased in the pediatric cohort, but not in either adult study. Genes encoding calcium channel subunits and myosin subunits were significantly down‐regulated in pediatric patients with NAFLD compared with controls, most of which were not differentially regulated in adults (Figure 5C; calcium signaling). This is consistent with “calcium signaling pathway” only being down‐regulated in pediatric NAFLD (Figure 5A).
In terms of liver fibrosis progression, up‐regulated cytokine and immune activation genes are a common feature in both adult and pediatric patients with NAFLD (Figure 5D; cytokine expression), although a subset of genes was only found significantly changed in the pediatric and not the adult cohorts (CD69, CXCL2, FCER2 [Fc epsilon receptor II], and FOS). IGFBP7 significantly increased in advanced fibrosis across all studies. Finally, we identified significant changes in oxidative phosphorylation and eukaryotic initiation factor 2 (EIF2) signaling in this cohort, but these changes were not present in the adult studies (Figure 5D; OXPHOS [oxidative phosphorylation]/EIF2 signaling). Comparing the gene‐expression profiles of this cohort with similar adult NAFLD/NASH studies identified common as well as unique genes in NAFLD progression and fibrotic development. Further studies are required to investigate their potential mechanistic implications and prospective utility as noninvasive biomarkers.
DISCUSSION
Considering the alarming prevalence of NAFLD/NASH among children and adolescents, as well as its progressive and chronic nature, there is an imperative need to understand whether there are unifying and unique mechanisms in children and adolescents. Analyzing the global liver transcriptome profiles of patients with NAFLD could facilitate the identification of biomarkers and therapeutic targets for the detection, quantification, and treatment of NAFLD as well as its associated fibrosis. Furthermore, by combining the findings from pediatric and adult patients, it facilitates evaluating whether there are biomarkers and therapies unique to pediatrics, adults, or equally suitable for both age groups.
Previous attempts to study transcriptomic changes in NAFLD predominantly used adult cohorts, composed primarily of Caucasian populations.[ 12 , 13 , 14 ] In this study, we used liver biopsy samples from a total of 52 pediatric patients in the northeast United States with an average age of 13.6 years representing multiple ethnicities and both genders. Most of the cohort was Hispanic (69.2%). We did not detect a difference in a variety of biochemical markers between Hispanics and the remainder of our cohort (Table 1), although Hispanics tend to exhibit an overall higher NAS score compared with non‐Hispanic patients. Transcriptional profiles between our Hispanic and non‐Hispanic patients were not significantly different (data not shown). Although Hispanics represent most NAFLD cases in the general pediatric population,[ 10 , 11 ] the large proportion of Hispanic participants in this cohort may make the findings less generalizable to non‐Hispanic children.
We compared our cohort to a convenience sample of adolescents with a liver biopsy at the time of bariatric surgery.[ 24 ] By cross‐referencing this study with our cohort, we identified seven commonly regulated genes (Table S7). Several are involved in lipid metabolism, and, notably, PLA2G7 (phospholipase A2 group VII) is directly linked to calorie‐related inflammation.[ 38 ] The modest number of differentially expressed genes between these studies limited our ability to perform extensive bioinformatic analysis. This is partially explained by the significant sex, ethnic, and referral differences between these studies, highlighting the need to expand efforts to recruit pediatric and adolescent patients. These genes, as highlighted by PLA2G7’s recently described role, deserve consideration as drivers of cardiometabolic disease.
Children often present with different histopathological features of NAFLD from adults. For example, hepatocellular ballooning is less common. Inflammation and fibrosis are often present in the portal tracts (Zone 1), while adult disease more typically involves inflammation and fibrosis of the central vein (Zone 3).[ 39 ] A second explanation for these potential differences is the chronic nature of liver fibrosis. From adult studies, it has been estimated that fibrosis progresses at a pace of 14.3 years in NAFLD and 7.1 years in NASH.[ 40 ] In our cohort, we found bridging fibrosis (S3) present in subjects between 10 to 15 years of age, suggesting that more aggressive mechanisms of inflammation and fibrosis are present in at least a subset of this population.
In our study, we examined the progression of NAFLD (control to steatosis to NASH) and liver fibrosis separately. The transcriptomic profiles of controls are dramatically different between patients with steatosis and NASH. In comparison, livers with steatosis versus NASH show minimal differences in gene‐expression profiles (Figure 2). Dysregulation of many well‐recognized signaling pathways involved in NAFLD/NASH development such as glucose/insulin signaling, IL‐17 signaling, and RXR activation occurs early in the disease course, even before pathologic identification of inflammation and fibrosis. Similarly, dysregulation of metabolic programs such as carbohydrate, fatty acid, and retinol metabolism occur early in the development of steatosis, before the clinical development of diabetes or dyslipidemia (Figure 2), which is consistent with its association to metabolic syndrome.[ 41 ]
In this patient cohort, NAS does not correlate well with fibrosis, unlike adult studies in which patients with high NAS scores are more likely to have advanced fibrosis.[ 17 , 20 ] Although fibrosis stage is not calculated in NAS, it is thought to be the major predictor of clinical outcomes in these patients. Advanced fibrosis is associated with a higher mortality rate in adults,[ 42 ] although its applicability to the pediatric population is unclear. The development of inflammation and recruitment of inflammatory cells is generally presumed to be a precursor to fibrotic development. As expected, inflammatory cytokine and immune marker activation were enriched during fibrotic progression in both adults[ 17 , 20 ] and in our pediatric NALFD cohort (Figure 5B,D). We examined the expression of a recently described NAFLD fibrotic gene signature identified from adults[ 12 ] to determine its potential applicability in pediatrics. A subset (4 of 25; IL‐32, CFAP221, HEC21, RGS4) of these genes were significantly regulated in our pediatric cohort (Figure S3C), suggesting that fibrotic regulation in adult and pediatric patients with NAFLD share some similarities, but are not identical.
From our data set, we identified a core 25‐gene signature associated with fibrotic progression in the pediatric cohort with NAFLD (Figure 3D). Among them, NOTCH4 (notch receptor 4) has been well characterized in liver tissue repair and injury [ 43 , 44 ]; however, its involvement in fibrotic NAFLD progression remains unclear. IGF2BP1, one of the three mRNA‐binding proteins in the IGF2BP family, is linked to HCC.[ 45 , 46 ] These signature genes warrant further functional studies in vivo and in vitro for their evaluation as potential biomarkers and therapeutic targets.
Currently, liver biopsy remains the gold standard for staging of NAFLD and evaluation of fibrosis. Identifying peripherally accessible liver‐specific biomarkers could facilitate the diagnosis and staging of NAFLD. It is also well recognized that many members of the population likely have hepatic steatosis and fibrosis, but clinical evaluation is not considered because of a normal serum ALT. In this study, we propose IGFBP1/2/3/7 as potential NAFLD biomarkers. This protein family can bind, transport, prolong the half‐life, and regulate the availability of insulin‐like growth factors. They are primarily expressed in the liver and can be detected and measured in the bloodstream, making them ideal potential biomarkers for liver‐associated diseases.[ 47 ] With regard to their relationship with NAFLD, recent studies indicated that patients with NAFLD show lower serum levels of IGFBP1[ 48 ] and IGFBP2,[ 49 ] and increased serum level of IGFBP7.[ 34 ]
Our data support the notion that IGFBP1/2 levels decrease as NAFLD progresses (Figure 5C), and IGFBP7 levels increase in S3/S4 fibrosis (Figure 5D). These markers were consistent across the pediatric and adult cohorts we examined. Furthermore, we found that IGFBP3 increased in our pediatric cohort as NAFLD progressed, which was not reflected in the adult studies (Figure 5C). Directly examining the IGFBP family members in the serum and specifically, whether IGFBP3 is a unique biomarker for pediatric patients with NAFLD warrants further investigation.
We also want to acknowledge some limitations of our study. First, we were unable to obtain liver samples from healthy children or adolescents. As a result, we used RNA and liver biopsies from healthy adults as control. There may be baseline differences between pediatric and adult livers, although it is notable that the transcriptional profile of some of our subjects more closely aligned with the adult controls (Figure 1). Larger future studies are required to determine whether the differences we identified as unique to this pediatric population are due to developmental versus disease‐related differences.
Second, our pediatric cohort lacks patients with advanced disease phenotypes: Only 1 patient has a NAS score larger than 6; 10% of patients have stage 3 fibrosis; and none have S4 fibrosis. Generally, these proportions of disease are like a larger pediatric histopathologic study.[ 50 ] Despite these limitations, our findings provide reference and guidance for future studies on pediatric NAFLD. We propose several gene signatures and biological pathways that are potentially critical to pediatric NAFLD.
CONFLICT OF INTEREST
R.F. consults for Mirum and advises Albireo. D.Y. received grants from Kadmon Corp, LLC.
AUTHOR CONTRIBUTIONS
Experiment design and manuscript draft: Kangning Yao, Elena Tarabra, Silvia Liu, and Dean Yimlamai. Bioinformatic analysis supervision: Silvia Liu. Liver samples and analysis: Daniela Sia. All authors provided intellectual input, and vetted and approved the final manuscript.
Supporting information
Fig S1
Fig S2
Fig S3
Supplementary Material
Yao K, Tarabra E, Sia D, Morotti R, Fawaz R, Valentino P, et al. Transcriptomic profiling of a multiethnic pediatric NAFLD cohort reveals genes and pathways associated with disease. Hepatol Commun. 2022;6:1598–1610. 10.1002/hep4.1940
Kangning Yao and Elena Tarabra contributed equally to this work.
Funding information
National Institute on Minority Health and Health Disparities (R01 MD015974); National Institute of Diabetes and Digestive and Kidney Diseases (P30 DK034989, P30 DK045735, P30 DK120531, R01 DK111038, R01 DK129552, and R03 DK124743); Eunice Kennedy Shriver National Institute of Child Health and Human Development (R01 HD028016); and National Center for Advancing Translational Sciences (UL1 TR001863)
Contributor Information
Silvia Liu, Email: shl96@pitt.edu, Email: dean.yimlamai@yale.edu.
Dean Yimlamai, Email: dean.yimlamai@yale.edu.
DATA AVAILABILITY STATEMENT
Data were deposited into the NCBI GEO database (GSE185051).
REFERENCES
- 1. Caprio S, Santoro N, Weiss R. Childhood obesity and the associated rise in cardiometabolic complications. Nat Metab. 2020;2:223–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease‐meta‐analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64:73–84. [DOI] [PubMed] [Google Scholar]
- 3. Anderson EL, Howe LD, Jones HE, Higgins JP, Lawlor DA, Fraser A. The prevalence of non‐alcoholic fatty liver disease in children and adolescents: a systematic review and meta‐analysis. PLoS One. 2015;10:e0140908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schwimmer JB, Deutsch R, Kahen T, Lavine JE, Stanley C, Behling C. Prevalence of fatty liver in children and adolescents. Pediatrics. 2006;118:1388–93. [DOI] [PubMed] [Google Scholar]
- 5. Feldstein AE, Charatcharoenwitthaya P, Treeprasertsuk S, Benson JT, Enders FB, Angulo P. The natural history of non‐alcoholic fatty liver disease in children: a follow‐up study for up to 20 years. Gut. 2009;58:1538–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Goyal NP, Schwimmer JB. The progression and natural history of pediatric nonalcoholic fatty liver disease. Clin Liver Dis. 2016;20:325–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Anstee QM, Reeves HL, Kotsiliti E, Govaere O, Heikenwalder M. From NASH to HCC: current concepts and future challenges. Nat Rev Gastroenterol Hepatol. 2019;16:411–28. [DOI] [PubMed] [Google Scholar]
- 8. Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. 2018;15:11–20. [DOI] [PubMed] [Google Scholar]
- 9. Loomba R, Sirlin CB, Schwimmer JB, Lavine JE. Advances in pediatric nonalcoholic fatty liver disease. Hepatology. 2009;50:1282–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Goran MI, Ventura EE. Genetic predisposition and increasing dietary fructose exposure: the perfect storm for fatty liver disease in Hispanics in the U.S. Dig Liver Dis. 2012;44:711–3. [DOI] [PubMed] [Google Scholar]
- 11. Marzuillo P, Miraglia del Giudice E, Santoro N. Pediatric fatty liver disease: role of ethnicity and genetics. World J Gastroenterol. 2014;20:7347–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Govaere O, Cockell S, Tiniakos D, Queen R, Younes R, Vacca M, et al. Transcriptomic profiling across the nonalcoholic fatty liver disease spectrum reveals gene signatures for steatohepatitis and fibrosis. Sci Transl Med. 2020;12:aba4448. [DOI] [PubMed] [Google Scholar]
- 13. Hoang SA, Oseini A, Feaver RE, Cole BK, Asgharpour A, Vincent R, et al. Gene expression predicts histological severity and reveals distinct molecular profiles of nonalcoholic fatty liver disease. Sci Rep. 2019;9:12541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Arendt BM, Comelli EM, Ma DWL, Lou W, Teterina A, Kim TH, et al. Altered hepatic gene expression in nonalcoholic fatty liver disease is associated with lower hepatic n‐3 and n‐6 polyunsaturated fatty acids. Hepatology. 2015;61:1565–78. [DOI] [PubMed] [Google Scholar]
- 15. Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–21. [DOI] [PubMed] [Google Scholar]
- 16. Brunt EM, Kleiner DE, Wilson LA, Belt P, Neuschwander‐Tetri BA, Network NCR. Nonalcoholic fatty liver disease (NAFLD) activity score and the histopathologic diagnosis in NAFLD: distinct clinicopathologic meanings. Hepatology. 2011;53:810–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Andrews S. FastQC: a quality control tool for high throughput sequence data, Version 0.11.7. Babraham Bioinformatics; [cited 2019 Jan 8]. Available from: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ [Google Scholar]
- 18. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA‐seq aligner. Bioinformatics. 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thevenot EA, Roux A, Xu Y, Ezan E, Junot C. Analysis of the human adult urinary metabolome variations with age, body mass index, and gender by implementing a comprehensive workflow for univariate and OPLS statistical analyses. J Proteome Res. 2015;14:3322–35. [DOI] [PubMed] [Google Scholar]
- 21. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA‐seq data. BMC Bioinform. 2013;14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Okuda S, Yamada T, Hamajima M, Itoh M, Katayama T, Bork P, et al. KEGG Atlas mapping for global analysis of metabolic pathways. Nucleic Acids Res. 2008;36:W423–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xanthakos SA, Jenkins TM, Kleiner DE, Boyce TW, Mourya R, Karns R, et al. High prevalence of nonalcoholic fatty liver disease in adolescents undergoing bariatric surgery. Gastroenterology. 2015;149:623–34.e628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–9. [DOI] [PubMed] [Google Scholar]
- 26. Taksali SE, Caprio S, Dziura J, Dufour S, Calí AMG, Goodman TR, et al. High visceral and low abdominal subcutaneous fat stores in the obese adolescent: a determinant of an adverse metabolic phenotype. Diabetes. 2008;57:367–71. [DOI] [PubMed] [Google Scholar]
- 27. Liang JQ, Teoh N, Xu L, Pok S, Li X, Chu ESH, et al. Dietary cholesterol promotes steatohepatitis related hepatocellular carcinoma through dysregulated metabolism and calcium signaling. Nat Commun. 2018;9:4490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Arruda AP, Hotamisligil GS. Calcium homeostasis and organelle function in the pathogenesis of obesity and diabetes. Cell Metab. 2015;22:381–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ali ES, Petrovsky N. Calcium signaling as a therapeutic target for liver steatosis. Trends Endocrinol Metab. 2019;30:270–81. [DOI] [PubMed] [Google Scholar]
- 30. Loomba R, Friedman SL, Shulman GI. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell. 2021;184:2537–64. [DOI] [PubMed] [Google Scholar]
- 31. Parthasarathy G, Revelo X, Malhi H. Pathogenesis of nonalcoholic steatohepatitis: an overview. Hepatol Commun. 2020;4:478–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Widjaja AA, Singh BK, Adami E, Viswanathan S, Dong J, D’Agostino GA, et al. Inhibiting interleukin 11 signaling reduces hepatocyte death and liver fibrosis, inflammation, and steatosis in mouse models of nonalcoholic steatohepatitis. Gastroenterology. 2019;157:777–92.e714. [DOI] [PubMed] [Google Scholar]
- 33. Harley ITW, Stankiewicz TE, Giles DA, Softic S, Flick LM, Cappelletti M, et al. IL‐17 signaling accelerates the progression of nonalcoholic fatty liver disease in mice. Hepatology. 2014;59:1830–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stanley TL, Fourman LT, Zheng I, McClure CM, Feldpausch MN, Torriani M, et al. Relationship of IGF‐1 and IGF‐binding proteins to disease severity and glycemia in nonalcoholic fatty liver disease. J Clin Endocrinol Metab. 2021;106:e520–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. López‐Bermejo A, Khosravi J, Corless CL, Krishna RG, Diamandi A, Bodani U, et al. Generation of anti‐insulin‐like growth factor‐binding protein‐related protein 1 (IGFBP‐rP1/MAC25) monoclonal antibodies and immunoassay: quantification of IGFBP‐rP1 in human serum and distribution in human fluids and tissues. J Clin Endocrinol Metab. 2003;88:3401–8. [DOI] [PubMed] [Google Scholar]
- 36. Nobili V, Mantovani A, Cianfarani S, Alisi A, Mosca A, Sartorelli MR, et al. Prevalence of prediabetes and diabetes in children and adolescents with biopsy‐proven non‐alcoholic fatty liver disease. J Hepatol. 2019;71:802–10. [DOI] [PubMed] [Google Scholar]
- 37. Newton KP, Hou J, Crimmins NA, Lavine JE, Barlow SE, Xanthakos SA, et al. Prevalence of prediabetes and type 2 diabetes in children with nonalcoholic fatty liver disease. JAMA Pediatr. 2016;170:e161971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Spadaro O, Youm Y, Shchukina I, Ryu S, Sidorov S, Ravussin A, et al. Caloric restriction in humans reveals immunometabolic regulators of health span. Science. 2022;375:671–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, et al. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology. 2018;67:328–57. [DOI] [PubMed] [Google Scholar]
- 40. Singh S, Allen AM, Wang Z, Prokop LJ, Murad MH, Loomba R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: a systematic review and meta‐analysis of paired‐biopsy studies. Clin Gastroenterol Hepatol. 2015;13:643–54.e641–649; quiz e639–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Eslam M, Sanyal AJ, George J, International Consensus Panel . MAFLD: a consensus‐driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology. 2020;158:1999–2014.e1991. [DOI] [PubMed] [Google Scholar]
- 42. Dulai PS, Singh S, Patel J, Soni M, Prokop LJ, Younossi Z, et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: systematic review and meta‐analysis. Hepatology. 2017;65:1557–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ding W‐J, Wu W‐J, Chen Y‐W, Chen H‐B, Fan J‐G, Qiao L. Expression of Notch family is altered in non‐alcoholic fatty liver disease. Mol Med Rep. 2020;22:1702–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Romeo S. Notch and nonalcoholic fatty liver and fibrosis. N Engl J Med. 2019;380:681–3. [DOI] [PubMed] [Google Scholar]
- 45. Gutschner T, Hämmerle M, Pazaitis N, Bley N, Fiskin E, Uckelmann H, et al. Insulin‐like growth factor 2 mRNA‐binding protein 1 (IGF2BP1) is an important protumorigenic factor in hepatocellular carcinoma. Hepatology. 2014;59:1900–11. [DOI] [PubMed] [Google Scholar]
- 46. Zhou X, Zhang CZ, Lu S‐X, Chen GG, Li L‐Z, Liu L‐L, et al. miR‐625 suppresses tumour migration and invasion by targeting IGF2BP1 in hepatocellular carcinoma. Oncogene. 2015;34:965–77. [DOI] [PubMed] [Google Scholar]
- 47. Firth SM, Baxter RC. Cellular actions of the insulin‐like growth factor binding proteins. Endocr Rev. 2002;23:824–54. [DOI] [PubMed] [Google Scholar]
- 48. Savastano S, Di Somma C, Pizza G, De Rosa A, Nedi V, Rossi A, et al. Liver‐spleen axis, insulin‐like growth factor‐(IGF)‐I axis and fat mass in overweight/obese females. J Transl Med. 2011;9:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fahlbusch P, Knebel B, Hörbelt T, Barbosa DM, Nikolic A, Jacob S, et al. Physiological disturbance in fatty liver energy metabolism converges on IGFBP2 abundance and regulation in mice and men. Int J Mol Sci. 2020;21:4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Carter‐Kent C, Yerian LM, Brunt EM, Angulo P, Kohli R, Ling SC, et al. Nonalcoholic steatohepatitis in children: a multicenter clinicopathological study. Hepatology. 2009;50:1113–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Supplementary Material
Data Availability Statement
Data were deposited into the NCBI GEO database (GSE185051).