

Abstract
Hepatitis B virus (HBV) infection is a major risk factor of liver cirrhosis and hepatocellular carcinoma. Clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR‐associated protein 9 (Cas9) has been used to precisely edit the HBV genome and eliminate HBV through non‐homologous end‐joining repair of double‐stranded break (DSB). However, the CRISPR/Cas9‐mediated DSB triggers instability of host genome and exhibits low efficiency to edit genome, limiting its application. CRISPR cytidine base editors (CBEs) could silence genes by generating a premature stop codon. Here we developed a CRISPR base editor approach to precisely edit single nucleotide within the HBV genome to impair HBV gene expression. Specifically, a single‐guide RNA (sgRNA) was designed to edit the 30th codon of HBV S gene, which encodes HBV surface antigen (HBsAg), from CAG (glutamine) to stop codon TAG. We next used human hepatoma PLC/PRF/5 cells carrying the HBV genome to establish a cell line that expresses a CBE (PLC/PRF/5‐CBE). Lentivirus was used to introduce sgRNA into PLC/PRF/5‐CBE cells. Phenotypically, 71% of PLC/PRF/5‐CBE cells developed a premature stop codon within the S gene. Levels of HBs messenger RNA were significantly decreased. A 92% reduction of HBsAg secretion was observed in PLC/PRF/5‐CBE cells. The intracellular HBsAg was also reduced by 84% after treatment of gRNA_S. Furthermore, no off‐target effect was detected in predicted off‐target loci within the HBV genome. Sequencing confirmed that 95%, 93%, 93%, 9%, and 72% S gene sequences of HBV genotypes B, C, F, G, and H had the binding site of sgRNA. Conclusion: Our findings indicate that CRISPR‐mediated base editing is an efficient approach to silence the HBV S gene, suggesting its therapeutic potential to eliminate HBV.
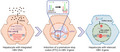
Silencing of hepatitis B virus (HBV) S gene by clustered regularly interspaced short palindromic repeats (CRISPR) base editors. HBV surface antigen (HBsAg) can be produced either from covalently closed circular DNA or from integrated HBV DNA. Current antiviral treatment exhibits low efficiency to suppress HBsAg production. Our data show that cytidine base editor (CBE) could introduce a premature stop codon (PTC) in the HBV S gene under the guidance of a single‐guide RNA (sgRNA). Levels of HBs messenger RNA were significantly decreased. Intracellular and excreted HBsAg were remarkably reduced by the CBE treatment. Created with BioRender.com.
INTRODUCTION
It is estimated that over 250 million people are chronically infected with the hepatitis B virus (HBV).[ 1 ] Chronic HBV infection is a major global cause of liver cirrhosis and hepatocellular carcinoma (HCC), accounting for about 650,000 deaths every year.[ 2 ] Two categories of drugs have been approved to treat HBV infection: the interferon‐alpha and nucleos(t)ide analogues. However, only a small proportion of patients could achieve a functional cure, which is defined as hepatitis B surface antigen (HBsAg) loss with undetectable serum HBV DNA, allowing cessation of therapy without risk of viral rebound.[ 3 ] In addition to low effectiveness of current therapeutic approaches, most patients with HBV need a long‐term therapy. Therefore, effective therapies for chronic HBV infection are urgently needed.
The clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR‐associated protein 9 (Cas9) system is a powerful tool for genome editing. The CRISPR/Cas9 system includes two components: Cas9 nuclease and a single‐guide RNA (sgRNA). The Cas9 nuclease is directed to the target DNA sequence, which is complementary to the sgRNA and followed by a protospacer adjacent motif (PAM). Cas9 nuclease could cut both strands of the target DNA, forming a double‐strand DNA break (DSB). The DSB is repaired through the error‐prone non‐homologous end‐joining pathway, which introduces a frameshift indel mutation and knocks out gene function.[ 4 ] Targeting HBV with CRISPR/Cas9 efficiently suppresses HBV replication and viral gene expression both in vitro and in vivo.[ 5 , 6 , 7 , 8 , 9 ] However, repair of DSB induced by Cas9 nuclease could also generate large deletions and complex rearrangements in the target site.[ 10 , 11 ] CRISPR/Cas9 editing of HBV covalently closed circular DNA (cccDNA) generated deletions longer than 100 base pairs in 10%–15% of the edited cells. The length of deletion ranged from several hundred base pairs to over 2000 base pairs.[ 6 ] Because the integration of HBV DNA into host genome is common in chronic HBV infection,[ 12 ] editing of the integrated HBV DNA by CRISPR/Cas9 may lead to chromosomal instability of host cells and limit its clinical application.
Recently, CRISPR base editors have been engineered to convert one DNA base to another without DSB. Base editors can be divided into two categories: the cytidine base editor (CBE) and the adenine base editor.[ 13 , 14 , 15 ] CBE is a fusion protein of a cytidine deaminase and a partially inactive nickase Cas9 protein. CBE is directed to the target DNA through base pairing between the sgRNA and DNA. Deamination of the target cytidine by cytidine deaminase results in uridine and guanosine in the nonedited strand is then converted to adenosine by cellular mismatch repair. Following DNA replication or repair, the original C·G base pair is permanently substituted by the T·A base pair.[ 15 ] CBE can also silence genes through induction of premature stop codons.[ 16 , 17 , 18 ] Targeting coding strand of genes containing CGA, CAG, CAA, and TGG codons by base editor create TGA, TAG, or TAA stop codons. The efficiency of base editing in silencing endogenous genes has been proven to be comparable with wild‐type Cas9.[ 16 ] CBE has been used to introduce nonsense mutations in 25% of Pcsk9 (proprotein convertase subtilisin/kexin type 9) gene allies in hepatocytes, resulted in reduced plasma PCSK9 protein levels and cholesterol levels.[ 19 ] Similarly, delivery of a CBE and sgRNA targeting Angptl3 (angiopoietin‐like protein 3) into the liver generated mature stop codons in 35% of target alleles, causing a reduction of plasma ANGPTL3, triglycerides, and cholesterol by 49%, 31%, and 19%, respectively.[ 20 ]
A major characteristic of patients with HBV is the integration of HBV DNA into the genome of hepatocytes. As an effective approach to edit genes, the feasibility and efficiency of CBE in silencing genes of integrated HBV within the genome of hepatocytes has not been investigated. In this study, we developed a CRISPR/CBE approach to silence the S gene of HBV and assessed its efficiency to reduce the level of HBsAg and possible off‐target effects.
MATERIALS AND METHODS
Vector construction and HBV‐specific sgRNA design
The sequence of a CBE with high base editing efficiency in mammalian cells, AncBE4max, was amplified from plasmid pCMV_AncBE4max_P2A_GFP (Addgene, #112100). LentiAncBE4max‐Blast expression vector was constructed by insertion of the AncBE4max sequence into plasmid LentiCas9‐Blast (Addgene, #52962) digested by both XbaI and BamHI using an In‐fusion HD Cloning Kit (Takara, Mountain View, CA).
To identify a conserved site within the S gene of different HBV genotypes, 20,974 HBV S gene sequences of HBV genotypes from A to H acquired from the HBVdb database (https://hbvdb.lyon.inserm.fr/HBVdb/) were aligned using Clustal Omega 1.2.4.[ 21 , 22 ] Multiple sequence alignment was visualized by Jalview 2.10.[ 23 ] A total of 230 S gene sequences containing ambiguous bases (other than A, C, G, T) at nucleotides 72 to 118, which was found to be a conserved sequence, were excluded from further analysis. A sequence logo of this conserved sequence was created using the ggseqlogo package.[ 24 ]
To introduce a stop codon in the HBV S gene through CBE, a sgRNA of the form 5′‐N(3 to 7)‐(CGA/CAG/CAA)‐N(14 to 10)‐NGG(PAM)‐3′ on the coding strand of the conserved sequence of S gene was selected and named as gRNA_S (5′‐TACCACAGAGTCTAGACTCG‐3′).[ 16 ] An alternative sgRNA, gRNA_S2 (5′‐TACCGCAGAGTCTAGACTCG‐3′), was also designed to expand the targeting scope. Another sgRNA based on the 5′‐N(2 to 7)‐(CCA)‐N(15 to 10)‐NGG(PAM)‐3′ on the noncoding strand was also designed and referred as to gRNA_S3 (5′‐CACCACGAGTCTAGACTCTG‐3′), which was assumed to introduce a stop codon at a different locus of the S gene.
Lentiviral expression vectors encoding gRNA_S (LentiGuide‐gRNA_S), gRNA_S3 (LentiGuide‐gRNA_S3) and a nonspecific sgRNA (LentiGuide‐NS‐gRNA), were constructed by cloning gRNA_S, gRNA_S3 or the nonspecific sgRNA (5′‐GCCTGCCCTAAACCCCGGAA‐3′) into the BsmB1 site of plasmid LentiGuide‐Puro (Addgene, #52963). Because gRNA_S and gRNA_S3 did not start with G, which is favorable for efficient U6 promoter‐driven transcription, an additional G was added to the 5′ end.[ 25 ] To construct a dual sgRNA expression vector (LentiGuide‐gRNA_S/S3), a DNA segment encoding gRNA_S3, sgRNA scaffold, human U6 promoter, and gRNA_S was synthesized by GENEWIZ (Suzhou, China) and cloned into the BsmB1 site of LentiGuide‐Puro.
Sequence analysis of S gene of different HBV genotypes
A BLAST database containing 20,744 S gene sequences mentioned previously were generated by the BLAST command‐line application (ncbi‐blast‐2.7.1+).[ 26 ] gRNA_S and gRNA_S2 with NGG PAM were subjected to the blastn search against the S gene‐sequence database. The targetable sequences of gRNA_S and gRNA_S2 were identified using a cutoff score of 46.1 Bits.
Cell culture, lentiviral vector production, and transduction
Human embryonic kidney HEK293T and hepatoma PLC/PRF/5 cell lines were purchased from ATCC and maintained in Dulbecco’s modified Eagle’s medium (Life Technologies, Grand Island, NY) supplemented with 10% fetal bovine serum (Life Technologies), 100 U/ml penicillin, and 100 μg/ml streptomycin (Life Technologies) at 37°C and 5% CO2. Lentiviral vectors expressing AncBE4max (LV‐AncBE4max), Cas9 (LV‐Cas9), nonspecific sgRNA (LV‐NS‐gRNA), gRNA_S (LV‐gRNA_S), gRNA_S3 (LV‐gRNA_S3), gRNA_S/S3 (LV‐gRNA_S/S3), and an empty lentiviral vector (LV‐empty) were produced by transient co‐transfection of HEK293T cells with the lentiviral expression vector constructs and packaging plasmids pMDLg/pRRE (Addgene, #12251), pMD2.G (Addgene, #12259), and pRSV‐Rev (Addgene, #12253) using lipofectamine 3000 (Life Technologies) according to the manufacturer’s instruction. Briefly, 7 × 106 293T cells were plated in a 10‐cm dish. Cells were grown to 90%–95% confluence and transfected with 8 μg of LentiAncBE4max‐Blast, LentiCas9‐Blast, LentiGuide‐NS‐gRNA, LentiGuide‐gRNA_S, LentiGuide‐gRNA_S3, LentiGuide‐gRNA_S/S3 or LentiGuide‐Puro, 4 μg of pMDLg/pRRE, 4 μg of pMD2.G, and 4 μg of pRSV‐Rev. Twenty‐four hours and 52 hour following transfection, cell supernatant was collected, passed through a 0.45‐μm pore filter, and stored at −80°C.
To establish PLC/PRF/5 cell line stably expressing AncBE4max (PLC/PRF/5‐CBE) or Cas9 (PLC/PRF/5‐Cas9), 4.5 × 105 PLC/PRF/5 cells were seeded in a 6‐cm dish. Cultures were grown to 70%–80% confluence and transduced with LV‐AncBE4max or LV‐Cas9 in the presence of polybrene (8 μg/ml). Forty‐eight hours following transduction, cells were selected with 10 μg/ml blasticidin (InvivoGen, San Diego, CA) for 7 days. PLC/PRF/5‐CBE cells were then transduced with LV‐empty, LV‐NS‐gRNA, LV‐gRNA_S, LV‐gRNA_S3 and LV‐gRNA_S/S3, respectively. Transduced cells were selected with 2 μg/ml puromycin (InvivoGen) for 7 days before harvest.
Direct sequencing for on‐target and off‐target editing
To assess the off‐target effects of base editing, we identified 11 potential off‐target sites of gRNA_S with up to 4‐nucleotide mismatches using a web‐based tool Cas‐Offinder (Table S1).[ 27 ] PLC/PRF/5‐CBE cells were harvested after transduction with empty lentiviral vector or lentiviral vectors encoding different sgRNAs and puromycin selection. Genomic DNA was isolated by the DNeasy blood and tissue kit (Qiagen, Hilden, Germany). The target site of gRNA_S and possible off‐target sites were amplified by polymerase chain reaction (PCR) using Q5 high‐fidelity DNA polymerase (New England Biolabs, Beverly, MA) with specific primers (Table S2). Amplicons were purified using the QIAquick PCR purification kit (Qiagen), and the PCR products were analyzed by Sanger sequencing on an ABI 3730xl DNA Analyzer (Applied Biosystems, Foster City, CA). The sequencing chromatograms were analyzed by EditR V1.0.8 software to determine the base editing efficiency.[ 28 ]
Northern blot analysis
Total RNA was extracted from PLC/PRF/5‐CBE cells transduced with LV‐empty, LV‐NS‐gRNA, or LV‐gRNA_S using TRIZol reagent (Invitrogen, Carlsbad, CA). A total of 10 μg RNA were separated by electrophoresis on a 2% agarose‐formaldehyde gel and transfered to a positively charged Nylon membrane (Roche Diagnostics, Mannheim, Germany). The membrane was probed with a DIG‐labeled HBV‐RNA probe corresponding to nucleotides 138 to 472 of the HBV genome. The glyceraldehyde‐3‐phosphate dehydrogenase transcript was used as an internal control. The DIG‐RNA probe preparation and hybridization were conducted with the DIG Northern starter kit (Roche Diagnostics) according to the manufacturer’s instruction (Table S2).
Detection of HBsAg by enzyme‐linked immunosorbent assay
To access the effect of base editing on HBsAg production, PLC/PRF/5‐CBE cells transduced with LV‐empty, LV‐NS‐gRNA, LV‐gRNA_S, gRNA_S3, or LV‐gRNA_S/S3 were seeded in a 12‐well plate (1.2 × 105 cells per well) after puromycin selection. Cells were cultured for 24 h, and the medium was changed to fresh medium. Forty‐eight hours after medium exchange, culture supernatants were collected and cells lysates were prepared as described subsequently. The concentrations of HBsAg were determined using the QuickTiter HBsAg enzyme‐linked immunosorbent assay (ELISA) kit according to the manufacture’s instruction (Cell Biolabs, San Diego, CA).
Immunofluorescence
PLC/PRF/5‐CBE cells transduced with LV‐empty, LV‐NS‐gRNA, or LV‐gRNA_S (40,000 cells per well) were seeded in four‐chamber wells (Lab‐Tek II chamber slide; Nunc, Rochester, NY). On the following day, cells were washed 3 times in phosphate buffered saline (PBS), fixed in 4% paraformaldehyde for 15 min, permeabilized in 0.1% Triton X‐100 for 10 min, and incubated with blocking solution (5% bovine serum albumin in PBS) for 30 min. The cells were then incubated with primary rabbit anti‐HBsAg (Novus biological) overnight at 4°C, washed 3 times with buffer (0.1% Tween 20 in PBS), and incubated with TRITC goat anti‐rabbit IgG (Earthox, Millbrae, CA) for 1 h at room temperature. After 3 times of washing, the slide was mounted using 4′,6‐diamidino‐2‐phenylindole Fluoromount‐GTM (Yeason, Shanghai, China). Images were captured on an Olympus FV1000 confocal microscope (Tokyo, Japan).
Reverse‐transcription PCR assay
Total RNA was extracted from the cells using QIAzol lysis reagent (Qiagen) according to the manufacturer’s instructions. The isolated RNA was transcribed reversely into its complementary DNA (cDNA) using a high‐capacity cDNA reverse‐transcription kit (Applied Biosystems). This cDNA was then amplified by PCR (Table S2). The PCR product was subject to gel electrophoresis and visualized.
Statistical analysis
Data are presented as means ± SD. One‐way analysis of variance was performed using the Prism 7 software (GraphPad Software, San Diego, CA). A two‐sided p < 0.05 was considered statistically significant.
All procedures were conducted in accordance with the appropriate ethics and/or institutional review committee(s).
RESULTS
Design of a sgRNA capable of inducing a premature stop codon in HBV S gene
HBV has three envelope proteins, the small, middle and large HBsAg, that are encoded by a single PreS/S open reading frame (ORF).[ 29 ] The PreS/S ORF contains three start codons, which divide it into preS1, preS2, and S domain (Figure S1A). The S sequence consisting of 226 amino acids is present in all HBsAg proteins. Thus, a nonsense mutation in the S gene is expected to direct the synthesis of truncated, nonfunctional HBsAg. Indeed, a naturally occurring T‐to‐A mutation at the 207th nucleotide of the S gene, which generates a premature stop codon of the 69th codon, could completely cease the HBsAg expression and reduce the secretion of virions.[ 30 ] It has been reported that the region between amino acid 119 and 128 of small HBsAg is critical for the infectivity of the virus.[ 31 ] To ensure that the resulting truncated HBsAg is devoid of functional domains, the first 120 bp from 5′ end of S gene is selected as the target region for base editing.
HBV is genetically diverse, and at least eight distinct genotypes (A to H) have been identified with over 8% genetic divergence among the viral genome.[ 32 ] To design a sgRNA that can target HBV of different genotypes, we first performed a multiple‐sequence alignment of 20,974 HBV S gene sequences obtained from the HBVdb database to identify the conserved sites. We identified a conserved sequence that is located at nucleotides 72 to 118 of S gene (Figure S1A). A sequence logo for this region was generated, which shows extraordinarily high sequence conservation (~2 bits) (Figure S1B). sgRNAs must contain CGA, CAG, and CAA codon and be located within the editing window of CBE (position 4–8 of sgRNA, counting the 5′ end as position 1).[ 13 ] Based on these criteria, we selected a sgRNA that is able to modify the 30th codon of HBV S gene from CAG (glutamine) to stop codon TAG, referred as to gRNA_S (Figure 1A).
FIGURE 1.
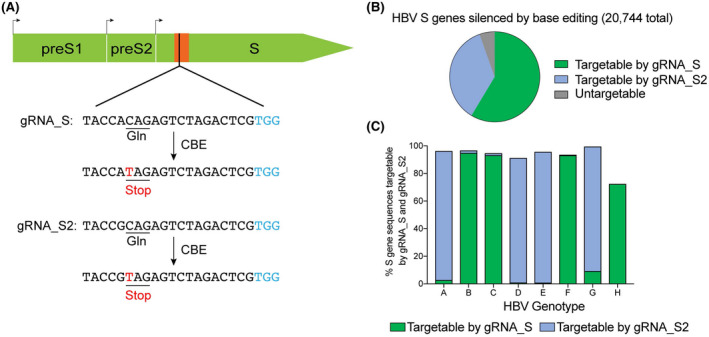
Design of single‐guide RNAs (sgRNAs) capable of introducing a premature stop codon in the hepatitis B virus (HBV) S gene. (A) The target sequences of gRNA_S and gRNA_S2 within the HBV S gene. The target sequence of sgRNA and the protospacer adjacent motif (PAM) sequence are shown in black and blue, respectively. The substituted bases are marked in red. The conserved sequence of S gene is shown in orange. (B) Fifty‐nine percent of HBV S gene sequences in the HBVdb database were silenced by base editing. The sequences targetable by gRNA_S and gRNA_S2 are shown in green and blue, respectively. A BLAST database containing S gene sequences was generated and used to identify sequences containing gRNA_S and gRNA_S2 with NGG PAM. (C) Percentage of S gene sequences of HBV genotypes A to H containing gRNA_S and gRNA_S2 with NGG PAM. (See Table S3 for a detailed conservation analysis of gRNA_S and gRNA_S2)
The conservation of gRNA_S was evaluated by local blast analysis of the S gene sequences obtained from the HBVdb database. The result showed that 59% of S gene sequences could be edited by gRNA_S (Figure 1B and Table S3). To increase the targeting scope of base editing, an alternative sgRNA with one nucleotide difference with gRNA_S, named as gRNA_S2, was also designed. Local blast analysis revealed that 36% of S gene sequences could be edited by gRNA_S2 (Figure 1B and Table S3). The subgroup analysis was also performed based on virus genotype. We found that 95%, 93%, 93%, 9%, and 72% of S gene sequences of HBV genotypes B, C, F, G, and H are targetable by gRNA_S, whereas 94%, 90%, 95%, and 90% of S gene sequences of HBV genotypes A, D, E, and G are targetable by gRNA_S2 (Figure 1C and Table S3). Thus, gRNA_S is highly conservative across different HBV genotypes.
Establishment of HBV‐positive human hepatoma cell line that stably expresses a CBE
Lentivirus is a highly efficient delivery approach to introduce a gene product into in vitro systems or animal models. To examine the efficiency of CRISPR‐mediated base editing in silencing of the HBV S gene, we first constructed lentiviral vectors expressing a CBE AncBE4max (LV‐AncBE4max), which has high base‐editing efficiency in mammalian cells.[ 33 ] AncBE4max was inserted into a lentiviral expression vector, referred as to LentiAncBE4max‐Blast, and lentiviral vectors were packaged in HEK293T cells (Figure 2A). PLC/PRF/5 is a human hepatoma cell line that carries the HBV genome and is able to produce HBsAg. To establish a stable cell line that expresses AncBE4max and Cas9, PLC/PRF/5 cells were transduced with LV‐AncBE4max and lentiviral vectors expressing Cas9 (LV‐Cas9). After selection with blasticidin, we established two stable PLC/PRF/5 cell lines that stably express CBE (PLC/PRF/5‐CBE) or Cas9 (PLC/PRF/5‐Cas9), respectively. High levels of AncBE4max was observed in PLC/PRF/5‐CBE cells (Figure 2B,C). In summary, we established a human hepatoma cell line that stably expresses AncBE4max.
FIGURE 2.
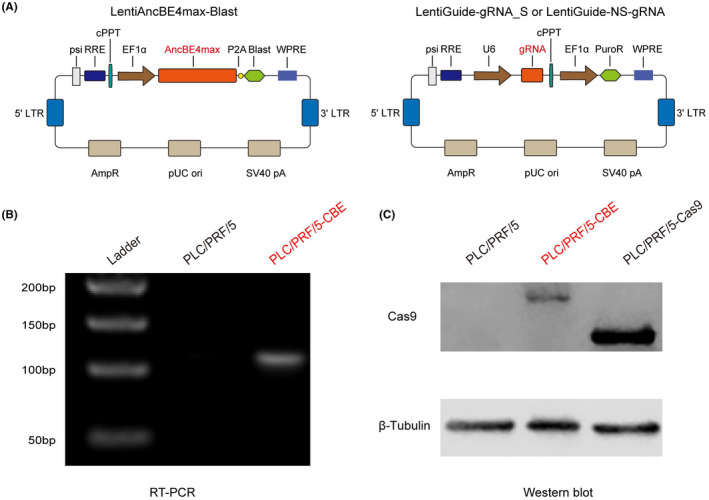
Establishment of a hepatic cell line that stably expresses a cytidine base editor (CBE). (A) Schematic illustration of three lentiviral expression vectors containing AncBE4max (left, LentiAncBE4max‐Blast), gRNA_S (LentiGuide‐gRNA_S), and nonspecific sgRNA (LentiGuide‐NS‐gRNA). The sequences of AncBE4max and sgRNAs are marked in red. (B) High level of AncBE4max messenger RNA (mRNA) was detected in PLC/PRF/5‐CBE cells by reverse‐transcription polymerase chain reaction (RT‐PCR). Wild‐type PLC/PRF/5 cells served as a control. (C) High level of AncBE4max protein in PLC/PRF/5‐CBE cells as revealed by western blot. PLC/PRF/5–CRISPR‐associated protein 9 (Cas9) was used as a positive control. The molecular weight of AncBE4max protein in PLC/PRF/5‐CBE cells was larger than that of Cas9 protein in PLC/PRF/5‐Cas9 cells. PLC/PRF/5 cells were transduced with lentivectors expressing a CBE (LV‐AncBE4max) or Cas9 (LV‐Cas9) and selected with blasticidin for 7 days. Total RNA and total protein were extracted from the cell lines and then subjected to reverse‐transcription PCR and western blot analysis
CRISPR‐mediated base editor efficiently disrupts HBV S gene in vitro
The PLC/PRF/5‐CBE cell line was then used to test the efficiency of base editing on silencing HBV S gene. Specifically, lentivirus was used to introduce nonspecific sgRNA or gRNA_S into PLC/PRF/5‐CBE cells, and puromycin was used to select the PLC/PRF/5‐CBE cells with nonspecific sgRNA or gRNA_S. Genomic DNA was isolated from nonspecific sgRNA or gRNA_S positive cells. Genomic DNA containing the binding site of gRNA_S was amplified for sequencing analysis. In cells transduced with the empty lentiviral vector or lentiviral vector expressing nonspecific sgRNA, no change in the 30th codon of the HBV S gene was observed in PLC/PRF/5‐CBE cells without the treatment of gRNA_S (Figure 3A). In contrast, gRNA_S treatment facilitated the conversion of CAG to a stop codon TAG in PLC/PRF/5‐CBE cells (Figure 3A). The editing efficiency was then quantified by EditR, an innovative program for analysis of Sanger sequencing data.[ 28 ] As expected, 71% of cells treated with gRNA_S developed a stop codon in the target site within the HBV genome (Figure 3B,C), indicating that CRISPR‐mediated CBE is an efficient approach to silence HBV that integrates into the genome of hepatocytes.
FIGURE 3.
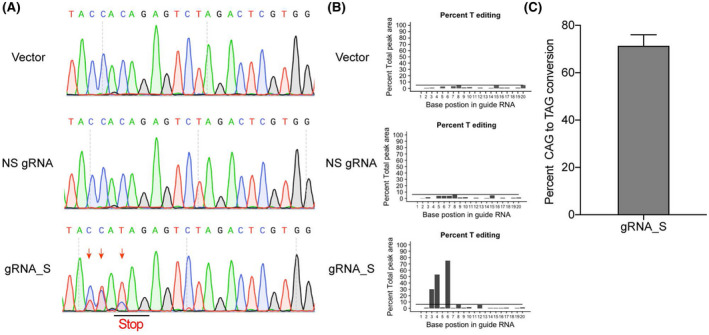
Silencing of the HBV S gene by CBE‐mediated base editing. (A) CAG to TAG editing was obtained in PLC/PRF/5‐CBE cells transduced with LV‐gRNA_S. Representative sequencing chromatogram of the amplicon obtained from PLC/PRF/5‐CBE cells transduced with either an empty lentiviral vector (LV‐empty) or lentiviral vectors expressing nonspecific sgRNA (LV‐NS‐gRNA) or gRNA_S (LV‐gRNA_S). Red arrows indicate the edited nucleotides. (B) The output plots of Sanger sequencing traces by EditR. Percentage of T editing at each position was depicted. C‐to‐T editing was only observed in the gRNA_S group. (C) The percentage of TAG in the 30th codon of HBV S gene was shown for treatment with LV‐gRNA_S
Base editing of S gene dramatically inhibits HBsAg production
Serum HBsAg levels reflect the transcriptional activity of intrahepatic HBV cccDNA, an indicator of viral protein synthesis. The ultimate aim for the treatment of HBV infection is the loss of HBsAg. To investigate the impact of base editing on HBV RNA, HBV transcripts in PLC/PRF/5‐CBE cells treated with nonspecific RNA and gRNA_S were analyzed by northern blot. As shown in Figure 4A, gRNA_S treatment significantly reduced levels of the 2.4‐kb and 2.1‐kb RNAs. Moreover, the 2.1‐kb RNA was suppressed more efficiently. The results demonstrated that the premature stop codon generated in the S gene would lead to massive degradation of HBs mRNA.
FIGURE 4.
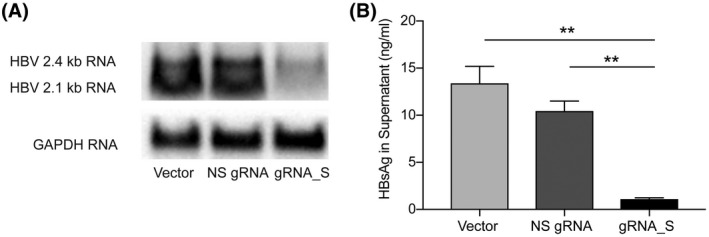
CBE‐mediated base editing led to reduced HBs mRNA and HBsAg secretion. (A) Levels of 2.4‐kb and 2.1‐kb HBV RNA in PLC/PRF/5‐CBE cells transduced with LV‐empty, LV‐NS‐gRNA or LV‐gRNA_S, as revealed by northern blot. (B) Transduction of LV‐gRNA_S significantly reduced the HBV surface antigen (HBsAg) level in the cell supernatant. Values and error bars represent the mean and SD of three independent biological replicates performed on different days (**p < 0.01; two‐way analysis of variance [ANOVA] test). Abbreviation: GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase
To determine the effect of CBE‐mediated S gene silencing on HBsAg production, PLC/PRF/5‐CBE cells treated with gRNA_S were cultured for 48 h, and the HBsAg concentration in the medium supernatants was measured by ELISA. Creation of stop codon led to a 92% reduction of HBsAg secretion in PLC/PRF/5‐CBE cells treated with gRNA_S (Figure 4B). Next, we investigated whether base editing of S gene has a similar effect on intracellular HBsAg. PLC/PRF/5‐CBE cells treated with empty vector, nonspecific sgRNA and gRNA_S were cultured for 48 h. Western blot analysis revealed a significant decrease of intracellular HBsAg (Figure 5A). The intracellular HBsAg was reduced by 84% after transduction of lentivectors expressing gRNA_S, as quantitated by ELISA (Figure 5B). HBsAg expression in PLC/PRF/5‐CBE cells treated with nonspecific sgRNA and gRNA_S were also analyzed by immunofluorescence. The result showed that intracellular HBsAg was remarkably reduced by gRNA_S treatment (Figure 5C). To evaluate whether other viral proteins are affected by gRNA_S, we analyzed levels of hepatitis B x protein (HBx) level in PLC/PRF/5‐CBE cells treated with gRNA_S and control vectors. Western blot revealed that levels of HBx protein was not affected by treatment of gRNA_S (Figure 5D). In summary, CBE not only generated the stop codon in the HBV genome that harbored within the genome of hepatocytes, but also led to a favorable phenotype of reduced HBsAg.
FIGURE 5.
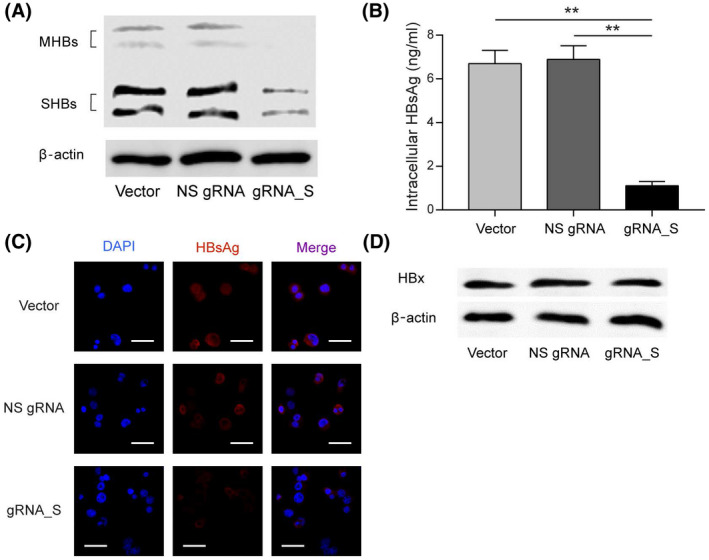
Base editing of the S gene resulted in a reduction of intracellular HBsAg. (A) Levels of intracellular HBsAg in PLC/PRF/5‐CBE cells transduced with LV‐empty, LV‐NS‐gRNA or LV‐gRNA_S, as revealed by western blot. (B) Enzyme‐linked immunosorbent assay (ELISA) measurement of HBsAg in the cell lysate of PLC/PRF/5‐CBE cells transduced with LV‐empty, LV‐NS‐gRNA, or LV‐gRNA_S. (C) Immunostaining of HBsAg reveal a significant reduction in intracellular HBsAg in PLC/PRF/5‐CBE cells treated with gRNA_S. Scale bar: 50 μm. (D) Levels of HBx protein in PLC/PRF/5‐CBE cells treated with empty lentivector, NS gRNA or gRNA‐S, as revealed by western blot. Values and error bars represent the mean and SD of three independent biological replicates performed on different days (**p < 0.01; (two‐way ANOVA test). Abbreviations: DAPI, 4',6‐diamidino‐2‐phenylindole; MHBs, middle HBsAg; SHBs, small HBsAg
We next determined whether a dual sgRNA was able to increase the efficiency of silencing the S gene. For this purpose, we designed another sgRNA, gRNA_S3, which was assumed to modify the 35th codon of the HBV S gene from TGG to a stop codon. A dual lentiviral vector containing gRNA_S and gRNA_S3 was constructed (Figure S2A). gRNA_S3 treatment was able to induce a stop codon at the target loci in about 15% of cells (Figure S2B). Combined treatment of gRNA_S and gRNA_S3 was able to drive a C‐to‐T and a G‐to‐A conversion in the target sites of gRNA_S and gRNA_S3 simultaneously. Unexpectedly, the editing efficiency of gRNA_S/S3 was significantly reduced compared with treatment of gRNA_S or gRNA_S3 alone (Figure S2B). As ELISA revealed, gRNA_S/S3 treatment was less effective than gRNA_S treatment in suppression of HBsAg secretion (Figure S2C).
Analysis of the off‐target effect of base editing
Off‐target effects of the CRISPR/Cas9 approach are a major hurdle for its clinical application. We next examined the potential off‐target mutagenesis on the host genome by gRNA_S. Bioinformatic analysis and database mining identified 11 potential off‐target sites within the genomes of hepatocytes. To determine whether these potential off‐targets sites can be edited by gRNA_S, 300 bp DNA fragments flanking each off‐target site were amplified using genomic DNA extracted from PLC/PRF/5‐CBE cells treated with gRNA_S. Sequencing revealed that no editing of cytidine or guanosine on the predicted off‐target sites of the host genome was detected (Figure S3). Together, our CBE is a high‐fidelity approach to edit the specific loci with low off‐target effects.
DISCUSSION
HBsAg is the envelope protein of HBV and the primary marker of ongoing HBV infection. Loss of HBsAg, together with undetectable HBV DNA, is termed a functional cure of HBV infection, allowing safe cessation of antiviral treatment.[ 34 ] The small and large HBsAg are essential for virion formation, whereas the middle HBsAg is not necessary.[ 35 ] All three HBsAg proteins are encoded by one open‐reading frame and share the S gene. HBsAg can be produced either from HBV cccDNA or from integrated HBV DNA.[ 36 , 37 , 38 ] A recent study found that about 80% of the HBsAg transcripts were generated by integrated viral DNA.[ 37 ] To reduce or eliminate HBsAg production, both HBV cccDNA and the integrated HBV gene should be targeted.
Although nucleos(t)ide analogues are highly effective in suppressing HBV replication, the serum HBsAg level is only modestly reduced after prolonged antiviral treatment. Treatment with nucleos(t)ide analogues for 6–12 years resulted in a 71% reduction of serum HBsAg level. In comparison, the level of HBV cccDNA was reduced by 99.84%. RNA interference (RNAi) therapy and nucleic acid polymers have shown the ability to reduce serum HBsAg.[ 39 , 40 ] However, because the template of HBsAg transcription is not eradicated, treatment cessation might cause viral reactivation. In a phase 2 clinical trial, 10 of 24 patients who achieved HBsAg loss after triple treatment with a nucleic acid polymer, tenofovir disoproxil, and pegylated interferon alfa‐2a underwent HBsAg reappearance after treatment cessation for 1 year.[ 40 ]
As a genome editing tool, CRISPR/Cas9 system has the potential for permanent elimination of viral genome. CRISPR/Cas9 could effectively disrupt HBV replication and suppress viral gene expression.[ 5 , 6 , 7 , 8 , 9 ] Nonetheless, many studies proved that repair of DSB induced by Cas9 nuclease also generates large deletions extending over many kilobases at the targeted site.[ 10 , 41 , 42 ] As integration of HBV DNA into host genome occurs in the early phase of HBV infection,[ 12 ] CRISPR/Cas9 targeting an HBV sequence raises concerns about host chromosomal instability. In contrast to Cas9 nuclease, CRISPR base editors can convert one DNA base to another without DSB.[ 13 , 33 ] In particular, CBEs could mediate C→T (or G→A) substitution in the genome and can be used to knock out gene functions by introducing premature stop codons.
Based on the structure of HBV genome, we selected nucleotides 72 to 118 of the S gene to design our gRNA_S that can generate a premature stop codon in the HBV S gene and subsequently impair expression of all three HBV envelope proteins. CBE‐mediated silencing of HBV genes has several advantages. First, viral genes in both of the cccDNA and integrated DNA could be permanently silenced through base editing, allowing a finite term treatment; second, editing with a base editor does not generate DSB, and there is no risk of affecting host chromosomal stability with the existence of integrated HBV DNA; third, the consequence of base editing is predictable, as effects on the overlapping HBV gene can be evaluated during the design of sgRNA, which is impossible in CRISPR/Cas9 system based on DSB; and finally, multiple virus genes can be silenced simultaneously with a multiplexed base editing system.
In the present study, we designed a sgRNA targeting the beginning of the S gene, gRNA_S. gRNA_S could direct a CBE to generate a premature stop codon in the 30th codon of S gene, thus terminating expression of all envelope proteins. The conservativeness of sgRNA is critical for clinical application, as a 2‐bp mismatch might completely abolish the binding of the sgRNA.[ 43 ] gRNA_S is highly conservative among HBV genotypes B, C, F, and H. Regarding genotypes A, D, E, and G, another sgRNA was designed and a combination of two sgRNAs can be subcloned into the same vector, allowing us to target all different genotypes of HBV.
We have proven that gRNA_S efficiently created a C‐to‐T substitution, which introduced a stop codon in the target site of the HBV S gene in vitro. The resulting HBs mRNA was substantially reduced, which is probably mediated by nonsense‐mediated mRNA decay. Consequently, the amount of secreted and intracellular HBsAg was dramatically reduced in the cells edited by gRNA_S. Among the six cytidines of the gRNA_S binding site, C‐to‐T conversion only occurs in positions 3, 4, and 6, counting the 5′ end of gRNA_S as position 1 (Figure 3A,B). Noticeably, the target cytidine at position 6 had the highest editing efficacy, which is consistent with reports of other groups.[ 13 , 33 ] The narrow editing window is preferred, to minimize undesirable nontarget mutations. C‐to‐T mutation in positions 3 and 4 of gRNA_S might lead to leucine or serine substitution of proline at the 29th codon of the HBV S gene. However, because a stop codon is generated at the 30th codon, these genetic code changes fail to generate a functional HBsAg.
The editing efficiency of CBE at the target site was about 71% in our study, which was high but might be improved further. In a recent study, a phage‐assisted continuous evolution system was developed to evolve base editors with better editing efficiency and sequence compatibility.[ 44 ] Although base editors with higher deaminase activity were successfully evolved, the base editing efficacy was only improved at the sites that showed poor editing by unevolved CBEs. The plateau editing level (~60%–80%) at the well‐edited sites could not be improved, suggesting that deaminase‐extrinsic factors such as cell state and DNA repair processes limited the efficiency of base editing.[ 44 ] Intriguingly, a recent study proved that fusion of a single‐stranded DNA‐binding protein domain could increase the activity of CBE by 1.7‐fold to 15.2‐fold.[ 45 ] These engineered CBEs could be used to produce an enhanced suppression of HBsAg expression.
Gene‐silencing efficiency can also be improved by targeting two or more sites to generate more than one premature stop codon. However, in this study, a dual sgRNA system failed to generate more premature stop codons in the S gene. This observation might be caused by the proximity of the two sgRNAs targeting the S gene. Considering the safety concern, the target site of gRNA_S3 was considered as the best choice. Within the 227 codons of the S gene, five codons could be targeted by CBE to generate premature stop codons, including the 30th codon encoding glutamine, and the 35th, 156th, 172th, and 223th codons encoding arginine. Although nonsense mutations at the 5' end of the S gene such as codon 36 and codon 69 could abolish expression of large, middle, and small HBsAg completely,[ 30 ] nonsense mutations at the 3' end of the S gene may generate C‐terminal truncated HBsAg, which possesses diverse biological properties. It has been reported that HBV variants with premature stop codon at position 172, 182, and 196 of the S gene could promote apoptosis of the cell and may enhance the progression to HCC.[ 46 ] Moreover, because the S gene is overlapped by the HBV polymerase gene, stop codon at position 172 of the S gene will also lead to rtA181T nucleos(t)ide drug‐resistant mutation. Therefore, the 35th codon of the S gene was the best choice as the second target site of CBE. However, there are 15 consecutive nucleotides in gRNA_S3 that are fully complementary to gRNA_S1. As a consequence, a sense‐antisense RNA duplex may form at the overlapping region of these two sgRNAs, resulting in a reduced level of each sgRNA and compromised editing efficiency at both target sites. Considering these limitations, the single sgRNA system should be considered to generate more premature stop codons in the S gene.
Because of the overlapping nature of HBV genome, editing on the HBV S gene also changed the genetic code of HBV reverse transcriptase. The on‐target editing of the S gene by gRNA_S will lead to substitution of the 38th amino acid of HBV reverse transcriptase from alanine to valine (A38V). The effect of this mutation on the function of reverse transcriptase has not been reported. However, the HBV nucleocapsid could not be released from cells without the expression of envelope proteins.[ 35 ]
At the 11 possible off‐target sites, we did not detect any off‐target editing. Nevertheless, some off‐target mutations might be overlooked. An unbiased whole‐genome sequencing is preferred to evaluate the off‐target effect of gRNA_S. Another limitation of the study is that on‐target editing was estimated only by Sanger sequencing, whereas next‐generation sequencing can provide more accurate measurements. The third limitation is that the Cas9‐independent off‐target mutations in the genome and transcriptome were not evaluated in this study. Whole genome or transcriptome sequencing is needed to clarify this concern. Newly developed CBE variants with minimized Cas9‐independent off‐target DNA editing is probably a better alternative to the canonical CBE used in this study.[ 47 ] Finally, HBV cccDNA does not exist in PLC/PRF/5 cells. Although several studies have proven that HBV cccDNA can be targeted by Cas9 protein,[ 6 , 7 , 9 , 41 ] the editing efficacy of base editors on cccDNA remains to be elucidated.
In addition to CBE‐mediated gene silencing, RNAi using small interfering RNA is another potent strategy to suppress HBsAg.[ 39 ] One advantage of CBE‐mediated gene silencing is its ability to permanently knock out the S gene, while RNAi‐induced inhibition of HBsAg probably could not be maintained once the therapy is stopped. Another advantage is that the off‐target effect of the CRISPR system is far less effective than RNAi. A head‐to‐head comparison of RNAi and CRISPR technologies revealed that RNAi frequently produces systemic off‐target effects, whereas CRISPR technology has negligible off‐target effects.[ 48 ] Nonetheless, the expression of exogenous CBE protein is required to attain the desired editing, and the large size of the gene encoding CBE poses a challenge for the delivery of the therapeutic agent. Recently, a split base editor dual‐adeno‐associated virus (AAV) strategy has been developed to bypass the packing‐size limit of AAV vectors. A single injection of dual AAV split‐intein CBE resulted in 21% of target C•G‐to‐T•A editing in the liver.[ 49 ] This split‐intein CBE delivery system could be used to introduce premature stop codons in the HBV genome in vivo.
In conclusion, our data show that CRISPR‐mediated base editing is an efficient approach to silence the HBV S gene, suggesting its potential in curing chronic HBV infection. However, further studies are needed to evaluate the editing efficiency of gRNA_S in vivo.
CONFLICT OF INTEREST
Nothing to report.
AUTHOR CONTRIBUTIONS
Data acquisition: Zhou H and Wang X. Funding acquisition, study supervision, study concept and design, and manuscript draft: Song G, Steer C, and Niu J.
Supporting information
Supplementary Material
Zhou H, Wang X, Steer CJ, Song G, Niu J. Efficient silencing of hepatitis B virus S gene through CRISPR‐mediated base editing. Hepatol Commun. 2022;6:1652–1663. 10.1002/hep4.1933
Funding information
American Cancer Society (ISG‐16‐210‐01‐RMC)
Contributor Information
Guisheng Song, Email: gsong@umn.edu.
Junqi Niu, Email: junqiniu@jlu.edu.cn.
REFERENCES
- 1. Polaris Observatory Collaborators . Global prevalence, treatment, and prevention of hepatitis B virus infection in 2016: a modelling study. Lancet Gastroenterol Hepatol. 2018;3:383–403. [DOI] [PubMed] [Google Scholar]
- 2. Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2095–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Terrault NA, Lok ASF, McMahon BJ, Chang K‐M, Hwang JP, Jonas MM, et al. Update on prevention, diagnosis, and treatment of chronic hepatitis B: AASLD 2018 hepatitis B guidance. Hepatology. 2018;67:1560–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Komor AC, Badran AH, Liu DR. CRISPR‐based technologies for the manipulation of eukaryotic genomes. Cell. 2017;168:20–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lin S‐R, Yang H‐C, Kuo Y‐T, Liu C‐J, Yang T‐Y, Sung K‐C, et al. The CRISPR/Cas9 system facilitates clearance of the intrahepatic HBV templates in vivo . Mol Ther Nucleic Acids. 2014;3:e186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Seeger C, Sohn JA. Targeting hepatitis B virus with CRISPR/Cas9. Mol Ther Nucleic Acids. 2014;3:e216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schiwon M, Ehrke‐Schulz E, Oswald A, Bergmann T, Michler T, Protzer U, et al. One‐vector system for multiplexed CRISPR/Cas9 against hepatitis B virus cccDNA utilizing high‐capacity adenoviral vectors. Mol Ther Nucleic Acids. 2018;12:242–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jiang C, Mei M, Li B, Zhu X, Zu W, Tian Y, et al. A non‐viral CRISPR/Cas9 delivery system for therapeutically targeting HBV DNA and pcsk9 in vivo . Cell Res. 2017;27:440–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kostyushev D, Brezgin S, Kostyusheva A, Zarifyan D, Goptar I, Chulanov V, et al. Orthologous CRISPR/Cas9 systems for specific and efficient degradation of covalently closed circular DNA of hepatitis B virus. Cell Mol Life Sci. 2019;76:1779–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kosicki M, Tomberg K, Bradley A. Repair of double‐strand breaks induced by CRISPR‐Cas9 leads to large deletions and complex rearrangements. Nat Biotechnol. 2018;36:765–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shin HY, Wang C, Lee HK, Yoo KH, Zeng X, Kuhns T, et al. CRISPR/Cas9 targeting events cause complex deletions and insertions at 17 sites in the mouse genome. Nat Commun. 2017;8:15464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mason WS, Gill US, Litwin S, Zhou Y, Peri S, Pop O, et al. HBV DNA integration and clonal hepatocyte expansion in chronic hepatitis B patients considered immune tolerant. Gastroenterology. 2016;151:986–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double‐stranded DNA cleavage. Nature. 2016;533:420–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nishida K, Arazoe T, Yachie N, Banno S, Kakimoto M, Tabata M, et al. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science. 2016;353:aaf8729. [DOI] [PubMed] [Google Scholar]
- 15. Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, et al. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature. 2017;551:464–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kuscu C, Parlak M, Tufan T, Yang J, Szlachta K, Wei X, et al. CRISPR‐STOP: gene silencing through base‐editing‐induced nonsense mutations. Nat Methods. 2017;14:710–2. [DOI] [PubMed] [Google Scholar]
- 17. Billon P, Bryant EE, Joseph SA, Nambiar TS, Hayward SB, Rothstein R, et al. CRISPR‐mediated base editing enables efficient disruption of eukaryotic genes through induction of STOP codons. Mol Cell. 2017;67:1068–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim K, Ryu S‐M, Kim S‐T, Baek G, Kim D, Lim K, et al. Highly efficient RNA‐guided base editing in mouse embryos. Nat Biotechnol. 2017;35:435–7. [DOI] [PubMed] [Google Scholar]
- 19. Chadwick AC, Wang X, Musunuru K. In vivo base editing of PCSK9 (proprotein convertase subtilisin/kexin type 9) as a therapeutic alternative to genome editing. Arterioscler Thromb Vasc Biol. 2017;37:1741–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chadwick AC, Evitt NH, Lv W, Musunuru K. Reduced blood lipid levels with in vivo CRISPR‐Cas9 base editing of ANGPTL3. Circulation. 2018;137:975–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, et al. Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hayer J, Jadeau F, Deleage G, Kay A, Zoulim F, Combet C. HBVdb: a knowledge database for Hepatitis B Virus. Nucleic Acids Res. 2013;41:D566–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wagih O. ggseqlogo: a versatile R package for drawing sequence logos. Bioinformatics. 2017;33:3645–7. [DOI] [PubMed] [Google Scholar]
- 25. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR‐Cas9 system. Nat Protoc. 2013;8:2281–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bae S, Park J, Kim JS. Cas‐OFFinder: a fast and versatile algorithm that searches for potential off‐target sites of Cas9 RNA‐guided endonucleases. Bioinformatics. 2014;30:1473–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kluesner MG, Nedveck DA, Lahr WS, Garbe JR, Abrahante JE, Webber BR, et al. EditR: a method to quantify base editing from Sanger sequencing. CRISPR J. 2018;1:239–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Selzer L, Zlotnick A. Assembly and release of hepatitis B virus. Cold Spring Harb Perspect Med. 2015;5:a021394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xiang K‐H, Michailidis E, Ding H, Peng Y‐Q, Su M‐Z, Li Y, et al. Effects of amino acid substitutions in hepatitis B virus surface protein on virion secretion, antigenicity, HBsAg and Viral DNA. J Hepatol. 2017;66:288–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jaoude GA, Sureau C. Role of the antigenic loop of the hepatitis B virus envelope proteins in infectivity of hepatitis delta virus. J Virol. 2005;79:10460–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lin CL, Kao JH. Hepatitis B virus genotypes and variants. Cold Spring Harb Perspect Med. 2015;5:a021436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Koblan LW, Doman JL, Wilson C, Levy JM, Tay T, Newby GA, et al. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat Biotechnol. 2018;36:843–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Durantel D, Zoulim F. New antiviral targets for innovative treatment concepts for hepatitis B virus and hepatitis delta virus. J Hepatol. 2016;64:S117–31. [DOI] [PubMed] [Google Scholar]
- 35. Bruss V, Ganem D. The role of envelope proteins in hepatitis B virus assembly. Proc Natl Acad Sci U S A. 1991;88:1059–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wooddell CI, Yuen MF, Chan HL, Gish RG, Locarnini SA, Chavez D, et al. RNAi‐based treatment of chronically infected patients and chimpanzees reveals that integrated hepatitis B virus DNA is a source of HBsAg. Sci Transl Med. 2017;9:eaan0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Podlaha O, Wu G, Downie B, Ramamurthy R, Gaggar A, Subramanian M, et al. Genomic modeling of hepatitis B virus integration frequency in the human genome. PLoS One. 2019;14:e0220376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Freitas N, Lukash T, Gunewardena S, Chappell B, Slagle BL, Gudima SO. Relative abundance of integrant‐derived viral RNAs in infected tissues harvested from chronic hepatitis B virus carriers. J Virol. 2018;92:e02221–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gane EJ, Locarnini S, Lim TH, Strasser SI, Sievert W, Cheng W, et al. Dose response with the RNA interference (RNAi) therapy JNJ‐3989 combined with nucleos(t)ide analogue (NA) treatment in expanded cohorts of patients (PTS) with chronic hepatitis B (CHB). Hepatology. 2019;70(Suppl 1):434A–5A.30724374 [Google Scholar]
- 40. Bazinet M, Pântea V, Placinta G, Moscalu I, Cebotarescu V, Cojuhari L, et al. Safety and efficacy of 48 weeks REP 2139 or REP 2165, tenofovir disoproxil, and pegylated interferon alfa‐2a in patients with chronic HBV infection naïve to nucleos(t)ide therapy. Gastroenterology. 2020;158:2180–94. [DOI] [PubMed] [Google Scholar]
- 41. Seeger C, Sohn JA. Complete spectrum of CRISPR/Cas9‐induced mutations on HBV cccDNA. Mol Ther. 2016;24:1258–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Adikusuma F, Piltz S, Corbett MA, Turvey M, McColl SR, Helbig KJ, et al. Large deletions induced by Cas9 cleavage. Nature. 2018;560:e8–9. [DOI] [PubMed] [Google Scholar]
- 43. Jiang F, Doudna JA. CRISPR‐Cas9 structures and mechanisms. Annu Rev Biophys. 2017;46:505–29. [DOI] [PubMed] [Google Scholar]
- 44. Thuronyi BW, Koblan LW, Levy JM, Yeh W‐H, Zheng C, Newby GA, et al. Continuous evolution of base editors with expanded target compatibility and improved activity. Nat Biotechnol. 2019;37:1070–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang X, Chen L, Zhu B, Wang L, Chen C, Hong M, et al. Increasing the efficiency and targeting range of cytidine base editors through fusion of a single‐stranded DNA‐binding protein domain. Nat Cell Biol. 2020;22:740–50. [DOI] [PubMed] [Google Scholar]
- 46. Colledge D, Soppe S, Yuen L, Selleck L, Walsh R, Locarnini S, et al. Stop codons in the hepatitis B surface proteins are enriched during antiviral therapy and are associated with host cell apoptosis. Virology. 2017;501:70–8. [DOI] [PubMed] [Google Scholar]
- 47. Doman JL, Raguram A, Newby GA, Liu DR. Evaluation and minimization of Cas9‐independent off‐target DNA editing by cytosine base editors. Nat Biotechnol. 2020;38:620–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Smith I, Greenside PG, Natoli T, Lahr DL, Wadden D, Tirosh I, et al. Evaluation of RNAi and CRISPR technologies by large‐scale gene expression profiling in the Connectivity Map. PLoS Biol. 2017;15:e2003213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Levy JM, Yeh W‐H, Pendse N, Davis JR, Hennessey E, Butcher R, et al. Cytosine and adenine base editing of the brain, liver, retina, heart and skeletal muscle of mice via adeno‐associated viruses. Nat Biomed Eng. 2020;4:97–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material